
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 11 January 2022
Sec. Infectious Agents and Disease
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.802404
 Willi Quino1*†
Willi Quino1*† Junior Caro-Castro1
Junior Caro-Castro1 Verónica Hurtado1
Verónica Hurtado1 Diana Flores-León1,2
Diana Flores-León1,2 Narjol Gonzalez-Escalona3
Narjol Gonzalez-Escalona3 Ronnie G. Gavilan1,2†
Ronnie G. Gavilan1,2†
Campylobacter is the leading cause of human bacterial gastroenteritis worldwide and has a major impact on global public health. Whole Genome Sequencing (WGS) is a powerful tool applied in the study of foodborne pathogens. The objective of the present study was to apply WGS to determine the genetic diversity, virulence factors and determinants of antimicrobial resistance of the populations of C. jejuni and C. coli in Peru. A total of 129 Campylobacter strains (108 C. jejuni and 21 C. coli) were sequenced using Illumina Miseq platform. In silico MLST analysis identified a high genetic diversity among those strains with 30 sequence types (STs), several of them within 11 clonal complexes (CC) for C. jejuni, while the strains of C. coli belonged to a single CC with 8 different STs. Phylogeny analysis showed that Peruvian C. jejuni strains were divided into 2 clades with 5 populations, while C. coli formed a single clade with 4 populations. Furthermore, in silico analyses showed the presence of several genes associated with adherence, colonization and invasion among both species: cadF (83.7%), jlpA (81.4%), racR (100%), dnaJ (83.7%), pebA (83.7%), pldA (82.1%), porA (84.5%), ceuE (82.9%), ciaB (78.3%), iamB (86.8%), and flaC (100%). The majority (82.9%) of the Campylobacter strains carried the cdtABC operon which code for cytolethal distending toxin (CDT). Half of them (50.4%) carried genes associated with the presence of T6SS, while the frequency of genes associated with T4SS were relatively low (11.6%). Genetic markers associated with resistance to quinolones, tetracycline (tetO, tetW/N/W), beta-lactamases (blaoxa–61), macrolides (A2075G in 23S rRNA) were found in 94.5, 21.7, 66.7, 6.2, 69.8, and 18.6% of strains, respectively. The cmeABC multidrug efflux operon was present in 78.3% of strains. This study highlights the importance of using WGS in the surveillance of emerging pathogens associated with foodborne diseases, providing genomic information on genetic diversity, virulence mechanisms and determinants of antimicrobial resistance. The description of several Campylobacter genotypes having many virulence factors and resistance to quinolones and tetracyclines circulating in Peru provides important information which helps in the monitoring, control and prevention strategies of this emerging pathogen in our country.
Campylobacter is a zoonotic pathogen that causes foodborne diarrheal diseases globally (Sheppard and Maiden, 2015). Campylobacter jejuni and Campylobacter coli are the most important pathogenic species which causes approximately 90% of human infection (Newell and Fearnley, 2003). Although the most important source of infection worldwide is the consumption of undercooked poultry contaminated with Campylobacter or the mishandling of raw poultry products (Mullner et al., 2009), other potential sources such as wild and domestic animals have been demonstrated (Rukambile et al., 2019). The infectious dose of Campylobacter is 104 bacterial cells (Mardones and López, 2017); however, lower infectious doses have been shown (Wieczorek and Osek, 2013), which after multiplying in the small intestine, produce inflammation with infiltration of leukocytes in the intestinal lamina, resulting in the presence of leukocytes in the stool in 25–80% of cases.
Campylobacteriosis constitutes an important public health problem due to the increase in its occurrence, new forms of transmission, increased resistance of microorganisms to antibiotics and the socioeconomic impact they cause. Furthermore, it is associated with multiple gastrointestinal conditions, including inflammatory bowel diseases (IBD), Barrett’s esophagus, and colorectal cancer (Mardones and López, 2017). In some cases, extra gastrointestinal manifestations may develop, including bacteremia, lung infections, brain abscess, meningitis, and reactive arthritis (Kaakoush et al., 2015). In some severe cases, C. jejuni infections contribute to post-infectious risks of acquiring neurological complications such as Guillain-Barré syndrome and Miller-Fisher syndrome (Humphrey et al., 2007), and have also been reported to lead to IBD, such as Crohn’s disease (Horrocks et al., 2009).
Few studies about the prevalence of Campylobacter that causing gastroenteritis are available in Peru. The most recent report indicates a 13.3% of Campylobacter spp. Isolated from children under 12 years (Perales et al., 2002). The frequency rate is similar in Latin American countries such as Chile, which reaches 12.7% (Valenzuela et al., 2018) or Argentina, with a value of 15.2% (Tamborini et al., 2012). On the other hand, the prevalence in animals for human consumption is much higher, reporting 16.7% of positive carcasses and 26.7% of positive cecum in Peru (Lucas et al., 2013), values even higher than them are reported in Chile and Argentina, reaching 70% of contaminated carcasses (Figueroa et al., 2009; Lopez et al., 2017).
Due to the great diversity of Campylobacter, the pathogenic mechanisms that cause clinical symptoms are not yet well defined (Silva et al., 2011). cadF, racR, flaA, and dnaJ genes are associated with the adherence, colonization and thermotolerance of this bacterium, and ciaB and pldA genes play a role in invasion and survival inside the host (Ramires et al., 2020; Wysok et al., 2020). Another important virulence factor is the Cytolethal distending toxin (CDT) formed by three subunits encoded by cdtA, cdtB, and cdtC genes, leading to apoptosis of the immune and epithelial cells of the intestine (Silva et al., 2011). Additionally, cst-II/cst-III, cgtA, and cgtB genes involved in sialic acid production are associated with the synthesis of lipo-oligosaccharides (LOS) of human ganglioside type that can induce Guillain Barré syndrome (Zhang et al., 2015). Two different type IV secretion systems (T4SS) have been described in Campylobacter, one codified by the plasmid pVir, associated with the adherence and invasion (Bravo et al., 2021), and the second present on the plasmids pTet and pCC31, which contribute to bacterial conjugation (Batchelor et al., 2004). Moreover, a type VI secretion system (T6SS) probably involved in virulence in Campylobacter is poorly understood despite its increasing prevalence in Campylobacter jejuni isolates (Wieczorek and Osek, 2013). T6SS plays a role in host colonization and consists of 12 central components (TssA-TssM and tagH) and accessory proteins codified by hcp genes associated with T6SS (Liaw et al., 2019).
Often the disease does not require any specific antimicrobial treatment. However, antimicrobial therapy is necessary in patients with severe and immunocompromised diseases (Wieczorek and Osek, 2013). Macrolides are the most widely used antimicrobial agents, and fluoroquinolones, tetracyclines and aminoglycosides are treatment options (Payot et al., 2006). Nevertheless, resistance to these antimicrobials in Campylobacter is increasing (Center for Disease Control Prevention [CDC], 2019b). The unreasonable use of antibiotics in humans and animals has contributed to increased resistance to different group of antibiotics in Campylobacter strains (Wieczorek and Osek, 2013). For example, a study made in a Peruvian public hospital reported 87% of strains resistant to quinolones like ciprofloxacin (Moya-Salazar et al., 2018). Also, a recent report detected 53% of animal strains resistant to macrolides (Anampa et al., 2020).
Currently, the application of WGS in the surveillance of foodborne pathogens of public health concern has become commonplace, allowing improved epidemiological surveillance and the detection of molecular markers of virulence and antimicrobial resistance in emerging genotypes (Jagadeesan et al., 2019; Su et al., 2019). This enhanced surveillance allows strengthening strategies for the prevention and control of foodborne diseases, by improving the tracking of those pathogens and targeting or eliminating the possible sources from the active commerce (Zhao et al., 2016; Painset et al., 2020). In the case of Peru, little is known of the genetic types circulating in the second decade of the twenty-first century and there is the need to increase our resolution by using a more comprehensive technique such as WGS. To test and fine tune our WGS analysis approach, and to start building our own in-country database, Campylobacter spp. strains from the following period (2010–2020) were selected to be subjected to WGS analysis. The aim of this study was to apply WGS analysis to determine the genetic diversity, virulence factors and antimicrobial resistance determinants of C. jejuni and C. coli populations in Peru.
A total of 523 strains were obtained from 2010 to 2020 as part of the surveillance of the Laboratorio de Referencia Nacional de Enteropatogenos of Instituto Nacional de Salud, Peru, sequencing 200 strains of this group. However, only 129 good quality genomes were obtained (C. jejuni n = 108 and C. coli n = 21) and all of them were included in the analysis. Most of the strains were recovered from human clinical samples (n = 123) and six were recovered from poultry (n = 6). The number of strains from each year is indicated in Table 1.
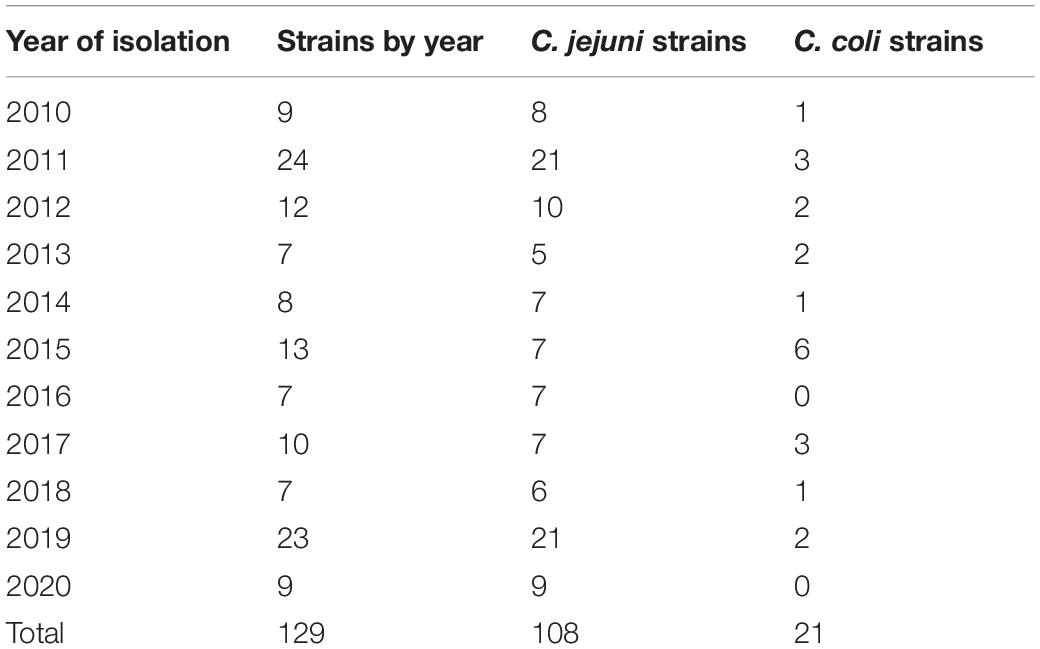
Table 1. Number of strains of C. jejuni and C. coli used in the study by year of isolation.
