 Tingting XiangWei ZhouCailing XuJing XuRui LiuNuo WangLiang XuYu ZhaoMinhui LuoXiaoxin MoZeyang Mao
Tingting XiangWei ZhouCailing XuJing XuRui LiuNuo WangLiang XuYu ZhaoMinhui LuoXiaoxin MoZeyang Mao Yongji Wan*
Yongji Wan*- Laboratory of Invertebrate Pathology and Applied Microbiology, College of Sericulture, Textile and Biomass Sciences, Southwest University, Chongqing, China
Serratia marcescens is a common bacterium well-known for the red secondary metabolite prodigiosin. However, color mutants have long been described. Non-pigmented strains can be found to exist both naturally and under laboratory conditions. It is unclear why S. marcescens loses prodigiosin synthesis capacity in certain conditions. In the present study, we find that the spontaneous color mutants arise within a few generations (about five passages) and rapidly replace the wild-type parent cells (about 24 passages), which indicates a growth advantage of the former. Although, the loss of prodigiosin synthesis genes (pigA-N) is frequently reported as the major reason for pigment deficiency, it was unexpected that the whole gene cluster is completely preserved in the different color morphotypes. Comparative transcriptomic analysis indicates a dramatic variation at the transcriptional level. Most of the pig genes are significantly downregulated in the color morphotypes which directly lead to prodigiosin dyssynthesis. Besides, the transcriptional changes of several other genes have been noticed, of which transcriptional regulators, membrane proteins, and nearly all type VI secretion system (T6SS) components are generally downregulated, while both amino acid metabolite and transport systems are activated. In addition, we delete the transcription regulator slyA to generate a non-pigmented mutant. The ΔslyA strain loses prodigiosin synthesis capacity, but has a higher cell density, and surprisingly enhances the virulence as an entomopathogen. These data indicate that S. marcescens shuts down several high-cost systems and activates the amino acid degradation and transport pathways at the transcriptional level to obtain extra resources, which provides new insights into the competitive growth advantage of bacterial spontaneous color mutants.
Introduction
Serratia marcescens, a bacterium of the Enterobacteriaceae family which produces a red secondary metabolite prodigiosin, is reported with multiple color phenotypes (Zhou et al., 2016b). This bacterium is widely distributed in diverse environments, including water, soil, plants, and animals (Grimont and Grimont, 1978). In addition to the saprophytic life cycle, both mutualistic and parasitic roles of S. marcescens have been reported. Serratia marcescens may live in mutualistic symbiosis with fungi, plants, or animals. The mutualistic interactions between S. marcescens UENF-22GI and a biocontrol fungus Trichoderma longibrachiatum are shown not only beneficial for plant growth but also increased ecological fitness of the bacterium (de Andrade Reis et al., 2021). The S. marcescens strain NBRI1213 is beneficial to host plants by promoting growth and controlling a typical soil-borne plant pathogen Phytophthora nicotianae (Lavania et al., 2006). Several S. marcescens strains can colonize in the gut of mosquitoes and render host resistance to Plasmodium infection (Chen et al., 2017; Bai et al., 2019). Serratia marcescens is also a well-known opportunistic pathogen, which infects plants, invertebrates, and vertebrates. In plants, S. marcescens causes cucurbit yellow vine disease after invading into host phloem elements (Rascoe et al., 2003). Many reports indicate that the bacterium infects various invertebrates, mainly insects (Burritt et al., 2016; Zhou et al., 2016b), which causes deadly septicemia. Serratia marcescens has also been frequently reported as a common nosocomial infection pathogen (Schuppener et al., 2017). Like other bacterial pathogens, S. marcescens produces abundant virulence factors, such as extracellular enzymes including proteases, nucleases, lipases, lecithinase, hydrolytic enzymes, and toxins, etc. (Hines et al., 1988; Hejazi and Falkiner, 1997; Aucken and Pitt, 1998), which are controlled by transcriptional regulators, such as rssAB, slyA, and eepR, etc. (Ellison and Miller, 2006; Lin et al., 2010; Shanks et al., 2017). Previous reports have suggested that bacterial pigments are probably related to the pathogenic process (Liu and Nizet, 2009). However, unlike other pathogens, the red pigment prodigiosin seems not to be a necessary virulence factor that is required for S. marcescens pathogenesis (Zhou et al., 2016b), and its physiological or ecological roles remain largely unclear.
The prodigiosin produced by S. marcescens has a linear tripyrrole skeleton (2-methyl-3-pentyl-6-methoxyprodiginine), which is reported with antimicrobial, antialgal, antiprotozoal, antimalarial, antitumor, immunosuppressive, and antiviral activities (Williamson et al., 2007; Genes et al., 2011; Stankovic et al., 2014; Zhou et al., 2016c). The biosynthesis pathway of prodigiosin in S. marcescens has been well-studied (Williamson et al., 2006). Generally, the pathway can be divided into two parts which correlate with two major components, the 2-methyl-3-n-amyl-pyrrole (MAP) and 4-methoxy-2,2′-bipyrrole-5-carboxyaldehyde (MBC). In the presence of the enzyme PigC, MAP, and MBC are finally condensed to form the red pigment prodigiosin. The prodigiosin biosynthesis gene cluster (Pig gene cluster) consists of 14 opening reading frames (pigA-N), of which pigB, pigD, and pigE are responsible for the biosynthesis of MAP, while pigA and pigF-N are involved in the biosynthesis of MBC (Harris et al., 2004). Based on previous reports, all the pig genes are essential for prodigiosin synthesis. Williamson et al. (2005) have constructed several in-frame mutants of pigA-N in Serratia, and most of them (except for pigK) show reduced prodigiosin production. It has been proved that non-pigmented bacterial strains, such as S. marcescens Sma12 or other species, e.g., Escherichia coli XL-1, acquire prodigiosin-producing ability by gene transfer of the whole Pig gene cluster (Harris et al., 2004; Williamson et al., 2005; Coulthurst et al., 2006).
The biosynthesis of prodigiosin is modulated by various genetic and environmental factors. Several transcription factors have been revealed to involve in the prodigiosin biosynthesis process. The transcription regulators pigP, slyA (alternative: rap), pigR, pigV, pigS, eepR, and hfq show positive effects, while CRP, pigX, pigU, hexS, pigT, rpoS, pigZ, copA, rsmA, rsmC, rcsB, metR, and cpxR show negative effects (Williamson et al., 2006; Gristwood et al., 2008; Wilf et al., 2011; Wilf and Salmond, 2012; Hampton et al., 2016; Shanks et al., 2017; Pan et al., 2020a,b; Sun et al., 2020). Quorum sensing system, e.g., SpnIR and LuxS/AI-2, a global regulatory system, is also involved in prodigiosin biosynthesis in S. marcescens. SpnR usually acts as a negative regulator which is inhibited by AHLs that are synthesized by SpnI (Horng et al., 2002). LuxS/AI-2 often acts as a second quorum sensing system to control certain phenotypes (Sun et al., 2016). The two-component systems RssB/RssA, PigQ/W, and EnvZ/OmpR also regulate prodigiosin production in Serratia species (Fineran et al., 2005; Horng et al., 2010; Jia et al., 2021). It is worth noting that all these regulators may result in multiple unexpected phenotypic changes in Serratia, including biosurfactants, antibiotics and exoenzymes production, cell motility, and virulence. In addition, by using transposon insertion sequencing technology, several potential gene targets have been provided which might be involved in the prodigiosin synthesis of S. marcescens (Pan et al., 2019; Jia et al., 2021). Similar to other secondary metabolites, the biosynthesis of prodigiosin is extremely sensitive to environmental conditions. Many environmental factors, such as temperature, pH, oxygen concentration, media composition, ionic strength, light, and inorganic phosphate availability have been proved to affect prodigiosin production (Williamson et al., 2006). However, non-pigmented spontaneous mutants also arise, while the wild-type S. marcescens strains are subjected to continuous incubation under relatively stable laboratory conditions (Zhou et al., 2016b). It is interesting why S. marcescens loses pigment synthesis capacity in certain environments.
Both pigmented and non-pigmented S. marcescens strains naturally exist. According to the genetic data in the GenBank database, the majority of sequenced S. marcescens strains are non-pigmented which naturally lack the essential Pig gene cluster. Indeed, of all the 773 genome sequences of S. marcescens by July 7, 2021, there are only 51 strains contain the complete Pig gene cluster. Moreover, the largest proportion of non-pigmented isolates are obtained from clinical settings. We previously reported that the spontaneous non-pigmented mutants were derived from a wild-type S. marcescens strain during continuous cultivations in vitro (Zhou et al., 2016b). Except for the pigmentation features, the virulence of the mutants was very similar to each other. In the present study, we have found a growth difference among different color morphotypes. Transcriptomic and homologous-recombination-based gene knockout approaches were applied to explore why S. marcescens spontaneous non-pigmented strain has a growth advantage in laboratory conditions. According to our findings, S. marcescens both shuts down and activates several systems, including suppressing pig genes at the transcriptional level, to gain a growth advantage in certain conditions.
Materials and Methods
Microorganism, Culture, and Sub-Culture Conditions
In this study, the wild-type S. marcescens strain SCQ1 (CCTCC AB 2010221) was isolated from the hemolymph of diseased silkworm larva (Bombyx mori) previously, which was stored at −80°C (Zhou et al., 2016b). Unless mentioned specifically, all S. marcescens strains were routinely inoculated on Luria-Bertani (LB) agar (yeast extract 5 g/L, peptone 10 g/L, NaCl 10 g/L, and agar 20 g/L) or LB broth inoculated at 28°C. Where required, the medium was supplemented with ampicillin (50 μg/ml), chloramphenicol (50 μg/ml), and rifampicin (10 μg/ml).
The continuous sub-culture of the wild-type strain SCQ1 was performed. A single colony of SCQ1 was transferred to a 50 ml LB medium to prepare a seed culture with an OD600 of 0.6. The 1% of the seed culture was then transferred into a liquid LB medium (pH 7.0), and incubated at 28°C for 24 h with shaking (200 rpm) which was defined as the first passage of continuous sub-culture. Every 24 h, 500 μl of culture was transferred to 50 ml fresh LB broth. Before each sub-culture step, the samples were collected and 50 μl of diluted cultures were plated onto LB agar, incubated at 28°C for 48 h. The phenotypes of newly formed colonies were recorded and the proportions of color morphotypes were statistically analyzed. Triplicate experiments have been performed.
Growth Curves and Prodigiosin Production Assays
The growth of all S. marcescens strains was measured in a Bioscreen C instrument (Growth Curves United States). The overnight cultures were adjusted to an OD600 of 0.6 using fresh LB medium, and 2 μl of which were inoculated in a 100-well honeycomb plate that each contained 198 μl of LB medium. In addition, 200 μl of the LB medium was added to three wells as blank controls. The honeycomb plate was placed in the Bioscreen C instrument (Growth Curves United States) incubated at 28°C and the OD600 was automatically measured every 2 h. The experiment was performed with three technical replicates and the growth curves were finally plotted with the growth time as the abscissa and the OD600 as the ordinate.
The assay of prodigiosin production was performed as follows: the colonies of S. marcescens were inoculated in 50 ml of LB medium and incubated at 28°C for 16 h with shaking (200 rpm). The 1 ml of each culture was harvested by centrifugation at 12,000 rpm for 5 min. The pellet was resuspended in 1 ml distilled water to measure the OD600 value and the supernatant was collected to measure the A534 value (Slater et al., 2003). The prodigiosin production was calculated and plotted as (A534/OD600). Triplicate experiments have been performed.
Transcriptome Sequencing and Comparative Analysis of SCQ1 and SCQ1-3M
A single colony of SCQ1 and SCQ1-3M was transferred to an LB medium to prepare a seed culture with an OD600 value of 0.6. The 1% of seed culture was inoculated into 50 ml of LB medium and cultured at 28°C, 180 rpm for 8 h. Total RNA was extracted by the TRIzol-based method (Life Technologies, CA, United States). RNA degradation degree and potential contamination were monitored on 1% agarose gels. RNA purity and integrity were checked using NanoPhotometer® spectrophotometer (IMPLEN, CA, United States) and Bioanalyzer 2100 (Agilent, Santa Clara, CA, United States). The sequencing library was constructed using NEBNext® Poly (A) mRNA Magnetic Isolation Module (New England Biolabs, Ipswich, MA, United States). The clustering of the index-coded samples was performed on a cBot Cluster Generation System according to the manufacturer’s instructions. After cluster generation, sequencing was performed using the Illumina HiSeq™ 2500 platform with paired-end 150 base reads. The RNA-seq reads with adapters, with all A bases, with an N ratio > 10% and with low quality (number of bases with Q ≤ 20 was >50% among the entire reads) were removed to obtain the clean data. Then, the clean data were compared with ribosomal RNA in the S. marcescens genome to remove the corresponding ribosomal RNA, and the remaining reads were aligned to the SCQ1 reference genome using Bowtie2 (Version 2.2.8). Fragments per Kilobase of transcript per Million fragments (FPKM) mapped was used to present the quantification of gene expression and to eliminate the impacts of different gene lengths and sequencing depth. The edgeR package1 was used to identify differentially expressed genes (DEGs) across samples with fold changes (FC) ≥ 2 and a false discovery rate-adjusted p (q-value) < 0.05. DEGs were then subjected to enrichment analysis of GO function and KEGG pathways, and q-values were corrected using <0.05 as the threshold. Each sample was sequenced in three replications.
