
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 12 January 2022
Sec. Microbe and Virus Interactions with Plants
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.815911
This article is part of the Research TopicPopulation and Comparative Genomics of Plant Pathogenic BacteriaView all 16 articles
 Prem P. Kandel1*
Prem P. Kandel1* Marina Naumova1Chad Fautt1
Marina Naumova1Chad Fautt1 Ravikumar R. Patel2
Ravikumar R. Patel2 Lindsay R. Triplett2
Lindsay R. Triplett2 Kevin L. Hockett1,3*
Kevin L. Hockett1,3*
Bacterial toxin-antitoxin (TA) systems consist of two or more adjacent genes, encoding a toxin and an antitoxin. TA systems are implicated in evolutionary and physiological functions including genome maintenance, antibiotics persistence, phage defense, and virulence. Eight classes of TA systems have been described, based on the mechanism of toxin neutralization by the antitoxin. Although studied well in model species of clinical significance, little is known about the TA system abundance and diversity, and their potential roles in stress tolerance and virulence of plant pathogens. In this study, we screened the genomes of 339 strains representing the genetic and lifestyle diversity of the Pseudomonas syringae species complex for TA systems. Using bioinformatic search and prediction tools, including SLING, BLAST, HMMER, TADB2.0, and T1TAdb, we show that P. syringae strains encode 26 different families of TA systems targeting diverse cellular functions. TA systems in this species are almost exclusively type II. We predicted a median of 15 TA systems per genome, and we identified six type II TA families that are found in more than 80% of strains, while others are more sporadic. The majority of predicted TA genes are chromosomally encoded. Further functional characterization of the predicted TA systems could reveal how these widely prevalent gene modules potentially impact P. syringae ecology, virulence, and disease management practices.
The Pseudomonas syringae species complex consists of a monophyletic group of plant-pathogenic, plant-commensal, and environmental isolates within the major clade of the genus Pseudomonas (Xin et al., 2018). The strains are grouped into 13 phylogroups by multilocus sequence analysis (Berge et al., 2014) and phylogenomics (Gomila et al., 2017; Dillon et al., 2019a; Newberry et al., 2019). Based on host range and symptomatology, P. syringae strains have been also classified into over 60 pathovars that together affect almost all known crop plants of economic importance. Periodic outbreaks, including a recent devastating epidemic of bacterial canker in kiwifruit in New Zealand (McCann et al., 2013), establish these plant pathogens as a serious threat to global food production. In addition to being a pathogen, P. syringae survives well in the environment and is commonly found in precipitation and natural bodies of water (Morris et al., 2008, 2013). Epiphytic and environmental populations face extreme fluctuations in UV light, temperature, humidity, and nutrient availability, as well as antimicrobial compounds produced by plants and their microbiota (Hirano and Upper, 2000; Lindow and Brandl, 2003; Craig et al., 2021). Previous studies have reported induction of viable but not culturable (VBNC) state and a concomitant alteration in the gene expression profile upon exposure of P. syringae cells to conditions that mimic oxidative burst of early stage plant infection (Mock et al., 2015; Postnikova et al., 2015). We recently reported that a plant pathogenic P. syringae can survive bacteriocin and antibiotics exposure by entering another dormant, tolerant state known as persistence (Kandel et al., 2020; Patel et al., 2021). Although several stress response pathways have been identified, it is still not well understood how P. syringae can cope with environmental stresses in epiphytic and free-living conditions or maintain virulence genes in the extended absence of host selection pressure.
Toxin-antitoxin (TA) systems are small, self-regulating genetic elements, comprised of two to three adjacent genes. The toxin component interferes with an essential cellular function such as translation, DNA replication, and cell wall biosynthesis, whereas the cognate antitoxin neutralizes toxin activity (Unterholzner et al., 2013b). Because the antitoxin is typically less stable than the toxin, conditions that affect antitoxin expression or degradation can increase the pool of unbound toxin, inhibiting cellular processes (Unterholzner et al., 2013b). TA systems have been broadly classified into eight types depending on the nature of antitoxin and its mode of interaction with the toxin (Song and Wood, 2020b). Most well-studied TA systems are Type II, characterized by protein antitoxins that directly bind and neutralize toxin active sites (unlike the type I and III systems, where antitoxins are non-coding RNAs). TA systems were initially implicated as a plasmid maintenance mechanism that suppressed the growth of plasmid-free daughter cells in the absence of antitoxin expression (Ogura and Hiraga, 1983). Subsequent studies have identified TA genes in bacterial chromosomes, which can similarly prevent loss of genomic islands (Wozniak and Waldor, 2009; Yao et al., 2015). TA loci have been implicated in a wide range of roles in addition to plasmid and genome maintenance, including physiological tolerance to antibiotics (Moyed and Bertrand, 1983), phage defense (Sberro et al., 2013; Song and Wood, 2020a), biofilm formation, and virulence (Wood and Wood, 2016). Recent studies have demonstrated that TA systems can impact the ecology, plasmid maintenance, chemical control tolerance, and virulence capabilities of plant pathogens. For example, in Acidovorax citrulli, which causes bacterial fruit blotch of cucurbits, expression of a TA operon was significantly induced during plant infection (Shavit et al., 2016). In Xylella fastidiosa, overexpression of a TA toxin decreased virulence and increased persistence to copper exposure (Merfa et al., 2016), while in a separate study, deletion of another TA system caused a hypervirulence phenotype suggestive of a regulatory role governing systemic infection (Burbank and Stenger, 2017). In Xanthomonas oryzae pv. oryzicola, the toxin of one TA system functions as a virulence effector delivered into host cells via the type III secretion system (Triplett et al., 2016). Moreover, in Erwinia amylovora, a 10-fold greater tolerance to streptomycin was reported when a TA toxin gene was expressed (Peng et al., 2021). TA systems were also shown to play a significant role in maintenance of P. syringae virulence plasmids (Bardaji et al., 2019). While TA systems have been predicted in several plant pathogenic species, including in Xanthomonas citri and Erwinia amylovora (Martins et al., 2016; Shidore et al., 2019), TA system composition and diversity has not been described in the P. syringae species complex.
In this study, we used genome mining to predict TA systems in 339 P. syrinage strains representing the broad diversity of the species complex, examined their abundance, diversity, and association with strain phylogeny, source of isolation, and virulence determinants. We show that homologs of well-characterized TA systems are ubiquitous and present in multiple copies in P. syringae genomes. We identified six major TA families conserved across species, and others that have more sporadic distributions. Most TA systems occurred in the chromosome and were likely associated to mobile elements and virulence factors. Further experimental characterization of the predicted TA system will reveal their significance in the context of Pseudomonas syringae biology, ecology, and virulence, and can provide important insights on how these bacteria can survive in very diverse and harsh environments. These insights could be important in designing new management options for the pathogens.
We retrieved a set of 2,345 genomes identified as Pseudomonas syringae complex in the NCBI database as of October 2020, and generated a preliminary phylogenetic tree using the evolutionary distance estimation method of andi with default settings (Haubold et al., 2014). From this tree, we manually extracted 357 genomes representing the genetic diversity within the P. syringae complex. We then assessed the completeness of the assembly and annotation with the benchmarking tool BUSCO (Simão et al., 2015), using the Pseudomonadales single-copy gene ortholog dataset. Genomes with a BUSCO score of less than 95% complete genes were filtered out, and 339 genomes were retained for further analysis (Supplementary Table 1).
A phylogenomic tree of the 339 strains was constructed using GToTree v1.5.46 using Gammaproteobacteria gene profiles (Lee, 2019). Sequences of 172 homologous gene sets were used to construct the tree. The clade containing P. fuscovaginae SE-1 was used as an outgroup to root the tree. Clustering of strains with previously known phylogroup designation was used as the basis for assigning phylogroups to unknown strains, after confirming that strains of the same phylogroup clustered together. Together, we included 77, 104, 38, 31, 11, 2, 61, 2, 4, 4, 2 strains from phylogroups 1, 2, 3, 4, 5, 6, 7, 9, 10, 11, and 13, respectively. The P. fuscovaginae group clade was not assigned to any phylogroup. Our genome list did not have any representatives from phylogroups 8 and 12 due to unavailability of quality genomes in the public database.
Type II and IV TA systems were predicted for whole genomes of all 339 strains using SLING 2.0.1 (Horesh et al., 2018), a program that identifies gene pairs based on Hidden Markov Models (HMM) alignments to annotated genes or unannotated putative ORFs, as well as gene pair structural requirements. Nucleotide sequence, annotations, and toxin profile HMMs included with the program were used as inputs with default parameters. SLING was run on Roar high-performance computing platform using 16 processors at Penn State. SLING output consisted of 260 hits that contained a gene with a known toxin domain (Supplementary Figure 1). A hit is a CDS with a HMMER bit score of at least 20 for overall sequence/profile comparison with a previously characterized and/or predicted toxin CDS included in the program. Hits with the same toxin gene domain were given separate designations using an e-value cutoff of 0.01 and an identity score of 30% as reported previously (Horesh et al., 2018). Toxin hits sharing common domain names were considered homologs and combined into one category to simplify the results. For example, the parE toxin gene included 47 hits that were combined to form a single ParDE TA system. The homologs with the same hit label (eg., 4H-parE) across the strains (i.e., orthologs) were highly conserved (> 70% amino acid identity) in their amino acid sequence, while hits within a strain (i.e., paralogs) with different labels (eg., 4H-parE and 11H-parE) were divergent (< 30% identity) (Supplementary Figure 2). To check for completeness of the SLING prediction, we acquired the TA systems of the three model strains (DC3000, B728a, and 1448A) predicted in TADB2.0, an updated database of bacterial type II TA loci (Xie et al., 2018). The TAFinder tool of TADB2.0 was also run with default parameters using the Refseq accession numbers of the model strains.
