 Han Wang1†
Han Wang1† Ali Mohamed Elyamine
Ali Mohamed Elyamine Qixuan Chen
Qixuan Chen Zhong Hu
Zhong Hu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 31 January 2022
Sec. Microbial Symbioses
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.788709
This article is part of the Research Topic Plant Microbiome: Interactions, Mechanisms of Action, and Applications, Volume II View all 20 articles
Green algae are photosynthetic organisms and play an important role in coastal environment. The microbial community on the surface of green algae has an effect on the health and nutrition of the host. However, few species of epiphytic microbiota have been reported to play a role in promoting the growth of algae. In this study, 16S rDNA sequencing was used to study the changes of microbial composition on the surface of Ulva fasciata at different growth stages. Some growth promoting bacteria were identified. The possible growth-promoting behavior of the strains were verified by co-culture of pure bacteria obtained from the surface of U. fasciata with its sterile host. Among the identified species, a new bacterial species, Hyunsoonleella sp. HU1-3 (belonging to the family Flavobacteriaceae) significantly promoted the growth of U. fasciata. The results also showed that there were many genes involved in the synthesis of growth hormone and cytokinin in the genome of Hyunsoonleella sp. HU1-3. This study identified the bacterium Hyunsoonleella sp. HU1-3 for the first time, in which this bacterium has strong growth-promoting effects on U. fasciata. Our findings not only provide insights on the establishment of the surface microbiota of U. fasciata, but also indicate that Hyunsoonleella sp. HU1-3 is one of the important species to promote the growth of U. fasciata.
Epiphytic microbiota refers to a specific community harboring the surface of plants and does not parasitize or interfere with the growth and development of its host (Gopal and Gupta, 2016). The microbiota can be found on terrestrial plants, marine macroalgae (Short et al., 2015) and intertidal macroalgae (Hovel et al., 2016). For example, in marine ecosystems, microorganisms such as epiphytic bacteria, fungi and archaea gather on the surface of algae, making this micro-habitat an extremely active interface between host and microbes (Wahl et al., 2012).
Long-term coexistence enables microorganisms to evolve adaptive characteristics closely relating to plants (Vorholt, 2012). Many epiphytes may have a mensal, commensal or parasitic relationships with plants, including macroalgae (Gopal and Gupta, 2016). Microbiota can help plants better absorb nutrients necessary for growth and development (Doty et al., 2016). Recently, the significant role of epiphytic microbiota in assisting plants maintain normal growth and metabolism has attracted scientists attention (Barott et al., 2011). Host-adapted microorganisms are more resistant to abiotic stresses such as harmful ultraviolet radiation (Kamo et al., 2018), oxidative stress and desiccation (Vorholt, 2012), and affect plant health by reducing biotic or abiotic stresses.
The surface of seaweed also provides habitat for microbial communities (Wiese et al., 2009). The continuous colonization of epiphytic microbiota on healthy algae indicates that there is a positive interaction between them and the host. For example, the growth and development of some green algae depend on specific bacteria (Provasoli and Pintner, 1980). Some microorganisms can provide vitamins (Droop, 2007), fatty acids (Karthick and Mohanraju, 2018), and other active substances to regulate the growth of algae. Furthermore, microorganisms can even produce antimicrobial agents for algae to enable the hosts to adapt to environmental stresses (Miranda et al., 2013; El Shafay et al., 2016). Studies have shown that their abundance is also related to availability of nutrients on the host surface (Paix et al., 2020).
Marine green macroalgae Ulva is widely distributed in coastal areas all over the world. It is commonly used as a model to study the environmental adaptability of biological life cycle (Juhmani et al., 2020). It has been reported that high nutrient content in seawater leads to an increase in the number of Ulva, which may lead to green tide (Zhang et al., 2019). The development of high-throughput sequencing technology makes it possible to characterize the composition of Ulva epiphytic microbiota. To our knowledge, the current research on the epiphytic microbiota of Ulva mainly focuses on the functional characteristics of microorganisms that responsible for Ulva morphogenesis and the different species composition that responsible for Ulva surface morphogenesis during the green tide (Nakanishi et al., 1996; Dobretsov and Qian, 2002; Spoerner et al., 2012; Ghaderiardakani et al., 2017, 2019; Weiss et al., 2017; Alsufyani et al., 2020; Chen et al., 2020; Gianmaria et al., 2020; Inaba et al., 2020; Juhmani et al., 2020; Liu et al., 2020; Qu et al., 2020). However, the biodiversity and functional relationship of epiphytic microbiota harbored by Ulva have not been fully explored. Previous studies have shown that bacterial communities located in different genotypes of the same algae usually have small differences. In addition to the influences of genotype, the composition of epiphytic bacterial community is also affected by seasons, environment and nutrients (El-Said and El-Sikaily, 2013).
Most studies show that α-Proteobacteria and γ-Proteobacteria are the main bacterial phyla colonizing the surface of algae (Bondoso et al., 2014). Different plants, such as rice (Edwards et al., 2015), grape (Zarraonaindia et al., 2015), sugarcane (De Souza et al., 2016), and citrus (Xu et al., 2018), have the same core microbiota (at the genus level), among which Pseudomonas, Methylobacterium, and Sphingomonas are the most common. This suggests that the core microbiome is selectively recruited and usually has the necessary functions to co-existence with the host. The identification methods and techniques of core microorganisms constitute the basis for studying how to construct a synthetic microbiota, revealing plant-microbiome interaction and possibly promoting plant growth.
Ulva fasciata are widely distributed in the rocky intertidal zone along the coast of China. Bacteria on algae vary with season and the life cycle of host (Lachnit et al., 2011). The purpose of this study was to determine (1) whether the epiphytic microbial community on the surface of U. fasciata varies at different growth stages, (2) whether the epiphytic microbiota is related to the growth stage of U. fasciata, and (3) whether the selected bacteria can promote the growth of U. fasciata.
The application of the bacterial treatment method in a large number of studies showed that the bacterial treatment method not only reveals how plants affect their microbial communities and how microbial communities affect plant growth and health but also has high operability and application value for agricultural production such as promoting plant growth and resisting plant diseases. It is believed that the application of the bacterial treatment method will also bring light to the solution of ecological problems such as green tide. Future studies will focus on the detailed identification of the nutrient fixation and promotion abilities of these isolates from the surface of U. fasciata.
Ulva fasciata was randomly sampled from Nanao Island, Shantou, Guangdong, China (about 100 m from the coast) (116.53°E 23.11°N). In the seedling stage, U. fasciata were sampled once a week on November 9th, 16th, and 23th, that corresponding to seedling stages 1, 2, and 3, respectively. During the growth stage, U. fasciata were sampled on December 27, February 2, and March 28, which corresponding to growth stages 1, 2, and 3, respectively. At maturity, U. fasciata were collected on April 2, May 3, and June 3, corresponding to maturity stages 1, 2, and 3, respectively (Figure 1A). The samples were collected in three replicates, washed with sterile seawater, and stored in 50-mL sterilized centrifuge tubes.
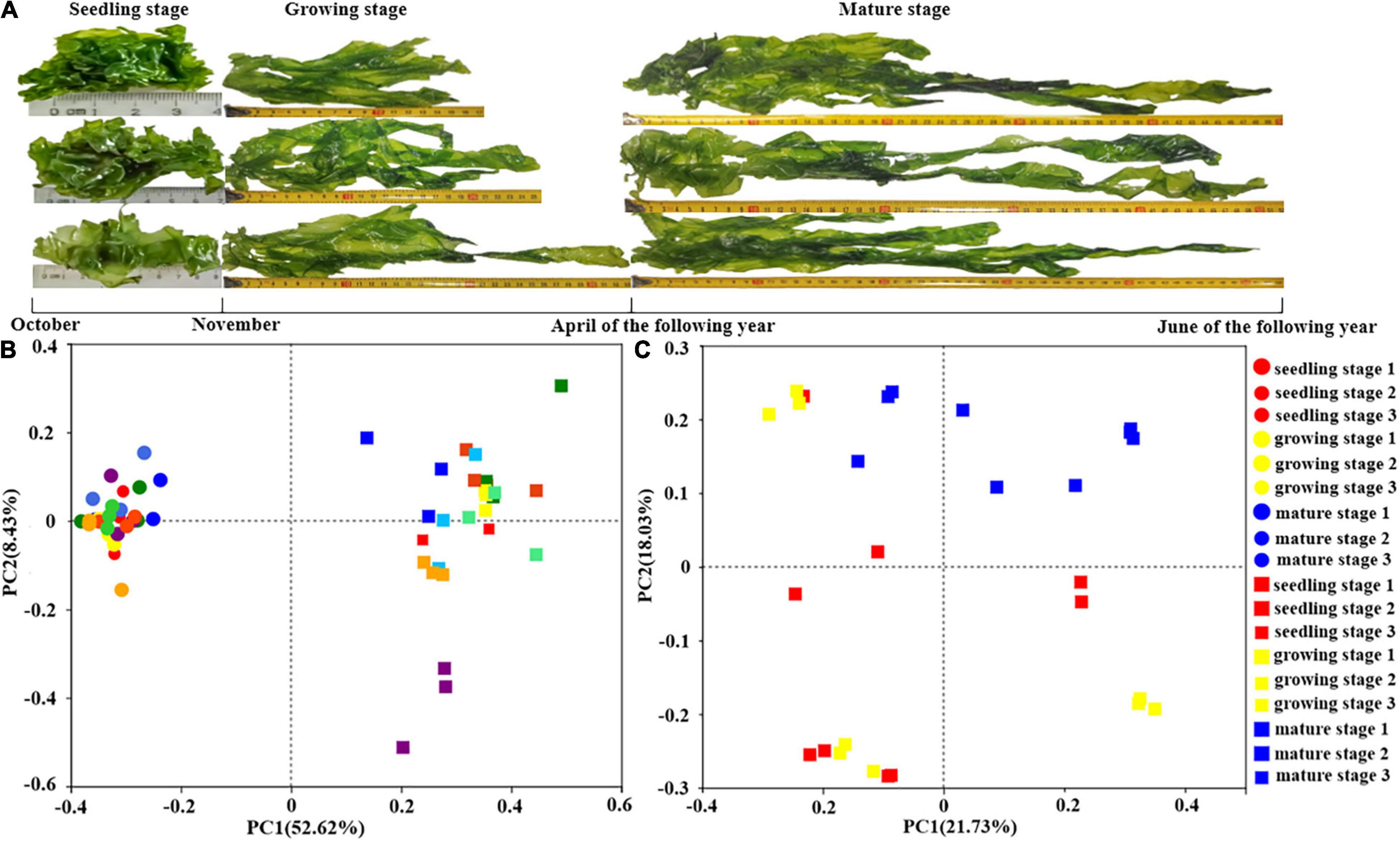
Figure 1. The surface microbiota of Ulva fasciata variation at different growth stages. (A) Photographs of U. fasciata samples in different life cycles (B) Principal Coordinate Analysis of microbiota changes on the surface of U. fasciata and seawater samples of different growth stages at Genus Level (C) Principal coordinate analysis of microbiota changes on the surface of U. fasciata in different growth stages at Genus Level (the circles represent the seawater sample and the squares represent the surface of the U. fasciata sample).