Quantitative Real-Time RT-PCR
The total RNA was extracted using RNAiso Plus (Takara, China) according to the manufacturer’s protocol. The complementary DNA (cDNA) synthesis was carried out with RevertAid First Strand cDNA Synthesis Kit (Vazyme Biotechnology, Nanjing, China) according to the manufacturer’s instructions. Primer Premier 5.0 software was used to design primers, and rpoB was selected as the internal control gene (Supplementary Table S1). Reactions were performed using ChamQ SYBR qPCR Master Mix (Vazyme Biotechnology, Nanjing, China) in a 20 μl final volume containing 10 μl SYBR premix Ex Taq II, 0.8 μl of each primer, 0.4 μl ROX Reference Dye, 2 μl of diluted cDNA, and 6 μl sterile distilled water. Quantitative real-time RT-PCR (qRT-PCR) was performed on a StepOne™ Real-Time PCR System (48-well format; Applied Biosystem, United States) with the following program: 95°C for 10 min, followed by 40 cycles at 95°C for 15 s, at 60°C for 1 min. A melting curve analysis was performed to confirm product specificity. The relative expression levels were calculated using the 2-ΔΔCT method. The FC in mRNA level were determined using the −ΔΔCt data analysis method. All samples were analyzed in three replications. The primers used in this study were listed in Supplementary Table S1.
Strain Construction, Phenotypic and Virulence Investigation
The knockout plasmid of slyA was constructed by seamless cloning technology. The primers were designed using online software2 and were listed in Supplementary Table S1. Using S. marcescens SCQ1 genomic DNA as a template, the right-hand fragment and the left-hand fragment were amplified using primer pairs slyA-U-F/R and slyA-D-F/R, respectively. Amp-resistance fragment was amplified using Amp-F/R with pMD19-T as a template. The three fragments were ligated into SacI/SalI digested suicide plasmid pDM4 using ClonExpress MultiS One Step Cloning Kit (Vazyme Biotechnology, Nanjing, China) and then transformed into E. coli S17-1 to generate E. coli S17-1 (pDM4-slyA). The conjugative transfer was used to transform the knockout plasmid pDM4-slyA into S. marcescens SCQ1. Briefly, the E. coli S17-1 (pDM4-slyA) and SCQ1 were cultured in LB medium at 37°C and 28°C, respectively, to OD600 ≈ 0.6. The fermentation of E. coli S17-1 and SCQ1 were mixed at a ratio of 3:1 and the bacterial pellet was harvested by centrifugation at 12,000 rpm for 5 min. Then, the bacterial pellet was resuspended in 50 μl LB and 5 μl was transferred to LB plate containing a 0.22 μm membrane filter in the surface, which was cultured at 37°C for 4 h, followed by 28°C for 12–16 h. The colonies were spread on LB plates containing ampicillin, rifampicin, and chloramphenicol to obtain the single crossover recombinants SCQ1-pDM4-slyA which was verified by PCR using slyA-1F/R, slyA-2F/R. Overnight fermentation broth of SCQ1-pDM4-slyA was spread on LB plates containing 10% of sucrose and ampicillin without NaCl at 28°C for 24 h. The ΔslyA mutant was sucrose-resistant, ampicillin-resistant, and chloramphenicol intolerant and was further verified by PCR using slyA-1F/R, slyA-3F/R, and slyA-F/R and sequencing. To complement the mutants, the fragment containing the upstream non-coding region and the CDS of slyA was amplified using primer pairs slyA-hb-F/R and ligated into XbaI/XhoI digested vector pJQ200SK to generate complement plasmid pJQ200SK-slyA. Then, the plasmids pJQ200SK and pJQ200SK-slyA were transformed into E. coli S17-1 to producing E. coli S17-1-pJQ200SK and E. coli S17-1-pJQ200SK-slyA, respectively. As mentioned above, conjugative transfer was used to transform the pJQ200SK plasmid into SCQ1 and ΔslyA to generate SCQ1-pJQ200SK and ΔslyA-pJQ200SK, and the pJQ200SK-slyA plasmid was transformed into ΔslyA to producing ΔslyA-pJQ200SK-slyA. PCR (M13F/R) and sequencing were used for further verification.
The colony morphotypes, prodigiosin production, and growth of the ΔslyA mutant were investigated according to the methods mentioned above.
The healthy silkworm fifth-instar larvae reared with fresh mulberry leaves at 26°C were prepared for the experiments. Bacteria were harvested in the early exponential phase and counted by serial dilution methods which were expressed as colony-forming units (CFUs). The bacterial concentrations were adjusted to 5, 10, 25, and 50 CFU/μl with sterile insect saline (150 mM NaCl, 5 mM KCl, and 1 mM CaCl2), respectively. To assess the virulence of ΔslyA and SCQ1, 2 μl of each dilution was injected into silkworm hemocoel using a micro-injector, and sterile insect saline was used as control. Each group contained 30 larvae and the experiments were performed in triplicate. The dead larvae were continuously cultured at 26°C and the characteristics were recorded. The median lethal dose (LD50), median lethal time (LT50) values, and the statistical significance were evaluated using the SPSS software version 21.0 (SPSS, Inc./IBM, Armonk, NY, United States).
Statistical Analyses
Data were presented as mean ± SD or mean ± SE as needed. Statistical analysis was performed using the SPSS software version 21.0 (SPSS, Inc./IBM, Armonk, NY, United States). When necessary, the student t-test was performed to compare the differences between the two groups. The p-value lower than 0.05 and 0.01 were designated as statistically significant (*) and highly significant (**), respectively.
Results
The Features of Different Morphotypes Derived From the S. marcescens SCQ1
The secondary metabolite prodigiosin endowed the S. marcescens wild-type strain SCQ1 with a red colony characteristic. Continuous cultivation of the wild-type strain led to different color morphotypes. As shown in Figure 1, the different color morphotypes were firstly detected in the broth at the 5th passage. From the 5–14th passage, the numbers of non-pigmented morphotypes increased dramatically which reached as high as 78% of the population. After the 24 passages, the population of the non-pigmented morphotypes exceeded 99% which indicated that the wild-type strains have been generally replaced (Figures 1A,B). During the passage processes, the spontaneous mutants with multiple color morphotypes were observed on the LB plate, including the colonies with a purple, pink, red center, or ring-shaped red color. However, most of the mutants displayed white colonies (Figure 1A).
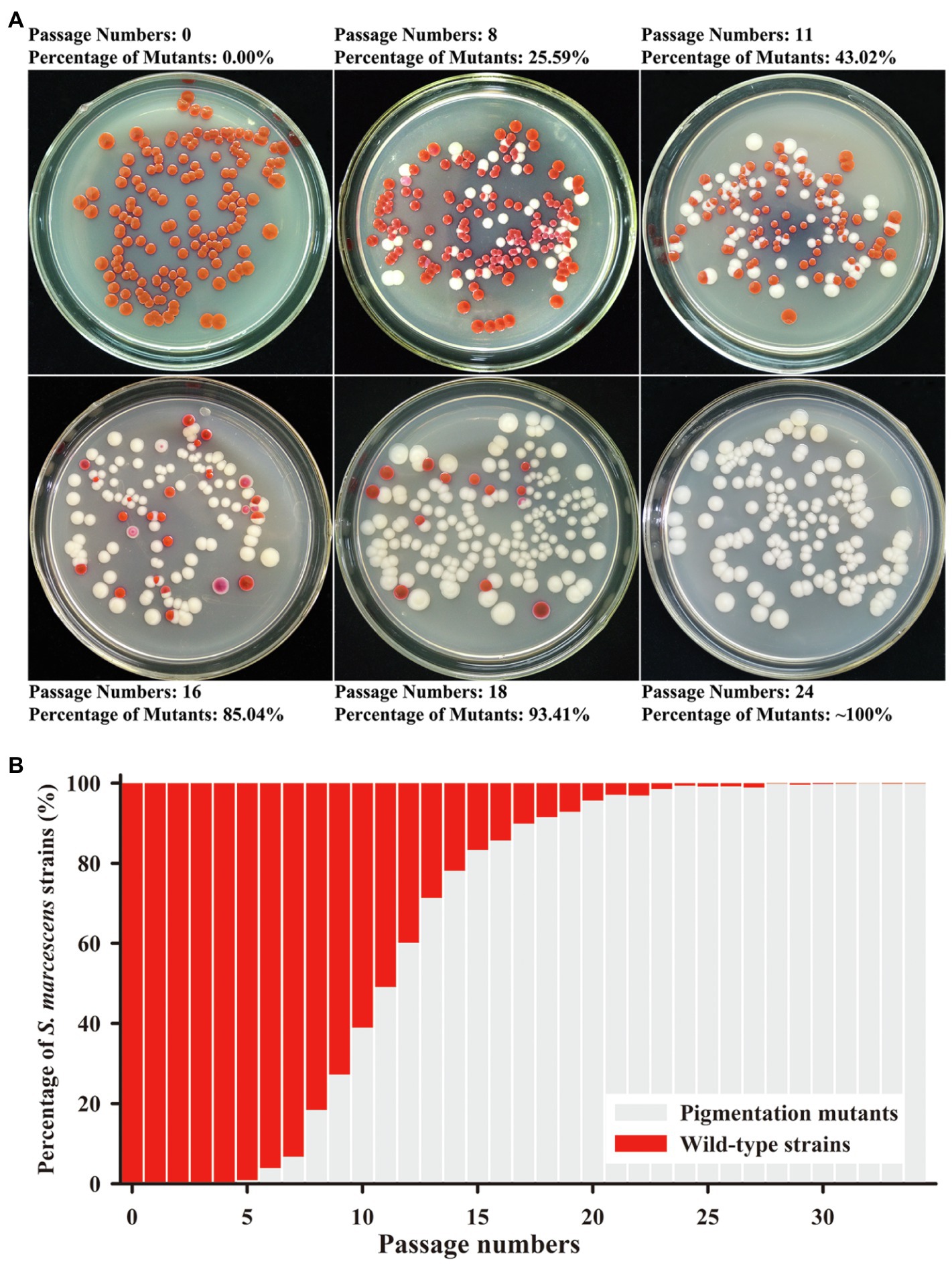
Figure 1. Mutation trend of Serratia marcescens during continuous passages. (A) Colonies on Luria-Bertani (LB) plate at the representative generations of passage. The percentage of color mutants is labeled upside. (B) The percentage of wild-type strains and mutants in each generation of passage.
Growth and Prodigiosin Production of Different Color Morphotypes
By further separation and purification, we have obtained four phenotypically stable mutants which were named SCQ1-1M (pink), SCQ1-2M (white), SCQ1-3M (white), and SCQ1-4M (red center; Figure 2A). The growth curves of the four morphotypes were distinctly different from the wild-type strain SCQ1 (Figure 2B). All tested strains entered the logarithmic growth phase after 2 h of inoculation. Within the first 50 h of culture, the growth trend of the pink strain was identical to the red wild-type strain, in which the OD600 of the SCQ1-1M reached the maximum value of 0.984 ± 0.011. The time point that the strain SCQ1 entered the stationary phase is about 52 h of cultivation, and the OD600 reached the highest value of 0.985 ± 0.01. SCQ1-1M entered the stationary phase a bit earlier than SCQ1 because the OD600 value was slightly decreased to 0.978 ± 0.014 at 52 h (Figure 2C). In contrast, the OD600 values of the other three color morphotypes were significantly lower than the strain SCQ1 at the period from 6 to 46 h (Figure 2C), but continuously increased and reached a higher value than that of the wild-type strain. Although, the cell densities of the three color morphotypes were eventually higher than the wild-type strain SCQ1, the time point was different which was delayed in the SCQ1-2M at 64 h (Figure 2C). Besides, the OD600 of the SCQ1-2M reached the maximum value of 1.007 ± 0.012 at 76 h, and then gradually decreased to 0.935 ± 0.017 at 100 h (Figure 2C). The OD600 of SCQ1-3M and SCQ1-4M were continuously increased within the cultivation period (Figure 2C). The prodigiosin productions of all the color morphotypes SCQ1-1M, SCQ1-2M, SCQ1-3M, and SCQ1-4M were significantly lower than the wild-type strain SCQ1, which decreased for 8.84, 45.76, 42.98, and 9.47-fold, respectively (Figure 2D). These results indicated that both bacterial growth and pigment production of the S. marcescens spontaneous mutants were changed, which suggested a complicated relationship between the two aspects.
