 Matheus Augusto Calvano Cosentino1Mirela D’arc1Filipe Romero Rebello Moreira1,2Liliane Tavares de Faria Cavalcante1Ricardo Mouta1Amanda Coimbra1Francine Bittencourt Schiffler1Thamiris dos Santos Miranda1Gabriel Medeiros1Cecilia A. Dias3Antonizete R. Souza3Maria Clotilde Henriques Tavares3Amilcar Tanuri4Marcelo Alves Soares1,5André Felipe Andrade dos Santos1*
Matheus Augusto Calvano Cosentino1Mirela D’arc1Filipe Romero Rebello Moreira1,2Liliane Tavares de Faria Cavalcante1Ricardo Mouta1Amanda Coimbra1Francine Bittencourt Schiffler1Thamiris dos Santos Miranda1Gabriel Medeiros1Cecilia A. Dias3Antonizete R. Souza3Maria Clotilde Henriques Tavares3Amilcar Tanuri4Marcelo Alves Soares1,5André Felipe Andrade dos Santos1*- 1Laboratório de Diversidade e Doenças Virais, Departamento de Genética, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 2Department of Infectious Diseases Epidemiology, Imperial College London, London, United Kingdom
- 3Centro de Primatologia, Universidade de Brasília, Brasília, Brazil
- 4Laboratório de Virologia Molecular, Departamento de Genética, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 5Programa de Oncovirologia, Instituto Nacional de Câncer, Rio de Janeiro, Brazil
The development of high-throughput sequencing (HTS) technologies and metagenomics protocols deeply impacted the discovery of viral diversity. Moreover, the characterization of novel viruses in the Neotropical primates (NP) is central for the comprehension of viral evolution dynamics in those hosts, due to their evolutionary proximity to Old World primates, including humans. In the present work, novel anelloviruses were detected and characterized through HTS protocols in the NP Callithrix penicillata, the common black-tufted marmoset. De novo assembly of generated sequences was carried out, and a total of 15 contigs were identified with complete Anelloviridae ORF1 gene, two of them including a flanking GC-rich region, confirming the presence of two whole novel genomes of ~3 kb. The identified viruses were monophyletic within the Epsilontorquevirus genus, a lineage harboring previously reported anelloviruses infecting hosts from the Cebidae family. The genetic divergence found in the new viruses characterized two novel species, named Epsilontorquevirus callithrichensis I and II. The phylogenetic pattern inferred for the Epsilontorquevirus genus was consistent with the topology of their host species tree, echoing a virus-host diversification model observed in other viral groups. This study expands the host span of Anelloviridae and provides insights into their diversification dynamics, highlighting the importance of sampling animal viral genomes to obtain a clearer depiction of their long-term evolutionary processes.
Introduction
The development of high-throughput sequencing (HTS) technologies and metagenomics protocols deeply impacted the discovery of viral diversity (Greninger, 2018; Pérez-Losada et al., 2020). Once a matter of contention, the establishment of novel viral species and higher taxonomic groups based solely on sequencing data became commonplace. Studies describing multiple previously unknown viral groups are increasingly common (Datta, 2015; Shi et al., 2016; Atoni et al., 2019; Zhang et al., 2019), as are studies expanding the diversity of well-established viral families (Smeele et al., 2018; de Sabato et al., 2019; Sawaswong et al., 2019; dos Santos et al., 2020). While these efforts have broad implications on the comprehension of virus diversity, ecology, evolution, and epidemiology, large fractions of the viruses infecting non-model organisms remain to be discovered (Geoghegan and Holmes, 2017). In this regard, Neotropical primates (NP; infraorder Platyrrhini) stand out as a group with great ecological importance, whose viral diversity has not been fully explored. Only 18% (4/22) and 3.4% (6/178) of NP genera and species, respectively, have been previously assessed through viral metagenomics (Ng et al., 2015; Troncoso et al., 2015; Silvestre et al., 2016; Muniz et al., 2018; D’arc et al., 2020; dos Santos et al., 2020).
Currently, NP comprehends 178 species and 22 genera that occupy a myriad of ecological niches in Central and South Americas (IUCN/SSC Primate Specialist Group, 2021). As it is the sister group to Old World primates (OWP; infraorder Catarrhini), including humans, the comprehension of their evolution, immunology, and susceptibility to infectious diseases is central to the characterization of the scope of etiological agents that may impact global health (Devaux et al., 2019). In this sense, previous investigations have shown that multiple viral families might be found in the virome of NP: novel papillomaviruses were found infecting Callithrix penicillata (D’arc et al., 2020) and Alouatta guariba (Silvestre et al., 2016), anellovirus and genomovirus in Sapajus nigritus (dos Santos et al., 2020), smacovirus in Alouatta caraya (Ng et al., 2015), and foamy virus in Brachyteles arachnoides (Muniz et al., 2018) and Sapajus xanthosternos (Troncoso et al., 2015).
The Anelloviridae family is one of the most frequently found in metagenomic surveys of NP. This family is composed of non-enveloped virions with circular single-stranded negative sense DNA (ssDNA), whose genomes range from 1.6 to 3.9 kb. Three major ORFs are found in their genomes—named ORF1, ORF2, and ORF3—with putative additional ORFs among isolates. Overlay between ORFs is common and their estimated sizes differ widely among isolates. ORF1 is believed to encode the putative capsid and replication-associated protein, while ORF2 may encode a protein with phosphatase activity or a peptide capable of suppressing NF-kB pathways (Varsani et al., 2021). Currently, the group is composed of 30 classified genera by the International Committee of Virus Taxonomy (Nishizawa et al., 1997). The Torque Teno Virus was first identified in the 1990s in a post-transfusion hepatitis case of unknown etiology in humans (Bendinelli et al., 2001) and was posteriorly discovered in healthy human populations with prevalence ranging from 10 to 96% (Hino and Miyata, 2007; Freer et al., 2018). No pathology has been confirmed by its infection, although an association with pulmonary and liver diseases has been suggested (Ninomiya et al., 2009). Furthermore, the Anelloviridae family is proposed to be a common part of the human virome, with major importance to the immune system maturation (Hrazdilová et al., 2016). A high prevalence in healthy populations and the lack of clinical signs have also been shown in free-living and captive populations of chimpanzees and gorillas (Inami et al., 2000; Okamoto et al., 2000; de Souza et al., 2018). Although new anelloviruses species have been discovered in NP—such as in Saguinus oedipus, Aotus trivirgatus (Ng et al., 2011), and Sapajus nigritus (dos Santos et al., 2020)—an extensive knowledge gap for anelloviruses infecting NP still remains.
While the diversity and host span of Anelloviridae is constantly being updated (Okamoto et al., 2000; Noppornpanth et al., 2001; Pujol et al., 2005; Hon et al., 2008; Holmes, 2010; Ng et al., 2011; Sawaswong et al., 2019; dos Santos et al., 2020; Weber et al., 2020; Kraberger et al., 2021), little is known about the factors driving their diversification dynamics. Specifically, whether the generation of novel viral species is driven by zoonotic spillovers to new host species or co-divergence with long-term host lineages has not been formally assessed. Anelloviridae phylogenetics topography suggests a co-divergence within some host lineages, as seen in felids (Noppornpanth et al., 2001) and rodents (Wei et al., 2019; Lam et al., 2020).This hypothesis has been addressed by phylogenetic methods for other groups in large scale meta-analysis (de Souza et al., 2018) as well as in studies focusing on specific viral families (McGeoch et al., 2006; Buck et al., 2016; Silvestre et al., 2016; Smeele et al., 2018; Bandoo et al., 2021), revealing that the mechanisms that generate viral diversity are often determined by viruses genomic architecture and variability. As the discovery of novel viruses allows a broader evaluation of these mechanisms, metagenomic surveys on non-model organisms have allowed the clarification of the evolutionary dynamics of diverse viral families (Niewiadomska and Gifford, 2013; Kohl et al., 2015; Troncoso et al., 2015; Muniz et al., 2018).
In this study, we characterized the virome of captive Callithrix penicillata, the black-tufted marmoset, by HTS and discovered two novel species of anelloviruses, expanding the scope of primate hosts infected by these viruses. Phylogenetic analysis of the novel sequences suggests anellovirus evolution might be driven by the within-species diversity followed by co-divergence model, as suggested for other viral groups (Buchfink et al., 2014; Nurk et al., 2017; Chen et al., 2018; D’arc et al., 2020). Overall, our results shed light on previously unexplored anellovirus diversity and address a hypothesis regarding their diversification dynamics.
Materials and methods
Sample collection
The data analyzed herein was generated on a larger virome project using samples from NP housed at the Centro de Primatologia of Universidade de Brasília (CP/UnB; Brasília—Brazil). General information on the viral diversity characterized can be found in the first publication of the larger project that the present study is part of D’arc et al. (2020). For this study, the data analyzed were sequenced from anal swab samples from 16 black-tufted marmosets (C. penicillata), immersed in phosphate-buffered saline (PBS) and stored at −20°C (Table 1). Samples were collected following the national guidelines and provisions of National Council for Animal Experimentation Control (CONCEA), which included animal welfare standard operating procedures. The procedure was conducted under permission of the Brazilian Institute of Environment and Renewable Natural Resources (IBAMA) under license number 1/53/1999/000006-2 and the project was approved by Ethics Committee on the Use of Animals of Universidade Federal do Rio de Janeiro (reference number 037/14).
