
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 23 September 2022
Sec. Microorganisms in Vertebrate Digestive Systems
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1006285
This article is part of the Research Topic Animal Social Behaviour and Gut Microbiome View all 19 articles
 Xugang Yang1,2†
Xugang Yang1,2† Xueni Fan1†Hui Jiang3Qiang Zhang3Basangwangdui3Qunying Zhang2Siqi Dang2
Xueni Fan1†Hui Jiang3Qiang Zhang3Basangwangdui3Qunying Zhang2Siqi Dang2 Ruijun Long2*
Ruijun Long2* Xiaodan Huang1,3*
Xiaodan Huang1,3*Yak is the only ruminant on the Qinghai-Tibetan Plateau that grazes year-round. Although previous research has shown that yak rumen microbiota fluctuates in robust patterns with seasonal foraging, it remains unclear whether these dynamic shifts are driven by changes in environment or nutrient availability. The study examines the response of yak rumen microbiota (bacteria, fungi, and archaea) to simulated seasonal diets, excluding the contribution of environmental factors. A total of 18 adult male yaks were randomly divided into three groups, including a nutrition stress group (NSG, simulating winter pasture), a grazing simulation group (GSG, simulating warm season pasture), and a supplementation group (SG, simulating winter pasture supplemented with feed concentrates). Volatile fatty acids (VFAs) profiling showed that ruminal acetate, propionate and total VFA contents were significantly higher (p < 0.05) in GSG rumen. Metagenomic analysis showed that Bacteroidetes (53.9%) and Firmicutes (37.1%) were the dominant bacterial phyla in yak rumen across dietary treatments. In GSG samples, Actinobacteriota, Succinivibrionaceae_UCG-002, and Ruminococcus albus were the most abundant, while Bacteroides was significantly more abundant in NSG samples (p < 0.05) than that in GSG. The known fiber-degrading fungus, Neocallimastix, was significantly more abundant in NSG and SG samples, while Cyllamyces were more prevalent in NSG rumen than in the SG rumen. These findings imply that a diverse consortium of microbes may cooperate in response to fluctuating nutrient availability, with depletion of known rumen taxa under nutrient deficiency. Archaeal community composition showed less variation between treatments than bacterial and fungal communities. Additionally, Orpinomyces was significantly positively correlated with acetate levels, both of which are prevalent in GSG compared with other groups. Correlation analysis between microbial taxa and VFA production or between specific rumen microbes further illustrated a collective response to nutrient availability by gut microbiota and rumen VFA metabolism. PICRUSt and FUNGuild functional prediction analysis indicated fluctuation response of the function of microbial communities among groups. These results provide a framework for understanding how microbiota participate in seasonal adaptations to forage availability in high-altitude ruminants, and form a basis for future development of probiotic supplements to enhance nutrient utilization in livestock.
The Qinghai-Tibetan Plateau is the highest and largest plateau in the world, and is utilized for ruminant grazing year-round (Long et al., 1999), besides playing vital ecological roles as a watershed and ruminant habitat (Yu et al., 2012). Yak (Bos grunniens), referred to in Chinese as the “boat of the plateau,” is the only large mammal endemic to the Qinghai-Tibet Plateau, and yak husbandry is tightly interwoven in the lifestyle of the Tibetan people, providing indispensable goods, such as dairy, meat, textiles, and fuel (Long et al., 1999). As a result of their prolonged development and adaptation to survival on the plateau, yaks can withstand extreme cold, high ultraviolet radiation, hypoxia, and seasonally low nutrient availability (Qiu et al., 2012; Zhang et al., 2016). This adaptation to harsh environments raises numerous questions about the function and dynamics of their resident rumen microbiota, and especially the role of rumen microbiota in nutrient utilization efficiency under wide variation of pasture conditions.
Rumen microbiota perform integral functions in nutrient utilization, metabolism, immune function, animal health, and even host developmental processes (Heijtza et al., 2011; Sonnenburg and Backhed, 2016; Islam et al., 2019). Dietary and feeding regimens can also reportedly affect the composition of bacterial and archaeal communities in yak rumen (Xue et al., 2017; Zhou et al., 2017; Liu et al., 2019), and their microbial symbionts have been shown to ferment plant proteins and polysaccharides (Deusch et al., 2017), and provide the host with essential nutrients, volatile fatty acids (VFAs), and microbial proteins obtained from relatively recalcitrant plant fibers (Ishaq and Wright, 2012). Previous studies in yak have found that rumen microbiota composition is not only related to feed efficiency (Henderson et al., 2015), but also subject to co-evolution with the host genome as part of the adaptive response to extreme natural environments (Zhang et al., 2016). Guo et al. (2021) and Huang et al. (2022) demonstrated that seasonal constraints on forage availability led to restructuring of the yak rumen microbiota, enriching for fiber-degrading microbiota in the winter season when low-nutrient, high-fiber plant residue comprises the majority of the food supply. However, the contribution of rumen fungi, which are well-established microbial fiber degraders, has been largely overlooked in previous investigations of seasonal nutrient variation. Moreover, environmental factors can also impact rumen microbiota composition and diversity, and therefore, experiments simulating different nutrient availability conditions can promote a more robust comprehension of yak rumen community adaptation to seasonal changes in forage conditions, and identify functionally relevant microorganisms that are enriched under different conditions.
In the present study, we investigated the influence of seasonal changes in nutrient availability on yak rumen microbiota (including bacteria, fungi, and archaea) by simulating grazing conditions in winter pasture, warm season pasture, and winter pasture with supplementation. To this end, high throughput metagenomic rRNA gene sequencing was used to characterize rumen community composition and diversity under different grazing conditions. Then, to test the hypothesis that specific microbes are enriched under different grazing conditions, leading to differences in rumen fermentation, we characterized the volatile fatty acid (VFA) profiles of all rumen samples and conducted correlation analysis between microbial taxa (ASVs) and different VFAs. This study extends our understanding of the role of rumen microbes in metabolic adaptation to extreme seasonal changes in forage availability.
Procedures of the current study were approved by the Animal Welfare and Ethics Committee of Lanzhou University (Approval Reference Number: EAF2021033). The study was conducted in August 2021, at the Yak Breeding Cooperative in Nimu County, Lhasa City (29°36′N, 90°6′E 4,230 m a.s.l.).The experiment selected 18 male yaks (350 ± 26 kg live weight, aged 4–5 years) and randomly divided into three different diet groups (shown in Table 1). The experiment was carried out over a period of 35 days, of which the first 14 days were for adaptation and the next 21 days for experiment and sampling. The test was divided into three groups: Grazing simulation group (GSG), Nutrition stress group (NSG) and Supplementation group (SG) according to the nutrient intake and average daily gain of yaks grazing in different seasons (Xue et al., 2005, 2007), the Feeding Standards of Beef Cattle (NY-T815-2004) and the nutrient changes of forage grass in different seasons on the Qinghai-Tibet Plateau (Long et al., 1999; Guo et al., 2021). In which, Nutrient stress group (NSG) was considered as fed deficiency group according to the nutrient level of local winter pasture; Grazing simulation group (GSG) simulated the nutrient intake of yak while grazing in warm-season pasture; Supplementation group (SG) simulated the nutrient intake of yak while grazing in the winter pasture with supplementation. The yaks were fed twice a day at 8:30 and 16:30 h at 2% BW on a dry matter basal diet and allowed ad libitum access to water. The remaining feed was collected and weighed before morning feeding to calculate the actual feed intake.
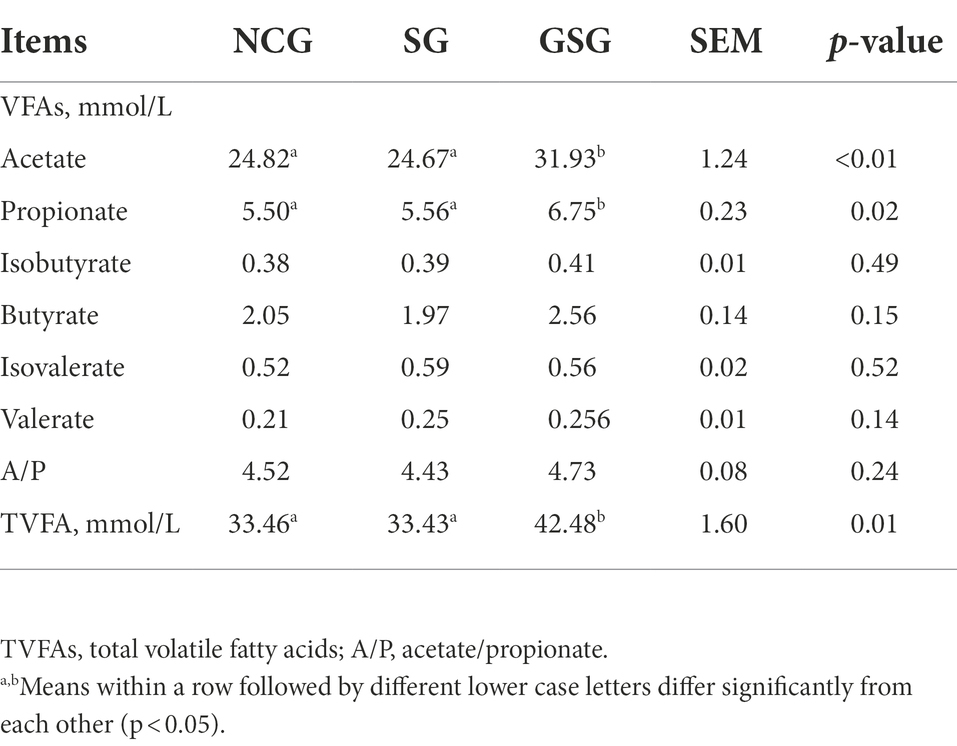
Table 1. Diet composition of three treatment groups.
On the last day (day 35) of the experiment, a stomach tube (Kelibo Co. Ltd., Wuhan, China) was used to collect rumen fluid from each yak before morning feeding. The initial 50 ml of fluid were discarded to avoid contamination by saliva. Then, 40 ml of rumen fluid was taken and stored in a sterile enzyme-free cryopreservation tube with liquid nitrogen freezer and transferred immediately to the laboratory and stored in a –80°C refrigerator. The collected samples were mainly used for the determination of volatile fatty acids (VFAs) and microbial community analysis.