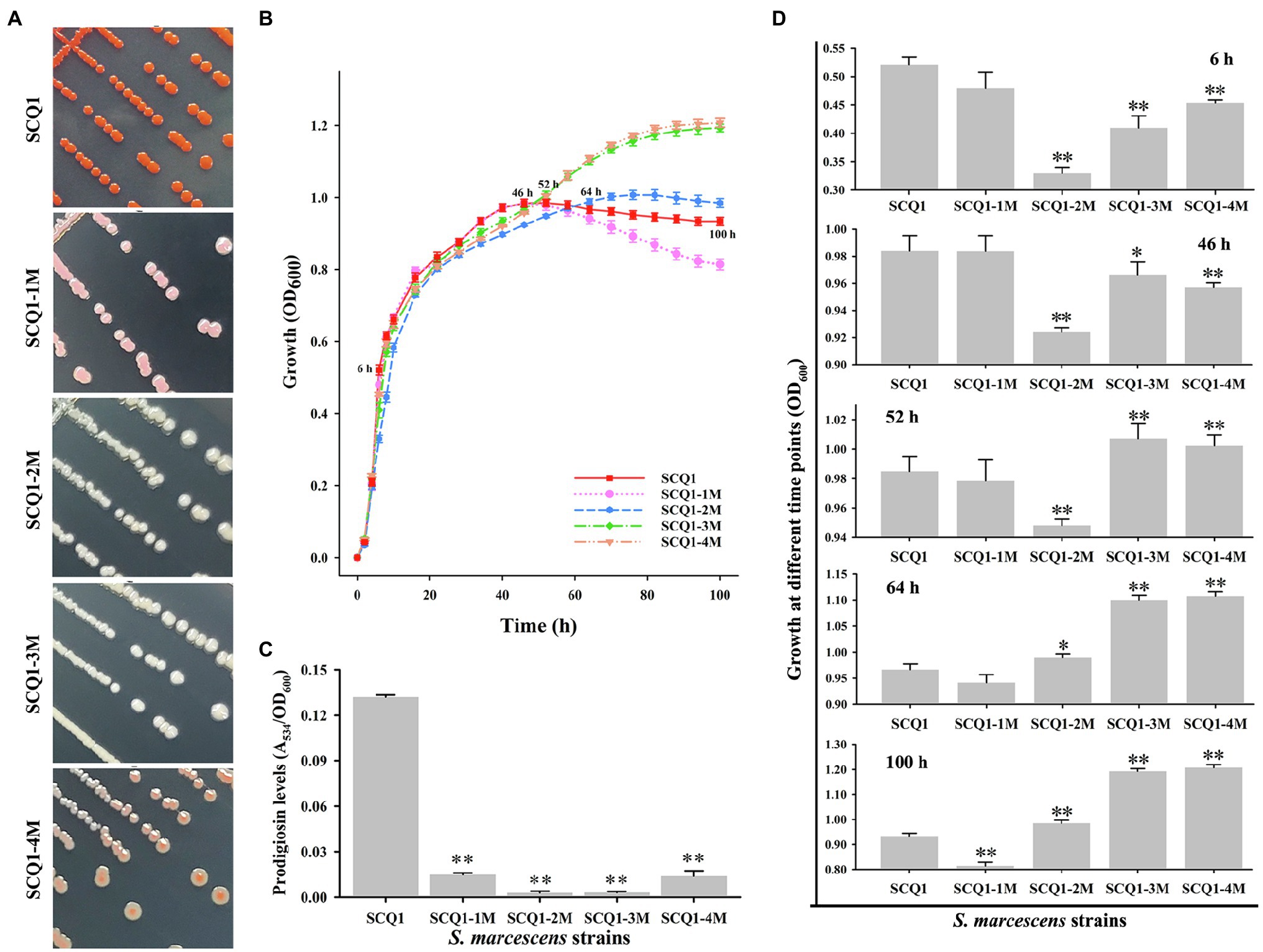
Figure 2. The phenotypic patterns of wild-type S. marcescens and its spontaneous mutants. (A) The colony of the wild-type strain SCQ1 and the four mutants on LB plate. (B) Growth of different S. marcescens strains in LB medium. The OD600 values are monitored every 2 h for 100 h using the Bioscreen C instrument with three technical replicates. Different lines and symbols represent different bacterial strains. The data are expressed as mean ± SD. Several time points are labeled and the details are shown in (C). (D) The relative prodigiosin production in different S. marcescens strains is measured after 16 h incubation at 28°C. The values are calculated and expressed as mean ± SD (n = 3, *p < 0.05, **p < 0.01).
Expression Levels of pigA-N in the Four Mutants
To determine the expression levels of the pig genes in the mutants, qRT-PCR analysis was performed (Figure 3). In SCQ1-1M, the expression levels of pigA and pigC were 1.954 ± 0.0313 and 1.747 ± 0.012, respectively. The values were even significantly higher than that of the wild-type strain SCQ1. However, the expression levels of pigG, pigI, pigJ, and pigK were significantly lower than that of in SCQ1, which were 0.372 ± 0.095, 0.454 ± 0.115, 0.413 ± 0.0258, and 0.548 ± 0.161, respectively. The expression levels of the other eight pig genes of SCQ1-1M were similar to SCQ1. In the SCQ1-2M, SCQ1-3M, and SCQ1-4M, the expression levels of pigA-N were all remarkably downregulated. The pigD is involved in the initial step of MAP biosynthesis, which the relative expression levels in the strains SCQ1-2M, SCQ1-3M, and SCQ1-4M were as low as 0.020 ± 0.0004, 0.001 ± 0.001, and 0.022 ± 0.001, respectively. The pigI is involved in the first step of MBC biosynthesis, which the relative expression levels in strains SCQ1-2M, SCQ1-3M, and SCQ1-4M were decreased to 0.195 ± 0.161, 0.003 ± 0.001, and 0.049 ± 0.004, respectively. The pigC is involved in the final step of prodigiosin biosynthesis, which the relative expression levels in strains SCQ1-2M, SCQ1-3M, and SCQ1-4M were 0.038 ± 0.009, 0.005 ± 0.004, and 0.051 ± 0.022, respectively. These results suggested that the pig genes of the spontaneous mutants were varied at the transcriptional level, and most of them were downregulated which was consistent with the phenotypic changes.
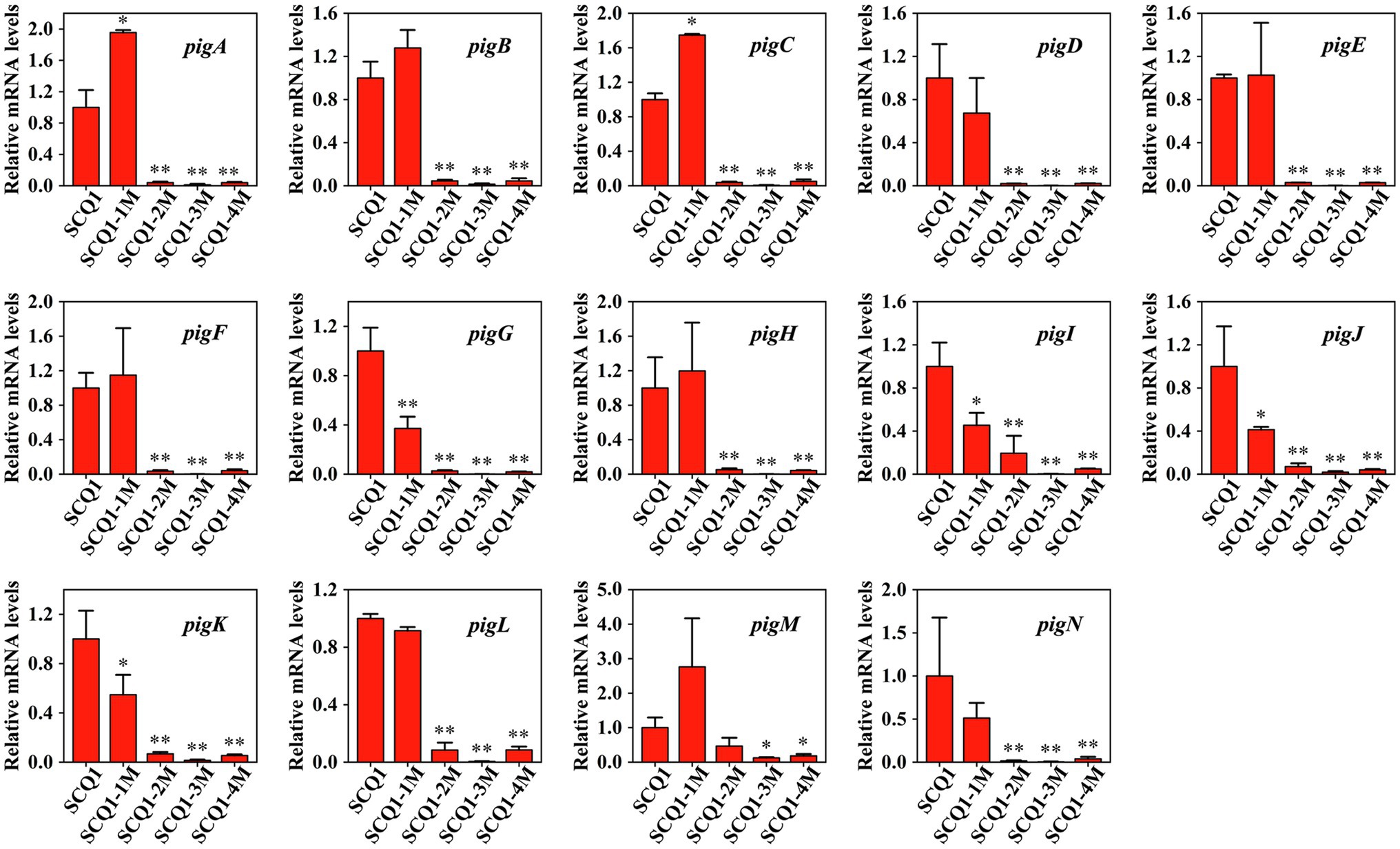
Figure 3. The mRNA expression levels of pigA-pigN in the wild-type strain SCQ1 and the four spontaneous mutants by quantitative real-time RT-PCR (qRT-PCR) analysis. Relative mRNA levels are quantified using rpoB as internal control and the data are displayed as mean ± SE (n = 3, *p < 0.05, **p < 0.01).
Transcriptome Sequencing and Mapping Data
Pig genes are regulated by various factors. Transcriptome sequencing was used to characterize the expression pattern of the whole genome, which may provide useful data for exploring the spontaneous pigmentation mutants. RNA-Seq data were generated from the pigmented wild-type strain SCQ1 and the non-pigmented mutant SCQ1-3M in biological triplicates. As the results, a total of 12,363,419,512 bp and 13,666,792,091 bp clean data were generated from Illumina HiSeq™ 2500 platform in SCQ1 and SCQ1-3M samples, respectively. The values of Q30 and the GC% were approximately 94 and 53% in each sample. After quality control and removing the rRNA, we obtained 68,491,508 and 77,474,952 clean reads in samples SCQ1 and SCQ1-3M (Table 1). The total reads of each sample were then mapped to the SCQ1 reference genome, and the ratio was higher than 96% (Table 1). After calculating and normalizing for FPKM, DEGs between SCQ1-3M and SCQ1 (SCQ1-3M vs. SCQ1) were identified. The volcano plot indicated the DEGs of SCQ1-3M vs. SCQ1 (Figure 4A). Of all the 4,692 genes, 582 DEGs with fold changes |log2(FC)| ≥ 1 and value of q < 0.05 were identified in SCQ1-3M when compared to SCQ1, in which the upregulated and downregulated DEGs were half and a half (Supplementary Table S2).
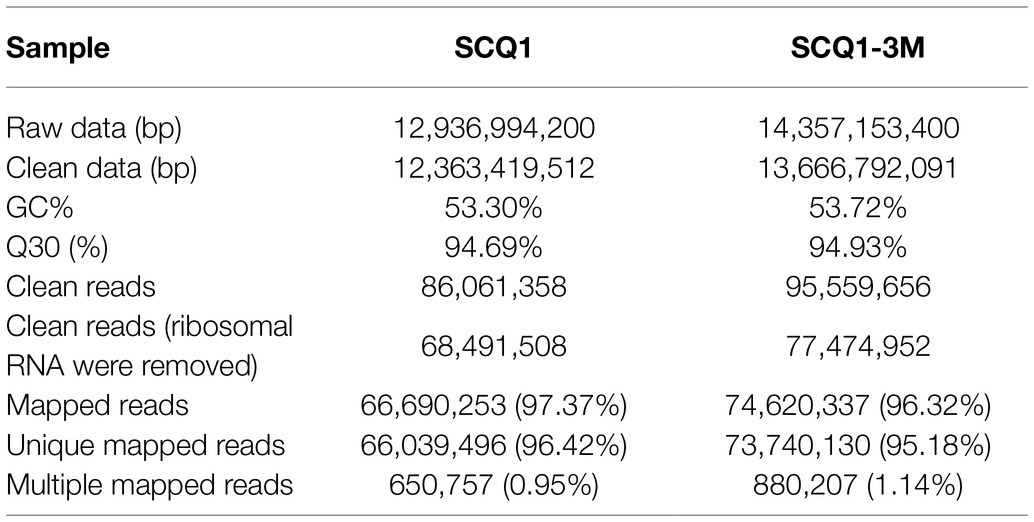
Table 1. Summary of transcriptome sequencing for SCQ1 and SCQ1-3M.