During nine sampling, the in situ environmental parameters (temperature, salinity, pH, and dissolved oxygen concentration) were recorded (Supplementary Table 1). At the same time, 1 L seawater samples were collected, filter through 0.45 and 0.22 μm membranes to remove impurities and collect bacteria. The filtrates (0.22 μm filter membrane) were stored in sterile sampling bags and the filtered water was cryopreserved for the determination of physical and chemical parameters, including dissolved organic carbon (TOC), inorganic carbon (TIC), total nitrogen (TN), and total carbon (TC) according to the methods described by Urgun-Demirtas et al. (2008). All samples were placed in a foam box with ice cubes and transferred to the laboratory within 4 h. In the laboratory, the microbiota harboring the surface of U. fasciata was then immediately collected. The U. fasciata samples in a 50-mL sterilized centrifuge tube was mixed with an appropriate amount of sterile seawater and treated with ultrasonic (1 min, 50 W, 5s/5s). Algae were removed, and then the suspension was centrifuged at 4,000 × g for 10 min to collect the pallet, which was then used for bacterial DNA extraction.
Bacterial DNA was extracted using the CTAB method (Zhou et al., 1996). DNA quality and integrity were evaluated by gel electrophoresis. Samples with high-quality DNA were stored at −80°C until further use. The DNA of all samples was sent to Shanghai Majorbio Bio-Pharm Technology Company for sequencing and data analyses.
To determine the species and abundance of bacteria, the V3–V4 region of the 16S rRNA gene was amplified using primers 806R (5′-GGACTACHVGGGTWTCTAAT-3′) and 338F (5′-ACTCCTACGGGAGGCAGCAG-3′), and the PCR products were sequenced using Illumina MiSeq sequencing platform. The sequencing data were stitched by Flash V1.2.11, and then the optimized Fastq files were obtained after quality control by Fastp V0.19.6. The sequences were preprocess using the software Usearch 7 (parameter setting: minsize 2), and the sequences were grouped into operational classification units (OTUs) for species level classification using 97% sequence similarity. After obtaining the OTU classification table, those OTUs assigned to chloroplasts or mitochondria and eukaryotes were excluded from the data set. OTU counts were normalized, the original count was divided by the total number of each sample, and then multiplied by the median to calculate the relative abundance. Using QIIME V1.9.1, the taxonomic information was assigned to each representative OTU sequence and filtered OTUs were used to calculate alpha diversity indexes, including Shannon, Chao1, and Simpson. Calculate the Bray-Curtis distance for all pairs of samples to determine the amount of diversity shared between communities (β-diversity) (Liu et al., 2021). Principal Co-ordinates Analysis (PCoA) sorting was performed according to Bray-Curtis distance. Adonis based on Bray distance was used to calculated the significant difference between bacterial community and the null hypothesis (Anderson, 2001). Dispersion of microbial communities was evaluated using the pairwise permutation tests with Tukey’s HSD. The Kruskal–Wallis test was used to determine whether there were significant differences in the abundance of bacterial classes. Bacterial taxonomic biomarkers for bacterial classification were obtained by referring to the random forest algorithm (Zhang et al., 2018). The Cytoscape software package was used for visualization. All of the above software used default parameters.
Samples of U. fasciata were collected at different times, rinsed and immediately transferred to the laboratory. The blade surface was gently scraped with a sterile knife, and the scraped materials were gradually diluted and spread on the marine agar plate 2216E, and placed in an incubator at 25°C for 3–4 days. Single colonies were selected, streaked, purified, and inoculated in marine broth 2216E. The 16S rRNA gene of bacteria was amplified with universal primers 27F and 1492R, and the samples were sent to Bioengineering (Shanghai) for sequencing. The phylogenetic names of the isolates were identified by EzBioCloud database. A total of 228 pure strains were obtained. These strains were cultured on marine broth 2216E to logarithmic growth phase. The bacterial suspension was mixed with 40% sterilized glycerol in the ratio of 1:1. These strains were selected as candidate strains for bacterial treatment. To test the promoting effect of bacterial treatment on plant growth, U. fasciata without surface bacteria (WSB) was cultured with isolates.
Ulva fasciata without surface bacteria was prepared as follow:
The surface of U. fasciata was washed with sterile seawater and ultrasonic waves (1 min, 50 KHz, 5s/5s) for three times to remove bacteria. The mixture of antibiotics (including 100 μg/mL streptomycin sulfate, 0.25 μg/mL amphotericin, 100 μg/mL ampicillin, and 0.1 μg/mL imidazole) was added to the culture tube and incubated for 2 days; the same dose of antibiotics was added on the third day. The surface of U. fasciata was quickly scrubbed with a sterile cotton swab dipped in a multi-enzyme cleaning solution (3M), and then rinsed with sterile seawater. The obtained SBR was evaluated through three methods: (1) PCR detection of seawater collected from the last rinsing of U. fasciata and algal abrasive solution in the range of about 1500 bp using primers 27F and 1492R, (2) the seawater collected from the last time rinsing of U. fasciata was used to culture on marine agar 2216E to confirm the growth of bacteria in 7 days, (3) the sterile surface of U. fasciata was stained with 1% (v/v) of DAPI for 20 min, the excess DAPI was removed with sterile water, and then the confocal laser scanning microscopy (CLSM) was used to check whether there were bright blue bacterial spots (bacteria) in the sample. Samples were imaged with CLSM (LSM 700, Zeiss, Germany) and x100 oil immersion objective. Select DAPI channel 400–630 nm. Four random locations were scanned on each sample.
The experiment consisted of three bacterial treatment groups, sterile seawater (C group), Bacillus cereus U5-30 (P group) and Hyunsoonleella sp. HU1-3 (H group). The bacteria were incubated in marine broth 2216E at 25°C for 2 days, centrifuged, washed twice with sterile seawater, and then resuspended to a density of 107 cells per mL. The volume of bacterial liquid added to each bacterial treatment was 5 mL. The mixed bacteria were added to a bottle connected to filtered air and contained 800 mL of sterile seawater, and then co-cultured with U. fasciata (WSB) for 7 days. At the end of the experiment, the co-cultured U. fasciata was washed with sterile seawater, and the surface bacterial strains of U. fasciata were obtained by ultrasonic shock. The 2216E marine agar plate was used to test whether the bacterial treatment strains survived after 7 days.
To determine the growth rate, at the beginning of the co-culture experiment, young U. fasciata with similar growth trend and size were selected, weighed (set up as Wt) and sterilized. The vase was placed in a light incubator for 7 days (temperature, light intensity, photoperiod and air supply time were 20°C, 4000 LX, 12 h:12 h, and 24 h, respectively). After 7 days of co-culture, U. fasciata was taken out, the surface seawater was removed and weighed (W0). Growth rate was calculated by the formula of [(Wt/W0)1/t-1] × 100% (t is days of culture) (Yong et al., 2013). During the experiment, each bacterial treatment group was guaranteed at least three biological replicates. The contents of soluble protein, soluble sugar, phycocyanin, and chlorophyll-a (Schwenzfeier et al., 2011) were determined to characterize the biomass of U. fasciata.
The bacteria strains were incubated in 2216E liquid medium at 25°C for 3 days, with or without 100 μg/mL of L-tryptophan, and then centrifuged. After that, the supernatant was diluted to OD600 = 0.8 with sterile seawater, and 10 mL of culture were added to a bottle containing 800 mL of sterile seawater with U. fasciata (WSB) and inoculated for 7 days. In the experiments, six supernatant were used, including sterile seawater (A group), 2216E liquid medium (B group), supernatant of B. cereus strain U5-30 living in 2216E liquid medium (C group), supernatant of Hyunsoonleella sp. strain HU1-3 living in 2216E liquid medium (D group), supernatant of B. cereus strain U5-30 living in 2216E liquid medium and L-tryptophan (E group), supernatant of Hyunsoonleella sp. strain HU1-3 living in 2216E and L-tryptophan (F group). Six groups of supernatants were separately cocultivated with U. fasciata (WSB), and the growth rate of U. fasciata was measured by the same method as above.
In order to illustrate how Hyunsoonleella strain HU1-3 promotes the growth of U. fasciata, the extraction of its genome was sent to BGI Corporation for genome test, and double-terminal sequencing was performed based on Illumina PE150 platform. Raw data is processed by low-quality filtering. Low quality bases of more than 40 bp were removed (quality value 38 or less), N bases were up to 10 bp, and the overlap between junctions was more than 15 bp. The repeated contamination is then removed to obtain valid data. The genome size was estimated by K-mer statistical analysis, and the raw sequencing reads were trimmed and assembled by SOAPdenovo v2.04 software1. The FastQC software was used to analyze sequence quality (Brown et al., 2017). GeneMark software (V.4.17) (Besemer et al., 2001) was used for comparison and genomic protein-coding genes were obtained. These gene sequences were compared with the known Nr database (non-redundant protein database) (Li et al., 2002). For the comparison results of each sequence, the highest score (identity ≥40%, coverage ≥40%) was selected for gene function annotation.
The bacteria strains were incubated in 2216E liquid medium at 25°C for 3 days. 1-mL incubation fluid was centrifuged, the cell precipitates were washed twice with PBS, and then resuspended to 107 cells/mL. 1 mL of the suspension was mixed with 10 mL of liquid medium (with and without 100 μg/mL of tryptophan). After 3 days, 50 μL of supernatant was collected and mixed with 50 μL of Salkowski reagent (50 mL 35% HClO4 + 1 mL 0.5 mol/L FeCl3). After 25 min, the absorbance value was read at 530 nm (Bric et al., 1991) to measure the production of bacterial IAA. Bacteria were inoculated in 50 mL sterilized liquid NBRIP medium at a dose of 0.2% and cultured in a flask at 25°C for 7 days. The culture was centrifuged at 5,000 RPM for 20 min, and the precipitation of P2O5 was determined by molybdenum blue method according to Galhardo and Masini (2000) to measure the solubility of phosphate. All measurements were conducted in triplicate.