The strains were cultured on blood-based agar (BD BBL™, United States) with 5% defibrinated sheep blood and incubated at 42°C for 48 h under microaerobic conditions (85% nitrogen, 10% carbon dioxide and 5% oxygen). The strains were confirmed as C. jejuni or C. coli using Lior’s biotyping scheme (Lior, 1984) and the PCR technique based on the highly conserved gene glyA (serine hydroxymethyltransferase) (Farace and Viñas, 2007).
Antimicrobial susceptibility to ciprofloxacin (CIP), nalidixic acid (NA), tetracycline (TE), and erythromycin (E) was determined by agar microdilution method (Clinical Laboratory Standards Institute [CLSI], 2018). Minimum inhibitory concentrations (MIC) were interpreted according to the Clinical and Laboratory Standards Institute (Clinical Laboratory Standards Institute [CLSI], 2016), with the cutoff points being erythromycin (R ≥ 32μg/ml), ciprofloxacin (R ≥ 4 μg/ml) and tetracycline (R ≥ 16 μg/ml). The cut-off point for nalidixic acid (≥32 μg/ml) was established according to The National Antimicrobial Resistance Monitoring System for Enteric Bacteria (NARMS) (Center for Disease Control Prevention [CDC], 2018). C. jejuni strain ATCC 33560 was used as quality control of the method. The strains were cultured in Mueller Hinton Agar (BD BBL™, United States) with 5% defibrinated sheep blood and incubated at 42°C under micro-anaerobiosis conditions (85% Nitrogen, 10% carbon dioxide and 5% oxygen) for 24 h. Multidrug resistance was defined as the resistance of three or more classes of antimicrobials.
Genomic DNA extraction was performed using Pure Link Genomic DNA Mini Kit (Invitrogen, United States) and was quantified by fluorometry (Qubit 3.0 Invitrogen, Malaysia). The elaboration of the sequencing libraries was carried out using Nextera XT Library Preparation Kit (Illumina, Inc., United States), and genomic sequencing using MiSeq next generation sequencer (Illumina, Inc., United States). The quality control of each sequence was evaluated using Fastqc v0.11.5 (Andrews, 2010). Low quality adapters and nitrogenous bases were removed using Trimmomatic v0.38 (Bolger et al., 2014). The sequences were assembled de novo using A5-miseq v20160825 (Coil et al., 2015). The species and serotype of each strain were identified using Multilocus Sequence Typing (MLST) v2.10 (Larsen et al., 2012). To review the frequency of STs detected in this work, a minimum spanning tree (MST) was constructed using the MLST profiles obtained in this work and from other genomics studies previously carried out in Peru, using Bionumerics (AppliedMaths, bioMérieux). The reference genomes NCTC11351 (C. jejuni) and CFSAN054106 (C. coli) (Genbank ID: LN831025 and CP028187, respectively) were used to identify the chromosome contigs of each strain sequenced using Bandage v0.8.1 (Wick et al., 2015), based on BLAST algorithm with parameters of 80% identity and 20 base pairs (bp) of minimal coverage. The central genome alignment was performed using HarvestTools (Treangen et al., 2014). The maximum likelihood phylogeny was constructed using RaxML v8.0 (Stamatakis, 2014), applying the GTR + G model and 1,000 of bootstrap, defining the clades by branch length. Recombinant regions were removed by ClonalFrameML (Didelot and Wilson, 2015). The analysis of the population structure was evaluated using hierBAPS algorithm implemented in R language v3.2.3, using following parameters: (a) Include singleton SNPs, b) maximum depth = 2 and c) number of populations = 10 (Tonkin-Hill et al., 2018). The results were visualized using Grapetree (Zhou et al., 2018), associating MLST data for each evaluated species, and Microreact (Argimón et al., 2016) associating virulence and resistance genes detected.
The detection of virulence genes was carried out using a local BLAST alignment (Altschul et al., 1990): Adherence, colonization and invasion (cadF, jlpA, racR, dnaJ, pebA, pldA, porA, ceuE, ciaB, iamB, and flaC), CDT (cdtABC operon), T6SS and T4SS genes. The prediction of coding sequences for each library was performed using Prodigal v2.6.3 (Hyatt et al., 2010). Sequence homologous genes were identified from a reference genome, with a BLAST algorithm with > 90% identity and > 60% coverage alignment with the reference. The code used for the annotation uses a Python script developed by Mestanza (2019). All the sequences obtained during this study were deposited in the GenBank database.1
The detection of resistance markers related to tetracycline [tetO and tet(W/N/W)], macrolides [ermB, rplD, rplV, and Mef (En2)], aminoglycosides [APH(2″)-Iia, APH(3′)-IIIa, and aad(6)], beta-lactams (OXA-61, OXA-184, and cepA), lincosamides (LsaE) and the multi-drug efflux pump (cmeABC) was performed using the comprehensive antibiotic resistance database (CARD) (Jia et al., 2017) and ResFinder 4.0 (Bortolaia et al., 2020). Point mutations such as T86I and V149I mutations in the DNA gyrase A (gyrA) gene associated with resistance to quinolones was checked using the output alignment of CARD and ResFinder to verify the corresponding mutation (C. jejuni ACA > ATA, C. coli ACT > ATT). Also, the A2074G/C and A2075G mutation in the 23S rRNA gene, associated with resistance to macrolides was analyzed and verified using the alignment. The results of resistance genes were associated with the previously obtained phylogeny and were also visualized using GrapeTree (Zhou et al., 2018) to evaluate the association of these genes with the genotypes found. Additionally, the correlation rate of the resistance phenotypes obtained by Antimicrobial Susceptibility Test (AST) and the genotypes by analysis was determined. Each interpretation (resistance or susceptible) for an AST result was compared to the presence or absence of a corresponding known resistance gene(s) and/or specific mutations, calculated as the sum of true positives and true negatives divided by all strains tested.
This study was developed within the framework of the project “Genomic Characterization of the molecular mechanisms of antimicrobial resistance and virulence factors in Clinical strains of Campylobacter spp. In Peru, 2010–2019,” approved by D.R. N°0154-2018-OGITT-OPE/INS. All procedures and methods were performed in accordance with ethical standards of the Declaration of Helsinki or comparable relevant guidelines and regulation. The approval of an informed consent was waived due to the retrospective nature of this study by the Institutional Committee of Research and Ethics (IRB) of the Instituto Nacional de Salud of Peru, in accordance with the national legislation and the institutional requirements for Public Health Surveillance.
The draft genome sequences for all Campylobacter spp. Strains sequenced in this study are available in GenBank under the accession numbers listed in Supplementary Table 1.
The phenotypic antimicrobial resistance to four antimicrobial agents, determined by agar microdilution method (CLSI, VET01-S2.) is presented in Table 2. The antimicrobial resistance rate in C. jejuni (n = 108) were: ciprofloxacin (CIP) 94.4% (n = 102), nalidixic acid (NA) 94.4% (n = 102), tetracycline (TE) 71.3% (n = 77), and erythromycin (E) 18.5% (n = 20). Nineteen strains (17.59%) were resistant to all four (CIP, E, TE, and NA), while 57 (52.8%) were resistant to 3 of them (CIP, TE and NA). In the case of C. coli (n = 21), the antimicrobial resistance rate was: CIP 100% (n = 21), NA 100% (n = 21), TE 85.7% (n = 18), and E 52.4% (n = 11). Resistance to all four antimicrobials was found in 11 of the strains (52.4%), while seven strains (33.3%) were resistant to CIP, TE, and NA.
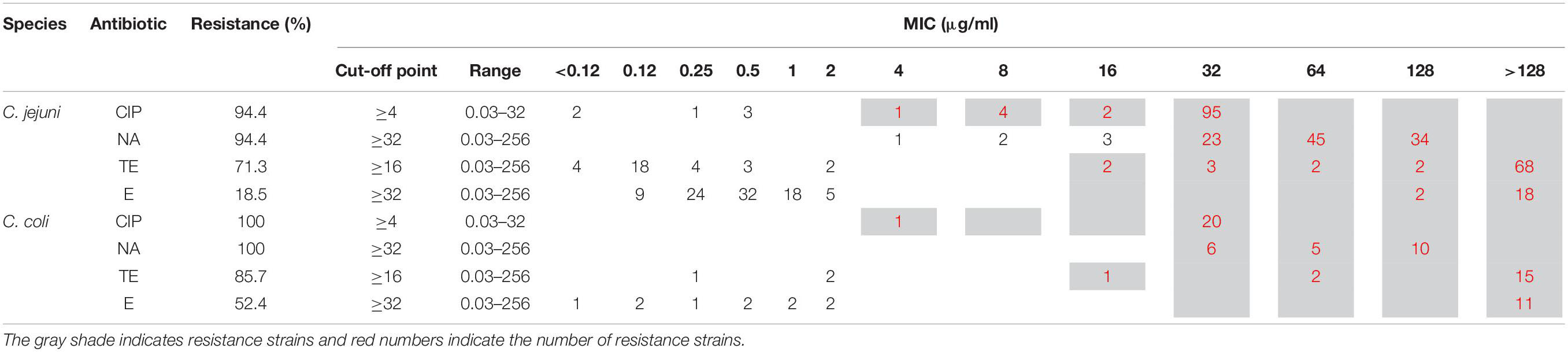
Table 2. Minimum inhibitory concentration (MIC) results observed among C. jejuni (n = 108) and C. coli (n = 21) strains.
