 Prasad Thomas1,2†
Prasad Thomas1,2† Mostafa Y. Abdel-Glil1,3,4*†
Mostafa Y. Abdel-Glil1,3,4*† Anbazhagan Subbaiyan2
Anbazhagan Subbaiyan2 Anne Busch5
Anne Busch5 Inga Eichhorn6
Inga Eichhorn6 Lothar H. Wieler6,7
Lothar H. Wieler6,7 Heinrich Neubauer1
Heinrich Neubauer1 Mathias Pletz3
Mathias Pletz3 Christian Seyboldt1
Christian Seyboldt1- 1Institute of Bacterial Infections and Zoonoses, Friedrich-Loeffler-Institut, Jena, Germany
- 2Division of Bacteriology and Mycology, ICAR-Indian Veterinary Research Institute, Izatnagar, India
- 3Institute for Infectious Diseases and Infection Control, Jena University Hospital – Friedrich Schiller University, Jena, Germany
- 4Department of Pathology, Faculty of Veterinary Medicine, Zagazig University, Zagazig, Egypt
- 5Department of Anaesthesiology and Intensive Care Medicine, University Hospital Jena, Jena, Germany
- 6Department of Veterinary Medicine, Institute of Microbiology and Epizootics, Freie Universität Berlin, Berlin, Germany
- 7Robert Koch Institute, Berlin, Germany
Clostridium septicum is a Gram-positive, toxin-producing, and spore-forming bacterium that is recognized, together with C. perfringens, as the most important etiologic agent of progressive gas gangrene. Clostridium septicum infections are almost always fatal in humans and animals. Despite its clinical and agricultural relevance, there is currently limited knowledge of the diversity and genome structure of C. septicum. This study presents the complete genome sequence of C. septicum DSM 7534T type strain as well as the first comparative analysis of five C. septicum genomes. The taxonomy of C. septicum, as revealed by 16S rRNA analysis as well as by genomic wide indices such as protein-based phylogeny, average nucleotide identity, and digital DNA–DNA hybridization indicates a stable clade. The composition and presence of prophages, CRISPR elements and accessory genetic material was variable in the investigated genomes. This is in contrast to the limited genetic variability described for the phylogenetically and phenotypically related species Clostridium chauvoei. The restriction-modification (RM) systems between two C. septicum genomes were heterogeneous for the RM types they encoded. C. septicum has an open pangenome with 2,311 genes representing the core genes and 1,429 accessory genes. The core genome SNP divergence between genome pairs varied up to 4,886 pairwise SNPs. A vast arsenal of potential virulence genes was detected in the genomes studied. Sequence analysis of these genes revealed that sialidase, hemolysin, and collagenase genes are conserved compared to the α-toxin and hyaluronidase genes. In addition, a conserved gene found in all C. septicum genomes was predicted to encode a leucocidin homolog (beta-channel forming cytolysin) similar (71.10% protein identity) to Clostridium chauvoei toxin A (CctA), which is a potent toxin. In conclusion, our results provide first, valuable insights into strain relatedness and genomic plasticity of C. septicum and contribute to our understanding of the virulence mechanisms of this important human and animal pathogen.
Introduction
Clostridium septicum, the first anaerobic pathogen described (MacLennan, 1962) is the major causative agent of a pathologic fatal condition in animals and humans named as malignant edema or gas gangrene that may also involve other Clostridia species such as C. perfringens type A, C. novyi, C. chauvoei, and C. sordellii (MacLennan, 1962). Malignant edema in animals arises predominantly from exogenous wound contamination. This is in contrast to blackleg disease that is non-traumatic endogenous infection mainly reported in cattle and caused by C. chauvoei (Silva et al., 2016). Clostridium septicum induced malignant edema has a broad host range with disease reported in ruminants, horses, pigs, elephants, and birds (Sasaki et al., 2001; Odani et al., 2009; Rahman et al., 2009; Almeida E Macêdo et al., 2013). Animals of all ages can be affected and the disease occurs worldwide. Rare outbreaks in animals due to injection of contaminated vaccines or other medical interventions have also been reported (Morris et al., 2002). Clostridium septicum infections leading to vulvovaginitis and metritis after parturition (post parturient malignant oedema) in cattle (Odani et al., 2009; Junior et al., 2020) and necrotizing abomasitis (braxy) in lambs and calves were reported (Schamber et al., 1986; Glenn Songer, 2009). In humans, C. septicum was reported to cause atraumatic gas gangrene in immune-compromised patients (Barnes et al., 2004). Association of C. septicum-mediated myonecrosis in human patients with colonic (Nanjappa et al., 2015) or hematologic malignancy resulting in high mortality rates (79%) was also described (Kornbluth et al., 1989; Powell et al., 2008). In cases of fatal gas gangrene with multiorgan involvement and in a rare aortitis in human patients with colonic malignancy, C. septicum was detected in blood and tissue cultures (Seder et al., 2009; Curtis-Martínez and Sánchez-Guillén, 2019; Kousa et al., 2020). Table 1 lists selected human and animal disease reports and conditions primarily associated with C. septicum, indicating its relevance in the medical and veterinary settings. Despite this relevance, current information on genomic features and diversity of the species is limited to strain P1044, isolated from human intestine, whose genome is represented by a draft sequence and briefly described in a genome announcement (Benamar et al., 2016). Genetic information on the diversity of the pathogen is also limited, with few studies focusing on the genetic diversity of the α-toxin gene (Amimoto et al., 2006). Multi-locus sequence typing (MLST) analysis of poultry C. septicum strains associated with gangrenous dermatitis revealed a high conservation among selected MLST genes (Neumann and Rehberger, 2009).