Volatile fatty acids (VFAs) were determined by modified method from Erwin et al. (1961). The rumen fluid was firstly thawed at 4°C and centrifuged at 3850×g at 4°C for 15 min, metaphosphoric acid solution(0.2 ml) containing 2-ethyl butyric acid as internal standard was added to supernatant(1 ml). After mixing well and standing for 30 min at 4°C. The mixed solution was centrifuged at 3850×g at 4°C for 15 min, and the supernatant was collected and filtered (0.22 μm) for VFAs determination using a gas chromatograph (GC, SP-3420A; Beijing Beifen-Ruili Analytical Instrument Co., Ltd., Beijing, China) with an AT-FFAP type capillary column (30 m × 0.32 mm × 0.5 μm) and a flame ionization detector, with the temperature regime as: 90°C for 1 min, increased to 120°C at 10°C/min for 1 min, then increased from 120 to 150°C at 10°C/min, and maintained at 150°C for 3 min. The injection hole temperature was 250°C, and the auxiliary chamber temperature was 250°C.
The collected feed samples were dried in a forced-air oven at 65°C for 48 h. After crushed and then passed through a 1 mm sieve, feed samples were weighed and dried at 105°C to a constant weight to thus subjected to dry matter (DM) and crude protein (CP) determination by the Kjeldahl method (Thiex et al., 2012), while neutral detergent fiber (NDF) and acid detergent fiber (ADF) were measured by an automatic fiber analyzer (ANKOM 2000i, ANKOM Technology, Macedon, NY, USA) (Vansoest et al., 1991). Determination of crude ash in diet was carried out according to the method described by Thiex et al. (2012) and ether extract (EE) was measured according to the method described by Cunniff and Association of Official Analytical Chemists (1995).
Rumen fluid stored at −80°C was subjected to genomic DNA extraction using TIANamp Stool DNA Kit (Tiangen Biotech Co., Ltd., Beijing, China). DNA spectrophotometer (ND-1000; NanoDrop, Wilmington, DE, United States) was used for DNA yield and purity screening. Normalized genomic DNA was applied to perform barcode PCR with a set of primers (338\u00B0F and 806R for bacteria; ITS1-1F-F-ITS1-1F-R for Fungi and 1106F-1378R for Archaea). Phusion® High-Fidelity PCR Master Mix (New England Biolabs) was used in the PCR reactions, cycling conditions were as follows: 1. denaturation step at 98°C, 1 min; 2. 30 cycles at 98°C for 10s, 50°C for 30 s, 72°C for 30 s; and 3. a final 5 min extension at 72°C. The PCR products were thus subjected to agarose gel electrophoresis for separation and visualization. The final products were sequenced on an Illumina NovaSeq platform and 250 bp paired-end reads were generated.
Paired-end reads were assigned to each sample by barcodes identification. After cutting off the barcodes and primer sequences, Paired-end reads were merged and spliced by FLASH v 1.2.11 (Magoč and Salzberg, 2011). Quality filtering was performed using the fastp (Version 0.20.0), after getting rid of noise and chimeric reads by QIIME2 (Version QIIME2-202006) DADA2 and Vsearch (Version 2.15.0; Li et al., 2020), sequences are then clustered into ASVs (Amplicon Sequence Variants) (default: DADA2) based on the database. The database used for the taxonomic assignment comes from Silva database1 for 16S and Unite database2 for ITS (Haas et al., 2011).
Alpha and beta diversity were calculated from different indices in QIIME2 (Version QIIME2-202006). Principal Coordinate Analysis (PCoA) was used to obtain principal coordinates and visualize differences of rumen samples in complex multi-dimensional data. The results were plotted using ade4 and ggplot2 packages in R. LEfSe analysis was performed through the Huttenhower Lab Galaxy Server3. The PICRUSt software (Version 2.1.2-b) was used to predict the function of bacteria and archaea (Douglas et al., 2020). The FUNGuild software was conducted to assign ecological functions to each ASVs (Nguyen et al., 2016). The original 16S rRNA/ITS data were available at the NCBI SRA database with accession number PRJNA8631314.
The association between rumen microbiota and VFA through Spearman’s correlation analysis. One-way ANOVA by SAS version 9.2 was used to compare rumen VFAs. Non-parametric factorial Kruskal–Wallis sum-rank tests were performed to test for differences among groups at the bacterial phylum and genus levels, and Dunn’s test was performed to separate means where significance was found. Tukey-adjusted p values were used to separate means and statistical significance was accepted at p < 0.05.
In order to better understand the differences in microbial-associated metabolic processes in yak rumen under varying nutrient availability, volatile fatty acid (VFA) composition was determined in yak rumen fluid of animals treated with simulated nutrient deficiency (NSG), simulated warm season grazing (GSG), or supplemented cold season grazing (SG) (n = 6 animals per treatment; Table 2). The results suggested that diet (i.e., nutrient levels) affected the amounts of rumen acetate, propionate, and total VFAs, which were significantly higher (p < 0.05) in GSG than in NSG or SG, while the concentrations of rumen butyrate and valerate, as well as the acetate: propionate ratio showed no difference among the three experimental groups. These results suggested that metabolic processes in yak rumen indeed varied with nutrient intake, leading us to hypothesize that rumen microbiota contributed to this process.
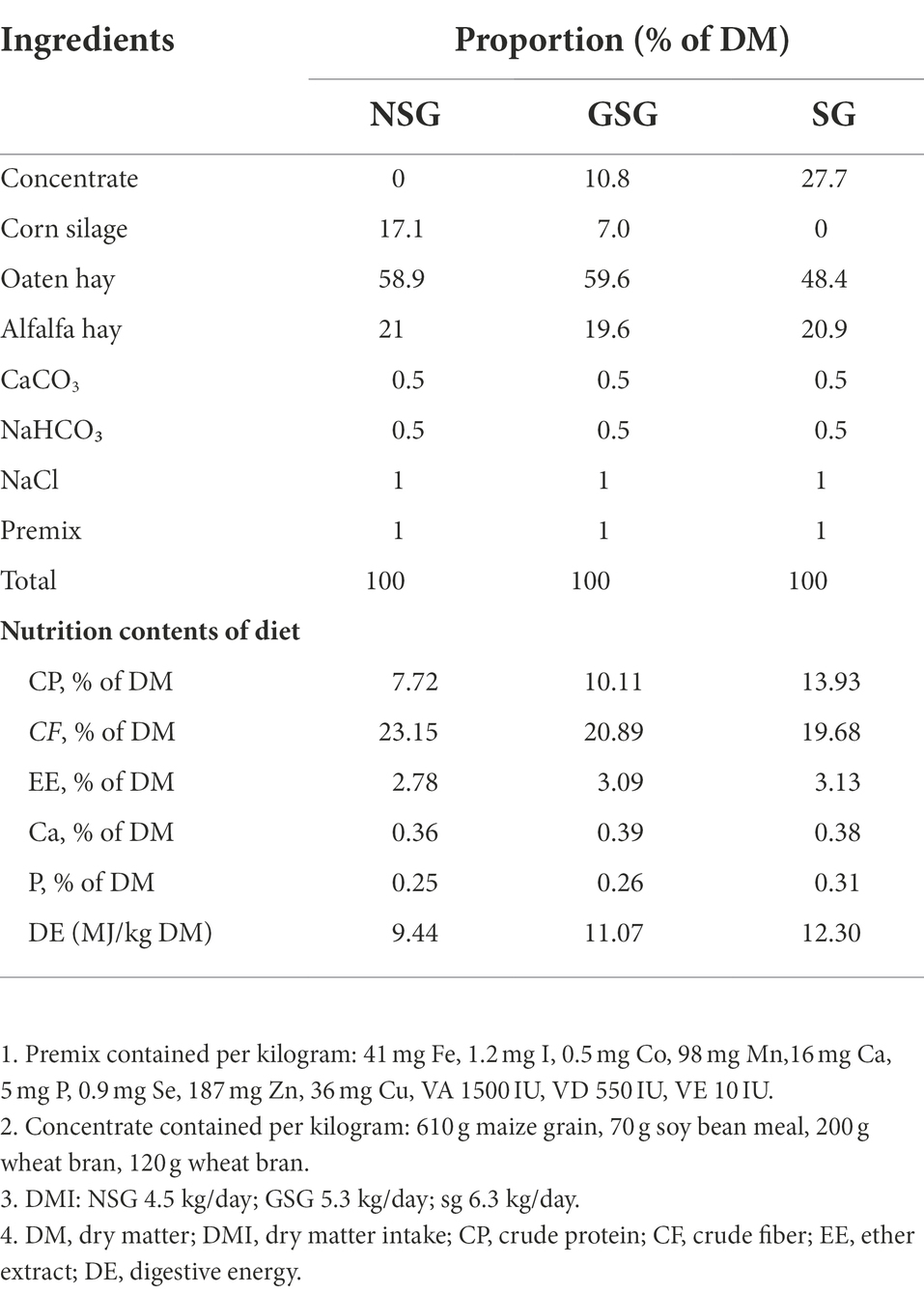
Table 2. Volatile fatty acids (VFAs) composition affected by different nutrient simulations.
To identify differences in yak rumen microbiota associated with nutrient fluctuation, metagenomic sequencing of the 16S rRNA gene was conducted in rumen samples of yaks fed under NSG, GSG and SG conditions. In total, 1,959,743 cleaned reads were obtained from 18 samples after quality control and filtering, with an average of 69,564 valid sequences per sample. Denoising with DADA 2A revealed that 3,341 ASVs were present in the three groups combined, among which 2068 ASVs were detected in all three groups, while 245 were unique to NSG, 192 were exclusively detected in SG, and 265 were only present in the GSG rumen (Supplementary Figure S1A).
Analysis of the alpha diversity of rumen bacteria indicated that species richness (Chao 1 index) was significantly higher in the GSG rumen (1611) than in the SG rumen (1360) (p < 0.05), while NSG had intermediate richness between the other treatments (Figure 1A). Shannon diversity indices were not significantly different among groups (p > 0.05) (Figure 1B). Principal coordinates analysis (PCoA) based on an unweighted UniFrac distance matrix to visualize overall structural changes in rumen bacterial communities showed obvious separation of the three groups (Figure 2A), with GSG rumen bacteria exhibiting the tightest clustering. LEfSe analysis to identify indicator taxa under different nutrient conditions revealed 10 (Bacteroides, Roseburia, etc.), 12 (Oribacterium, Pasteurellales, etc.) and 3 (Lachnospiraceae_ND3007_group, Roseburia_intestinalis, etc.) bacterial taxa significantly associated with NSG, GSG and SG yak rumen, respectively (Figure 3).