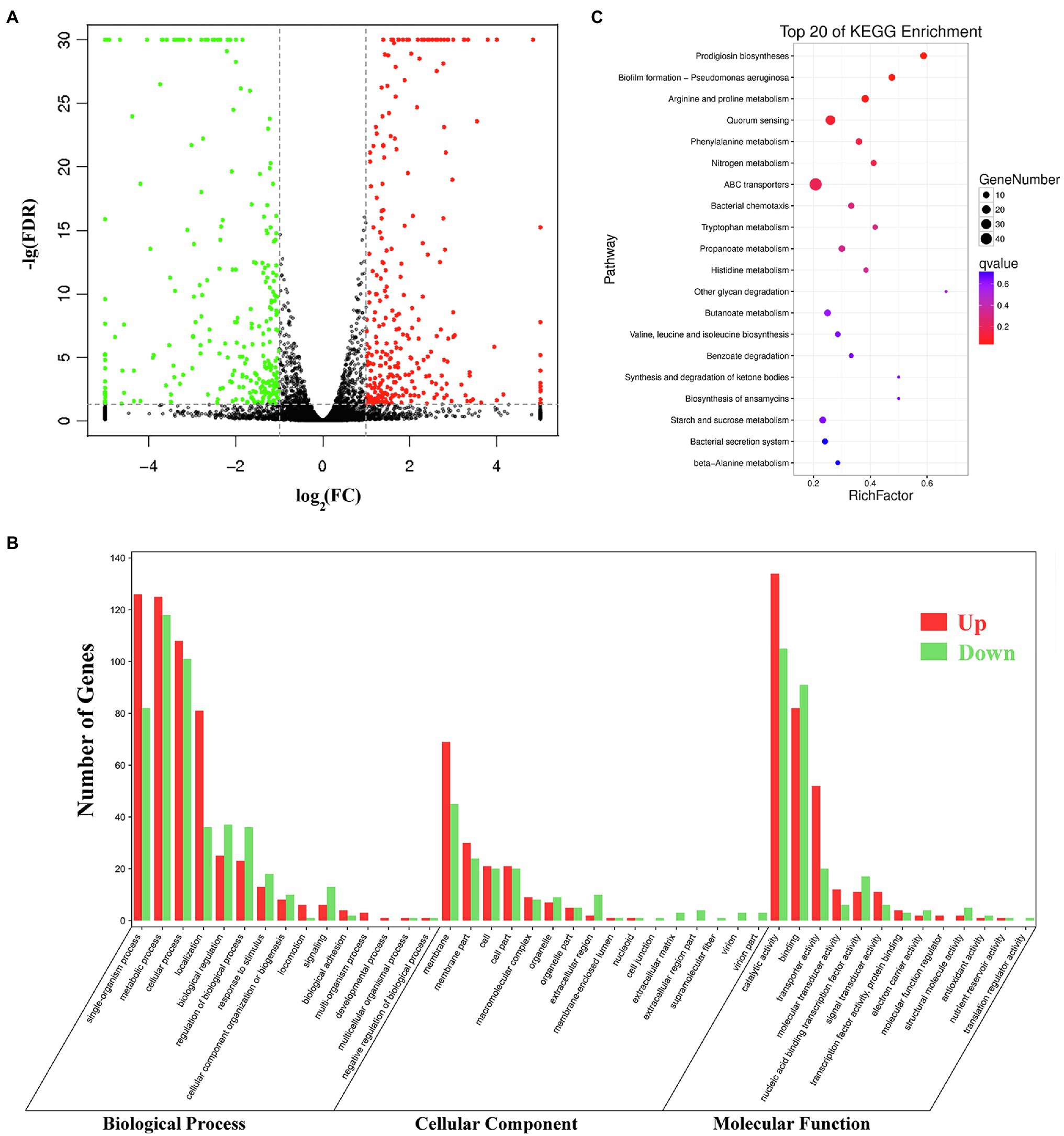
Figure 4. Transcriptomic analysis of the wild-type strain SCQ1 and the non-pigmented spontaneous mutant SCQ1-3M. (A) The volcano plot shows a general gene expression pattern of the SCQ1-3M. The red, green, and black dots represent significant upregulated, significant downregulated and, non-significant genes, respectively. The |log2(FC)| ≥ 1; FDR < 0.05 is set as the threshold for significantly DEGs. (B) Gene Ontology (GO) annotations of DEGs of the mutant SCQ1-3M. The DEGs are grouped into three main categories which are “cellular component,” “molecular function,” and “biological process” according to the GO data library. The red and green bars represent significant upregulated and downregulated DEGs, respectively. The Y-axis indicates the number of DEGs in each category. (C) The top 20 enriched KEGG pathways of DEGs. X- and Y-axes represent the Rich factor and the KEGG pathways, respectively.
qRT-PCR Validation
Quantitative real-time RT-PCR analysis was performed to validate the RNA-sequencing results. As shown in Supplementary Figure S1, 11 downregulated genes, including 4′-phosphopantetheinyl transferase (pho), O-methyltransferase (pigF), BsmB family protein BsmB (BsmB), 7-keto-8-aminopelargonate synthetase (pigH), Porin OmpC (ompC), ImpA family type VI secretion-associated protein (vgrG1), type VI secretion system (T6SS) effector, Hcp1 family (hcp1), type IV secretion protein Rhs (vgrG), transcriptional regulator slyA (slyA), transcriptional regulator (tran), KDP operon response regulator KdpE (kdpE), and 12 upregulated genes, including phenylacetate-CoA oxygenase subunit PaaB (paaB), succinylglutamate desuccinylase (astE), D-threitol dehydrogenase (dthD), periplasmic protein CpxP (cpxP), binding-dependent transport system inner membrane component family protein (dppB), phosphonate-transporting ATPase (livF), phenylacetic acid degradation protein (paaE), PTS system maltose and glucose-specific IICB components (malX), D-amino-acid oxidase (mnmC), ABC transporter substrate-binding protein (dppA), transcriptional regulator LrhA (pecT), and periplasmic substrate-binding component of an ABC superfamily oligopeptide transporter (oppA), were selected. In general, the expression characteristics of the tested genes were consistent with RNA-Seq transcriptomic analysis.
Functional Annotation of DEGs
Gene Ontology (GO) assignments were used to classify the functions of the DEGs. A total of 3,328 reference genes assigned with GO terms were used as the background for enrichment analysis. The DEGs were assigned into 1,253 GO terms consisting of 698 biological process terms (353 DEGs), 413 molecular function terms (152 DEGs), and 142 cellular component terms (365 DEGs; Figure 4B). Most of the DEGs were annotated with more than one GO term. In the biological process group, the DEGs are enriched in “metabolic process” (68.84%), “cellular process” (59.21%), “single-organism process” (58.92%), and “organic substance metabolic process” (51.84%). In the molecular function group, the expression ratio of the DEGs in “catalytic activity” (65.48%) was the highest, followed by “binding” (47.4%) and “organic cyclic compound binding” (31.23%). In the cellular component group, the higher proportion of the DEGs involved in the “membrane” (75%) and “membrane part” (35.53%). KEGG assignments were performed to categorize gene functions. A total of 253 DEGs were mapped to 99 level-3 KEGG pathways, which were assigned into five KEGG level-1 groups, including “metabolism,” “cellular processes,” “environmental information processing,” “genetic information processing,” and “organismal systems.” The top 20 enriched KEGG pathways were shown in Figure 4C. Most of the DEGs were enriched in the “metabolic pathways” (39.53%), “ABC transporters” (19.37%), “microbial metabolism in diverse environments” (16.21%), “biosynthesis of antibiotics” (13.83%), “biosynthesis of secondary metabolites” (13.44%), and “quorum sensing” (10.28%). Based on the q-value, three pathways were significantly enriched, including “prodigiosin biosynthesis,” “biofilm formation,” and “arginine and proline metabolism” (Supplementary Table S3). These results suggested that the DEGs involved in metabolism, membrane proteins, biosynthesis of secondary metabolites, and transporters might play important roles in spontaneous non-pigmented mutants.
Transcription Patterns of Several Pathways in Spontaneous Non-pigmented Mutant
Transcription Factors
Numerous studies have shown that the biosynthesis of prodigiosin was regulated by a variety of transcription factors. In this study, a total of 22 transcriptional factors were detected (Table 2). There were five transcription regulators, including hexS, cdaR, lrp, malT, and feaR, which were upregulated with the fold change values a biter higher than 1.0. In contrast, the remaining 17 regulators were significantly downregulated (Table 2). SMR_GM001072 and SMR_GM004456 showed the highest fold changes that were downregulated as −13.34 and −12.70, respectively. Besides, the fold changes of the most downregulated genes were above −1.5, which included two previously reported prodigiosin-synthesis-related regulators, slyA and pigP, and the values were −2.09 and −1.66, respectively. These transcriptional regulators might possess direct or indirect effects related to the prodigiosin biosynthesis pathway in the spontaneous mutant SCQ1-3M.
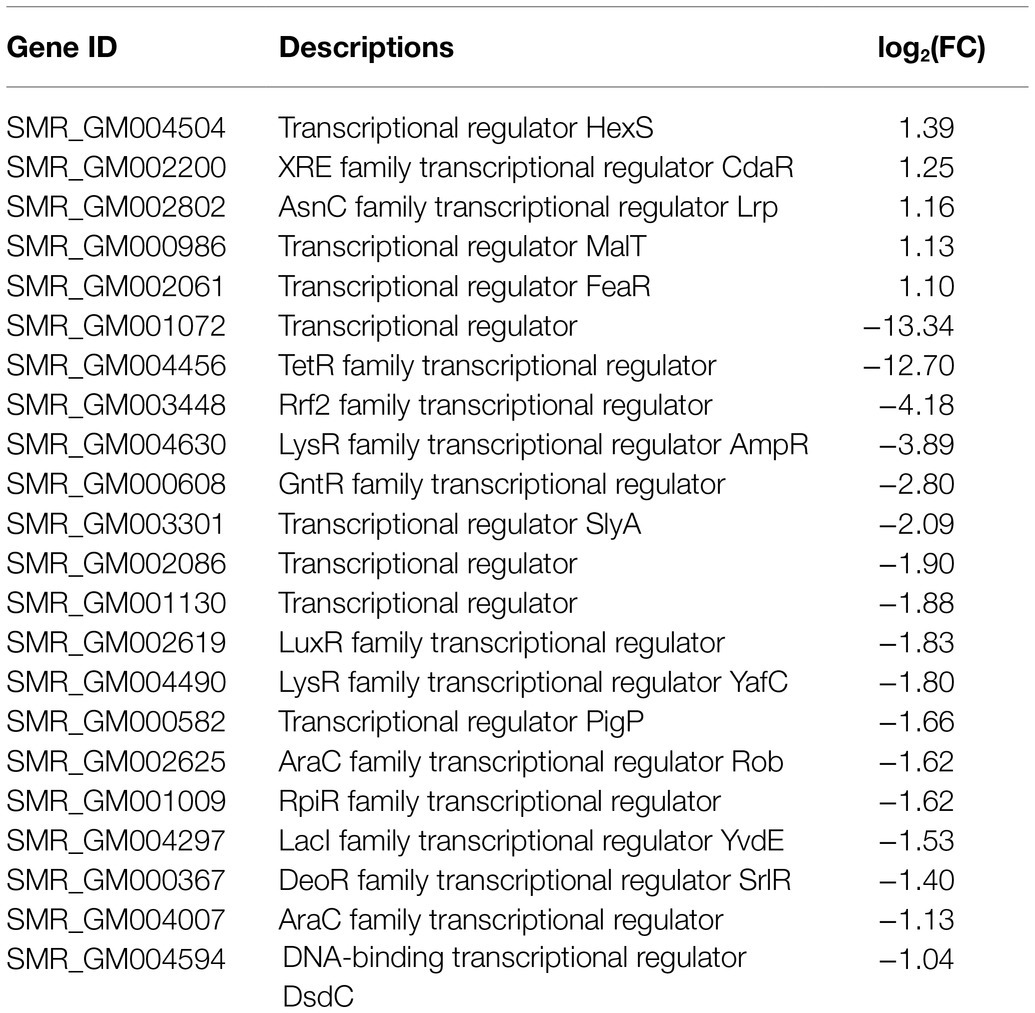
Table 2. Transcriptional regulators in differentially expressed genes (DEGs).
Membrane Proteins
It has been investigated that the membrane vesicles which consisted of essential membrane proteins were prodigiosin storage and secretion device. In transcriptome sequencing, a total of 26 membrane proteins were identified (Table 3). There are five DEGs, including ompF (SMR_GM002838, SMR_GM002556, and SMR_GM003927), ompW (SMR_GM003755), membrane protein (SMR_GM002333), and ydcZ (SMR_GM004201) were upregulated with fold changes ranging from 1.1 to 2.61. The other 21 membrane proteins were downregulated in SCQ1-3M with fold changes ranging from −1.02 to −4.56. The membrane proteins SMR_GM002390 and SMR_GM001605 were downregulated with maximum fold changes of −13.20 and −13.02, respectively. The changes of membrane proteins might be related to the storage and transportation of bacterial prodigiosin.
