
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 29 November 2021
Sec. Infectious Agents and Disease
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.784856
This article is part of the Research Topic The Deadly Secrets of C. Difficile - Insights into Host-Pathogen Interaction, Volume II View all 12 articles
 Panagiotis Papatheodorou1*
Panagiotis Papatheodorou1* Selina Kindig1
Selina Kindig1 Adriana Badilla-Lobo2
Adriana Badilla-Lobo2 Stephan Fischer1
Stephan Fischer1 Ebru Durgun1†Tharani Thuraisingam1†Alexander Witte3†
Ebru Durgun1†Tharani Thuraisingam1†Alexander Witte3† Shuo Song3†
Shuo Song3† Klaus Aktories3
Klaus Aktories3 Esteban Chaves-Olarte2
Esteban Chaves-Olarte2 César Rodríguez2
César Rodríguez2 Holger Barth1
Holger Barth1The intestinal pathogen Clostridioides (C.) difficile is a major cause of diarrhea both in hospitals and outpatient in industrialized countries. This bacterium produces two large exotoxins, toxin A (TcdA) and toxin B (TcdB), which are directly responsible for the onset of clinical symptoms of C. difficile-associated diseases (CDADs), such as antibiotics-associated diarrhea and the severe, life-threatening pseudomembranous colitis. Both toxins are multidomain proteins and taken up into host eukaryotic cells via receptor-mediated endocytosis. Within the cell, TcdA and TcdB inactivate Rho and/or Ras protein family members by glucosylation, which eventually results in cell death. The cytotoxic mode of action of the toxins is the main reason for the disease. Thus, compounds capable of inhibiting the cellular uptake and/or mode-of-action of both toxins are of high therapeutic interest. Recently, we found that the sterol regulatory element-binding protein 2 (SREBP-2) pathway, which regulates cholesterol content in membranes, is crucial for the intoxication of cells by TcdA and TcdB. Furthermore, it has been shown that membrane cholesterol is required for TcdA- as well as TcdB-mediated pore formation in endosomal membranes, which is a key step during the translocation of the glucosyltransferase domain of both toxins from endocytic vesicles into the cytosol of host cells. In the current study, we demonstrate that intoxication by TcdA and TcdB is diminished in cultured cells preincubated with the compound U18666A, an established inhibitor of cholesterol biosynthesis and/or intracellular transport. U18666A-pretreated cells were also less sensitive against TcdA and TcdB variants from the epidemic NAP1/027 C. difficile strain. Our study corroborates the crucial role of membrane cholesterol for cell entry of TcdA and TcdB, thus providing a valuable basis for the development of novel antitoxin strategies in the context of CDADs.
Clostridioides difficile is a (nosocomial) pathogen of the human gut and the major cause of antibiotics-associated diarrhea and pseudomembranous colitis. This bacterium produces two exotoxins, toxin A (TcdA) and toxin B (TcdB), that belong to the family of the clostridial glucosylating toxins (CGTs), also referred to as large clostridial cytotoxins (LCCs). The onset of clinical symptoms after C. difficile infections (CDIs) is strictly dependent on the toxins’ actions on target cells in the human gut (Kelly and LaMont, 2008; Aktories et al., 2017; Chandrasekaran and Lacy, 2017).
TcdA and TcdB are glucosylating toxins that specifically modify host cell target proteins by covalent attachment of a glucose moiety (Just et al., 1995). Both toxins utilize UDP-glucose for the mono-O-glucosylation of GTPases from the Rho and/or Ras family, which renders them inactive. Rho family members are master regulators of the actin cytoskeleton and numerous other cellular processes (Hall, 1994). Consequently, the intoxication of cultured mammalian cells with TcdA or TcdB leads amongst other effects to cell rounding due to the destruction of the actin cytoskeleton (Aktories and Just, 2005).
In order to reach their target proteins in the host cell cytosol, TcdA and TcdB need to enter cells via receptor-mediated, clathrin-dependent endocytosis (Papatheodorou et al., 2010). Both toxins are single-chain, multidomain toxins, which bind to host cell surface receptors via at least two independent, C-terminally located receptor-binding domains (Gerhard, 2017; Papatheodorou et al., 2018). After internalization of the toxin/receptor complexes, the pH of the endosomal lumen decreases, triggering membrane insertion and pore formation by a central region within TcdA and TcdB, denoted as translocation domain (TD) (Barth et al., 2001; Orrell et al., 2017). The glucosyltransferase domain (GTD) and the adjacent cysteine protease domain (CPD) are located at the N-terminus of both toxins (Jank and Aktories, 2008). The TD enables the translocation of the GTD and CPD across the endosomal membrane. Eventually, binding of cytosolic inositol hexakisphosphate to CPD triggers autocatalytic cleavage and release of the GTD into the cytosol (Giesemann et al., 2008; Egerer et al., 2009).
Earlier findings indicated that membrane cholesterol is crucial for the generation of translocation pores formed by TcdA and TcdB in endosomal membranes (Giesemann et al., 2006). More recently, we were able to show that an active sterol regulatory element–binding protein 2 (SREBP-2) pathway, which regulates cholesterol content in membranes, is required for efficient uptake of TcdA and TcdB into target cells (Papatheodorou et al., 2019). In addition, we demonstrated that the cholesterol-lowering drug simvastatin significantly reduces the intoxication of mouse embryonic fibroblasts (MEF cells) with TcdB in vitro (Papatheodorou et al., 2019).
Prompted by these observations and based on the recognized inhibitory role of the amphipathic steroid U18666A (3-β-[2-(diethylamino)ethoxy]androst-5-en-17-one) in cholesterol biosynthesis and/or intracellular transport (Cenedella, 2009), we investigated whether preincubation with the compound U18666A turns green monkey kidney epithelial cells (Vero), human cervix- (HeLa) and colon- (CaCo-2) cancer cells less prone to intoxication by TcdB. Importantly, U18666A exhibited a protective effect against TcdB in the tested cell lines. In addition, U18666A also protected cells from TcdA as well as from TcdA and TcdB from the epidemic NAP1/027 C. difficile strain, which is of particular clinical relevance.
Overall, the results corroborate the crucial role of membrane cholesterol for cell entry of TcdA and TcdB and should pave the way for the development of novel pharmacological antitoxin strategies for the supportive therapy after CDI.
Minimum Essential Medium (MEM; Fisher Scientific GmbH, Schwerte, Germany; #11524426) supplemented with 10% fetal calf serum (FCS), 1% sodium pyruvate, 1% non-essential amino acids, and 1% penicillin/streptomycin was used for the cultivation of HeLa and Vero cells. CaCo-2 cells (CLS Cell Line Service GmbH, Eppelheim, Germany; #300137) were cultivated in Dulbecco’s Modified Eagle’s Medium (DMEM; Fisher Scientific GmbH, Schwerte, Germany; #11594486) supplemented with 10% FCS, 1% sodium pyruvate, 1% non-essential amino acids, and 1% penicillin/streptomycin. Alternatively, HeLa cells were grown in DMEM (Fisher Scientific GmbH, Schwerte, Germany; #13345364) supplemented with 10% FCS and 1% penicillin/streptomycin. Cells were maintained in the incubator under humidified conditions at 37°C and 5% CO2.
TcdA from C. difficile VPI 10463 as well as TcdA and TcdB from a NAP1/027 strain were purified from 72 h culture supernatants using dialysis culture in Brain Heart Infusion broth as previously described (Lyerly et al., 1988; Pruitt et al., 2010). Toxin cross-contaminations were excluded by MALDI-TOF analysis. TcdB from C. difficile VPI 10463 was purified as described before (Just et al., 2008).
U18666A was ordered from Biomol (Hamburg, Germany; Cay10009085) or from Merck/Sigma-Aldrich (Darmstadt, Germany; U3633), dissolved in dimethyl sulfoxide (DMSO) to obtain a 20 mM stock solution and aliquoted prior to storage at −20°C.
If not otherwise stated, incubation of cells with C. difficile toxins and/or the U18666A compound occurred at 37°C.
The following microscopes were used for monitoring toxin-induced cell rounding: Axiovert 40 CFL microscope (Carl Zeiss Microscopy, Jena, Germany) equipped with a ProgRes C10 plus camera (Jenoptik, Jena, Germany); Leica DMi1 equipped with a Leica MC170 HD camera (Leica, Wetzlar, Germany); Primovert (Carl Zeiss Microscopy, Jena, Germany); Lionheart FX Automated Microscope (BioTek, Vermont, United States). Toxin-induced cell rounding was manually quantified in microscopic images and, optionally, facilitated by the Neuralab online tool (https://neuralab.de).