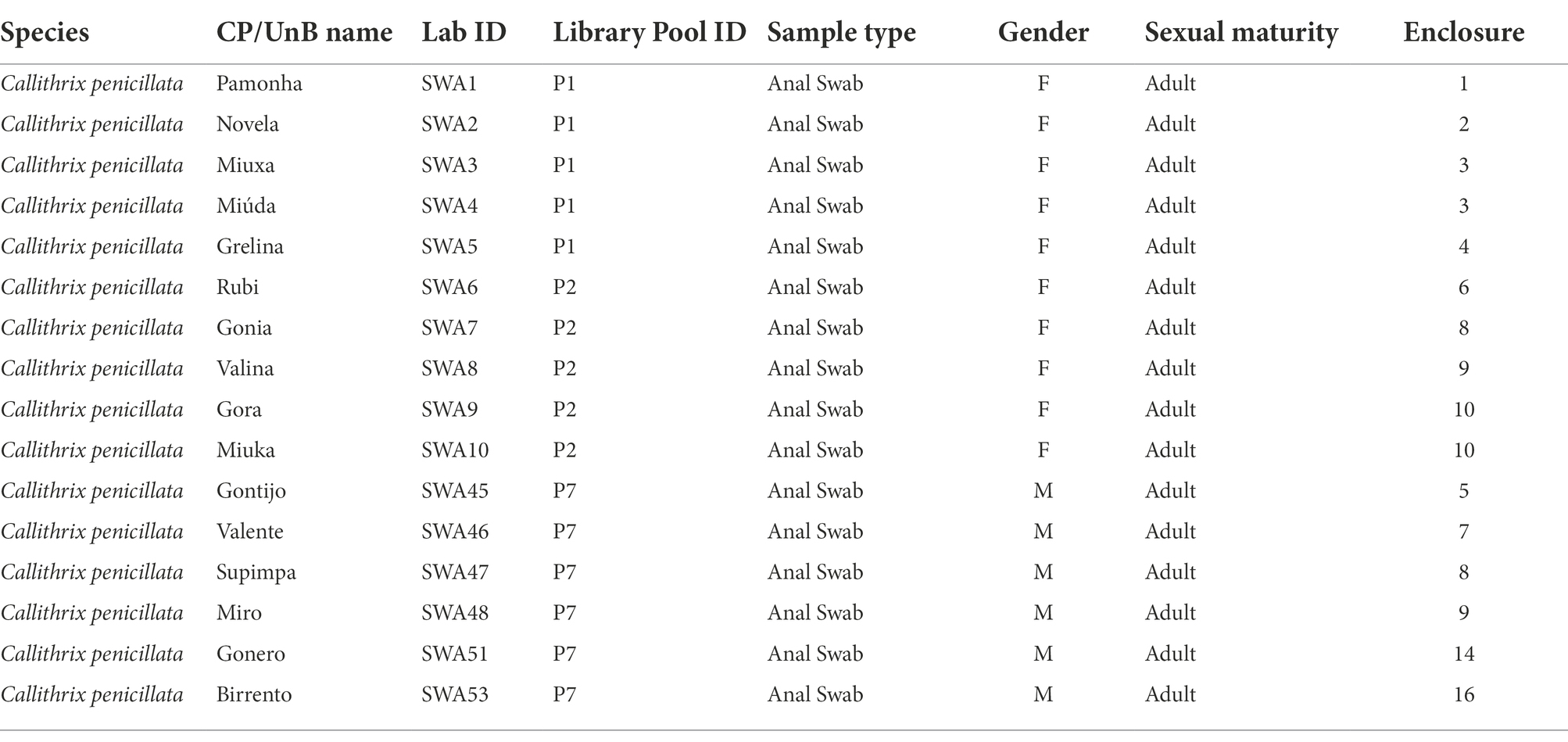
Table 1. General information of all samples from Callithrix penicillata housed at Centro de Primatologia of Universidade de Brasília (CP/UnB).
High-throughput sequencing
The samples were shipped to the Laboratório de Diversidade e Doenças Virais of Universidade Federal do Rio de Janeiro (LDDV/UFRJ; Rio de Janeiro—Brazil) and then the homogenized PBS aliquots were pooled into three arbitrarily sampled groups (arbitrary named P1, P2, and P7) to maximize sequencing efficiency with 4–6 samples each (Table 1). Pooled samples were filtered using Millex-HV 0.45 μm SLHV0135L (Merck, German) and sacarose gradient by ultracentrifugation at 35,000 g, 4°C for 90 min was conducted to separate the enriched viral bottom fraction. Supernatants were discarded and the remaining 0.2 ml were digested with DNases (Promega and Epicentre, United States), RNase A (Ambion, United States), and Benzonase (Sigma, United States). The QIAamp MinElute Virus Spin Kit (QIAGEN, Germany) was used for viral nucleic acid extraction (DNA + RNA), following the manufacturer’s protocol with slight modifications to improve the recovery of total nucleic acids in accordance to the CDC protocol methodology (Ondov et al., 2011): (i) the “Carrier RNA” was not used in the AL Buffer; (ii) the protease was resuspended with AVE Buffer, instead of Protease Resuspension Buffer; (iii) the washing step with AW1 was not performed; and (iv) the final elution was done in 20 μl of ultra-pure water. The material was then subjected to a complementary DNA (cDNA) synthesis using the Superscript III First Strand Synthesis Supermix Kit (Thermo Fisher Scientific, United States). The second strand cDNA synthesis was performed using Klenow fragment 3′-5′ exo (New England Biolabs Inc., United States), following the manufacturer’s instructions. Sequencing libraries were constructed using mixed DNA (DNA + cDNA) with Nextera XT DNA Library Preparation Kit (Illumina, United States), following the standard protocol. In a MiSeq V2 500-cycle cartridge (Illumina), 4 pM of each library were loaded and massive sequencing was conducted on the Illumina MiSeq platform of the Department of Genetics of UFRJ.
Bioinformatic analysis
Sequencing data were processed with a custom pipeline. Raw sequencing reads were filtered with Fastp v.0.20.1 (Kearse et al., 2012), removing short reads (<50 bp) and low-quality (Phred score < 30) bases. Reads for each sample were then de novo assembled with the Meta-spades software v.3.15.3 (Peter et al., 2000). Diamond v.2.0.14 (Edgar, 2004) was used to taxonomically assign both reads and their respective contigs, with an e-value cut-off of 10−5 and parameters “--more-sensitive” and “--max-target 1.” A similarity search was performed against the complete non-redundant protein (nr) database from NCBI (as of July 27, 2021). Krona v.2.7.1 (Capella-Gutiérrez et al., 2009) was used to generate interactive visualization plots, allowing the fast identification of viral families of interest. To avoid false positive results due to sequencing index-hoping (incorrect assignment of a viral read to a given sample), we considered that samples presenting reads for a viral family with a read count of less than 1% than the highest count among all libraries sequenced in the same MiSeq run to be product of contamination and hence excluded from further analyses.
Given that Anelloviridae was the major viral family identified across the samples (see the section “High-throughput sequencing and taxonomic assignments” in Results), we were prompted to further explore the structure and diversity of the novel viral genomes. The genome sequences were annotated by the Transfer annotations tool in Geneious Prime v.2021.2.2 (Minh et al., 2020) with a 40% similarity threshold, using as template TTV tamarin (NCBI accession number NC_014085.1) from S. oedipus, as well as identifying ORFs through the Find Orf tool in Geneious. Complete genomes were circularized also in Geneious by identifying the GC-rich region in Anelloviridae.
Phylogenetic analysis of novel Anelloviridae genomes
To contextualize the novel genomes within the Anelloviridae diversity, all annotated contigs were searched with the getorf/EMBOSS v.6.6.0.0 (Le et al., 2012) for the presence of ORF1 fragments with at least 1,500 nucleotides. These sequences were aligned to a dataset previously assembled containing all species from the viral family (Nishizawa et al., 1997), composing a comprehensive dataset of 618 ORF1 sequences. These sequences were translated and aligned with MUSCLE v.3.8.425 (Kalyaanamoorthy et al., 2017). The alignment was trimmed with TrimAl v.1.4 (Guindon et al., 2010) with the “--gappyout” option. For quality control, a complete recombination scan was performed with RDP4 (Rodriguez et al., 1990). A maximum likelihood tree was then inferred with IQ-Tree v.2.1.4 (Yang, 1994) under the LG + F + G4 model (R Core Team. R: A, 2018), suggested by ModelFinder (Paradis and Ape, 2019), and with the implementation of the SH-like approximate likelihood ratio test (SH-aLRT; Suchard et al., 2018). A complete list of used sequences, as well as the described alignment and tree are available in Supplementary File S1.