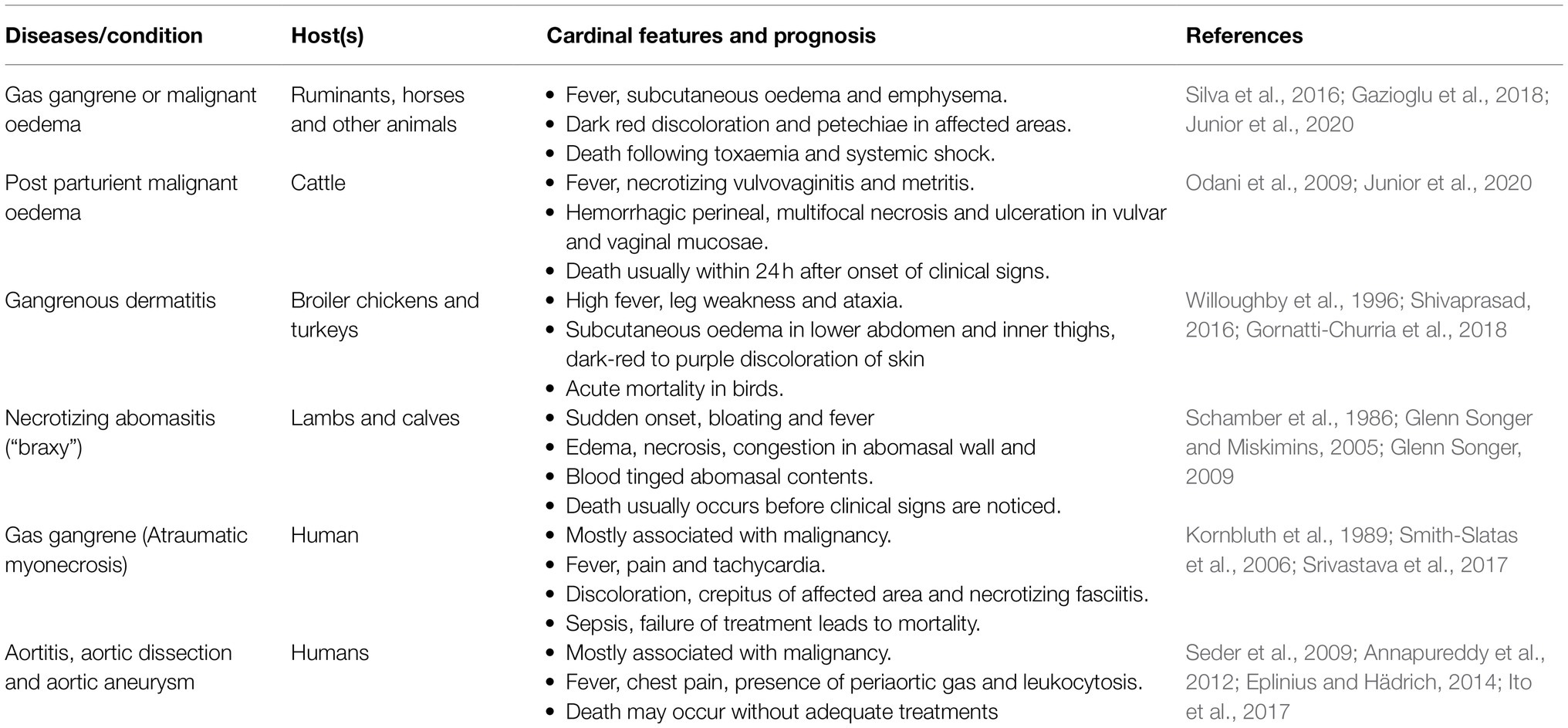
Table 1. List of selected disease reports and conditions in humans and animals that are mainly linked to C. septicum.