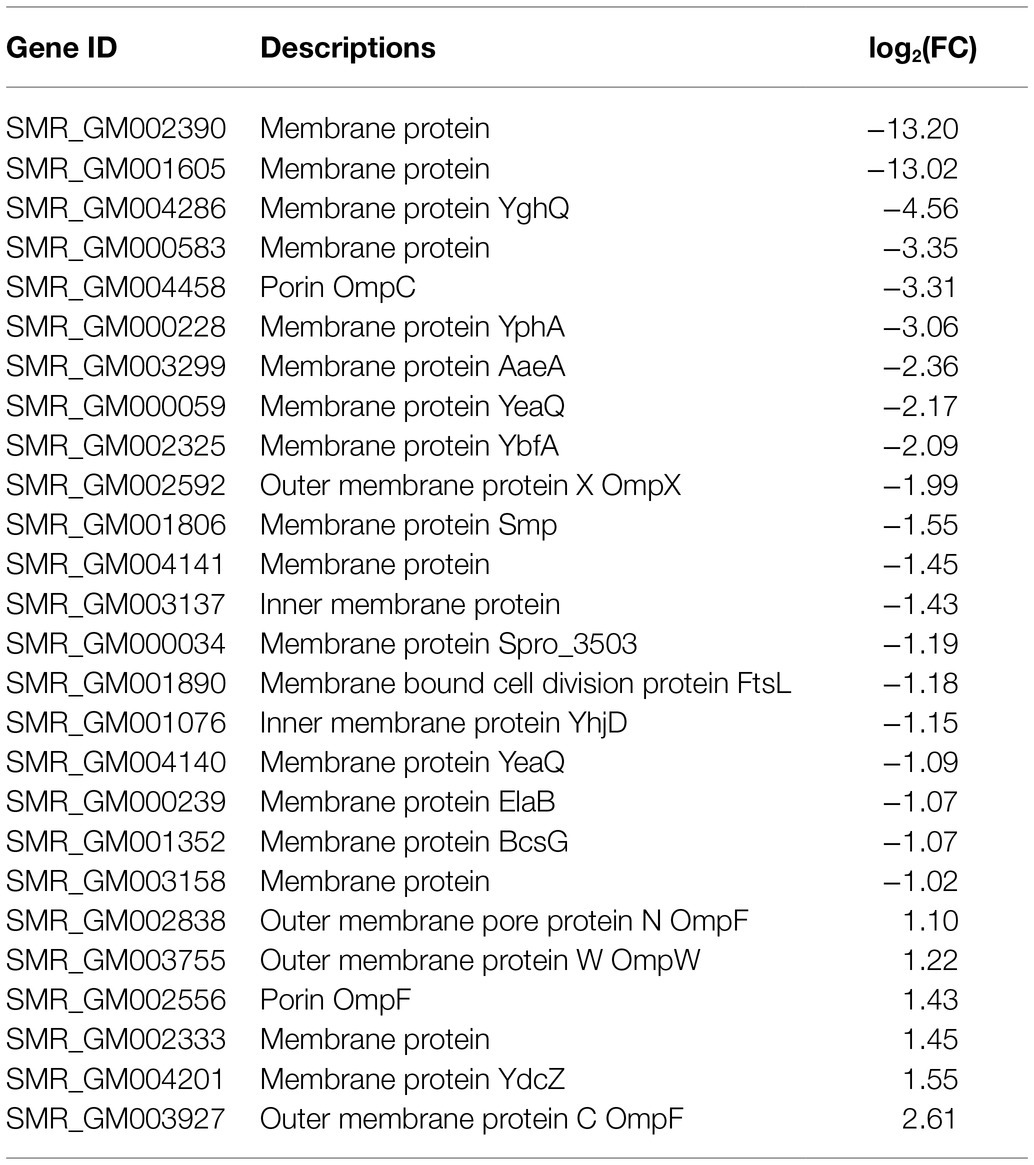
Table 3. Membrane proteins in DEGs.
Type VI Secretion System
Type VI secretion system is a weapon used by Gram-negative bacteria to fight against other bacterial competitors. As in the wild-type strain SCQ1, S. marcescens possessed a T6SS that contains 13 core proteins. In the transcriptome sequencing results, all the DEGs related to T6SS, including 11 core components, were downregulated in SCQ1-3M with fold changes ranging from −1.02 to −5.16 (Table 4). The membrane-anchoring complex subunit TssM was downregulated in SCQ1-3M with a fold change of −4.57. DotU, TssK, and TssF which were the main components of the T6SS baseplate complex showed the fold changes of −4.19, −2.80, and −2.65, respectively. The tssA which encoded a dodecamer complex had a fold change value of −2.97. The results suggested a metabolic tradeoff between a high-cost system and growth in S. marcescens. Under artificial conditions which lacked competitors, cutting down T6SS would save resources that benefit bacterial growth.

Table 4. Differentially expressed genes involved in type VI secretion system (T6SS).
Amino Acid Metabolism and Transport
In the DEGs of SCQ1-3M vs. SCQ1, we have found several enzymes which could be classified into 11 amino acid metabolic pathways, including “Arginine and proline metabolism (ko00330),” “Phenylalanine metabolism (ko00360),” “Tryptophan metabolism (ko00380),” “Histidine metabolism (ko00340),” “beta-Alanine metabolism (ko00410),” “Alanine, aspartate and glutamate metabolism (ko00250),” “Valine, leucine and isoleucine degradation (ko00280),” “Tyrosine metabolism (ko00350),” “Lysine degradation (ko00310),” “Glycine, serine and threonine metabolism (ko00260),” and “Cysteine and methionine metabolism (ko00270)” (Table 5; Supplementary Table S4). Most DEGs in these pathways were upregulated. The enhanced amino acid degradation ability reminded us to analyze the closely related pathways, e.g., amino acid import pathways. Several DEGs involved in amino acid and peptide transport pathways were generally upregulated, including “oligopeptide transport (Opp) system,” “dipeptide transport (Dpp) system,” “histidine transport system,” “arginine transport system,” and “branched chain amino acid transport system” (Table 6). These results suggested that the spontaneous mutant SCQ1-3M imported extra amino acids for further degradation.

Table 5. Summary of DEGs involved in amino acid metabolism.
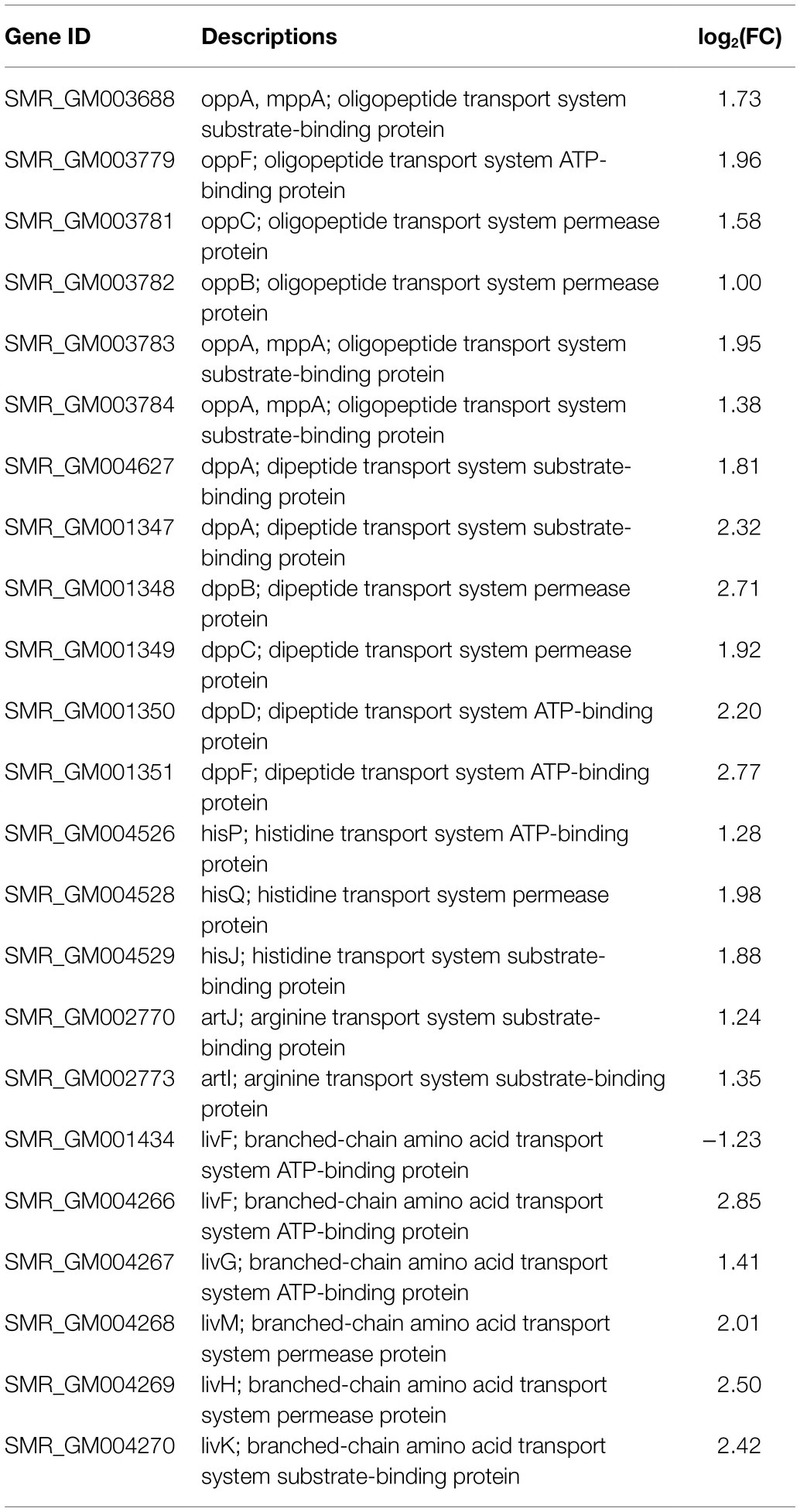
Table 6. Differentially expressed genes involved in amino acid and peptide transport system.
slyA Affected the Prodigiosin Biosynthesis, Bacterial Virulence, and Growth
The growth advantage of color mutants is related to several genes. The transcriptional regulator slyA was significantly downregulated in the non-pigmented mutant SCQ1-3M. However, different strains had distinct expression patterns of slyA (Figure 5A), which suggested changes in the regulation of prodigiosin biosynthesis. Nonetheless, slyA was reported as a relative downstream positive regulator of the pig gene cluster. Therefore, we have constructed a slyA knockout mutant to investigate whether downregulated pig genes directly led to a similar growth advantage. The ΔslyA mutant showed a white colony, increased virulence and growth rate. Just like the SCQ1-3M, the ΔslyA mutant could not synthesize prodigiosin which was distinct from the wild-type strain SCQ1 (Figure 5B). The relative prodigiosin production of the ΔslyA mutant was 0.0015 ± 0.0002 after 16 h cultivation, which was decreased by about 70-fold compared with the SCQ1 (Figure 5C). The ΔslyA mutant was complemented by the wild-type slyA gene on the recombinant plasmid pJQ200SK-slyA. An empty plasmid pJQ200SK was transformed into both SCQ1 and ΔslyA as controls (Figure 5D). The prodigiosin production in the complemented mutant was largely restored (Figure 5C). The results indicated that slyA was responsible for prodigiosin dyssynthesis in SCQ1-3M but might not be responsible for that in other spontaneous mutants.
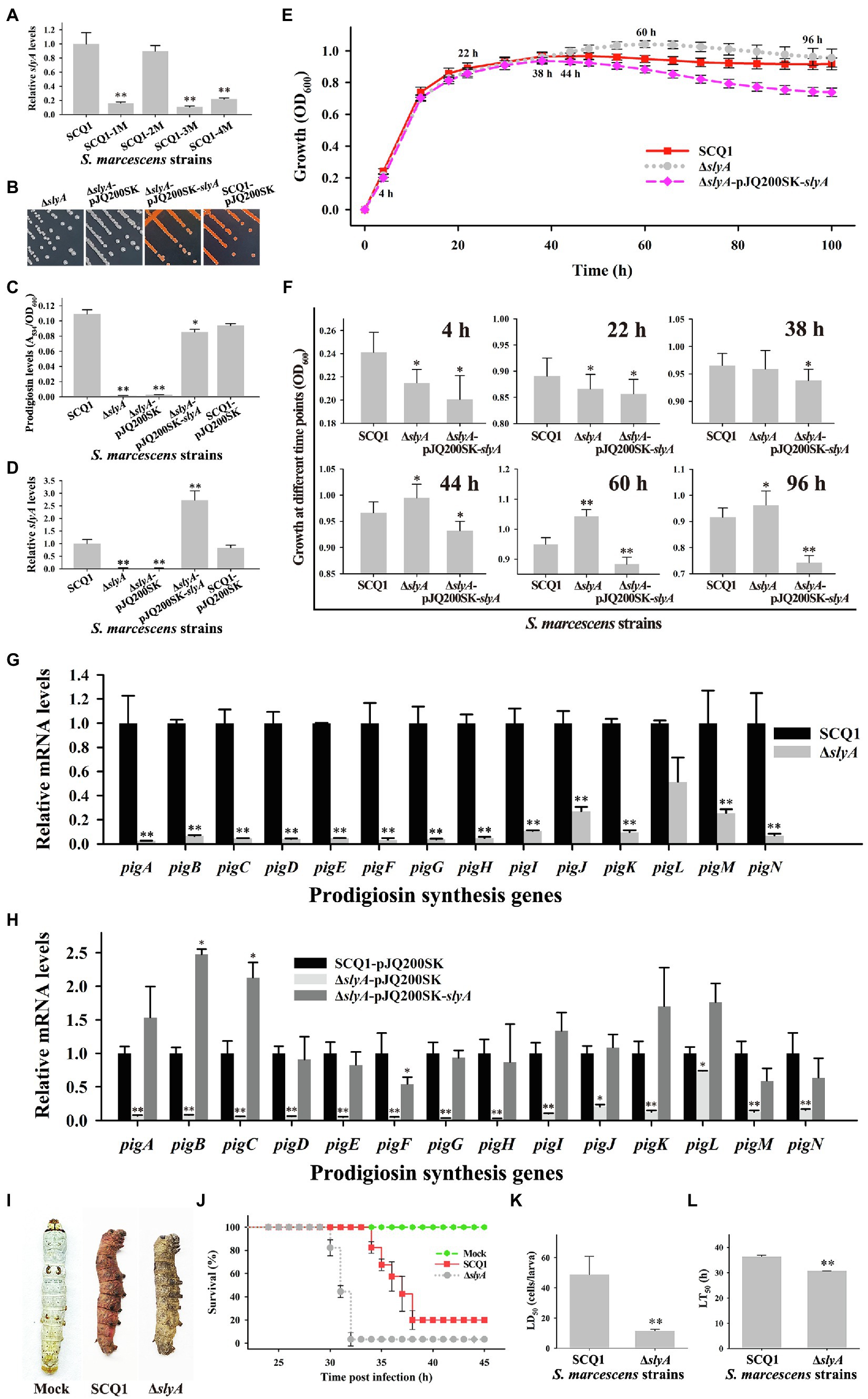
Figure 5. The characteristics of the ΔslyA mutant. (A) The mRNA expression level of slyA in different spontaneous mutants. (B) Colonies of the mutant ΔslyA, the control strains ΔslyA-pJQ200SK and SCQ1-pJQ200SK, and the complemented strain ΔslyA-pJQ200SK-slyA on LB plate inoculated at 28°C for 36 h. (C) The relative prodigiosin production levels in the strain SCQ1, ΔslyA, ΔslyA-pJQ200SK, ΔslyA-pJQ200SK-slyA, and SCQ1-pJQ200SK are measured after 16 h incubation at 28°C. The A534/OD600 values are calculated and expressed as mean ± SD. (D) The mRNA expression level of slyA in the recombinant strains. (E) The growth curves of the strain SCQ1, ΔslyA, and ΔslyA-pJQ200SK-slyA. The OD600 values are monitored every 2 h for 100 h using the Bioscreen C instrument with three technical replicates. Different lines and symbols represent different bacterial strains. The data are expressed as mean ± SD. Several time points are labeled and the details are shown in (F). (G) The mRNA expression levels of pigA-pigN in the wild-type strain SCQ1 and the mutant ΔslyA. (H) The mRNA expression levels of pigA-pigN in ΔslyA-pJQ200SK-slyA and the control strains with an empty plasmid pJQ200SK. (I) Symptom of silkworm larvae infected by the strain SCQ1 and ΔslyA. The mock group represents silkworm larvae injected with sterile saline as the negative control. (J) Cumulative survival of silkworm larvae injected with SCQ1 [100 colony-forming units (CFUs) per larva], ΔslyA (100 CFUs per larva), and sterile saline. (K) The LD50 values of the strain SCQ1 and ΔslyA in the silkworm larvae. (L) The LT50 of the strain SCQ1 and ΔslyA in the silkworm larvae. The data were expressed as mean ± SD (n = 3, *p < 0.05, **p < 0.01).