In order to investigate the variations of microbes harboring the surface of U. fasciata under natural conditions, seawater samples and U. fasciata samples, a total of 2,969,125 high-quality sequences were obtained from all samples. The bacterial community structure over time in different samples is illustrated in Figures 1B,C. There was a significant difference (P < 0.01) between the microbial community living in seawater and the U. fasciata surface community (Figure 1B). PCoA analysis also showed that there were different microbiota harboring the surface of U. fasciata at different growth stages (R2 = 0.68, P < 0.01, adonis) (Figure 1C). This indicated that the growth stage plays a significant role in the establishment of microbiota inhabiting the surface of U. fasciata. Mature stage samples were close to one another, while seedling stage samples were relatively far away from one another (Supplementary Figure 1). The microbial community in mature stage overlapped with that in seedling stage and growth stage, indicating that the microbial community is in a transitional state between mature and seedling/growth stages. Similar results can be obtained by calculating the community diversity index (Shannon) and community richness index (Chao and ace) (Table 1), in which the alpha-diversity index of surface bacteria was significantly lower than that of seawater bacteria, while the diversity index of U. fasciata surface bacteria remained relatively stable.
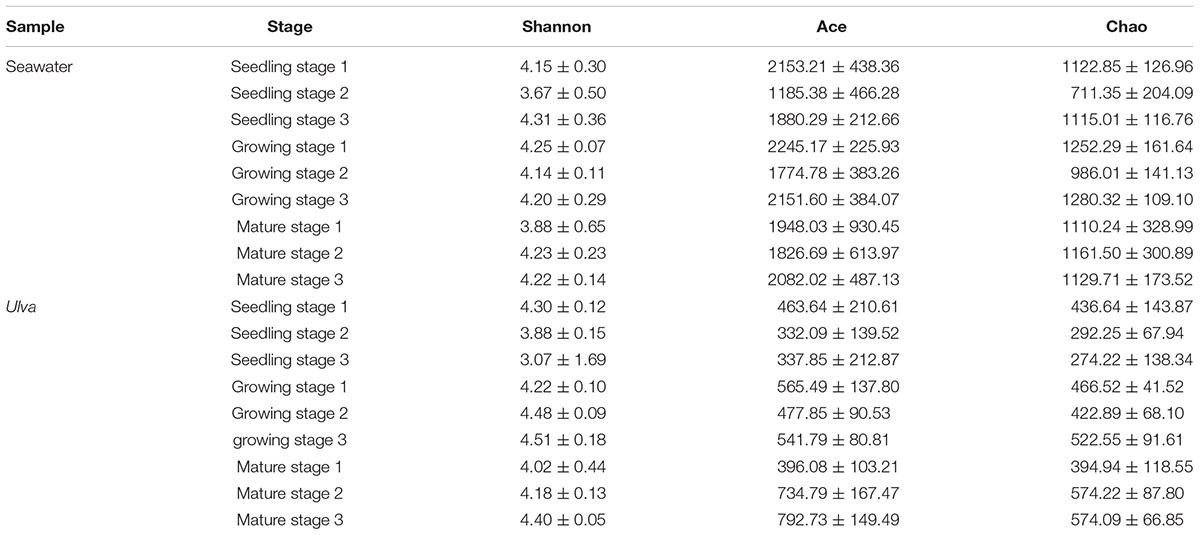
Table 1. Microbiodiversity indices of U. fasciata surface and seawater at different growth stages.
The relative abundance of epiphytic microbiota of U. fasciata and seawater is illustrated in Supplementary Figures 2A,B. Seawater samples were dominated by marine bacteria, such as HIMB11 and NS5 marine groups (Supplementary Figure 2A). Microbial community abundance were significant differences in the surface of U. fasciata with the seawater (Supplementary Figure 2B). The relative abundance of Proteobacteria and Bacteroidetes on U. fasciata surface was significantly higher that in seawater. The results of Kruskal-Wallis analysis (Supplementary Figure 3A) showed that although the relative abundance of these species increased or decreased throughout the growth period, there was no significant difference. Actinobacteria and Firmicutes changed greatly throughout the growth stage. At the genus level, there were significant differences in the relative abundance of some species (Supplementary Figure 2C). For instance, at the seedling stage, the relative abundances of Rhodobacteraceae (13.0%, P < 0.01, Kruskal–Wallis), Acinetobacter (6.2%, P < 0.01, Kruskal–Wallis), and Vibrio (1.2%, P < 0.01, Kruskal–Wallis) was significantly lower, while the relative abundance of Ralstonia (2.3%, P < 0.01, Kruskal–Wallis), Erythrobacter (2.8%, P < 0.01, Kruskal–Wallis), and Rhodococcus (2.4%, P < 0.01, Kruskal–Wallis) were significantly higher. The composition of microbiota on the surface of U. fasciata varied greatly throughout the growth, but the dominant species of microbiota were Rhodobacteraceae, Ralstonia, Erythrobacter, Rhodococcus, Acinetobacter, Alteromonas, Vibrio, Halomonas, and Granulosicoccus.
The regression analysis between the relative abundance of bacteria on the surface of U. fasciata species and the growth stage was established and revealed that the model explained 56% of the surface of U. fasciata microbiota variance related to U. fasciata growth stage (Supplementary Figure 4). At the class level, the class number (n = 30) was stable and relative to the cross-validation error curve. Therefore, these 30 bacterial classes, especially Rhodococcus and Hyunsoonleella, were defined as biomarker groups in the model.
The relationship between environmental factors and the abundance of different phyla is shown in Figure 2. At the phylum level, the relative abundance of Patescibacteria, Firmicutes and Bacteroidetes was positively correlated with the physiological parameters of different carbon forms. As mentioned above, the U. fasciata growth stage is the best explanation for the variation of microbial community. However, the environmental and physiological parameters of U. fasciata at different growth stages have different explanations for the variation of microbial community (Table 2). Dissolved oxygen (DO) and Phycocyanin had no effect on the microbial community on the surface of U. fasciata (R2 = 0.04 and 0.05, respectively, P > 0.05, Adonis). TOC, pH, soluble protein and TN had moderate effects on the microbial community on the surface of U. fasciata. IC and TC had the greatest effect on the microbial community on the surface of U. fasciata (R2 = 0.13, 0.14, respectively, P < 0.05, Adonis). The results showed that environmental factors and physiological parameters of U. fasciata also affected the structure of surface bacteria.
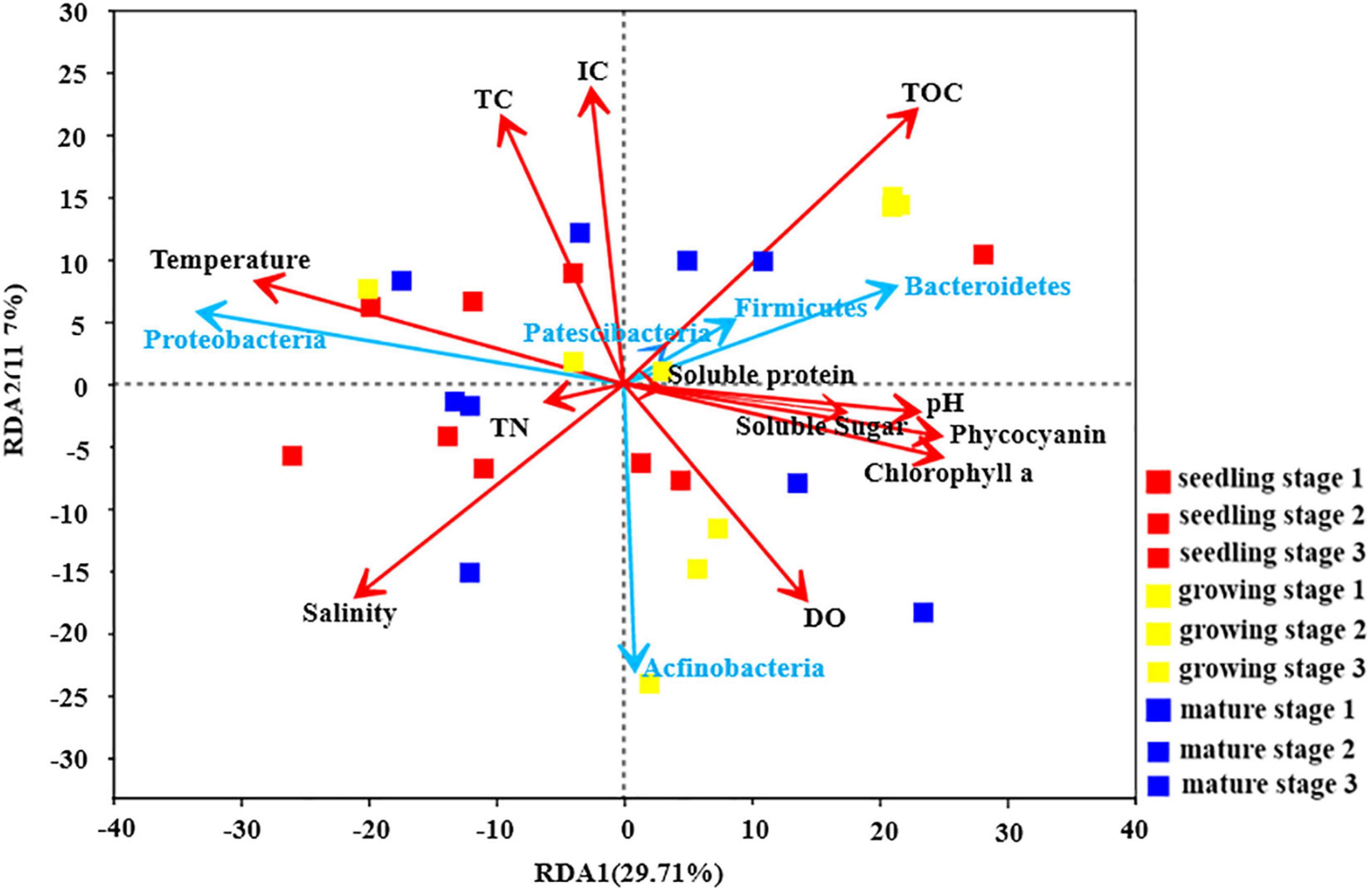
Figure 2. Redundancy Analysis of U. fasciata surface microorganisms with environmental factors and plant physiological indexes at Phylum level (The red arrows represented different environmental parameters and physiological indicators of U. fasciata and the blue arrows represented the top five groups of bacteria in abundance).