To predict TA systems not included in the training sets of above programs, an HMM alignment approach was used to screen genomes for type I, III, V, and VI systems. For each family of toxin and protein antitoxin to be searched, a minimum of 20 amino acid sequences representing that family across diverse bacterial species were retrieved from UniProt. Sequences were aligned with MAFFT v 7.450 using default settings and an HMM profile was built using default settings of the hmmbuild function in the HMMER webserver (Finn et al., 2011). A locally created target database of 339 P. syringae genomes translated into six reading frames was used in HMMER’s HMMsearch. The TA systems screened this way were type I (hok/sok, ldrD/rdlD, tisB/istR-1, symE/symR, ibsC/sibC, txpA/ratA, fst/RNAI-RNAII, pndA/pndB, shoB/ohsC); type III (toxN/toxI, abiQ/antiQ, cptI/cptN, tenpN/tenpI); type V (ghoT/ghoS); and type VI (socB/socA). A type I TA system predicted for strain DC3000 in the type I toxin-antitoxin database T1TAdb (Tourasse and Darfeuille, 2021) was included in the analysis after confirming its presence in our genomes by nucleotide and protein BLAST.
Heatmaps were created using the Interactive Tree of Life (iTOL) v5 (Letunic and Bork, 2021) using the phylogenomic tree and TA system abundance used as annotation. Predicted TA systems were categorized based on their frequency among strains, as prevalent (if at least one homolog of the given toxin domain is present in ≥ 80% of strains), common (present in 20–79% of strains), or rare (present in < 20% of the strains).
Differences between phylogroups in TA system abundance were determined from the SLING output and uniquely predicted TA system of TADB2.0 (psyrTA), and T1TAdb (hok-sok). Genome size was defined as the assembly size and was recorded for all strains, while plasmid counts were based on analysis of 28 closed genomes with resolved plasmids. Chromosome or plasmid localization of genes was determined by manually examining the start and end nucleotide positions of predicted toxin genes. Isolation sources were obtained from strain information within NCBI annotation files, with all isolates from non-plant sources classified as “other” (Supplementary Table 1). To identify correlations between TA system abundance and the abundance of predicted type III effectors, we used bioinformatically predicted effector repertoires of 140 of the 339 of our genomes as computed in a previous study (Dillon et al., 2019b). Putative secretion signals in toxin and antitoxin genes of P. syringae strain DC3000 was predicted using EffectiveDB (Eichinger et al., 2016).
To identify signs of TA system acquisition through horizontal transfer, GC content of top 20 abundant toxin hits was extracted and was compared to that of the genomic GC content. Codon usage was extracted from the Codon Usage Database.1 The gene neighborhood of the most ubiquitous three TA system homologs (pasTI, RES-Xre, and vapBC) for the three strains DC3000, B728a, and 1448A was visualized using gene graphics.2 Grouping of strains based on similar TA system content was performed using SLING outputs of the toxin hits using the average linkage method for clustering and the Euclidean method for distance measurement in heatmapper (Babicki et al., 2016).
Sequences of the orthologs and paralogs of the same toxin hits (e.g., parE had 47 total homologs with up to 12 paralogs predicted in a single genome) were retrieved from the model strains DC3000, B728a, and 1448A. Alignment of the sequences was performed in Muscle 3.8.425 within Geneious using 10 maximum iterations. A phylogenetic tree of the homologs was constructed using FastTree 2.1.11 using default parameters.
We identified 10 papers for which RNA-Seq or microarray data were reported for strains DC3000, B728a, and 1448A under environmental or mutant conditions. Fold change or log2 fold change data was obtained from supplementary materials, and locus IDs corresponding to SLING-predicted TA systems were manually extracted using the start and end nucleotide positions of TA genes in the genome. Significantly differentially expressed loci were reported here as reported by the authors; no new data analysis was performed. Moreover, we used data from a recent study on genome-wide identification of fitness conferring genes in strain B728a and analyzed the fitness profiles of TA genes from their supplementary data (Helmann et al., 2019).
Statistical analysis of difference in TA counts by phylogroup was performed in JMP Pro 15 using the non-parametric Kruskal-Wallis test (P < 0.05) followed by each pair multiple comparisons after confirming that assumptions of parametric tests are not satisfied as per the goodness of fit test and unequal variance test in JMP. Phylogroups having fewer than 10 strains were excluded from this analysis. Correlation analysis between the TA counts and genome size, plasmid content, and type III effector count was also performed in JMP Pro 15 using the multivariate option and Spearman’s ρ. GC content differences were assessed using Kruskal-Wallis test with a genomic GC used as control group in Steel method.
As a basic step in understanding the roles of TA systems in P. syringae ecology, we aimed to obtain a comprehensive list and abundance patterns of TA systems across the species complex using bioinformatics prediction tools including SLING, BLAST, and HMMER. We predicted 26 distinct TA system families throughout the P. syringae genomospecies (Figure 1 and Supplementary Figure 3). Of these, 24 belonged to the type II TA system families, and one each belonged to type I (hok-sok) and type IV system (abiEii) families. The majority of TA system toxins predicted are orthologs of toxins in other species, which have been previously reported to inhibit translation through mRNA cleavage or through modification of translation factors, while others are reported to inhibit replication, cell wall biosynthesis, NAD + homeostasis, and other essential functions (Table 1). To confirm the comprehensiveness of the SLING method, predictions from three P. syringae model strains were compared to that from the database TADB2.0. The majority of the SLING predictions were also identified in TADB2.0 (7 of 12 in B728a, 14 of 19 in DC3000, and 1448A), including five of the six most prevalent systems described below. Six TA systems, including the highly conserved pasTI module, were only predicted using SLING. A TA system from a validated family, psyrTA (Sberro et al., 2013), was predicted in TADB2.0 but not SLING. Few TA genes were uniquely predicted in TADB2.0 and TAFinder (Supplementary Table 2).
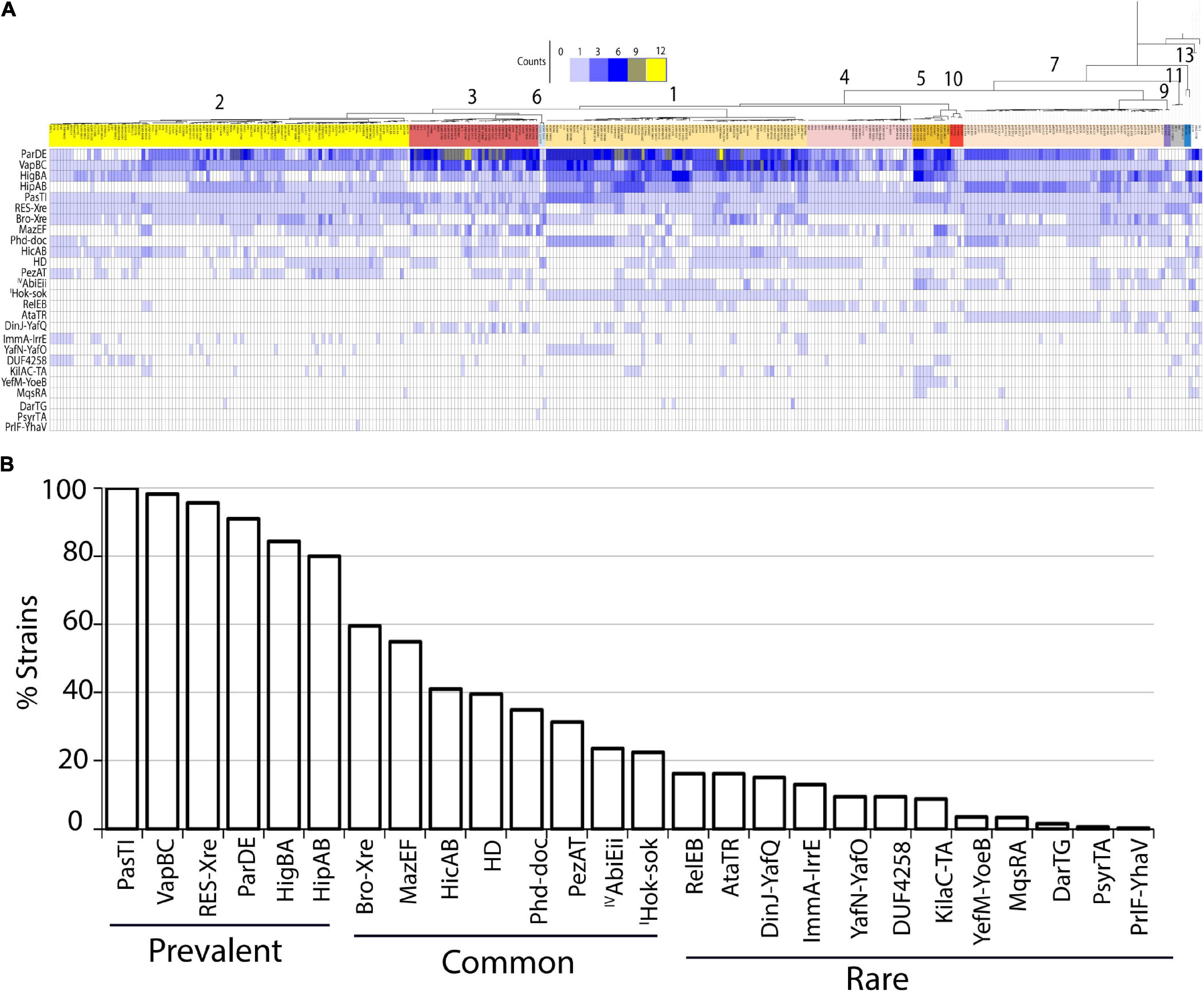
Figure 1. (A) Heatmap showing TA system content and distribution across P. syringae species complex. Names of TA systems are indicated for each row. Number of homologs of each TA systems are indicated by the color key at the top. Phylogenetic tree color coded according to phylogroups are indicated by numerals. (B) Percentage of strains containing the TA systems. List of all the TA systems found in P. syringae are shown in the x-axis, and the fraction of strains carrying a given TA system out of the 339 strains is shown in the y-axis. TA systems were categorized as prevalent, common, and rare based on their abundance as shown. Except abiEii (type IV), and hok-sok (type I) all belong to type II TA families.