Molecular clock analysis of TTVs from NP
A separate nucleotide dataset comprising all TTVs from NP was assembled and a maximum likelihood tree was inferred under the GTR + F + I + G4 model (Perelman et al., 2011; Baele et al., 2012). The tree and alignment were used for identity analysis with the R software (Baele et al., 2013), using the APE package v.5.5 (Griffiths and Tavare, 1994). As the phylogenetic pattern observed at the genus level was consistent with the ancient within-species diversity model, we were prompted to infer a time-scaled phylogenetic tree for Episolontorquevirus genus, which comprehends TTVs from capuchin (S. nigritus), tamarin (S. oedipus), and the new marmoset (C. penicillata) viruses. This analysis was performed with BEAST v1.10.4 (Drummond et al., 2005). Following Perelman et al. (2011) and (Gernhard, 2008), estimated host divergence dates were used to calibrate internal nodes of the viral tree. A calibration time point between TTV capuchin and other Epsilontorquevirus was set by a normal distribution of mean 19.95 million years ago (mya; 95% CI = 15.66–24.03 mya), while the calibration time point between TTVs tamarin and marmoset was set by a normal distribution of mean 14.89 mya (95% CI = 11.04–18.48 mya). Marginal likelihoods for eight different combinations of tree priors and clock models have been evaluated with path-sampling and stepping-stone sampling algorithms (Drummond et al., 2006; Ferreira and Suchard, 2008). The evaluated tree priors were: speciation yule and coalescent constant, exponential and skyline (Hino and Prasetyo, 2009; Rambaut et al., 2018; Li et al., 2019). Both the strict and uncorrelated relaxed clock models have been assessed (Bédarida et al., 2017; Deb et al., 2021). For each model combination, a Markov chain Monte Carlo run with 50 million generations was executed, sampling every 5,000 steps. Mixing (effective sample size > 200) for all models parameters was verified on Tracer v.1.7.0 (Arze et al., 2021), and a maximum clade credibility tree was inferred with TreeAnnotator v1.10.4 (Arze et al., 2021) for the best fit model. All BEAST xmls, logs and maximum clade credibility are available in Supplementary File S2.
Results
High-throughput sequencing and taxonomic assignments
The HTS protocol generated a total of 8,352,114 reads, harboring a total of 1,487,358 raw reads for P1 library, 3,319,936 for P2, and 3,544,820 for P7. After reads were filtered, a total of 363,161 (24.4%), 696,397 (21%), and 544,058 (15.3%) reads were obtained for libraries P1, P2, and P7, respectively. Reads were taxonomically assigned through similarity search against the complete nr database from NCBI. Viral reads corresponded to approximately 0.142% of the total generated data (1,109 viral reads in P1, 9,200 in P2, and 1,547 in P7). The meta-spades software de novo assembled more than 54,000 contigs, 15,519 contigs assembled in P1, 17,500 in P2, and 21,671 in P7. In the assembled contigs, 312 (0.57% of total) were taxonomically assigned as viruses, from which 78 (0.5%), 186 (1%), and 48 (0.2%) belonged to the sequenced libraries P1, P2, and P7, respectively.
Several viral families have been identified among the evaluated libraries. A complete table with viral taxonomic assignments can be found in Supplementary Table S1. Among the vertebrate viral families identified, Anelloviridae stands out as the one presenting the vast majority of viral reads and contigs (3,816 reads and 164 contigs in total), being hence the main group explored throughout this study. Anelloviridae characterization.
The Anelloviridae family was identified in all evaluated pools, including 216 assigned reads for library P1, 3,514 for P2, and 86 for P7. All of them had a read count greater than the contamination cut-off previously established (n = 35), supporting the bona fide presence of the viral family. A total of 27, 129, and eight assembled contigs were identified as Anelloviridae (Table 2) for P1, P2, and P7, respectively.
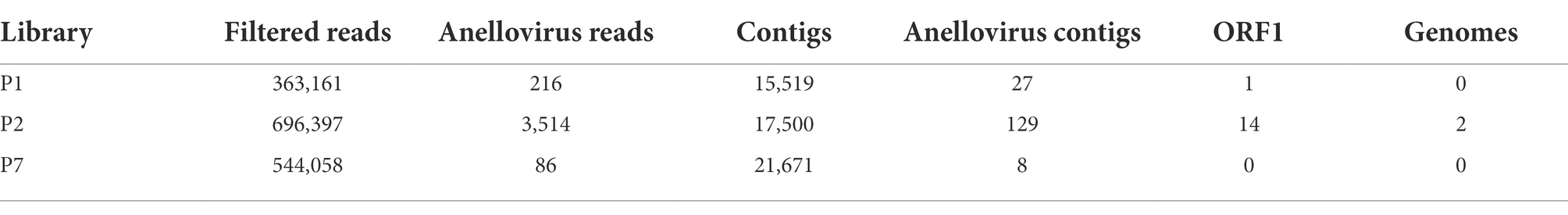
Table 2. Summary of the number of total reads and contigs identified as Anelloviridae and number of ORF1 regions and complete genomes obtained.
To further contextualize the novel sequences within the Anelloviridae diversity, all contigs were searched for the presence of ORF1 fragments with at least 1,500 nucleotides. We found a total of one contig with an ORF1-like in P1 and 14 contigs in P2. No contig with ORF1-like was identified in P7. Among those 15 contigs, two of them possessed a flanking GC-rich region and all other anellovirus ORFs could be completely annotated, confirming the presence of two whole novel genomes of ~3 kb in P2 library (Figure 1; Tables 2, 3).
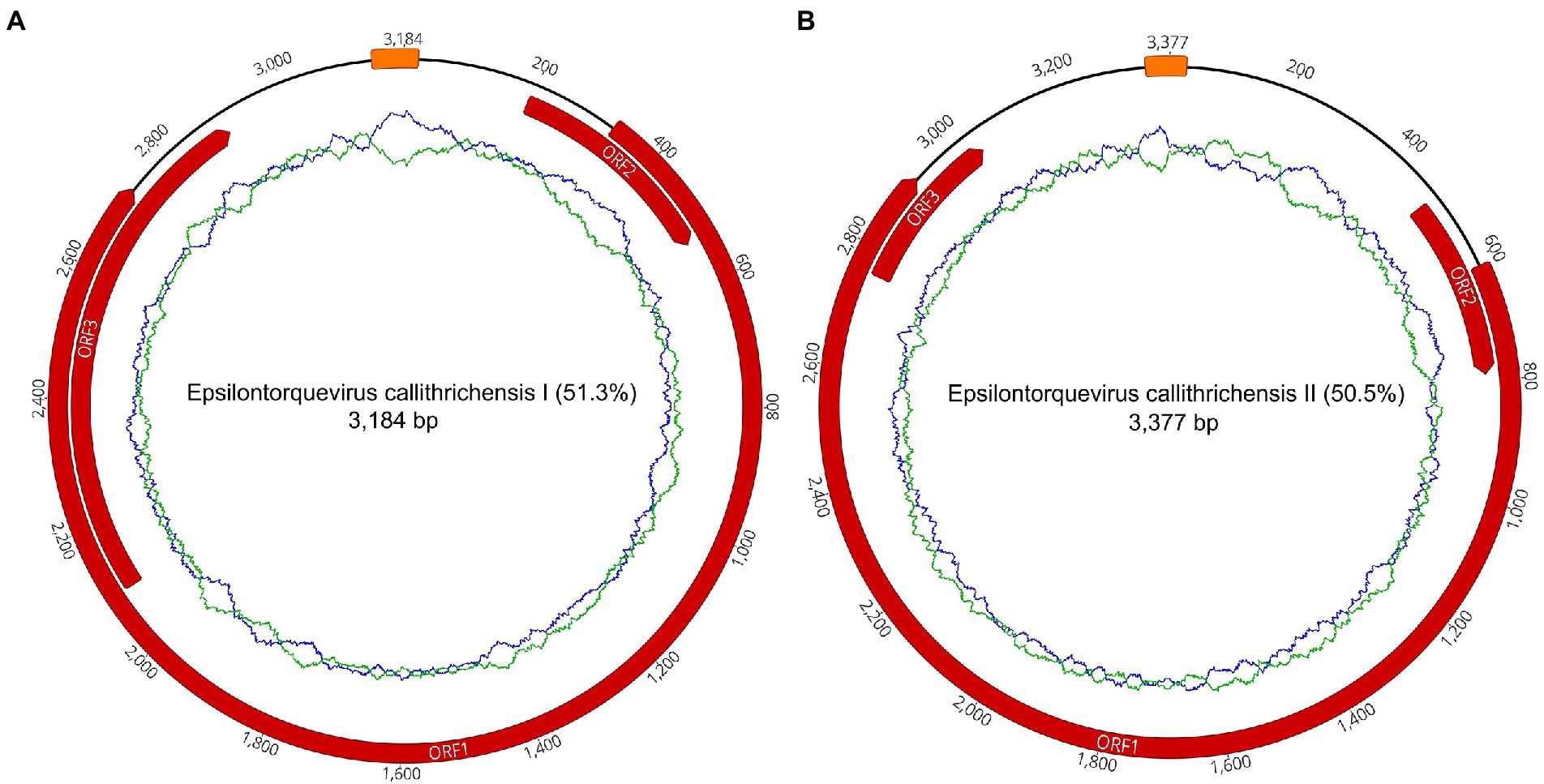
Figure 1. Complete gene maps and genome of the Callithrix penicillata torque teno marmoset virus types 1 and 2. Anelloviruses provisionally named Epsilontorquevirus callithrichensis I (A) and Epsilontorquevirus callithrichensis II (B). Gene annotations are represented in red. Both genomes contained three open reading frames: ORF1, ORF2, and ORF3. The putative non-coding region, Repeat Region, was located between the ORF1 start codon and ORF3 stop codon. The green ring represents the A + T content and the blue ring indicates C + G content.