Whole-cell lysates from cells growing in the wells from a 24-well plate were generated directly in wells by removing the growth medium and resuspending cell monolayers in 2.5-fold pre-heated Laemmli buffer (typically 30 μl per well). Lysate samples were heated at 95°C for 5 min, prior to SDS-PAGE and Western blotting of lysate proteins onto a nitrocellulose membrane. For the immunodetection of non-glucosylated Rac1 and GAPDH, primary mouse anti-Rac1 (clone 102; BD Biosciences, Heidelberg, Germany; #610651) and mouse anti-GAPDH (G-9; Santa Cruz Biotechnology, Dallas, United States; sc-365062) antibodies were used, respectively. Horseradish peroxidase (HRP)-coupled mouse IgG kappa binding protein (m-IgGκ BP-HRP; Santa Cruz Biotechnology, Dallas, United States; sc-516102) was used for developing antibody signals by the enhanced chemiluminescence (ECL) reaction.
The EVOMX apparatus equipped with the STX2 electrode (World Precision Instruments, Sarasota, United States) was used for measuring the transepithelial electrical resistance (TEER) of CaCo-2 cells. At day 0, 1.2 × 105 cells were seeded into 24-well hanging cell culture inserts (Brand GmbH, Wertheim, Germany; polyester membrane with pore size 0.4 μm) and incubated at 37°C until day 4. Optionally, at day 3, the U18666A compound was added basolaterally to the cells at a final concentration of 10 μM. Prior to TEER measurement at day 4, cell culture inserts were transferred into new wells containing only pre-warmed medium and further incubated at 37°C until TEER values (∼2,000 Ω) were stabilized. Then, TcdB (100 pM) was added apically to the cells and TEER measured every 30 min for up to 6 h. TEER values were normalized to time point 0 (t0, addition of the toxin), which was set to 100%.
Microsoft Excel (Student’s) t-test was used for calculating the significance of differences between mean values. Resulting p-values were indicated by asterisks as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
We initiated our study by using TcdB (derived from the historical C. difficile VPI 10463 strain) as representative for both C. difficile large glucosylating toxins and the human HeLa cervical carcinoma cell line as established model system for in vitro intoxication experiments. At first, we confirmed the biological activity of the prepared TcdB and identified an operable toxin concentration by analyzing cell rounding, which is a typical, specific, and well-established hallmark of intoxication of cells with TcdB or TcdA. Supplementary Figure 1A shows that TcdB (10 pM) directly added to HeLa cells lead to obvious cell rounding after 90 min of incubation and after 210 min, almost all cells were round. In contrast, without toxin treatment, the cells maintained their flat morphology (Supplementary Figure 1A). Thus, 10 pM of TcdB were used for the further experiments with HeLa cells.
U18666A is a pharmacological inhibitor of cholesterol biosynthesis and/or its intracellular transport (Cenedella, 2009). Therefore, a long-term incubation of cells with this compound is required to achieve a recognizable decrease in membrane cholesterol levels without causing cell toxicity. In this context, HeLa cells were incubated for 24 h with increasing concentrations of U18666A and their morphology was monitored microscopically for the same time period to identify the maximal tolerable concentration of U18666A. While HeLa cells tolerated U18666A concentrations up to 20 μM after overnight treatment (Supplementary Figure 1B), 50 μM U18666A was moderately toxic, as indicated by rounding up of a small proportion of cells, and 100 μM U18666A lead to complete disruption of the cell layer and detachment of the cells. Therefore, overnight treatment with up to 20 μM U18666A appears to be tolerable for HeLa cells. Nevertheless, we decided to limit the U18666A concentration at 10 μM in subsequent experiments.
Next, we incubated HeLa cells with increasing concentrations of U18666A (0, 0.1, 1, and 10 μM) for 24 h, followed by the direct addition of 10 pM TcdB into the medium. Microscopic analysis of the cell morphology was performed at different time points after toxin application for up to 300 min. Figure 1A shows representative images at time points 0 and 180 min after TcdB addition. In direct comparison, preincubation of cells with 10 μM U18666A, but not with 0.1 μM or 1 μM U18666A, decreased the number of rounded cells in TcdB-treated cells (Figure 1A). Quantification of TcdB-induced cell rounding over time confirmed that HeLa cells pretreated with 10 μM U18666A were less affected by the toxin (∼25–30% reduced cell rounding after 180 min and 240 min), when compared to mock-pretreated cells and cells pretreated with 1 μM U18666A (Figure 1B).
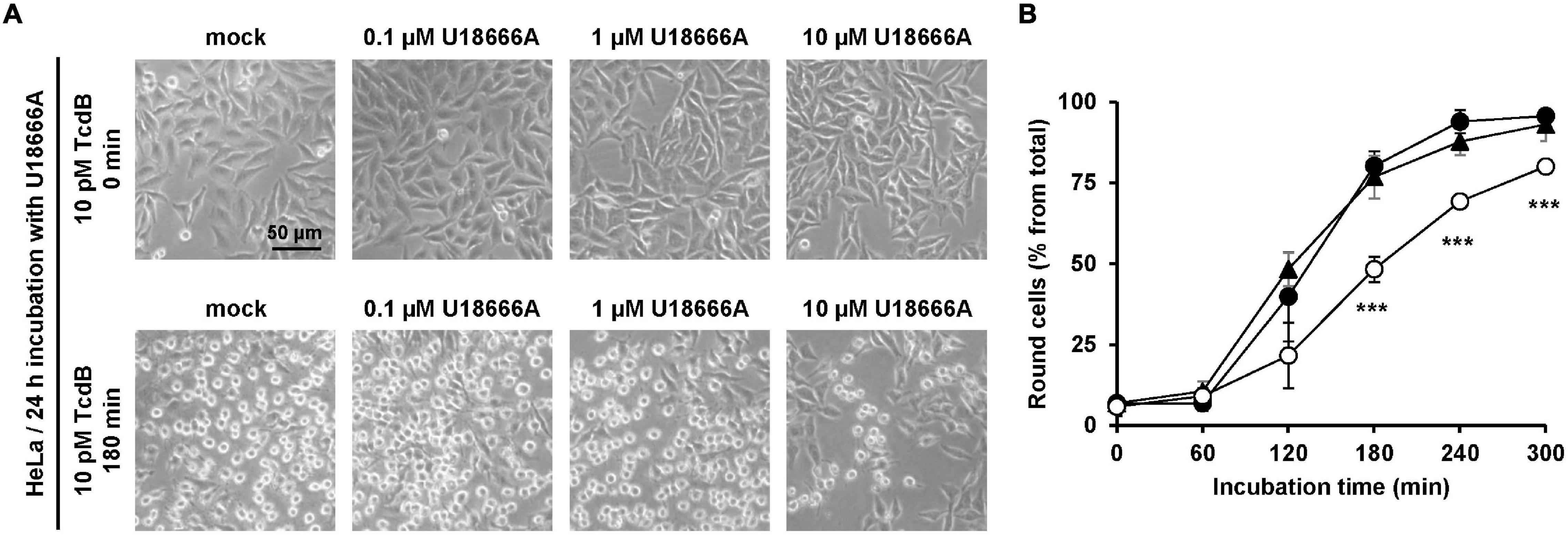
Figure 1. Effect of U18666A on intoxication of HeLa cells by TcdB. (A) HeLa cells were pretreated for 24 h with increasing concentrations of U18666A as indicated or were left untreated (mock), prior to addition of 10 pM TcdB. Microscopic images shown were taken after time point 0 min (upper row) and 180 min (lower row). (B) Quantification of the TcdB-induced cell rounding over time in mock-treated (black filled circles) and 24 h U18666A-pretreated HeLa cells (1 μM, black filled triangles; 10 μM, white filled circles). Graph shows mean (percentage values of round cells) from three experiments performed in parallel in independent wells. Error bars represent ± SD. Asterisks indicate statistical significance compared to control (mock-treated HeLa cells) with ***p < 0.001.