The growth pattern of the ΔslyA mutant was also varied. During 4–22 h of cultivation, the OD600 of the ΔslyA mutant was significantly lower than that of the SCQ1, but the value increased quickly and exceeded that of the SCQ1 in the further 44–100 h with a maximum of 1.043 ± 0.022 at 60 h (Figures 5E,F). The growth pattern of the control strain SCQ1-pJQ200SK and ΔslyA-pJQ200SK was similar to the SCQ1 and ΔslyA (data not shown), respectively. The complemented mutant ΔslyA-pJQ200SK-slyA showed a lower growth rate compared with the wild-type strain (Figures 5E,F). The maximum value of OD600 in ΔslyA-pJQ200SK-slyA was 0.938 ± 0.021 at 38 h (Figure 5F), and it was gradually decreased. The reason was unclear, but possibly due to the varied expression level of the transcriptional regulator slyA. qRT-PCR analysis indicated that the decreased prodigiosin yield was caused by suppressed pig genes (Figure 5G). However, in ΔslyA-pJQ200SK-slyA, the expression level of slyA was higher than the wild-type strain SCQ1 and the negative control SCQ1-pJQ200SK, which might cause unexpected effects. As shown in Figure 5H, ΔslyA-pJQ200SK-slyA showed a relatively high expression level of pig genes, in which the pigB and pigC were even higher than the negative control SCQ1-pJQ200SK. Nonetheless, ΔslyA mutant had a higher cell density than the wild-type strain SCQ1 for a long period, which indicated a better growth at laboratory conditions.
Then, we evaluated the virulence of the ΔslyA mutant and found it was increased compared with the wild-type strain SCQ1. Both the ΔslyA and SCQ1 caused the rapid death of silkworms (Bombyx mori), but with a different symptom in which SCQ1-infected larvae showed red color, while ΔslyA-infected ones exhibited dark brown color (Figure 5I). Briefly, most of the inoculated larvae were dead within 38 h, and the lethal time of the ΔslyA mutant was shorter than that of the SCQ1 (Figure 5J). The LD50 value of the ΔslyA mutant was 9.977 ± 3.105 (cells/larva) which was decreased by 4.89-fold compared with 48.83 ± 11.56 (cells/larva) of SCQ1 (Figure 5K). The LT50 value of ΔslyA (30.68 h) was also lower than SCQ1 (36.34 h; Figure 5L). Despite the higher virulence suggesting a higher cost of resources, ΔslyA mutant still possessed a growth advantage compared with the wild-type strain under laboratory conditions.
Discussion
The bacterial prodigiosin is synthesized by the biosynthetic gene cluster that contains 14 pig genes (Harris et al., 2004). In S. marcescens, both pigmented and non-pigmented strains are widely distributed in the environment. What happens to the pig genes in non-pigmented or less pigmented strains? Based on the sequence data from the GeneBank database, several non-pigmented strains are lacking one or more pig genes, even the entire gene cluster.3 In laboratory conditions, we have observed that spontaneous non-pigmented mutants of S. marcescens frequently occurred during the continuous propagation or prolonged incubation (Figure 1). This phenomenon of the S. marcescens SCQ1 is similar to the previous reports of S. marcescens 274 and HY (Bunting, 1940; Labrum and Bunting, 1953). It seems very easy for S. marcescens strains to lose prodigiosin synthesis capacity. However, the spontaneous reversion was not observed under the conditions tested, which was quite different compared with the previous studies (Bunting, 1946). Previously, we have sequenced the four color morphotypes and the parent strain SCQ1 (Xiang et al., 2021). Surprisingly, the Pig gene cluster (both sequences and positions) is completely preserved among all sequenced strains, and neither insertions nor deletions are detected. The results reminded us that the changes might occur at the transcriptional level. The qRT-PCR results strongly supported this hypothesis that the expression levels of pig genes in the mutants SCQ1-2M, SCQ1-3M, and SCQ1-4M were significantly downregulated (Figure 3). The SCQ1-1M was an exception because it expressed some pig genes at a relatively high level, e.g., the expression levels of pigA and pigC are even higher than that of the parent strain (Figure 3). The highly expressed genes suggested a higher cost of resources. However, downregulation or loss-of-function variant in a single pig gene can cause prodigiosin dyssynthesis (Williamson et al., 2006). In SCQ1-1M, four out of 14 pig genes were significantly downregulated which would decrease prodigiosin production. Thus, under laboratory conditions, the reduced pigment production of S. marcescens can be directly ascribed to the suppressed expression of the pig genes.
The different spontaneous color morphotypes of S. marcescens bring the question of whether they have any physiological differences. During the sub-culturing processes, we found that the non-pigmented morphotypes rapidly replaced nearly all the wild-type cells within 24 passages (Figure 1B). The results indicated that the non-pigmented morphotypes possessed a competitive growth advantage over the parent strain. The growth curves showed a higher cell density of the mutants SCQ1-2M, SCQ1-3M, and SCQ1-4M at the stationary phase compared with the red parent strain. The result was correlated with the lower expression level of pig genes as mentioned above. The biosynthesis of the red pigment prodigiosin is controlled by transcriptional regulators. In the present study, 22 differentially expressed regulators were identified in the strain SCQ1-3M (Table 2). However, only three of them, including the repressor hexS, the positive regulators pigP and slyA, were proved involved in prodigiosin synthesis (Williamson et al., 2006; Shanks et al., 2013). According to previous reports, the hexS regulated pigP (Shanks et al., 2013), and the latter regulated slyA (Williamson et al., 2006). The slyA is a relative downstream regulator in prodigiosin biosynthesis; however, it shows different expression patterns among spontaneous mutants (Figure 5A). The results suggested different color mutants might have different regulatory features. Nonetheless, in SCQ1-3M, it is possible that downregulated slyA directly led to prodigiosin dyssynthesis, and might bring growth advantage. To test the hypothesis, we have constructed a slyA deletion mutant. The ΔslyA mutant showed greatly reduced prodigiosin production as previously reported (Fineran et al., 2005; Pan et al., 2020a). Within a long period from 44 to 96 h, the cell density of the ΔslyA mutant was significantly higher than that of the wild-type strain SCQ1, which indicated a growth advantage. However, transcription regulators usually have multiple roles, e.g., slyA is involved in many physiological activities (Ellison and Miller, 2006). In this study, the growth advantage was not observed in the complemented strain ΔslyA-pJQ200SK-slyA (Figures 5E,F). Although the details were not clear, it might be caused by differentially expressed slyA (Figure 5D), which also led to the varied expression level of pig genes (Figure 5H). The phenomenon that highly expressed or overexpressed pig genes correlated with quickly decreased cell density was quite similar to that in the SCQ1-1M (Figures 2B, 3). Because slyA is also involved in the virulence in many bacterial species (Libby et al., 1994; Haque et al., 2009; Michaux et al., 2011; Zhou et al., 2016a), we also used silkworm larvae as the animal model to assess the virulence of the ΔslyA mutant. Unexpectedly, the virulence of ΔslyA mutant was increased, which suggested a higher cost of resources. Despite that, the ΔslyA mutant showed better growth than the wild-type strain as mentioned above. In general, the downregulated pig genes that resulted in non-pigmented mutants would bring a competitive growth advantage.
Pigment dyssynthesis was not the only factor that contributed to bacterial growth because the growth differences existed among the tested strains. The transcriptomic analysis also indicated that membrane proteins, T6SS, amino acid metabolism, and amino acid and peptide transport pathways were significantly varied. The membrane proteins are high-cost components that are involved in prodigiosin synthesis, storage, and exocytosis processes (Williamson et al., 2006; Chawrai et al., 2012; McMahon et al., 2012). McMahon et al. (2012) have found at least 13 outer membrane vesicle (OMV) proteins in S. marcescens. In the present study, downregulated membrane proteins, e.g., OmpX and OmpC, would hinder OMV formation which might cause feedback inhibition of prodigiosin synthesis (Pan et al., 2019). The T6SS is a high-cost nanomachine used by Gram-negative bacteria with multiple functions (Silva et al., 2020). In this study, we have found 18 downregulated T6SS-related genes in the spontaneous mutant SCQ1-3M, including most of the core components (Figure 6). Numerous studies indicate that T6SS generally implicates virulence in pathogenic bacteria (Miyata et al., 2011; Repizo et al., 2015; Hu et al., 2019). However, it has been proved that T6SS is not a virulence factor in S. marcescens (Murdoch et al., 2011). Previously, we have reported that the spontaneous non-pigmented mutants showed similar pathogenicity to silkworm larvae as the wild-type strain S. marcescens (Zhou et al., 2016b). In addition, a mutation in T6SS was suggested involving S. marcescens pigment production by using Tn5-mutagenesis technology (Jia et al., 2021). The downregulated T6SS might relate to the reduced prodigiosin production, or at least did not affect bacterial virulence. In addition to the suppressed high-cost systems, both amino acid degradation and import pathways were activated. The biosynthesis of prodigiosin relies on several amino acids, including proline, serine, alanine, and methionine (Williamson et al., 2006). Because the non-pigmented mutant is unable to synthesize the red pigment prodigiosin, the redundant amino acids certainly have another destination. We have found 68 DEGs were enriched in 11 amino acid degradation pathways, and most of them were upregulated (Table 5). Therefore, the amino acids which were originally prepared for prodigiosin synthesis or other high-cost systems were probably degraded. Activated bacterial amino acid degradation pathways also suggested extra substrate for over-consuming. In this study, we have found at least five gene clusters related to the amino acid and peptide import pathways that were generally upregulated in the mutant SCQ1-3M (Table 6). The upregulated amino acid transport pathways provided potential routes for non-pigmented mutants to uptake extra amino acids. Taken together, the non-pigmented mutant shut down several high-cost systems and activated amino degradation and transport pathways to obtain extra nutrients, which contributed to bacterial growth.
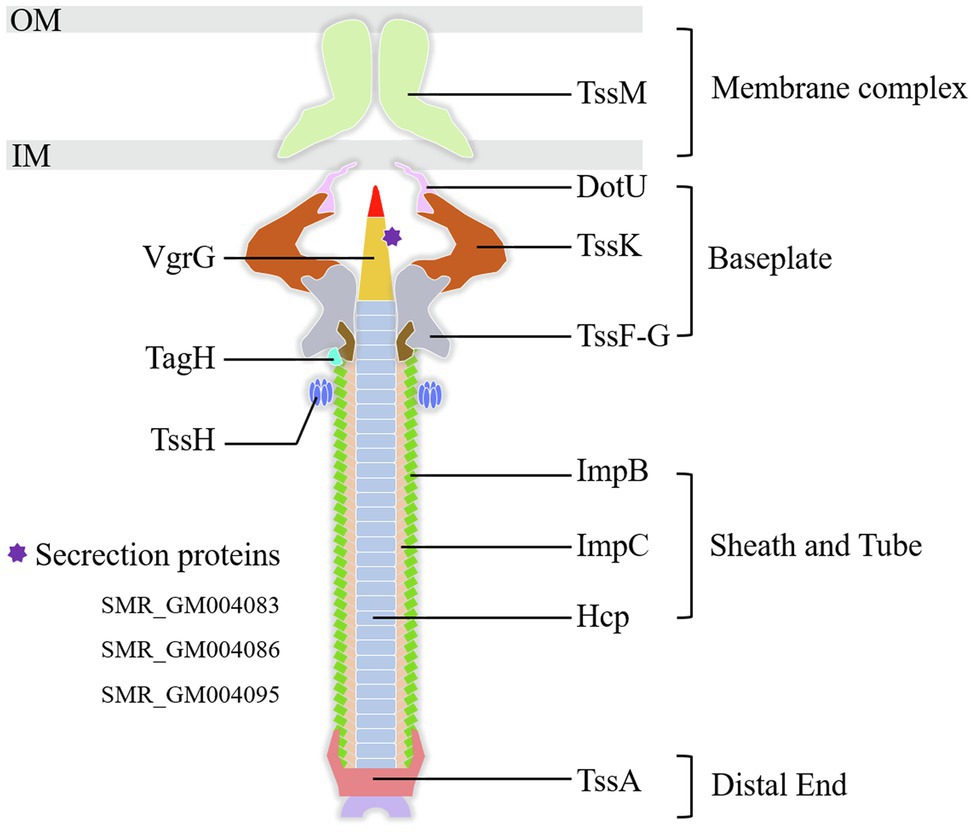
Figure 6. The components of the T6SS detected in the present study. All the labeled genes are significantly downregulated in the spontaneous non-pigmented mutant SCQ1-3M. The DEGs of T6SS related secretion proteins in the present study were listed at lower-left. OM, outer membrane; IM, inner membrane.