Furthermore, the toxin repertoire of C. septicum has not been studied in detail. C. septicum is known to produce four types of toxins: alpha, beta, gamma, and delta toxin (Moussa, 1958; Ballard et al., 1992; Amimoto et al., 2006). The best studied of these is α-toxin, a potent pore-forming toxin that is released extracellularly (Bernheimer, 1944; Kennedy et al., 2005, 2009). Studies have shown the importance of α-toxin in virulence and development of myonecrosis (Kennedy et al., 2005; Hickey et al., 2008), and a recent study has shown that α-toxin could modulate the host innate immune response (Chakravorty et al., 2015).
The aim of the current study was to (1) generate a high-quality complete genome sequence of the type strain (DSM 7534T = ATCC 12464) using a combination of short and long reads and (2) perform a comparative genome analysis of C. septicum strains, using genome data sets from public databases. Analysis of the genomes with respect to species taxonomy, repeat regions, prophage elements and virulence factors was carried out for all genomes. In addition, methylation motifs (one genome) and restriction-modification (RM) systems of two finished C. septicum genomes were investigated and described.
Materials and Methods
Genome Sequencing and Assembly (DSM 7534T)
C. septicum type strain DSM 7534T (ATCC 12464, CIP 61.10, NCIMB 547, and NCTC 547) was sequenced. Genomic DNA was extracted using Qiagen Genomic-tip 100/Q (Qiagen, Germany) with minor modifications as the washing step was repeated five times and the DNA was incubated at 37°C until dissolved. DNA quality was examined by using Qubit 2.0 fluorometer (Life Technologies, Germany) and species confirmation was made with PCR (Sasaki et al., 2000). Genome sequencing was carried out by Pacific Biosciences (PacBio) sequencing using PacBio RSII sequencer at GATC Biotech (Germany). Additional sequencing for the same strain was carried out using MiSeq™ System (Illumina, United States) paired-end sequencing technology (2 × 300-bp) at the Institute of Microbiology and Epizootics (IMT), Freie Universität Berlin. Genome assembly was carried out using Unicycler, run under the bold mode (Wick et al., 2017). The unfinished contigs generated by Unicycler were circularized using Circlator (Hunt et al., 2015).
Retrieval and Processing of Publicly Available Sequence Data
For the analysis, genomes (whole-genome assemblies and raw sequencing reads) available in NCBI for C. septicum (taxonomy number 1504) were downloaded (March 2021). They comprise genome assemblies of strain VAT12 (accession NZ_CP034358) isolated in 2012 in Virginia, the United States from a wild turkey and strain P1044 (accession NZ_FLTT00000000.1) isolated from a human stool in Marseille, France. A duplicate genome of strain P1044 was available under the accession no. NZ_CABMIZ000000000.1 (genome MGYG-HGUT-02373; Almeida et al., 2021). In addition, the sequence read archive database of NCBI included raw sequence data for only three C. septicum strains. Strain RVDL ALI_Clost_septicum_01 (hereafter RVDL_ALI; accession SRR10484857) sequenced using Illumina MiSeq was isolated in 2017 from a calf in Saudi Arabia. Strains DRS014147 (accession, DRR016039) and ERS2884028 (accession ERR3283734) were sequenced using Illumina HiSeq™ system. Epidemiological data were not available for the latter two strains. Initial taxonomy analysis using Kranken2 (Wood et al., 2019) classified the genome ERS2884028 to the species Streptococcus pneumonia, hence it was excluded. Paired-end Illumina reads were assembled with Shovill v1.0.4 using the option “-trim” enabling adapter and quality trimming using Trimmomatic (Wood et al., 2019).1 Assembly statistics were reported using QUAST v5.0.2 (Gurevich et al., 2013). In total, five genome sequences of C. septicum strains representing different regions and hosts were included in our analysis. Genome annotations were carried out using Prokka v1.14.6 (Seemann, 2014) in Galaxy server (Afgan et al., 2018).
Taxonomic Classification
Taxonomic classification of C. septicum genomes was done first using the classical 16S rRNA gene sequence. The rRNA genes were predicted using barrnap v0.93 with default settings.2 In the case of genomes with multiple hits for the 16S rRNA gene, only one representative sequence (1.4-kb) was used and for partial 16S rRNA representations, the sequence with the highest alignment coverage was chosen. The 16S rRNA from all genomes was aligned using MAFFT v7.221 (Katoh et al., 2002). IQ-TREE v1.5.5.3 was used for maximum-likelihood (ML) phylogenetic analysis (Katoh et al., 2002) using ultrafast bootstrap approximation approach (UFBoot) and with 1,000 bootstraps (Minh et al., 2013; Nguyen et al., 2015). Tree visualization was done using iTOL v6 (Letunic and Bork, 2019).
