 Ling Wang*†
Ling Wang*† Qing Liu†Shuailing GeWenhao LiangWeiyang LiaoWen Li
Qing Liu†Shuailing GeWenhao LiangWeiyang LiaoWen Li Guiai Jiao
Guiai Jiao Xiangjin Wei
Xiangjin Wei Gaoneng ShaoLihong Xie
Gaoneng ShaoLihong Xie Zhonghua ShengShikai HuShaoqing Tang*
Zhonghua ShengShikai HuShaoqing Tang* Peisong Hu*
Peisong Hu*- State Key Laboratory of Rice Biology, China National Center for Rice Improvement, China National Rice Research Institute, Hangzhou, China
Fusarium proliferatum is the principal etiological agent of rice spikelet rot disease (RSRD) in China, causing yield losses and fumonisins contamination in rice. The intraspecific variability and evolution pattern of the pathogen is poorly understood. Here, we performed whole-genome resequencing of 67 F. proliferatum strains collected from major rice-growing regions in China. Population structure indicated that eastern population of F. proliferatum located in Yangtze River with the high genetic diversity and recombinant mode that was predicted as the putative center of origin. Southern population and northeast population were likely been introduced into local populations through gene flow, and genetic differentiation between them might be shaped by rice-driven domestication. A total of 121 distinct genomic loci implicated 85 candidate genes were suggestively associated with variation of fumonisin B1 (FB1) production by genome-wide association study (GWAS). We subsequently tested the function of five candidate genes (gabap, chsD, palA, hxk1, and isw2) mapped in our association study by FB1 quantification of deletion strains, and mutants showed the impact on FB1 production as compared to the wide-type strain. Together, this is the first study to provide insights into the evolution and adaptation in natural populations of F. proliferatum on rice, as well as the complex genetic architecture for fumonisins biosynthesis.
Introduction
Rice spikelet rot disease (RSRD), also called pecky rice or kernel spotting, is one of the fastest spreading diseases in China, particularly along the middle and lower reaches of the Yangtze River (Huang et al., 2011a). It was categorized as a minor disease that occurred only sporadically before 2006. After that, RSRD epidemics became more frequent and serious due to the extensive cultivation of japonica varieties and indica-japonica hybrids (Wang L. et al., 2022). Fusarium proliferatum (teleomorph: Gibberella fujikuroi) is the most predominant pathogenic fungus associated with RSRD (Huang et al., 2011b; Wang et al., 2021). Rice is highly susceptible to F. proliferatum during the flowering stage (Sun et al., 2019a). After spore germination, the fungal hyphae enter glumes through the anthers at the pollen-filling stage, proceed to colonize pistils and endosperms, and finally infect the whole floral tissues (Sun et al., 2019b). The initial lesions of spikelet rot symptom are reddish-brown or rust-red on rice spikelets at the flowering and milking stage, eventually become blackish-brown during the ripening stage. High precipitation, relative humidity, and warm temperature during anthesis in rice favor the development of RSRD. The percentage of diseased panicles can reach up to 80% under the severe situation of the epidemic (Li et al., 2015). To date, RSRD has been estimated to occur in one third of rice-growing regions in China, resulting in yield losses of 5–30% (Wang L. et al., 2022). How and why the disease emerged in rice is still a matter of speculation. Examination of the spatial and temporal dynamics of disease progression that is prerequisite to develop risk assessments and provide the effective management approaches.
F. proliferatum is well known for its pathogenicity to a wide range of cereals, such as wheat, rice, barley, maize, rye and oats. Moreover, the pathogen is notorious for producing multiple mycotoxins, including fumonisins, moniliformin, fusaric acid, fusarin C, and beauvericin (Studt et al., 2012). Fumonisins are associated with multiple human and animal diseases, as they are potent inhibitors of sphingolipid metabolism in eukaryotes (Braun and Wink, 2018). Fumonisins induce equine leukoencephalomalacia in horses, pulmonary edema in pigs, as well as liver and kidney toxicities in rats (Riley and Merrill, 2019). Epidemiological evidences indicated a causal relationship between high levels of fumonisins exposure and neural tube defects and esophageal cancer in human beings (Yu et al., 2021). A widespread survey of fumonisins contamination on a global scale in the post-harvest grain foods ranged from 39% in Europe to 95% in North and South America (Lee and Ryu, 2017). Fumonisins comprise a group of 28 analog, which can be separated into four main groups, fumonisin A, B, C, and P (Braun and Wink, 2018). Fumonisin B (FB) analogs include FB1, FB2, and FB3, with FB1 being the most prevalent component and usually being found at the highest levels (Kamle et al., 2019). International Agency for Research on Cancer (IARC) classified FB1 as a class 2B possible carcinogen for human (International Agency for Research on Cancer [IARC], 1993). The European Union had set maximum limits for fumonisins in food and feedstuffs intended for human consumption (European Commission [EC], 2006). In order to prevent the reproduction and contamination of F. proliferatum as efficiently as possible, the control methods should be implemented in dependence on the fungi growth, reproduction, infection and mycotoxins formation in the agricultural fields.
Currently, it is known that the activation of biosynthesis pathways of secondary metabolites (SMs) not only depends on pathway-specific regulators, but also on global transcriptional complexes, signal transduction regulation, and transcriptional and epigenetic manipulation (Brakhage, 2013; Keller, 2019). Fumonisins are a family of polyketide synthases (PKS) derived SMs, consisting of a linear 20-carbon aminopolyhydroxyalkyl chain backbone. The 42-kb long cluster of fumonisin biosynthetic genes (FUM) had been well characterized in Fusarium verticillioides (Brown et al., 2007; Proctor et al., 2013). FUM genes are usually silent or expressed at very low levels under non-inducing conditions. Several external triggers, such as nutritional input (Kim and Woloshuk, 2008), light regulation (Wiemann et al., 2010), ambient pH (Flaherty et al., 2003), and water activity (Cendoya et al., 2018), had been characterized to be involved in fumonisins production. It is therefore necessary to decipher the key molecular players regulating the biosynthesis of fumonisins, as well as the interplay between the lifestyle of toxigenic fungi and genetic constitution.
To date, no reports have been elucidated about the biology and genetics of F. proliferatum population from rice. In this study, we reported the genome sequences of 67 F. proliferatum strains collected from major rice-growing regions in China, in order to characterize its genetic diversity and population structure, and dissect the genetic determinants involved in fumonisins biosynthesis. The findings of this study provided a genome-wide perspective on the population of F. proliferatum and clarified their genetic relationships with fumonisins production.
Materials and methods
Fungal strains and growth conditions
The F. proliferatum strains were isolated from naturally infected spikelets of rice at different geographical locations in China (Figure 1 and Table 1). The monoconidial culture was grown on potato dextrose agar (PDA, 20% potato extract, 2% dextrose, 1.5% agar) and minimal media [MM: 1% (w/v) carbon source, 50 ml of a 20 × salt solution (120 g/l NaNO3, 10.4 g/l KCl, 30 g/l KH2PO4, and 10.4 g/l MgSO4), and 1 ml of 5 × trace elements (22.0 g/l ZnSO4, 11 g/l boric acid, 5 g/l MnCl2, 5 g/l FeSO4, 1.6 g/l CoCl2, 1.6 g/l CuSO4, 1.1 g/l (NH4)2MoO4, and 50 g/l EDTA), pH 6.5] at 28°C. The test of the evaluation of different carbon sources included the adding to the MM of glucose, sucrose, fructose, maltose and galactose. PDA media supplemented with H2O2 (3 mM), sodium dodecyl sulfonate (SDS, 0.025%) and calcofluor white (CW, 50 μg/mL) were used for stress response assays. Conidia were obtained in YEPD liquid media (0.5% yeast extract, 1% peptone and 2% glucose) and stored into 20% glycerol at −80°C for long term storage. Species identification was performed based on colony morphology and PCR amplification as previously described (Mulè et al., 2004). Mating-type (MAT) idiomorph was determined by blasting the known MAT gene sequences (Steenkamp et al., 2000). The chitin content was measured by percentage of N-acetylglucosamine (GlcNAc) relative to the dry mycelial weight (Kong et al., 2012).
Figure 1. Map of China showing the location (dots) where Fusarium proliferatum strains were collected.
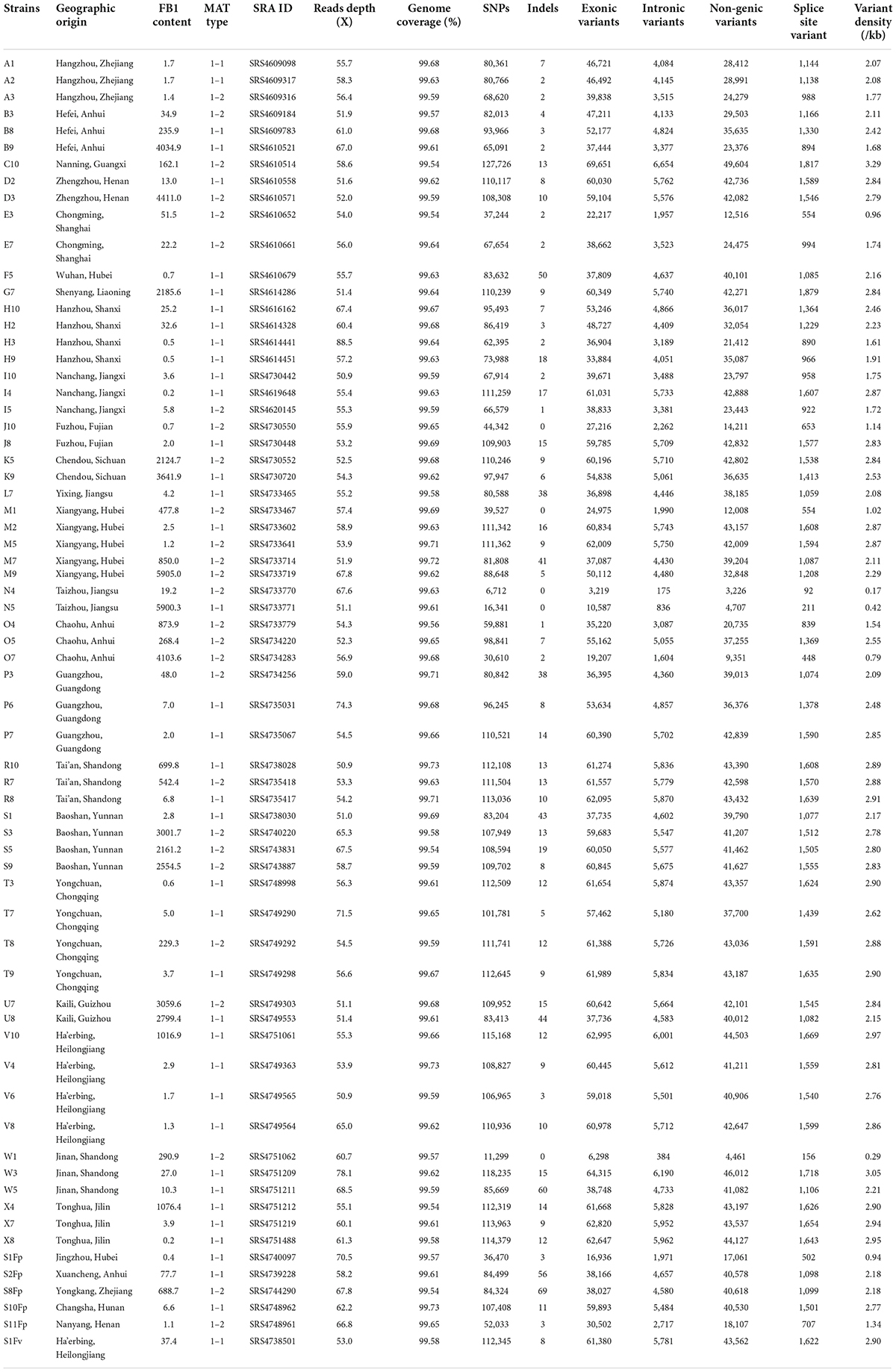
Table 1. Fumonisin FB1 production and variant calling statistics of Fusarium proliferatum.
Determination of fumonisin B1 production and fungal biomass
To investigate FB1 content, F. proliferatum strains were inoculated on cracked rice kernels or defined liquid (DL, 10 mM glutamine, 22 mM KH2PO4, 2.5 mM MgSO4, 85 mM NaCl, 117 mM sucrose, pH 5.9) media as described with some modification (Shim and Woloshuk, 1999). FB1 production was quantified by high-performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS) (Li et al., 2012). Briefly, samples were extracted with acetonitrile: water (1:1, v/v) and separated on HPLC (Thermo Fisher Scientific Waltham, MA, United States) equipped with Zorbax Extend-C18 column (100 mm × 2.1 mm, 3.5 μm). Methanol–water–formic acid (75:25:0.2, v/v/v) was used as the mobile phases at flow rate of 0.2 mL/min. The settings were used for MS/MS detection equipped with electrospray ionization (ESI) mode as follows: capillary voltage, 3,500 V; source temperature, 120°C; desolvation temperature, 350°C; and flow rate of desolvation gas, 600 L/h. Ergosterol content was measured as quantitative assessment of fungal growth with HPLC as described previously (Kim and Woloshuk, 2008). Samples were extracted overnight in chloroform: methanol (2:1, v/v). After centrifugation, the supernatants were filtered and injected into HPLC system with Eclipse XDB-C18 column (4.6 mm × 250 mm, 5 μm) and UV detector (Agilent Technologies, Inc., Santa Clara, CA, United States) set to monitor at 282 nm using 100% methanol as the mobile phases at a flow rate of 1.0 mL/min. Finally, FB1 content per fungal biomass was evaluated by the ratio of FB1 production and ergosterol content.