Campylobacter strains sequenced in this study (108 C. jejuni and 21 C. coli) were grouped by species. C. jejuni strains (n = 108) showed a great variety of STs. They were divided into 30 sequence types (STs) and many of them belonged to 11 clonal complexes (CC) (Supplementary Figures 1,4A). The most frequent STs were ST-2993 (n = 24), ST-8310 (n = 12), ST-6091 (n = 9), and ST-5789 (n = 7). On the other hand, C. coli strains were represented by 8 STs and grouped within the same CC (Supplementary Figures 2, 4B). The most frequent C. coli ST was ST-8317 (n = 4). Frequency data for each ST is detailed in Supplementary Table 2.
A minimum spanning tree (MST) built using the ST information for the 129 Campylobacter strains from this study and 62 strains from another Peruvian study (Pascoe et al., 2020; Supplementary Table 3) illustrated the high diversity of C. jejuni strains not previously detected in Peru (Figure 1). The presence of some new allelic profiles not previously described in the pubMLST database2 (Supplementary Figure 3) are highlighted, whose allelic combinations are detailed in Supplementary Table 1.
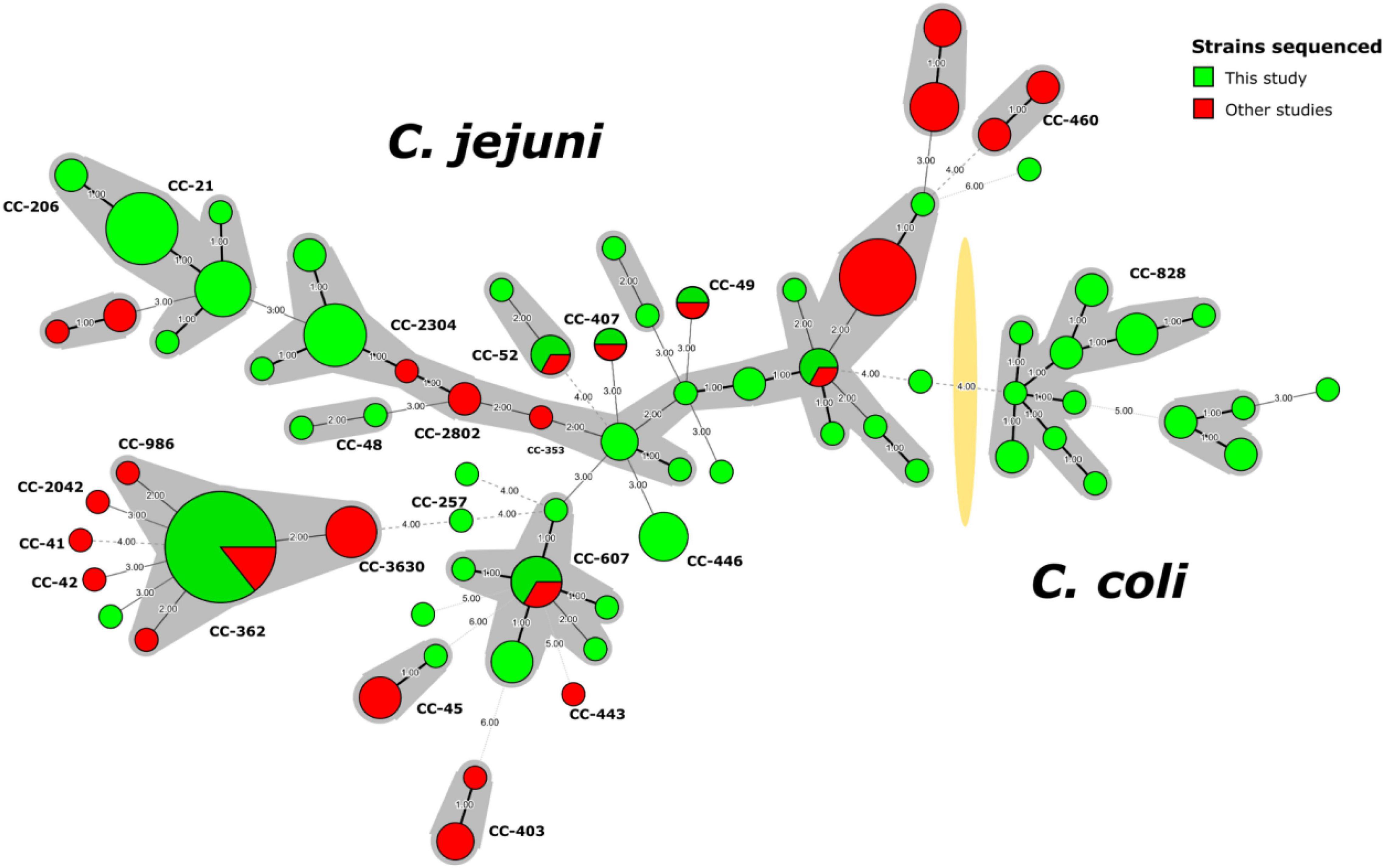
Figure 1. Minimum spanning tree of C. jejuni and C. coli strains sequenced in Peru. The nodes indicate the detected genotypes (STs) and are proportional to the number of strains. Clonal complexes (CC) are indicated with shadows around the corresponding nodes and with bold letters closed to the nodes. The yellow shadow divided Campylobacter spp. into C. jejuni and C. coli. Strains from this study (n = 129) are marked in green, while strains from other studies (n = 62) are indicated in red.
The origin of the samples, isolation dates, WGS lineages, sequencing quality, and basic assembly metrics of all genomes are detailed in Supplementary Table 1.
Phylogenetic analysis using whole genomes of C. jejuni strains from this study showed two main clades (I and II) and 5 populations (A-E) (Figure 2), which include strains from the same CC or with little allelic variation based on the MLST. All strains from clade I were recovered in Lima. On the other hand, no geographic association was observed between strains from clade II; human strains came from seven Peruvian regions (Cajamarca, Callao, Junin, La Libertad, Lambayeque, Lima and Piura) and poultry strains came from a single region (Junin). However, most strains were isolated in 2019, belonging to an outbreak that happened that year. In fact, both human and poultry strains belong to the same genotype, ST-2993. Moreover, lineages A, B, and E appear to have a distribution according to the presence and absence of resistance and virulence determinants related to lipopolysaccharides (LOS).
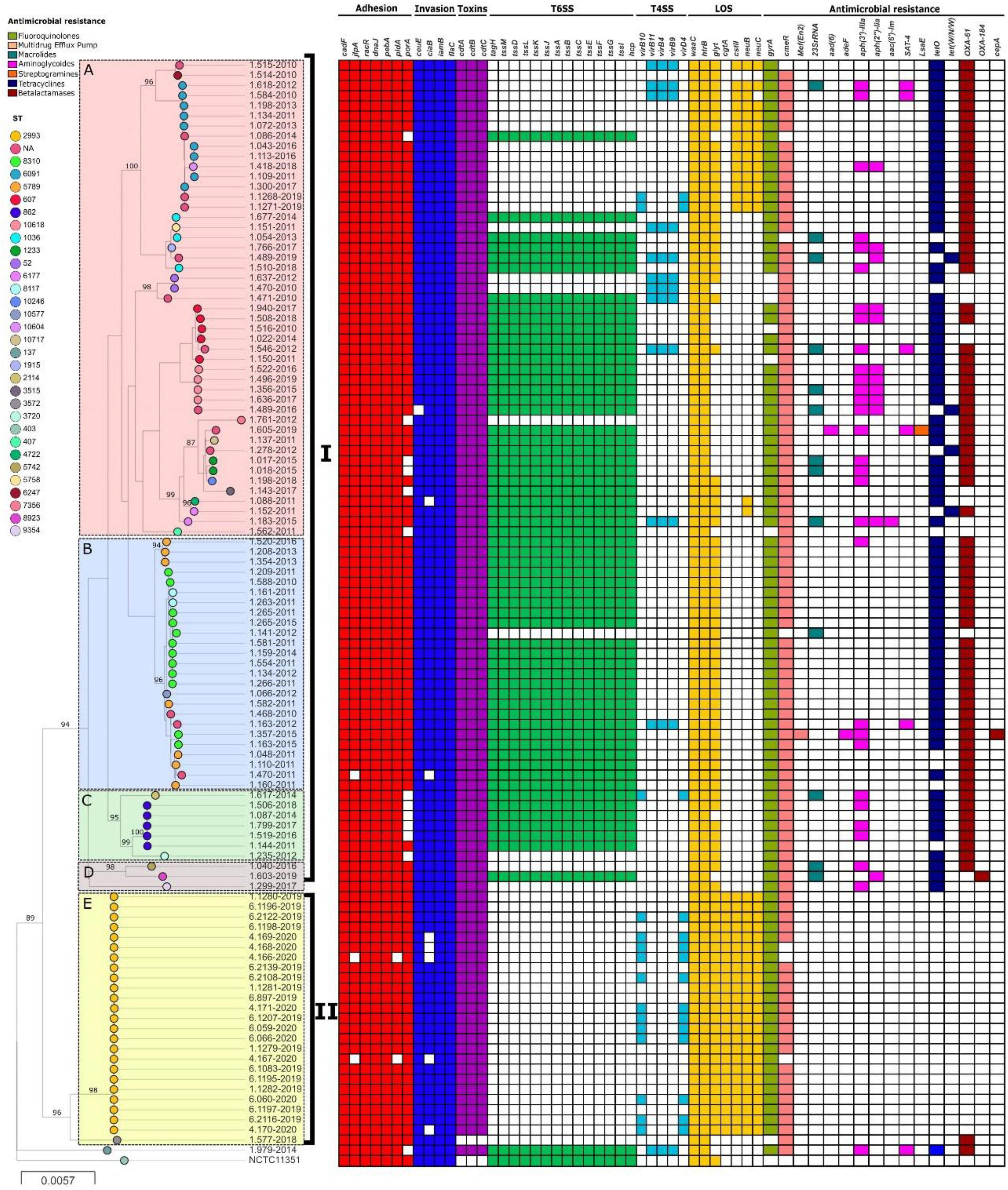
Figure 2. Phylogeny of C. jejuni (n = 108) constructed by maximum likelihood using strain NCTC11351 as reference. The strains are represented by circles connected by branches proportional to the phylogenetic distance. The code of each strain is indicated as a label parallel to the corresponding circles. The genotypes (ST) among the studied population are denoted by different colors. The clades are denoted by the numbers I and II. The HierBAPS populations are highlighted by colored boxes and named A–E. The presence of virulence genes is denoted by blocks in color based on six categories: Adhesion (Red), Invasion (Blue), Toxins (Purple), T6SS (Green), T4SS (Light Blue), and LOS (Yellow). The presence of resistance genes is denoted by blocks in color according to the spectrum of action: Fluoroquinolones (green), Multidrug Efflux pump (Red), Macrolides (Blue), Aminoglycosides (Pink), Streptogramins (Orange), Tetracycline (Blue) and Beta-Lactamases (Brown). The names of detected genes are indicated above the blocks. The absence of a gene is indicated by a white block.