Since U18666A interferes with the cholesterol biosynthesis and/or intracellular transport, the incubation time with the compound correlates with the extent of decrease in membrane cholesterol levels. Thus, we aimed to analyze the consequences of various incubation periods during pretreatment of HeLa cells with the U18666A compound on the subsequent intoxication of the cells by TcdB. To this end, we pretreated HeLa cells with 10 μM U18666A for 0, 2, 4, and 24 h, and exchanged the U18666A-containing medium with fresh medium including 10 pM TcdB. We then analyzed microscopically and quantified TcdB-induced cell rounding over time up to 300 min. In comparison to the cells without U18666A pretreatment, TcdB-induced cell rounding was inhibited only moderately (∼10% reduced cell rounding at time point 180 min) in cells pretreated for 2 and 4 h, respectively, and most prominent inhibition (∼25% reduced cell rounding at time point 180 min) was observed with a 24 h U18666A pretreatment (Figure 2A). Thus, comparatively short incubation periods of cells with the U18666A compound correlate with less efficient protection against TcdB intoxication. Since TcdB intoxication took place after removal of the U18666A compound by medium exchange, direct inhibitory effects of U18666A on the activity of the toxin in the cell culture supernatant can be excluded.
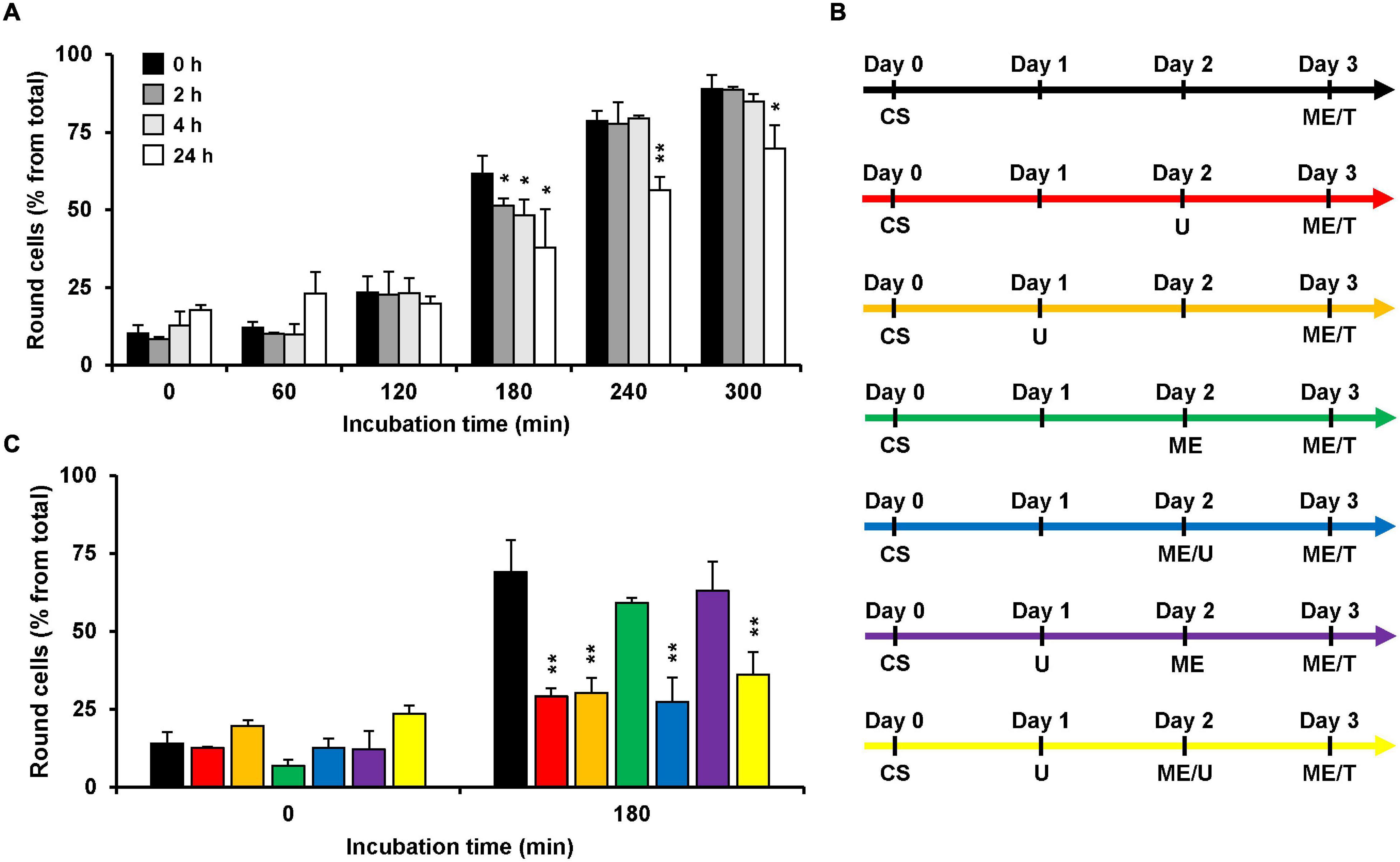
Figure 2. Effect of U18666A preincubation conditions on the TcdB intoxicaton of HeLa cells. (A) HeLa cells were preincubated for 0 h (black bars), 2 h (dark gray bars), 4 h (light gray bars), and 24 h (white bars) with U18666A, followed by TcdB intoxication (10 pM) and microscopic analysis of TcdB-induced cell rounding over time. The bar chart shows mean percentages of round cells at the indicated incubation times after addition of the toxin. Error bars represent ± SD. Asterisks indicate statistical significance at each time point compared to control (black bar) with *p < 0.05 and **p < 0.01. (B,C) The various U18666A pretreatment- and TcdB-intoxication conditions shown in (C) are schematically depicted with different colors in (B). Cell seeding (CS) of HeLa cells at day 0 was followed by the addition of 10 μM U18666A (U) at day 1 (orange, purple and yellow) and/or at day 2 without (red) or with medium exchange (ME; blue, yellow) and TcdB intoxication (10 pM) by medium exchange (ME/T) at day 3. The bar chart in (C) shows the percentages of round cells at the indicated incubation times after addition of the toxin. Error bars represent ± SD. Asterisks indicate statistical significance at each time point compared to control (black bar for red and orange bar; green bar for blue, purple and yellow bar) with **p < 0.01. In all cases, data was obtained in three parallel experiments performed in independent wells.
Next, we wanted to identify the best condition for the pretreatment of HeLa cells with U18666A, which are summarized schematically in Figure 2B, regarding the inhibition of TcdB intoxication in HeLa cells. For all conditions, cells were seeded into wells at day 0 and intoxicated at day 3 for 180 min with TcdB by exchanging the medium with fresh medium including 10 pM TcdB. In the control condition (Figure 2B, black arrow), cells were not pretreated with the U18666A inhibitor and cell rounding occurred in ∼70% of the cells (Figure 2C, black bar). Direct addition of 10 μM U18666A at day 2 (Figure 2B, red arrow; 24 h pretreatment) or day 1 (Figure 2B, orange arrow; 48 h pretreatment), reduced cell rounding equally to ∼30% (Figure 2C, red vs. orange bar). Thus, 48 h pretreatment of HeLa cells with U18666A was not superior to the 24 h pretreatment, in terms of TcdB inhibition. We also tested the sequential addition of 10 μM U18666A to cells, first by direct addition at day 1 and then by medium exchange at day 2 (Figure 2B, yellow arrow). Although cells were pretreated under this condition for a total of 48 h with the U18666A compound (but with U18666A renewal at day 2), TcdB inhibition was not superior to the 48 h pretreatment with U18666A without repeated application of the compound (Figure 2C, orange vs. yellow bar). Addition of 10 μM U18666A to cells at day 2 by medium exchange (Figure 2B, blue arrow) or by direct transfer into the wells (Figure 2B, red arrow), inhibited TcdB-induced cell rounding to equal extents (Figure 2C, red vs. blue bar). Interestingly, when U18666A (10 μM) was added to the cells at day 1 and removed again by medium exchange at day 2 (Figure 2B, purple arrow), TcdB-induced cell rounding was not inhibited and indistinguishable from the corresponding control without U18666A pretreatment at day 1 and medium exchange at day 2 (Figure 2C, green vs. purple bar). This finding suggests that membrane cholesterol levels recovered when U18666A was removed 24 h before toxin addition, either by cellular uptake from the medium or by biosynthesis.