For genome-wide approaches, we used PhyloPhlAn v0.43 to investigate species taxonomy by constructing a phylogenetic tree based on conserved ubiquitous proteins in bacteria (Segata et al., 2013). For that, an ML phylogeny was performed with RAxML v8.2.12 using the PROTCATLG model and 100 bootstrap replicates (Stamatakis, 2014). We also estimated the whole-genome-based average nucleotide identity (ANI) with pyani v0.2.3 (module: ANIm; Pritchard et al., 2016). For all the above-mentioned taxonomy analyses, representative genomes of all species of the genus cluster Clostridium sensu stricto were included for comparison (Supplementary Table S1). In addition, we calculated the genome-to-genome distances by using an in silico DNA–DNA hybridization (isDDH) approach as implemented in Genome-to-Genome Distance Calculator (GGDC) 2.1 webserver (Meier-Kolthoff et al., 2013; Jain et al., 2018). For that, the query genome assembly files were subjected to local alignments with BLAST+ tool (Camacho et al., 2009) against the reference C. septicum genome DSM 7534T and estimates independent of genome lengths were used for distance calculations as recommended for draft genomes (Formula 2; Auch et al., 2010). For the isDDH analysis, C. chauvoei genome sequences (accessions NZ_CP018624 and NZ_LT799839.1) were kept as outgroups.
Restriction-Modification Systems and Methylation Analysis
Single-molecule real-time (SMRT) sequencing by PacBio RSII allows the detection of base modifications in the genomes (Clark et al., 2012; Meier-Kolthoff et al., 2014). The RM systems information for C. septicum DSM 7534T and VAT12 strains were retrieved from the REBASE website (Schadt et al., 2013).3 The methylated bases and methylation-associated motifs (DSM 7534T) were identified using the RS_Modification_and_Motif_analysis.1 tool in SMRT portal v2.3.0.
Genome Comparison
Schematic representations of complete circular chromosomes and plasmids were generated using the CGView Server (Grant and Stothard, 2008). Comparative genomics involved five genome datasets. These genomes were investigated for CRISPR (clustered regularly interspaced short palindromic repeats) loci using the CRISPRDetect v2.4 (Biswas et al., 2016) with a minimum number of repeats > = 5. The CRISPR spacers were visualized using CRISPRStudio (Dion et al., 2018) in the Galaxy server (Afgan et al., 2018). Prophage elements were predicted using PHASTER (Arndt et al., 2016). Orthologous clustering of genes (pangenome analysis) was carried out using Prokka v1.14.6 (Seemann, 2014) annotated genomes with Panaroo pipeline v1.2.7 (Tonkin-Hill et al., 2020). Curves for the core and pangenome were calculated and plotted using PanGP v1.0.1 (Zhao et al., 2014). The pangenome plots were created also using GView Server with BLAST options (expect value 1-e 10, alignment length 100 and percent identity 80) and the GView v7.1 (Stothard et al., 2019).4 Prediction of antimicrobial resistance (AMR) genes was perfromed using ABRicate v1.0.1 and ResFinder (Zankari et al., 2012), CARD (McArthur et al., 2013) and AGR-Annot (Gupta et al., 2014) databases.5 Functional annotation of the core and accessory genes was carried out for Clusters of Orthologous Groups (COGs) using the eggnog-mapper v2 (Huerta-Cepas et al., 2017) based on eggNOG 5.0 orthology data (Huerta-Cepas et al., 2019) specifying the genus Clostridia. Additonally, the metabolic pathways were assessed using the BlastKOALA annotation server that use Kyoto Encyclopedia of Genes and Genomes (KEGG) Tools for functional characterization of protein coding sequences (Kanehisa et al., 2016). Positional homology multiple genome alignments of genome sequences were done using progressiveMauve (Darling et al., 2010). Core genome alignment generated using Panaroo was used for an ML phylogenetic analysis in IQ-TREE v1.5.5.3 using UFBoot as mentioned earlier (Minh et al., 2013; Nguyen et al., 2015). Tree visualization was done using iTOL v6 (Letunic and Bork, 2019). Pairwise core genome SNP variations between strains were determined using snp-dists v0.6.3.6
Virulence Factors
Primary virulence factors for C. septicum were identified with BLASTP v2.9.0+ (Camacho et al., 2009) using a custom database constructed based on the virulence factor database (Liu et al., 2019) and putative virulence factors previously reported in the C. chauvoei type strain DSM 7528T (NZ_CP018624). From the BLAST output results, we only kept BLAST hits with e-value of less than 1e-20, protein sequence identity more than 40%, coverage more than 70%, and total query gene length more than 90% of the total reference gene length. Nucleotide and protein alignments were performed for identified virulence factors using MAFFT v7.221 (Katoh et al., 2002). The amino acid (aa) variations between strains were visualized from alignments using Geneious Prime® 2019.2.3 (Kearse et al., 2012). Phylogenetic analysis was carried using IQ-TREE as mentioned above.