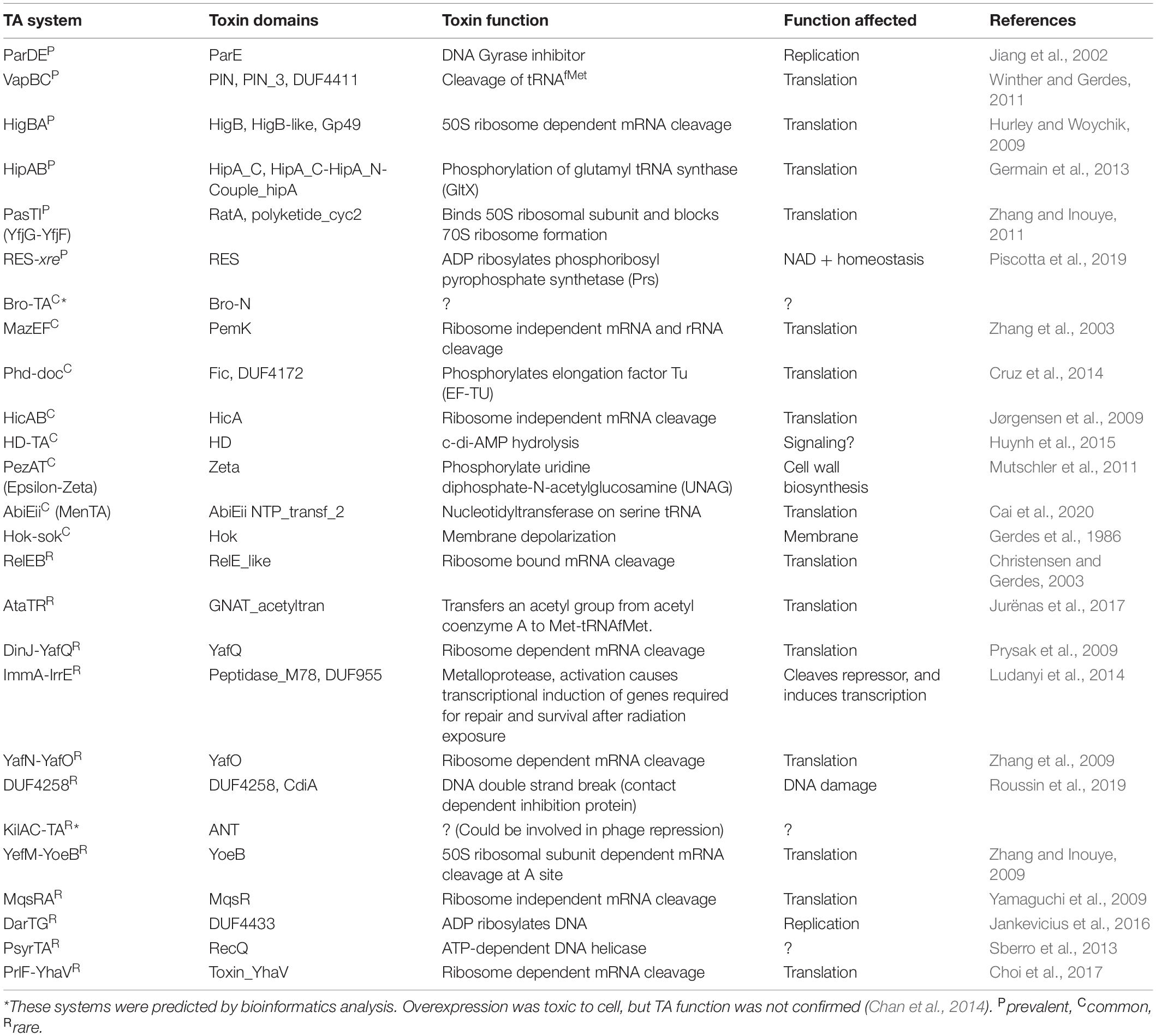
Table 1. Toxin-antitoxin systems predicted in P. syringae, toxin function and physiological effect.
Genomes varied substantially in TA system repertoires, and only six TA system families were highly prevalent (i.e., present in ≥ 80% of the genomes analyzed (Figures 1A,B). These included systems encoding putative translation inhibitor toxins (pasTI, vapBC, hipAB, and higBA), a replication inhibitor (parDE), and a RES domain toxin proposed to interfere with cellular NAD+ homeostasis (Table 1). Eight additional predicted systems present in more than 20% of the strains were categorized as common TA systems, and the remaining 12 were rare (Figure 1B). Prevalent TA system families included sets of toxin orthologs that diverged across strains despite sharing the common toxin domain, and some included multiple distinct paralogs within a genome. For example, for the parE toxin gene of the parDE system, 47 variants were predicted (Supplementary Figure 1). Similarly, the vapBC family had 21 distinct variants. A conserved copy of pasTI, RES-xre, and vapBC systems were predicted in all or most genomes (100, 94, and 87%, respectively), suggesting an important conserved function.
A hok-sok module of type I TA system was predicted in the strain DC3000 in the T1TAdb database (Tourasse and Darfeuille, 2021). Nucleotide BLAST of the predicted sequence in the locally constructed database of our 339 genomes confirmed presence of full complement of this system in strains of phylogroup 1 (Figure 1A). Other genomes contained orthologs of only the toxin component. Alignment of the predicted Hok sequence to previously characterized Hok homologs showed low identity (15% to that of pR1 plasmid and 13% to that of Erwinia amylovora). No TA toxin genes for any of the type I, III, V, and VI systems were detected using BLAST and HMMER alignment methods. All strains contained a homolog to the proposed type IV TA system cptB/sdhE antitoxin that is encoded in an operon with a hypothetical protein of unknown function (DC3000 locus ID: PSPTO_4227), but no cptA toxin was identified, and sdhE is an essential flavinylation factor in bacteria, so this is not a likely TA system. A few potential homologs were detected for antitoxins abiGii (type IV), ghoS (type V), and socA (type VI). Since these did not occur in the type strains and no toxin homolog was detected, no further assessment was performed.
As TA system abundance greatly varied across genomes with a minimum of 5 to a maximum of 35 TA systems (median of 15) predicted per genome of P. syringae, we aimed to understand the determinants that influence TA system abundance. TA system abundance varied significantly across phylogroups (Figure 2A). Phylogroups 1, 3, and 5 had significantly more TA systems per genome than other phylogroups (Figure 2A). These differences were mainly driven by a large variation in the within-genome abundance of three of the prevalent TA systems, parDE, vapBC, and higBA (Figure 2B). In addition to the copy number differences, phylogroups also differed in composition of TA system families. For example, putative bro-xre systems were common in phylogroup 2, but absent in phylogroups 4, 5, and 10. The mazEF system was ubiquitously predicted in phylogroup 7 strains but was sporadic in others (Figures 1A, 2B). Similarly, full complement of the type I hok-sok system was predicted only in phylogroup 1 (Figure 1A). Phylogroups could also be distinguished by divergent orthologs of the same families; for example, the HipA-5H homologs were predominantly found in phylogroups 7, while the HipA-6H group was concentrated in phylogroups 1 and 4 (Supplementary Figure 1).
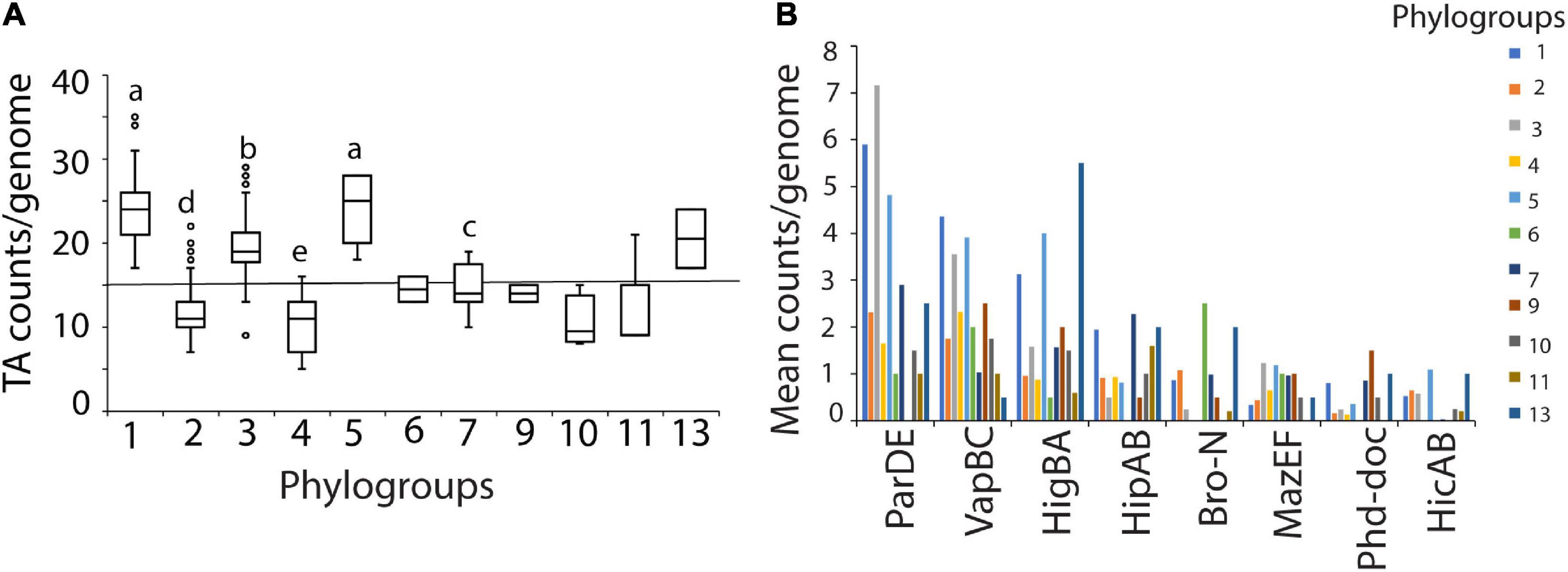
Figure 2. (A) TA counts per genome by phylogroups. The central line shows median number of TA system genes from the 339 strains used. Different letters indicate significant difference in counts as analyzed by Kruskal-Wallis test at P < 0.05. Phylogroups with less than 10 strains were not used in the analysis. (B) Differences in major TA system content among phylogroups. Mean counts per genome by phylogroup are shown. Phylogroups differed in copy number per genome of parDE, vapBC and higBA families (also refer heatmap in Figure 1A).