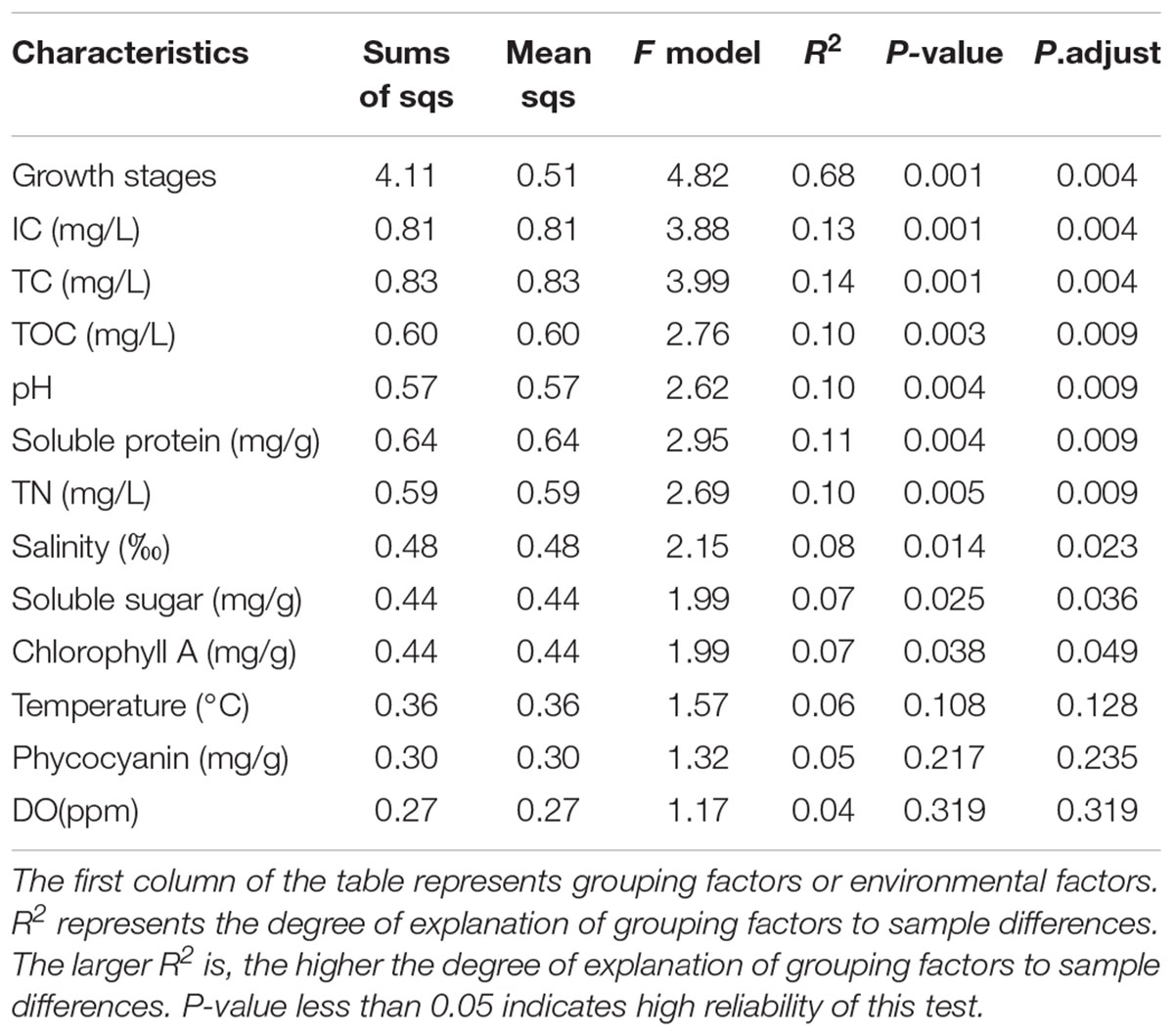
Table 2. Effects of growth stage, environmental factors, and physiological parameters of U. fasciata on U. fasciata surface microbiota.
A total of 228 bacterial strains were isolated from the surface of U. fasciata at different growth stages (Supplementary Figure 5). These strains were classified into three phyla, including Firmicutes, Bacteroidetes, and Proteobacteria. Although bacteria were still present in the U. fasciata (WSB) lapped samples (Supplementary Figure 6C) and intercellular bacteria were detected by CLSM (Supplementary Figure 6P), the quality of U. fasciata (WSB) was comparable to that of wild U. fasciata (Supplementary Figure 7) (P > 0.05, ANOVA).
The prediction results of machine forest model suggest that members of Rhodobacteraceae and Flavobacteriaceae may play an important role in the growth of U. fasciata. Therefore, bacterial treatment experiments were carried out to investigate the growth-promoting effect of Rhodobacteraceae and Flavobacteriaceae on U. fasciata. The role of endophytic bacteria was ignored in the experiment bacterial treatments. Among the isolates, Hyunsoonleella sp. HU1-3 demonstrated the greatest growth promoting potential on U. fasciata. Compared with other bacteria of Hyunsoonleella, its 16S rRNA gene sequence similarity and average nucleotide identity values were less than 98.7 and 95%, respectively (Chun et al., 2018), so it is considered as a new species of the genus.
The four physiological indices and growth rate of U. fasciata of group H [co-culture of U. fasciata (WSB) and Hyunsoonleella sp. HU1-3] were significantly higher than those of group C (only seawater was added as the negative control) and group P [co-culture of U. fasciata (WSB) and B. cereus U5-30 as the positive control] (P < 0.05, ANOVA) (Figure 3). In fact, compared with group P, the positive effect of Hyunsoonleella HU1-3 on soluble protein, soluble sugar and phycocyanin of U. fasciata showed that the strain significantly increased the biomass of sterile U. fasciata (P < 0.05, ANOVA) (Figures 3A–D). The growth rate of group H (5.49% day–1) and group P (4.05% day–1) was significantly higher than that of group C (1.10% day–1) (Figure 3E).
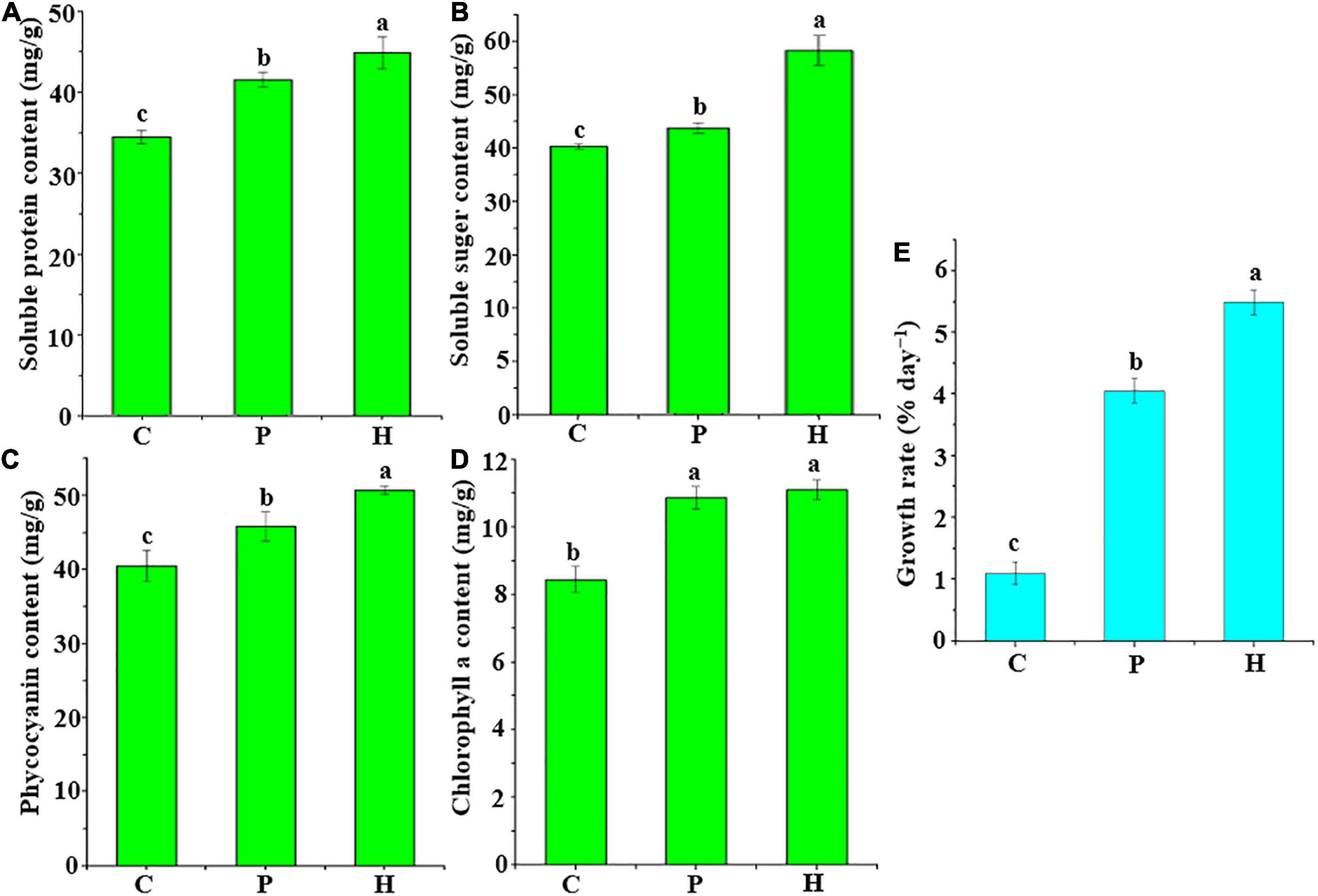
Figure 3. Changes of growth rate and four physiological indices of bacterial treatment and U. fasciata (WSB). (A) Soluble protein content, (B) Soluble sugar content, (C) Phycocyanin content, (D) Chlorophyll a content, and (E) Growth rate. C group used sterile seawater for the negative control, P group only contains Bacillus cereus U5-30 strain was used to be the positive control, H group contained only one strain of Hyunsoonleella sp. HU1-3, the difference coefficient represents the difference between C, P, and H group.