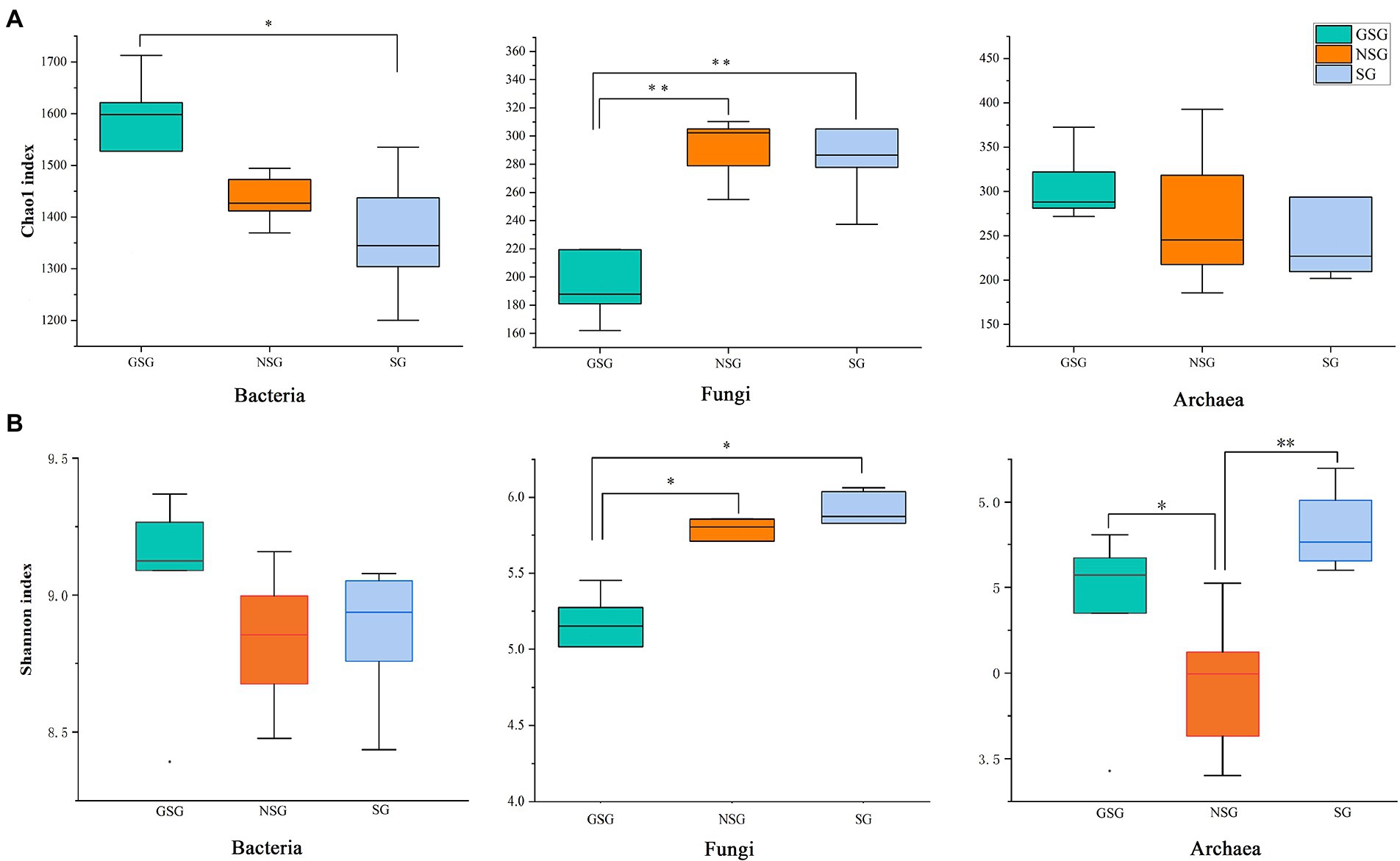
Figure 1. Differences in Yak rumen microbial diversity and richness between the GSG, NSG, and SG. Diversity was estimated by Shannon index. Richness estimated by the Chao1 value. (A) Chao 1 index. (B) Shannon index. Asterisks indicate significant difference between the Concentrate Group and the Forage Group (*p ≤ 0.05; **p ≤ 0.01).
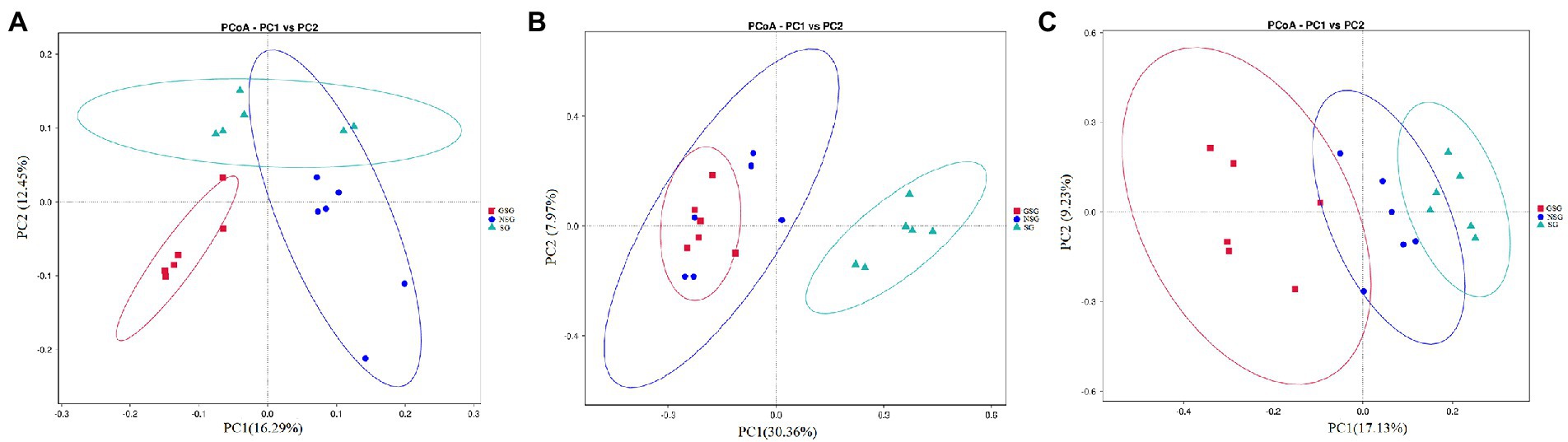
Figure 2. Principal coordinate analysis (PCoA) of rumen microbial communities. (A) Bacteria. (B) Fungi. (C) Archaea.
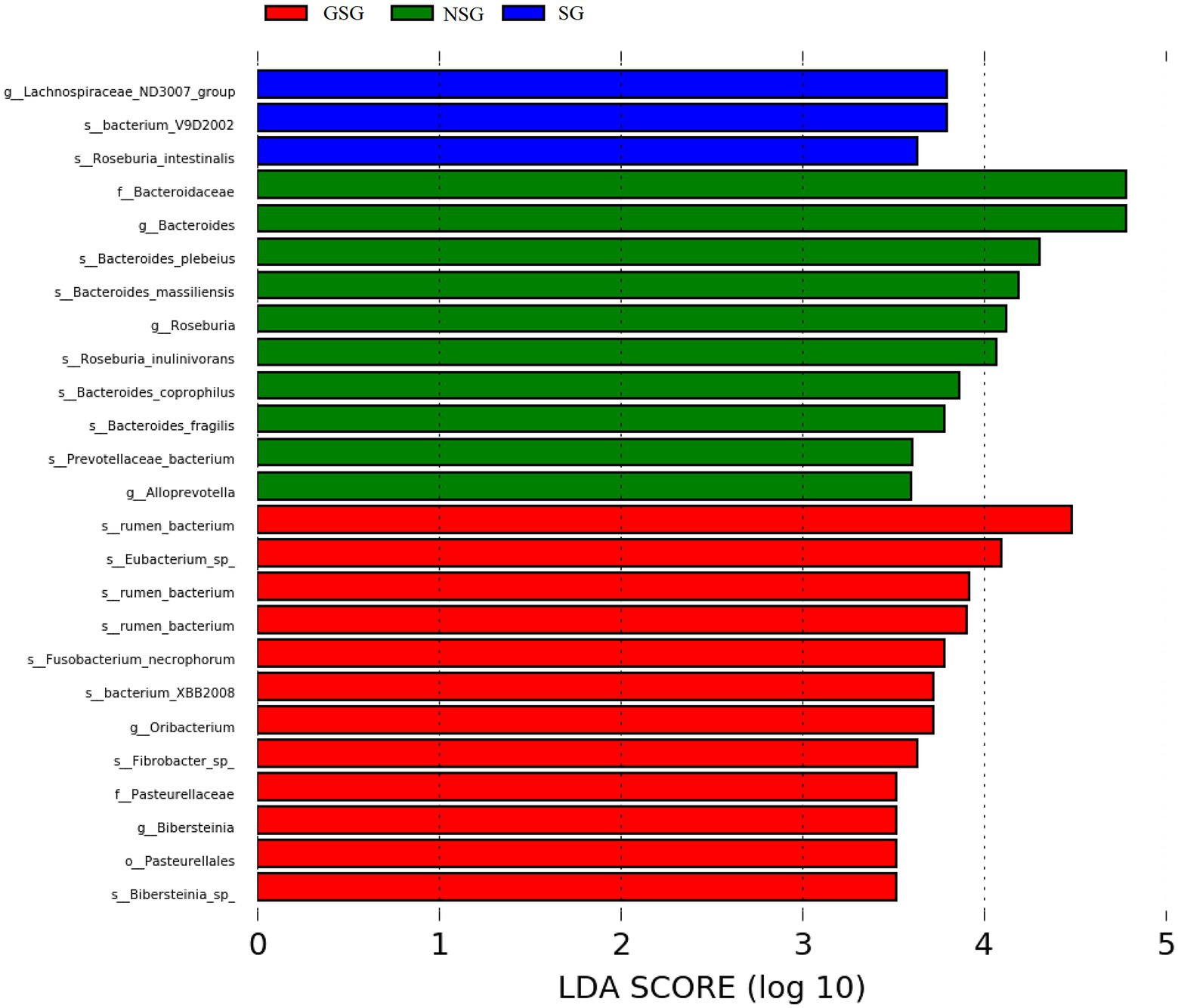
Figure 3. Linear discriminant analysis (LDA) of rumen bacteria. (LDA cutoff of +/−2.0).