Next, the protective effect of U18666A against TcdB intoxication in a second cell line was investigated. African green monkey kidney (Vero) cells were used. Due to their flat morphology, Vero cells are a well-established model cell line for studying TcdB effects. Vero cells were pretreated for 24 h with increasing U18666A concentrations (0, 1, 2, 5, 10 μM) and, subsequently, TcdB was added to the cells at a final concentration of 10 and 100 pM, respectively. TcdB-induced cell rounding was analyzed microscopically for up to 300 min (10 pM TcdB) or 240 min (100 pM TcdB). Figures 3A,C show representative images of mock- and U18666A-pretreated Vero cells at time points zero and 300 min (10 pM TcdB) and at time points zero and 240 min (100 pM), respectively, after toxin addition. Diagrams in Figures 3B,D show the corresponding quantification of TcdB-induced cell rounding over time for 10 pM or 100 pM TcdB, respectively. After 300 min, cell rounding induced by 10 pM TcdB was strongly reduced to ∼10% in Vero cells pretreated with 5 and 10 μM U18666A vs. mock-pretreated cells (∼60% cell rounding) (Figure 3B). In cells intoxicated with 100 pM TcdB, nearly 100% cell rounding was observed after 210 min, which was decreased to ∼50% by pretreatment with 5 μM U18666A and to ∼25% by pretreatment with 10 μM U18666A. Thus, U18666A confers robust resistance in Vero cells toward 10 pM TcdB and moderate resistance toward 100 pM TcdB. Notably, the protective effect of U18666A against TcdB was more pronounced in Vero than in HeLa cells.
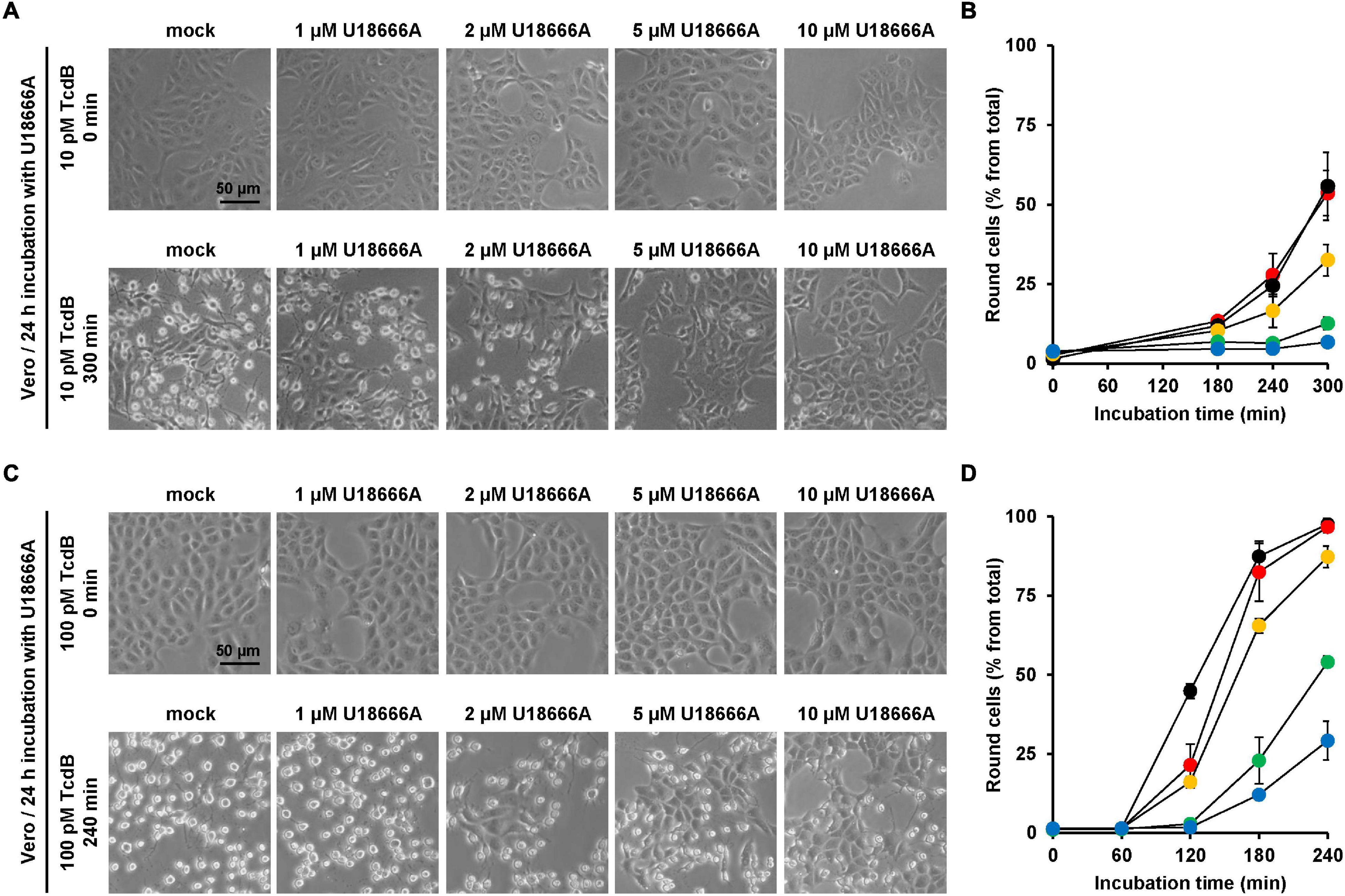
Figure 3. Effect of U18666A on the TcdB-induced cell rounding of Vero cells. (A,C) Vero cells pretreated for 24 h with increasing concentrations of U18666A as indicated or without pretreatment (mock) were intoxicated with (A) 10 pM or (C) 100 pM TcdB. Representative images were taken microscopically after time point 0 min [upper row in (A,C)] and 300 min [lower row in (A)] or 240 min [lower row in (C)]. (B,D) Quantification of the TcdB-induced cell rounding over time in mock-treated (black filled circles) and 24 h U18666A-pretreated HeLa cells (1 μM, red filled circles; 2 μM, orange filled circles; 5 μM, green filled circles, 10 μM, blue filled circles). Graphs in (B,D) show mean percentage values of round cells as indicated by three parallel experiments performed in independent wells. Error bars represent ± SD.
The influence of the pretreatment time of U18666A on the intoxication of Vero cells with TcdB was analyzed. To this end, Vero cells were pretreated with 10 μM U18666A for 0, 2, 4, and 24 h, prior to intoxication with 10 pM TcdB. Eventually, TcdB-induced cell rounding was analyzed microscopically and quantified after 0, 300, and 360 min. Importantly, 24 h pretreatment with the U18666A compound strongly inhibited TcdB-induced cell rounding (from ∼60% in mock-pretreated cells to ∼10% after 360 min of TcdB intoxication) (Figure 4A, black bar vs. white bar). Pretreatment with 10 μM U18666A for 2 and 4 h did not reduce TcdB-induced cell rounding significantly after 300 and 360 min of intoxication, when compared to mock-pretreated cells (Figure 4A, black bar vs. dark gray bar and black bar vs. light gray bar). Thus, also in Vero cells the incubation time has a major impact on the U18666A-mediated inhibition of TcdB. Similarly as observed with HeLa cells, a short incubation of Vero cells with the U18666A compound inefficiently protects these cells against TcdB intoxication.