Six different groups of bacterial supernatants were cocultured with U. fasciata (WSB) (Figure 4). There was no significant difference in the growth rate of U. fasciata between group A (sterile seawater) and group B (sterile 2216E liquid medium) (P > 0.05, ANOVA). Interestingly, the supernatant of group C (B. cereus strain U5-30) and group D (Hyunsoonleella sp. HU1-3) significantly increased the growth rate of U. fasciata (1.52% day–1 and 2.45% day–1, respectively; P < 0.05, ANOVA). Surprisingly, the U. fasciata growth rate of group E and group F (2.77% day–1 and 3.95% day–1, respectively; P < 0.05, ANOVA) was significantly higher than that of group C.
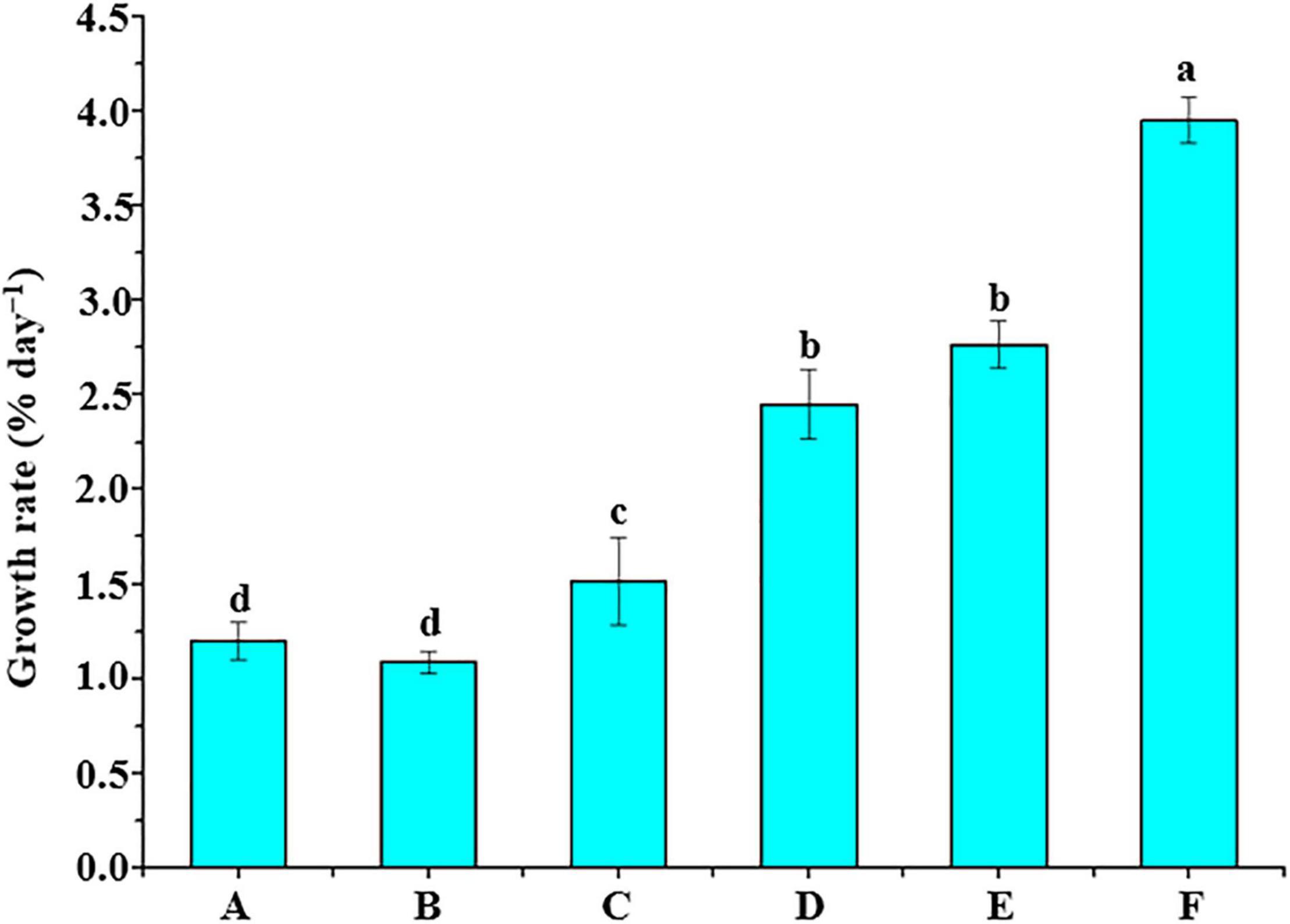
Figure 4. Supernatant of bacterial culture and U. fasciata (WSB) growth promotion test. A group added sterile seawater, B group added sterile 2216E liquid medium, C group added the supernatant of the strain B. cereus U5-30 living in 2216E, D group added the supernatant of the strain Hyunsoonleella sp. HU1-3 living in 2216E, E group added the supernatant of the strain B. cereus U5-30 living in 2216E + L-tryptophan, F group added the supernatant of the strain Hyunsoonleella sp. HU1-3 living in 2216E + L-tryptophan. The difference coefficient represents the difference between A and F group.
Based on the genome of Hyunsoonleella HU1-3 (Figure 5 and Supplementary Table 2), several amidases (such as N-acetylmuramoyl-L-alanine amidase LytC and 2-oxoglutaramate amidase) and tryptophan related enzymes (such as tryptophan 2,3-dioxygenase and tryptophan-tRNA ligase) were identified, indicating that Hyunsoonleella sp. HU1-3 may produce IAA through IpyA and IAM pathways. Genes involved in the synthesis of cytokinins (CK) have also been annotated, such as tRNA-dimethylallyltransferase, tRNA-2-methylthio-N6-dimethylallyladenosine, and cytokinin-nucleoside 5-monophosphate ribose hydrolase. These results indicate that Hyunsoonleella sp. HU1-3 has the potential to synthesize a variety of CKs. Furthermore, seven phosphate synthases genes were found in the genome of Hyunsoonleella sp. HU1-3. Interestingly, Hyunsoonleella sp. HU1-3 contains genes involved in vitamin synthesis (thiamine-phosphate synthase and thiamine-monophosphate kinase). Other genes annotated as iron carrier enzymes (such as iron-sulfur cluster carrier protein and iron-dependent repressor IdeR) were also found in the genome.
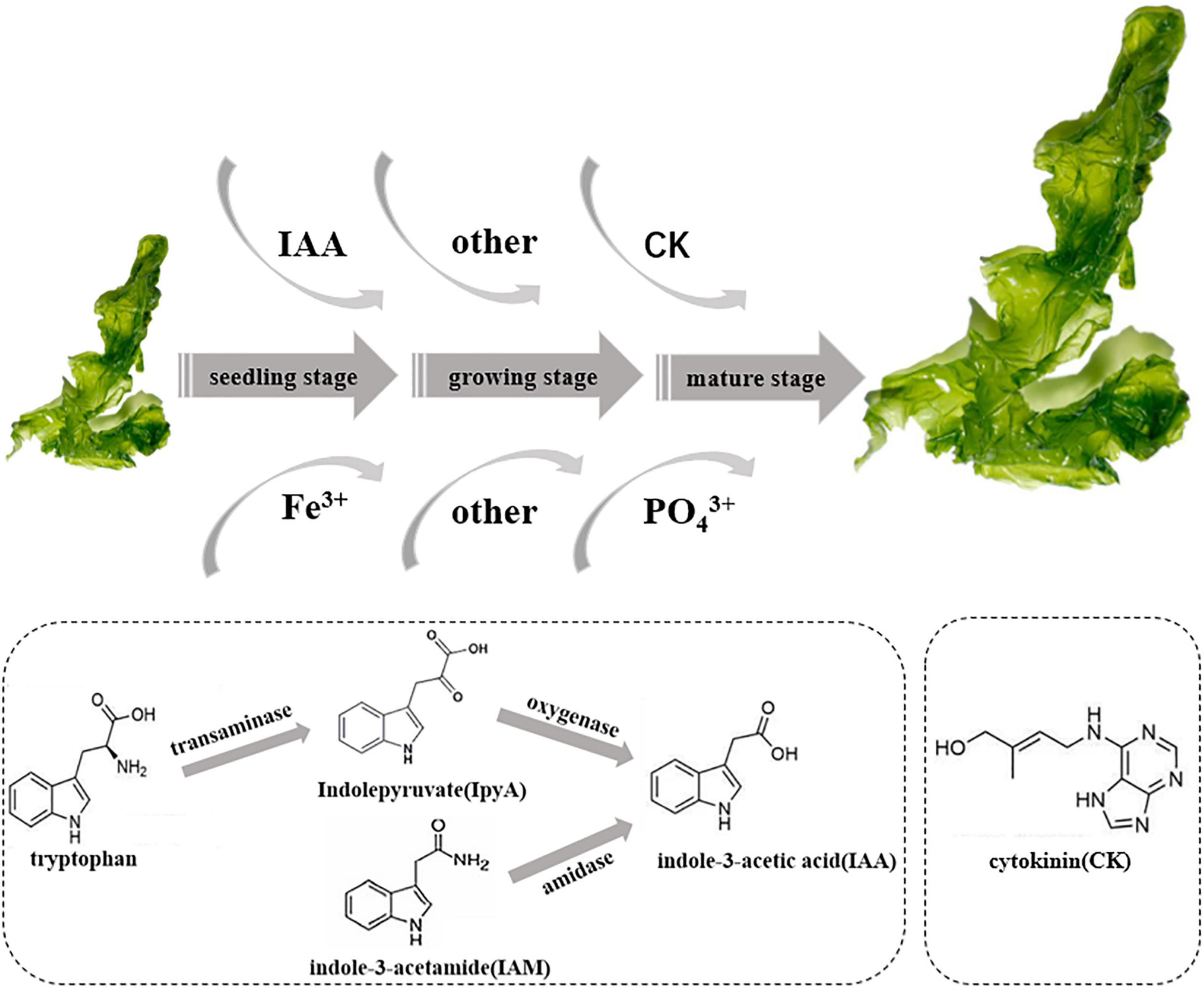
Figure 5. Schematic diagram of potential promoting growth of U. fasciata of strain Hyunsoonleella sp. HU1-3.
Plant growth is known to depends on several transaminase proteins, especially growth hormones, such as IAA. In the absence of tryptophan, the IAA yield of B. cereus U5-30 was 209.70 ± 3.64 μg/mL. The addition of tryptophan increased the IAA production to 375.16 ± 3.59 μg/mL. The phosphate solubility of B. cereus U5-30 was 191.18 ± 2.87 μg/mL. On the other hand, the IAA yield and phosphate solubility of Hyunsoonleella sp. HU1-3 without tryptophan were 227.64 ± 0.82 μg/mL, and 213.98 ± 1.15 μg/mL, respectively. It is speculated that the IAA yield and phosphate solubility of Hyunsoonleella sp. HU1-3 are better than B. cereus U5-30 (Table 3).