A total of 21 bacterial phyla and 336 bacterial genera were identified in the combined experimental samples. Bacteroidetes (53.97%) and Firmicutes (37.16%) were the dominant phyla in all three treatment groups (Supplementary Figure S2A), with Proteobacteria and Actinobacteriota (5.40% and 1.08%, respectively) comprising the next most abundant taxa. These four phyla with highest relative abundance accounted for >90% of all bacterial ASVs. At the genus level, the predominant genera included Rikenellaceae_RC9_gut_group (24.25%), uncultured_bacterium_f_F082 (8.01%), and Prevotella_1 (5.12%) in all samples (Figure 4A). Less prevalent but relatively abundant genera included p-251-o5, Bacteroides, Papillibacter, Quinella, and Lachnospiraceae_ND3007_group, respectively accounting for 2.81%, 2.71%, 2.41%, 2.36% and 1.97% of detected ASVs.
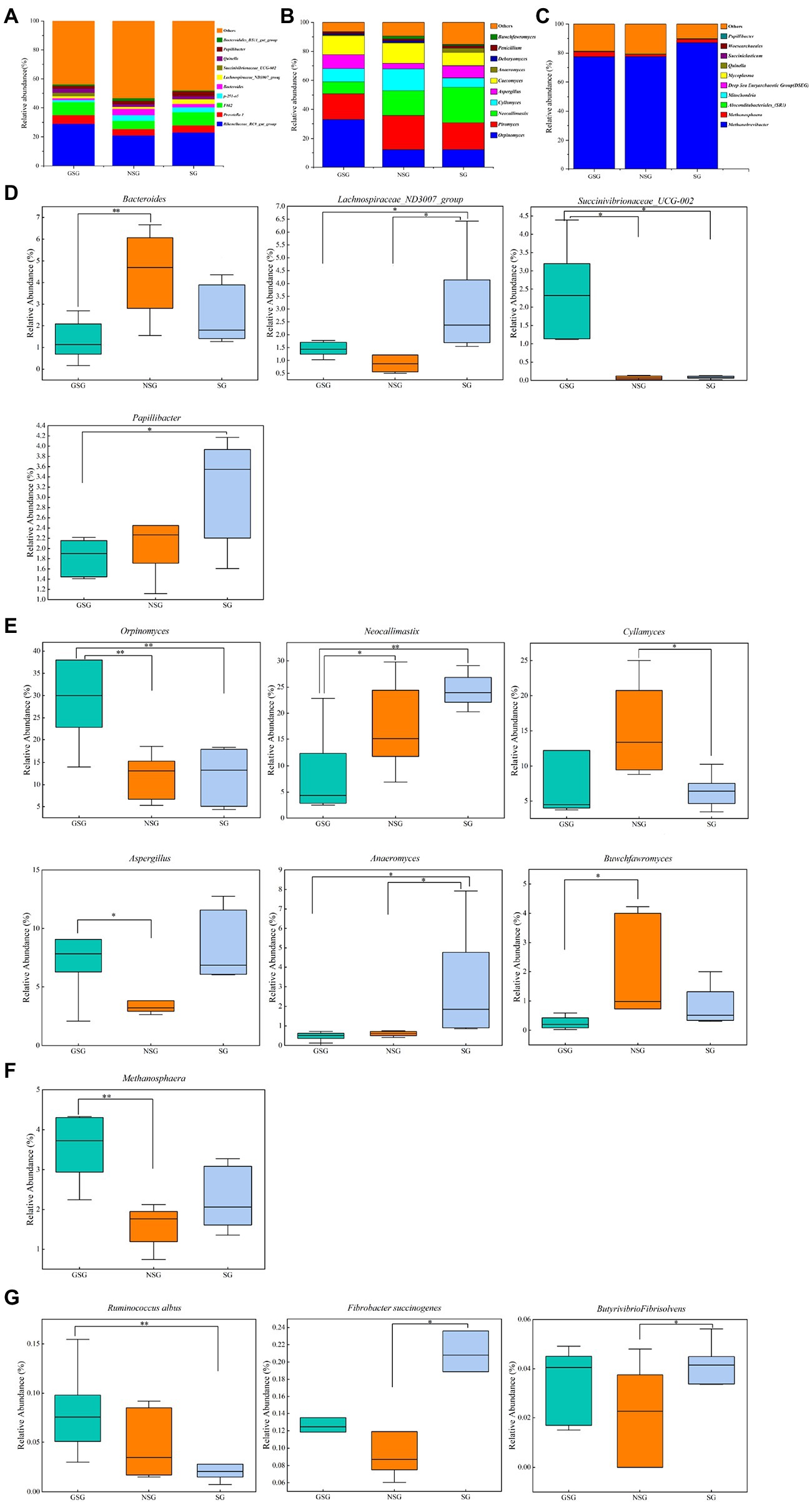
Figure 4. Classification of the rumen microbial composition at the genus level across the different nutrient simulations in different grazing patterns. (A) Bacteria. (B) Fungi. (C) Archaea. (D) Bacterial genera with significant changes under various nutrient simulations. (E) Fungal genera with significant changes under various nutrient simulations. (F) Archaeal genera with significant changes under various nutrient simulations. (G) Major fiber-degrading bacteria with significant changes under various nutrient simulations. Asterisks indicate significant difference between the three groups (*p ≤ 0.05; **p ≤ 0.01).
At the phylum level (Supplementary Figure S2D), in comparisons with the NSG yak rumen, the GSG rumen had a higher abundance of Bacteroidetes (57.2% vs. 50.4%) but lower abundance of Firmicutes (33.4% vs. 39.4%; p < 0.05). Yak rumen from GSG also had the highest abundance of Actinobacteriota among all the groups (2.1% vs. 0.5% and 0.5%; p < 0.05). At the genus level, the relative abundance of Bacteroides in NSG was higher than that in the GSG rumen (4.4% vs. 2.4%; p < 0.05), while Lachnospiraceae_ND3007_group was significantly enriched in SG (3.1% vs. 1.3% and 1.4%; p < 0.05) and Succinivibrionaceae_UCG-002 was significantly higher in GSG rumen (2.2% vs. 0.06% and 0.08%; p < 0.05) compared with other groups (Figure 4D). In addition, the differences in nutrient availability significantly affected (p < 0.05) species level ASVs such as Fibrobacter succinogenes, Butyrivibrio fibrisolvens, and Ruminococcus albus. The relative abundance of Fibrobacter succinogenes and Butyrivibrio fibrisolvens in SG was significantly higher (0.2% vs. 0.1% and 0.04% vs. 0.02%; p < 0.05) than that in NSG, while GSG rumen had the higher abundance of Ruminococcus albus than that in SG group (0.08% vs. 0.02%, p < 0.05) (Figure 4G). These cumulative results suggested that the nutrient deficiency can inhibit proliferation of known rumen taxa, while selecting for nutrient stress-tolerant, fiber-degrading bacteria and fungi.
Based on the observed differences in bacterial communities between treatments, metagenomic sequencing of the fungal ITS hypervariable region was conducted to examine changes in rumen fungi, which are well known to contribute to cellulose degradation in ruminants. In total, 92,749 cleaned reads were obtained for each sample after splicing and filtering, with a total of 1,441 ASVs detected in the three groups combined after denoising. Among these fungal ASVs, 213 were common to all three groups, while 395 were unique to NSG rumen, 383 were unique to SG, and 200 were exclusively detected in GSG (Supplementary Figure S1B).
Examination of fungal diversity (Shannon index) and species richness (Chao 1) in yak rumen indicated that both indexes were lower in GSG than in the NSG and SG rumen (p < 0.01) (Figures 1A,B). PCoA analysis showed no obvious distinction between rumen fungal communities of the NSG and SG rumen, whereas GSG was clearly separated from the other two groups (Figure 2B). Subsequent LEfSe analysis identified 7 (Buwchfawromyces, Kazachstania, etc.), 17 (Eurotiales, Aspergillaceae, etc.), and 26 (Melanocarpus, Neocallimastix, etc.) fungal taxonomic groups that were significant indicator taxa in the NSG, GSG, and SG rumen, respectively (Supplementary Figure S3A).
A total of 7 fungal phyla and 122 fungal genera were identified in the three groups. In particular, Neocallimastigomycota (81.29%) and Ascomycota (12.91%) were the most abundant phyla in all rumen samples (Supplementary Figure S2B), followed by Basidiomycota, Mortierellomycota, and Mucoromycota (0.76, 0.10 and 0.03%, respectively). At the genus level, predominant genera included Piromyces (20.13%), Orpinomyces (19.01%), and Neocallimastix (16.56%) in all samples. Less prevalent but significantly abundant genera included Caecomyces, Cyllamyces, Aspergillus, Anaeromyces, and Penicillium, accounting for 12.00, 10.26, 7.16, 1.37, and 1.28% of fungal ASVs, respectively (Figure 4B).
Examination of phylum-level community composition showed that Mortierellomycota in SG was significantly higher than in GSG (p < 0.05 Supplementary Figure S1E), whereas, at the genus level, the relative abundance of Orpinomyces was higher in the GSG rumen than in the NSG and SG rumen (33.0% vs. 12.0% and 11.9%; p < 0.05) and Neocallimastix was lower (8.2% vs. 17.2% and 24.4%; p < 0.05). Yak rumen in the SG rumen had the highest abundance of Anaeromyces among the three groups (3.0% vs. 0.6% and 0.5%, p < 0.05), while Cyllamyces was enriched in NSG compared to SG (15.1% vs. 6.5%, p < 0.05), and Buwchfawromyces was more abundant in NSG than in the GSG rumen (p < 0.05) (Figure 4E). In summary, these results indicated that nutrient utilization is a complex process which requires a diverse consortium of microbes working together, and thus builds a plasticity to allow fungi to response to different substrates.