In a clustering analysis based on TA system repertoires, TA system composition followed phylogroup structure in P. syringae, with >90% of strains clustering with their phylogroup, suggesting that most TA system acquisition happened prior to phylogroup diversification (Supplementary Figure 4). However, at least 26 strains clustered outside their phylogroups, and phylogroups 1, 4, and 5 were significantly split into two or more clusters, potentially indicative of more recent TA system gain or loss in these strains. Overall, genetic similarity (i.e., phylogroup association) is a major factor governing similarities in TA system abundance.
TA systems frequently occur in mobile genetic elements (MGE), that are known to be horizontally transferred. As the genome sizes increase due to integration of MGEs or presence of plasmids, we hypothesize that the number of TA systems will also increase. We performed a correlation analysis on 339 strains to determine whether predicted TA system abundance is associated with genome size. Genome size was weakly, though significantly, correlated to TA system count (ρ = 0.52, P < 0.0001, Figure 3A). There was a similar positive correlation observed between TA system count and the number of plasmids (ρ = 0.44, P = 0.018, Figure 3B).
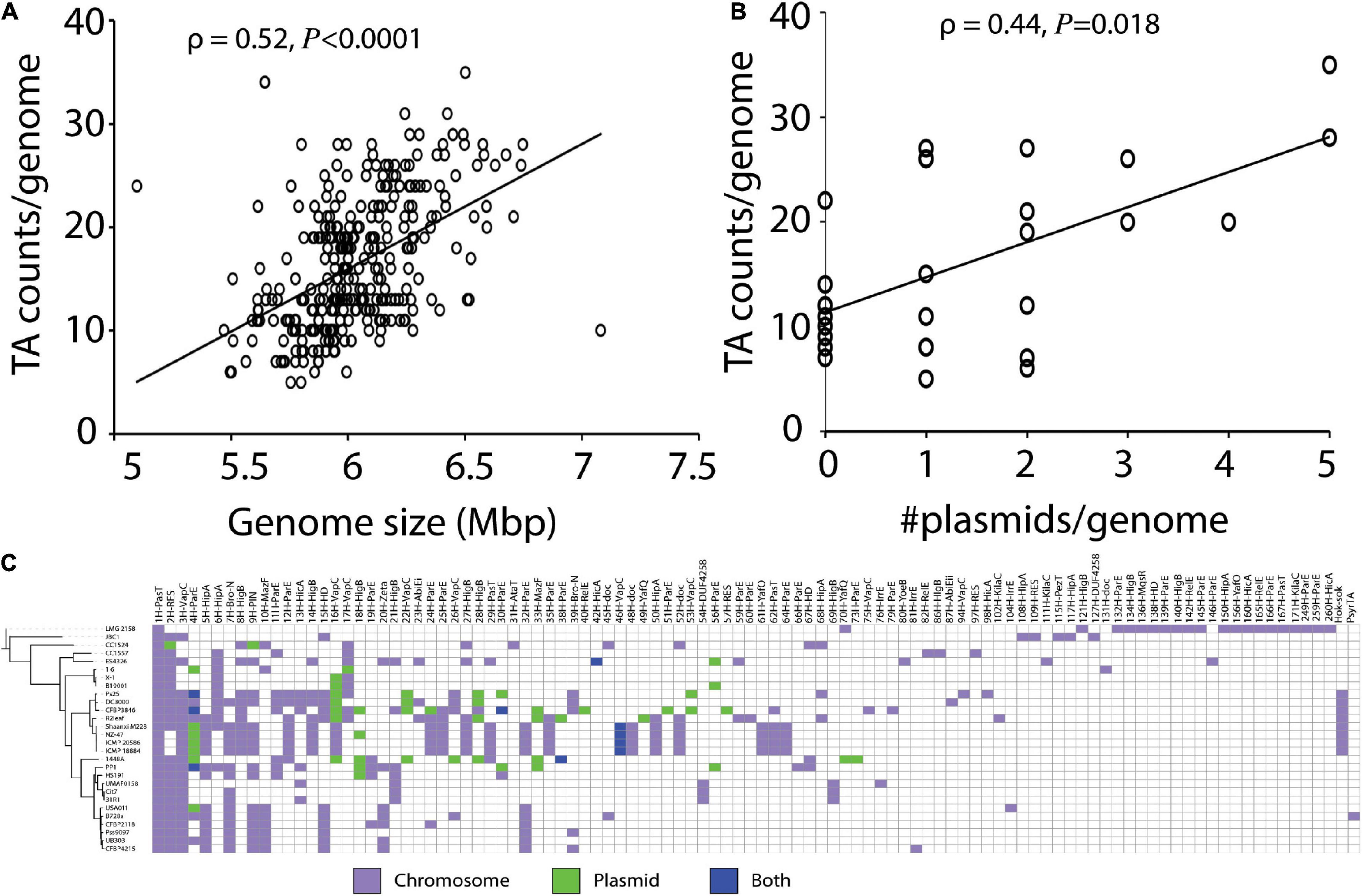
Figure 3. (A) Association of TA counts by genome size and (B) plasmid counts. (C) Heatmap showing occurrence of all 260 homologs of TA systems in the 28 completely sequenced strains in chromosome or plasmids. Most TA systems were encoded in the chromosome.
Next, we examined whether the TA systems predicted in the 28 complete P. syringae genomes were chromosome- or plasmid-borne. The majority (45–100%, mean = 87.7 ± 14.3) of predicted TA systems were encoded in the chromosome (Figure 3C and Supplementary Figure 3). The most widely distributed TA systems, with pasT, vapC, and RES domain toxins, were exclusively or almost exclusively detected on chromosomes, though the common and rare vapC homologs also occurred in plasmids (Figure 3C). Of the 40 plasmids analyzed, 33 contained at least one TA system with a mean count of 1.8 TA systems per plasmid, and 7 plasmids did not contain TA systems. A maximum of eight TA systems were predicted in the large plasmid of the strain 1448A.
We next analyzed GC content, codon usage variation, and genomic context of TA genes to detect if they contain signs of horizontal transfer. By comparing the GC content of top 20 toxin hits to that of the genome, we showed that GC content of 17 of the 20 toxin genes significantly differed from that of the genome. GC content varied by > 5% in 8 of the 20, and by > 3% in 12 of the 20 toxin genes (Supplementary Figure 5). Although we did not detect a noticeable codon usage variation compared to the genome in strain DC3000, few toxin genes such as parE, hicA and higB showed minor differences (Supplementary Figure 6). Upon examining the genomic context of the three most abundant and highly conserved TA system variants (i.e., pasT, RES, and vapC), we observed that these systems are immediately flanked by or lie within 20 kb region from MGE in at least one of the strains (Supplementary Figure 7). Together, these results suggest that most TA systems in P. syringae occur in mobile elements and are likely to be acquired by horizontal transfer.
A previous study reported higher abundance of TA systems in free-living than in host-associated isolates (Pandey and Gerdes, 2005). We therefore analyzed if the source of isolation of the strains influences the abundance of TA system in P. syringae. We did not detect any difference in TA abundance based on the environment from which strains were isolated (Figure 4A and Supplementary Figure 8). Notably, strain Psy642, which forms a separate subclade 2c within phylogroup 2, lacks the major virulence determinant of canonical type III secretion system, and was reported to be avirulent in a previous study (Demba Diallo et al., 2012), showed a different pattern of TA content than other strains of the same phylogroup (Supplementary Figure 1). Psy642 and two other strains in this clade did not contain the vapBC genes (Figure 1A). The highly prevalent vapC-3H, parE-4H, and hipA-5H homologs are missing in these strains (Supplementary Figure 1). Since TA systems are known to stabilize genomic islands that contain virulence and antibiotics resistant determinants (Wozniak and Waldor, 2009; Yao et al., 2015), we hypothesized that the predicted type III secretion system effector counts are correlated to TA gene counts. To this end, we examined the association between the number of TA loci and type III effectors, as predicted in a previous study (Dillon et al., 2019b). Although a similar trend was observed in the TA gene counts and type III effectors in the three phylogroups (1, 2, and 3) that are dominated by plant pathogens, overall, a weak positive correlation (ρ = 0.38, P < 0.0001) was detected between the two variables (Figure 4B). We also attempted to examine the association of TA genes to markers of copper and streptomycin resistance, bactericides that are frequently used in control of P. syringae diseases. However, these markers could be predicted only in a limited number of strains based on BLAST search of the resistant genes as query against our local database of 339 genomes.