In the case of C. coli, they were also grouped into two clades (I and II) which could be divided further into 4 populations with high support (Figure 3). There was no association between the lineages and the years of isolation (Supplementary Table 1). Population B is represented by strains that do not have a ST reported in the pubMLST database, and belong to an unknown not yet reported CC in C. coli. Strains belonging to lineage C were the only C. coli strains from this study carrying T6SS genes.
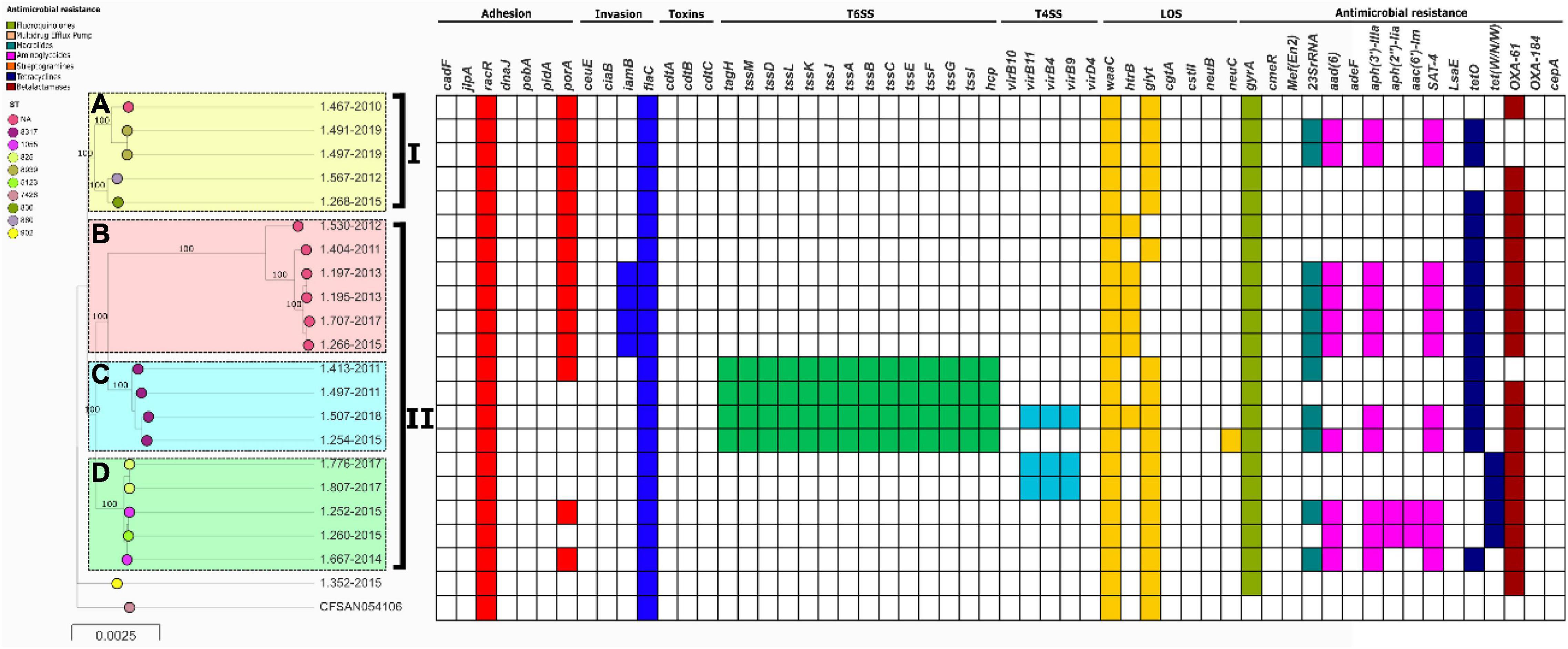
Figure 3. Phylogeny of C. coli (n = 21) constructed by the maximum likelihood using strain CFSAN054106 as reference. The strains are represented by circles connected by branches proportional to the phylogenetic distance. The code of each strain is indicated as a label parallel to the corresponding circles. The genotypes (ST) among the studied population are denoted by different colors. The clades are denoted by the numbers I and II. The HierBAPS populations lineages found are highlighted by colored boxes and named A–D. The presence of virulence genes is denoted by blocks in color based on six categories: Adhesion (Red), Invasion (Blue), Toxins (Purple), T6SS (Green), T4SS (Light Blue), and LOS (Yellow). The presence of resistance genes is denoted by blocks in color according to the spectrum of action: Fluoroquinolones (green), Multidrug Efflux pump (Red), Macrolides (Blue), Aminoglycosides (Pink), Streptogramins (Orange), Tetracycline (Blue), and Beta-Lactamases (Brown). The names of detected genes are indicated above the blocks. The absence of a gene is indicated by a white block.
In silico analysis for the presence of various genes associated with adherence and colonization were detected in all Campylobacter strains (n = 129): cadF 83.7% (n = 108), jlpA 81.4% (n = 105), racR 100% (n = 129), dnaJ 83.7% (n = 108), pebA 83.7% (n = 108), pldA 82.1% (n = 106), and porA 84.5% (n = 109). Presences of genes associated with cell invasion were detected: ceuE 82.9% (n = 107), ciaB 78.3% (n = 101), iamB 86.8% (n = 112) and flaC 100% (n = 129) of strains. The presence of CDT encoded by the cdtABC operon was detected in 82.9% (n = 107) of strains. T6SS encoded by TssA-TssM, tag and hcp gene were detected in 50.4% (n = 65) of the strains. A smaller number of Campylobacter strains carried T4SS genes: virB10 and virD4 genes in 11.6% (n = 15); virB11, virB4 and virB9 in 11.6% (n = 15). Additionally, genes associated with the synthesis of LOS were detected as follows: waaC 99.2% (n = 128), htrB 87.6% (n = 113), glycosyltransferase (glyt) 77.5% (n = 100), cgtA 18.6% (n = 24), cstII 29.5% (n = 38), neuB 31% (n = 40), and neuC 29.5% (n = 38). The percentage of virulence genes detected by species are detailed in Table 3 and Figures 2, 3.
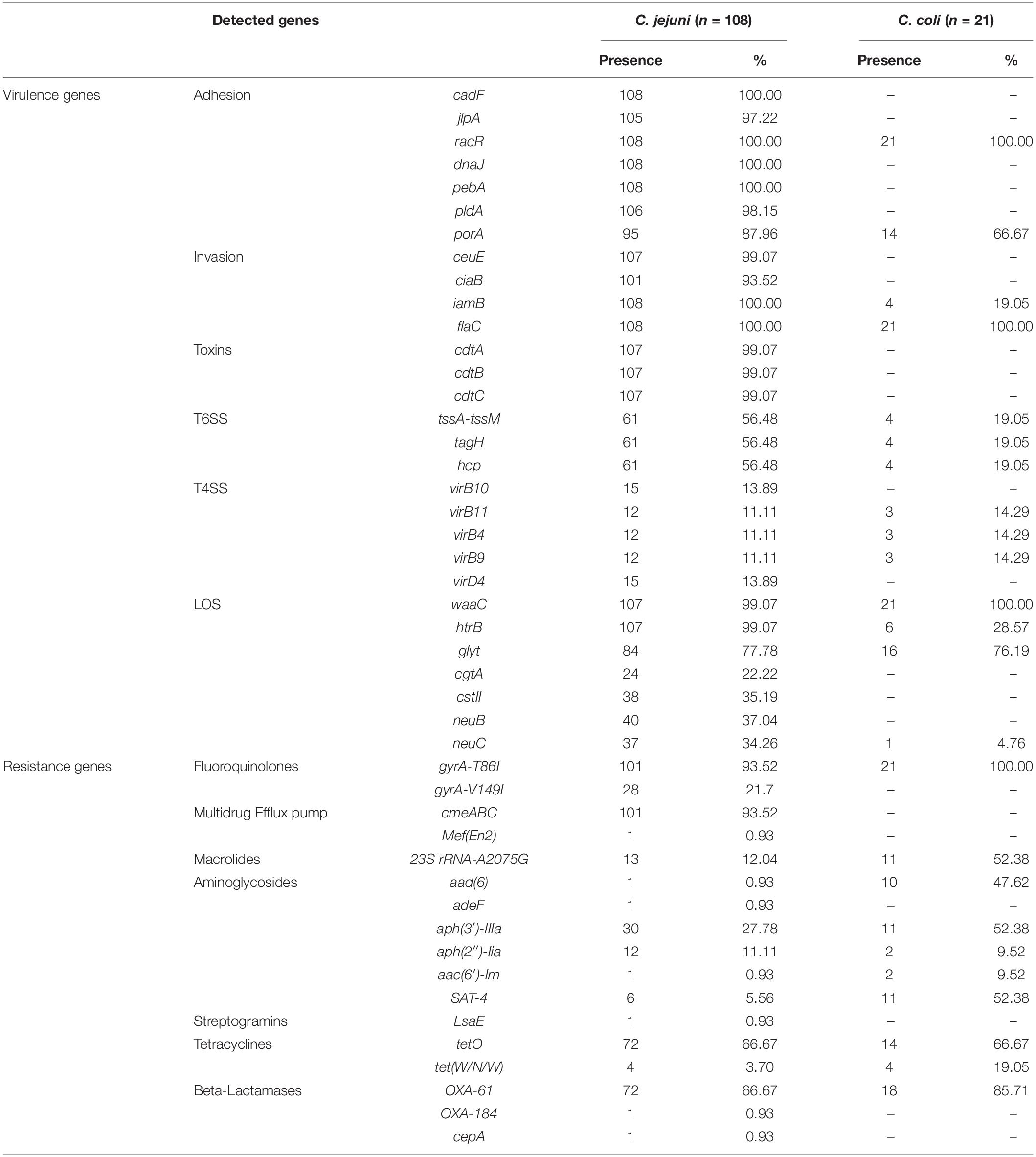
Table 3. Detection of virulence and resistance genes in Campylobacter strains (n = 129) according to the studied species.