In addition to bacterial and fungal contribution to rumen metabolism, we hypothesized that rumen archaea may also vary under different nutrient conditions. To test this possibility, metagenomic sequencing of the archaeal was conducted in rumen of yaks from each treatment group, which resulted in 1,699,212 clean reads, after quality control and filtering, from all samples combined. On average, 83,546 valid sequences were obtained for each sample, which contained 1,997 total ASVs. There were 164 ASVs shared by all three groups, while much larger proportions were unique to each grazing simulation, including 545 ASVs exclusive to NSG rumen, 428 ASVs unique to SG rumen, and 481 ASVs only found in GSG rumen (Supplementary Figure S1C).
Comparison of rumen archaeal diversity (Shannon index) indicated that SG (4.8) had significantly higher diversity than in GSG (4.4) and NSG (3.9) (p < 0.05), although species richness (Chao 1) was not significantly different among the three groups (Figures 1A,B). PCoA analysis showed that archaeal communities clustered into distinct groups based on nutrient conditions, with little overlap (Figure 2C). LeFse analysis of indicator taxa for each nutrient condition group identified Methanobrevibacter in NSG, Methanosphaera and Methanobrevibacter ruminantium in GSG rumen, and Methanobrevibacter millerae in the SG rumen (Supplementary Figure S3B). Examination archaeal community composition identified four distinct phyla and six distinct genera that were present in all groups, including Euryarchaeota, which was the most abundant phylum in all samples, accounting for 84.19% of archaeal ASVs (Supplementary Figure S2C). Significant, but less prevalent phyla included Halobacterota (0.15%) and Aenigmarchaeota (0.02%). At the genus level, Methanobrevibacter (81.03%) and Methanosphaera (2.47%) were predominant across samples (Figure 4C). Further analysis by one-way ANOVA showed that the relative abundance of Methanosphaera in NSG was significantly lower than in GSG (p < 0.05), but did not differ from that in SG (Figure 4F). Collectively, these results found that sufficient nutrient conditions could enhance VFA production by enrich less hydrogen consumer-Methanosphaera.
A total of 69 enriched metabolic pathways were predicted based on identified bacteria and archaea amplicon sequences using the KEGG pathway database, and thus were mainly divided into three categories as metabolism, biosynthesis, and cellular processes (Supplementary Figures S4A,C). We found that the Calvin–Benson–Bassham cycle and 5-aminoimidazole ribonucleotide biosynthesis were significantly enriched in GSG when compared with that in NSG (p < 0.05). In addition, fungal function prediction using FUNGuild software showed that pathotrophs, symbiotrophs, and saprotrophs were the major components. Plant_Pathogen and Endophyte-Plant_Pathogen were significantly enriched in SG than that in NSG (p < 0.05), and Animal_Pathogen-Endophyte-Plant_Pathogen-Wood_Saprotroph was significantly more abundant in SG than that in GSG (p < 0.05). Moreover, the relative abundance of Plant_Pathogen-Soil_Saprotroph-Wood_Saprotroph was significantly higher in GSG than that in NSG (p < 0.05) (Supplementary Figure S4B).
Based on the above detection of microbial taxa significantly associated with different grazing simulation treatments, we next conducted Spearman’s correlation analysis to determine whether and which yak rumen microbes shared an association with significant VFAs (Figure 5). This analysis indicated that the relative abundance of p.251.o5 was negatively correlated with acetate and propionate concentrations (|r| > 0.5, p < 0.05); Lachnospiraceae_ND3007_group showed a positive correlation with valerate and isovalerate concentrations (|r| > 0.5, p < 0.05), and Christensenellaceae_R-7_group was negatively correlated with butyrate, valerate and isovalerate concentrations (|r| > 0.5, p < 0.05). In addition, the Ruminococcaceae_NK4A214_group bacterial ASV was significantly negatively correlated with all VFAs except isovalerate (|r| > 0.5, p < 0.05). Among fungal ASVs, the relative abundance of Orpinomyces was significantly positively correlated with acetate concentration (r = 0.43, p < 0.05), while Piromyces was negatively correlated with acetate and isobutyrate concentrations (r = −0.54 and −0.51, p < 0.05). At the genus level, Penicillium and Candida were positively correlated with valerate and acetate concentrations, respectively (|r| > 0.5, p < 0.05).
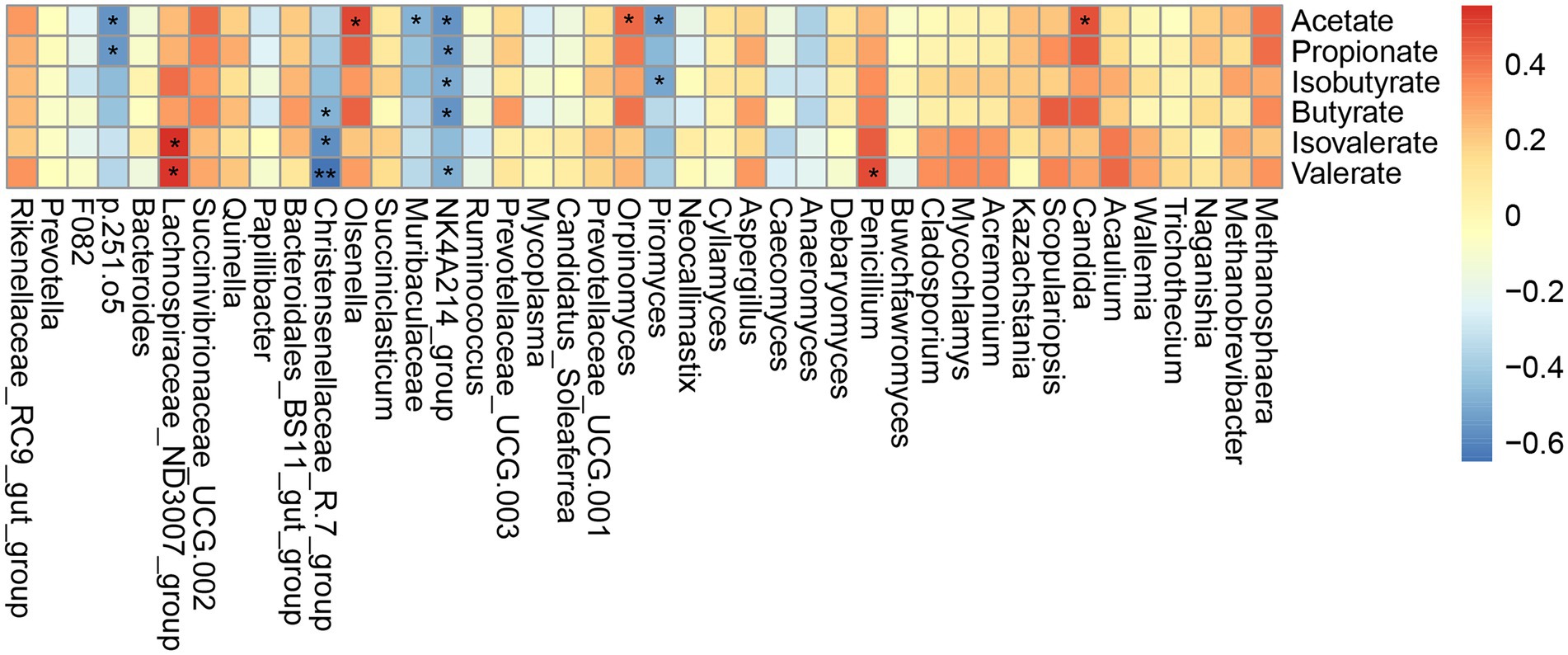
Figure 5. Correlations between the higher abundant genera of rumen microbial and VFAs. Each row in the graph represents a genus, each column represents a metabolite, and each lattice represents a Spearman’s correlation coefficient between a component and a metabolite. Red represents a positive correlation, while blue represents a negative correlation. *p < 0.05, **p < 0.01.
Analysis of mutual correlations among high-abundance rumen microbiota (bacteria, fungi and archaea) at the genus level (Figure 6) showed that Deep_Sea_Euryarchaeotic_Group (DSEG) was positively correlated with Rikenellaceae_RC9_gut_group (r = 0.60, p < 0.05) and Cyllamyces (r = 0.59, p < 0.05). Methanosphaera ASV counts were positively correlated with Succinivibrionaceae_UCG-002, Quinella, Orpinomyces, and Aspergillus (|r| > 0.5, p < 0.05); Methanosphaera was negatively correlated with Neocallimastix and Cyllamyces (|r| > 0.5, p < 0.05). Rikenellaceae_RC9_gut_group, Prevotella, and F082 were negatively correlated with Bacteroides and Buwchfawromyces, Papillibacter and Methanobrevibacter, Bacteroides, and Cyllamyces, respectively (|r| > 0.5, p < 0.05). There were also significant positive correlations between Lachnospiraceae_ND3007_group and Penicillium, between Orpinomyces and Succinivibrionaceae_UCG-002, between Papillibacter and Anaeromyces, and between Cyllamyces and Buwchfawromyces. By contrast, negative correlations in ASV counts were identified between Piromyces and Methanobrevibacter, between Cyllamyces and Aspergillus, between Succinivibrionaceae_UCG-002 and Neocallimastix, and between Quinella and Cyllamyces (|r| > 0.5, p < 0.05).