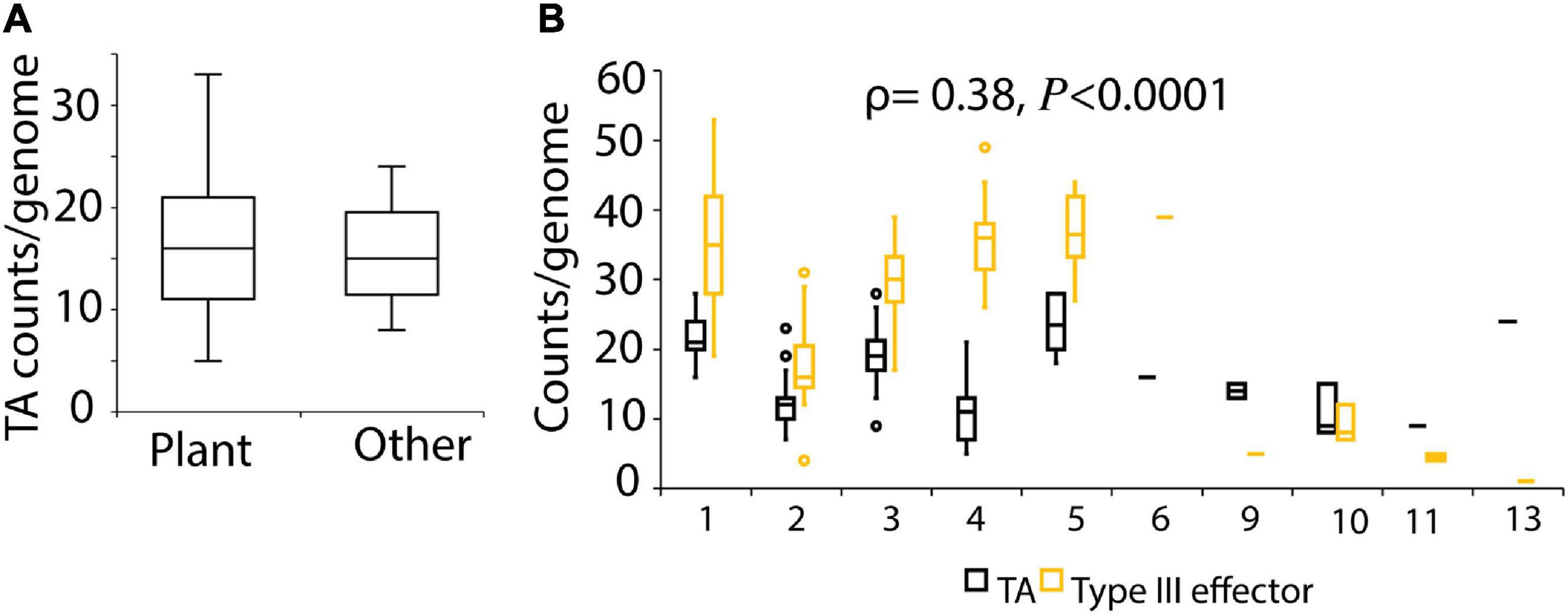
Figure 4. (A) TA counts by isolation source of strains. Strains isolated outside of plant environment (rain, snow, stream, lake water, epilithon) were categorized as other (n = 13). (B) TA counts and type III effector counts by phylogroups (x-axis). Type III secretion system effector counts predicted in 140 genomes from a previous study (Dillon et al., 2019b) were used. Strains of phylogroup 7 were not used in this analysis as the strains used here were not used in Dillon et al. (2019b).
EffectiveT3 predicted four TA gene products of DC3000 as type III secreted proteins. These included antitoxins of vapBC-3H (PSPTO_2000), vapBC-26H (PSPTO_1058), and toxins of HD-15H (PSPTO_2479), and ataTR (PSPTO_RS175570).
Although TA systems are posttranslationally regulated, some antitoxins are repressors of the TA operon, and conditions leading to antitoxin degradation and excess free toxin might lead to induction of the TA operon (Chan et al., 2016). Therefore, expression patterns could point to conditions in which certain toxins might be functional. We compiled reported transcriptional differences from nine RNA-seq studies and one well-replicated microarray study, all performed on one of the model strains DC3000, B728a, or 1448A. Seven of the studies reported significant differential expression of one or more of the four highly conserved TA system genes (pasTI, RES-xre, vapBC, and parDE) under host or abiotic stress, or in virulence regulatory mutants (Table 2), while three studies found no difference. In microarray analysis of strain B728a, epiphytic growth and nitrogen starvation induced 3 TA systems. vapC toxin genes were reported to be substantially induced during nitrogen starvation (Supplementary Table 3). The expression of RES-Xre was suppressed during early infection of DC3000 on Arabidopsis plants and was conditionally affected by several regulatory gene deletions in 1448A (Table 2). While these expression patterns do not provide conclusive evidence of a function or lack thereof, they indicate that the different TA system families are regulated in distinct and complex pathogen and environmental contexts.

Table 2. Reported expression patterns of TA system genes in three model strains B728a, 1448A, DC3000.
In a recent Rb-TnSeq study aimed at identifying genome-wide fitness profile of B728a genes, three antitoxin gene mutants (that of two parDE and a vapBC) showed below threshold abundance in the BarSeq library (Supplementary Figure 9). The authors suggested that these genes could be important for in vitro growth and survival. Similarly, although not reported to be significant in the paper, the plant fitness score was lower for antitoxin mutants of RES-xre, hipAB, psyrTA, and a vapBC system (Supplementary Figure 9). We speculate that the disruption of antitoxin genes by transposon insertion could have freed their cognate toxins. The freed toxins subsequently caused growth inhibition or death of the mutants lowering their abundance in the library. Interestingly, toxin gene mutants of the pasTI system showed below threshold abundance in the library, which could suggest a different regulation of this TA system or multiple cellular functions of the predicted toxin gene. Moreover, in the study that validated TA system function of psyrTA mentioned above, a parDE system is reported to be functional based on cloning success of predicted TA genes during Sanger-based whole-genome shotgun sequencing (Sberro et al., 2013).
Using in silico prediction from a broad-scale genomic data, we show that TA systems are prevalent and occur in diverse patterns in Pseudomonas syringae species complex. We found that 100% of screened genomes contain at least five TA systems with a median count of 15 per genome across the species. We predicted 26 distinct TA families that almost exclusively belong to the type II group, except for a type IV system that was sporadically detected in some strains and a type I system detected in phylogroup 1. Of the 26 TA systems, six are highly abundant across the species implying potentially conserved roles in P. syringae ecology. The most prevalent among the TA systems was that of the pasTI system, occurring mostly as a single copy in all strains with a high degree of sequence conservation. Although limited data exists regarding the functional characterization, a previous study in Escherichia coli CFT073 showed that this TA system is involved in stress tolerance under nutrient limitation, oxidative stress, and antibiotic persistence (Norton and Mulvey, 2012). Another recent study has confirmed its role in stress survival but contradicted its function as a TA system (Fino et al., 2020). Interestingly, The pasT gene was recently shown to be important for in vitro fitness in P. syringae, indicating a functional role (Helmann et al., 2019).
Orthologs of other prevalent TA loci (vapBC, RES-xre, parDE, higBA, hipAB) have been widely described to function as TA systems. We predicted multiple paralogs of parDE, vapBC, and higBA systems in some phylogroups and strains. We determined that sequences of paralogs are highly divergent. Whether all paralogs are active and if they are also functionally diverse is a subject of further research. The parDE and vapBC homologs were also among the most prevalent in another plant pathogen, Erwinia amylovora (Shidore et al., 2019). A vapBC ortholog was characterized as a TA system in the plant pathogen Acidovorax citruli, and was shown to be overexpressed during plant infection (Shavit et al., 2016). We predicted that some of the vapB antitoxin sequences contain a type III secretion signal, indicating a potential role in virulence. The role of vapBC in stress responses is reported in several other species (Arcus et al., 2010). The parDE system has been implicated in persistence against DNA gyrase inhibiting antibiotics in P. aeruginosa (Muthuramalingam et al., 2019). Homologs are also known for plasmid and genome maintenance in several other species including plant pathogens (Roberts et al., 1994; Yuan et al., 2011; Unterholzner et al., 2013a). Similarly, TA systems containing the RES domain toxin was also prevalent, and a copy was highly conserved across species. Although this system is previously characterized as a bona fide TA system including in P. putida (Skjerning et al., 2019), its physiological role remains to be determined. higBA system, another prevalent TA system in P. syringae, was shown to regulate virulence factor production in P. aeruginosa (Wood and Wood, 2016). Similarly, hipAB system has been shown to function in antibiotics persistence in vitro (Moyed and Bertrand, 1983; Germain et al., 2013) and also in the natural host environment (Schumacher et al., 2015). Taken together, the prevelance of these TA systems in P. syringae and their characterized roles in other taxa indicate that they may play multiple roles in the ecology of this phytopathogen. Further experiments will be able to reveal more specific details on their roles and the conditions under which they are activated.
TA genes that were less prevelant but were commonly found in P. syringae included toxin families of well characterized TA systems in other species. Some notable examples in this category were mazEF, hicAB, phd-doc, pezAT, abiEii, and hok-sok. Previous studies have reported stress and virulence associated function of these TA genes. For example, MazF toxin of the mazEF system was shown to contribute Mycobacterium tuberculosis cells survive oxidative stress, nutrient depletion, and virulence in the host (Tiwari et al., 2015). Rare TA systems also contained TA candidates that have been experimentally verified in other systems, including toxin families RelE, YafO, YafQ, AtaT (GNAT), YoeB, MqsR, and YhaV. Some candidates of both common and rare TA systems predicted in this study are less well studied and have not been demonstrated to function as TA systems experimentally, such as the Bro-N and KilaC(ANT) toxins (Chan et al., 2014). Only further experimental research can validate if they function as a TA system in P. syringae or are involved in other functions.
Our results show that TA system abundance differs by phylogroups. In addition to the genetic similarity, strains belonging to the same phylogroup are generally expected to have similar phenotypic traits consistent with their unique ecology. For instance, phylogroup 2 strains were shown to be more aggressive in cantaloupe seedling, were most consistently ice-nucleation active, and more likely to produce syringomycin-like toxin than strains of other phylogroups. Similarly, phylogroup 3 strains are rarely known to possess ice-nucleation activity and are negative for syringomycin-like toxins (Berge et al., 2014). Previous studies have also reported phylogroup dependent differences in the content of virulence factors (Baltrus et al., 2012; Dillon et al., 2019b). While it is impossible to speculate the significance of these difference at this point, similar sporadic distribution of TA system linked to genetic similarity has been also described previously both in clinical (Horesh et al., 2020) and plant pathogens (Shavit et al., 2016; Shidore et al., 2019).