Genes for multidrug resistance were found in Campylobacter strains, with a high frequency of resistance to ciprofloxacin due to a mutation in the gyrA gene (T86I) in 94.5% (n = 122), while another mutation (V149I) was detected only in 21.7% (n = 28). All strains with V149I mutation presented T86I mutation. In addition, the presence of the tetO gene, which is associated with resistance to tetracycline, was detected in 66.7% (n = 86). Resistance to macrolides by the A2075G mutation of 23S rRNA gene was observed in 18.6% (n = 24). The presence of multidrug efflux pump encoded by the cmeABC operon was also detected in 78.3% (n = 101) of the strains. Furthermore, the presence of blaOXA–61 gene that confers resistance to beta-lactams was observed in 69.8% (n = 90) of strains, as well as the presence of genes for resistance to aminoglycosides; aph(3′)- IIIa, aph(2″)-Iia, aad(6) and aac(6′)-Im in 31.8% (n = 41), 10.9% (n = 14), 8.5% (n = 11), and 2.3% (n = 3), respectively. The presence of the resistance genes described above, as well as other genes which code for beta-lactams and streptogramins, evaluated by strains and species, are detailed in Table 3 and Figures 2, 3.
Among the strains with a quinolone-resistant phenotype (CIP, NA), 99.2% (n = 122) presented the mutation in the gyrA gene (T86I). In these strains resistant to quinolones, 77.2% (n = 95) presented genes of cmeABC multi-efflux pump operon; likewise, the presence of this operon was observed in 100% (n = 6) of the sensitive strains. Of the 31 strains that were erythromycin-resistant phenotypes, 77.4% (n = 24) had a mutation in the 23S rRNA gene (A2075G). Eighteen of these erythromycin-resistant phenotypes carried the cmeABC operon. The presence of this operon was also detected in 84.6% (n = 83) of the antimicrobial sensitive strains for erythromycin. Of the 95 strains with tetracycline resistant phenotype, 90.5% (n = 86) carried the tetO gene and 8.4% (n = 8) carried the tet(W/N/W) gene. Additionally, 77.9% of the strains resistant to tetracycline carried the cmeABC operon. In strains sensitive to tetracycline, the presence of this operon was detected in 79.4% (n = 27) of the strains (Table 4).
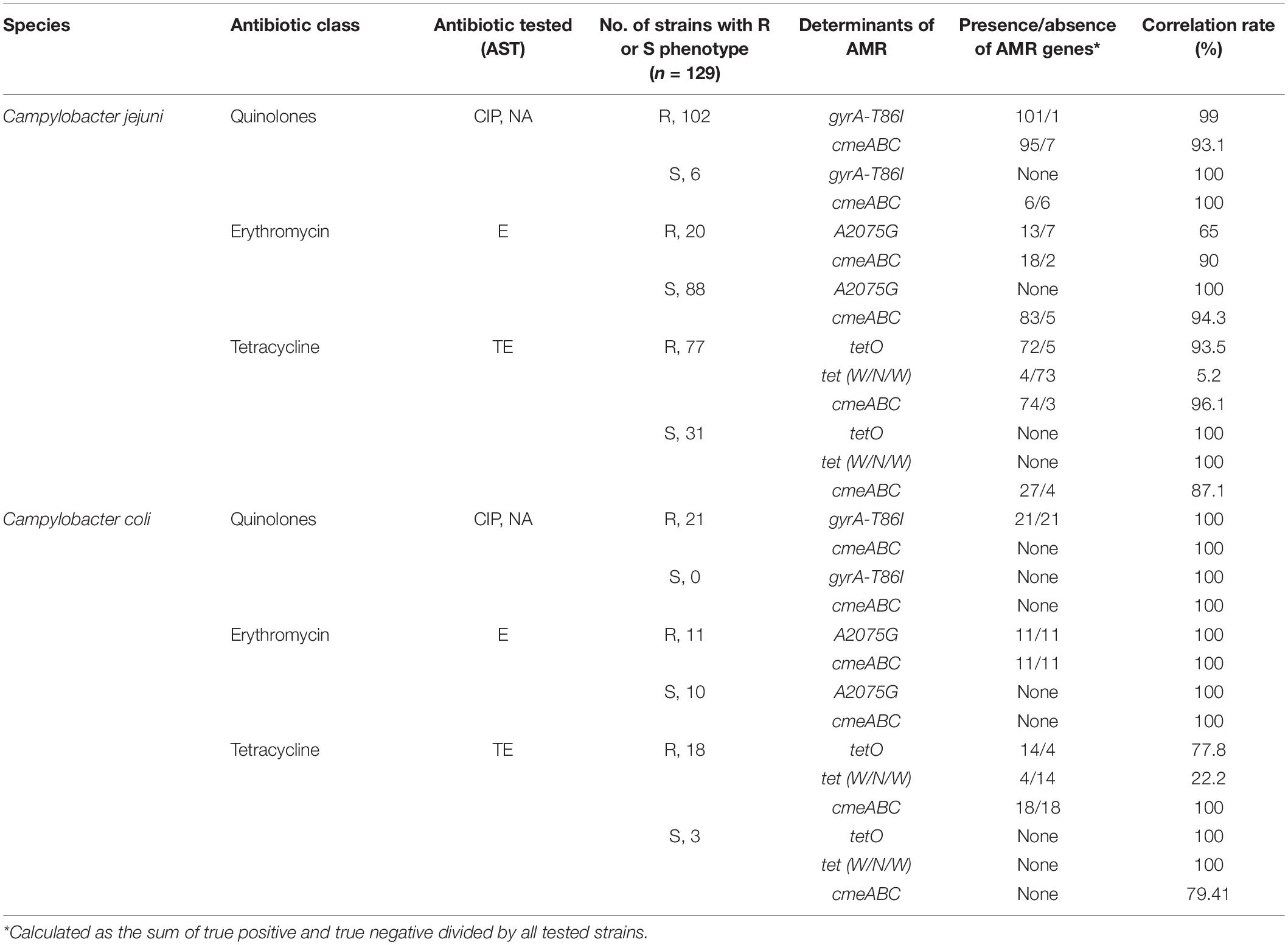
Table 4. Correlations of resistance phenotype and genotype in C. jejuni (n = 108) and C. coli (n = 21) strains.
In recent decades, the increase of antimicrobial resistance in Campylobacter has gradually become a major public health, political, social and economic problem in the world (Ramon et al., 2018). Likewise, the dramatic increase in the emergence of antimicrobial resistance observed worldwide in C. jejuni, specifically ciprofloxacin and tetracycline, has prompted research on the prevalence and molecular determinants of resistance. The present study is one of the largest genomic surveys of Campylobacter spp. Isolated in Peru. The use of whole genome sequencing (WGS) provided relevant information on the prevalence of virulence genes, antimicrobial resistance markers, as well as the phylogenetic relationships of the Campylobacter species and genotypes circulating in the period 2010–2020.
In this study, an important increase of antimicrobial resistance for the 129 Campylobacter strains was determined. During the last decade, C. jejuni reported 94.4% of resistance to quinolones, rates higher than reports from the National Antimicrobial Resistance Monitoring System for Enteric Bacteria (NARMS) of the United States (28%) (Center for Disease Control Prevention [CDC], 2019a). However, the results obtained here are comparable with Portugal (96.5%), Lithuania (91.5%), Spain (88.6%), Estonia (84.0%), and Cyprus (80%) (European Food Safety Authority [EFSA] and European Centre for Disease Prevention and Control [ECDC], 2019). Erythromycin resistance from C. jejuni also was higher (18.5%) than the United States (4.0%) (Center for Disease Control Prevention [CDC], 2019a) and European Union (2.0%) (European Food Safety Authority [EFSA] and European Centre for Disease Prevention and Control [ECDC], 2019). On the other hand, resistance level to quinolones in C. coli (100%) were comparable with 13 countries of the European Union (EU) for presenting extremely high levels (70.5–100%) of resistance to ciprofloxacin (European Food Safety Authority [EFSA] and European Centre for Disease Prevention and Control [ECDC], 2019). Also, resistance to macrolides obtained here for C. coli (52.4%) were very close to those obtained in Cyprus (12.8%). Portugal (59.5%), Malta (31.9%), and Spain (27.7%) (European Food Safety Authority [EFSA] and European Centre for Disease Prevention and Control [ECDC], 2019).