Genome resequencing and variant calling
Genomic DNA of F. proliferatum was extracted from the mycelium using DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Paired-end (PE) libraries with insert fragment around 350 bp were constructed and sequenced by Illumina Hiseq4000 platform. Library construction and sequencing were performed at the Beijing Novogene Bioinformatics Technology Co., Ltd., Beijing, China. Raw sequences were pre-processed by the removal of sequencing adapters and low-quality reads by Fastq-mcf with default parameters. Filtered sequencing reads were aligned to the reference genome of F. proliferatum strain Fp9 (GenBank accession no. WKFO00000000) using BWA v0.7.8 (Li and Durbin, 2010), and de novo genome assembly of the reference strain was previously performed (Wang L. et al., 2022). Spurious reads were filtered with SAMtools v0.1.19 (Li, 2011). Local realignment, duplicate marking and base quality recalibration were further processed using Picard v1.88.1 Variant calling for single nucleotide polymorphism (SNPs) and insertions and deletions (Indels) were calculated and identified by GATK v3.6.0 (McKenna et al., 2010) with default parameters. HaplotypeCaller and GenotypeGVCFs, followed by hard filtering with VariantFiltration (generic filter recommendations of GATK plus DP > 200.0, DP < 10.0). All the SNPs retrieved from sequencing reads were mapped to the reference genomes and functionally annotated with SnpEff program (Cingolani et al., 2012).
The genes of F. proliferatum strain Fp9 were originally identified through homology searches of F. proliferatum strain ET1 sequence (GenBank accession no. FJOF00000000). The SNPs were categorized as occurring in exonic regions (overlapping with a coding exon), intronic regions (overlapping with an intron), splicing sites (within 2 bp of a splicing junction) or intergenic regions. Based on the putative effects of SNPs, the genes were classified as follows, ‘‘non-functional genes’’ with loss of the function, ‘‘modified genes’’ with change of one or few amino acid sequences without major disruption of protein function, ‘‘conserved genes’’ with only change of nucleotide but no change of amino composition, and ‘‘highly conserved genes’’ with no variant in nucleotide sequence. Multigene families were chosen to analyze variants of SNPs. Among them, putative carbohydrate-active enzymes (CAZymes) were identified and classified with the CAZyme database.2 Cytochrome P450 monooxygenases (CYPs) were predicted with the fungal cytochrome P450 database.3 The homologs of known pathogen-host interaction (PHI-base) genes were predicted using the PHI database.4 Secreted proteins with N-terminal signal peptides were predicted by SignalP 5.0 (Almagro et al., 2019). Membrane transporters were predicted by Phobius5 and TMHMM (Krogh et al., 2001), and classified based on the fungal transcription factor database.6 The annotation of biosynthesis genes of secondary-metabolite (SM) were performed using antiSMURF 4.0 with the Hidden Markov Model (Blin et al., 2017).
Phylogenetic analysis and population structure
Differences of pairwise SNPs were used to calculate the genetic distances among strains of F. proliferatum. The phylogenetic tree was constructed by neighbor-joining method using MEGA 7.0 program (Kumar et al., 2016) based on the genetic differences of strains with 1,000 bootstrap replicates. According to the geographical origin of the strains, three groups including nine strains (G7, X4, X7, X8, V4, V6, V8, V10, and S1Fv), ten strains (A1, A2, A3, E3, E7, N4, N5, J8, J10, and S8Fp) and six strains (C10, K5, U7, S3, S5, and S9) were defined as northeast population, eastern population, and southern population, respectively. Nucleotide diversity (π) within populations, as well as genetic differentiation (fixation index, FST) and gene flow (Nm) between populations were calculated by VCFtools v.0.1.14 (Danecek et al., 2011) with a 50-kb slide window. Population structure was performed using STRUCTURE v.2.3.4 (Pritchard et al., 2000) with the admixture model for the most likely number of clusters (K) ranging from 2 to 5. The length of burn-in period and the number of Monte Carlo Markov Chain (MCMC) replications after burn-in were set to 50,000 and 100,000, respectively.
Genome-wide association study
FB1 production of F. proliferatum strains were used as a phenotype for the association analysis. The genome-wide association study (GWAS) was performed by linear regression analysis using PLINK v1.90 (Purcell et al., 2007) with the following parameters: minor allele frequency (>0.05), proportion of missing genotypes (<0.05) and the Hardy-Weinberg equilibrium (> 1e–5). P-value of each site was estimated and graphically displayed using Haploview v3.2 (Barrett et al., 2005). Raw p-values were adjusted for multiple testing with Benjamini-Hochberg correction to control false discovery rate. Sequences for predicted genes at associated loci were retrieved from the reference genome of F. proliferatum. Genes with Q-value (adjusted p-value < 0.05) as cut-off were considered for significance. The regional plots were generated using the asplot function in the R package “gap” (Zhao, 2007). The functional annotations were performed on the annotated genes with Trinotate pipeline (Haas et al., 2013).
Gene deletion and complementation
Deletion mutants of target genes were constructed by homologous recombination. For each gene, upstream and downstream flanking sequences were amplified with the primer pairs 5F/5R and 3F/3R, then cloned into the pFGL821 vector with hygromycin resistance cassette (hph). The deletion cassettes were transformed into protoplasts of wild-type strain Fp9 using the polyethylene glycol (PEG) method as previously described (Sun et al., 2019a). Positive transformants were screened with 100 μg/ml hygromycin B (Calbiochem, LaJolla, CA, United States) as selection marker, and verified using PCR with the primer pairs P1/P2 and P3/P4 to confirm gene replacement events. The single copy mutants were checked by the real-time genomic PCR analysis as described before (Wang G. et al., 2022). For complementation assays, the target gene with its full-length promoter region was amplified with primer pairs P5/P6 and co-transformed with XhoI-digested plasmid pDL2 into yeast strain XK1-25. The fusion constructs rescued from Trp+ yeast transformants were confirmed by sequencing and transformed into the respective knockout mutant as described (Yin et al., 2020). Transformants of complementation were screened with 100 μg/ml geneticin (Sigma, St. Louis, MO, United States) and were identified using primer pairs P5/P6. Primers used for fragments amplification were listed in Supplementary Table 1.
RNA extraction and relative gene expression analysis
Total RNA was isolated from mycelia of F. proliferatum using Trizol reagent (Invitrogen, Carlsbad, CA, United States). RNA quantity was measured on the NanoDrop 2000 spectrophotometer (Thermo Fischer Scientific, Waltham, United States). The cDNA was carried out with the ReverTraAce qPCR RT Master Mix with gDNA Remover Kit (Toyobo, Osaka, Japan) following the manufacture’s protocol. Quantitative real-time reverse transcription PCR (qRT-PCR) was performed in a volume of 20 μl containing 100 ng template cDNA, 10 μl THUNDERBIRD SYBR qPCR Mix (Toyobo, Japan), and 200 nM forward and reverse primers. Reactions were performed on ABI 7300 Real-Time System (Applied Biosystems, Foster City, CA, United States). The β-tubulin encoding gene (TUB2) was used as the internal control. Relative transcript level of each gene was calculated by the 2–Δ Δ Ct method (Livak and Schmittgen, 2001). qRT-PCR assays were repeated with three biological replicates. Primers utilized for qRT-PCR were shown in Supplementary Table 1.
Virulence assay and infectious growth
To assay virulence, conidia were harvested from 5-day-old YEPD cultures and re-suspended to 3.0 × 105 conidia per ml in sterile distilled water. Rice spikelets of cultivar Xiushui 134 were inoculated with conidial suspension and examined disease symptoms as previously described (Sun et al., 2019b). Typical symptoms of spikelets were observed at 14 days post-inoculation. For assaying infectious growth, glumes of rice were collected from inoculated spikelets at 24 h post-inoculation and photographed under scanning electron microscope. All the infection assays were repeated at least three times.
Statistical analysis
The data were presented as means and standard deviations with three biological replicates. Statistical analysis was performed using Student’s t-test implemented in the SAS software package (SAS Institute). The level of p < 0.05 was considered statistically significant.
Results
Genome sequencing and variation discovery
The entire collection of 67 F. proliferatum strains covered the regions with frequent outbreaks of RSRD (Figure 1 and Table 1). To investigate the genomic variability of 67 strains, we carried out the whole-genome resequencing using Illumina platform. Sequencing data was submitted to NCBI Short Read Archive (SRA) under the BioProject PRJNA517364 (Table 1). Approximately 500 million PE reads of the 67 sequenced strains were aligned to the reference genome of strain Fp9. The effective mapped read depth ranged from 50- to 100-fold coverage, and the average genome coverage was 99.63%. A total of 5,908,467 SNPs and 883 Indels were observed at non-redundant polymorphic sites (Table 1). Out of these, 3,206,711 variants were located in exonic regions, 307,113 in intronic regions, 2,311,110 in non-genic regions, and 85,426 in splice sites. The average of SNP density was 2.3 per kb in the whole genome of F. proliferatum. A little more than half of the SNPs (59.45%) were located in coding domains, with the number of synonymous SNPs amounting to 114,185 vs. 53,401 of that of non-synonymous SNPs (Supplementary Figure 1A). Genes were classified based on functional annotation of variants, and the majority (81.8%) of them were regarded as “modified genes” with non-synonymous effects (Supplementary Figure 1B). The “modified genes” were overrepresented in categories of CAZymes, CYPs, PHI-base, secreted proteins, membrane transporters and SM-encoding genes (Supplementary Figure 2).
Population genetic structure
Phylogenetic tree in radial pattern was constructed based on the genome-wide SNPs of F. proliferatum strains using the neighbor-joining method, which indicated that the strains were genetically independent of each other and not congruent with geographical distribution of the strains (Figure 2A). With minor exceptions, the phylogenetic relationship of strains based on genome-wide SNPs was inconsistent with the characteristics revealed by SNPs variations of FUM gene cluster (R = 0.007, p = 0.957) (Figure 2B). We investigated the genetic structure of three geographical populations. The eastern population of F. proliferatum exhibited the highest heterogeneity as indicated by nucleotide diversity (π = 0.213%) relative to that of southern population (π = 0.064%) and northeastern population (π = 0.047%). The eastern population showed low levels of genetic differentiation and moderate levels of gene flow against southern population (FST = 0.171, Nm = 0.962) and northeastern population (FST = 0.178, Nm = 0.908), respectively (Table 2). Genetic differentiation between northeastern and southern populations was fairly high (FST = 0.334) due to restricted gene flow (Nm = 0.248). Structure analysis indicated that F. proliferatum population was optimally partitioned with two ancestral components (equivalent to K = 2) (Figure 3). The genetic admixture at the individual level (K ranging from 3 to 5) revealed the significant signature of within-species introgression in recent evolutionary history. The sexual reproduction of the ascomycetes is determined by MAT loci, and an intact MAT1 locus contains one of the two idiomorphs typical for a heterothallic bipolar mating system. We observed that the frequency (59.7%) of MAT1-1 idiomorph predominated over that of MAT1-2 idiomorph within F. proliferatum population (Table 1). Two idiomorphs at the MAT1 locus were identified in the eastern and southern populations, supporting the expectation of footprints of sexual development. Conversely, only single MAT1-1 idiomorph was found in the northeastern population, showing the probability of clonal reproduction during the process of population establishment.
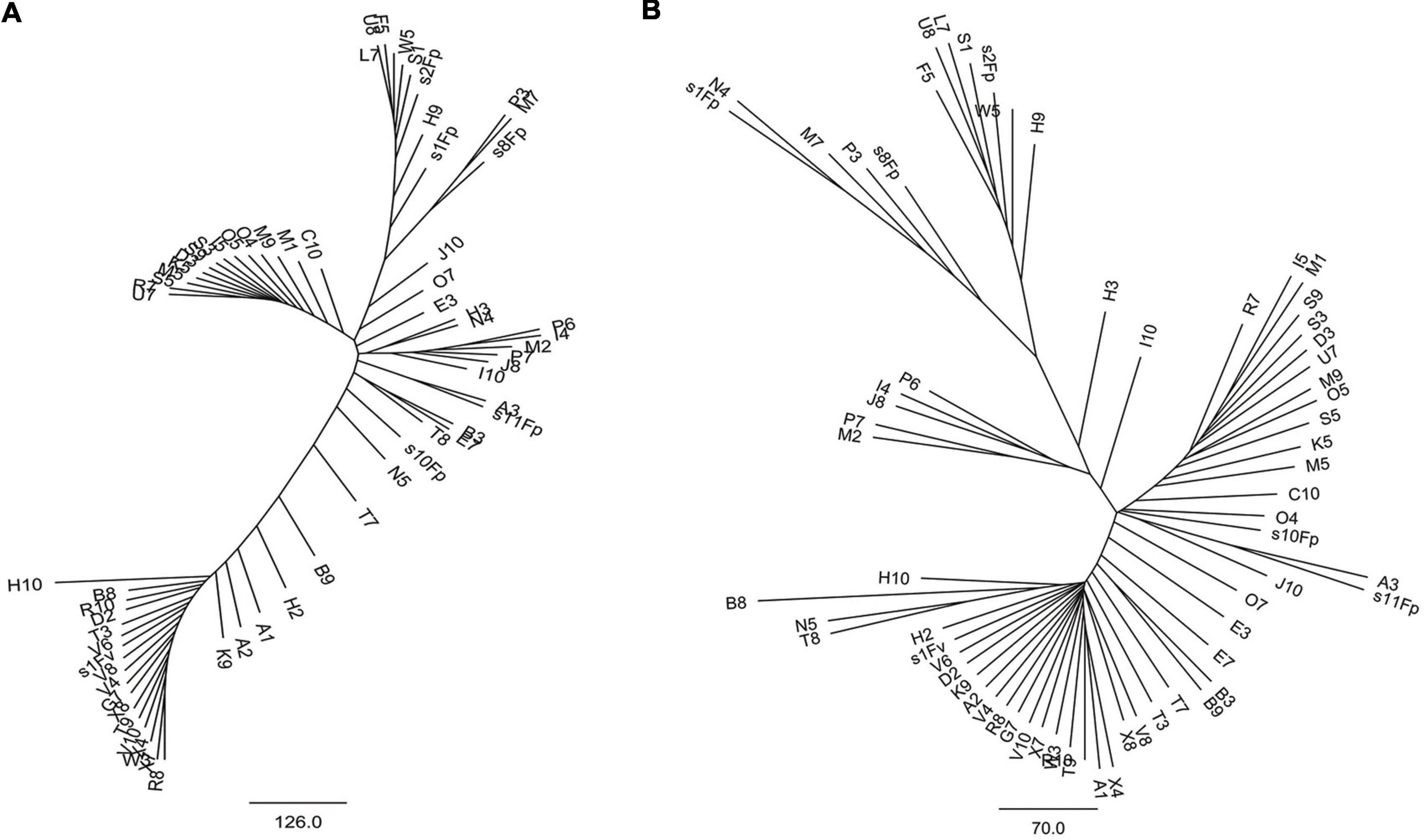
Figure 2. Radial neighbor-joining phylogenetic tree of genetic diversity within Fusarium proliferatum. (A) Genomic phylogeny determined based on SNP differences using whole-genome alignments of 67 strains to the reference strain Fp9. (B) Genomic phylogeny constructed based on SNP differences using FUM-cluster alignments of 67 strains to the reference strain Fp9. Branch lengths were proportional to the number of segregating sites that differentiated each pair of strains.