TA systems are mostly known to occur in the accessory genome and are associated to MGEs or plasmids (Fraikin et al., 2020). Although we predicted that 82.5% (33 out of 40) of plasmids contain at least one TA system, a vast majority of TA systems are encoded in the chromosome in P. syringae. Chromosomally encoded TA systems are also reported to contribute to stability of genomic islands (Wozniak and Waldor, 2009; Yao et al., 2015). Moreover, virulence and antimicrobial resistance determinants are known to occur in laterally acquired genomic islands including in P. syringae (Kim et al., 1998; Feil et al., 2005). A comparison of the counts of type III effectors and TA loci showed similar trends in three of the phylogroups that are dominated by plant pathogens. Phylogroup 2 strains, which are known to be broad-host range and contain relatively smaller repertoire of type III effectors compared to other phylogroups (Baltrus et al., 2012), also contained lower counts of TA genes in our study. Moreover, strain CC1557 that was shown to contain unusually lower counts of type III effectors in a previous study (Hockett et al., 2014) was predicted to encode fewer of TA system genes (8 TA systems encoded compared to a median of 15). It could be possible that the TA systems occur in same genomic islands as the virulence and resistant determinants and could prevent their loss, imparting adaptational benefits. Alternatively, these results might indicate that strains that rely on larger repertoires of type III effectors, also rely on larger TA system repertoires in a manner related to their ecology. Testing these alternative hypotheses will require a combination of additional bioinformatic analyses and experimentation.
Taken together, we show that P. syringae genomes encode multiple TA systems that target diverse cellular functions upon activation. Based on our results of the abundance patterns and evidence of the ecological roles discussed above, we speculate that TA systems could be important as virulence effectors, in maintaining genes of virulence and antimicrobial resistance, in antimicrobial and environmental stress tolerance, in defending phage predation among others in P. syrinage. Further research focusing on the experimental characterization and ecological roles of the predicted TA systems can provide important insights in the epidemiology and management of plant disease caused by P. syringae.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
PK, LT, and KH: conceptualization and writing manuscript drafts. PK, MN, CF, and RP: data acquisition and analysis. LT and KH: supervision and funding acquisition. All authors: revision of drafts.
This research was supported by the USDA NIFA AFRI grant (2019-67013-29353) awarded to LT and KH. KH was also supported by the USDA National Institute of Food and Agriculture and Federal Hatch appropriations under project PEN04648 and accession no. 1015871.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank David S. Guttman from University of Toronto for sharing the raw data on the predicted type III secretion system effectors of Pseudomonas syringae.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.815911/full#supplementary-material
Supplementary Table 1 | Pseudomonas syringae strain genomes used in this study.
Supplementary Table 2 | Sequences and other details on toxin antitoxin systems of the three model strains DC3000, B728a, and 1448A.
Supplementary Table 3 | Data on transcriptomics study analyzed for TA systems in this study.
Arcus, V. L., McKenzie, J. L., Robson, J., and Cook, G. M. (2010). The PIN-domain ribonucleases and the prokaryotic VapBC toxin–antitoxin array. Protein Eng. Des. Select. 24, 33–40. doi: 10.1093/protein/gzq081
Babicki, S., Arndt, D., Marcu, A., Liang, Y., Grant, J. R., Maciejewski, A., et al. (2016). Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 44, W147–W153. doi: 10.1093/nar/gkw419
Baltrus, D. A., Nishimura, M. T., Dougherty, K. M., Biswas, S., Mukhtar, M. S., Vicente, J., et al. (2012). The molecular basis of host specialization in bean pathovars of Pseudomonas syringae. Mol. Plant Microbe Interact 25, 877–888. doi: 10.1094/MPMI-08-11-0218
Bardaji, L., Añorga, M., Echeverría, M., Ramos, C., and Murillo, J. (2019). The toxic guardians — multiple toxin-antitoxin systems provide stability, avoid deletions and maintain virulence genes of Pseudomonas syringae virulence plasmids. Mobile DNA 10:7. doi: 10.1186/s13100-019-0149-4
Berge, O., Monteil, C. L., Bartoli, C., Chandeysson, C., Guilbaud, C., Sands, D. C., et al. (2014). A user’s guide to a data base of the diversity of Pseudomonas syringae and its application to classifying strains in this phylogenetic complex. PLoS One 9:e105547. doi: 10.1371/journal.pone.0105547
Bronstein, P. A., Filiatrault, M. J., Myers, C. R., Rutzke, M., Schneider, D. J., and Cartinhour, S. W. (2008). Global transcriptional responses of Pseudomonas syringae DC3000 to changes in iron bioavailability in vitro. BMC Microbiol. 8:209. doi: 10.1186/1471-2180-8-209
Burbank, L. P., and Stenger, D. C. (2017). The DinJ/RelE toxin-antitoxin system suppresses bacterial proliferation and virulence of Xylella fastidiosa in grapevine. Phytopathology 107, 388–394. doi: 10.1094/PHYTO-10-16-0374-R
Cai, Y., Usher, B., Gutierrez, C., Tolcan, A., Mansour, M., Fineran, P. C., et al. (2020). A nucleotidyltransferase toxin inhibits growth of Mycobacterium tuberculosis through inactivation of tRNA acceptor stems. Sci. Adv. 6:eabb6651. doi: 10.1126/sciadv.abb6651
Chan, W. T., Espinosa, M., and Yeo, C. C. (2016). Keeping the wolves at bay: antitoxins of prokaryotic type II toxin-antitoxin systems. Front. Mol. Biosci. 3:9. doi: 10.3389/fmolb.2016.00009
Chan, W. T., Yeo, C. C., Sadowy, E., and Espinosa, M. (2014). Functional validation of putative toxin-antitoxin genes from the Gram-positive pathogen Streptococcus pneumoniae: phd-doc is the fourth bona-fide operon. Front. Microbiol. 5:677. doi: 10.3389/fmicb.2014.00677
Choi, W., Yamaguchi, Y., Lee, J.-W., Jang, K.-M., Inouye, M., Kim, S.-G., et al. (2017). Translation-dependent mRNA cleavage by YhaV in Escherichia coli. FEBS Lett. 591, 1853–1861. doi: 10.1002/1873-3468.12705
Christensen, S. K., and Gerdes, K. (2003). RelE toxins from bacteria and Archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol. Microbiol. 48, 1389–1400. doi: 10.1046/j.1365-2958.2003.03512.x
Craig, K., Johnson, B. R., and Grunden, A. (2021). Leveraging Pseudomonas stress response mechanisms for industrial applications. Front. Microbiol. 12:660134. doi: 10.3389/fmicb.2021.660134
Cruz, J. W., Rothenbacher, F. P., Maehigashi, T., Lane, W. S., Dunham, C. M., and Woychik, N. A. (2014). Doc toxin is a kinase that inactivates elongation factor Tu. J. Biol. Chem. 289, 7788–7798. doi: 10.1074/jbc.M113.544429
Demba Diallo, M., Monteil, C. L., Vinatzer, B. A., Clarke, C. R., Glaux, C., Guilbaud, C., et al. (2012). Pseudomonas syringae naturally lacking the canonical type III secretion system are ubiquitous in nonagricultural habitats, are phylogenetically diverse and can be pathogenic. ISME J. 6, 1325–1335. doi: 10.1038/ismej.2011.202
Dillon, M. M., Thakur, S., Almeida, R. N. D., Wang, P. W., Weir, B. S., and Guttman, D. S. (2019a). Recombination of ecologically and evolutionarily significant loci maintains genetic cohesion in the Pseudomonas syringae species complex. Genome Biol. 20:3. doi: 10.1186/s13059-018-1606-y
Dillon, M. M., Almeida, R. N. D., Laflamme, B., Martel, A., Weir, B. S., Desveaux, D., et al. (2019b). Molecular evolution of Pseudomonas syringae type III secreted effector proteins. Front. Plant Sci. 10:418. doi: 10.3389/fpls.2019.00418
Eichinger, V., Nussbaumer, T., Platzer, A., Jehl, M.-A., Arnold, R., and Rattei, T. (2016). EffectiveDB–updates and novel features for a better annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Res. 44, D669–D674. doi: 10.1093/nar/gkv1269
Feil, H., Feil, W. S., Chain, P., Larimer, F., DiBartolo, G., Copeland, A., et al. (2005). Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000. Proc. Natl. Acad. Sci. U. S. A. 102, 11064–11069. doi: 10.1073/pnas.0504930102
Filiatrault, M. J., Stodghill, P. V., Bronstein, P. A., Moll, S., Lindeberg, M., Grills, G., et al. (2010). Transcriptome analysis of Pseudomonas syringae identifies new genes, noncoding RNAs, and antisense activity. J. Bacteriol. 192, 2359–2372. doi: 10.1128/JB.01445-09
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Fino, C., Vestergaard, M., Ingmer, H., Pierrel, F., Gerdes, K., and Harms, A. (2020). PasT of Escherichia coli sustains antibiotic tolerance and aerobic respiration as a bacterial homolog of mitochondrial Coq10. Microbiol. Open 9:e1064. doi: 10.1002/mbo3.1064
Fraikin, N., Goormaghtigh, F., Melderen, L. V., and Margolin, W. (2020). Type II toxin-antitoxin systems: evolution and revolutions. J. Bacteriol. 202, e763–e719. doi: 10.1128/JB.00763-19
Gerdes, K., Bech, F. W., Jørgensen, S. T., Løbner-Olesen, A., Rasmussen, P. B., Atlung, T., et al. (1986). Mechanism of postsegregational killing by the hok gene product of the parB system of plasmid R1 and its homology with the relF gene product of the E. coli relB operon. Embo J. 5, 2023–2029.