This study highlighted the wide diversity of Peruvian Campylobacter strains. Our findings revealed that 108 Peruvian C. jejuni strains were divided into 30 STs, with strains belonging to CC-362 being the most predominant (22%), followed by strains belonging to CC-21 (18%) (Supplementary Table 2 and Supplementary Figure 4). All strains from CC-362 belonged to ST-2993, which were associated with an outbreak of Guillain-Barré Syndrome in Peru (Ramos et al., 2021). Also, it is important to highlight that the frequency of this ST is very low within the PubMLST database (0.019%) (Pascoe et al., 2020). Strains belonging to CC-21 are widely distributed in the world, representing 18.9% of all strains logged to the PubMLST database. Our study reported the presence of strains in Peru belonging to CC-353, 607, and 464, which have also been found in several countries (Bravo et al., 2021; Zbrun et al., 2021), showing the transmission of these strains across very distant geographical countries, but in general, STs diversity is variable among countries, especially when comparing developed and developing countries (Noormohamed and Fakhr, 2014). Also, in this study,14 strains of C. jejuni that have a novel allelic profile or ST which were not previously reported in the MLST database were identified, an information corroborated by other diversity studies using MSLT, which detected usually strains that were not assigned STs (Noormohamed and Fakhr, 2014).
In the case of the C. coli strains, the in silico MLST analysis revealed that 67% (14/21) belonged to CC-828, while 33% (7/21) presented MLST profiles that were novel and not previously reported in the MLST database. The high prevalence of CC-828 strains in this study is consistent with its wide distribution throughout the world (Sheppard and Maiden, 2015; Bravo et al., 2021), being sometimes the unique CC detected by investigators (Noormohamed and Fakhr, 2014). The MST and phylogenetic analysis revealed the same composition; strains belonged to the same ST and/or CC grouped together into single clusters (Ramos et al., 2021). The reciprocity between these results is also reflected when applying tools such as Bayesian analysis (Epping et al., 2021).
Regarding the clades and populations found, previous studies have shown that it is common to find associations between clusters and the presence of resistance and virulence determinants, but not all the clades found in a phylogenetic tree are usually defined by other characteristics like geographic distribution or year of isolation, even among strains that correspond to a single country (Fiedoruk et al., 2019). In Peru, there are few previous studies of phylogenetic analysis of Campylobacter populations, and only one evaluated different STs Pascoe et al. (2020), which described lineages associated with the host (farm animals or humans), as well as clusters defined by their ability to cause disease (asymptomatic or symptomatic). In this study, all strains were isolated from humans and were recovered from symptomatic cases. Few strains isolated from poultry (ST-2993) grouped within the same clade (II) with other ST-2993 strains isolated from humans, complementing the study of Pascoe et al. (2020), but there is not epidemiological connection between strains of both studies. Both studies showed a wide diversity of reported genotypes in Peru, regardless of the isolation source.
The pathogenesis in Campylobacter is still not well defined. Several genes have been associated with the virulence of this pathogen; however, their functions are not fully understood in the development of Campylobacter gastroenteritis (Silva et al., 2011). In this study, a large part of the Campylobacter strains carried genes that have previously been associated with adherence, colonization, and invasion. The observed results for the presence of virulence genes were consistent with findings previously reported by several researchers where the cadF, racR, flaA, dnaJ, pebA, pldA, porA, jlpA, ceuE, ciaB, iaB, and flaC genes were also present in most of the strains that were associated with clinical cases (Ramires et al., 2020). The cdtABC operon, responsible for the presence of the cytolethal distending cytotoxin (CDT) in Campylobacter, was also present in most of the Peruvian strains from this study. Previous studies in Chile, Brazil, Korea, Poland and Ireland (Redondo et al., 2019; Ramires et al., 2020; Wysok et al., 2020; Bravo et al., 2021) have reported a high prevalence of CDT in Campylobacter strains isolated from patients with life-threatening diarrhea.
There are two secretion systems that have been described for Campylobacter: T4SS and T6SS. T4SS is encoded by genes found in the plasmid pVir, which are important for adhesion and invasion of intestinal epithelial cells (Wysok et al., 2020). In this study, the frequency of the genes present in pVir were low; these findings correlate with previous studies from Chile, Brazil, Korea, Poland and Ireland (Redondo et al., 2019; Ramires et al., 2020; Wysok et al., 2020; Bravo et al., 2021), where they also found a low prevalence of these genes, which suggests that they may not be important virulence factors in the mechanisms of pathogenesis in humans, but could facilitate the events of horizontal gene transfer that could lead to increased fitness and virulence (Redondo et al., 2019). On the other hand, the presence of genes associated with T6SS was detected in 50.4% of all Campylobacter strains, consistent with previously reports, which suggest that these genes might contribute to adherence, invasion and resistance to bile salts and to host cell oxidative stress (Wysok et al., 2020). Some authors connect T6SS with megaplasmids which might facilitate survival and virulence of the C. jejuni inside the host, enhancing hemolytic activity to lyse blood cells and then use their components as a nutrient source (Marasini et al., 2020). However, the presence of plasmids in this work was not confirmed.
The function of individual lipo-oligosaccharide (LOS) genes (waaC, htrB, glyt, cgtA, cstII, neuB, and neuC) is not very well defined; however, previous studies suggested that these genes are essential for the formation of human ganglioside-like LOS structures which can induce Guillain Barré syndrome (Zhang et al., 2015). In this study, most strains carried at least two of the genes associated with LOS biosynthesis, while all strains from ST-2993 carrying all seven genes and have been described as causing Guillain Barré in Peru in 2019 (Ramos et al., 2021).
The advantage of knowing which molecular markers for antimicrobial resistance are present in the bacterial population is crucial for the future development of prevention and control strategies to combat antimicrobial resistance in the world (Su et al., 2019; Painset et al., 2020). In our study, resistance to quinolones has generally been associated with two different markers: point mutations in the QRDR region of gyrA gene and the presence of cmeABC operon. In this study, 93.52% of C. jejuni and 100% of C. coli presented a mutation in the C257T position, which leads to the substitution of T86I in gyrA gene, a mutation frequently observed that confers high-level resistance to quinolones (Wieczorek and Osek, 2013; Liaw et al., 2019). On the other hand, the substitution of V149I was less frequently observed, which was according to other studies (Espinoza et al., 2020), emphasizing that only C. jejuni strains presented this mutation. Using the presence or absence of markers like gyrA could influence the clustering of strains inside a country (Leekitcharoenphon et al., 2018); however, due to the high rate of resistance strains, this observation was not possible. Besides, the cmeABC operon, detected only in C. jejuni (93.52%), encodes the most common multidrug efflux pump found in Campylobacter and contributes to resistance to quinolones and macrolides by decreasing the amount of the drug in cells (Bravo et al., 2021).
Regarding macrolides, the most common class of antibiotics for the treatment of Campylobacter infections, two markers of antimicrobial resistance used for many studies, A2075G mutation in the gene encoding 23S rRNA and ermB gene (Liaw et al., 2019; Caldwell et al., 2020) were analyzed. Among the strains studied here, 12.04% of C. jejuni and 52.38% of C. coli carried the mutation of 23S rRNA gene, which is the main marker associated with resistance to macrolides (Bravo et al., 2021). The presence of the ermB gene was not detected.
Currently, Campylobacter resistance to tetracycline is widespread in the world (Yan et al., 2006; Bravo et al., 2021). In this study, tetO gene, which confers resistance to tetracycline by preventing it from binding to the primary binding site on the ribosome (Wu et al., 2014), was detected in C. jejuni (66.67%) and C. coli (66.67%), data consistent with previously reported studies (Rokney et al., 2020). This represents a significant threat to public health, due to the use of antimicrobials in the poultry industry around the world, spreading drug-resistant pathogens in animals and humans.
It was also determined that C. jejuni (66.67%) and C. coli (85.71%) harbor the blaOXA–61 gene, which confers resistance to beta-lactams. This genetic marker has a high prevalence in Campylobacter worldwide (Alfredson and Korolik, 2005). The prevalence of blaOXA–61 has also been reported in Campylobacter of veterinary origin (Casagrande et al., 2020). However, beta-lactam antibiotics have limited application for the treatment of Campylobacter human infections (Yan et al., 2006).
Other genes detected confer resistance to aminoglycosides by enzymatic modification (Rukambile et al., 2019), which can be grouped based on their enzymatic activities: aminoglycosides phosphotransferases (aph), aminoglycosides nucleotidyltransferases (ant) also described as aminoglycosides adenylyltransferase (aad), and aminoglycosides acetyltransferases (aac) (Alfredson and Korolik, 2005). In this study, the genes aph(3′)-IIIa, aph (2′)-Iia, aad(6), and aac(6′)-Im were detected, with the aph(3′)-IIIa gene being the most prevalent. The prevalence of these genes is variable, but aph and ant genes are the most widely described (Yao et al., 2017; Painset et al., 2020).
Additionally, resistance discrepancies between phenotypes and genotypes were observed. A strain with phenotypic resistance to quinolones did not present a mutation in the gyrA gene (T86I); however, the presence of cmeABC operon was observed, which is also associated with resistance to quinolones, macrolides and tetracyclines, so the efflux pumps would be conferring resistance to quinolones in this strain (Yan et al., 2006). Also, 7 strains with phenotypic resistance to erythromycin did not present a mutation in the gene encoding 23S rRNA (A2075G); however, the presence of cmeABC operon was observed in 18 strains, which would be responsible for the resistance to erythromycin in strains that did not present mutation in the 23S rRNA (Caldwell et al., 2020). Finally, 9 strains with phenotypic resistance to tetracycline did not present tetO gene; however, the presence of tet(W/N/W) gene was observed in 8 strains; likewise, the presence of cmeABC operon was observed in 74 strains, which would also be responsible for the resistance to tetracycline in strains that do not present tetO or tet(W/N/W) genes.