Table 2. Pairwise comparisons of gene flow (Nm, above diagonal) and genetic differentiation (FST, below diagonal) of Fusarium proliferatum populations.
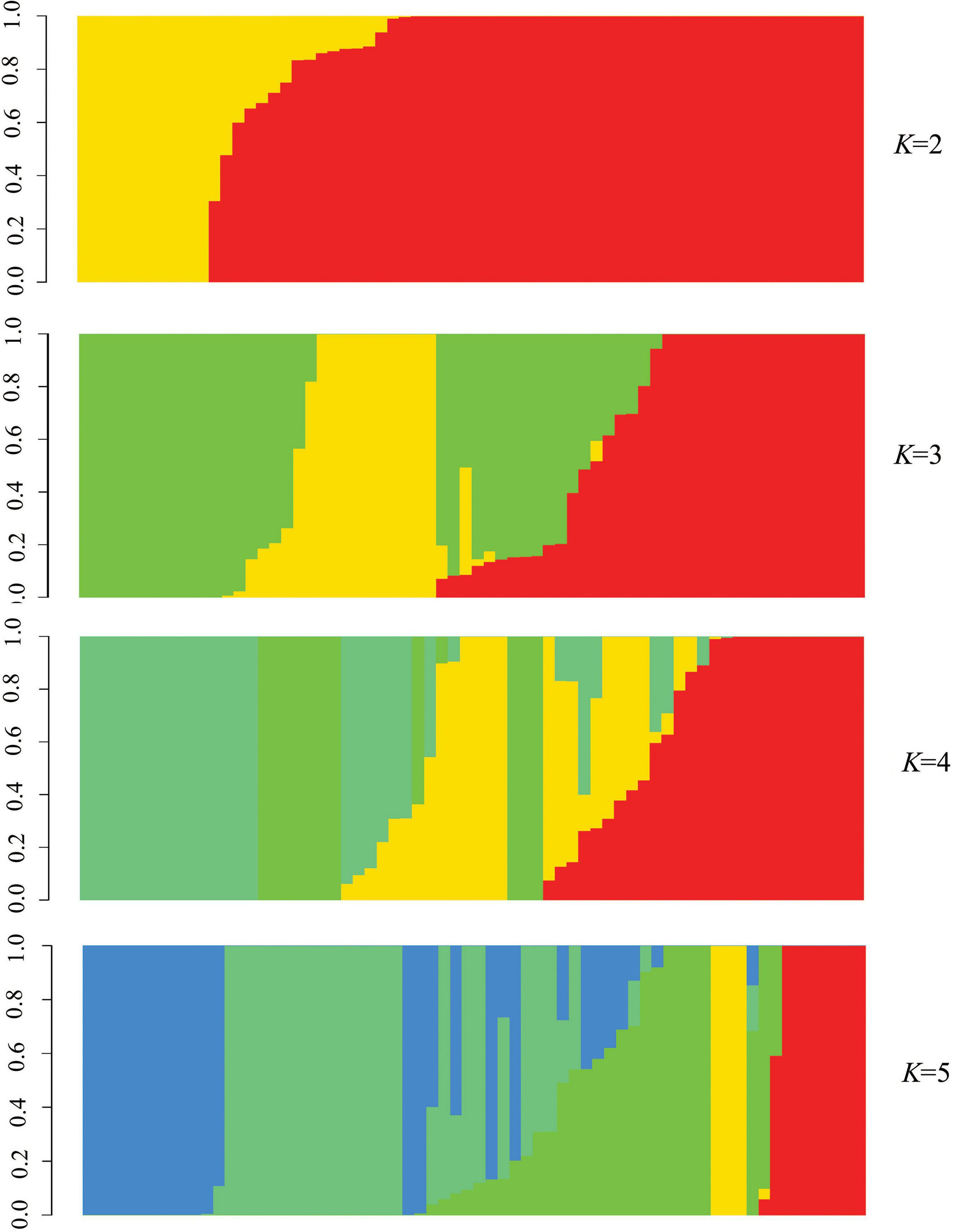
Figure 3. Population structure of Fusarium proliferatum strains. Each strain was represented by a vertical bar. Each color represented one ancestral population, and the length of each colored segment in vertical bar represented the proportion contributed by ancestral populations.
Genetic elements associated with fumonisins production
FB1 amounts of 67 strains of F. proliferatum were determined with rice kernels, ranging from 6.0 to 390,790.9 mg/kg with a mean amount of 46,187.2 mg/kg. Ergosterol was used as an indicator of the fungal biomass. There was the notable difference for relative ratios of FB1 production and ergosterol content among the strains of F. proliferatum (Table 1). GWAS was performed to investigate genetic variants associated with phenotypic variation of FB1 production. A total of 121 SNPs was found to be significantly associated with variation of FB1 content per unit of fungal biomass (Figure 4 and Supplementary Table 2). The identified associations were located in 85 candidate genes, and the majority (56.5%) of them were implicated the exon regions. The top 35 candidate genes significantly associated with FB1 production were depicted in Table 3. Functional enrichment analysis with GO term indicated that the genic SNPs were significantly associated with oxidation-reduction processes (GO:0016705, GO:0016491), transmembrane transport (GO:0055085), carbohydrate metabolism (GO:0005975), amine metabolism (GO:0009308), lipid metabolic process (GO:0006629) and chitin biosynthetic process (GO:0006031). Subsequently, we selected five associations in coding regions of genes with FB1 production to validate their functions in the reference strain Fp9.
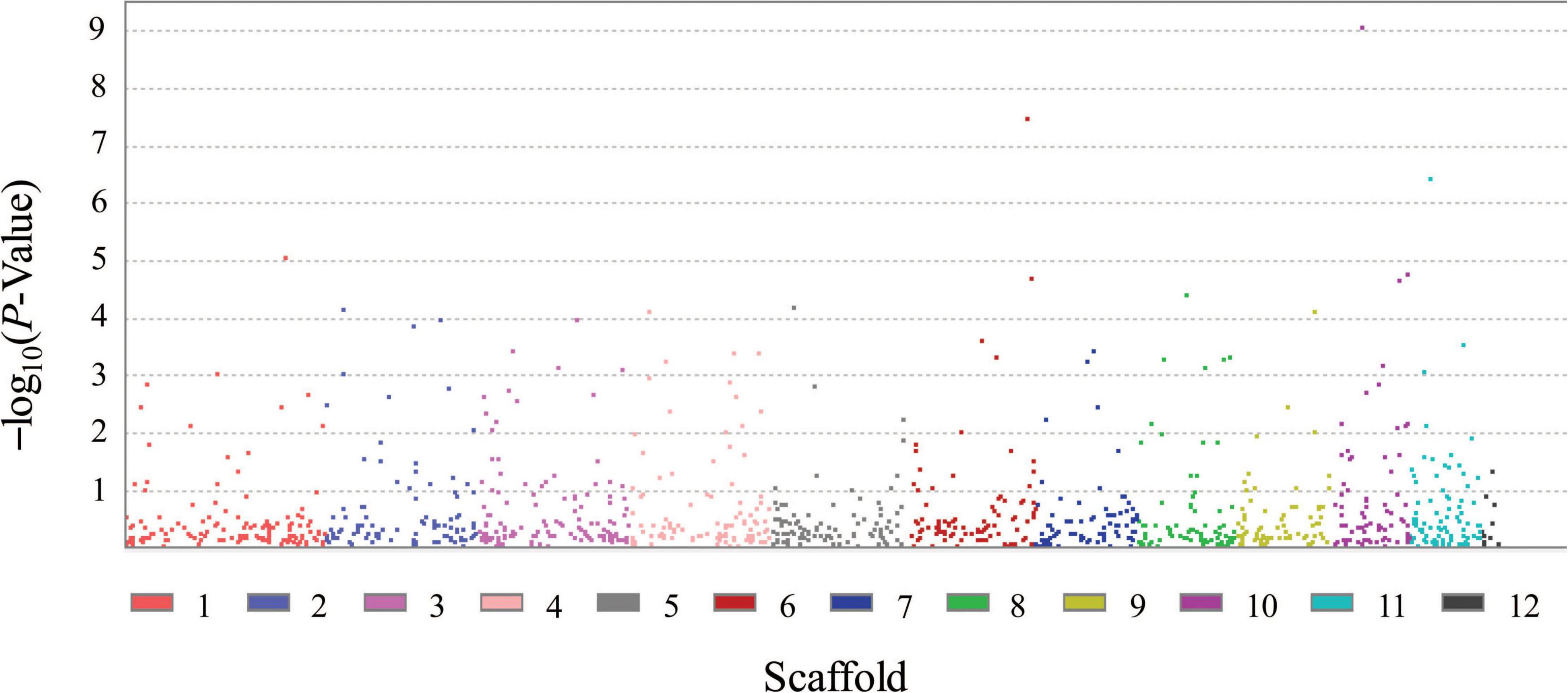
Figure 4. Genomic polymorphisms associated with FB1 production amongst 67 strains of Fusarium proliferatum.
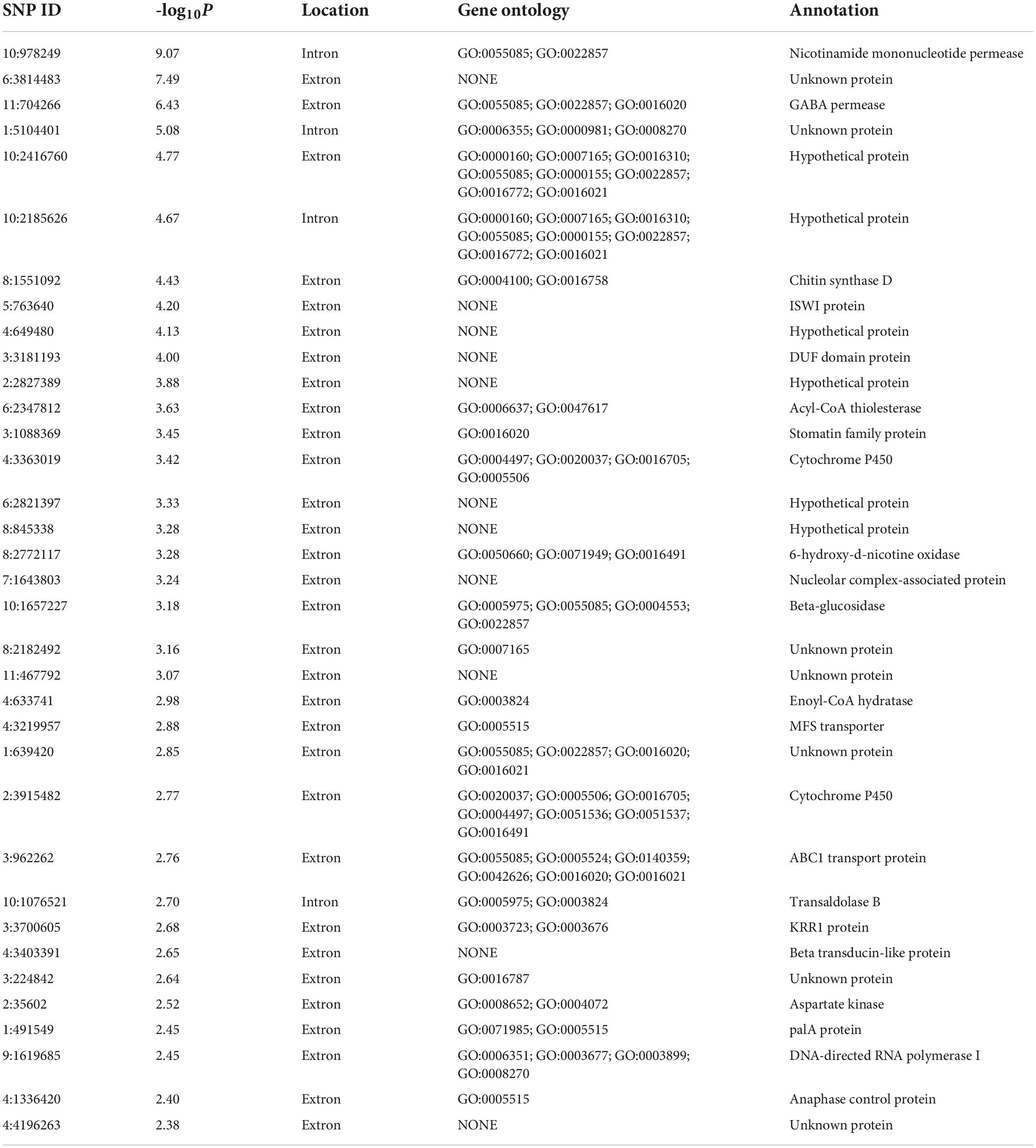
Table 3. Top 35 candidate genes significantly associated with FB1 production in Fusarium proliferatum.