Conclusion
We use transcriptomic approaches to analyze the non-pigmented spontaneous mutant of S. marcescens, which rapidly replaces the wild-type parent strain within a few passages under laboratory conditions. Unexpectedly, the Pig gene cluster responsible for prodigiosin biosynthesis is completely preserved in the non-pigmented mutants, but significantly changed at the transcriptional level. In addition, qRT-PCR and growth analysis indicate that the downregulated pig genes are correlated with the growth advantage. The RNA-seq data suggests that the transcriptional regulators play important roles in prodigiosin dyssynthesis of the non-pigmented mutant SCQ1-3M. It is supported by deletion of the relative downstream transcriptional regulator slyA, in which the pigment production of ΔslyA mutant is greatly reduced because of the strongly repressed pig genes. However, the different bacterial growth patterns and slyA expression levels of different color morphotypes suggest other factors may contribute to bacterial growth. In SCQ1-3M, the transcriptomic analysis reveals that the high-cost systems, including membrane proteins and T6SS, are suppressed, which may affect prodigiosin synthesis indirectly according to previous reports. In contrast, both amino acid degradation and import pathways are activated, which provides extra substances for bacterial growth (Figure 7). Generally, S. marcescens shuts down high-cost systems (including prodigiosin biosynthesis) but activates amino acid metabolite and transport systems for extra resources which contribute to growth advantage under laboratory conditions.
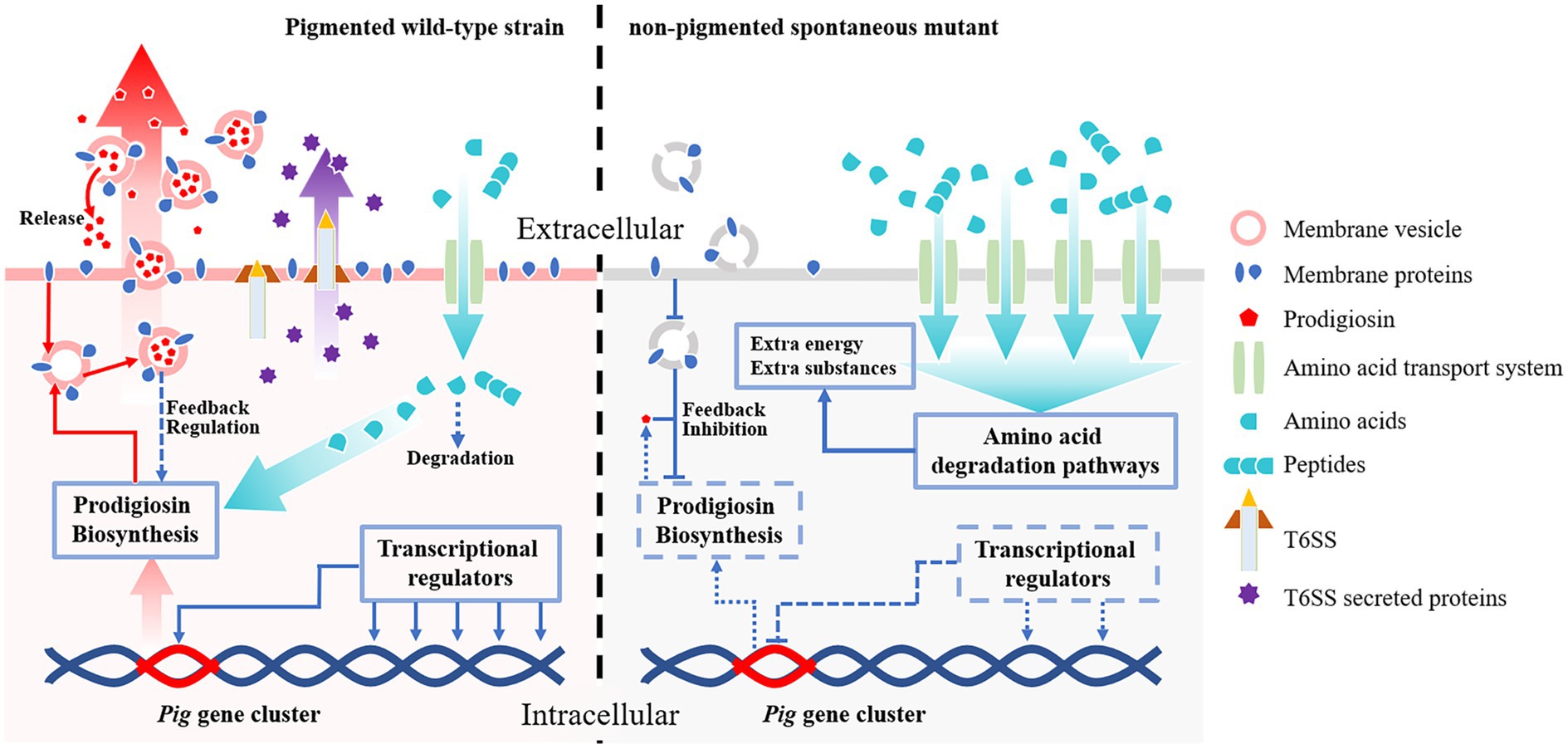
Figure 7. The potential mechanisms contribute to competitive growth advantage in S. marcescens spontaneous mutants under laboratory conditions. Both the wild-type strain and its spontaneous mutants preserve the complete prodigiosin synthesis gene cluster (Pig gene cluster). In the pigmented wild-type strain (left side), the pig genes are highly expressed under the control of transcriptional regulators, and the pigment is massively produced. The abundant prodigiosin is stored in membrane vesicles and secreted outside the cell by vesicular transport pathways to maintain lower intracellular concentrations which avoid feedback inhibition. The wild-type strain also consumes necessary resources to maintain some high-cost systems, e.g., T6SS. The amino acids required for prodigiosin synthesis and other systems are imported via amino acid transport systems, in which only a small amount of amino acids is degraded. In the non-pigmented spontaneous mutant (right side), the expression of the pig genes is highly suppressed by several factors. The transcriptional regulators, e.g., the positive regulator slyA, are down-expressed, which results in downregulated pig genes. Lack of essential membrane proteins for vesicle formation also potentially contribute to prodigiosin dyssynthesis. In this scenario, prodigiosin is neither correctly stored nor secreted outside the cells, which leads to strong feedback inhibition effects. Besides the pigment synthesis system, the non-pigmented mutant also shuts down T6SS or other high-cost systems to save resources. In addition, both amino acid transport- and degradation-pathways associated proteins are over-expressed in the non-pigmented mutant, which supplies extra nutrients and energies for bacterial growth. Therefore, the non-pigmented mutant shows a competitive growth advantage over the wild-type strain, which rapidly replaces the latter in laboratory conditions.
Data Availability Statement
The datasets generated for this study can be found in the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/). The accession number of the S. marcescens SCQ1 chromosome is CP063354. Raw sequence read data for Genome-seq of SCQ1, SCQ1-1M, SCQ1-2M, SCQ1-3M, and SCQ1-4M has been deposited in the NCBI SRA with accessions SRR12825186, SRR12737688, SRR12825185, SRR12825184, and SRR12825183, respectively, under BioProject PRJNA386063. Raw sequence read data for RNA-seq has been deposited in the NCBI SRA with BioSample accessions from SAMN21893314 to SAMN21893319 under BioProject PRJNA767201.
Author Contributions
TX, WZ, and YW designed the study and drafted the manuscript. TX, CX, JX, RL, and WZ performed the experiments and data analysis. NW, LX, YZ, ML, XM, and ZM edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by China Agriculture Research System (No. CARS-22), the Fund of China Qujing Academician and Experts Workstation (grant number WYJ20170413), and the Fundamental Research Funds for the Central Universities (XDJK2020C047).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.793202/full#supplementary-material
Abbreviations
Pig gene cluster, Prodigiosin biosynthesis gene cluster; qRT-PCR, Quantitative real-time RT-PCR; FC, Fold changes; T6SS, Type VI secretion system; OMV, Outer membrane vesicle.
Footnotes
2. ^https://crm.vazyme.com/cetool/singlefragment.html
3. ^https://www.ncbi.nlm.nih.gov/genome/browse/#!/prokaryotes/1112/
References
Aucken, H. M., and Pitt, T. (1998). Antibiotic resistance and putative virulence factors of Serratia marcescens with respect to O and K serotypes. J. Med. Microbiol. 47, 1105–1113. doi: 10.1099/00222615-47-12-1105
Bai, L., Wang, L., Vega-Rodríguez, J., Wang, G., and Wang, S. (2019). A gut symbiotic bacterium Serratia marcescens renders mosquito resistance to Plasmodium infection through activation of mosquito immune responses. Front. Microbiol. 10:1580. doi: 10.3389/fmicb.2019.01580
Bunting, M. I. (1940). The production of stable populations of color variants of Serratia marcescens,# 274 in rapidly growing cultures. J. Bacteriol. 40, 69–81. doi: 10.1128/jb.40.1.69-81.1940
Bunting, M. I. (1946). The inheritance of color in bacteria, with special reference to Serratia marcescens. Cold Spring Harb. Symp. Quant. Biol. 11, 25–32. doi: 10.1101/SQB.1946.011.01.005
Burritt, N. L., Foss, N. J., Neeno-Eckwall, E. C., Church, J. O., Hilger, A. M., Hildebrand, J. A., et al. (2016). Sepsis and hemocyte loss in honey bees (Apis mellifera) infected with Serratia marcescens strain sicaria. PLoS One 11:e0167752. doi: 10.1371/journal.pone.0167752
Chawrai, S. R., Williamson, N. R., Mahendiran, T., Salmond, G. P., and Leeper, F. J. (2012). Characterisation of PigC and HapC, the prodigiosin synthetases from Serratia sp. and Hahella chejuensis with potential for biocatalytic production of anticancer agents. Chem. Sci. 3, 447–454. doi: 10.1039/C1SC00588J
Chen, S., Blom, J., and Walker, E. D. (2017). Genomic, physiologic, and symbiotic characterization of Serratia marcescens strains isolated from the mosquito Anopheles stephensi. Front. Microbiol. 8:1483. doi: 10.3389/fmicb.2017.01483
Coulthurst, S. J., Williamson, N. R., Harris, A. K., Spring, D. R., and Salmond, G. P. (2006). Metabolic and regulatory engineering of Serratia marcescens: mimicking phage-mediated horizontal acquisition of antibiotic biosynthesis and quorum-sensing capacities. Microbiology 152, 1899–1911. doi: 10.1099/mic.0.28803-0
de Andrade Reis, R. J., Alves, A. F., dos Santos, P. H. D., Aguiar, K. M., da Rocha, L. O., da Silveira, S. F., et al. (2021). Mutualistic interaction of native Serratia marcescens UENF-22GI with Trichoderma longibrachiatum UENF-F476 boosting seedling growth of tomato and papaya. World J. Microbiol. Biotechnol. 37:211. doi: 10.1007/s11274-021-03179-z
Ellison, D. W., and Miller, V. L. (2006). Regulation of virulence by members of the MarR/SlyA family. Curr. Opin. Microbiol. 9, 153–159. doi: 10.1016/j.mib.2006.02.003
Fineran, P. C., Slater, H., Everson, L., Hughes, K., and Salmond, G. P. (2005). Biosynthesis of tripyrrole and β-lactam secondary metabolites in Serratia: integration of quorum sensing with multiple new regulatory components in the control of prodigiosin and carbapenem antibiotic production. Mol. Microbiol. 56, 1495–1517. doi: 10.1111/j.1365-2958.2005.04660.x
Genes, C., Baquero, E., Echeverri, F., Maya, J. D., and Triana, O. (2011). Mitochondrial dysfunction in Trypanosoma cruzi: the role of Serratia marcescens prodigiosin in the alternative treatment of Chagas disease. Parasit. Vectors 4, 1–8. doi: 10.1186/1756-3305-4-66
Grimont, P. A., and Grimont, F. (1978). The genus serratia. Annu. Rev. Microbiol. 32, 221–248. doi: 10.1146/annurev.mi.32.100178.001253
Gristwood, T., Fineran, P. C., Everson, L., and Salmond, G. P. (2008). PigZ, a TetR/AcrR family repressor, modulates secondary metabolism via the expression of a putative four-component resistance-nodulation-cell-division efflux pump, ZrpADBC, in Serratia sp. ATCC 39006. Mol. Microbiol. 69, 418–435. doi: 10.1111/j.1365-2958.2008.06291.x
Hampton, H. G., McNeil, M. B., Paterson, T. J., Ney, B., Williamson, N. R., Easingwood, R. A., et al. (2016). CRISPR-Cas gene-editing reveals RsmA and RsmC act through FlhDC to repress the SdhE flavinylation factor and control motility and prodigiosin production in Serratia. Microbiology 162, 1047–1058. doi: 10.1099/mic.0.000283
Haque, M. M., Kabir, M. S., Aini, L. Q., Hirata, H., and Tsuyumu, S. (2009). SlyA, a MarR family transcriptional regulator, is essential for virulence in Dickeya dadantii 3937. J. Bacteriol. 191, 5409–5418. doi: 10.1128/JB.00240-09
Harris, A. K., Williamson, N. R., Slater, H., Cox, A., Abbasi, S., Foulds, I., et al. (2004). The Serratia gene cluster encoding biosynthesis of the red antibiotic, prodigiosin, shows species-and strain-dependent genome context variation. Microbiology 150, 3547–3560. doi: 10.1099/mic.0.27222-0
Hejazi, A., and Falkiner, F. (1997). Serratia marcescens. J. Med. Microbiol. 46, 903–912. doi: 10.1099/00222615-46-11-903