For the identified putative novel toxin homolog (beta-channel forming cytolysin: Locus tag-CP523_11160), feature and genetic structure predictions of signal peptide and Leukocidin/Hemolysin domain were carried out using SignalP v. 5.0 (Almagro Armenteros et al., 2019) and HMMER web server (Potter et al., 2018) searches to Pfam database (Mistry et al., 2021), respectively. An ML tree involving putative C. septicum toxin homolog with representative toxins from related species were carried out from protein alignment. Comparison of B cell epitopes of C. septicum cytolysin and CctA was carried out using ABCpred webserver (Saha and Raghava, 2006) with the settings threshold score 0.75; window length 20; overlapping filter –ON.
Results
Genome Features
The finished genome of the C. septicum type strain DSM 7534T was determined, revealing a circular 3.3-Mb chromosome (NZ_CP023671) and a 5.2-kb plasmid (NZ_CP023672.1; Figure 1). Strain VAT 12 was also represented by a completed genome containing a circular 3.45-Mb chromosome (Figure 1). The other three genomes were in draft form (Table 2). Strains DRS014147 and RVDL_ALI had 125 and 578 contigs, representing 3.26 and 2.88-Mb genomes, respectively. The genomes of strain P1044 was represented by 79 contigs with a total genome size of 3.29-Mb. The overall genome size and GC composition of all assembled genomes were in similar ranges, except for strain RVDL_ALI. This strain had a smaller genome with a relatively higher GC content (28.2% compared with 27.5–27.9% for the other strains) and a lower number of predicted protein-coding genes (2,581 compared with 3,028–3,376 for the other genomes). These results could indicate poor sequencing or assembly of the genome which is also reflected in the genome high fragmentation: 578 contigs compared to less than 125 contigs for the other genomes (Table 2).
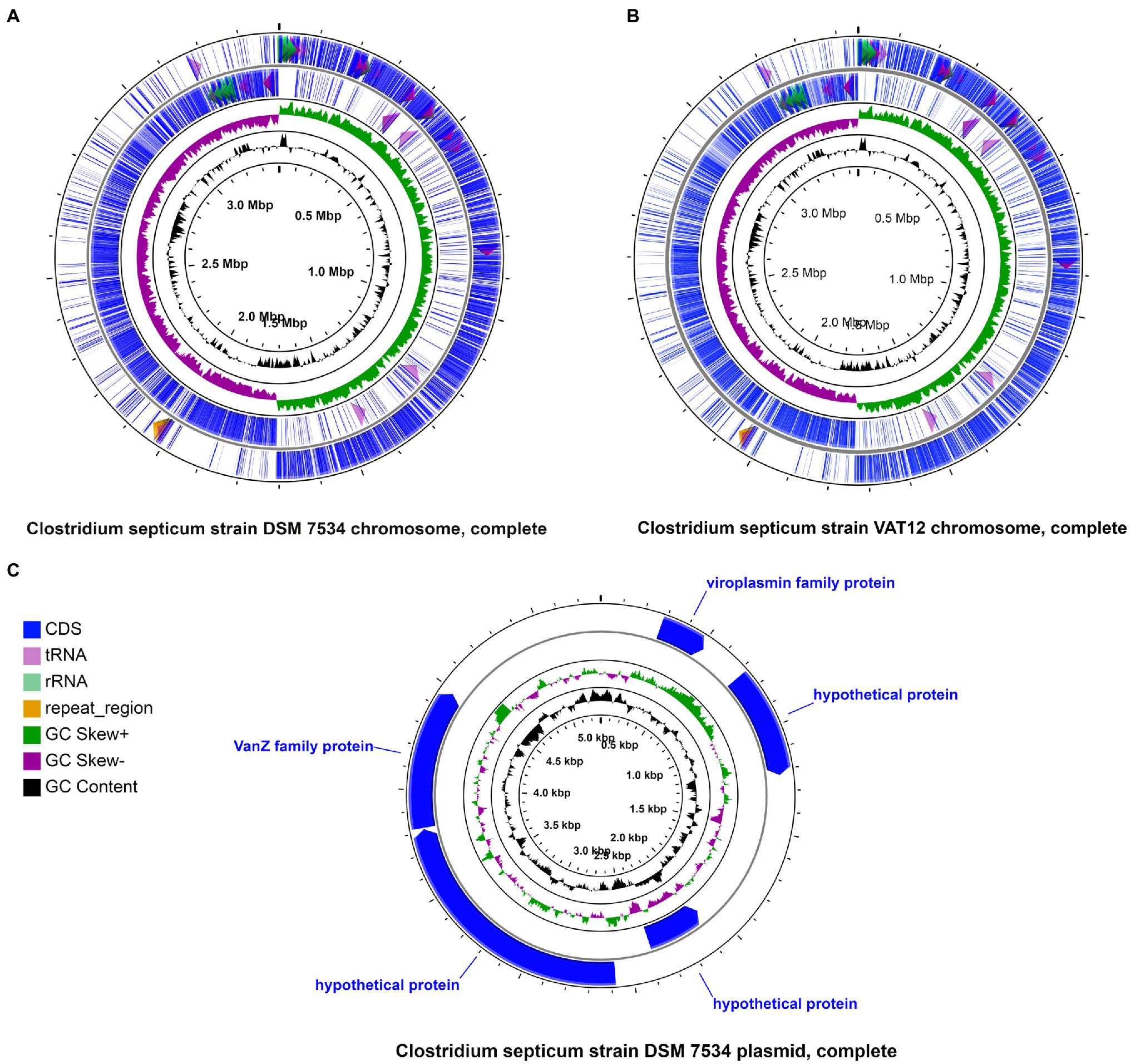
Figure 1. Schematic representation of circular chromosomes and plasmid. Circular chromosomes of DSM 7534T (A) and VAT12 (B) strains and the circular plasmid of DSM 7534T (C) strain are shown highlighting CDS, tRNA, rRNA, CRISPR elements, GC content and GC Skew.
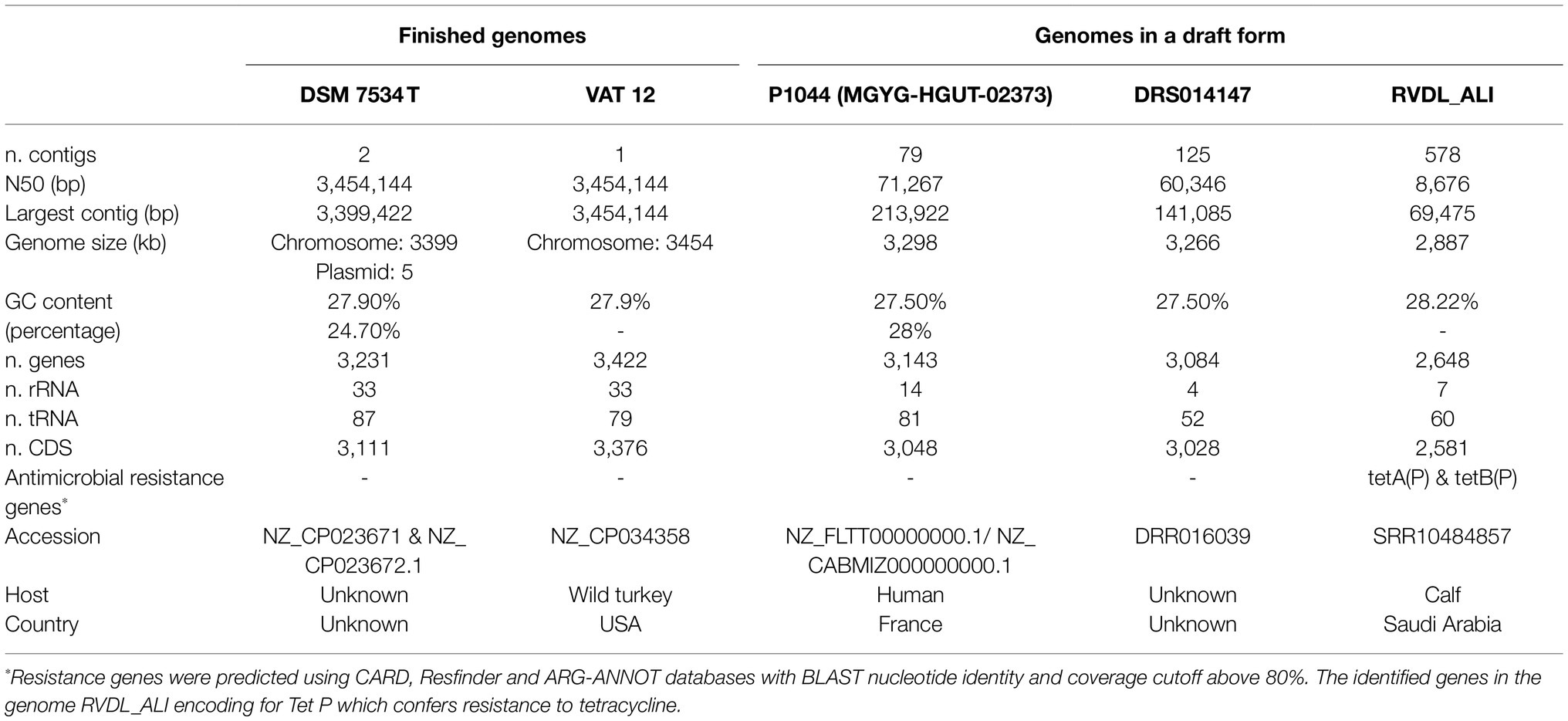
Table 2. Genome assembly, annotation, and metadata summary.