Examination of possible genetic elements from genome-wide association study
The γ-aminobutyric acid (GABA) shunt is a bypass of tricarboxylic acid (TCA) cycle. A strongest association (11:704266) of the gene encoding GABA permease (GABAP) was found to be involved in FB1 production (Table 3). Compared with wide type, the growth morphology of deletion mutant Δgabap appeared unaffected in MM media (Figure 5A). While GABA was supplied as the sole nitrogen source rather than nitrate, the vegetative growth of the mutant Δgabap was retarded as expected. The mutant Δgabap produced only 34.7% as much FB1 amount as that of wild type (Figure 5B). The addition of glutamate, the precursor to GABA, had no impact on the growth and sporulation, but down-regulated gene expression of TCA cycle in the mutant Δgabap (Figure 5C). The transcriptional levels of FUM1 encoding polyketide synthase responsible for fumonisin biosynthesis and nitrogen metabolite activator AreA virtually declined in Δgabap (Figure 5D).
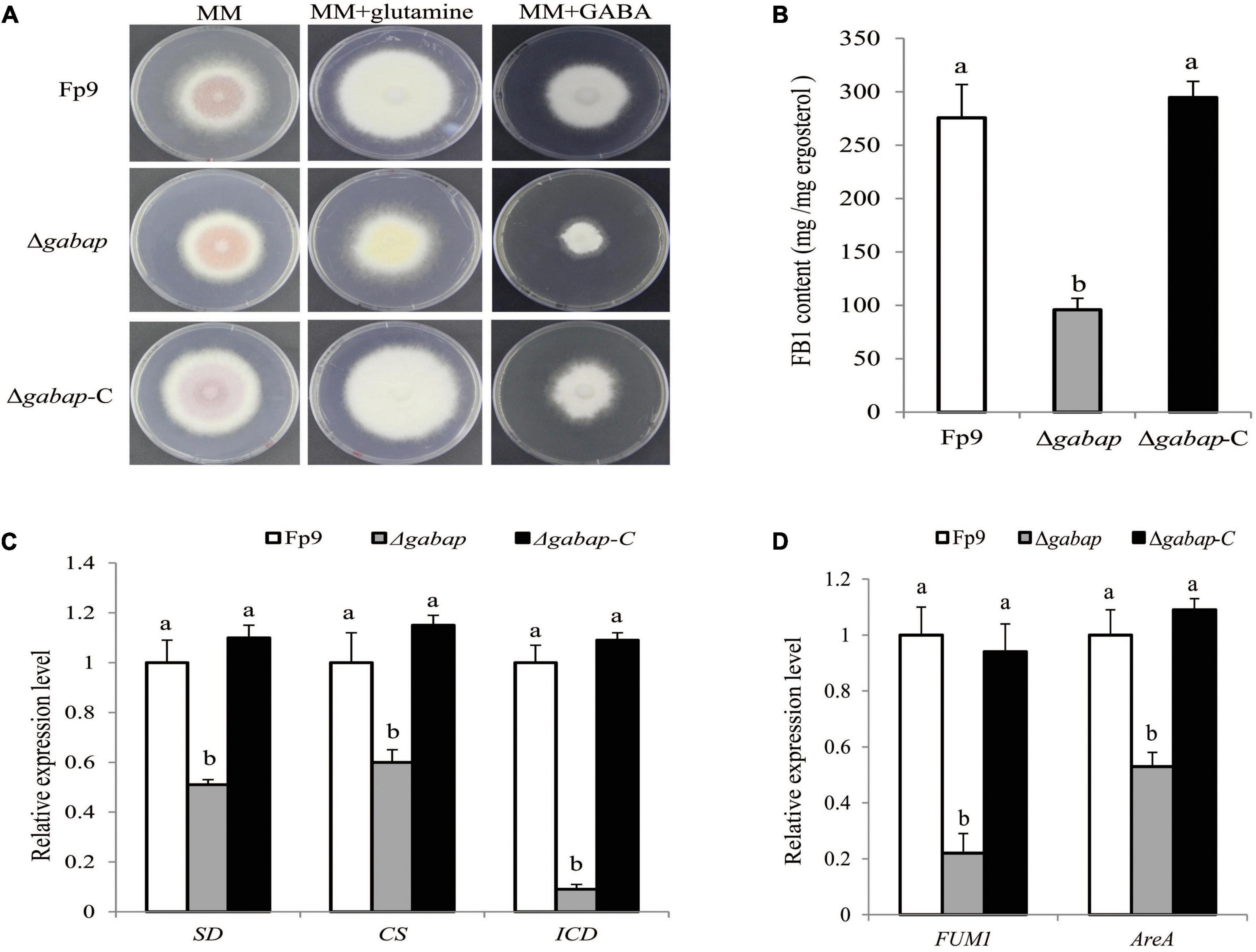
Figure 5. Impact of GABAP on the TCA cycle and FB1 production. (A) Five-day-old colony of Δgabap mutant grown on minimal media (MM) containing 30 mM glutamine or GABA as the sole nitrogen source. (B) FB1 production of Δgabap cultured with 15-day-old rice kernels. (C) The expression levels of SD encoding succinate dehydrogenase, CS encoding citrate synthase and ICD encoding isocitric dehydrogenase of Δgabap grown in YEPD media for 72 h before switching to DL cultures for 3 h. Its relative expression level in wild-type strain Fp9 was arbitrarily set to 1. Different letters on bars indicated a significant difference (p < 0.05) with three biological replicates. (D) The expression levels of FUM1 and AreA genes of Δgabap grown in YEPD media for 72 h before switching to DL cultures for 48 h. Its relative expression level in wild-type strain Fp9 was arbitrarily set to 1.
Chitin is an essential component of cell wall in filamentous fungi. A SNP (8:1551092) located in the chitin synthase gene (chsD) was detected to be correlated with FB1 production (Table 3). Deletion of chsD gene led to reduction, but not significant, of FB1 production by 16.7% (Figure 6A). Compared to wild type, the mutant ΔchsD displayed a decrease of 56.7% in chitin content (Figure 6B) and an increase of sensitivity to cell wall stressing agents (Figure 6C). Moreover, pathogenicity assays showed that ΔchsD mutant lost virulence on rice (Figure 6D). Perhaps not surprisingly, chsD was necessary for cell wall sensitivity and pathogenicity in F. proliferatum.
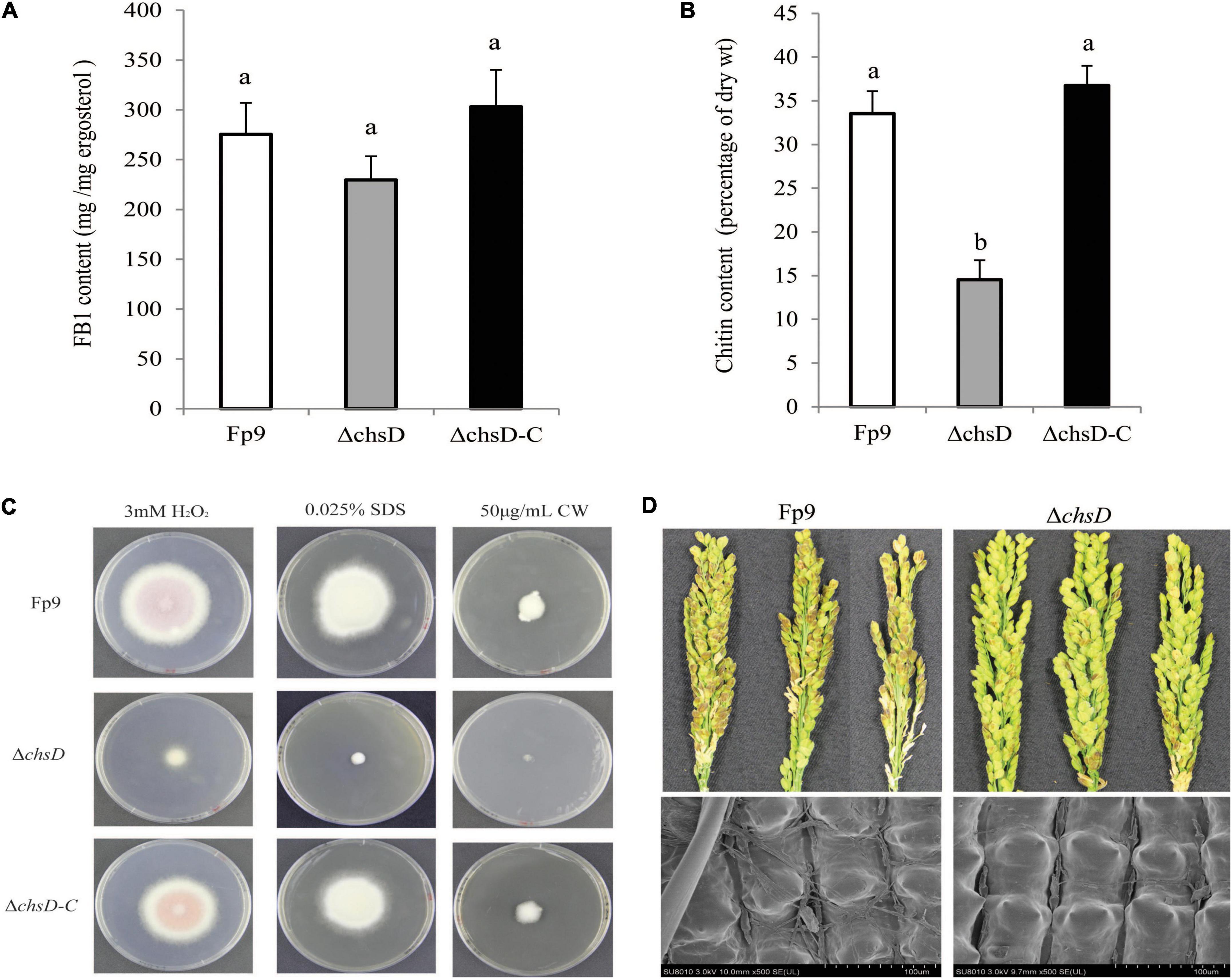
Figure 6. Defects of ΔchsD mutant in response to cell wall stress and virulence. (A) FB1 production of ΔchsD mutant cultured with 15-day-old rice kernels. (B) Chitin content of vegetative hyphae of ΔchsD harvested from 5-day-old PDA cultures. Different letters on bars indicated a significant difference (p < 0.05) with three biological replicates. (C) Sensitivity to cell wall perturbing agents of ΔchsD on PDA media with 3 mM H2O2, 0.025% SDS and 50 μg/mL calcofluor white (CW) for 5 days, respectively. (D) Disease severity of infected spikelets (upper) and mycelial growth on the outer surface of infected glumes (lower) inoculated with ΔchsD strain. Scale bars = 100 μm.
PAL signal transduction pathway is known to adapt fungi to sense and respond the changes in the external pH (Peñalva et al., 2008). The pathway relies on the endosomal sorting complex required for transport (ESCRT) complex assembled by PalA and PalB on the endosome membrane, which mediates cytoplasmic proteolysis of transcription factor PacC that activates or represses transcription of acidic- or alkaline-pH responsive genes (Lucena-Agell et al., 2015). We found an associated SNP (1:491549) that was located in the gene encoding PalA (Table 3). FB1 amount of disruption mutant ΔpalA was similar to that of wild type under acidic-inducing conditions, but drastically increased under alkaline-repressing conditions (Figures 7A,B). The transcriptional level of FUM1 gene was positively correlated with FB1 accumulation (Figure 7C). Accordingly, we concluded that PalA might be involved in FB1 biosynthesis in pH-dependent manner.
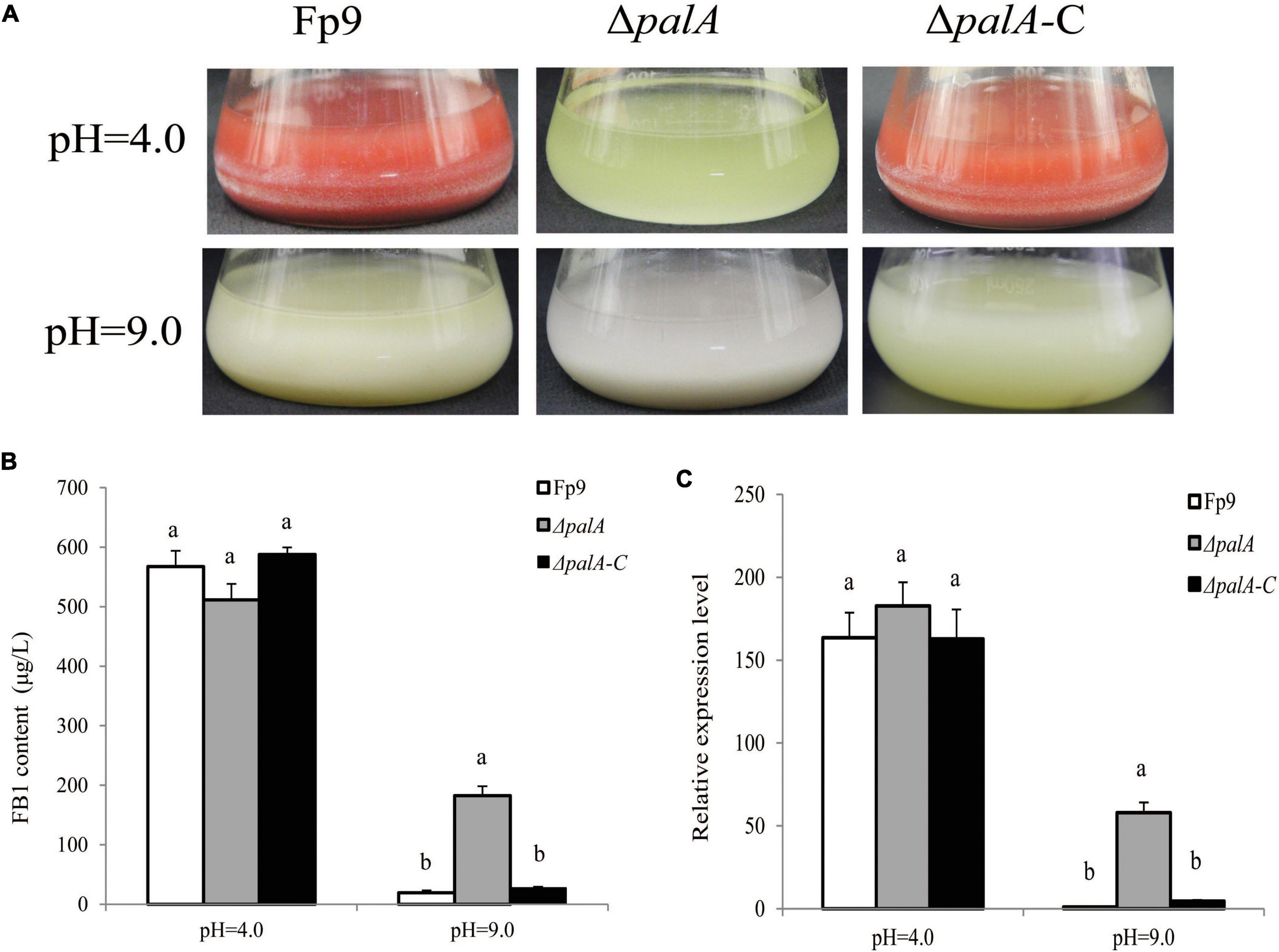
Figure 7. FB1 production activated by PalA in pH-dependent manner. (A) Seven-day-old DL cultures of ΔpalA mutant. (B) FB1 production of ΔpalA grown in YEPD media for 72 h before switching to DL cultures for 7 days at pH = 4.0 and pH = 9.0, respectively. Different letters on bars indicated a significant difference (p < 0.05) with three biological replicates. (C) The expression levels of FUM1 gene of ΔpalA grown in YEPD media for 72 h before switching to DL cultures for 48 h at pH = 4.0 and pH = 9.0, respectively.