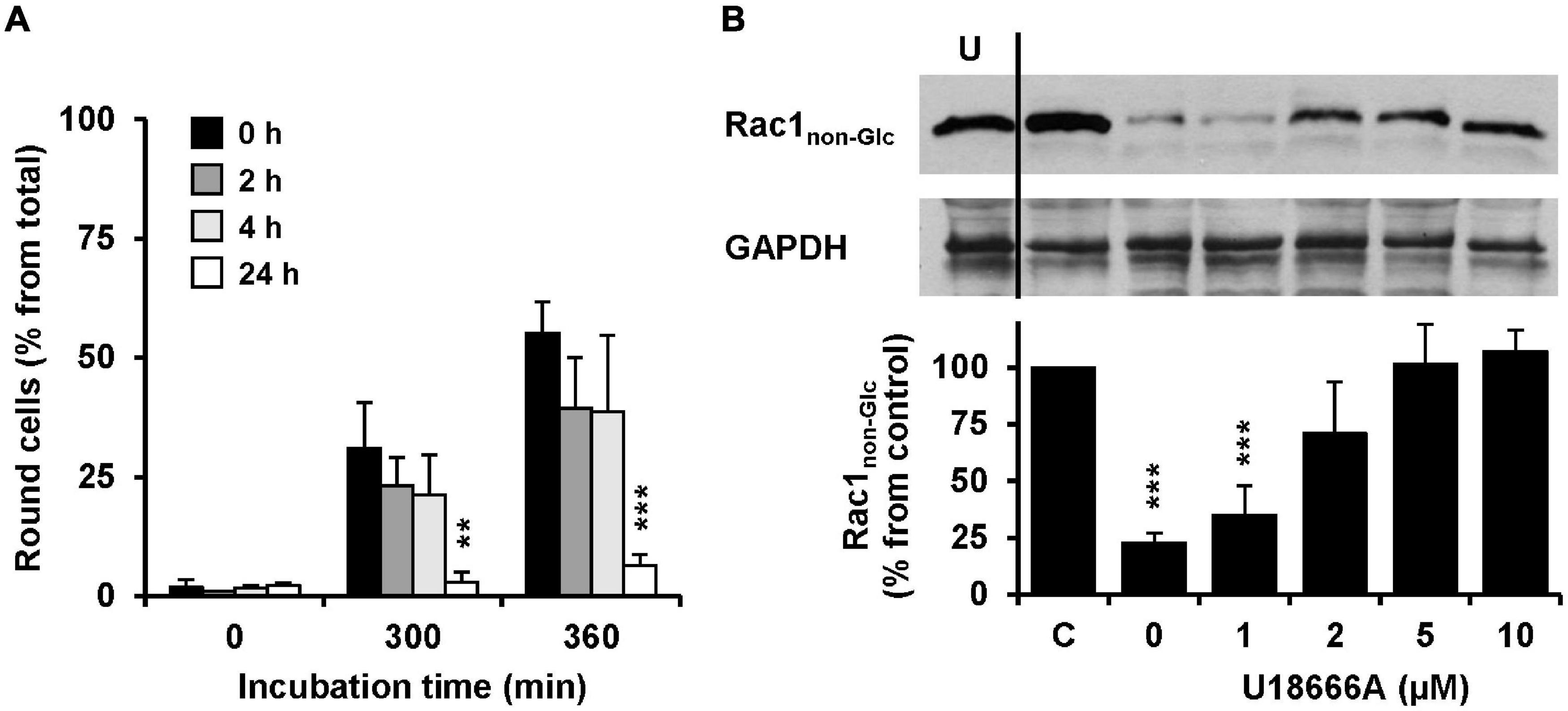
Figure 4. Effect of U18666A on the TcdB-induced Rac1 glucosylation and role of U18666A pretreatment duration on TcdB-induced cell rounding in Vero cells. (A) Vero cells were preincubated for 0 h (black bars), 2 h (dark gray bars), 4 h (light gray bars), and 24 h (white bars) with the U18666A compound, prior to TcdB intoxication (10 pM) and microscopic analysis of TcdB-induced cell rounding over time. The mean percentage values of round cells at the indicated incubation times after addition of the toxin and from three parallel experiments performed in independent wells are plotted in the bar chart. Error bars represent ± SD. Asterisks indicate statistical significance at each time point compared to control (black bar) with **p < 0.01 and ***p < 0.001. (B) Immunoblots against non-glucosylated Rac1 (Rac1non–Glc) and GAPDH (loading control) are shown (upper panel), obtained with whole-cell lysates that were generated from Vero cells pretreated for 24 h with increasing concentrations of U18666A as indicated and after intoxication for 120 min with 100 pM TcdB. In a control sample (C) cells were left without U18666A pretreatment and toxin addition. In another control (U) cells were preincubated with 10 μM U18666A but were left without toxin treatment. The bar chart (lower panel) shows the quantification of the Rac1 signals (normalized with the signals of the GAPDH loading control) relative to the control sample (C), which was set to 100%. Error bars represent ± SEM, calculated from six experiments performed in independent wells (three experiments in parallel on two different days). Asterisks indicate statistical significance at each time point compared to control (C) with ***p < 0.001.
To confirm our findings with another experimental approach, we generated whole-cell lysates from Vero cells intoxicated for 120 min with 100 pM TcdB, followed by immunoblotting against the TcdB target protein Rac1 with an antibody that detects only the non-glucosylated form of Rac1. Prior to TcdB intoxication the cells were pretreated for 24 h with increasing concentrations of U18666A (0, 1, 2, 5, and 10 μM). Whole-cell lysates obtained from untreated cells were used as control for maximal non-glucosylated Rac1 signal in the immunoblot. As expected, the Rac1 signal in whole-cell lysates obtained from TcdB-intoxicated cells without U18666A pretreatment was decreased to ∼10–20% from control (100%) (Figure 4B). However, the Rac1 signal was present almost to the level of the control in TcdB-intoxicated cells pretreated either with 5 or 10 μM U18666A. Lower U18666A concentrations (1 or 2 μM) protected cells less effectively toward TcdB intoxication, because the Rac1 signal was found to be reduced to ∼30% (1 μM U18666A) and ∼70% (2 μM U18666A), respectively, in these samples. Notably, pretreatment of cells with U18666A had no influence on the Rac1 expression level or on the binding by the glucosylation-sensitive anti-Rac1 antibody.
Since C. difficile is a pathogen of the human intestine, it was relevant to test whether U18666A confers resistance toward TcdB intoxication in the medically and physiologically more relevant human colon carcinoma cell line CaCo-2. To this end, CaCo-2 cells were preincubated for 24 h with increasing concentrations of U18666A (0, 1, 2, 5, and 20 μM), prior to addition of 100 pM TcdB and subsequent microscopic analysis of the cell morphology over time. Pretreatment of CaCo-2 cells with increasing U18666A concentrations correlated with less microscopically observable cell rounding after incubation with TcdB for 300 min, when compared to mock-pretreated cells (Figure 5A). The quantification of TcdB-induced cell rounding over time of mock- and 10 μM U18666A-pretreated cells confirmed the protective effect of U18666A against TcdB also in CaCo-2 cells (Figure 5B). Pretreatment of CaCo-2 cells with 10 μM U18666A decreased TcdB-induced cell rounding to ∼25% after 300 min of intoxication.
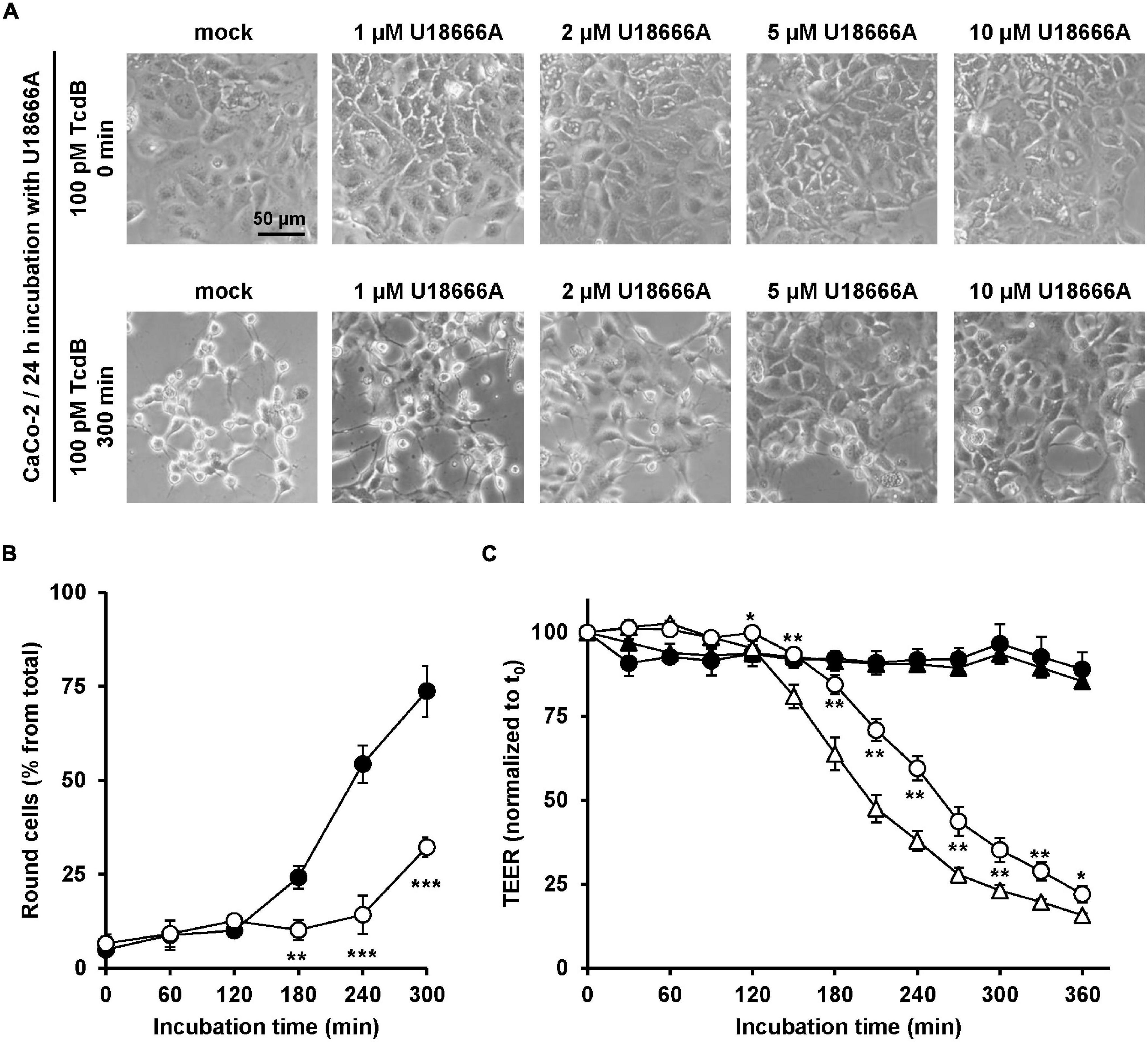