Species Taxonomy
Phylogenetic analysis of the 16S rRNA gene revealed the clustering of C. septicum genomes in a monophyletic lineage. Nevertheless, grouping was observed between individual genomes of the species, for example, the genomes VAT 12 and P1044 formed a subgroup that was distinct from the other subgroup of the type strain (DSM 7534T) and the genomes of strains DRS014147 and RVDL_ALI (Supplementary Figure S1). The closely related species from Clostridium sensu stricto was C. chauvoei with 100% bootstrap support.
The results of 16S rRNA analysis were further upheld by analysis of the whole genome, in which the phylogenetic tree based on the concatenated protein alignment of 400 universal bacterial genes placed the genomes of C. septicum in a separate group with close association to the species C. chauvoei (Figure 2A). ANI values between pairs of the five C. septicum genome datasets were more than 99% indicating that all five different genomes met the single species criteria. Pairwise ANI values were ~85% when C. septicum is compared with representative strains of the neighboring species, indicating clear species demarcation (Figure 2B). Finally, the C. septicum genomes were highly similar based on the DDH values that ranged from 96.7 to 97.9% for the five genomes in comparison to the C. septicum type strain DSM 7534T. On the other hand, the isDDH value of C. septicum DSM 7534T compared to C. chauvoei genomes (DSM 7528Tand JF4335) was 28.8. The GC content variability between the C. septicum genomes to the reference genome ranged from 0.09 to 0.35 whereas to C. chauvoei genomes, it was 0.43 and 0.44 (Supplementary Table S2). Taken together, these results indicate a clear species delimitation of C. septicum and confirm the stable phylogenetic relationship of C. septicum with C. chauvoei.
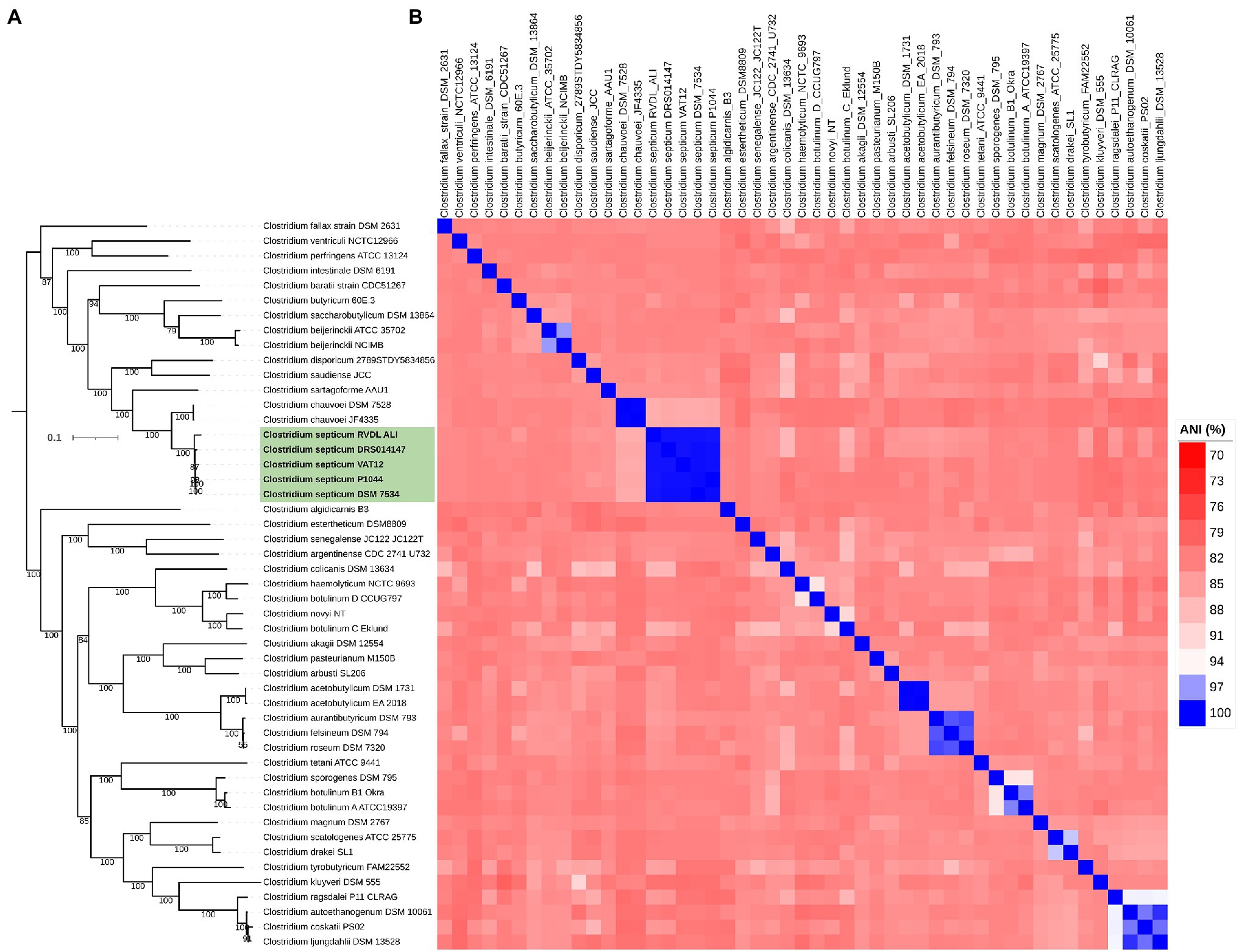