Hexokinases (HXK) are known to be involved in carbon catabolism and glucose sensing in fungi (Santangelo, 2006). We found a significant association (6:1695154) of the gene encoding hexokinase1 (HXK1) with FB1 production (Supplementary Table 2). Gene disruption showed that Δhxk1 was unable to grow in MM media with fructose as the sole carbon source, and its morphological growth was repaired when glucose was provided (Figure 8A). The transcriptional levels of FBP1 encoding fructose-1,6-biophosphatase and ICL1 encoding isocitrate lyase in gluconeogenesis were elevated in mutant Δhxk1 relative to wild type, while PFK1 encoding phosphofructokinase in the glycolytic pathway performed the reverse reaction (Figure 8B). The deletion of HXK1 exhibited obvious reduction in the expression of FUM genes and FB1 production (Figures 8C,D). One could simply speculate that the activity of HXK1 was more likely to be responsible for FB1 production via sugar metabolism.
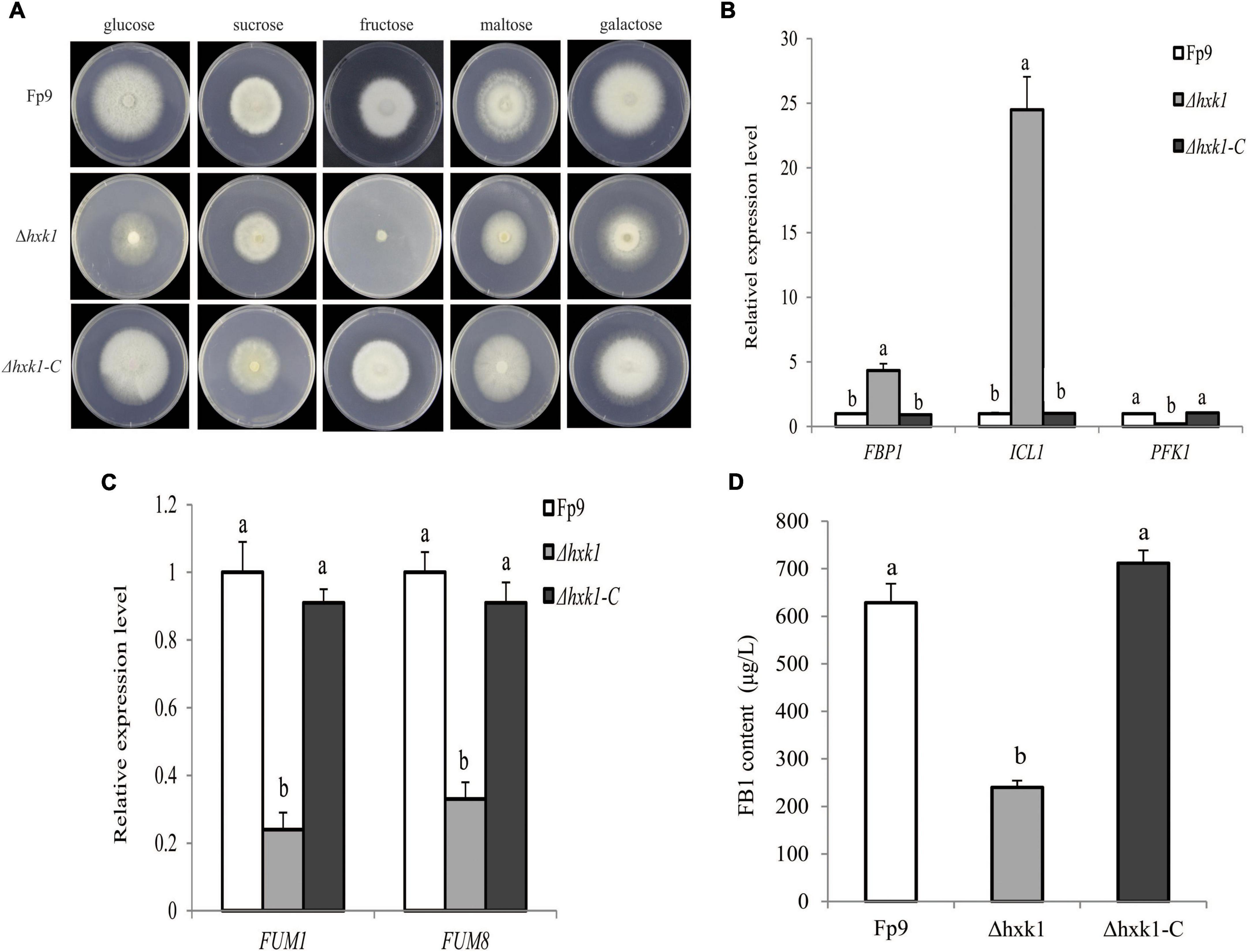
Figure 8. Defects of Δhxk1 mutant in fructose assimilation and FB1 production. (A) Five-day-old colonies of Δhxk1 mutant grown on minimal media (MM) containing glucose, sucrose, fructose, maltose and galactose as the sole carbon source at 55 mM concentration, respectively. (B) The expression levels of FBP1 encoding fructose-1,6-biophosphatas, ICL1 encoding isocitrate lyase and PFK1 encoding phosphofructokinase of Δhxk1 grown in YEPD media for 72 h before switching to DL cultures for 3 h. Its relative expression level in wild-type strain Fp9 was arbitrarily set to 1. Different letters on bars indicated a significant difference (p < 0.05) with three biological replicates. (C) The expression levels of FUM1 and FUM8 encoding aminotransferase of Δhxk1 grown in YEPD media for 72 h before switching to DL cultures for 48 h, respectively. Its relative expression level in wild-type strain Fp9 was arbitrarily set to 1. (D) FB1 production of Δhxk1 grown in YEPD media for 72 h before switching to DL cultures for 7 days.
The initiation switch (ISWI) is a subfamily of ATP-dependent chromatin remodeling complexes. It can alter nucleosome positioning, catalyze chromatin assembly, and lead to chromosome condensation (Clapier and Cairns, 2012). Of particular interest was the serendipitous observation that a SNP (5:763640) located in the gene encoding ISWI catalytic subunit (ISW2) that was associated with FB1 production (Table 3). Although ISW2 was not required for hyphal growth and pathogenicity, the deletion mutant Δisw2 was substantially impaired in FB1 production. Unlike wild type, the lack of ISW2 exhibited down-regulation of the expression levels of SMs cluster genes responsible for fumonisins, fusarubin and bikaverin under nitrogen-starved conditions (Figure 9A), as well as that of the cluster genes responsible for fusarin C and fusaric acid under nitrogen-sufficient conditions (Figure 9B).
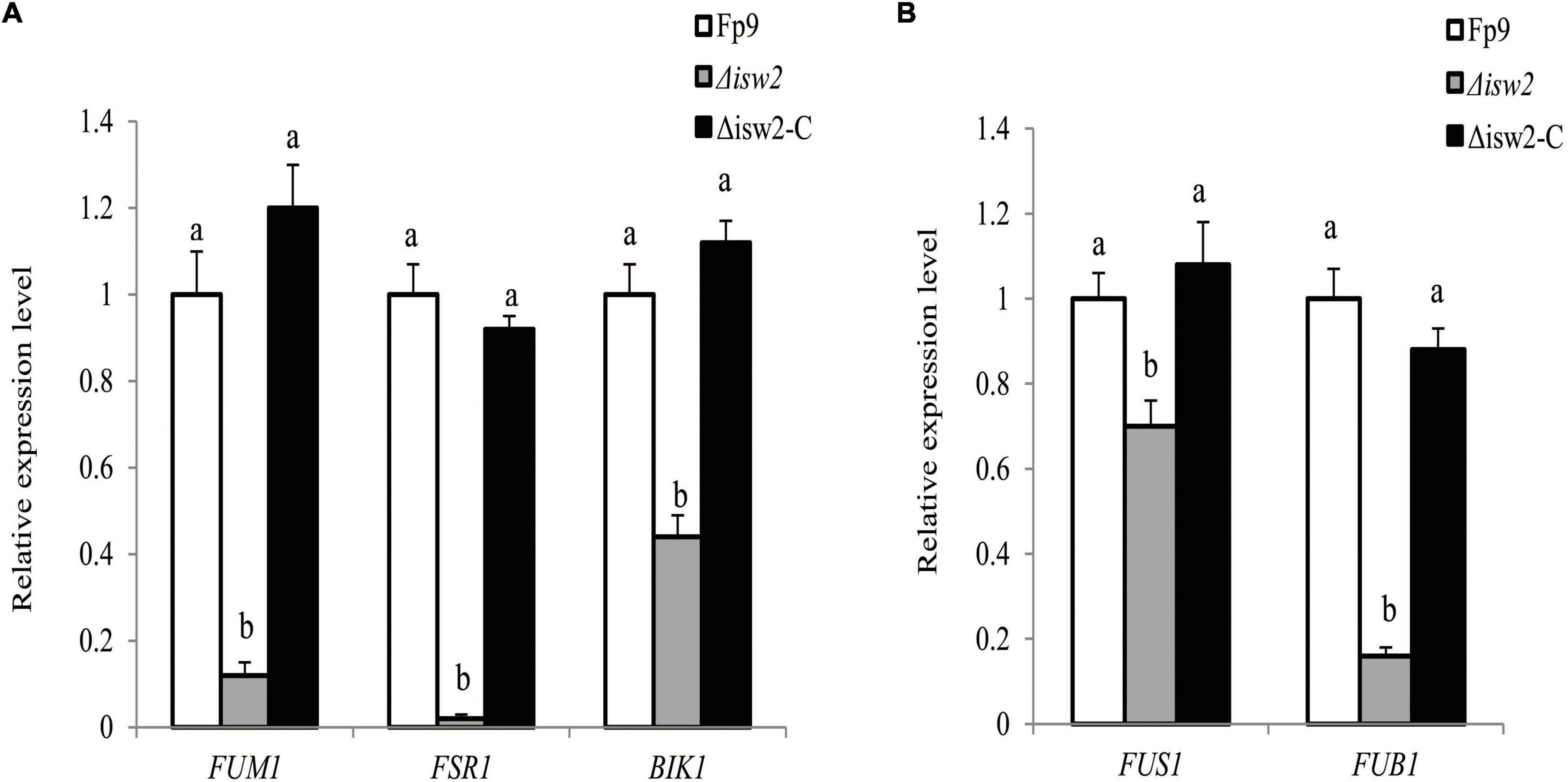
Figure 9. ISW2 affected the expression of SM biosynthetic gene clusters. (A) Expression of FUM1 encoding fumonisin cluster-polyketide synthase, FSR1 encoding fusarubin cluster-polyketide synthase, and BIK1 encoding bikaverin cluster-polyketide synthase of Δisw2 mutant grown in YEPD media for 72 h before switching to DL cultures for 48 h with 6 mM glutamine as the sole nitrogen source. Its relative expression level in wild-type strain Fp9 was arbitrarily set to 1. Different letters on bars indicated a significant difference (p < 0.05) with three biological replicates. (B) Expression of FUS1 encoding fusarin C cluster-polyketide synthase, FUB1 encoding fusaric acid cluster-polyketide synthase of Δisw2 grown in YEPD media for 72 h before switching to DL cultures for 48 h with 60 mM glutamine as the sole nitrogen source. Its relative expression level in wild-type strain Fp9 was arbitrarily set to 1.
Discussion
In the last decade, RSRD has been one of the most widespread diseases in China, causing severe reduction in rice yield and grains contamination by producing toxic metabolites that are hazardous to humans and livestocks. More importantly, the application of fungicides was not fully operative in controlling accumulation of mycotoxins. Few technologies and approaches are available to prevent fumonisins contamination. Given its economic and public health impacts, an efficient way is to gain the perspective of population biology of F. proliferatum before the control is implemented. To the best of our knowledge, this was the first report of the genomic features of F. proliferatum population obtained from the major rice-growing region in China (Figure 1 and Table 1). These findings provided new insights into its epidemiologic characteristics and evolutionary processes of this fungus. Such information could contribute to future prospects in designing disease management for RSRD.