Table 3. Genome characteristics of the two novel TTV marmosets characterized.
The two novel genomes sequenced were submitted to ICTV and were provisionally named as Epsilontorquevirus callithrichensis (E. callithrichensis) types I and II. The E. callithrichensis I is 3,184 bp long with a guanine-cytosine (GC) content of 51.3%, while E. callithrichensis II is 3,337 bp long with a GC content of 50.5%. Both genomes contain the three major ORFs found in Anelloviridae. The complete ORF1 gene, a fundamental gene to species determination in the family, had an estimated length of 2,403 and 2,313 bp for the E. callithrichensis I and II, respectively, and shared less than 53.49% identity to previously known Epsilontorquevirus. Particularly, the ORF3 gene from E. callithrichensis I had an estimated length of 801 bp, superior to the estimated ORF3 in E. callithrichensis II with 282 bp and to the TTV tamarin (Accession Number NC_014085.1) ORF3, with 497 bp.
A viable ORF4 was not found in any E. callithrichensis, but the Transfer annotations tool in Geneious software found regions with moderate similarity (52.89%, nucleotide positions 2,189–- 2,896) to TTV capuchin ORF4 in E. callithrichensis I. The same region similarity was also found in E. callithrichensis II (49.26%, nucleotide positions 2,356–3,040), as well as regions with moderate similarity to TTV tamarin ORF4 (48.15%, nucleotide positions: 2,978–3,145). Those hypothetical ORF4 found in E. callithrichensis do not possess the typical start codons from other annotated ORFs in Anelloviridae.
Evolutionary analysis
To contextualize the novel Anelloviridae genome sequences, a maximum likelihood phylogeny was reconstructed with a comprehensive dataset, following Varsani et al. (2021). No recombination signal was detected in the dataset. The obtained tree presented phylogenetic consistency with the previously proposed Anelloviridae phylogeny, being all genera inferred as monophyletic with moderate to high support values (SH-aLRT = 67.4–100). The marmoset anelloviruses composed a monophyletic group within Epsilontorquevirus (SH-aLRT = 100), a genus comprehending virus species described for the NP. The external group of Epsilontorquevirus was the Zetatorquevirus genus, which comprehends the TTV douroucouli sampled in Aotus trivirgatus (Ng et al., 2011; Figure 2). This major clade comprised all characterized NP TTVs.
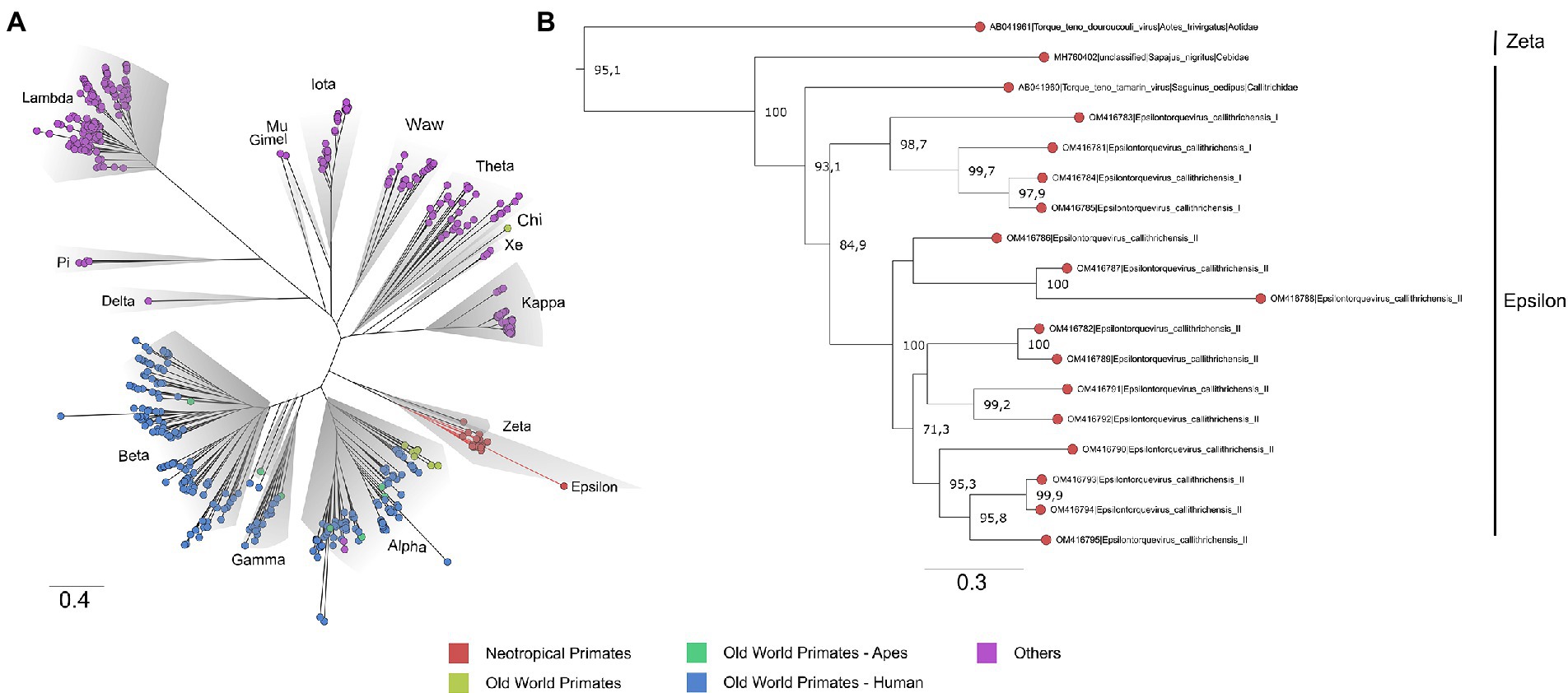
Figure 2. Phylogenetic tree based on complete ORF1 dataset of the Anelloviridae family. (A) The Anelloviridae tips were colored according to the virus host: strains infecting NP have tips highlighted in red, OWP in yellow, and OWP from the Ape lineage in green and Humans in blue. The main representative genera are highlighted in gray. The novel viruses herein described are provisionally named Epsilontorquevirus callithrichensis I and Epsilontorquevirus callithrichensis II and formed a monophyletic group within Epsilontorquevirus. Branches in red are the sequences obtained in this work. (B) The Zetatorquevirus genus is the external group from Epsilontorquevirus, which is divided in three clades according to host species, with great genetic divergence in the Epsilontorquevirus callithrichensis lineage.
To verify whether the newly identified marmoset viruses belonged to a new species or genus, a separate dataset for NP TTVs was assembled and analyzed. Pairwise nucleotide sequence distances were obtained and visualized through heatmap (Figure 3). The ORF1 sequences from the novel viruses clustered in two monophyletic groups, presenting pairwise identities between 55.89 and 59.35% to the previously described Epsilontorquevirus in tamarin. For the TTV capuchin, distances ranged from 53.68 to 56.58%. The ICTV criteria for TTV classification establishes genetic distances thresholds for species (69%; Nishizawa et al., 1997). When compared to the single Zetatorquevirus described, the marmoset anelloviruses demonstrated diminished identity, ranging from 42.19 to 44.96%. The first marmoset anellovirus monophyletic lineage, named E. callithrichensis I, had a pairwise nucleotide sequence distance ranging from 65.68 to 86.92% among its representatives. The second, named E. callithrichensis II, had a pairwise nucleotide sequence distance ranging from 60.66 to 88.89% among its representatives. In this sense, the results presented in this study indicate the existence of two novel viral species from the genus Episolontorquevirus—with a 57.28 to 61.3% pairwise nucleotide distance. Additionally, we report considerable intra-species genetic differences, suggesting that diverse viral strains co-circulate. The pairwise identity matrix of NP Anellovirus can be found in the Supplementary File S1.