In conclusion, this study highlights once more the importance of using WGS in the surveillance of emerging pathogens associated with foodborne diseases, providing genomic information on genetic diversity, possible virulence mechanisms, and determinants of antimicrobial resistance. In this study, genomic data on diversity, virulence, and resistance profiles in C. jejuni and C. coli strains circulating in Peru from 2010 to 2020 were provided. Furthermore, the reasons for a high rate of resistance to quinolones and tetracyclines observed among those C. jejuni and C. coli strains were described, as well the low rate of resistance to macrolides. The information provided in this study is of paramount importance in monitoring, control and prevention strategies to counteract the growing threat of resistance of this pathogen in our country. Finally, this study shows a possible source for the strains belonging to ST-2993 causing GBS in Peru during the past 5 years. The use of WGS for our future surveillance of Campylobacter circulating in the country could support the implementation of more effective One Health intervention strategies focused on the prevention and control of campylobacteriosis cases.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
WQ and RG performed the design of the work, analysis, interpretation of data, writing, and revision of the manuscript. JC-C contributed to the analysis, interpretation of data, writing, and revision of the manuscript. VH performed the experimental procedures. DF-L contributed to formal analysis and data curation. NG-E contributed to the critical revision of the article. All authors contributed to the article and approved the submitted version.
This study was developed within the framework of the project “Genomic Characterization of the molecular mechanisms of antimicrobial resistance and virulence factors in Clinical strains of Campylobacter spp. In Peru, 2010–2019,” supported by the Instituto Nacional de Salud, Lima, Peru (D.R. No. 0154-2018-OGITT-OPE/INS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Gustavo Bellido for his technical support, and all the team of the Laboratorio de Referencia Nacional de Enteropatogenos of the Instituto Nacional de Salud, Lima, Peru for their help provided during the performance of this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.802404/full#supplementary-material
Alfredson, D. A., and Korolik, V. (2005). Isolation and expression of a novel molecular class D beta-lactamase, OXA-61, from Campylobacter jejuni. Antimicrob. Agents Chemother. 49, 2515–2518. doi: 10.1128/AAC.49.6.2515-2518.2005
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anampa, D., Benites, C., Lazaro, C., Espinoza, J., Angulo, P., Diaz, D., et al. (2020). Detección del gen ermB asociado a la resistencia a macrólidos en cepas de Campylobacter aisladas de pollos comercializados en Lima, Perú. Rev. Panam. Salud Publ. 44, 1–7. doi: 10.26633/RPSP.2020.60
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed May 07, 2021).
Argimón, S., Abudahab, K., Goater, R., Fedosejev, A., Bhai, J., Glasner, C., et al. (2016). Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb. Genom. 2:e000093. doi: 10.1099/mgen.0.000093
Batchelor, R. A., Pearson, B. M., Friis, L. M., Guerry, P., and Wells, J. M. (2004). Secuencias de nucleótidos y comparación de dos grandes plásmidos conjugativos de diferentes especies de Campylobacter. Microbiology 150, 3507–3517. doi: 10.1099/mic.0.27112-0
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bortolaia, V., Kaas, R. F., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345
Bravo, V., Katz, A., Porte, L., Weitzel, T., Varela, C., Gonzales-Escalona, N., et al. (2021). Genomic analysis of the diversity, antimicrobial resistance and virulence potential of clinical Campylobacter jejuni and Campylobacter coli strains from Chile. PLoS Negl. Trop. Dis. 15:e0009207. doi: 10.1371/journal.pntd.0009207
Caldwell, D. B., Wang, Y., and Lin, J. (2020). Development, stability, and molecular mechanisms of macrolide resistance in Campylobacter jejuni. Antimicrob. Agents Chemother. 52:3947. doi: 10.1128/AAC.00450-08
Casagrande, P., Guelfi, G., Bellucci, S., De Luca, S., Di Gregorio, S., Pieramati, C., et al. (2020). Beta-lactam resistance in Campylobacter coli and Campylobacter jejuni chicken isolates and the association between blaOXA-61 gene expression and the action of β-lactamase inhibitors. Vet. Microbiol. 241:108553. doi: 10.1016/j.vetmic.2019.108553
Center for Disease Control Prevention [CDC] (2018). National Antimicrobial Resistance Monitoring System for Enteric Bacteria (NARMS): Human Isolates Surveillance Report for 2015 (Final Report). Georgia: U.S. Department of Health and Human Services, CDC.
Center for Disease Control Prevention [CDC] (2019a). Antibiotic Resistance Threats in the United States. Georgia: U.S. Department of Health and Human Services, CDC.
Center for Disease Control Prevention [CDC] (2019b). Campylobacter (Campylobacteriosis). Questions and Answers. Available online at: https://www.cdc.gov/campylobacter/faq.html (accessed November 21, 2020).
Clinical Laboratory Standards Institute [CLSI] (2016). Methods for Antimicrobial Dilution and Disk Susceptibility Testing of Infrequently Isolated or Fastidious Bacteria, 3rd Edn. Pennsylvania: Clinical and Laboratory Standards Institute.
Clinical Laboratory Standards Institute [CLSI] (2018). Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated from Animals, 5th Edn. Pennsylvania: Clinical and Laboratory Standards Institute.
Coil, D., Jospin, G., and Darling, A. (2015). A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinform. Oxf. Engl. 31, 587–589. doi: 10.1093/bioinformatics/btu661
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
Epping, L., Walther, B., Piro, R. M., Knüver, M., Huber, C., Thürmer, A., et al. (2021). Genome-wide insights into population structure and host specificity of Campylobacter jejuni. Sci. Rep. 11:10358. doi: 10.1038/s41598-021-89683-6
Espinoza, N., Rojas, J., Pollett, S., Meza, R., Patiño, L., Leiva, M., et al. (2020). Validation of the T86I mutation in the gyrA gene as a highly reliable real time PCR target to detect Fluoroquinolone-resistant Campylobacter jejuni. BMC Infect. Dis. 20:518. doi: 10.1186/s12879-020-05202-4
European Food Safety Authority [EFSA], and European Centre for Disease Prevention, and Control [ECDC] (2019). The European Union summary report on antimicrobial resistance in zoonotic and indicator bacteria from humans, animals and food in 2017. EFSA J. 17:e05598. doi: 10.2903/j.efsa.2019.5598
Farace, M., and Viñas, M. (2007). Manual de Procedimientos Para el Aislamiento y Caracterización de Campylobacter spp. Buenos Aires: Instituto Nacional de Enfermedades Infecciosas (INEI).
Fiedoruk, K., Daniluk, T., Rozkiewicz, D., Oldak, E., Prasad, S., and Swiecicka, I. (2019). Whole-genome comparative analysis of Campylobacter jejuni strains isolated from patients with diarrhea in northeastern Poland. Gut Pathog. 11:32. doi: 10.1186/s13099-019-0313-x
Figueroa, G., Troncoso, M., López, C., Rivas, P., and Toro, M. (2009). Occurrence and enumeration of Campylobacter spp. during the processing of Chilean broilers. BMC Microbiol. 9:94. doi: 10.1186/1471-2180-9-94
Horrocks, S. M., Anderson, R. C., Nisbet, D. J., and Ricke, S. C. (2009). Incidence and ecology of Campylobacter jejuni and coli in animals. Anaerobe 15, 18–25. doi: 10.1016/j.anaerobe.2008.09.001
Humphrey, T., O’Brien, S., and Madsen, M. (2007). Campylobacters as zoonotic pathogens: a food production perspective. Int. J. Food Microbiol. 117, 237–257. doi: 10.1016/j.ijfoodmicro.2007.01.006
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M., Larimer, F., and Hauser, L. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Jagadeesan, B., Gerner-Smidt, P., Allard, M. W., Leuillet, S., Winkler, A., Xiao, Y., et al. (2019). The use of next generation sequencing for improving food safety: translation into practice. Food Microbiol. 79, 96–115. doi: 10.1016/j.fm.2018.11.005
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Kaakoush, N. O., Castaño-Rodríguez, N., Mitchell, H. M., and Man, S. M. (2015). Global epidemiology of Campylobacter infection. Clin. Microbiol. Rev. 28, 687–720. doi: 10.1128/CMR.00006-15
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Leekitcharoenphon, P., Garcia-Graells, C., Botteldoorn, N., Dierick, K., Kempf, I., Olkkola, S., et al. (2018). Comparative genomics of quinolone-resistant and susceptible Campylobacter jejuni of poultry origin from major poultry producing European countries (GENCAMP). EFSA Support. Publ. 15:1398E. doi: 10.2903/sp.efsa.2018.EN-1398
Liaw, J., Hong, G., Davies, C., Elmi, A., Sima, F., Stratakos, A., et al. (2019). The Campylobacter jejuni type VI secretion system enhances the oxidative stress response and host colonization. Front. Microbiol. 10:2864. doi: 10.3389/fmicb.2019.02864
Lior, H. (1984). New, extended biotyping scheme for Campylobacter jejuni, Campylobacter coli, and Campylobacter laridis. J. Clin. Microbiol. 20, 636–640. doi: 10.1128/jcm.20.4.636-640.1984
Lopez, C., Giacoboni, G., and Sommerfelt, I. (2017). Resistencia a antimicrobianos de Campylobacter jejuni aislados de pollos, provincia de Buenos Aires, Argentina. Rev. Med. Vet. 98, 08–12.
Lucas, J., Vilca, M., and Ramos, D. (2013). Presencia de campylobacter spp en canales y ciegos de pollos de engorde en Lima, Perú. Rev. Investig. Vet. Perú. 24, 346–352.
Marasini, D., Karki, A. B., Bryant, J. M., Sheaff, R. J., and Fakhr, M. K. (2020). Molecular characterization of megaplasmids encoding the type VI secretion system in Campylobacter jejuni isolated from chicken livers and gizzards. Sci. Rep. 10, 1–10. doi: 10.1038/s41598-020-69155-z
Mardones, P. G., and López, M. J. (2017). Implicancias de Campylobacter spp. como patógeno alimentario. Chil. J. Agricanim. Sci. 33, 73–83. doi: 10.4067/S0719-38902017005002005
Mestanza, O. (2019). Like Blast Score Ratio (BSR). Available online at: https://github.com/OrsonMM/Blast-score-ratio-for-genomics (accessed May 01, 2021).