The contemporary patterns of pathogen composition and population structure were shaped by historical evolutionary processes over time and space. However, the early history of F. proliferatum on cultivated rice, especially the impact of rice domestication on pathogen population, remains still unknown. Here, high level of genetic variation and recombinant mode were found in eastern population of F. proliferatum located in the middle and lower valley of the Yangtze River in China, where traditional varieties of indica and japonica rice were domesticated and co-cultivated with a long history. A hypothetical but straightforward scenario was that the eastern population might be the origin center from where F. proliferatum migrate toward the rest of regions, whereas the southern and northeastern populations were the founder populations that may undergo genetic bottlenecks or recent population introduction. There was evidence of gene flow between eastern population and southern or northeastern populations (Table 2), probably as a consequence of human activities like trade and transportation of F. proliferatum–contaminated rice seeds.
Intriguingly, southern and northeast populations of F. proliferatum showed geographically habitat preferences at the regional scale. Northeast population was represented in areas where japonica varieties were cultivated. Conversely, southern population was prevalent in regions where indica varieties were grown. The geographical populations of F. proliferatum followed the unique evolutionary trajectory, which suggested the signature of rice domestication as the major driving force for population divergence. In Microbotryum fungi, species-specific genes corresponded to putative adaptations responding to host-mediated selection (Badouin et al., 2017). Such epidemiological implications, probably accompanied by the host specialization, exemplified that the deployment of resistance cultivars must keep up with co-evolutionary “arm races.” By introducing the genes, such as major resistance genes and defense-responsive or defense-related genes specific for one subspecies into the other subspecies, F. proliferatum might confront selection pressures imposed by host resistance that it has never met before.
Sexual reproduction plays the fundamental roles in maintaining the genetic diversity and enhancing the adaptation to environments in fungi. Nonetheless, the stage of teleomorph of F. proliferatum has not been well described. One reason for the widespread spread of F. proliferatum was its ability to produce vast numbers of mitotically derived asexual spores, namely conidia. Our results supported that heterothallism was likely to occur frequently in the eastern and southern populations demonstrated by existence of two MAT idiomorphs (Table 1). The opposite mating types were needed for the perithecium formation of F. proliferatum. Epidemic of RSRD was possibly aggravated by the ability of the pathogen to spread through airborne ascospores. The phenomenon of F. proliferatum strains containing both MAT idiomorphs over large regions was congruent to the previous observations of that collected from durum wheat and garlic bulb (Palacios et al., 2015; Gálvez et al., 2017). The evidences implied that there was probability that F. proliferatum propagate asexually by conidia and sexually by ascospores allowing both selfing and outcrossing. The strains with strong competition exhibit dominance in geographical areas, which enhanced its potentiality to respond to disease management, e.g., fungicides modes, resistant cultivars, agronomic practices, or even the climate change. In addition, a single clonal lineage of mating type MAT1-1 was detected in northeast population (Table 1). From the practical standpoint, the finding highlighted the need for persistent vigilance of northeast population, to prevent the invasion of MAT1-2 idiomorph that might initiate sexual component to the rice-F. proliferatum pathosystem.
On a broader scale, the genome-wide level of polymorphism in F. proliferatum was 2.3 variants per kb (Table 1), which was similar to that in wheat powdery mildew Blumeria graminis (1.4–2.0 SNPs/kb) (Wicker et al., 2013), larger than blackleg Leptosphaeria maculans (0.5 SNPs/kb) (Zander et al., 2013), but much lower than poplar rust Melampsora larici-populina (6 SNPs/kb) (Persoons et al., 2014). Naturally, plant pathogens with higher diversity have relative advantages of the ecological fitness. Combining effects of natural selection, sexual recombination and genome-wide polymorphism, F. proliferatum was considered as the high-adaptive pathogen with the ability to occupy ecological niches or infect broad range of hosts during fungal population expansion. In this regard, continued surveillance of pathogen will be critical for evaluation of resistant cultivars. Resistance breeding can benefit from the inclusion of representative strains of F. proliferatum rather than a single strain when screening resistance varieties to RSRD.
There was the remarkable difference in capability of fumonisins biosynthesis among F. proliferatum strains from rice (Table 1). Similar phenotypic differences were observed among F. proliferatum strains from asparagus (von Bargen et al., 2009), pineapple (Stêpieñ et al., 2013), pea (Waśkiewicz et al., 2013), and garlic (Gálvez et al., 2017). It has been proved that the regulatory networks for fungal SMs are modulated by hierarchical interconnection with diverse cellular processes from cluster-specific pathways to global transcriptional complexes (Macheleidt et al., 2016). Biogenesis of fumonisins at the levels of pathway steps and their regulation remains a challenging task. There is a knowledge gap in the genetic diversity and fumonisins production capacity of F. proliferatum. Several attempts have been made to decipher certain interested genes located outside FUM gene cluster, it is not sufficient to elucidate the unexplored biomolecules responsible for fumonisins formation. This work aimed to depict the regulation factors involved in fumonisins production in F. proliferatum using GWAS based on whole-genome resequencing. Currently, GWAS is a preferable method for detection of the genetic determinants associated with phenotypic variation for fungal pathogens at species level, such as cold tolerance in Clonostachys rosea (Broberg et al., 2018), virulence in Zymoseptoria tritici (Hartmann et al., 2017), as well as aggressiveness, deoxynivalenol (DON) production and azole sensitivity in Fusarium graminearum (Talas et al., 2016). As far as we know, this is the first study to apply GWAS to link polymorphism in the genome to functional variability associated with fumonisins production among natural populations of F. proliferatum strains.
We identified that oxidation-reduction processes (GO:0016705, GO:0016491) were connected with fumonisins biosynthesis in F. proliferatum (Table 3). Similarly, the activation roles of cellular redox stress were previously found on aflatoxin biosynthesis in Aspergillus flavus (Fountain et al., 2019) and trichothecene production in F. graminearum (Montibus et al., 2013). The majority of oxidation, reduction and hydrolysis reactions are performed by CYPs. CYPs enzymes have unique ability to catalyze regio-, chemo-, and stereospecific conversions of lipophilic compounds to hydrophilic derivatives by introducing an oxygen atom. Three members from CYPs (FPRO_06718, FPRO_05147, FPRO_05649) were identified to be related with fumonisins production (Supplementary Table 2). Several CYPs participating in SMs biosynthesis pathways had been characterized in other fungi. Among 26 genes located in aflatoxin biosynthesis gene cluster, aflG, aflQ, aflU, and aflV encoded CYPs in A. flavus (Yu et al., 2004). In F. graminearum, three CYPs (TRI1, TRI4, and TRI11) were responsible for trichothecene biosynthesis, and three CYPs encoded ergosterol biosynthetic enzymes (Shin et al., 2017). In Verticillium dahliae, VdCYP1 had a pronounced effect on 14 kinds of fungal metabolites that likely contributed to pathogenic process (Zhang D. D. et al., 2016). Therefore, it was conceivable that the catalytic regulation of fungal CYPs was essential for cell viability and secondary metabolism.
Transmembrane transport (GO:0055085) was found to be involved in fumonisins production (Table 3). Transporters are responsible for nutrient uptake, metabolite extrusion, multidrug resistance and signal exchange. ATP-binding cassette (ABC) transporters are a superfamily of proteins that use the energy from ATP hydrolysis to transport substances across the cell membrane. Our results showed that fumonisins production was affected by an ABC transport (FPRO_03446) (Supplementary Table 2). This seemed to be the case for Bacillus cereus as well because ABC transporters were involved in the biosynthesis of metabolites, such as toxins, antibiotics and siderophores (Gacek-Matthews et al., 2020), but the underlying mechanisms remain unclear. Peptide transporters belonging to the major facilitator superfamily (MFS) transporters use the proton-motive force to translocate di- and tripeptides. Internalized peptides are rapidly hydrolyzed and the resulting amino acids are used for protein synthesis or alternative sources of nitrogen and carbon. We found that a peptide transporter (FPRO_13944) was associated with fumonisins biosynthesis (Supplementary Table 2). In F. graminearum, deletion of peptide transporters (FgPTR2A, FgPTR2C, and FgPTR2D) resulted in a sharp increase in DON and zearalenone and a decrease in fusarielin H. The three mycotoxins fell under the regulation of nitrogen regulator AreA because that the transportation activity of peptide transporters was influenced by the quality of the nitrogen source (Droce et al., 2017). Further studies are required to understand the exact nature of how the peptide transporters modulate the genetic regulatory network underlying the SM biosynthesis.
Carbohydrate metabolic process (GO:0005975) was shown the association with fumonisins production in F. proliferatum (Table 3). The sources of carbon available were reported to play a critical role in induction of SM biosynthesis, such as fumonisins biosynthesis in F. verticillioides (Malapi-Wight et al., 2013), aflatoxin production in A. flavus (Fasoyin et al., 2018), ochratoxin A biosynthesis in Aspergillus ochraceus (Wang et al., 2020). Glucose is the preferred carbon source for the filamentous fungi. Several genes encoding enzymes (e.g., xylanases, cellulases, and arabinases) required for the use of alternative carbon sources, such as lignocellulose, are repressed by carbon catabolite repression (CCR) when glucose is present. From the GWAS, three significant SNPs were located in genes encoding β-glucosidase (FPRO_14041, FPRO_03671, FPRO_15192) (Supplementary Table 2). β-glucosidase mainly hydrolyze β-glucosidic bonds to release glucose from the non-reducing end of β-glucoologosaccharides or glucosides. This effect was mediated in part by sucrose non-fermenting (SNF1) kinase that was regarded as an important regulator in cellulose degradation and sterigmatocystin biosynthesis in Podospora anserina (Li et al., 2020). Several fungal and bacterial β-glucosidases had the abilities to hydrolyze the Fusarium mycotoxins, such as DON, nivalenol and HT-2 toxin (Michlmayr et al., 2015). Thus, this would be reasonable that fungi have evolved mechanisms to balance the operation of substrate feeding (enzyme production) and secondary metabolism during life cycle.
Amine metabolic process (GO:0009308) was found to exert influences on fumonisins biosynthesis (Supplementary Table 2). Nitrogen status directly affected the ability of filamentous fungi to biosynthesize SMs (Tudzynski, 2014). Biogenic amines are low molecular weight organic nitrogen compounds in living cells. Amine oxidases catalyze the oxidative deamination of amine substrates to their corresponding aldehydes, including aliphatic and aromatic monoamines, diamines, tertiary amines, polyamines, and amino acids. Amine oxidases were known to be involved in primary amine metabolism, but the effects of amine oxidases on SMs production is rare. Two genes encoding amine oxidase (FPRO_13752, FPRO_06592) were identified to be related to fumonisins production in F. proliferatum (Supplementary Table 2). Amine oxidases of Aspergillus niger and Cochliobolus victoriae were capable of enzymatic conversion of fumonisins and victorin into deaminated and detoxified counterparts, respectively (Garnham et al., 2020; Kessler et al., 2020), which demonstrated relaxed substrate specificity of amine oxidases in mycotoxins biosynthesis.
The gabap gene encoding GABA permease was found to be significantly associated with FB1 production (Figure 5B). The gabap mutant failed to synthesize FB1 and inactivated the expression of FUM1 gene responsible for fumonisin biosynthesis. There were several indications for the connectivity between GABA shunt and the regulation of FB1 production. GABA metabolism had been previously described to link MFS transporters that pumped out DON mycotoxin in F. graminearum (Wang et al., 2018). The addition of GABA-inducible agmatine promoted the production of starting substrate for DON biosynthesis in Fusarium asiaticum (Suzuki et al., 2013). Deletion of gabap impaired GABA utilization in F. proliferatum, which was partially regulated by the global nitrogen regulator AreA. The mutant Δgabap showed downregulation of the key enzymes of the TCA cycle. Thus, we proposed that GABA as a nitrogen source positively regulated FB1 accumulation in F. proliferatum.
We identified a correlation between chsD gene encoding chitin synthase and FB1 production (Figure 6A). Disruption of chsD resulted in reduced accumulation of chitin and enhanced sensitivity against cell wall perturbing agents. In addition, our study found that chitin synthase played an important role in pathogenicity in F. proliferatum as previous studies on Magnaporthe oryzae (Kong et al., 2012) and Botrytis cinerea (Cui et al., 2013). The reduced FB1 production in the ΔchsD deletion mutant further verified the involvement of chitin synthase in aggressiveness since FB1 played an important role in the infection of F. proliferatum in rice tissue (Sun et al., 2019a). The effect of chsD gene on FB1 production in F. proliferatum was similar to that on DON production in F. graminearum (Liu et al., 2016). Our results demonstrated that essentiality of chitin biosynthesis in vegetative growth, virulence and FB1 production in F. proliferatum, which allow the development of novel inhibitory agents in the control of this pathogen by targeting the chitin synthase.
Fungal microorganisms must be able to sense and respond to ambient pH for mycelial growth, host colonization and toxin production. The ambient pH signaling pathway in filamentous fungi was medicated by six Pal proteins (PalA, PalB, PalC, PalF, PalH, and PalI) and transcription factor PacC (Peñalva et al., 2008). It was clear from previous studies that PacC was a key factor for biosynthesis of various SMs, such as ochratoxin A in A. ochraceus (Barda et al., 2020), fumonisins in F. verticillioides (Flaherty et al., 2003), dipicolinic acid in Beauveria bassiana (Luo et al., 2017), patulin in Penicillium expansum (Chen et al., 2018). The activation of PacC by proteolysis required interactions of with PalA and PalB in an alkaline pH dependent manner. In the present study, the palA gene was involved in the association of FB1 production (Figure 7B). Genetic disruption of palA rendered enhanced FB1 accumulation under external pH alkalinization in F. proliferatum. To further characterize the mechanism of the environmental pH-sensing complex on fumonisins, studies are underway to dissect this intriguing phenomenon.
Our findings suggested a relationship between hxk1 gene encoding hexokinase and FB1 biosynthesis in F. proliferatum (Figure 8D). The deletion mutant of hxk1 failed to grow on fructose and down-regulated the expression level of glycolytic genes. More importantly, Δhxk1 produced a dramatically low level of FB1, indicating that hxk1 was involved in FB1 biosynthesis in F. proliferatum. Pyruvate, the end-product of glycolysis, was controlled by hexokinase as the main substrate of acetyl-CoA for the biosynthesis of many SMs (Gao et al., 2016), including fumonisins, aflatoxin, trichothecene, DON, and penicillin. HXK1 had been shown to regulate DON production in F. graminearum (Zhang L. et al., 2016) and FB1 production in F. verticillioides (Kim et al., 2011). Our observations demonstrated that HXK1 was required for FB1 biosynthesis by establishing genetic link between primary and secondary metabolism in F. proliferatum.