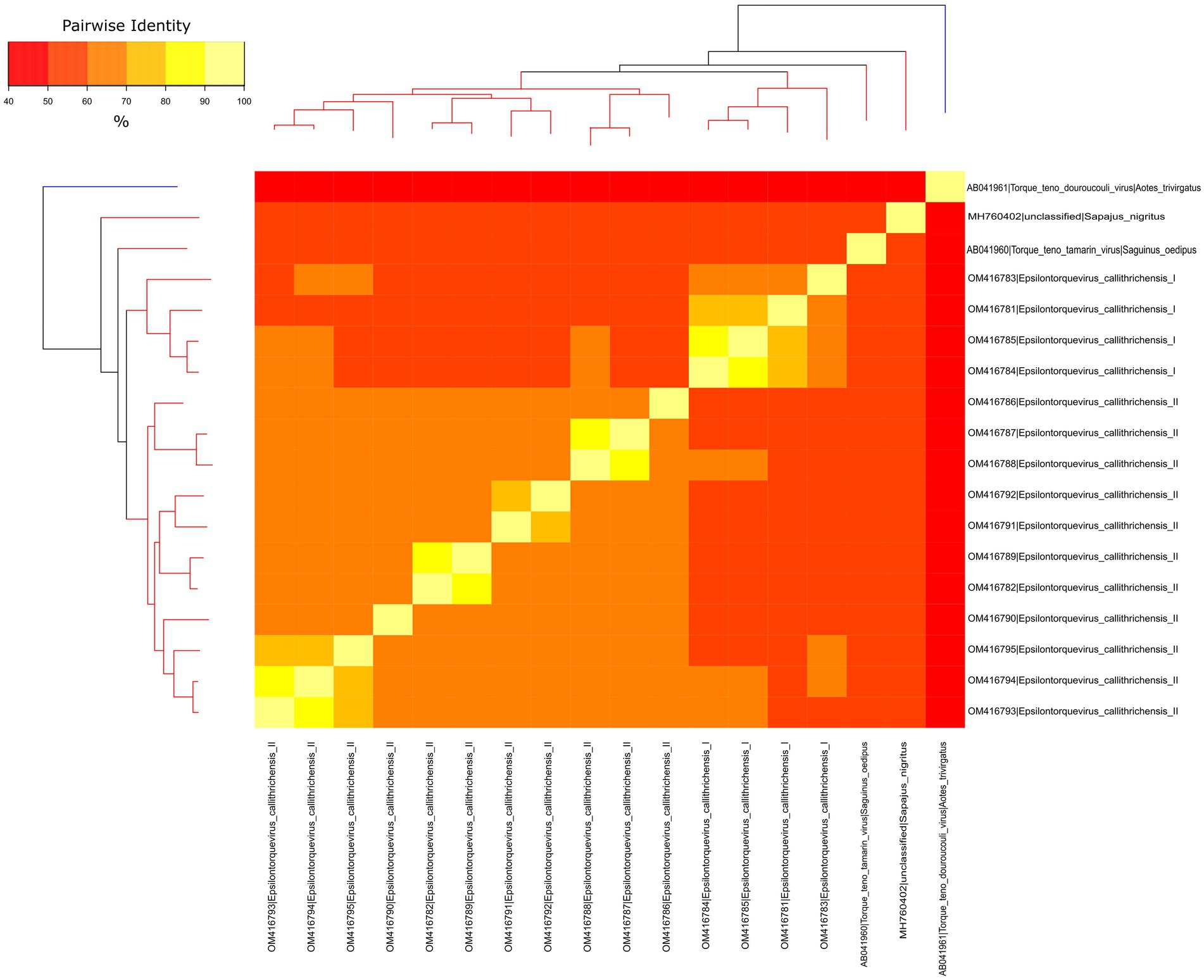
Figure 3. Phylogenetic tree and heatmap displaying the pairwise nucleotide sequence distances of NP anelloviruses. The phylogeny represented both on the left and upper corners is a Maximum likelihood tree of the known anelloviruses in NP. Branches in red are classified as Epsilontorquevirus, while branches in blue are classified as Zetatorquevirus. Sequence distances are indicated by different colors in the central plot. The Epsilontorquevirus was composed of four main lineages, one previously discovered in Sapajus nigritus and one in Saguinus oedipus, with the two newly discovered in Callithrix penicillata in this work. A great genetic diversity may be observed among C. penicillata anelloviruses, such that they can be classified as belonging to two new species, named: Epsilontorquevirus callithrichensis I and Epsilontorquevirus callithrichensis II.
Timescale analyses
The phylogenetic pattern inferred for the genus Epsilontorquevirus (Figure 2B) was consistent with the topology of their host species tree, echoing the within-species diversity followed by co-divergence model of viral diversification. The TTV capuchin was the first lineage to diverge within Epsilontorquevirus, followed by TTV tamarin, which is sister to the clade comprehending all TTVs marmoset. This branching pattern is consistent with the Cebidae molecular phylogeny inferred (Gernhard, 2008). In this sense, we inferred a time-scaled phylogenetic tree for Episolontorquevirus genus using host divergence dates to calibrate internal nodes of the viral tree. Among all models tested, the one with the best statistical fit was characterized by the strict clock and the Yule speciation prior (Supplementary File S2, Path and Stone Sampling table).
Overall, our time scale inference found that the most recent common ancestor of the Episolontorquevirus genus was estimated around 18.7 mya (95% HPD = 15.31–21.88), in accordance with the prior established for the analysis. In addition, the divergence across the TTV tamarin and E. callithrichensis was estimated around 15.48 mya (95% HPD = 12.73–18.17). The divergence between the E. callithrichensis I and E. callithrichensis II was estimated around 13.1 mya (95% HPD = 10.73–15.50 mya). The ancestor for E. callithrichensis I was dated around 7.9 mya (95% HPD = 6.37–9.54 mya) and the one for E. callithrichensis II around 9.6 mya (95% HPD = 7.71–11.2 mya) (Figure 4).
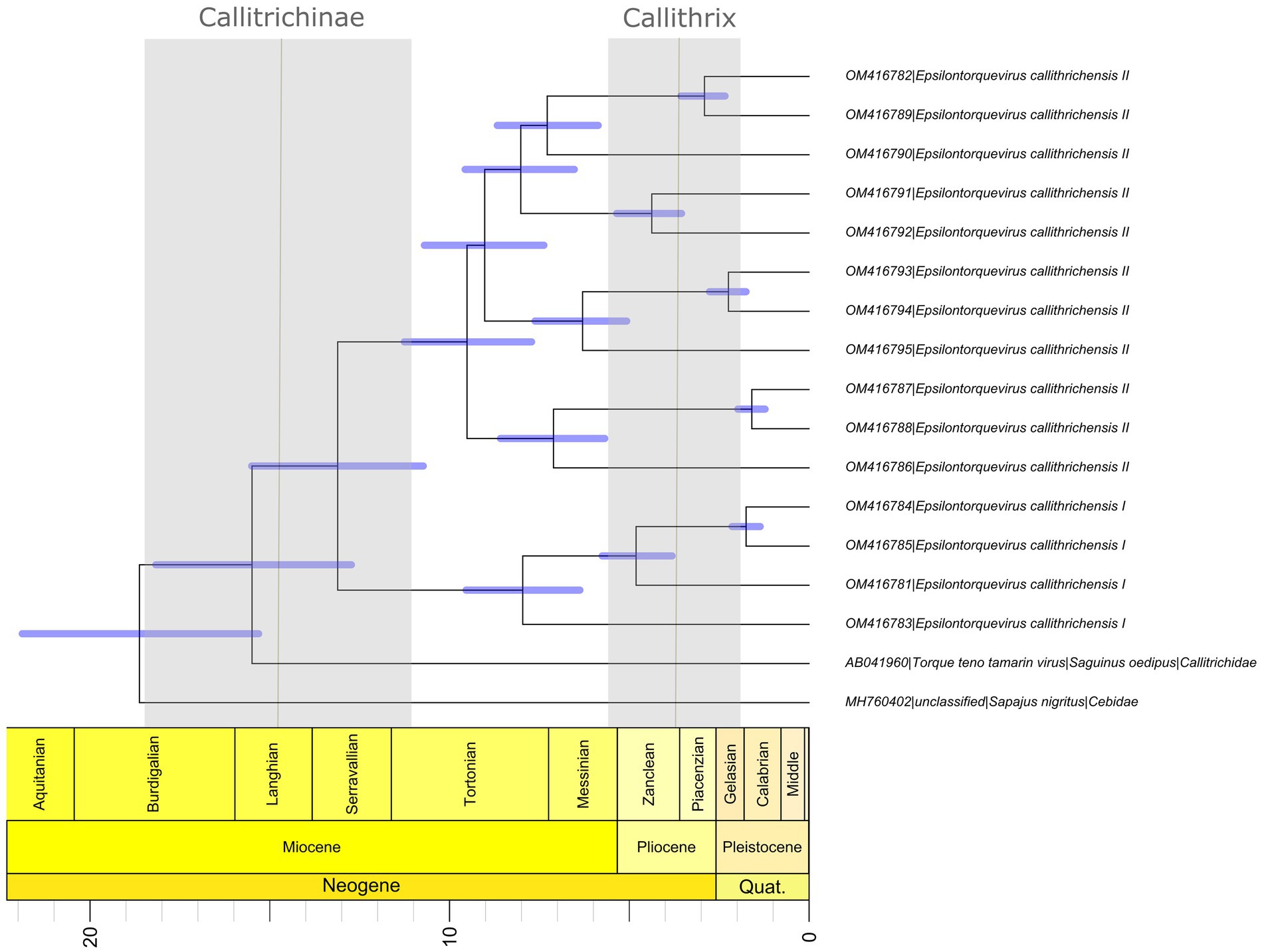
Figure 4. Divergence time estimates for Epsilontorquevirus based on host divergence times used for calibrating the viral molecular clock. Branch lengths are proportional to divergence times and blue bars represent the 95% HPD interval of virus divergence times. The black line represents the divergence times of Callithrix and Callitrichidae, while the blue columns represent its 95% HPD interval, as proposed by Perelman et al. (2011).