Figure 5. Effect of U18666A on the intoxication of CaCo-2 cells by TcdB. (A) After pretreatment of CaCo-2 cells for 24 h with increasing concentrations of U18666A as indicated or without pretreatment (mock), cells were intoxicated with 100 pM TcdB for 300 min and cell morphology analyzed microscopically. Representative images after time point 0 min (upper row) and 300 min (lower row) are shown. (B) Quantification of the TcdB-induced cell rounding over time in mock-treated (black filled circles) and 24 h U18666A (10 μM)-pretreated CaCo-2 cells (white filled circles). Graph shows mean values (percentage of round cells) from three parallel experiments performed in independent wells. Error bars represent ± SD. Asterisks indicate statistical significance compared to control (mock-treated CaCo-2 cells) with **p < 0.01 and ***p < 0.001. (C) CaCo-2 cells grown in inserts were pretreated for 24 h with 10 μM U18666A (circles) or were left without pretreatment (triangles), prior to apical addition of 100 pM TcdB (white filled circles and triangles) at time point 0 (t0) and measurement of the transepithelial electrical resistance (TEER) at indicated incubation times. Parallel samples were not treated with TcdB (black filled circles and triangles). Diagram shows relative TEER values normalized to time point 0 (t0). Error bars represent ± SD. Asterisks indicate statistical significance at each time point between white filled triangles (–U18666A/+TcdB) and white filled circles (+U18666A/+TcdB), with *p < 0.05 and **p < 0.01.
To investigate the protective effect of U18666A on the intoxication of CaCo-2 cells by TcdB by a further end point, the integrity of the barrier of function of CaCo-2 cells, grown as polarized monolayers in hanging cell culture inserts, was analyzed in terms of the transepithelial electrical resistance (TEER). Accordingly, CaCo-2 cell monolayers were pretreated with 10 μM U18666A for 24 h or were left untreated (mock) before apical addition of 100 pM TcdB. Then, TEER was measured every 30 min over a period of 6 h after intoxication. In direct comparison, the TcdB-induced TEER decrease over time was delayed in U18666A-pretreated CaCo-2 cells when compared to mock-pretreated cells (Figure 5C). Thus, U18666A pretreatment reduced the TcdB-induced disruption of the epithelial integrity of CaCo-2 cells.
Finally, it was tested whether U18666A also protects cells against TcdA from the historical C. difficile strain VPI 10463 and against TcdA and TcdB from the epidemic C. difficile strain NAP/027. For that purpose, HeLa cells were pretreated for 24 h with increasing concentrations of U18666A (0, 1.25, and 10 μM), prior to addition of 10 pM VPI-derived TcdA (TcdAVPI), 10 pM NAP1/027-derived TcdA (TcdANAP1) or 1 pM NAP1/027-derived TcdB (TcdBNAP1). Subsequently, microscopic analysis of the cell morphology was performed. As summarized in Figure 6, a 24 h pretreatment of HeLa cells with 10 μM U18666A effectively diminished cell rounding induced by TcdAVPI (Figure 6A), TcdANAP1 (Figure 6B) or TcdBNAP1 (Figure 6C). It is of note, however, that the reduction of TcdBNAP1 intoxication by U18666A was less effective when compared to TcdAVPI and TcdANAP1, respectively, both of which were inhibited to an equal extent by the U18666A pretreatment.
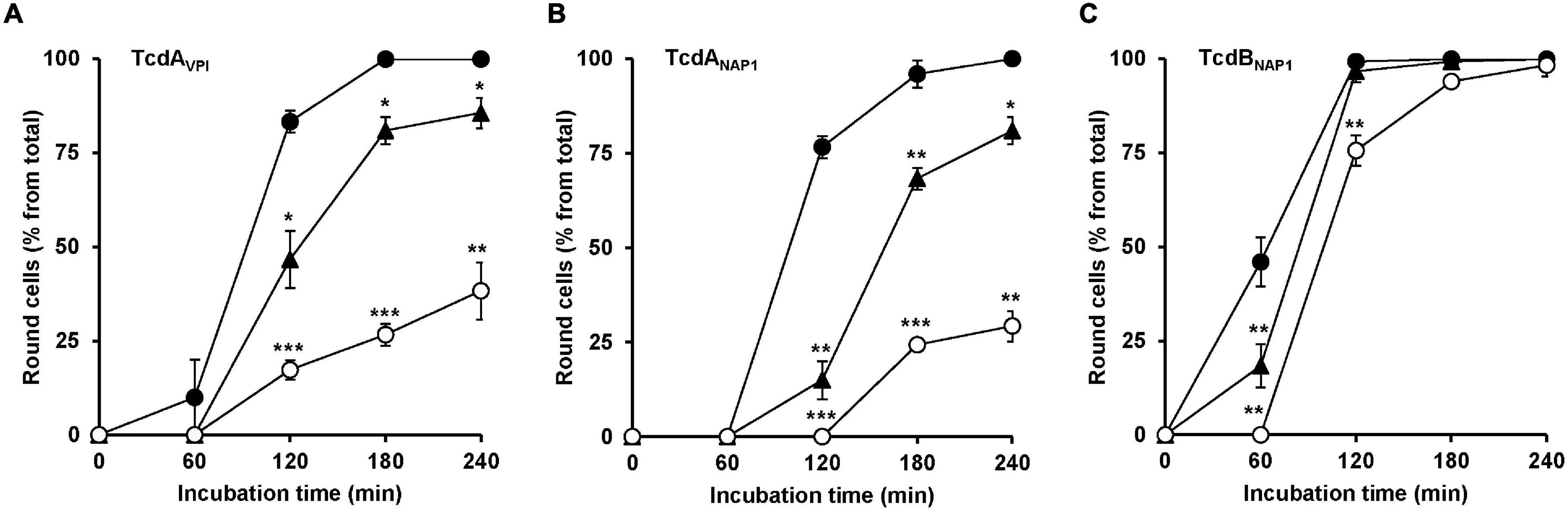
Figure 6. Effect of U18666A on the intoxication of HeLa cells by TcdAVPI, TcdANAP1 and TcdBNAP1. HeLa cells preincubated with increasing concentrations of U18666A (0 μM, black filled circles; 1.25 μM, black filled triangles and 10 μM, white filled circles) were intoxicated with (A) 10 pM VPI-derived TcdA (TcdAVPI), (B) 10 pM NAP1/027-derived TcdA (TcdANAP1) or (C) 1 pM NAP1/027-derived TcdB (TcdBNAP1), prior to microscopic analysis of the cell morphology over time. Graphs in (A–C) show means (percentage values of round cells) from three experiments performed in parallel in independent wells. Error bars represent ± SD. Asterisks indicate statistical significance compared to control (mock-treated HeLa cells) with *p < 0.05, **p < 0.01, and ***p < 0.001.