ISWI catalytic subunit (ISW2) belonging to a subfamily of chromatin remodeling complexes was associated with FB1 production (Figure 9A). Perhaps the more striking observation was that the effects of ISW2 on biosynthesis of multiple SMs were regulated by nitrogen availability in F. proliferatum. The concept of ISW2 triggering SMs biosynthesis in filamentous fungi was almost without precedent. The chromatin remodeling complex RSC1 had been well characterized in Saccharomyces cerevisiae, which was required for TOR protein kinase (Yu et al., 2015). TOR signaling pathway is a central regulatory hub that connects nitrogen availability with fungal development and metabolic processes (Dobrenel et al., 2016). Therefore, it was quite likely that ISW2 played an indirect role for secondary metabolism by establishing the genetic link between nutrient-signaling regulation and SMs production.
As noted above, five candidate genes (gabap, chsD, palA, hxk1, and isw2) from the GWAS that were all required for FB1 production, possibly reflecting pleiotropic effects in coordinating the processes of fumonisins biosynthesis. It was very likely that, in such cases, SM production of filamentous fungi was controlled by a complex regulatory and biosynthesis pathways in a manner related to fungal development or in response to abiotic and biotic stressors. However, it was worthy to note that some associations were poorly annotated with unknown contributions to SM production. Further research should be intensified to identify and validate the physiological functions of the genetic components. The top hit from our association analysis will serve as excellent targets for genetic and biochemical efforts to detoxify or decontaminate mycotoxins of F. proliferatum.
Conclusion
In this study, we firstly characterized the intraspecific genomic variation of F. proliferatum populations from rice in China, to highlight rapid evolution shaped by high genetic diversity and sexual recombination, and elucidate selection pressures imposed by host domestication on population structure. These findings provided the evidences that the host-specific selection left signatures of genetic differentiation in F. proliferatum populations. Additionally, this work indicated the capability of association mapping to pinpoint genetic determinants involved in fumonisins formation in F. proliferatum. The regulatory factors related with secondary metabolism need to be validated for their potential functions. The sustainability of resistant cultivars and fungicides used to control RSRD should be geared toward information on the population characteristics of F. proliferatum, which contribute to monitor the influx of pathogen with novel adaptations, as well as discover pharmaceutical chemicals to impact positively on food safety and security.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA517364.
Author contributions
LW, ST, and PH contributed to the idea and design of the research. LW, GJ, XW, GS, and LX collected the strains. LW, SG, WHL, WYL, and WL conducted the experiments. QL was responsible for the whole-genome sequencing and analysis work. ZS and SH collected the data and performed statistics. LW wrote the first draft of the manuscript. ST and PH performed the revision and editing of the final manuscript. All authors contributed to revisions of the manuscript, read, and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (32188102 and 31800133), the Zhejiang Provincial Natural Science Foundation of China (LQ18C140005), the Key Research and Development Program of Zhejiang Province (2021C02063, 2021C02056, and 2022C02011), the Science and Technology Major Project of Anhui Province (202203c08020006), and the Agricultural Sciences and Technologies Innovation Program of Chinese Academy of Agricultural Sciences (CAAS-ZDRW202001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1004454/full#supplementary-material
Footnotes
- ^ https://github.com/broadinstitute/picard
- ^ http://www.cazy.org/
- ^ http://drnelson.uthsc.edu/cytochromeP450.html
- ^ http://www.phi-base.org/
- ^ https://phobius.sbc.su.se/
- ^ http://ftfd.snu.ac.kr/
References
Almagro, A. J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/s41587-019-0036-z
Badouin, H., Gladieux, P., Gouzy, J., Siguenza, S., Aguileta, G., Snirc, A., et al. (2017). Widespread selective sweeps throughout the genome of model plant pathogenic fungi and identification of effector candidates. Mol. Ecol. 26, 2041–2062. doi: 10.1111/mec.13976
Barda, O., Maor, U., Sadhasivam, S., Bi, Y., Zakin, V., Prusky, D., et al. (2020). The pH-responsive transcription factor PacC governs pathogenicity and ochratoxin A biosynthesis in Aspergillus carbonarius. Front. Microbiol. 11:210. doi: 10.3389/fmicb.2020.00210
Barrett, J. C., Fry, B., Maller, J., and Daly, M. J. (2005). Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265. doi: 10.1093/bioinformatics/bth457
Blin, K., Wolf, T., Chevrette, M. G., Lu, X., Schwalen, C. J., Kautsar, S. A., et al. (2017). antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 45, W36–W41. doi: 10.1093/nar/gkx319
Brakhage, A. A. (2013). Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 11, 21–32. doi: 10.1038/nrmicro2916
Braun, M. S., and Wink, M. (2018). Exposure, occurrence, and chemistry of fumonisins and their cryptic derivatives. Compr. Rev. Food Sci. Food Saf. 17, 769–791. doi: 10.1111/1541-4337.12334
Broberg, M., Dubey, M., Sun, M. H., Ihrmark, K., Schroers, H., Li, S., et al. (2018). Out in the cold: Identification of genomic regions associated with cold tolerance in the biocontrol fungus Clonostachys rosea through genome-wide association mapping. Front. Microbiol. 9:2844. doi: 10.3389/fmicb.2018.02844
Brown, D. W., Butchko, R. A. E., Busman, M., and Proctor, R. H. (2007). The Fusarium verticillioides FUM gene cluster encodes a Zn(II)2Cys6 protein that affects FUM gene expression and fumonisin production. Eukaryot. Cell 6, 1210–1218. doi: 10.1128/EC.00400-06
Cendoya, E., Monge, M. D. P., Chiacchiera, S. M., Farnochi, M. C., and Ramirez, M. L. (2018). Influence of water activity and temperature on growth and fumonisin production by Fusarium proliferatum strains on irradiated wheat grains. Int. J. Food Microbiol. 266, 158–166. doi: 10.1016/j.ijfoodmicro.2017.12.001
Chen, Y., Li, B., Xu, X., Zhang, Z., and Tian, S. (2018). The pH-responsive PacC transcription factor plays pivotal roles in virulence and patulin biosynthesis in Penicillium expansum. Environ. Microbiol. 20, 4063–4078. doi: 10.1111/1462-2920.14453
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Clapier, C. R., and Cairns, B. R. (2012). Regulation of ISWI involves inhibitory modules antagonized by nucleosomal epitopes. Nature 492, 280–284. doi: 10.1038/nature11625
Cui, Z., Wang, Y., Lei, N., Wang, K., and Zhu, T. (2013). Botrytis cinerea chitin synthase BcChsVI is required for normal growth and pathogenicity. Curr. Genet. 59, 119–128. doi: 10.1007/s00294-013-0393-y
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Dobrenel, T., Caldana, C., Hanson, J., Robaglia, C., Vincentz, M., Veit, B., et al. (2016). TOR Signaling and Nutrient Sensing. Annu. Rev. Plant Biol. 67, 261–285. doi: 10.1146/annurev-arplant-043014-114648
Droce, A., Sørensen, J. L., Sondergaard, T. E., Rasmussen, J. J., Lysøe, E., and Giese, H. (2017). PTR2 peptide transporters in Fusarium graminearum influence secondary metabolite production and sexual development. Fungal Biol. 121, 515–527. doi: 10.1016/j.funbio.2017.02.003
European Commission [EC] (2006). Commission Recommendation of 17 August 2006 on the presence of deoxynivalenol, zearalenone, ochratoxin A, T-2 and HT-2 and fumonisins in products intended for animal feeding. Off. J. Eur. Union 229, 7–9.
Fasoyin, O. E., Wang, B., Qiu, M., Han, X., Chung, K. R., and Wang, S. (2018). Carbon catabolite repression gene creA regulates morphology, aflatoxin biosynthesis and virulence in Aspergillus flavus. Fungal Genet. Biol. 115, 41–51. doi: 10.1016/j.fgb.2018.04.008
Flaherty, J. E., Pirttila, A. M., Bluhm, B. H., and Woloshuk, C. P. (2003). PAC1, a pH-regulatory gene from Fusarium verticillioides. Appl. Environ. Microbiol. 69, 5222–5227. doi: 10.1128/AEM.69.9.5222-5227.2003
Fountain, J. C., Yang, L., Pandey, M. K., Bajaj, P., Alexander, D., Chen, S., et al. (2019). Carbohydrate, glutathione, and polyamine metabolism are central to Aspergillus flavus oxidative stress responses over time. BMC Microbiol. 19:209. doi: 10.1186/s12866-019-1580-x
Gacek-Matthews, A., Chromiková, Z., Sulyok, M., Lücking, G., Barák, I., and Ehling-Schulz, M. (2020). Beyond toxin transport: novel role of ABC transporter for enzymatic machinery of cereulide NRPS assembly line. mBio 11:e01577-20. doi: 10.1128/mBio.01577-20
Gálvez, L., Urbaniak, M., Waśkiewicz, A., Stêpień, L., and Palmero, D. (2017). Fusarium proliferatum - causal agent of garlic bulb rot in Spain: genetic variability and mycotoxin production. Food Microbiol. 67, 41–48. doi: 10.1016/j.fm.2017.05.006
Gao, T., Chen, J., and Shi, Z. (2016). Fusarium graminearum pyruvate dehydrogenase kinase 1 (FgPDK1) is critical for conidiation, mycelium growth, and pathogenicity. PLoS One 2016:e0158077. doi: 10.1371/journal.pone.0158077
Garnham, C. P., Butler, S. G., Telmer, P. G., Black, F. E., Renaud, J. B., and Sumarah, M. W. (2020). Identification and characterization of an Aspergillus niger amine oxidase that detoxifies intact fumonisins. J. Agric. Food Chem. 68, 13779–13790. doi: 10.1021/acs.jafc.0c04504
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hartmann, F. E., Sánchez-Vallet, A., McDonald, B. A., and Croll, D. (2017). A fungal wheat pathogen evolved host specialization by extensive chromosomal rearrangements. ISME J. 11, 1189–1204. doi: 10.1038/ismej.2016.196
Huang, S. W., Wang, L., Liu, L. M., Tang, S. Q., Zhu, D. F., and Savary, S. (2011a). Rice spikelet rot disease in China: 2. Pathogenicity tests, assessment of the importance of the disease, and preliminary evaluation of control options. Crop Prot. 30, 10–17. doi: 10.1016/j.cropro.2010.06.008
Huang, S. W., Wang, L., Liu, L. M., Tang, S. Q., Zhu, D. F., and Savary, S. (2011b). Rice spikelet rot disease in China: 1. Characterization of fungi associated with the disease. Crop Prot. 30, 1–9. doi: 10.1016/j.cropro.2010.07.010
International Agency for Research on Cancer [IARC] (1993). Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines And Mycotoxins, IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Lyon: IARC, doi: 10.1016/0278-6915(89)90053-7
Kamle, M., Mahato, D. K., Devi, S., Lee, K. E., Kang, S. G., and Kumar, P. (2019). Fumonisins: impact on agriculture, food, and human health and their management strategies. Toxins 11:328. doi: 10.3390/toxins11060328
Keller, N. P. (2019). Fungal secondary metabolism: regulation, function and drug discovery. Nat. Rev. Microbiol. 17, 167–180. doi: 10.1038/s41579-018-0121-1
Kessler, S. C., Zhang, X. H., McDonald, M. C., Gilchrist, C. L. M., Lin, Z., Rightmyer, A., et al. (2020). Victorin, the host-selective cyclic peptide toxin from the oat pathogen Cochliobolus victoriae, is ribosomally encoded. Proc. Natl. Acad. Sci. U.S.A. 117, 24243–24250. doi: 10.1073/pnas.2010573117
Kim, H., Smith, J. E., Ridenour, J. B., Woloshuk, C. P., and Bluhm, B. H. (2011). HXK1 regulates carbon catabolism, sporulation, fumonisin B(1) production and pathogenesis in Fusarium verticillioides. Microbiology 157, 2658–2669. doi: 10.1099/mic.0.052506-0
Kim, H., and Woloshuk, C. P. (2008). Role of AREA, a regulator of nitrogen metabolism, during colonization of maize kernels and fumonisin biosynthesis in Fusarium verticillioides. Fungal Genet. Biol. 45, 947–953. doi: 10.1016/j.fgb.2008.03.007
Kong, L. A., Yang, J., Li, G. T., Qi, L. L., Zhang, Y. J., Wang, C. F., et al. (2012). Different chitin synthase genes are required for various developmental and plant infection processes in the rice blast fungus Magnaporthe oryzae. PLoS Pathog. 8:e1002526. doi: 10.1371/journal.ppat.1002526
Krogh, A., Larsson, B., von Heijne, H. G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lee, H. J., and Ryu, D. (2017). Worldwide occurrence of mycotoxins in cereals and cereal-derived food products: public health perspectives of their co-occurrence. J. Agric. Food Chem. 65, 7034–7051. doi: 10.1021/acs.jafc.6b04847