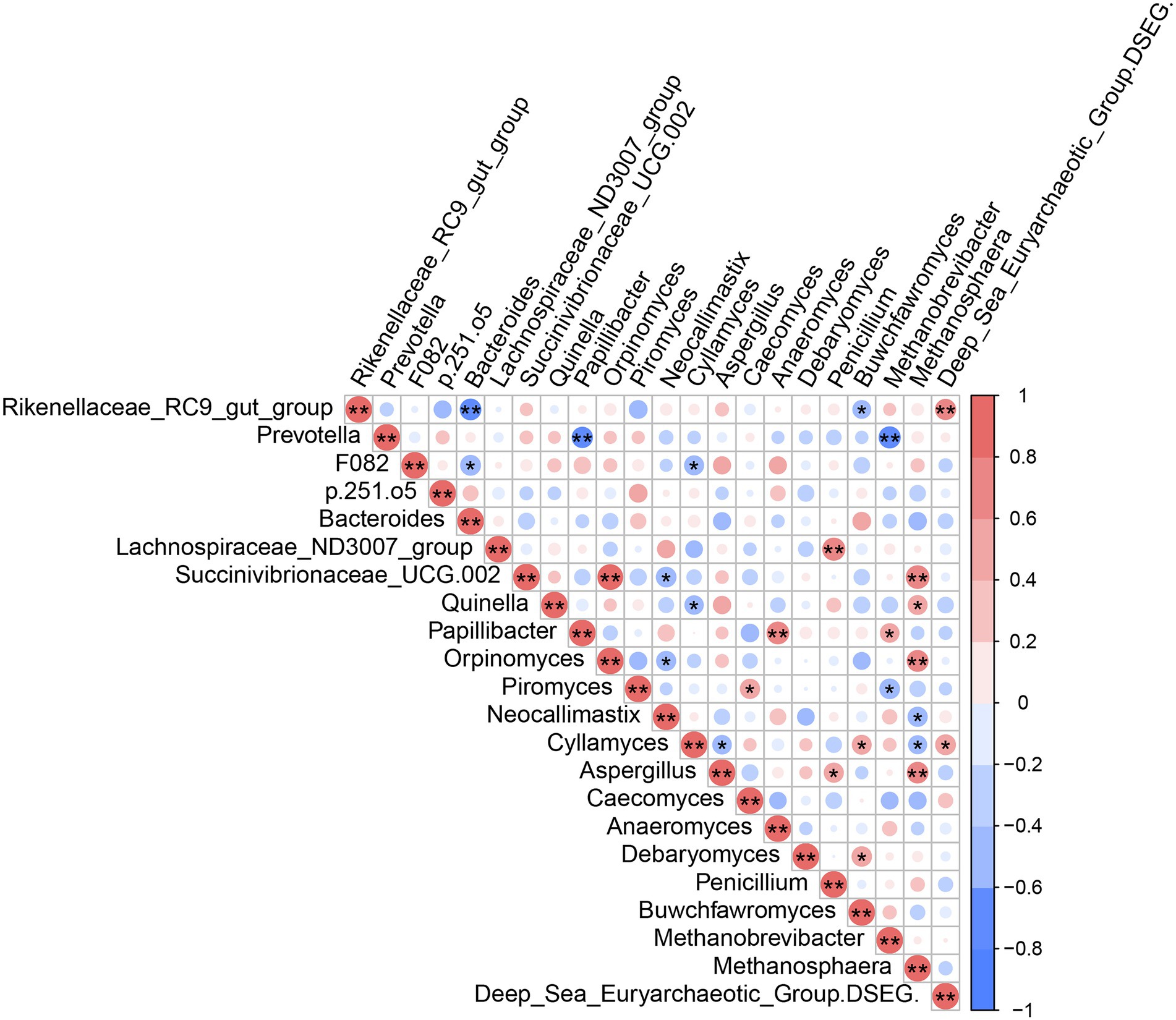
Figure 6. Mutual-correlation among the higher abundant genera of rumen microbial. Red color represents a positive correlation, while blue represents a negative correlation. *p < 0.05, **p < 0.01.
Yak is the only ruminant on the Qinghai Tibetan plateau that utilizes pasture year-round. Previous work has shown that yak rumen microbiota is robust to seasonal changes in forage availability, potentially enhancing the efficiency of fiber degradation during winter months (Huang et al., 2022). However, as important-fiber degrading microbes, the role of rumen fungi remains poorly understood in yak. In addition, environmental variability can strongly impact grazing studies (Wu et al., 2020), and current reports do not well-explain the role of rumen microbiota in the metabolic response to season-dependent changes in nutrient availability. Here, we explore the VFA profiles of rumen under simulated seasonal diet regiments, and characterize changes in rumen microbiota related to these seasonal diets by excluding environmental factors. Unlike grazing yaks, captive ruminants are subjected to less influence from complex seasonal shifts in environmental factors, and thus provide an ideal model for examining the metabolic effects of microbiota in high-altitude ruminants.
Volatile fatty acids reportedly participate in maintaining rumen homeostasis by providing an ideal acidic environment for the rumen microbiota (Bensadoun et al., 1962), and VFAs in rumen can provide approximately 75–80% of a host ruminant’s energy requirements (Lane and Jesse, 1997). Yaks have been shown to exhibit high VFA production in rumen but relatively low methane emission compared to cattle, possibly due to enrichment with energy metabolism-related genes, such as those involved in VFA production (Zhang et al., 2016). Ghimire et al. (2017) have reported various VFA profiles with different feed types. In the present study, rumen acetate, propionate, and total VFAs were higher in the GSG rumen (grazing simulation group, mimicking warm-season pasture) than in the SG (Supplementation group, mimicking winter grazing supplemented with concentrate) and NSG (nutrient stress group, mimicking winter grazing) rumen, possibly due to the availability of sufficient nutrients accompanied by a relatively high fiber/protein ratio (Liu et al., 2019). Previous work by Liu et al. (2019) showed that the forage group had higher acetate content and total VFA content than the supplemented group.
VFA production is closely related to rumen microbiota composition and diversity. R. flavefaciens and F. succinogenes are both known to produce large amounts of succinate through fiber degradation, which is increased by conversion to propionate (Fondevila and Dehority, 1996). Prevotella degrades starch and protein, producing propionate, succinate and acetate, which in turn affects VFA production (Strobel, 1992). Microorganisms can utilize acetate to produce butyrate via acetyl-CoA transferase and/or butyryl-CoA transferase (Hackmann and Firkins, 2015). In this study, we found that p.251.o5 was negatively correlated with acetate and propionate concentrations. Among fungal ASVs, Orpinomyces which was at the higher level in GSG was significantly positively correlated with acetate and propionate content, further suggesting that VFA-related microbes are enriched in GSG, leading to functional differences in acetate metabolism in rumen. Besides, acetate and propionate account for a large proportion of total VFA, and their enrichment was aligned with an increase in total VFA content. Piromyces was significantly negatively correlated with isobutyrate concentration, which has been previously reported to affect the growth of rumen fungi (Elliott et al., 1987), further supporting the effects of VFAs in determining microbial community structure.
Rumen microbiota can cooperatively degrade polysaccharides, starch, and fibers in the feed, producing VFAs that provide energy for the host, and play a crucial role in host growth and development. In this study, we found that the Bacteroidetes and Firmicutes were the dominant phyla among the three treatment groups, which is consistent with previous studies (Xue et al., 2016; Hu et al., 2021; Jiang et al., 2021; Liu et al., 2021). Bacteroidetes can degrade soluble polysaccharides and starch in the rumen to produce acetate, propionate and butyrate, which can be utilized by the host (Rosewarne et al., 2014; Ahmad et al., 2020). Belanche et al. (2019) showed that a grazing diet increases the abundance of Actinobacteriota, which aligns well with our finding of enrichment for Actinobacteriota in the GSG rumen. By contrast, Firmicutes were found in higher abundance in the NSG than in GSG rumen, suggesting a possible role in facilitating host survival during nutrient deficiency. Similarly in fungi, Neocallimastix were more abundant in NSG and SG samples, possibly due to their reported functions in cellulose utilization (Wang et al., 2011; Boots et al., 2013) and protease production for protein degradation (Michel et al., 1993). Hernandez et al. (2019) found that Cyllamyces also participated in fiber degradation, which was abundant in NSG samples, while well-known fiber degrading bacteria were less abundant under nutrient stress.
Other than host influence (Henderson et al., 2013; Hu et al., 2021), feed type strongly affects microbial diversity and species richness (Henderson et al., 2015; Cremonesi et al., 2018; Wang et al., 2020). In the present study, bacterial ASV richness was higher in GSG samples than in SG samples, further supporting that a concentrated diet can reduce bacterial diversity (McCann et al., 2014; Ku et al., 2021), while fungal richness and diversity were higher in the NSG and SG rumen. As cellulose and hemicellulose-degrading bacteria (Zened et al., 2013; Sha et al., 2020; Cui et al., 2022), Succinivibrionaceae_UCG-002 and Rikenellaceae_RC9_gut_group were enriched in GSG rumen, which had an intermediate fiber content (NSG > GSG > SG). At the species level, R. albus was more abundant in GSG rumen than that in SG rumen, while F. succinogenes, B. fibrisolvens, and R. flavefaciens were more abundant in SG samples than NSG. Together with the higher Firmicutes levels in NSG than GSG, enrichment with these known fiber-degrading bacteria (Forsberg et al., 1997) in GSG and even SG rumen, but their depletion in NSG samples, implies that nutrient deficiency can inhibit proliferation of known rumen taxa, while selecting for nutrient stress-tolerant, fiber-degrading bacteria and fungi.
Archaea account for ~2–4% of rumen microbes (Paul et al., 2015; Maman et al., 2020). Previous work by Liu et al. (2019) showed that archaeal rumen flora is affected by host factors and diet (Liu et al., 2019). In this work, Euryarchaeota was the dominant phylum, while Methanobrevibacter was the dominant genus, followed by Methanosphaera, which is consistent with other studies (Cunha et al., 2011; Sirohi et al., 2013). Compared to Methanobrevibacter, Methanosphaera spp. are remarkably enriched in “low hydrogen/methane” producing ruminants (Hoedt et al., 2018), further suggesting that hydrogen consumption by methanogens was decreased in GSG rumen, potentially enhancing VFA production.
PICRUSt analysis can be used to predict the metabolic function of bacterial communities. In the present study, superpathway of 5-aminoimidazole ribonucleotide biosynthesis was significantly enriched in GSG samples. 5-Aminoimidazole ribonucleotide was reported as a key intermediate in the biosynthesis of purine nucleotides and thiamine, as well as an important pathway in many cellular processes, such as cellular signaling and energy metabolism (Senecoff et al., 1996; Patterson et al., 1999). In addition, FUNGuild functional prediction analysis showed that the Plant_Pathogen, Endophyte−Plant_Pathogen, and Animal_Pathogen−Endophyte−Plant_Pathogen−Wood_Saprotroph were enriched in SG samples and indicated fluctuation response of the function of microbial communities among the different groups. Future validation towards understanding the metabolic function of microbial communities will require the integration of multi-omics analyses to identify the actual microbial and metabolic function response to various nutrient availability.