The crucial role of membrane cholesterol for pore formation of TcdA and TcdB during their endocytic uptake into target cells was first described by the Aktories group in 2006 (Giesemann et al., 2006). Along with this finding, we found more recently that an active and functional SREBP-2 pathway, which plays a major role in cholesterol metabolism, is required in target cells for the cytotoxicity of TcdA and TcdB and showed that the hypocholesterolemic drug simvastatin, which inhibits the HMG-CoA reductase, a key enzyme in cholesterol biosynthesis, efficiently protected cultured mammalian cells from TcdB intoxication (Papatheodorou et al., 2019). Since there is no structure of membrane-embedded TcdA or TcdB, the exact interaction between membrane cholesterol and TcdA/TcdB is not known. However, a recently discovered “mirror code” controlling protein-cholesterol interactions in the outer and inner membrane leaflets (Fantini et al., 2016) seems to be present also within a region of TcdA and TcdB (amino acids 830–990) (Gerhard, 2017), which is in charge of pore formation (Genisyuerek et al., 2011). Cholesterol might be crucial for the insertion of the translocation domain of the toxins into the endosomal membrane. Otherwise, membrane cholesterol could serve as a platform for the correct positioning of transmembrane segments of the toxins within the endosomal membrane for generating a translocation-competent pore.
Prompted by the above mentioned observations that membrane cholesterol is crucial for the uptake of TcdA and TcdB in target cells, the U18666A compound came into our focus, since it has been described by several studies as potent inhibitor of cholesterol biosynthesis and/or intracellular trafficking (Cenedella, 2009). At least three membrane-bound enzymes in the cholesterol biosynthetic pathway, namely 2,3-oxidosqualene cyclase, desmosterol reductase and cholestenol delta-isomerase, are known to be inhibited by the U18666A compound (Duriatti et al., 1985; Bae and Paik, 1997; Moebius et al., 1998). The activity of HMG-CoA reductase, another key enzyme in cholesterol biosynthesis, is decreased by addition of low concentrations of U18666A, but increased at higher drug levels (Panini et al., 1984). Another feature of U18666A is the inhibition of the intracellular transport of cholesterol, namely from the lysosome to the endoplasmic reticulum (ER) and to the plasma membrane (PM), respectively, and from the PM to the ER (Liscum and Dahl, 1992; Underwood et al., 1998). Effects of U18666A on intracellular cholesterol trafficking are explained by its direct interference with the lysosomal membrane protein Niemann-Pick type C1 (NPC1), which transports cholesterol from receptor-mediated uptake of low-density lipoproteins (LDL) to post-lysosomal destinations (Carstea et al., 1997; Blanchette-Mackie, 2000; Lange et al., 2000; Lu et al., 2015).
The present study revealed that the U18666A compound confers increased protection to various mammalian cell lines against the C. difficile toxin TcdB. In addition, TcdA-inhibition by U18666A-pretreatment was confirmed in HeLa cells and TcdA-inhibition by U18666A was more prominent when compared to TcdB, irrespective of the source strain of both toxins. Inhibition of TcdB is more challenging most likely due to the fact that TcdB preparations are up to 1,000-fold more toxic than TcdA preparations (Rothman et al., 1984). It is also feasible that TcdA membrane insertion and/or pore formation in endosomal membranes is more prone to changes of membrane cholesterol levels. Moreover, pretreatment with the U18666A compound was not equally protective against TcdB among the various tested cells. For instance, the inhibition of TcdB-intoxication by U18666A pretreatment was more prominent in Vero cells when compared to HeLa cells at equal inhibitor concentrations. Possible explanations for this observation could be that either the compound is more efficiently taken up into Vero cells or that the compound is more efficiently degraded or excreted from HeLa cells by drug transporters. It is also feasible that membrane cholesterol levels and/or cholesterol uptake, intracellular transport and biosynthesis capabilities are vastly differing between HeLa and Vero cells, thus influencing the inhibitory potential of the U18666A compound in each cell line.
The replication of many viruses is associated with intact cholesterol biosynthesis and intracellular transport. Thus, it has been reported that U18666A suppress the replication of several viruses, such as ebola virus, dengue virus, human hepatitis C virus, and type I feline coronavirus (Poh et al., 2012; Lu et al., 2015; Elgner et al., 2016; Takano et al., 2017). The mechanism underlying the U18666A-mediated inhibition of TcdB and TcdA is likely related to a blockage of cholesterol biosynthetic enzymes and of the intracellular transport of endocytosed cholesterol from the culture medium to the plasma membrane and/or to endocytic vesicles. Consistent with this hypothesis, cells were strongly protected from toxin intoxication, when they were pretreated for at least 24 h with U18666A, but not after shorter incubation periods with the compound. We widely excluded the possibilities of (i) an inactivating effect of U18666A by direct binding to the toxins or (ii) through alteration of endosomal membranes through U18666A incorporation, because there was no efficient protection from TcdB after short incubation periods with U18666A, but protection when the U18666A-containing medium on top of cells was removed by replacement with TcdB-containing medium during the intoxication step.
The cholesterol-dependent pore formation by TcdA or TcdB in endosomal membranes is a crucial step during cell entry of both toxins and this study corroborated that this specific step can be targeted by pharmacological tools such as U18666A. Importantly, U18666A also inhibited TcdA and TcdB variants from the epidemic NAP1/027 C. difficile strain. This finding indicates that cholesterol seems to be a conserved player in TcdA/TcdB intoxication. Thus, the development of cholesterol-lowering drugs, such as U18666A, as potential therapeutics against TcdA and TcdB might be of interest not only for the treatment of infections caused by non-epidemic C. difficile strains (such as VPI 10463), but also for the treatment of diseases associated with infections caused by the epidemic NAP1/027 C. difficile strain. However, the safety profile of U18666A for clinical use is not evaluated so far yet. Cataracts were described in rats that were treated daily with 10 mg/kg U18666A, but not when the rats were treated every fourth day with the U18666A compound (Cenedella and Bierkamper, 1979). Thus, the drug application form (enteral or rectal administration) and interval should be carefully considered in potential clinical studies on the efficacy and safety of U18666A and derivatives in the (supportive) treatment of severe CDI.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
PP, SF, EC-O, and HB designed the research. PP, SK, AB-L, SF, ED, TT, AW, and SS performed the research. PP, SK, AB-L, SF, KA, EC-O, CR, and HB analyzed the data. PP, KA, CR, and HB wrote the manuscript. All authors contributed to the article and approved the submitted version.
Research funding was received in the group of HB from the Deutsche Forschungsgemeinschaft (DFG; grant number Ba 2087/8-1, project no. 450938962) and in the group of EC-O and CR from the Vice-Presidency for Research of the University of Costa Rica (project no. B9450).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Anna Anastasia, Monika Berther and Jana Kuhn for excellent technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.784856/full#supplementary-material
Aktories, K., and Just, I. (2005). Clostridial Rho-inhibiting protein toxins. Curr. Top. Microbiol. Immunol. 291, 113–145.
Aktories, K., Schwan, C., and Jank, T. (2017). Clostridium difficile toxin biology. Annu. Rev. Microbiol. 71, 281–307.
Bae, S. H., and Paik, Y. K. (1997). Cholesterol biosynthesis from lanosterol: development of a novel assay method and characterization of rat liver microsomal lanosterol Δ24-reductase. Biochem. J. 326, 609–616. doi: 10.1042/bj3260609
Barth, H., Pfeifer, G., Hofmann, F., Maier, E., Benz, R., and Aktories, K. (2001). Low ph-induced formation of Ion channels by Clostridium difficile toxin B in target cells. J. Biol. Chem. 276, 10670–10676. doi: 10.1074/jbc.M009445200
Blanchette-Mackie, E. J. (2000). Intracellular cholesterol trafficking: role of the NPC1 protein. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1486, 171–183.