Germain, E., Castro-Roa, D., Zenkin, N., and Gerdes, K. (2013). Molecular mechanism of bacterial persistence by HipA. Mol. Cell. 52, 248–254. doi: 10.1016/j.molcel.2013.08.045
Gomila, M., Busquets, A., Mulet, M., García-Valdés, E., and Lalucat, J. (2017). Clarification of taxonomic status within the Pseudomonas syringae species group based on a phylogenomic analysis. Front. Microbiol. 8:2422. doi: 10.3389/fmicb.2017.02422
Greenwald, J. W., Greenwald, C. J., Philmus, B. J., Begley, T. P., and Gross, D. C. (2012). RNA-seq analysis reveals that an ECF σ Factor, AcsS, regulates achromobactin biosynthesis in Pseudomonas syringae pv. syringae B728a. PLoS One 7:e34804. doi: 10.1371/journal.pone.0034804
Haubold, B., Klötzl, F., and Pfaffelhuber, P. (2014). andi: fast and accurate estimation of evolutionary distances between closely related genomes. Bioinformatics 31, 1169–1175. doi: 10.1093/bioinformatics/btu815
Helmann, T. C., Deutschbauer, A. M., and Lindow, S. E. (2019). Genome-wide identification of Pseudomonas syringae genes required for fitness during colonization of the leaf surface and apoplast. Proc. Natl. Acad. Sci. 116, 18900–18910. doi: 10.1073/pnas.1908858116
Hirano, S. S., and Upper, C. D. (2000). Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae-a pathogen, ice nucleus, and epiphyte. Microbiol. Mol. Biol. Rev. 64, 624–653. doi: 10.1128/MMBR.64.3.624-653.2000
Hockett, K. L., Burch, A. Y., and Lindow, S. E. (2013). Thermo-regulation of genes mediating motility and plant interactions in Pseudomonas syringae. PLoS One 8:e59850. doi: 10.1371/journal.pone.0059850
Hockett, K. L., Nishimura, M. T., Karlsrud, E., Dougherty, K., and Baltrus, D. A. (2014). Pseudomonas syringae CC1557: a highly virulent strain with an unusually small type III effector repertoire that includes a novel effector. Mol. Plant Microbe Interact 27, 923–932. doi: 10.1094/MPMI-11-13-0354-R
Horesh, G., Fino, C., Harms, A., Dorman, M. J., Parts, L., Gerdes, K., et al. (2020). Type II and type IV toxin-antitoxin systems show different evolutionary patterns in the global Klebsiella pneumoniae population. Nucleic Acids Res. 48, 4357–4370. doi: 10.1093/nar/gkaa198
Horesh, G., Harms, A., Fino, C., Parts, L., Gerdes, K., Heinz, E., et al. (2018). SLING: a tool to search for linked genes in bacterial datasets. Nucleic Acids Res. 46:e128. doi: 10.1093/nar/gky738
Hua, C., Wang, T., Shao, X., Yingpeng, X., Hao, H., Jingui, L., et al. (2020). Pseudomonas syringae dual-function protein Lon switches between virulence and metabolism by acting as both DNA-binding transcriptional regulator and protease in different environments. Environ. Microbiol. 22, 2968–2988. doi: 10.1111/1462-2920.15067
Hurley, J. M., and Woychik, N. A. (2009). Bacterial toxin HigB associates with ribosomes and mediates translation-dependent mRNA cleavage at A-rich sites. J. Biol. Chem. 284, 18605–18613. doi: 10.1074/jbc.M109.008763
Huynh, T. N., Luo, S., Pensinger, D., Sauer, J. D., Tong, L., and Woodward, J. J. (2015). An HD-domain phosphodiesterase mediates cooperative hydrolysis of c-di-AMP to affect bacterial growth and virulence. Proc. Natl. Acad. Sci. U. S. A. 112, E747–E756. doi: 10.1073/pnas.1416485112
Jankevicius, G., Ariza, A., Ahel, M., and Ahel, I. (2016). The toxin-antitoxin system DarTG catalyzes reversible ADP-Ribosylation of DNA. Mol. Cell. 64, 1109–1116. doi: 10.1016/j.molcel.2016.11.014
Jiang, Y., Pogliano, J., Helinski, D. R., and Konieczny, I. (2002). ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol. Microbiol. 44, 971–979. doi: 10.1046/j.1365-2958.2002.02921.x
Jørgensen, M. G., Pandey, D. P., Jaskolska, M., and Gerdes, K. (2009). HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J. Bacteriol. 191, 1191–1199. doi: 10.1128/JB.01013-08
Jurënas, D., Chatterjee, S., Konijnenberg, A., Sobott, F., Droogmans, L., Garcia-Pino, A., et al. (2017). AtaT blocks translation initiation by N-acetylation of the initiator tRNA(fMet). Nat. Chem. Biol. 13, 640–646. doi: 10.1038/nchembio.2346
Kandel, P. P., Baltrus, D. A., and Hockett, K. L. (2020). Pseudomonas can survive tailocin killing via persistence-like and heterogenous resistance mechanisms. J. Bacteriol. 202:13. doi: 10.1128/JB.00142-20
Kim, J. F., Charkowski, A. O., Alfano, J. R., Collmer, A., and Beer, S. V. (1998). Sequences related to transposable elements and bacteriophages flank avirulence genes of Pseudomonas syringae. Mol. Plant Microbe Interact 11, 1247–1252. doi: 10.1094/mpmi.1998.11.12.1247
Lam, H. N., Chakravarthy, S., Wei, H.-L., BuiNguyen, H., Stodghill, P. V., Collmer, A., et al. (2014). Global analysis of the HrpL regulon in the plant pathogen Pseudomonas syringae pv. tomato DC3000 reveals new regulon members with diverse functions. PLoS One 9:e106115. doi: 10.1371/journal.pone.0106115
Lee, M. D. (2019). GToTree: a user-friendly workflow for phylogenomics. Bioinformatics 35, 4162–4164.
Letunic, I., and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Lindow, S. E., and Brandl, M. T. (2003). Microbiology of the phyllosphere. Appl. Environ. Microbiol. 69, 1875–1883. doi: 10.1128/aem.69.4.1875-1883.2003
Liu, J., Yu, M., Chatnaparat, T., Lee, J. H., Tian, Y., Hu, B., et al. (2020). Comparative transcriptomic analysis of global gene expression mediated by (p) ppGpp reveals common regulatory networks in Pseudomonas syringae. BMC Genomics 21:296. doi: 10.1186/s12864-020-6701-2
Lovelace, A. H., Smith, A., and Kvitko, B. H. (2018). Pattern-triggered immunity alters the transcriptional regulation of virulence-associated genes and induces the sulfur starvation response in Pseudomonas syringae pv. tomato DC3000. Mol. Plant Microbe Interact 31, 750–765. doi: 10.1094/MPMI-01-18-0008-R
Ludanyi, M., Blanchard, L., Dulermo, R., Brandelet, G., Bellanger, L., Pignol, D., et al. (2014). Radiation response in Deinococcus deserti: irrE is a metalloprotease that cleaves repressor protein DdrO. Mol. Microbiol. 94, 434–449. doi: 10.1111/mmi.12774
Markel, E., Stodghill, P., Bao, Z., Myers, C. R., and Swingle, B. (2016). AlgU controls expression of virulence genes in Pseudomonas syringae pv. tomato DC3000. J. Bacteriol 198, 2330–2344. doi: 10.1128/JB.00276-16
Martins, P. M. M., Machado, M. A., Silva, N. V., Takita, M. A., and de Souza, A. A. (2016). Type II Toxin-Antitoxin Distribution and Adaptive Aspects on Xanthomonas Genomes: focus on Xanthomonas citri. Front Microbiol. 7:652. doi: 10.3389/fmicb.2016.00652
McCann, H. C., Rikkerink, E. H. A., Bertels, F., Fiers, M., Lu, A., Rees-George, J., et al. (2013). Genomic analysis of the kiwifruit pathogen Pseudomonas syringae pv. actinidiae provides insight into the origins of an emergent plant disease. PLoS Pathog. 9:e1003503. doi: 10.1371/journal.ppat.1003503
Merfa, M. V., Niza, B., Takita, M. A., and De Souza, A. A. (2016). The MqsRA toxin-antitoxin system from Xylella fastidiosa plays a key role in bacterial fitness, pathogenicity, and persister cell formation. Front Microbiol. 7:904. doi: 10.3389/fmicb.2016.00904
Mock, N. M., Baker, C. J., and Aver’yanov, A. A. (2015). Induction of a viable but not culturable (VBNC) state in some Pseudomonas syringae pathovars upon exposure to oxidation of an apoplastic phenolic, acetosyringone. Physiol. Mol. Plant Pathol. 89, 16–24.