Yak rumen contain a complex network of symbiotic microorganisms that can synergistically ferment plant fibers, providing nutrients for themselves and the host (Ishaq and Wright, 2012). In this study, we identified interactions within and between rumen bacteria, fungi and archaea. Among fungi, Cyllamyces was negatively correlated with Aspergillus, while Orpinomyces was negatively correlated with Neocallimastix, suggesting that some rumen fungi may compete with each other. In addition, Penicillium (fungi) was significantly positively correlated with Lachnospiraceae_ND3007_group (Firmicutes), although further experiments and functional analysis based on metagenomic study are necessary to determine whether these clades can directly cooperate to utilize different components of the plant fiber.
This study comprehensively explores the response of yak rumen microbiota to simulated modes of seasonal feeding to exclude the impact of environmental factors. Our results show that different microbes in yak rumen gain dominance under (simulated) seasonal changes in nutrient availability, with relatively high nutrient availability promoting enrichment for known fiber degrading bacteria, and nutrient stress selecting for bacteria and fungi that are competitive under low nutrient availability and enhance the efficiency of host nutrient utilization. This study expands the scope of our understanding of microbial alteration and interactions under fluctuating nutrient conditions. Our findings provide a framework for future studies examining precise nutrient interventions or cold season probiotic treatments to enhance yak rumen function in nutrient utilization.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI SRA database with accession number PRJNA863131.
XH and RL conceived the study and designed the experiments. XY and XF performed DNA extraction and drafted the manuscript. XY, HJ, QZ, Basangwangdui, SD, and QZ coordinated in animal experiment and sample collection. XY, XF, and XH analyzed the data and contributed to data interpretation. All authors contributed to the article and approved the submitted version.
This work was supported by National Key R&D Program of China (2021YFD1200904), Special Item of Regional Collaborative Innovation in Tibet Autonomous Region (QYXTZXLS2021-01, XZ202101ZD002N, QYXTZX-NQ2022-01, and QYXTZX-RKZ2020-04) and the Open Project Program of State Key Laboratory of Barley and Yak Germplasm Resources and Genetic Improvement (XZNKY-2021-C-014-K06) and National Modern Agricultural Technology System (CARS-37).
We gratefully acknowledge Isaac V. Greenhut, ELS for advice with language and editorial assistance in manuscript preparation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1006285/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Amplicon sequence variants (ASV) of rumen microbiota in GSG, NSG and SG. (A) Bacteria. (B) Fungi. (C)Archaea.
SUPPLEMENTARY FIGURE S2 | Classification of the rumen microbial composition at the phylum level across the different nutrient simulations in different grazing patterns. (A) Bacteria. (B) Fungi. (C) Archaea. (D) Bacterial phylum with significant changes under various nutrient simulations. (E) Fungal phylum with significant changes under various nutrient simulations. Asterisks indicate significant difference between the three groups (*P ≤ 0.05; **P ≤ 0.01).
SUPPLEMENTARY FIGURE S3 | Linear discriminant analysis (LDA) of rumen microbial communities. (A) Fungi. (B) Archaea. (LDA cut--off of +/−2.0).
SUPPLEMENTARY FIGURE S4 | Bacterial, archaea and fungal function profiles of GSG, NSG, and SG by KEGG pathway database and the FUNGuild database. Heatmap plot showing significant differences among the three groups. (A) Bacteria. (B) Fungi. (C) Archaea. * P < 0.05, * * P < 0.01.
Ahmad, A. A., Yang, C., Zhang, J., Kalwar, Q., Liang, Z., Li, C., et al. (2020). Effects of dietary energy levels on rumen fermentation, microbial diversity, and feed efficiency of yaks (Bos grunniens). Front. Microbiol. 11:625. doi: 10.3389/fmicb.2020.00625
Belanche, A., Yanez-Ruiz, D. R., Detheridge, A. P., Griffith, G. W., Kingston-Smith, A. H., and Newbold, C. J. (2019). Maternal versus artificial rearing shapes the rumen microbiome having minor long-term physiological implications. Environ. Microbiol. 21, 4360–4377. doi: 10.1111/1462-2920.14801
Bensadoun, A., Paladines, O., and Reid, J. (1962). Effect of level of intake and physical form of the diet on plasma glucose concentration and volatile fatty acid absorption in ruminants. J. Dairy Sci. 45, 1203–1210. doi: 10.3168/jds.S0022-0302(62)89597-6
Boots, B., Lillis, L., Clipson, N., Petrie, K., Kenny, D., Boland, T., et al. (2013). Responses of anaerobic rumen fungal diversity (phylum N eocallimastigomycota) to changes in bovine diet. J. Appl. Microbiol. 114, 626–635. doi: 10.1111/jam.12067
Cremonesi, P., Conte, G., Severgnini, M., Turri, F., Monni, A., Capra, E., et al. (2018). Evaluation of the effects of different diets on microbiome diversity and fatty acid composition of rumen liquor in dairy goat. Animal 12, 1856–1866. doi: 10.1017/s1751731117003433
Cui, Y., Liu, H., Gao, Z., Xu, J., Liu, B., Guo, M., et al. (2022). Whole-plant corn silage improves rumen fermentation and growth performance of beef cattle by altering rumen microbiota. Appl. Microbiol. Biotechnol. 106, 4187–4198. doi: 10.1007/s00253-022-11956-5
Cunha, I. S., Barreto, C. C., Costa, O. Y., Bomfim, M. A., Castro, A. P., Kruger, R. H., et al. (2011). Bacteria and Archaea community structure in the rumen microbiome of goats (Capra hircus) from the semiarid region of Brazil. Anaerobe 17, 118–124. doi: 10.1016/j.anaerobe.2011.04.018
Cunniff, P., and Association of Official Analytical Chemists. (1995). Official methods of analysis of AOAC international. (Washington, DC: Association of Official Analytical Chemists).
Deusch, S., Camarinha-Silva, A., Conrad, J., Beifuss, U., Rodehutscord, M., and Seifert, J. (2017). A structural and functional elucidation of the rumen microbiome influenced by various diets and microenvironments. Front. Microbiol. 8:1605. doi: 10.3389/fmicb.2017.01605
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Elliott, R., Ash, A. J., Calderoncortes, F., Norton, B. W., and Bauchop, T. (1987). The influence of anaerobic fungi on rumen volatile fatty-acid concentrations in vivo. J. Agric. Sci. 109, 13–17. doi: 10.1017/s0021859600080928
Erwin, E., Marco, G. J., and Emery, E. (1961). Volatile fatty acid analyses of blood and rumen fluid by gas chromatography. J. Dairy Sci. 44, 1768–1771. doi: 10.3168/jds.S0022-0302(61)89956-6
Fondevila, M., and Dehority, B. A. (1996). Interactions between Fibrobacter succinogenes, Prevotella ruminicola, and Ruminococcus flavefaciens in the digestion of cellulose from forages. J. Anim. Sci. 74, 678–684. doi: 10.2527/1996.743678x
Forsberg, C. W., Cheng, K.-J., and White, B. A. (1997). “Polysaccharide degradation in the rumen and large intestine,” in Gastrointestinal microbiology. eds. R. I. Mackie and B. A. White (New York: Chapman and Hall, Springer), 319–379.