Moya-Salazar, J., Teran-Vasquez, A., and Salazar-Hernandez, H. (2018). Alta resistencia antimicrobiana a Fluoroquinolonas por Campylobacter en pacientes pediátricos de un hospital peruano. Rev. Peru. Med. Exp. Salud Publ. 35, 155–156. doi: 10.17843/rpmesp.2018.351.3607
Mullner, P., Spencer, S. E., Wilson, D. J., Jones, G., Noble, A. D., Midwinter, A. C., et al. (2009). Assigning the source of human campylobacteriosis in New Zealand: a comparative genetic and epidemiological approach. Infect. Genet. Evol. 9, 1311–1319. doi: 10.1016/j.meegid.2009.09.003
Newell, D. G., and Fearnley, C. (2003). Sources of Campylobacter colonization in broiler chickens. Appl. Environ. Microbiol. 69, 4343–4351. doi: 10.1128/AEM.69.8.4343-4351.2003
Noormohamed, A., and Fakhr, M. K. (2014). Molecular typing of Campylobacter jejuni and Campylobacter coli isolated from various retail meats by MLST and PFGE. Foods 3, 82–93. doi: 10.3390/foods3010082
Painset, A., Day, M., Doumith, M., Rigby, J., Jenkins, C., Grant, K., et al. (2020). Comparison of phenotypic and WGS-derived antimicrobial resistance profiles of Campylobacter jejuni and Campylobacter coli isolated from cases of diarrhoeal disease in England and Wales, 2015–16. J. Antimicrob. Chemother. 75, 883–889. doi: 10.1093/jac/dkx301
Pascoe, B., Schiaffino, F., Murray, S., Méric, G., Bayliss, S. C., Hitchings, M. D., et al. (2020). Genomic epidemiology of Campylobacter jejuni associated with asymptomatic pediatric infection in the Peruvian Amazon. PLoS Negl. Trop. Dis. 14:e0008533. doi: 10.1371/journal.pntd.0008533
Payot, S., Bolla, J. M., Corcoran, D., Fanning, S., Mégraud, F., and Zhang, Q. (2006). Mechanisms of fluoroquinolone and macrolide resistance in Campylobacter spp. Microbes Infect. 8, 1967–1971. doi: 10.1016/j.micinf.2005.12.032
Perales, M., Camiña, M., and Quiñones, C. (2002). Infección por Campylobacter y Shigella como causa de diarrea aguda acuosa en niños menores de dos años en el distrito de la Victoria, Lima-Perú. Rev. Peru. Med. Exp. Salud Publ. 19, 186–192.
Ramires, T., de Oliveira, M. G., Kleinubing, N. R., de Fátima, S., Mata, M. M., Iglesias, M. A., et al. (2020). Genetic diversity, antimicrobial resistance, and virulence genes of thermophilic Campylobacter isolated from broiler production chain. Braz. J. Microbiol. 51, 2021–2032. doi: 10.1007/s42770-020-00314-0
Ramon, P., Sati, H., and Galas, M. (2018). Enfoque de Una Salud en las acciones para enfrentar la resistencia a los antimicrobianos desde una óptica latinoamericana. Rev. Peru. Med. Exp. Salud Publ. 35, 103–109. doi: 10.17843/rpmesp.2018.351.3605
Ramos, A. P., Leonhard, S. E., Halstead, S. K., Cuba, M. A., Castañeda, C. C., Dioses, J. A., et al. (2021). Guillain-Barré syndrome outbreak in Peru 2019 associated with Campylobacter jejuni infection. Neurol. Neuroimmunol. Neuroinflamm. 8:e952. doi: 10.1212/NXI.0000000000000952
Redondo, N., Carroll, A., and McNamara, E. (2019). Molecular characterization of Campylobacter causing human clinical infection using whole-genome sequencing: virulence, antimicrobial resistance and phylogeny in Ireland. PLoS One 14:e0219088. doi: 10.1371/journal.pone.0219088
Rokney, A., Valinsky, L., Vranckx, K., Feldman, N., Agmon, V., Moran-Gilad, J., et al. (2020). WGS-based prediction and analysis of antimicrobial resistance in Campylobacter jejuni isolates from Israel. Front. Cell. Infect. Microbiol. 10:365. doi: 10.3389/fcimb.2020.00365
Rukambile, E., Sintchenko, V., Muscatello, G., Kock, R., and Alders, R. (2019). Infection, colonization and shedding of Campylobacter and Salmonella in animals and their contribution to human disease: a review. Zoonoses Public Health 66, 562–578. doi: 10.1111/zph.12611
Sheppard, S. K., and Maiden, M. C. (2015). The evolution of Campylobacter jejuni and Campylobacter coli. Cold Spring Harb. Perspect. Biol. 7:a018119. doi: 10.1101/cshperspect.a018119
Silva, J., Leite, D., Fernandes, M., Mena, C., Gibbs, P. A., and Teixeira, P. (2011). Campylobacter spp. as a foodborne pathogen: a review. Front. Microbiol. 2:200. doi: 10.3389/fmicb.2011.00200
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Su, M., Satola, S. W., and Read, T. D. (2019). Genome-based prediction of bacterial antibiotic resistance. J. Clin. Microbiol. 57, e01405–e01418. doi: 10.1128/JCM.01405-18
Tamborini, A., Casabona, L., Viñas, M., Asato, V., Hoffer, A., Farace, M., et al. (2012). Campylobacter spp.: prevalencia y caracterización feno-genotípica de aislamientos de pacientes con diarrea y de sus mascotas en la provincia de La Pampa, Argentina. Rev. Argent. Microbiol. 44, 266–271.
Tonkin-Hill, G., Lees, J., Bentley, S., Frost, S., and Corander, J. (2018). RhierBAPS: an R implementation of the population clustering algorithm hierBAPS. Wellcome Open Res. 3:93. doi: 10.12688/wellcomeopenres.14694.1
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Valenzuela, C., Legarraga, P., Peña, A., Arenas, A., Berkowitz, L., Ramírez, G., et al. (2018). Etiologic and clinical characterization of community acquired gastroenteritis in adult patients in a Chilean emergency room by the FilmArray GI panel. PLoS One 13:e0207850. doi: 10.1371/journal.pone.0207850
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wieczorek, K., and Osek, J. (2013). Antimicrobial resistance mechanisms among Campylobacter. Biomed. Res. Int. 2013:340605. doi: 10.1155/2013/340605
Wu, Z., Sippy, R., Sahin, O., Plummer, P., Vidal, A., Newell, D., et al. (2014). Genetic diversity and antimicrobial susceptibility of Campylobacter jejuni isolates associated with sheep abortion in the United States and Great Britain. J. Clin. Microbiol. 52:1853. doi: 10.1128/JCM.00355-14
Wysok, B., Wojtacka, J., Hänninen, M. L., and Kivistö, R. (2020). Antimicrobial resistance and virulence-associated markers in Campylobacter strains from diarrheic and non-diarrheic humans in Poland. Front. Microbiol. 11:1799. doi: 10.3389/fmicb.2020.01799
Yan, M., Sahin, O., Lin, J., and Zhang, Q. (2006). Role of the CmeABC efflux pump in the emergence of fluoroquinolone-resistant Campylobacter under selection pressure. J. Antimicrob. Chemother. 58, 1154–1159. doi: 10.1093/jac/dkl412
Yao, H., Liu, D., Wang, Y., Zhang, Q., and Shen, Z. (2017). High prevalence and predominance of the aph(2″)-If gene conferring aminoglycoside resistance in Campylobacter. Antimicrob. Agents Chemother. 61:e00112-17. doi: 10.1128/AAC.00112-17
Zbrun, M. V., Rossler, E., Soto, L. P., Rosmini, M., Sequeira, G., Frizzo, L., et al. (2021). Molecular epidemiology of Campylobacter jejuni isolates from the broiler production chain: first report of MLST profiles in Argentina. Rev. Argent. Microbiol. 53, 59–63. doi: 10.1016/j.ram.2020.06.005
Zhang, M., Gilbert, M., Yuki, N., Cao, F., Li, J., Liu, H., et al. (2015). Association of anti-GT1a antibodies with an outbreak of Guillain-Barré syndrome and analysis of ganglioside mimicry in an associated Campylobacter jejuni strain. PLoS One 10:e0131730. doi: 10.1371/journal.pone.0131730
Zhao, S., Tyson, G. H., Chen, Y., Li, C., Mukherjee, S., Young, S., et al. (2016). Whole-Genome Sequencing analysis accurately predicts antimicrobial resistance phenotypes in Campylobacter spp. Appl. Environ. Microbiol. 82, 459–466. doi: 10.1128/AEM.02873-15
Keywords: Campylobacter, whole-genome sequencing, genomic analysis, antimicrobial resistance, virulence genes
Citation: Quino W, Caro-Castro J, Hurtado V, Flores-León D, Gonzalez-Escalona N and Gavilan RG (2022) Genomic Analysis and Antimicrobial Resistance of Campylobacter jejuni and Campylobacter coli in Peru. Front. Microbiol. 12:802404. doi: 10.3389/fmicb.2021.802404
Received: 26 October 2021; Accepted: 14 December 2021;
Published: 11 January 2022.
Edited by:
Antonio Battisti, Experimental Zooprophylactic Institute of the Lazio and Tuscany Regions (IZSLT), ItalyReviewed by:
Ben Pascoe, University of Bath, United KingdomCopyright © 2022 Quino, Caro-Castro, Hurtado, Flores-León, Gonzalez-Escalona and Gavilan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Willi Quino, d3F1aW5vQGlucy5nb2IucGU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.