Discussion
With the development of HTS protocols to explore the diversity of existing viruses, previous works have identified new anelloviruses species in wildlife—bats, rodents (Niewiadomska and Gifford, 2013), seals (Weber et al., 2020), wild felids (Noppornpanth et al., 2001), canids (Pujol et al., 2005), and non-human primates (Okamoto et al., 2000; Hon et al., 2008; Holmes, 2010; Ng et al., 2011; Sawaswong et al., 2019; dos Santos et al., 2020). In this work, we sequenced and characterized two novel Anelloviridae species genomes in NP, increasing the host range for this family. To the best of our knowledge, this is the first report of this viral family in the black-tuffed marmoset (C. penicillata; Okamoto et al., 2000; Ng et al., 2011; de Souza et al., 2018), the known diversity in NP is yet remarkably limited. Until the present study, only two studies identified anelloviruses (Ng et al., 2011; dos Santos et al., 2020), though no effort to characterize their evolutionary dynamics has been presented. Hence, this is the first attempt to investigate in depth the evolution of novel NP anellovirus.
The anelloviruses sequenced in this work were unique to the known NP Anelloviridae diversity, comprehending two novel species by the ICTV species criterion (Figures 1, 2; Table 3). Both viruses’ complete genomes had the expected ORFs found in this viral family: the putative capsid protein and replication-associated protein (ORF1) and the ORF2 protein (75), as well as the putative ORF3. By particularly looking at some regions of the novel genomes, we could identify regions similar to TTV tamarin and TTV capuchin ORF4, though with numerous stop codons and frameshift mutations, implying lack of transcriptional function. This suggests ORF4 is not fundamental for E. callithrichensis infection and shows an instance of genomic structure plasticity in ssDNA viruses. The adoption of models and cell systems to study anelloviruses infections in vitro shall improve the knowledge regarding the function and structure of its proteins, deeply scarce at the present time.
Phylogenetic analysis of the new genome sequences suggests a topological pattern consistent with a previously proposed model for the diversification of multiple viral families, the within-species diversity followed by co-divergence model (Buchfink et al., 2014; Nurk et al., 2017; Chen et al., 2018; D’arc et al., 2020). Under this model, viral lineages diversify either alongside their host species or through a speciation process within a single host, occasionally caused by niche adaptation to specific cell types or tissues. This process culminates in a viral phylogenetic pattern where multiple clades reflect the host phylogeny, as is the case of primate TTVs (Ng et al., 2011). We explored the consequences of this model to calibrate the molecular clock and obtain, for the first time, a time-scaled phylogenetic tree for NP TTVs (Figure 4). This analysis suggests that the lineages that lead to both species described in this study diverged around 13.1 mya, while the time for the ancestors of E. callithrichensis I and II were dated around 7.9 and 9.6 mya, respectively. Both dates are much older than the origins of Callithrix, implying that these viral lineages circulated among the ancestors of Callitrichinae (around 14.9 mya; Gernhard, 2008). We thus hypothesize that its descendant extant species may harbor TTVs related to the described viral lineages (Figure 5). A central limitation of the presented analysis lies in the limited sampling of NP TTVs. Increasing the availability of anellovirus genomes from diverse NP species will allow a deeper evaluation of their diversification dynamics and a further verification of our hypothesis.
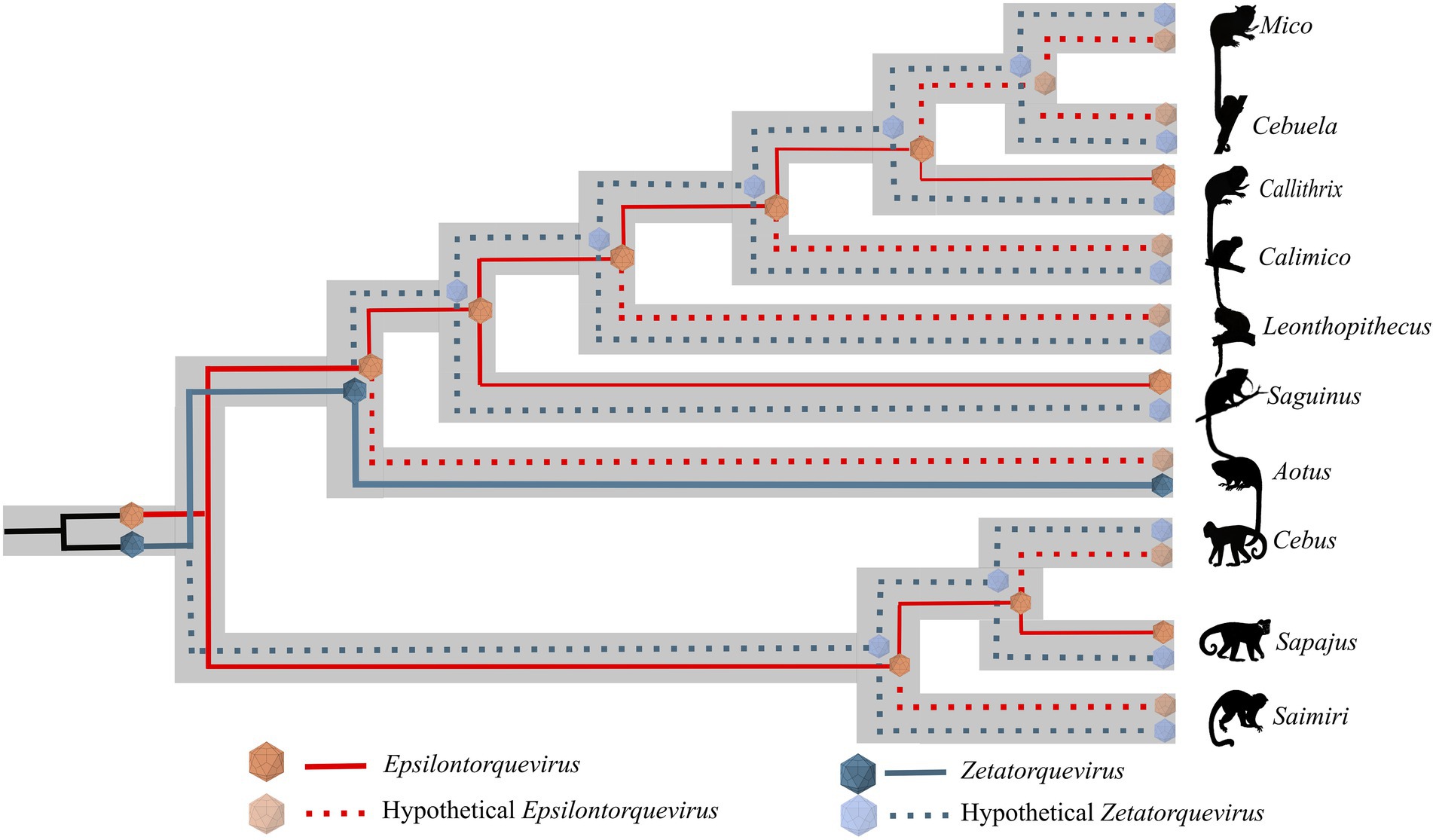
Figure 5. Schematic model of ancient within-species diversity model followed by co-divergence in NP from the Cebidae family. Continuous lines represent strains already sampled and dashed lines represent putative strains that might explain more likely ancient ancestors. The Epsilontorquevirus genus is represented by red lines, Zetatorquevirus by blue lines. We hypothesize the existence of an ancestral Anelloviridae in the Cebidae ancestor that posteriorly originated Epsilontorquevirus and Zetatorquevirus in a mirrored evolutionary process.
We observed a high genetic diversity between the E. callithrichensis characterized herein (Figure 3), a phenomenon already described in anelloviruses due to their high mutation rate (76–78). The presence of multiple lineages co-infecting a host has already been observed as three distinct Anelloviridae genera were described in humans (79). The same pattern is present in OWP, as chimpanzees are infected by Alpha, Beta, and Gammatorquevirus (Inami et al., 2000; Okamoto et al., 2000; Ng et al., 2011; de Souza et al., 2018). Once the NP Anelloviridae diversity is further explored, and the diversity of Epsilontorquevirus and Zetatorquevirus is thoroughly addressed, the pattern of coinfection by multiple TTV strains may be further verified in NP (Figure 5).