Hines, D., Saurugger, P., Ihler, G., and Benedik, M. (1988). Genetic analysis of extracellular proteins of Serratia marcescens. J. Bacteriol. 170, 4141–4146. doi: 10.1128/jb.170.9.4141-4146.1988
Horng, Y.-T., Chang, K.-C., Liu, Y.-N., Lai, H.-C., and Soo, P.-C. (2010). The RssB/RssA two-component system regulates biosynthesis of the tripyrrole antibiotic, prodigiosin, in Serratia marcescens. Int. J. Med. Microbiol. 300, 304–312. doi: 10.1016/j.ijmm.2010.01.003
Horng, Y. T., Deng, S. C., Daykin, M., Soo, P. C., Wei, J. R., Luh, K. T., et al. (2002). The LuxR family protein SpnR functions as a negative regulator of N-acylhomoserine lactone-dependent quorum sensing in Serratia marcescens. Mol. Microbiol. 45, 1655–1671. doi: 10.1046/j.1365-2958.2002.03117.x
Hu, T., Chen, R., Zhang, L., Wang, Z., Yang, D., Zhang, Y., et al. (2019). Balanced role of T3SS and T6SS in contribution to the full virulence of Edwardsiella piscicida. Fish Shellfish Immunol. 93, 871–878. doi: 10.1016/j.fsi.2019.08.014
Jia, X., Liu, F., Zhao, K., Lin, J., Fang, Y., Cai, S., et al. (2021). Identification of essential genes associated with prodigiosin production in Serratia marcescens FZSF02. Front. Microbiol. 12:705853. doi: 10.3389/fmicb.2021.705853
Labrum, E. L., and Bunting, M. I. (1953). Spontaneous and induced color-variation of the HY strain of Serratia marcescens. J. Bacteriol. 65, 394–404. doi: 10.1128/jb.65.4.394-404.1953
Lavania, M., Chauhan, P. S., Chauhan, S., Singh, H. B., and Nautiyal, C. S. (2006). Induction of plant defense enzymes and phenolics by treatment with plant growth–promoting rhizobacteria Serratia marcescens NBRI1213. Curr. Microbiol. 52, 363–368. doi: 10.1007/s00284-005-5578-2
Libby, S. J., Goebel, W., Ludwig, A., Buchmeier, N., Bowe, F., Fang, F. C., et al. (1994). A cytolysin encoded by Salmonella is required for survival within macrophages. Proc. Natl. Acad. Sci. U. S. A. 91, 489–493. doi: 10.1073/pnas.91.2.489
Lin, C.-S., Horng, J.-T., Yang, C.-H., Tsai, Y.-H., Su, L.-H., Wei, C.-F., et al. (2010). RssAB-FlhDC-ShlBA as a major pathogenesis pathway in Serratia marcescens. Infect. Immun. 78, 4870–4881. doi: 10.1128/IAI.00661-10
Liu, G. Y., and Nizet, V. (2009). Color me bad: microbial pigments as virulence factors. Trends Microbiol. 17, 406–413. doi: 10.1016/j.tim.2009.06.006
McMahon, K. J., Castelli, M. E., Vescovi, E. G., and Feldman, M. F. (2012). Biogenesis of outer membrane vesicles in Serratia marcescens is thermoregulated and can be induced by activation of the Rcs phosphorelay system. J. Bacteriol. 194, 3241–3249. doi: 10.1128/JB.00016-12
Michaux, C., Sanguinetti, M., Reffuveille, F., Auffray, Y., Posteraro, B., Gilmore, M. S., et al. (2011). SlyA is a transcriptional regulator involved in the virulence of Enterococcus faecalis. Infect. Immun. 79, 2638–2645. doi: 10.1128/IAI.01132-10
Miyata, S. T., Kitaoka, M., Brooks, T. M., McAuley, S. B., and Pukatzki, S. (2011). Vibrio cholerae requires the type VI secretion system virulence factor VasX to kill Dictyostelium discoideum. Infect. Immun. 79, 2941–2949. doi: 10.1128/IAI.01266-10
Murdoch, S. L., Trunk, K., English, G., Fritsch, M. J., Pourkarimi, E., and Coulthurst, S. J. (2011). The opportunistic pathogen Serratia marcescens utilizes type VI secretion to target bacterial competitors. J. Bacteriol. 193, 6057–6069. doi: 10.1128/JB.05671-11
Pan, X., Sun, C., Tang, M., Liu, C., Zhang, J., You, J., et al. (2019). Loss of serine-type D-Ala-D-Ala carboxypeptidase daca enhances prodigiosin production in Serratia marcescens. Front. Bioeng. Biotechnol. 7:367. doi: 10.3389/fbioe.2019.00367
Pan, X., Sun, C., Tang, M., You, J., Osire, T., Zhao, Y., et al. (2020a). LysR-type transcriptional regulator MetR controls prodigiosin production, methionine biosynthesis, cell motility, H2O2 tolerance, heat tolerance, and exopolysaccharide synthesis in Serratia marcescens. Appl. Environ. Microbiol. 86, e02241–e02219. doi: 10.1128/AEM.02241-19
Pan, X., Tang, M., You, J., Liu, F., Sun, C., Osire, T., et al. (2020b). Regulator RcsB controls prodigiosin synthesis and various cellular processes in Serratia marcescens JNB5-1. Appl. Environ. Microbiol. 87, e02052–e02020. doi: 10.1128/AEM.02052-20
Rascoe, J., Berg, M., Melcher, U., Mitchell, F., Bruton, B., Pair, S., et al. (2003). Identification, phylogenetic analysis, and biological characterization of Serratia marcescens strains causing cucurbit yellow vine disease. Phytopathology 93, 1233–1239. doi: 10.1094/PHYTO.2003.93.10.1233
Repizo, G. D., Gagné, S., Foucault-Grunenwald, M.-L., Borges, V., Charpentier, X., Limansky, A. S., et al. (2015). Differential role of the T6SS in Acinetobacter baumannii virulence. PLoS One 10:e0138265. doi: 10.1371/journal.pone.0138265
Schuppener, L. M., Pop-Vicas, A. E., Brooks, E. G., Duster, M. N., Crnich, C. J., Sterkel, A. K., et al. (2017). Serratia marcescens bacteremia: nosocomial cluster following narcotic diversion. Infect. Control Hosp. Epidemiol. 38, 1027–1031. doi: 10.1017/ice.2017.137
Shanks, R. M., Lahr, R. M., Stella, N. A., Arena, K. E., Brothers, K. M., Kwak, D. H., et al. (2013). Serratia marcescens PigP homolog controls prodigiosin biosynthesis, swarming motility and hemolysis and is regulated by cAMP-CRP and HexS. PLoS One 8:e57634. doi: 10.1371/journal.pone.0057634
Shanks, R. M., Stella, N. A., Lahr, R. M., Aston, M. A., Brothers, K. M., Callaghan, J. D., et al. (2017). Suppressor analysis of eepR mutant defects reveals coordinate regulation of secondary metabolites and serralysin biosynthesis by EepR and HexS. Microbiology 163, 280–288. doi: 10.1099/mic.0.000422
Silva, Y. R. D. O., Contreras-Martel, C., Macheboeuf, P., and Dessen, A. (2020). Bacterial secretins: mechanisms of assembly and membrane targeting. Protein Sci. 29, 893–904. doi: 10.1002/pro.3835
Slater, H., Crow, M., Everson, L., and Salmond, G. P. (2003). Phosphate availability regulates biosynthesis of two antibiotics, prodigiosin and carbapenem, in Serratia via both quorum-sensing-dependent and-independent pathways. Mol. Microbiol. 47, 303–320. doi: 10.1046/j.1365-2958.2003.03295.x
Stankovic, N., Senerovic, L., Ilic-Tomic, T., Vasiljevic, B., and Nikodinovic-Runic, J. (2014). Properties and applications of undecylprodigiosin and other bacterial prodigiosins. Appl. Microbiol. Biotechnol. 98, 3841–3858. doi: 10.1007/s00253-014-5590-1
Sun, S.-J., Liu, H.-J., Weng, C.-H., Lai, C.-F., Ai, L.-Y., Liu, Y.-C., et al. (2016). The response of Serratia marcescens JG to environmental changes by quorum sensing system. Arch. Microbiol. 198, 585–590. doi: 10.1007/s00203-016-1213-9
Sun, Y., Wang, L., Pan, X., Osire, T., Fang, H., Zhang, H., et al. (2020). Improved prodigiosin production by relieving CpxR temperature-sensitive inhibition. Front. Bioeng. Biotechnol. 8:344. doi: 10.3389/fbioe.2020.00344
Wilf, N. M., and Salmond, G. P. (2012). The stationary phase sigma factor, RpoS, regulates the production of a carbapenem antibiotic, a bioactive prodigiosin and virulence in the enterobacterial pathogen Serratia sp. ATCC 39006. Microbiology 158, 648–658. doi: 10.1099/mic.0.055780-0
Wilf, N. M., Williamson, N. R., Ramsay, J. P., Poulter, S., Bandyra, K. J., and Salmond, G. P. (2011). The RNA chaperone, Hfq, controls two luxR-type regulators and plays a key role in pathogenesis and production of antibiotics in Serratia sp. ATCC 39006. Environ. Microbiol. 13, 2649–2666. doi: 10.1111/j.1462-2920.2011.02532.x
Williamson, N. R., Fineran, P. C., Gristwood, T., Chawrai, S. R., Leeper, F. J., and Salmond, G. P. (2007). Anticancer and immunosuppressive properties of bacterial prodiginines. Future Microbiol. 2, 605–618. doi: 10.2217/17460913.2.6.605
Williamson, N. R., Fineran, P. C., Leeper, F. J., and Salmond, G. P. (2006). The biosynthesis and regulation of bacterial prodiginines.Nat. Rev. Microbiol. 4, 887–899. doi: 10.1038/nrmicro1531
Williamson, N. R., Simonsen, H. T., Ahmed, R. A., Goldet, G., Slater, H., Woodley, L., et al. (2005). Biosynthesis of the red antibiotic, prodigiosin, in Serratia: identification of a novel 2-methyl-3-n-amyl-pyrrole (MAP) assembly pathway, definition of the terminal condensing enzyme, and implications for undecylprodigiosin biosynthesis in Streptomyces. Mol. Microbiol. 56, 971–989. doi: 10.1111/j.1365-2958.2005.04602.x
Xiang, T., Liu, R., Xu, J., Xu, C., Wang, N., Zhou, W., et al. (2021). Complete genome sequence of the red-pigmented strain Serratia marcescens SCQ1 and its four spontaneous pigment mutants. Microbiol. Resour. Announc. 10, e01420–e01456. doi: 10.1128/MRA.01456-20
Zhou, W., Li, J., Chen, J., Liu, X., Xiang, T., Zhang, L., et al. (2016b). The red pigment prodigiosin is not an essential virulence factor in entomopathogenic Serratia marcescens. J. Invertebr. Pathol. 136, 92–94. doi: 10.1016/j.jip.2016.03.011
Zhou, W., Zeng, C., Liu, R., Chen, J., Li, R., Wang, X., et al. (2016c). Antiviral activity and specific modes of action of bacterial prodigiosin against Bombyx mori nucleopolyhedrovirus in vitro. Appl. Microbiol. Biotechnol. 100, 3979–3988. doi: 10.1007/s00253-015-7242-5
Keywords: Serratia marcescens, spontaneous mutation, color morphotypes, growth advantages, transcriptomic analysis
Citation: Xiang T, Zhou W, Xu C, Xu J, Liu R, Wang N, Xu L, Zhao Y, Luo M, Mo X, Mao Z and Wan Y (2022) Transcriptomic Analysis Reveals Competitive Growth Advantage of Non-pigmented Serratia marcescens Mutants. Front. Microbiol. 12:793202. doi: 10.3389/fmicb.2021.793202
Edited by:
Jai Justin Tree, University of New South Wales, AustraliaReviewed by:
Brian H. Kvitko, University of Georgia, United StatesMichael Benedik, Texas A&M University, United States
Copyright © 2022 Xiang, Zhou, Xu, Xu, Liu, Wang, Xu, Zhao, Luo, Mo, Mao and Wan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongji Wan, Y2FuYmwzMzEyQDEyNi5jb20=