Table 3. The production of IAA and the solubilization capacity of phosphate in the strain.
Epiphyte studies during plant growth are usually carried out under greenhouse conditions (Edwards et al., 2015). We conducted field sampling to represent the life cycle of U. fasciata. U. fasciata’s growth slowly in November (seedling stage) and reaches maturity in April of the following year. In June, with the increase of seawater temperature, the U. fasciata will bleached, rotten and immediately washed away by seawater, which makes sampling difficult. Therefore, we only focus on U. fasciata that grows well from seedling stage to decay stage. Roth-Schulze et al. (2018) demonstrated that the bacterial diversity on the surface of U. fasciata is different from the surrounding seawater. The PCoA analysis in this study revealed that the composition of epiphytic microbiota of U. fasciata was significantly different from that of surrounding seawater. In a previous study, Lachnit et al. (2011) reported that the microbial composition on the surface of Ulva infantis varied with the growth stage. Similarly, it was reported that the dynamic change of surface microbiota through the plant life cycle depends on the growth stage of plant (Chaparro et al., 2014; Dombrowski et al., 2016). The dominant bacteria on the surface of U. fasciata were Proteobacteria and Bacteroidota. This finding is consistent with Burke et al. (2011).
The core microbiome is defined as a group of shared members in microbial communities from similar habitats (Shade and Handelsman, 2012). Thus, the discovery of core microbiome is very important to understand the stable components of complex microbial assemblages. In this study, the core microbiome was consistent at different growth stages. Some Rhodobacteraceae, Ralstonia, Erythrobacter, Rhodococcus, Acinetobacter, Alteromonas, Vibrio, Halomonas, and Granulosicoccus were the dominant genera on the surface of U. fasciata. It is reported that Ulva australis are mainly composed of Alphaproteobacteria, Bacteroidetes, Planctomycetes, and unclassified Gammaproteobacteria (Burke et al., 2011), while the bacterial community associated with green seaweeds is mainly composed of Proteobacteria, Bacteroidetes, Verrucomicrobia, Cyanobacteria, Planctomycetes, Actinobacteria, and Firmicutes (Selvarajan et al., 2019). The species of core microbial communities on Ulva surface were similar. However, the differences of green algae epiphytic communities in different environments may be explained by compounds secreted by green algae surface that regulate the composition of microbial communities.
In addition, most of the genera level biomarker groups showed high relative abundance at the corresponding growth stages (e.g., Rhodobacteraceae and Flavobacteriaceae). It is reported that Flavobacteriaceae bacteria are beneficial to the growth of Ulva. For example, Spoerner et al. (2012) reported that the addition of MS6 from Maribacter sp. strain restored the morphogenesis of Ulva mutabilis. MS6 is similar to auxin it induces cell wall formation and promotes the growth of basal stem cells. Many other studies also have shown that Rhodobacteraceae promote plant growth (Stevens et al., 2017; Abraham and Silambarasan, 2018; Kuhl et al., 2019; Vergani et al., 2019).
Ambient environmental parameters are another important factor affecting the abundance and diversity of microbial community on the surface of U. fasciata. McDonough and Watmough (2015) reported that epiphytic microbiota has a better positive correlation with environmental factors, such as temperature and pH. The authors also emphasized that the of the specificity, abundance and diversity indices of epiphytes were negatively correlated with the concentration of ambient nutrients (McDonough and Watmough, 2015). Moreover, Stratil et al. (2014) revealed that temperature, salinity and DO are the main environmental factors affecting the structure of microbial community. However, these findings contradicted our results and those of Pei et al. (2021), which showed that temperature, salinity and DO have no significantly effects on algal epiphytic microbiota. In this study, IC and TC had a great influence on the surface microbiota of U. fasciata. It also reported that algae are more associated with nutrients in the surrounding environment (Aires et al., 2016). Therefore, the authors conclude that the species of algal epiphytic bacteria in different environments are the result the synergistic effects of sampling methods, growth indexes of algae and environmental factors (Selvarajan et al., 2019).
The results of this study revealed that the growth promoting effect of strain HU1-3 (belonging to Flavobacteriaceae) was the best on U. fasciata. B. cereus U5-30 has the closest genetic relationship with Bacillus cereus AR156, with 100% 16S rRNA gene sequence similarity. B. cereus AR156 was isolated from soil and is reported to promote plant growth (Jiang et al., 2017). In this study, B. cereus U5-30 can well promote the growth of U. fasciata, so it can be used as a positive control of bacterial treatment experiment. Compared with B. cereus U5-30, the biomass, phycocyanin, soluble sugar, soluble protein and growth rate of U. fasciata were greatly improved after co-culture with Hyunsoonleella, indicating that Hyunsoonleella sp. HU1-3 was a better growth promoter for U. fasciata. To our knowledge, this is the first study of Hyunsoonleella sp. as a growth promoter in plants.
The co-culture of different bacterial supernatants with U. fasciata (WSB) also indicated that Hyunsoonleella sp. HU1-3 increased the growth rate of U. fasciata. The genomic sequencing of Hyunsoonleella sp. HU1-3 revealed that the strain contained auxin related genes (an important phytohormone for the plant growth), involved in the production of IAA and CK, and several other genes encoding different enzymes (phosphate synthases genes and iron carrier enzymes genes). Plant growth depends on specific growth hormones, such as IAA and CK. These endogenous compounds present in plant tissues are considered to be signals to coordinate plant development (Kudo et al., 2010). This is consistent with previous reports that many Flavobacteriaceae produce auxin and play an important role in promoting plant growth (Elena et al., 2007; Kaul et al., 2018; Vukanti, 2020; Kalyanasundaram et al., 2021). Various phosphatases in the strain not only improve the phosphorus absorption capacity of plant, but also coordinate the transformation of insoluble, inorganic and organic forms of phosphorus into bioavailable forms of phosphate in the growing environment. Soluble phosphorus microorganisms can also improve the efficiency of nitrogen fixation, accelerate the accessibility of other trace elements, and promote the synthesis of iron chelating compounds (Kour et al., 2019). It is reported that inoculating different crops such as corn, tomato and mung bean with phosphorus-solubilizing bacteria can promote plant growth (Sandhya et al., 2010). The presence of genes encoding iron carrier and phosphatase further demonstrated that Hyunsoonleella sp. HU1-3 has the potential function of promoting the growth of U. fasciata. Further studies are highly recommended in the near future to determine which substances contribute to the growth of U. fasciata biomass.
Although U. fasciata grows in seawater, the microbial community on the surface of U. fasciata seems to be some specific algae-related communities, which are essentially different from the microbial community in the surrounding seawater (at the phylum level). This may be caused by different living environments, such as the nutrient concentration in seawater is relatively low, while the surface of algae emits organic carbon and nutrients (Pregnall, 1983). Previous studies have shown that although the community is variable, there is always a core population on the surface of the algae (Tujula et al., 2010). The results of this study also confirmed that the same bacteria were detected on the surface of U. fasciata samples at different growth stages, indicating that these bacteria can be colonized on the surface of algae as a core community. Besides, there were significant differences in epiphytic bacterial community in different U. fasciata samples, indicating the functional redundancy of U. fasciata epiphytic bacterial communities. This conclusion is consistent with the redundancy hypothesis (Naeem, 1998), which assumes that more than one species can play a specific role in an ecosystem, so as to endow the ecosystem with a certain degree of anti-disturbance ability. This hypothesis has also been confirmed to be applicable to laboratory microbial communities (Leflaive et al., 2008) and soil microbial communities (Yin et al., 2000; Persiani et al., 2008). Although functional redundancy can explain the variability of the epiphytic bacterial community of U. fasciata, it cannot explain the significant difference between this algae-associated community and the planktonic bacterial community living in the surrounding seawater. This suggests that there may be a selective mechanism that determines the environment in which bacterial communities exist.
The lottery hypothesis asserts that species with similar nutritional abilities will randomly replenish the ecosystem and occupy space (Sale, 1976). This hypothesis was proposed to explain the coexistence of corals and fish occupying the same ecological niche, but it is also consistent with the observations described in this study. It is assumed that various bacteria in algae-associated communities have the necessary metabolic capacity and colonize in the niches on the surface of algae. Due to functional redundancy, any bacterial species in the community will first colonized when they happen to encounter and occupy the surface of algae. This can explain the community variability between different U. fasciata samples. Bacterial species from the surface of the U. fasciata may not be able to adapt to plankton habitat in seawater, which is why the bacterial species found on the surface of U. fasciata are rarely detected in the surrounding seawater. The lottery hypothesis was conceived for dozens of species of fish that living in corals. It will be interesting to study whether this hypothesis also applies to communities containing thousands of bacterial species. It will also be of great interest to determine whether the isolated bacterial community is unique to the U. fasciata or is simply indicative of a surface-associated lifestyle.
In conclusion, a new bacterial species, Hyunsoonleella sp. HU1-3 was isolated from the surface of U. fasciata. This isolate significantly promoted the growth of U. fasciata. Genomic analysis revealed that the isolate process genes encoding the synthesis of growth hormones, phosphate synthases genes vitamins and iron carrier enzymes. The combination of high-throughput sequencing and bacterial treatment approaches helps to reveal the interaction between plants and microorganisms under the condition of reducing unknown variables.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MW527409; https://www.ncbi.nlm.nih.gov/genbank/, JAEPJQ000000000; https://www.ncbi.nlm.nih.gov/genbank/, PRJNA766413.
HW and ZH designed the study, wrote the manuscript, and contributed to the data analysis. AE, YL, WL, QC, YX, and TP performed the experiments and data acquisition. HW, TP, YL, WL, and ZH contributed to the treatment of the raw data. All authors contributed to the article and approved the submitted version.