While previous observations of co-divergence patterns have been made for other TTVs (Noppornpanth et al., 2001; Ng et al., 2011), no clear evolutionary model has been proposed to assess TTV diversity. Although this study focused on NP TTVs, we argue that the within-species diversity followed by co-divergence model might explain the drivers of diversification for the whole Anelloviridae family. Further characterization of TTVs diversity in non-model organisms through metagenomics might play a key role in unraveling the validity of the proposed model. Finally, this study showcases an instance of the importance of sampling wildlife viruses to obtain a better characterization of the long-term evolutionary processes driving the diversification of viral families.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, OM416781–OM416795; https://github.com/matheus-cosentino/Cosentino_2022_Epsilontorquevirus.
Ethics statement
The animal study was reviewed and approved by Ethics Committee on the Use of Animals of Universidade Federal do Rio de Janeiro (reference number 037/14).
Author contributions
The idea of characterizing anelloviruses originated from MD’s work on viral metagenomics. CD, AS, and MT were responsible for sample collection. Sequencing data by MD. Analysis and curation of generated data by MC, MD, FM, LC, RM, AC, FS, TM, and GM. Genome annotations, phylogenetic analyses, molecular dating, and images by MC, MD, and FM. Manuscript ideas and development by all authors. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the Conselho Nacional de Desenvolvimento Científico e Tecnológico/CNPq (grant 313005/2020–6 to ASa) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro/FAPERJ (grants E-26/112.647/2012 to MS and E26/202.738/2018, E26/210.122/2018, and E26/211.040/2019 to ASa) for funding this study.
Acknowledgments
We thank to all the workers of the Centro de Primatologia de Brasília, Universidade de Brasília (Distrito Federal, Brazil) for assistance with sample collection and to the fantastic LDDV members, in special to Déa Mota, for the wonderful teamwork. We are also grateful to Carolina Furtado and Nicole Scherer from Instituto Nacional de Câncer Jośe de Alencar (Rio de Janeiro, Brazil) for assistance with HTS and analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1002963/full#supplementary-material
References
Arze, C. A., Springer, S., Dudas, G., Patel, S., Bhattacharyya, A., Swaminathan, H., et al. (2021). Global genome analysis reveals a vast and dynamic anellovirus landscape within the human virome. Cell Host Microbe 29, 1305–1315.e6. doi: 10.1016/j.chom.2021.07.001
Atoni, E., Zhao, L., Karungu, S., Obanda, V., Agwanda, B., Xia, H., et al. (2019). The discovery and global distribution of novel mosquito-associated viruses in the last decade (2007-2017). Rev. Med. Virol. 29:e2079. doi: 10.1002/rmv.2079
Baele, G., Lemey, P., Bedford, T., Rambaut, A., Suchard, M. A., and Alekseyenko, A. V. (2012). Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 29, 2157–2167. doi: 10.1093/molbev/mss084
Baele, G., Li, W. L. S., Drummond, A. J., Suchard, M. A., and Lemey, P. (2013). Accurate model selection of relaxed molecular clocks in Bayesian phylogenetics. Mol. Biol. Evol. 30, 239–243. doi: 10.1093/molbev/mss243
Bandoo, R. A., Bautista, J., Lund, M., Newkirk, E., Squires, J., Varsani, A., et al. (2021). Identification of novel circovirus and anelloviruses from wolverines using a non-invasive faecal sampling approach. Infect. Genet. Evol. 93:104914. doi: 10.1016/j.meegid.2021.104914
Bédarida, S., Dussol, B., Signoli, M., and Biagini, P. (2017). Analysis of Anelloviridae sequences characterized from serial human and animal biological samples. Infect. Genet. Evol. 53, 89–93. doi: 10.1016/j.meegid.2017.05.017
Bendinelli, M., Pistello, M., Maggi, F., Fornai, C., Freer, G., and Vatteroni, M. L. (2001). Molecular properties, biology, and clinical implications of TT virus, a recently identified widespread infectious agent of humans. Clin. Microbiol. Rev. 14, 98–113. doi: 10.1128/CMR.14.1.98-113.2001
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Buck, C. B., van Doorslaer, K., Peretti, A., Geoghegan, E. M., Tisza, M. J., An, P., et al. (2016). The ancient evolutionary history of polyomaviruses. PLoS Pathog. 12:e1005574. doi: 10.1371/journal.ppat.1005574
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
D’arc, M., Moreira, F. R. R., Dias, C. A., Souza, A. R., Seuánez, H. N., Soares, M. A., et al. (2020). The characterization of two novel neotropical primate papillomaviruses supports the ancient within-species diversity model. Virus Evol. 6, 1–12. doi: 10.1093/ve/veaa036
Datta, S. (2015). Next-generation sequencing in clinical virology: Discovery of new viruses. World J. Virol. 4, 265–276. doi: 10.5501/wjv.v4.i3.265
de Sabato, L., Lelli, D., Faccin, F., Canziani, S., di Bartolo, I., Vaccari, G., et al. (2019). Full genome characterization of two novel Alpha-coronavirus species from Italian bats. Virus Res. 260, 60–66. doi: 10.1016/j.virusres.2018.11.007
de Souza, W. M., Fumagalli, M. J., de Araujo, J., Sabino-Santos, G., Maia, F. G. M., Romeiro, M. F., et al. (2018). Discovery of novel anelloviruses in small mammals expands the host range and diversity of the Anelloviridae. Virology 514, 9–17. doi: 10.1016/j.virol.2017.11.001
Deb, B., Uddin, A., and Chakraborty, S. (2021). Composition, codon usage pattern, protein properties, and influencing factors in the genomes of members of the family Anelloviridae. Arch. Virol. 166, 461–474. doi: 10.1007/s00705-020-04890-2
Devaux, C. A., Mediannikov, O., Medkour, H., and Raoult, D. (2019). Infectious disease risk across the growing human-non human primate interface: a review of the evidence. Front. Public Health 7:305. doi: 10.3389/fpubh.2019.00305
dos Santos, R. N., Campos, F. S., Finoketti, F., dos Santos, A. C., Campos, A. A. S., Wagner, P. G. C., et al. (2020). Viral diversity in oral cavity from Sapajus nigritus by metagenomic analyses. Braz. J. Microbiol. 51, 1941–1951. doi: 10.1007/s42770-020-00350-w
Drummond, A. J., Ho, S. Y. W., Phillips, M. J., and Rambaut, A. (2006). Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88–e710. doi: 10.1371/journal.pbio.0040088
Drummond, A. J., Rambaut, A., Shapiro, B., and Pybus, O. G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192. doi: 10.1093/molbev/msi103
Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ferreira, M. A. R., and Suchard, M. A. (2008). Bayesian analysis of elapsed times in continuous-time Markov chains. Can. J. Stat. 36, 355–368. doi: 10.1002/cjs.5550360302
Freer, G., Maggi, F., Pifferi, M., di Cicco, M. E., Peroni, D. G., and Pistello, M. (2018). The virome and its major component, Anellovirus, a convoluted system molding human immune defenses and possibly affecting the development of asthma and respiratory diseases in childhood. Front. Microbiol. 9:686. doi: 10.3389/fmicb.2018.00686
Geoghegan, J. L., and Holmes, E. C. (2017). Predicting virus emergence amid evolutionary noise. Open Biol. 7:170189. doi: 10.1098/rsob.170189
Gernhard, T. (2008). The conditioned reconstructed process. J. Theor. Biol. 253, 769–778. doi: 10.1016/j.jtbi.2008.04.005
Greninger, A. L. (2018). A decade of RNA virus metagenomics is (not) enough. Virus Res. 244, 218–229. doi: 10.1016/j.virusres.2017.10.014
Griffiths, R. C., and Tavare, S. (1994). Sampling theory for neutral alleles in a varying environment. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 344, 403–410. doi: 10.1098/rstb.1994.0079
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hino, S., and Miyata, H. (2007). Torque teno virus (TTV): Current status. Rev. Med. Virol. 17, 45–57. doi: 10.1002/rmv.524
Hino, S., and Prasetyo, A. A. (2009). Relationship of torque teno virus to chicken anemia virus. Curr. Top. Microbiol. Immunol. 331, 117–130. doi: 10.1007/978-3-540-70972-5_8
Holmes, E. C. (2010). The comparative genomics of viral emergence. Proc. Natl. Acad. Sci. U. S. A. 107, 1742–1746. doi: 10.1073/pnas.0906193106
Hon, C.-C., Lam, T.-Y., Shi, Z.-L., Drummond, A. J., Yip, C.-W., Zeng, F., et al. (2008). Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 82, 1819–1826. doi: 10.1128/jvi.01926-07