Li, C., Wu, Y. L., Yang, T., and Huang-Fu, W. G. (2012). Rapid determination of fumonisins B1 and B2 in corn by liquid chromatography-tandem mass spectrometry with ultrasonic extraction. J. Chromatogr. Sci. 50, 57–63. doi: 10.1093/chromsci/bmr009
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, L., Liu, L. M., Wang, G. R., Wang, A. J., Wang, L., Sun, L., et al. (2015). Research progress of spikelet rot disease and bacterial panicle blight of rice. Chin. J. Rice Sci. 29, 215–222. doi: 10.3969/j.issn.1001-7216.2015.02.014
Li, Y. J., Yan, P. F., Lu, X. J., Qiu, Y. L., Liang, S., Liu, G., et al. (2020). Involvement of PaSNF1 in fungal development, sterigmatocystin biosynthesis, and lignocellulosic degradation in the filamentous fungus Podospora anserina. Front. Microbiol. 11:1038. doi: 10.3389/fmicb.2020.01038
Liu, Z. Y., Zhang, X. P., Liu, X., Fu, C. Y., Han, X. Y., Yin, Y. N., et al. (2016). The chitin synthase FgChs2 and other FgChss co-regulate vegetative development and virulence in F. graminearum. Sci. Rep. 6:34975. doi: 10.1038/srep34975
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(T) (-Delta Delta C) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lucena-Agell, D., Galindo, A., Arst, H. N. Jr., and Peñalva, M. A. (2015). Aspergillus nidulans ambient pH signaling does not require endocytosis. Eukaryot. Cell 14, 545–553. doi: 10.1128/EC.00031-15
Luo, Z., Ren, H., Mousa, J. J., Rangel, D. E., Zhang, Y., Bruner, S. D., et al. (2017). The PacC transcription factor regulates secondary metabolite production and stress response, but has only minor effects on virulence in the insect pathogenic fungus Beauveria bassiana. Environ. Microbiol. 19, 788–802. doi: 10.1111/1462-2920.13648
Macheleidt, J., Mattern, D. J., Fischer, J., Netzker, T., Weber, J., Schroeckh, V., et al. (2016). Regulation and role of fungal secondary metabolites. Annu. Rev. Genet. 50, 371–392. doi: 10.1146/annurev-genet-120215-035203
Malapi-Wight, M., Smith, J., Campbell, J., Bluhm, B. H., and Shim, W. B. (2013). Sda1, a Cys(2)-His(2) zinc finger transcription factor, is involved in polyol metabolism and fumonisin B-1 production in Fusarium verticillioides. PLoS One 8:e67656. doi: 10.1371/journal.pone.0067656
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome. Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Michlmayr, H., Varga, E., Malachova, A., Nguyen, N. T., Lorenz, C., Haltrich, D., et al. (2015). Versatile family 3 glycoside hydrolase from bifidobacterium adolescentis hydrolyzes β-glucosides of the Fusarium mycotoxins deoxynivalenol, nivalenol, and HT-2 Toxin in cereal matrices. Appl. Environ. Microbiol. 81, 4885–4893. doi: 10.1128/AEM.01061-15
Montibus, M., Ducos, C., Bonnin-Verdal, M., Bormann, J., Ponts, N., Richard-Forget, F., et al. (2013). The bZIP transcription factor Fgap1 mediates oxidative stress response and trichothecene biosynthesis but not virulence in Fusarium graminearum. PLoS One 8:e83377. doi: 10.1371/journal.pone.0083377
Mulè, G., Susca, A., Stea, G., and Moretti, A. (2004). A species-specific PCR assay based on the calmodulin partial gene for identification of Fusarium verticillioides, F. proliferatum and F. subglutinans. Eur. J. Plant Pathol. 110, 495–502. doi: 10.1023/b:ejpp.0000032389.84048.71
Palacios, S. A., Susca, A., Haidukowski, M., Stea, G., Cendoya, E., Ramírez, M. L., et al. (2015). Genetic variability and fumonisin production by Fusarium proliferatum isolated from durum wheat grains in Argentina. Int. J. Food Microbiol. 201, 35–41. doi: 10.1016/j.ijfoodmicro.2015.02.011
Peñalva, M. A., Tilburn, J., Bignell, E., and Arst, H. N. Jr. (2008). Ambient pH gene regulation in fungi: making connections. Trends Microbiol. 16, 291–300. doi: 10.1016/j.tim.2008.03.006
Persoons, A., Morin, E., Delaruelle, C., Payen, T., Halkett, F., Frey, P., et al. (2014). Patterns of genomic variation in the poplar rust fungus Melampsora larici-populina identify pathogenesis-related factors. Front. Plant Sci. 5:450. doi: 10.3389/fpls.2014.00450
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
Proctor, R. H., Hove, F. V., Susca, A., Stea, G., Busman, M., van der Lee, T., et al. (2013). Birth, death and horizontal transfer of the fumonisin biosynthetic gene cluster during the evolutionary diversification of Fusarium. Mol. Microbiol. 90, 290–306. doi: 10.1111/mmi.12362
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Riley, R. T., and Merrill, A. H. Jr. (2019). Ceramide synthase inhibition by fumonisins: a perfect storm of perturbed sphingolipid metabolism, signaling, and disease. J. Lipid Res. 60, 1183–1189. doi: 10.1194/jlr.S093815
Santangelo, G. M. (2006). Glucose signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 70, 253–282. doi: 10.1128/MMBR.70.1.253-282.2006
Shim, W. B., and Woloshuk, C. P. (1999). Nitrogen repression of fumonisin B1 biosynthesis in Gibberella fujikuroi. FEMS Microbiol. Lett. 177, 109–116. doi: 10.1111/j.1574-6968.1999.tb13720.x
Shin, J. Y., Bui, D. C., Lee, Y., Nam, H., Jung, S., Fang, M., et al. (2017). Functional characterization of cytochrome P450 monooxygenases in the cereal head blight fungus Fusarium graminearum. Environ. Microbiol. 19, 2053–2067. doi: 10.1111/1462-2920.13730
Steenkamp, E. T., Wingfield, B. D., Coutinho, T. A., Zeller, K. A., Wingfield, M. J., Marasas, W. F. O., et al. (2000). PCR-based identification of MAT-1 and MAT-2 in the Gibberella fujikuroi species complex. Appl. Environ. Microbiol. 66, 4378–4382. doi: 10.1128/AEM.66.10.4378-4382.2000
Stêpieñ, Ł, Koczyk, G., and Waśkiewicz, A. (2013). Diversity of Fusarium species and mycotoxins contaminating pineapple. J. Appl. Genet. 54, 367–380. doi: 10.1007/s13353-013-0146-0
Studt, L., Troncoso, C., Gong, F., Hedden, P., Toomajian, C., Leslie, J. F., et al. (2012). Segregation of secondary metabolite biosynthesis in hybrids of Fusarium fujikuroi and Fusarium proliferatum. Fungal Genet. Biol. 49, 566–577. doi: 10.1016/j.fgb.2012.05.005
Sun, L., Chen, X., Gao, J., Zhao, Y., Liu, L. M., Hou, Y. X., et al. (2019a). Effects of disruption of five FUM genes on fumonisin biosynthesis and pathogenicity in Fusarium proliferatum. Toxins 11:327. doi: 10.3390/toxins11060327
Sun, L., Wang, L., Liu, L. M., Hou, Y. X., Xu, Y. H., Liang, M. Q., et al. (2019b). Infection and colonization of pathogenic fungus Fusarium proliferatum in rice spikelet rot disease. Rice Sci. 26, 60–68. doi: 10.1016/j.rsci.2018.08.005
Suzuki, T., Kim, Y. K., Yoshioka, H., and Iwahashi, Y. (2013). Regulation of metabolic products and gene expression in Fusarium asiaticum by agmatine addition. Mycotoxin Res. 29, 103–111. doi: 10.1007/s12550-013-0158-y
Talas, F., Kalih, R., Miedaner, T., and McDonald, B. A. (2016). Genome-wide association study identifies novel candidate genes for aggressiveness, deoxynivalenol production, and azole sensitivity in natural field populations of Fusarium graminearum. Mol. Plant Microbe Interact. 29, 417–430. doi: 10.1094/MPMI-09-15-0218-R
Tudzynski, B. (2014). Nitrogen regulation of fungal secondary metabolism in fungi. Front. Microbiol. 5:656. doi: 10.3389/fmicb.2014.00656
von Bargen, S., Martinez, O., Schadock, I., Eisold, A. M., Gossmann, M., and Büttner, C. (2009). Genetic variability of phytopathogenic Fusarium proliferatum associated with crown rot in Asparagus officinalis. J. Phytopathol. 157, 446–456. doi: 10.1111/j.1439-0434.2008.01525.x
Wang, G., Li, Y., Yang, B., Li, E., Wu, W., Si, P., et al. (2022). AwAreA regulates morphological development, ochratoxin A production, and fungal pathogenicity of food spoilage fungus Aspergillus westerdijkiae revealed by an efficient gene targeting system. Front. Microbiol. 13:857726. doi: 10.3389/fmicb.2022.857726
Wang, L., Ge, S., Liang, W., Liao, W., Li, W., Jiao, G., et al. (2022). Genome-wide characterization reveals variation potentially involved in pathogenicity and mycotoxins biosynthesis of Fusarium proliferatum causing spikelet rot disease in rice. Toxins 14:568. doi: 10.3390/toxins14080568
Wang, G., Wang, Y. L., Yang, B. L., Zhang, C. X., Zhang, H. Y., Xing, F. G., et al. (2020). Carbon catabolite repression gene AoCreA regulates morphological development and ochratoxin A biosynthesis responding to carbon sources in Aspergillus ochraceus. Toxins 12:697. doi: 10.3390/toxins12110697
Wang, L., Ge, S. L., Zhao, K. H., and Huang, S. W. (2021). First report of Fusarium incarnatum causing spikelet rot on rice in China. Plant Dis. 105:3306. doi: 10.1094/PDIS-12-20-2660-PDN
Wang, Q. H., Chen, D. P., Wu, M. C., Zhu, J. D., Jiang, C., Xu, J. R., et al. (2018). MFS transporters and GABA metabolism are involved in the self-defense against DON in Fusarium graminearum. Front. Plant Sci. 9:438. doi: 10.3389/fpls.2018.00438
Waśkiewicz, A., Stêpieñ, Ł, Wilman, K., and Kachlicki, P. (2013). Diversity of pea-associated F. proliferatum and F. verticillioides populations revealed by FUM1 sequence analysis and fumonisin biosynthesis. Toxins 5, 488–503. doi: 10.3390/toxins5030488
Wicker, T., Oberhaensli, S., Parlange, F., Buchmann, J. P., Shatalina, M., Roffler, S., et al. (2013). The wheat powdery mildew genome shows the unique evolution of an obligate biotroph. Nat. Genet. 45, 1092–1096. doi: 10.1038/ng.2704
Wiemann, P., Brown, D. W., Kleigrewe, K., Bok, J. W., Keller, N. P., Humpf, H., et al. (2010). FfVel1 and FfLae1, components of a velvet-like complex in Fusarium fujikuroi, affect differentiation, secondary metabolism and virulence. Mol. Microbiol. 77, 972–994. doi: 10.1111/j.1365-2958.2010.07263.x
Yin, J. R., Hao, C. F., Niu, G., Wang, W., Wang, G. H., Xiang, P., et al. (2020). FgPal1 regulates morphogenesis and pathogenesis in Fusarium graminearum. Environ. Microbiol. 22, 5373–5386. doi: 10.1111/1462-2920.15266
Yu, F. F., Imamura, Y., Ueno, M., Suzuki, S. W., Ohsumi, Y., Yukawa, M., et al. (2015). The yeast chromatin remodeler Rsc1-RSC complex is required for transcriptional activation of autophagy-related genes and inhibition of the TORC1 pathway in response to nitrogen starvation. Biochem. Biophys. Res. Commun. 464, 1248–1253. doi: 10.1016/j.bbrc.2015.07.114
Yu, J., Chang, P. K., Ehrlich, K. C., Cary, J. W., Bhatnagar, D., Cleveland, T. E., et al. (2004). Clustered pathway genes in aflatoxin biosynthesis. Appl. Environ. Microbiol. 70, 1253–1262. doi: 10.1128/AEM.70.3.1253-1262.2004
Yu, S., Jia, B. X., Liu, N., Yu, D. Z., Zhang, S., and Wu, A. B. (2021). Fumonisin B1 triggers carcinogenesis via HDAC/PI3K/Akt signaling pathway in human esophageal epithelial cells. Sci. Total Environ. 787:147405. doi: 10.1016/j.scitotenv.2021.147405
Zander, M., Patel, D. A., Wouw, A. V., Lai, K., Lorenc, M. T., Campbell, E., et al. (2013). Identifying genetic diversity of avirulence genes in Leptosphaeria maculans using whole genome sequencing. Funct. Integr. Genomics 13, 295–308. doi: 10.1007/s10142-013-0324-5
Zhang, D. D., Wang, X. Y., Chen, J. Y., Kong, Z. Q., Gui, Y. J., Li, N. Y., et al. (2016). Identification and characterization of a pathogenicity-related gene VdCYP1 from Verticillium dahliae. Sci. Rep. 6:27979. doi: 10.1038/srep27979
Zhang, L., Li, B., Zhang, Y., Jia, X., and Zhou, M. (2016). Hexokinase plays a critical role in deoxynivalenol (DON) production and fungal development in Fusarium graminearum. Mol. Plant Pathol. 17, 16–28. doi: 10.1111/mpp.12258
Keywords: Fusarium proliferatum, rice spikelet rot disease, fumonisins, genome resequencing, genetic variation, genome-wide association study
Citation: Wang L, Liu Q, Ge S, Liang W, Liao W, Li W, Jiao G, Wei X, Shao G, Xie L, Sheng Z, Hu S, Tang S and Hu P (2022) Genomic footprints related with adaptation and fumonisins production in Fusarium proliferatum. Front. Microbiol. 13:1004454. doi: 10.3389/fmicb.2022.1004454
Received: 27 July 2022; Accepted: 26 August 2022;
Published: 21 September 2022.
Edited by:
Pinmei Wang, Zhejiang University, ChinaReviewed by:
Lukasz Stepien, Institute of Plant Genetics, Polish Academy of Sciences, PolandAsadollah Ahmadikhah, Shahid Beheshti University, Iran
Hojjat Asadollahpour Nanaei, Northwest A&F University, China
Copyright © 2022 Wang, Liu, Ge, Liang, Liao, Li, Jiao, Wei, Shao, Xie, Sheng, Hu, Tang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling Wang, wangling03@caas.cn; Shaoqing Tang, tangshaoqing@caas.cn; Peisong Hu, peisonghu@126.com
†These authors have contributed equally to this work