Carstea, E. D., Morris, J. A., Coleman, K. G., Loftus, S. K., Zhang, D., Cummings, C., et al. (1997). Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277, 228–231. doi: 10.1126/science.277.5323.228
Cenedella, R. J. (2009). Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids 44, 477–487. doi: 10.1007/s11745-009-3305-7
Cenedella, R. J., and Bierkamper, G. G. (1979). Mechanism of cataract production by 3-β(2-diethylaminoethoxy) androst-5-en-17-one hydrochloride, U18666A: an inhibitor of cholesterol biosynthesis. Exp. Eye Res. 28, 673–688. doi: 10.1016/0014-4835(79)90068-x
Chandrasekaran, R., and Lacy, D. B. (2017). The role of toxins in clostridium difficile infection. FEMS Microbiol. Rev. 41, 723–750. doi: 10.1093/FEMSRE/FUX048
Duriatti, A., Bouvier-Nave, P., Benveniste, P., Schuber, F., Delprino, L., Balliano, G., et al. (1985). In vitro inhibition of animal and higher plants 2,3-oxidosqualene-sterol cyclases by 2-aza-2,3-dihydrosqualene and derivatives, and by other ammonium-containing molecules. Biochem. Pharmacol. 34, 2765–2777. doi: 10.1016/0006-2952(85)90578-7
Egerer, M., Giesemann, T., Herrmann, C., and Aktorles, K. (2009). Autocatalytic processing of Clostridium difficile toxin B: binding of inositol hexakisphosphate. J. Biol. Chem. 284, 3389–3395. doi: 10.1074/JBC.M806002200
Elgner, F., Ren, H., Medvedev, R., Ploen, D., Himmelsbach, K., Boller, K., et al. (2016). The Intracellular cholesterol transport inhibitor U18666A inhibits the exosome-dependent release of mature Hepatitis C Virus. J. Virol. 90, 11181–11196. doi: 10.1128/JVI.01053-16
Fantini, J., Di Scala, C., Evans, L. S., Williamson, P. T. F., and Barrantes, F. J. (2016). A mirror code for protein-cholesterol interactions in the two leaflets of biological membranes. Sci. Rep. 6:21907. doi: 10.1038/SREP21907
Genisyuerek, S., Papatheodorou, P., Guttenberg, G., Schubert, R., Benz, R., and Aktories, K. (2011). Structural determinants for membrane insertion, pore formation and translocation of Clostridium difficile toxin B. Mol. Microbiol. 79, 1643–1654. doi: 10.1111/J.1365-2958.2011.07549.X
Gerhard, R. (2017). Receptors and binding structures for clostridium difficile toxins A and B. Curr. Top. Microbiol. Immunol. 406, 79–96. doi: 10.1007/82_2016_17
Giesemann, T., Egerer, M., Jank, T., and Aktories, K. (2008). Processing of Clostridium difficile toxins. J. Med. Microbiol. 57, 690–696. doi: 10.1099/JMM.0.47742-0
Giesemann, T., Jank, T., Gerhard, R., Maier, E., Just, I., Benz, R., et al. (2006). Cholesterol-dependent pore formation of Clostridium difficile toxin A. J. Biol. Chem. 281, 10808–10815. doi: 10.1074/jbc.m512720200
Hall, A. (1994). Small GTP-binding proteins and the regulation of the actin cytoskeleton. Annu. Rev. Cell Biol. 10, 31–54. doi: 10.1146/annurev.cb.10.110194.000335
Jank, T., and Aktories, K. (2008). Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 16, 222–229. doi: 10.1016/J.TIM.2008.01.011
Just, I., Selzer, J., Hofmann, F., and Aktories, K. (2008). “Clostridium difficile Toxin B as a Probe for Rho GTPases,” in Bacterial Toxins - Tools in Cell Biology and Pharmacology, ed. K. Aktories (Weinheim: Chapman & Hall), 159–168.
Just, I., Selzer, J., Wilm, M., Von Eichel-Streiber, C., Mann, M., and Aktories, K. (1995). Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375, 500–503.
Kelly, C. P., and LaMont, J. T. (2008). Clostridium difficile - more difficult than ever. N. Engl. J. Med. 359, 1932–1940. doi: 10.1056/nejmra0707500
Lange, Y., Ye, J., Rigney, M., and Steck, T. (2000). Cholesterol movement in Niemann-Pick type C cells and in cells treated with amphiphiles. J. Biol. Chem. 275, 17468–17475. doi: 10.1074/jbc.M000875200
Liscum, L., and Dahl, N. K. (1992). Intracellular cholesterol transport. J. Lipid Res. 33, 1239–1254.
Lu, F., Liang, Q., Abi-Mosleh, L., Das, A., de Brabander, J. K., Goldstein, J. L., et al. (2015). Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. Elife 4:e12177. doi: 10.7554/eLife.12177
Lyerly, D. M., Krivan, H. C., and Wilkins, T. D. (1988). Clostridium difficile: its disease and toxins. Clin. Microbiol. Rev. 1, 1–18. doi: 10.1128/cmr.1.1.1-18.1988
Moebius, F. F., Reiter, R. J., Bermoser, K., Glossmann, H., Cho, S. Y., and Paik, Y. K. (1998). Pharmacological analysis of sterol Δ8=Δ7 isomerase proteins with [3H]fenprodil. Mol. Pharmacol. 54, 591–598. doi: 10.1124/mol.54.3.591
Orrell, K. E., Zhang, Z., Sugiman-Marangos, S. N., and Melnyk, R. A. (2017). Clostridium difficile toxins A and B: receptors, pores, and translocation into cells. Crit. Rev. Biochem. Mol. Biol. 52, 461–473. doi: 10.1080/10409238.2017.1325831
Panini, S. R., Sexton, R. C., and Rudney, H. (1984). Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase by oxysterol by-products of cholesterol biosynthesis. Possible mediators of low density lipoprotein action. J. Biol. Chem. 259, 7767–7771. doi: 10.1016/s0021-9258(17)42859-6
Papatheodorou, P., Barth, H., Minton, N., and Aktories, K. (2018). Cellular uptake and mode-of-action of Clostridium difficile toxins. Adv. Exp. Med. Biol. 1050, 77–96. doi: 10.1007/978-3-319-72799-8_6
Papatheodorou, P., Song, S., López-Ureña, D., Witte, A., Marques, F., Ost, G. S., et al. (2019). Cytotoxicity of Clostridium difficile toxins A and B requires an active and functional SREBP-2 pathway. FASEB J. 33, 4883–4892. doi: 10.1096/fj.201801440R
Papatheodorou, P., Zamboglou, C., Genisyuerek, S., Guttenberg, G., and Aktories, K. (2010). Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS One 5:e10673. doi: 10.1371/journal.pone.0010673
Poh, M. K., Shui, G., Xie, X., Shi, P. Y., Wenk, M. R., and Gu, F. (2012). U18666A, an intra-cellular cholesterol transport inhibitor, inhibits dengue virus entry and replication. Antiviral Res. 93, 191–198. doi: 10.1016/j.antiviral.2011.11.014
Pruitt, R. N., Chambers, M. G., Ng, K. K. S., Ohi, M. D., and Lacy, D. B. (2010). Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc. Natl. Acad. Sci. U. S. A. 107, 13467–13472. doi: 10.1073/pnas.1002199107
Rothman, S. W., Brown, J. E., Diecidue, A., and Foret, D. A. (1984). Differential cytotoxic effects of toxins A and B isolated from Clostridium difficile. Infect. Immun. 46, 324–331. doi: 10.1128/IAI.46.2.324-331.1984
Takano, T., Endoh, M., Fukatsu, H., Sakurada, H., Doki, T., and Hohdatsu, T. (2017). The cholesterol transport inhibitor U18666A inhibits type I feline coronavirus infection. Antiviral Res. 145, 96–102. doi: 10.1016/J.ANTIVIRAL.2017.07.022
Keywords: bacterial toxin, toxin inhibitor, cholesterol biosynthesis, cholesterol transport, cholesterol
Citation: Papatheodorou P, Kindig S, Badilla-Lobo A, Fischer S, Durgun E, Thuraisingam T, Witte A, Song S, Aktories K, Chaves-Olarte E, Rodríguez C and Barth H (2021) The Compound U18666A Inhibits the Intoxication of Cells by Clostridioides difficile Toxins TcdA and TcdB. Front. Microbiol. 12:784856. doi: 10.3389/fmicb.2021.784856
Received: 28 September 2021; Accepted: 01 November 2021;
Published: 29 November 2021.
Edited by:
Meina Neumann-Schaal, German Collection of Microorganisms and Cell Cultures GmbH (DSMZ), GermanyReviewed by:
Harald Genth, Hannover Medical School, GermanyCopyright © 2021 Papatheodorou, Kindig, Badilla-Lobo, Fischer, Durgun, Thuraisingam, Witte, Song, Aktories, Chaves-Olarte, Rodríguez and Barth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panagiotis Papatheodorou, cGFuYWdpb3Rpcy5wYXBhdGhlb2Rvcm91QHVuaS11bG0uZGU=
†Present address: Ebru Durgun, Institute of Anatomy and Cell Biology, Ulm University, Ulm, Germany; Tharani Thuraisingam, Nuvisan GmbH, Neu-Ulm, Germany; Alexander Witte, Pfizer Manufacturing Deutschland GmbH, Freiburg, Germany; Shuo Song, Central Laboratory, Shenzhen Samii International Medical Center, Shenzhen, China
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.