Hrazdilová, K., Slaninková, E., Brožová, K., Modrý, D., Vodička, R., and Celer, V. (2016). New species of Torque Teno miniviruses infecting gorillas and chimpanzees. Virology 487, 207–214. doi: 10.1016/j.virol.2015.10.016
Inami, T., Obara, T., Moriyama, M., Arakawa, Y., and Abe, K. (2000). Full-length nucleotide sequence of a simian TT virus isolate obtained from a chimpanzee: Evidence for a new TT virus-like species. Virology 277, 330–335. doi: 10.1006/viro.2000.0621
IUCN/SSC Primate Specialist Group (2021). Primates Diversity by Region. Available at: http://www.primate-sg.org/primate_diversity_by_region/
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kohl, C., Brinkmann, A., Dabrowski, P. W., Radonić, A., Nitsche, A., and Kurth, A. (2015). Protocol for metagenomic virus detection in clinical specimens. Emerg. Infect. Dis. 21, 48–57. doi: 10.3201/eid2101.140766
Kraberger, S., Serieys, L. E., Richet, C., Fountain-Jones, N. M., Baele, G., Bishop, J. M., et al. (2021). Complex evolutionary history of felid anelloviruses. Virology 562, 176–189. doi: 10.1016/j.virol.2021.07.013
Lam, T. T. Y., Jia, N., Zhang, Y. W., Shum, M. H. H., Jiang, J. F., Zhu, H. C., et al. (2020). Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 583, 282–285. doi: 10.1038/s41586-020-2169-0
Le, S. Q., Dang, C. C., and Gascuel, O. (2012). Modeling protein evolution with several amino acid replacement matrices depending on site rates. Mol. Biol. Evol. 29, 2921–2936. doi: 10.1093/molbev/mss112
Li, G., Zhang, W., Wang, R., Xing, G., Wang, S., Ji, X., et al. (2019). Genetic analysis and evolutionary changes of the torque teno sus virus. Int. J. Mol. Sci. 20:2881. doi: 10.3390/ijms20122881
McGeoch, D. J., Rixon, F. J., and Davison, A. J. (2006). Topics in herpesvirus genomics and evolution. Virus Res. 117, 90–104. doi: 10.1016/j.virusres.2006.01.002
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Muniz, C. P., Cavalcante, L. T. F., Dudley, D. M., Pissinatti, A., O’Connor, D. H., Santos, A. F., et al. (2018). First complete genome sequence of a simian foamy virus infecting the neotropical primate Brachyteles arachnoides. Microbiol. Res. Announc. 7:e00839-18. doi: 10.1128/mra.00839-18
Ng, T. F. F., Wheeler, E., Greig, D., Waltzek, T. B., Gulland, F., and Breitbart, M. (2011). Metagenomic identification of a novel anellovirus in Pacific harbor seal (Phoca vitulina richardsii) lung samples and its detection in samples from multiple years. J. Gen. Virol. 92, 1318–1323. doi: 10.1099/vir.0.029678-0
Ng, T. F. F., Zhang, W., Sachsenrö Der, J., Kondov, N. O., da Costa, A. C., Vega, E., et al. (2015). A diverse group of small circular ssDNA viral genomes in human and non-human primate stools. Virus. Evolution 1:vev017. doi: 10.1093/ve/vev017
Niewiadomska, A. M., and Gifford, R. J. (2013). The extraordinary evolutionary history of the reticuloendotheliosis viruses. PLoS Biol. 11:e1001642. doi: 10.1371/journal.pbio.1001642
Ninomiya, M., Takahashi, M., Hoshino, Y., Ichiyama, K., Simmonds, P., and Okamoto, H. (2009). Analysis of the entire genomes of Torque teno midi virus variants in chimpanzees: Infrequent cross-species infection between humans and chimpanzees. J. Gen. Virol. 90, 347–358. doi: 10.1099/vir.0.007385-0
Nishizawa, T., Okamoto, H., Konishi, K., Yoshizawa, H., Miyakawa, Y., and Mayumi, M. (1997). A novel DNA virus (TTV) associated with elevated transaminase levels in Posttransfusion Hepatitis of Unknown Etiology. Biochem. Biophys. Res. Commun. 241, 92–97. doi: 10.1006/bbrc.1997.7765
Noppornpanth, S, Chinchai, T, Ratanakorn, P, and Poovorawan, Y. (2001). TT Virus Infection in Gibbons. vol. 63.
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Okamoto, H., Nishizawa, T., Tawara, A., Peng, Y., Takahashi, M., Kishimoto, J., et al. (2000). Species-specific TT viruses in humans and nonhuman primates and their phylogenetic relatedness. Virology 277, 368–378. doi: 10.1006/viro.2000.0588
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385
Paradis, E., and Ape, S. K. (2019). 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Perelman, P., Johnson, W. E., Roos, C., Seuánez, H. N., Horvath, J. E., Moreira, M. A. M., et al. (2011). A molecular phylogeny of living primates. PLoS Genet. 7:e1001342. doi: 10.1371/journal.pgen.1001342
Pérez-Losada, M., Arenas, M., Galán, J. C., Bracho, M. A., Hillung, J., García-González, N., et al. (2020). High-throughput sequencing (HTS) for the analysis of viral populations. Infect. Genet. Evol. 80:104208. doi: 10.1016/j.meegid.2020.104208
Peter, R., Ian, L., and Bleasby, A. (2000). EMBOSS: The European MolecularBiology Open Software Suite. Trends Genet. 16, 276–277. doi: 10.1016/S0168-9525
Pujol, F. H., Mejías, E., Loureiro, C. L., Ludert, J. E., Liprandi, F., and Pernalete, J. M. (2005). Infection with transfusion-transmitted virus (TTV) in humans and other primates in Venezuela. Ann. Trop. Med. Parasitol. 99, 173–180. doi: 10.1179/136485905X24193
R Core Team. R: A (2018). Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna.
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Rodriguez, F, Olivert, JL, Marfn, A, and Med1na, J.R. (1990). ~t: The General Stochastic Model of Nucleotide Substitution. vol. 142.
Sawaswong, V., Fahsbender, E., Altan, E., Kemthong, T., Deng, X., Malaivijitnond, S., et al. (2019). High diversity and novel enteric viruses in fecal viromes of healthy wild and captive Thai cynomolgus macaques (Macaca fascicularis). Viruses 11:971. doi: 10.3390/v11100971
Shi, M., Lin, X. D., Tian, J. H., Chen, L. J., Chen, X., Li, C. X., et al. (2016). Redefining the invertebrate RNA virosphere. Nature 540, 539–543. doi: 10.1038/nature20167
Silvestre, R. V. D., de Souza, A. J. S., Júnior, E. C. S., Silva, A. K., de Mello, W. A., Nunes, M. R. T., et al. (2016). First New World primate papillomavirus identification in the Atlantic Forest, Brazil: Alouatta guariba papillomavirus 1. Genome Announc. 4:e00725-16. doi: 10.1128/genomeA.00725-16
Smeele, Z. E., Burns, J. M., van Doorsaler, K., Fontenele, R. S., Waits, K., Stainton, D., et al. (2018). Diverse papillomaviruses identified in weddell seals. J. Gen. Virol. 99, 549–557. doi: 10.1099/jgv.0.001028
Suchard, M. A., Lemey, P., Baele, G., Ayres, D. L., Drummond, A. J., and Rambaut, A. (2018). Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus. Evolution 4:vey016. doi: 10.1093/ve/vey016
Troncoso, L. L., Muniz, C. P., Siqueira, J. D., Curty, G., Schrago, C. G., Augusto, A., et al. (2015). Characterization and comparative analysis of a simian foamy virus complete genome isolated from Brazilian capuchin monkeys. Virus Res. 208, 1–6. doi: 10.1016/j.virusres.2015.05.022
Varsani, A., Opriessnig, T., Celer, V., Maggi, F., Okamoto, H., Blomström, A. L., et al. (2021). Taxonomic update for mammalian anelloviruses (family Anelloviridae). Arch. Virol. 166, 2943–2953. doi: 10.1007/s00705-021-05192-x
Weber, M. N., Mosena, A. C. S., da Silva, M. S., Canova, R., de Lorenzo, C., Olegário, J. C., et al. (2020). Virome of crab-eating (Cerdocyon thous) and pampas foxes (Lycalopex gymnocercus) from southern Brazil and Uruguay. Infect. Genet. Evol. 85:104421. doi: 10.1016/j.meegid.2020.104421
Wei, X., Chen, Y., Duan, G., Holmes, E. C., and Cui, J. (2019). A reptilian endogenous foamy virus sheds light on the early evolution of retroviruses. Virus. Evolution 5:vez001. doi: 10.1093/ve/vez001
Yang, Z. (1994). Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39, 306–314. doi: 10.1007/BF00160154
Keywords: metagenomic, marmoset, co-divergence, ssDNA viruses, virus evolution
Citation: Cosentino MAC, D’arc M, Moreira FRR, Cavalcante LTF, Mouta R, Coimbra A, Schiffler FB, Miranda TS, Medeiros G, Dias CA, Souza AR, Tavares MCH, Tanuri A, Soares MA and Santos AFA (2022) Discovery of two novel Torque Teno viruses in Callithrix penicillata provides insights on Anelloviridae diversification dynamics. Front. Microbiol. 13:1002963. doi: 10.3389/fmicb.2022.1002963
Edited by:
Peirong Jiao, South China Agricultural University, ChinaReviewed by:
Hudson Alves Pinto, Minas Gerais State University, BrazilGiovanni Franzo, University of Padua, Italy
Copyright © 2022 Cosentino, D’arc, Moreira, Cavalcante, Mouta, Coimbra, Schiffler, Miranda, Medeiros, Dias, Souza, Tavares, Tanuri, Soares and Santos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: André Felipe Andrade dos Santos, afsantos.ufrj@gmail.com