This study was funded by the Major Project of Talent Team introduction for Guangdong Provincial Laboratory of Southern Marine Science and Engineering (Guangzhou, GML2019ZD0606), Innovation Team Project of Guangdong Normal University (2018KCXTD012), Shantou University Scientific Research Foundation for Talents (NTF19013), and Guangdong Natural Science Foundation General Project (2021A1515010803).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge Tran Ngoc Tuan, Shantou University for the work of polishing the article. We gratefully acknowledge the laboratory staff members for their remarkable support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.788709/full#supplementary-material
Abraham, J., and Silambarasan, S. (2018). Biodegradation of carbendazim by Rhodococcus erythropolis and its plant growth-promoting traits. Biol. Environ. Proc. R. Irish Acad. 118B, 69–80. doi: 10.3318/bioe.2018.07
Aires, T., Serrão, E. A., and Engelen, A. H. (2016). Host and environmental specificity in bacterial communities associateds to two highly invasive marine species (genus Asparagopsis). Front. Microbiol. 7:559. doi: 10.3389/fmicb.2016.00559
Alsufyani, T., Califano, G., Deicke, M., Grueneberg, J., Weiss, A., Engelen, A. H., et al. (2020). Macroalgal–bacterial interactions: identification and role of Thallusin in morphogenesis of the seaweed Ulva (Chlorophyta). J. Exp. Bot. 71, 3340–3349. doi: 10.1093/jxb/eraa066
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Aust. Ecol. 26, 32–46. doi: 10.1046/j.1442-9993.2001.01070.x
Barott, K. L., Rodriguez-Brito, B., Janouškovec, J., Marhaver, K. L., Smith, J. E., Keeling, P., et al. (2011). Microbial diversity associated with four functional groups of benthic reef algae and the reef-building coral Montastraea annularis. Environ. Microbiol. 13, 1192–1204. doi: 10.3389/fmars.2021.627724
Besemer, J., Lamsadze, A., and Borodovsky, M. (2001). GeneMarkS: a self- training method for prediction of gene starts in microbial genomes: implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. doi: 10.1093/nar/29.12.2607
Bondoso, J., Balagué, V., Gasol, J. M., and Lage, O. M. (2014). Community composition of the Planctomycetes associated with different macroalgae. FEMS Microbiol. Ecol. 88, 445–456. doi: 10.1111/1574-6941.12258
Bric, J. M., Bostock, R. M., and Silverstone, S. E. (1991). Rapid in situ assay for indoleacetic acid production by bacteria immobilized on a nitrocellulose membrane. Appl. Environ. Microbiol. 57, 535–538. doi: 10.1128/aem.57.2.535-538.199
Brown, J., Pirrung, M., and McCue, L. A. (2017). FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139. doi: 10.1093/bioinformatics/btx373
Burke, C., Thomas, T., Lewis, M., Steinberg, P., and Kjelleberg, S. (2011). Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME J. 5, 590–600. doi: 10.1038/ismej.2010.164
Chaparro, J. M., Badri, D. V., and Vivanco, J. M. (2014). Rhizosphere microbiome assemblage is affected by plant development. ISME J. 8, 790–803. doi: 10.1038/ismej.2013.196
Chen, J., Li, H., Zhang, Z., He, C., Shi, Q., Jiao, N., et al. (2020). DOC dynamics and bacterial community succession during long-term degradation of Ulva prolifera and their implications for the legacy effect of green tides on refractory DOC pool in seawater. Water Res. 185:116268. doi: 10.1016/j.watres.2020.116268
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
De Souza, R. S. C., Okura, V. K., Armanhi, J. S., Jorrín, B., Lozano, N., da Silva, M. J., et al. (2016). Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Sci. Rep. 6:28774. doi: 10.1038/srep28774
Dobretsov, S. V., and Qian, P. Y. (2002). Effect of bacteria associated with the green alga Ulva reticulata on marine micro-and macrofouling. Biofouling 18, 217–228. doi: 10.1080/08927010290013026
Dombrowski, N., Schlaeppi, K., Agler, M. T., Hacquard, S., Kemen, E., Garrido-Oter, R., et al. (2016). Root microbiota dynamics of perennial Arabis alpina are dependent on soil residence time but independent of flowering time. ISME J. 11, 43–55. doi: 10.1038/ismej.2016.109
Doty, S. L., Sher, A. W., Fleck, N. D., Khorasani, M., Bumgarner, R. E., Khan, Z., et al. (2016). Variable nitrogen fixation in wild Populus. PLoS One 11:e0155979. doi: 10.1371/journal.pone.0155979
Droop, M. R. (2007). Vitamins, Phytoplankton and bacteria: symbiosis or scavenging? J. Plankton Res. 29, 107–113. doi: 10.1093/plankt/fbm009
Edwards, J., Johnson, C., Santos-Medellín, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, E911–E920. doi: 10.1073/pnas.1414592112
El Shafay, S. M., Ali, S. S., and El-Sheekh, M. M. (2016). Antimicrobial activity of some seaweeds species from Red sea, against multidrug resistant bacteria. Egypt. J. Aquat. Res. 42, 65–74. doi: 10.1016/j.ejar.2015.11.006
Elena, T. A., Cherdyntseva, T. A., Botina, S. G., and Netrusov, A. I. (2007). Bacteria associated with orchid roots and microbial production of auxin. Microbiol. Res. 162, 69–76. doi: 10.1016/j.micres.2006.07.014
El-Said, G. F., and El-Sikaily, A. (2013). Chemical composition of some seaweed from Mediterranean Sea coast, Egypt. Environ. Monit. Assess. 185, 6089–6099. doi: 10.1007/s10661-012-3009-y
Galhardo, C. X., and Masini, J. C. (2000). Spectrophotometric determination of phosphate and silicate by sequential injection using molybdenum blue chemistry. Anal. Chim. Acta 417, 191–200. doi: 10.1016/S0003-2670(00)00933-8
Ghaderiardakani, F., Califano, G., Mohr, J. F., Abreu, M. H., Coates, J. C., and Wichard, T. (2019). Analysis of algal growth- and morphogenesis-promoting factors in an integrated multi-trophic aquaculture system for farming Ulva spp. Aquac. Environ. Interact. 11, 375–391. doi: 10.3354/aei00319
Ghaderiardakani, F., Coates, J. C., and Wichard, T. (2017). Bacteria-induced morphogenesis of Ulva intestinalis and Ulva mutabilis (Chlorophyta): a contribution to the lottery theory. FEMS Microbiol. Ecol. 93:fix094. doi: 10.1093/femsec/fix094
Gianmaria, C., Kwantes, M., Abreu, M. H., Costa, R., and Wichard, T. (2020). Cultivating the macroalgal holobiont: effects of integrated multi-trophic aquaculture on the microbiome of Ulva rigida (Chlorophyta). Front. Mar. Sci. 7:52. doi: 10.3389/fmars.2020.00052
Gopal, M., and Gupta, A. (2016). Microbiome selection could spur next-generation plant breeding strategies. Front. Microbiol. 7:1971. doi: 10.3389/fmicb.2016.01971
Hovel, K. A., Warneke, A. M., Virtue-Hilborn, S. P., and Sanchez, A. E. (2016). Mesopredator foraging success in eelgrass (Zostera marina L.): relative effects of epiphytes, shoot density, and prey abundance. J. Exp. Mar. Biol. Ecol. 474, 142–147. doi: 10.1016/j.jembe.2015.10.014
Inaba, N., Kodama, I., Nagai, S., Shiraishi, T., Matsuno, K., Yamaguchi, A., et al. (2020). Distribution of harmful algal growth-limiting bacteria on artificially introduced Ulva and natural Macroalgal beds. Appl. Sci. 10:5658. doi: 10.3390/app10165658Song
Jiang, C. H., Chen, Y., Yan, F., Fan, Z. H., and Guo, J. H. (2017). Whole-genome sequence of Bacillus cereus AR156, a potential biocontrol agent with high soilborne disease biocontrol efficacy and plant growth promotion. Genome Announc. 5:e00886-17. doi: 10.1128/genomeA.00886-17
Juhmani, A.-S., Vezzi, A., Wahsha, M., Buosi, A., Pascale, F. D., Schiavon, R., et al. (2020). Diversity and dynamics of seaweed associated microbial communities inhabiting the lagoon of venice. Microorganisms 8:1657. doi: 10.3390/microorganisms8111657
Kalyanasundaram, T. G., Syed, N., and Subburamu, K. (2021). “Chapter 17 – Recent developments in plant growth-promoting rhizobacteria (PGPR) for sustainable agriculture,” in Recent Developments in Applied Microbiology and Biochemistry, ed. B. Viswanath (Cambridge, MA: Academic Press), 181–192. doi: 10.1016/B978-0-12-821406-0.00017-5
Kamo, T., Hiradate, S., Suzuki, K., Fujita, I., Yamaki, S., Yoneda, T., et al. (2018). Methylobamine, a UVA-absorbing compound from the plant-associated bacteria Methylobacterium sp. Nat. Product Commun. 13:1934578X1801300208. doi: 10.1177/1934578X1801300208
Karthick, P., and Mohanraju, R. (2018). Antimicrobial potential of epiphytic bacteria associated with seaweeds of little Andaman, India. Front. Microbiol. 9:611. doi: 10.3389/fmicb.2018.00611
Kaul, S., Gupta, S., Sharma, T., and Dhar, M. K. (2018). Unfolding the role of rhizomicrobiome toward sustainable agriculture. Root Biol. 52, 341–365. doi: 10.1016/j.gfs.2019.01.007
Kour, D., Rana, K. L., Yadav, N., Yadav, A. N., Kumar, A., Meena, V. S., et al. (2019). “Rhizospheric microbiomes: biodiversity, mechanisms of plant growth promotion, and biotechnological applications for sustainable agriculture,” in Plant Growth Promoting Rhizobacteria for Agricultural Sustainability: From Theory to Practices, eds A. Kumar and V. S. Meena (Singapore: Springer), 19–65. doi: 10.1007/978-981-13-7553-8_2
Kudo, T., Kiba, T., and Sakakibara, H. (2010). Metabolism and long-distance translocation of cytokinins. J. Integr. Plant Biol. 52, 53–60. doi: 10.1111/j.1744-7909.2010.00898.x
Kuhl, T., Felder, M., Nussbaumer, T., Fischer, D., Kublik, S., Chowdhury, S. P., et al. (2019). Genome assembly of a plant-associated Rhodococcus qingshengii strain (RL1) isolated from Eruca sativa mill and showing plant growth-promoting properties. Microbiol. Resour. Announc. 8:e01106-19. doi: 10.1128/MRA.01106-19
Lachnit, T., Meske, D., Wahl, M., Harder, T., and Schmitz, R. (2011). Epibacterial community patterns on marine macroalgae are host-specific but temporally variable. Environ. Microbiol. 13, 655–665. doi: 10.1111/j.1462-2920.2010.02371.x
Leflaive, J., Danger, M., Lacroix, G., Lyautey, E., Oumarou, C., and Ten-Hage, L. (2008). Nutrient effects on the genetic and functional diversity of aquatic bacterial communities. FEMS Microbiol. Ecol. 66, 379–390. doi: 10.1111/j.1574-6941.2008.00593.x
Li, W., Jaroszewski, L., and Godzik, A. (2002). Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18, 77–82.