Ghimire, S., Kohn, R. A., Gregorini, P., White, R. R., and Hanigan, M. D. (2017). Representing interconversions among volatile fatty acids in the Molly cow model. J. Dairy Sci. 100, 3658–3671. doi: 10.3168/jds.2016-11858
Guo, N., Wu, Q., Shi, F., Niu, J., Zhang, T., Degen, A. A., et al. (2021). Seasonal dynamics of diet-gut microbiota interaction in adaptation of yaks to life at high altitude. NPJ Biofilms Microbiomes 7. doi: 10.1038/s41522-021-00207-6
Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G., et al. (2011). Chimeric 16S rRNA sequence formation and detection in sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21, 494–504. doi: 10.1101/gr.112730.110
Hackmann, T. J., and Firkins, J. L. (2015). Maximizing efficiency of rumen microbial protein production. Front. Microbiol. 6:465. doi: 10.3389/fmicb.2015.00465
Heijtza, R. D., Wang, S., Anuar, F., Qian, Y., Bjorkholm, B., Samuelsson, A., et al. (2011). Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. U. S. A. 108, 3047–3052. doi: 10.1073/pnas.1010529108
Henderson, G., Cox, F., Ganesh, S., Jonker, A., Young, W., Janssen, P. H., et al. (2015). Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 5, 1–15. doi: 10.1038/srep14567
Henderson, G., Cox, F., Kittelmann, S., Miri, V. H., Zethof, M., Noel, S. J., et al. (2013). Effect of DNA extraction methods and sampling techniques on the apparent structure of cow and sheep rumen microbial communities. PLoS One 8:e74787. doi: 10.1371/journal.pone.0074787
Hernandez, C., Alamilla-Ortiz, Z. L., Escalante, A. E., Navarro-Díaz, M., Carrillo-Reyes, J., Moreno-Andrade, I., et al. (2019). Heat-shock treatment applied to inocula for H2 production decreases microbial diversities, interspecific interactions and performance using cellulose as substrate. Int. J. Hydrog. Energy 44, 13126–13134. doi: 10.1016/j.ijhydene.2019.03.124
Hoedt, E. C., Parks, D. H., Volmer, J. G., Rosewarne, C. P., Denman, S. E., McSweeney, C. S., et al. (2018). Culture- and metagenomics-enabled analyses of the Methanosphaera genus reveals their monophyletic origin and differentiation according to genome size. ISME J. 12, 2942–2953. doi: 10.1038/s41396-018-0225-7
Hu, C., Ding, L., Jiang, C., Ma, C., Liu, B., Li, D., et al. (2021). Effects of management, dietary intake, and genotype on rumen morphology, fermentation, and microbiota, and on meat quality in yaks and cattle. Front. Nutr. 8:755255. doi: 10.3389/fnut.2021.755255
Huang, X., Mi, J., Denman, S. E., Basangwangdui, P., Zhang, Q., Long, R.,, et al. (2022). Changes in rumen microbial community composition in yak in response to seasonal variations. J. Appl. Microbiol. 132, 1652–1665. doi: 10.1111/jam.15322
Ishaq, S. L., and Wright, A.-D. G. (2012). Insight into the bacterial gut microbiome of the north American moose (Alces alces). BMC Microbiol. 12. doi: 10.1186/1471-2180-12-212
Islam, M. M., Fernando, S. C., and Saha, R. (2019). Metabolic modeling elucidates the transactions in the rumen microbiome and the shifts upon virome interactions. Front. Microbiol. 10:2412. doi: 10.3389/fmicb.2019.02412
Jiang, C., Ding, L., Dong, Q., Wang, X., Wei, H., Hu, C., et al. (2021). Effects of root extracts of three traditional Chinese herbs as dietary supplements on dry matter intake, average daily gain, rumen fermentation and ruminal microbiota in early weaned yak calves. Anim. Feed Sci. Technol. 278:115002. doi: 10.1016/j.anifeedsci.2021.115002
Ku, M. J., Mamuad, L., Nam, K. C., Cho, Y. I., Kim, S. H., Choi, Y. S., et al. (2021). The effects of total mixed ration feeding with high roughage content on growth performance, carcass characteristics, and meat quality of hanwoo steers. Food Sci. Anim. Resour. 41, 45–58. doi: 10.5851/kosfa.2020.e73
Lane, M. A., and Jesse, B. W. (1997). Effect of volatile fatty acid infusion on development of the rumen epithelium in neonatal sheep. J. Dairy Sci. 80, 740–746. doi: 10.3168/jds.S0022-0302(97)75993-9
Li, M., Shao, D., Zhou, J., Gu, J., Qin, J., Chen, W., et al. (2020). Signatures within esophageal microbiota with progression of esophageal squamous cell carcinoma. Chin. J. Cancer Res. 32, 755–767. doi: 10.21147/j.issn.1000-9604.2020.06.09
Liu, C., Wu, H., Liu, S., Chai, S., Meng, Q., and Zhou, Z. (2019). Dynamic alterations in yak rumen bacteria community and metabolome characteristics in response to feed type. Front. Microbiol. 10, 1116. doi: 10.3389/fmicb.2019.01116
Liu, H., Jiang, H., Hao, L., Cao, X., Degen, A., Zhou, J., et al. (2021). Rumen bacterial community of grazing lactating yaks (Poephagus grunniens) supplemented with concentrate feed and/or rumen-protected lysine and methionine. Animals 11:2425. doi: 10.3390/ani11082425
Long, R. J., Apori, S. O., Castro, F. B., and Orskov, E. R. (1999). Feed value of native forages of the Tibetan plateau of China. Anim. Feed Sci. Technol. 80, 101–113. doi: 10.1016/s0377-8401(99)00057-7
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Maman, L. G., Palizban, F., Atanaki, F. F., Ghiasi, N. E., Ariaeenejad, S., Ghaffari, M. R., et al. (2020). Co-abundance analysis reveals hidden players associated with high methane yield phenotype in sheep rumen- microbiome. Sci. Rep. 10, 1–12. doi: 10.1038/s41598-020-61942-y
McCann, J. C., Wickersham, T. A., and Loor, J. J. (2014). High-throughput methods redefine the rumen microbiome and its relationship with nutrition and metabolism. Bioinform. Biol. Insights 8, BBI.S15389–BBI.S15125. doi: 10.4137/bbi.S15389
Michel, V., Fonty, G., Millet, L., Bonnemoy, F., and Gouet, P. (1993). In vitro study of the proteolytic activity of rumen anaerobic fungi. FEMS Microbiol. Lett. 110, 5–9. doi: 10.1016/0378-1097(93)90234-s
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Patterson, D., Bleskan, J., Gardiner, K., and Bowersox, J. (1999). Human phosphoribosylformylglycineamide amidotransferase (FGARAT): regional mapping, complete coding sequence, isolation of a functional genomic clone, and DNA sequence analysis. Gene 239, 381–391. doi: 10.1016/s0378-1119(99)00378-9
Paul, S., Deb, S., Dey, A., Somvanshi, S., Singh, D., Rathore, R., et al. (2015). 16S rDNA analysis of archaea indicates dominance of Methanobacterium and high abundance of Methanomassiliicoccaceae in rumen of Nili-Ravi buffalo. Anaerobe 35, 3–10. doi: 10.1016/j.anaerobe.2015.06.002
Qiu, Q., Zhang, G., Ma, T., Qian, W., Wang, J., Ye, Z., et al. (2012). The yak genome and adaptation to life at high altitude. Nat. Genet. 44:946. doi: 10.1038/ng.2343
Rosewarne, C. P., Pope, P. B., Cheung, J. L., and Morrison, M. (2014). Analysis of the bovine rumen microbiome reveals a diversity of Sus-like polysaccharide utilization loci from the bacterial phylum Bacteroidetes. J. Ind. Microbiol. Biotechnol. 41, 601–606. doi: 10.1007/s10295-013-1395-y
Senecoff, J. F., McKinney, E. C., and Meagher, R. B. (1996). De novo purine synthesis in Arabidopsis thaliana.2. The PUR7 gene encoding 5′-phosphoribosyl-4-(N-succinocarboxamide)-5-aminoimidazole synthetase is expressed in rapidly dividing tissues. Plant Physiol. 112, 905–917. doi: 10.1104/pp.112.3.905
Sha, Y., Hu, J., Shi, B., Dingkao, R., Wang, J., Li, S., et al. (2020). Characteristics and functions of the rumen microbial Community of Cattle-yak at different ages. Biomed. Res. Int. 2020, 1–9. doi: 10.1155/2020/3482692
Sirohi, S. K., Chaudhary, P. P., Singh, N., Singh, D., and Puniya, A. K. (2013). The 16S rRNA and mcrA gene based comparative diversity of methanogens in cattle fed on high fibre based diet. Gene 523, 161–166. doi: 10.1016/j.gene.2013.04.002
Sonnenburg, J. L., and Backhed, F. (2016). Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64. doi: 10.1038/nature18846
Strobel, H. J. (1992). Vitamin-b12-dependent propionate production by the ruminal bacterium prevotella-ruminicola-23. Appl. Environ. Microbiol. 58, 2331–2333. doi: 10.1128/aem.58.7.2331-2333.1992
Thiex, N., Novotny, L., and Crawford, A. (2012). Determination of Ash in animal feed: AOAC official method 942.05 revisited. J. AOAC Int. 95, 1392–1397. doi: 10.5740/jaoacint.12-129
Vansoest, P. J., Robertson, J. B., and Lewis, B. A. (1991). Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J. Dairy Sci. 74, 3583–3597. doi: 10.3168/jds.S0022-0302(91)78551-2
Wang, L., Zhang, G., Li, Y., and Zhang, Y. (2020). Effects of high forage/concentrate diet on volatile fatty acid production and the microorganisms involved in VFA production in cow rumen. Animals 10:223. doi: 10.3390/ani10020223
Wang, T.-Y., Chen, H.-L., Lu, M.-Y. J., Chen, Y.-C., Sung, H.-M., Mao, C.-T., et al. (2011). Functional characterization of cellulases identified from the cow rumen fungus Neocallimastix patriciarum W5 by transcriptomic and secretomic analyses. Biotechnol. Biofuels 4, 1–16. doi: 10.1186/1754-6834-4-24
Wu, D., Vinitchaikul, P., Deng, M., Zhang, G., Sun, L., Gou, X., et al. (2020). Host and altitude factors affect rumen bacteria in cattle. Braz. J. Microbiol. 51, 1573–1583. doi: 10.1007/s42770-020-00380-4
Xue, B., Liu, S. J., Zhao, X. Q., Hu, L. H., and Han, X. G. (2007). Protein supply and requirement of grazing yak in China. J. Anim. Feed Sci. 16, 86–91. doi: 10.22358/jafs/74461/2007
Xue, B., Zhao, X., and Zhang, Y. (2005). Seasonal changes in weight and body composition of yak grazing on alpine-meadow grassland in the Qinghai-Tibetan plateau of China. J. Anim. Sci. 83, 1908–1913. doi: 10.2527/2005.8381908x
Xue, D., Chen, H., Chen, F., He, Y., Zhao, C., Zhu, D., et al. (2016). Analysis of the rumen bacteria and methanogenic archaea of yak (Bos grunniens) steers grazing on the Qinghai-Tibetan plateau. Livest. Sci. 188, 61–71. doi: 10.1016/j.livsci.2016.04.009
Xue, D., Chen, H., Zhao, X., Xu, S., Hu, L., Xu, T., et al. (2017). Rumen prokaryotic communities of ruminants under different feeding paradigms on the Qinghai-Tibetan plateau. Syst. Appl. Microbiol. 40, 227–236. doi: 10.1016/j.syapm.2017.03.006
Yu, C., Zhang, Y., Claus, H., Zeng, R., Zhang, X., and Wang, J. (2012). Ecological and environmental issues faced by a developing Tibet. Environ. Sci. Technol. 46, 1979–1980. doi: 10.1021/es2047188
Zened, A., Combes, S., Cauquil, L., Mariette, J., Klopp, C., Bouchez, O., et al. (2013). Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets. FEMS Microbiol. Ecol. 83, 504–514. doi: 10.1111/1574-6941.12011
Zhang, Z., Xu, D., Wang, L., Hao, J., Wang, J., Zhou, X., et al. (2016). Convergent evolution of rumen microbiomes in high-altitude mammals. Curr. Biol. 26, 1873–1879. doi: 10.1016/j.cub.2016.05.012
Keywords: yak, volatile fatty acid, season, nutrient simulation, rumen microbiota, metagenomics
Citation: Yang X, Fan X, Jiang H, Zhang Q,, Zhang Q, Dang S, Long R and Huang X (2022) Simulated seasonal diets alter yak rumen microbiota structure and metabolic function. Front. Microbiol. 13:1006285. doi: 10.3389/fmicb.2022.1006285
Edited by:
Lifeng Zhu, Nanjing Normal University, ChinaReviewed by:
Jin Wei, Nanjing Agricultural University, ChinaCopyright © 2022 Yang, Fan, Jiang, Zhang, Basangwangdui, Zhang, Dang, Long and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruijun Long, bG9uZ3JqQGx6dS5lZHUuY24=; Xiaodan Huang, aHVhbmd4aWFvZGFuQGx6dS5lZHUuY24=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.