Morris, C. E., Monteil, C. L., and Berge, O. (2013). The life history of Pseudomonas syringae: linking agriculture to earth system processes. Annu. Rev. Phytopathol. 51, 85–104. doi: 10.1146/annurev-phyto-082712-102402
Morris, C. E., Sands, D. C., Vinatzer, B. A., Glaux, C., Guilbaud, C., Buffière, A., et al. (2008). The life history of the plant pathogen Pseudomonas syringae is linked to the water cycle. ISME J. 2, 321–334. doi: 10.1038/ismej.2007.113
Moyed, H. S., and Bertrand, K. P. (1983). hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 155, 768–775. doi: 10.1128/jb.155.2.768-775.1983
Muthuramalingam, M., White, J. C., Murphy, T., Ames, J. R., and Bourne, C. R. (2019). The toxin from a ParDE toxin-antitoxin system found in Pseudomonas aeruginosa offers protection to cells challenged with anti-gyrase antibiotics. Mol. Microbiol. 111, 441–454. doi: 10.1111/mmi.14165
Mutschler, H., Gebhardt, M., Shoeman, R. L., and Meinhart, A. (2011). A novel mechanism of programmed cell death in bacteria by toxin-antitoxin systems corrupts peptidoglycan synthesis. PLoS Biol. 9:e1001033. doi: 10.1371/journal.pbio.1001033
Newberry, E. A., Ebrahim, M., Timilsina, S., Zlatkovič, N., Obradović, A., Bull, C. T., et al. (2019). Inference of convergent gene acquisition among Pseudomonas syringae strains isolated from watermelon, cantaloupe, and squash. Front Microbiol. 10:270. doi: 10.3389/fmicb.2019.00270
Norton, J. P., and Mulvey, M. A. (2012). Toxin-antitoxin systems are important for niche-specific colonization and stress resistance of uropathogenic Escherichia coli. PLoS Pathog. 8:e1002954. doi: 10.1371/journal.ppat.1002954
Ogura, T., and Hiraga, S. (1983). Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc. Natl. Acad. Sci. U. S. A. 80, 4784–4788. doi: 10.1073/pnas.80.15.4784
Pandey, D. P., and Gerdes, K. (2005). Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 33, 966–976. doi: 10.1093/nar/gki201
Patel, R. R., Kandel, P. P., Traverso, E., Hockett, K. L., Triplett, L. R., and Vidaver, A. K. (2021). Pseudomonas syringae pv. phaseolicola uses distinct modes of stationary-phase persistence to survive bacteriocin and streptomycin treatments. Mbio 12, e00161–21. doi: 10.1128/mBio.00161-21
Peng, J., Triplett, L. R., and Sundin, G. W. (2021). Activation of metabolic and stress responses during subtoxic expression of the type I toxin hok in Erwinia amylovora. BMC Genomics 22:74. doi: 10.1186/s12864-021-07376-w
Piscotta, F. J., Jeffrey, P. D., and Link, A. J. (2019). ParST is a widespread toxin–antitoxin module that targets nucleotide metabolism. Proc. Natl. Acad. Sci. 116:826. doi: 10.1073/pnas.1814633116
Postnikova, O. A., Shao, J., Mock, N. M., Baker, C. J., and Nemchinov, L. G. (2015). Gene expression profiling in viable but nonculturable (VBNC) cells of Pseudomonas syringae pv. syringae. Front. Microbiol. 6:1419. doi: 10.3389/fmicb.2015.01419
Prysak, M. H., Mozdzierz, C. J., Cook, A. M., Zhu, L., Zhang, Y., Inouye, M., et al. (2009). Bacterial toxin YafQ is an endoribonuclease that associates with the ribosome and blocks translation elongation through sequence-specific and frame-dependent mRNA cleavage. Mol. Microbiol. 71, 1071–1087. doi: 10.1111/j.1365-2958.2008.06572.x
Roberts, R. C., Ström, A. R., and Helinski, D. R. (1994). The parDE operon of the broad-host-range plasmid RK2 specifies growth inhibition associated with plasmid loss. J. Mol. Biol. 237, 35–51. doi: 10.1006/jmbi.1994.1207
Roussin, M., Rabarioelina, S., Cluzeau, L., Cayron, J., Lesterlin, C., Salcedo, S. P., et al. (2019). Identification of a contact-dependent growth inhibition (CDI) system that reduces biofilm formation and host cell adhesion of Acinetobacter baumannii DSM30011 strain. Front. Microbiol. 10:2450. doi: 10.3389/fmicb.2019.02450
Santamaría-Hernando, S., Cerna-Vargas, J. P., Martínez-García, P. M., de Francisco-de Polanco, S., Nebreda, S., Rodríguez-Palenzuela, P., et al. (2020). Blue-light perception by epiphytic Pseudomonas syringae drives chemoreceptor expression, enabling efficient plant infection. Mol. Plant Pathol. 21, 1606–1619. doi: 10.1111/mpp.13001
Sberro, H., Leavitt, A., Kiro, R., Koh, E., Peleg, Y., Qimron, U., et al. (2013). Discovery of functional toxin/antitoxin systems in bacteria by shotgun cloning. Mol. Cell. 50, 136–148. doi: 10.1016/j.molcel.2013.02.002
Schumacher, M. A., Balani, P., Min, J., Chinnam, N. B., Hansen, S., Vulić, M., et al. (2015). HipBA-promoter structures reveal the basis of heritable multidrug tolerance. Nature 524, 59–64. doi: 10.1038/nature14662
Shao, X., Tan, M., Xie, Y., Yao, C., Wang, T., Huang, H., et al. (2021). Integrated regulatory network in Pseudomonas syringae reveals dynamics of virulence. Cell Rep. 34:108920. doi: 10.1016/j.celrep.2021.108920
Shavit, R., Lebendiker, M., Pasternak, Z., Burdman, S., and Helman, Y. (2016). The vapb–vapc operon of Acidovorax citrulli functions as a bona-fide toxin–antitoxin module. Front Microbiol. 6:1499. doi: 10.3389/fmicb.2015.01499
Shidore, T., Zeng, Q., and Triplett, L. R. (2019). survey of toxin–antitoxin systems in Erwinia amylovora reveals insights into diversity and functional specificity. Toxins 11:206. doi: 10.3390/toxins11040206
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Skjerning, R. B., Senissar, M., Winther, K. S., Gerdes, K., and Brodersen, D. E. (2019). The RES domain toxins of RES-Xre toxin-antitoxin modules induce cell stasis by degrading NAD+. Mol. Microbiol. 111, 221–236. doi: 10.1111/mmi.14150
Song, S., and Wood, T. K. (2020b). Toxin/antitoxin system paradigms: toxins bound to antitoxins are not likely activated by preferential antitoxin degradation. Adv. Biosyst. 4:1900290. doi: 10.1002/adbi.201900290
Song, S., and Wood, T. K. (2020a). A primary physiological role of toxin/antitoxin systems is phage inhibition. Front Microbiol. 11:1895. doi: 10.3389/fmicb.2020.0189
Tiwari, P., Arora, G., Singh, M., Kidwai, S., Narayan, O. P., and Singh, R. (2015). MazF ribonucleases promote Mycobacterium tuberculosis drug tolerance and virulence in guinea pigs. Nat. Commun. 6:6059. doi: 10.1038/ncomms7059
Tourasse, N. J., and Darfeuille, F. (2021). T1TAdb: the database of type I toxin-antitoxin systems. Rna 27, 1471–1481. doi: 10.1261/rna.078802.121
Triplett, L. R., Shidore, T., Long, J., Jiamin, M., Wu, S., Han, Q., et al. (2016). AvrRxo1 is a bifunctional type III secreted effector and toxin-antitoxin system component with homologs in diverse environmental contexts. PLoS One 11:e0158856. doi: 10.1371/journal.pone.0158856
Unterholzner, S. J., Poppenberger, B., and Rozhon, W. (2013b). Toxin-antitoxin systems: biology, identification, and application. Mob. Genet. Elements 3:e26219. doi: 10.4161/mge.26219
Unterholzner, S. J., Hailer, B., Poppenberger, B., and Rozhon, W. (2013a). Characterisation of the stbD/E toxin-antitoxin system of pEP36, a plasmid of the plant pathogen Erwinia pyrifoliae. Plasmid 70, 216–225. doi: 10.1016/j.plasmid.2013.04.002
Winther, K. S., and Gerdes, K. (2011). Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. U. S. A. 108, 7403–7407. doi: 10.1073/pnas.1019587108
Wood, T. L., and Wood, T. K. (2016). The HigB/HigA toxin/antitoxin system of Pseudomonas aeruginosa influences the virulence factors pyochelin, pyocyanin, and biofilm formation. Microbiol. Open 5, 499–511. doi: 10.1002/mbo3.346
Wozniak, R. A. F., and Waldor, M. K. (2009). A toxin–antitoxin system promotes the maintenance of an integrative conjugative element. PLoS Genet. 5:e1000439. doi: 10.1371/journal.pgen.1000439
Xie, Y., Wei, Y., Shen, Y., Li, X., Zhou, H., Tai, C., Deng, Z., et al. (2018). TADB 2.0: an updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 46, D749–D753. doi: 10.1093/nar/gkx1033
Xin, X.-F., Kvitko, B., and He, S. Y. (2018). Pseudomonas syringae: what it takes to be a pathogen. Nat. Rev. Microbiol. 16, 316–328. doi: 10.1038/nrmicro.2018.17
Yamaguchi, Y., Park, J. H., and Inouye, M. (2009). MqsR, a crucial regulator for quorum sensing and biofilm formation, is a GCU-specific mRNA interferase in Escherichia coli. J. Biol. Chem. 284, 28746–28753. doi: 10.1074/jbc.M109.032904
Yao, X., Chen, T., Shen, X., Zhao, Y., Wang, M., Rao, X., et al. (2015). The chromosomal SezAT toxin–antitoxin system promotes the maintenance of the SsPI-1 pathogenicity island in epidemic Streptococcus suis. Mol. Microbiol. 98, 243–257. doi: 10.1111/mmi.13116
Yu, X., Lund, S. P., Scott, R. A., Greenwald, J. W., Records, A. H., Nettleton, D., et al. (2013). Transcriptional responses of Pseudomonas syringae to growth in epiphytic versus apoplastic leaf sites. Proc. Natl. Acad. Sci. U. S. A. 110, E425–E434. doi: 10.1073/pnas.1221892110
Yuan, J., Yamaichi, Y., and Waldor, M. K. (2011). The three Vibrio cholerae chromosome II-encoded ParE toxins degrade chromosome I following loss of chromosome II. J. Bacteriol. 193, 611–619. doi: 10.1128/JB.01185-10
Zhang, Y., and Inouye, M. (2009). The inhibitory mechanism of protein synthesis by YoeB, an Escherichia coli toxin. J. Biol. Chem. 284, 6627–6638. doi: 10.1074/jbc.M808779200
Zhang, Y., and Inouye, M. (2011). RatA (YfjG), an Escherichia coli toxin, inhibits 70S ribosome association to block translation initiation. Mol. Microbiol. 79, 1418–1429. doi: 10.1111/j.1365-2958.2010.07506.x
Zhang, Y., Yamaguchi, Y., and Inouye, M. (2009). Characterization of YafO, an Escherichia coli toxin. J. Biol. Chem. 284, 25522–25531. doi: 10.1074/jbc.m109.036624
Keywords: Pseudomonas syringae, toxin-antitoxin systems, phylogroup, genomics, stress
Citation: Kandel PP, Naumova M, Fautt C, Patel RR, Triplett LR and Hockett KL (2022) Genome Mining Shows Ubiquitous Presence and Extensive Diversity of Toxin-Antitoxin Systems in Pseudomonas syringae. Front. Microbiol. 12:815911. doi: 10.3389/fmicb.2021.815911
Received: 16 November 2021; Accepted: 13 December 2021;
Published: 12 January 2022.
Edited by:
Ralf Koebnik, Plant Health Institute, FranceReviewed by:
Marie-Laure PINEL-MARIE, INSERM U1230 ARN Régulateurs Bactériens et Médecine (BRM), FranceCopyright © 2022 Kandel, Naumova, Fautt, Patel, Triplett and Hockett. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prem P. kandel, cHVrMTU1QHBzdS5lZHU=; Kevin L. Hockett, a2xoNDUwQHBzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.