Liu, L., Trevathan-Tackett, S. M., Lewis, C. J. E., Huang, X., and Peter, I. (2020). Beach-cast seagrass wrack contributes substantially to global greenhouse gas emissions. Macreadie Environ. Sci. Technol. 54, 14750–14760. doi: 10.1016/j.jenvman.2018.10.047
Liu, Y. X., Qin, Y., Chen, T., Lu, M., Qian, X., Guo, X., et al. (2021). A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 12:315. doi: 10.1007/s13238-020-00724-8
McDonough, A. M., and Watmough, S. A. (2015). Impacts of nitrogen deposition on herbaceous ground flora and epiphytic foliose lichen species in southern Ontario hardwood forests. Environ. Pollut. 196, 78–88. doi: 10.1016/j.envpol.2014.09.013
Miranda, L. N., Hutchison, K., Grossman, A. R., and Brawley, S. H. (2013). Diversity and abundance of the bacterial community of the red Macroalga Porphyra umbilicalis: did bacterial farmers produce macroalgae? PLoS One 8:e58269. doi: 10.1371/journal.pone.0058269
Naeem, S. (1998). Species redundancy and ecosystem reliability. Conserv. Biol. 12, 39–45. doi: 10.1111/j.1523-1739.1998.96379.x
Nakanishi, K., Nishijima, M., Nishimura, M., Kuwano, K., and Saga, N. (1996). Bacteria that induce morphogenesis in Ulva pertusa (Chlorophyta) grown under axenic conditions. J. Phycol. 32, 479–482. doi: 10.1111/j.0022-3646.1996.00479.x
Paix, B., Carriot, N., Barry-Martinet, R., Greff, S., Misson, B., Briand, J. F., et al. (2020). A multi-omics analysis suggests links between the differentiated surface metabolome and epiphytic microbiota along the thallus of a Mediterranean seaweed holobiont. Front. Microbiol. 11:494. doi: 10.3389/fmicb.2020.00494
Pei, P., Aslam, M., Du, H., Liang, H., Wang, H., Liu, X., et al. (2021). Environmental factors shape the epiphytic bacterial communities of Gracilariopsis lemaneiformis. Sci. Rep. 11:8671. doi: 10.1038/s41598-021-87977-3
Persiani, A. M., Maggi, O., Montalvo, J., Casado, M. A., and Pineda, F. D. (2008). Mediterranean grassland soil fungi: patterns of biodiversity, functional redundancy and soil carbon storage. Plant Biosyst. 142, 111–119. doi: 10.1080/11263500701872713
Pregnall, A. M. (1983). Release of dissolved organic carbon from the estuarine intertidal macroalga Enteromorpha prolifera. Mar. Biol. 73, 37–42. doi: 10.1007/BF00396283
Provasoli, L., and Pintner, I. J. (1980). Bacteria induced polymorphism in an axenic laboratory strain of Ulva lactuca (Chlorophyceae) 1. J. Phycol. 16, 196–201. doi: 10.1111/j.1529-8817.1980.tb03019.x
Qu, T., Zhao, X., Hao, Y., Zhong, Y., Guan, C., Hou, C., et al. (2020). Ecological effects of Ulva prolifera green tide on bacterial community structure in Qingdao offshore environment. Chemosphere 244:125477. doi: 10.1016/j.chemosphere.2019.125477
Roth-Schulze, A. J., Pintado, J., Zozaya-Valdés, E., Cremades, J., Ruiz, P., Kjelleberg, S., et al. (2018). Functional biogeography and host specificity of bacterial communities associated with the Marine Green Alga Ulva spp. Mol. Ecol. 27, 1952–1965. doi: 10.1111/mec.14529
Sandhya, V., Ali, S. Z., Grover, M., Reddy, G., and Venkateswarlu, B. (2010). Effect of plant growth promoting Pseudomonas spp. on compatible solutes, antioxidant status and plant growth of maize under drought stress. Plant Growth Regul. 62, 21–30. doi: 10.1007/s10725-010-9479-4
Schwenzfeier, A., Wierenga, P. A., and Gruppen, H. (2011). Isolation and characterization of soluble protein from the green microalgae Tetraselmis sp. Bioresour. Technol. 102, 9121–9127. doi: 10.1016/j.biortech.2011.07.046
Selvarajan, R., Sibanda, T., Venkatachalam, S., Ogola, H. J. O., Christopher Obieze, C., and Msagati, T. A. (2019). Distribution, interaction and functional profiles of epiphytic bacterial communities from the rocky intertidal seaweeds, South Africa. Sci. Rep. 9:19835. doi: 10.1038/s41598-019-56269-2
Shade, A., and Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12. doi: 10.1111/j.1462-2920.2011.02585.x
Short, M. F., Staniewski, M. A., Short, C. M., Long, A. M., Chaban, Y. V., and Short, S. M. (2015). Isolation and characterization of a virus infecting the freshwater algae Chrysochromulina parva. Virology 486, 105–115. doi: 10.1016/j.virol.2015.09.005
Spoerner, M., Wichard, T., Bachhuber, T., Stratmann, J., and Oertel, W. (2012). Growth and thallus morphogenesis of Ulva mutabilis (Chlorophyta) depends on a combination of two bacterial species excreting regulatory factors. J. Phycol. 48, 1433–1447. doi: 10.1111/j.1529-8817.2012.01231.x
Stevens, V., Thijs, S., McAmmond, B., Langill, T., Van Hamme, J., Weyens, N., et al. (2017). Draft genome sequence of Rhodococcus erythropolis VSD3, a diesel fuel-degrading and plant growth-promoting bacterium isolated from Hedera helix leaves. Genome Announc. 5:e01680-16. doi: 10.1128/genomeA.01680-16
Stratil, S. B., Neulinger, S. C., Knecht, H., Friedrichs, A. K., and Wahl, M. (2014). Salinity affects compositional traits of epibacterial communities on the brown macroalga Fucus vesiculosus. FEMS Microbiol. Ecol. 88, 272–279. doi: 10.1111/1574-6941.12292
Tujula, N., Crocetti, G., Burke, C., Thomas, T., Holmström, C., and Kjelleberg, S. (2010). Variability and abundance of the epiphytic bacterial community associated with a green marine Ulvacean alga. ISME J. 4, 301–311. doi: 10.1038/ismej.2009.107
Urgun-Demirtas, M., Sattayatewa, C., and Pagilla, K. R. (2008). Bioavailability of dissolved organic nitrogen in treated effluents. Water Environ. Res. 80, 397–406. doi: 10.2175/106143007X221454
Vergani, L., Mapelli, F., Suman, J., Cajthaml, T., Uhlik, O., and Borin, S. (2019). Novel PCB-degrading Rhodococcus strains able to promote plant growth for assisted rhizoremediation of historically polluted soils. PLoS One 14:e0221253. doi: 10.1371/journal.pone.0221253
Vorholt, J. A. (2012). Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828–840. doi: 10.1038/nrmicro2910
Vukanti, R. V. N. R. (2020). “Structure and function of rhizobiome,” in Plant Microbe Symbiosis, eds A. Varma, S. Tripathi, and R. Prasad (Cham: Springer). doi: 10.1007/978-3-030-36248-5_13
Wahl, M., Goecke, F., Labes, A., Dobretsov, S., and Weinberger, F. (2012). The second skin: ecological role of epibiotic biofilms on marine organisms. Front. Microbiol. 3:292. doi: 10.3389/fmicb.2012.00292
Weiss, A., Costa, R., and Wichard, T. (2017). Morphogenesis of Ulva mutabilis (Chlorophyta) induced by Maribacter species (Bacteroidetes, Flavobacteriaceae). Bot. Mar. 60, 197–206. doi: 10.1515/bot-2016-0083
Wiese, J., Thiel, V., Nagel, K., Staufenberger, T., and Imhoff, J. F. (2009). Diversity of antibiotic-active bacteria associated with the brown alga Laminaria saccharina from the Baltic Sea. Mar. Biotechnol. 11, 287–300. doi: 10.1007/s10126-008-9143-4
Xu, J., Zhang, Y., Zhang, P., Trivedi, P., Riera, N., Wang, Y., et al. (2018). The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 9:4894. doi: 10.1038/s41467-018-07343-2
Yin, B., Crowley, D., Sparovek, G., De Melo, W. J., and Borneman, J. (2000). Bacterial functional redundancy along a soil reclamation gradient. Appl. Environ. Microbiol. 66, 4361–4365. doi: 10.1128/AEM.66.10.4361-4365.2000
Yong, Y. S., Yong, W. T., and Anton, A. (2013). Analysis of formulae for determination of seaweed growth rate. J. Appl. Phycol. 25, 1831–1834. doi: 10.1007/s10811-013-0022-7
Zarraonaindia, I., Owens, S. M., Weisenhorn, P. B., West, K., Hampton-Marcell, J. T., Lax, S., et al. (2015). The soil microbiome influences grapevine-associated microbiota. mBio 6:e02527-14. doi: 10.1128/mBio.02527-14
Zhang, J., Zhang, N., Liu, Y. X., Zhang, X., Hu, B., Qin, Y., et al. (2018). Root microbiota shift in rice correlates with resident time in the field and developmental stage. Sci. China Life Sci. 61, 613–621. doi: 10.1007/s11427-018-9284-4
Zhang, Y., He, P., Li, H., Li, G., Liu, J., Jiao, F., et al. (2019). Ulva prolifera green-tide outbreaks and their environmental impact in the Yellow Sea. China Natl. Sci. Rev. 6, 825–838. doi: 10.1093/nsr/nwz026
Keywords: Ulva fasciata, growth stage, Hyunsoonleella sp. HU1-3, bacterial treatment, promoting-plant-growth-bacteria
Citation: Wang H, Elyamine AM, Liu Y, Liu W, Chen Q, Xu Y, Peng T and Hu Z (2022) Hyunsoonleella sp. HU1-3 Increased the Biomass of Ulva fasciata. Front. Microbiol. 12:788709. doi: 10.3389/fmicb.2021.788709
Received: 03 October 2021; Accepted: 09 December 2021;
Published: 31 January 2022.
Edited by:
Alok Kumar Srivastava, National Bureau of Agriculturally Important Microorganisms (ICAR), IndiaReviewed by:
Tal Luzzatto-Knaan, University of Haifa, IsraelCopyright © 2022 Wang, Elyamine, Liu, Liu, Chen, Xu, Peng and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhong Hu, aHpoQHN0dS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.