Figure 2. Taxonomic classification of C. septicum based on genome-wide approaches. Representative genomes of the Clostridium genus cluster 1 (Clostridium sensu stricto) were involved in the analysis. (A) Maximum likelihood tree based on PhyloPhlAn. (B) Pairwise average nucleotide identity (ANI) between the genomes.
CRISPR Regions
In strain DSM 7534T, we identified three CRISPR regions that were separated by a gene-encoding for IS256 family transposons. The CRISPR regions contained 28, 82, and 23 repeats, respectively, with the same repeat sequence (GTTTTATCTTAACTAGTGGAATGTAAAT). A CRISPR region containing 93 repeats was identified in the VAT 12 genome, while the draft genomes DRS014147 and RVDL_ALI carried 135 and 82 repeats, respectively. The CRISPR region of strain RVDL_ALI was split into two contigs. The genome P1044 harbored one CRISPR region with 79 repeats. Unique content of CRISPR spacer sequences was found in the all five genomes as depicted in Supplementary Figure S2. The repeat sequence (GTTTTATCTTAACTAGTGGAATGTAAAT) was present in all genomes.
Classification analysis of CRISPR-Cas systems revealed subtype I-B (Tneap–Hmari or CASS7) in all genomes, with Cas genes arranged as follow: cas2-cas1b-cas4-cas3-cas5b-cas7i-cas8a1-cas6 (Makarova et al., 2011).
Prophage Elements
Four different prophages (numbered 1 to 4) were predicted in the five genomes (Table 3, details Supplementary Table S3). The predicted prophages were less related to any reported prophages as indicted by the lower number and percentage values to first most common phage; 10.34% for Prophage 1, 16.66% for Prophage 2, 23.3% for Prophage 3, and 8% for Prophage 4. All prophages belonged to family Siphoviridae. Prophage 1 (47.7-kb) related to phiCD38-2/phiCD111/phiCD146 was present in all strains. Prophage vB_CpeS-CP51 (Prophage 2; 33.7-kb) was absent in VAT12. Prophage 3 (29.3-kb) related to phiCD6356 was present in the genome P1044 whereas prophage 4 (49.6-kb) related to Geobac_E2/Bacill_phIS3501/Coryne_StAB was shared among the genomes P1044 and RVDL_ALI. Schematic representation of prophages to C. septicum pangenome is depicted in Figure 3.
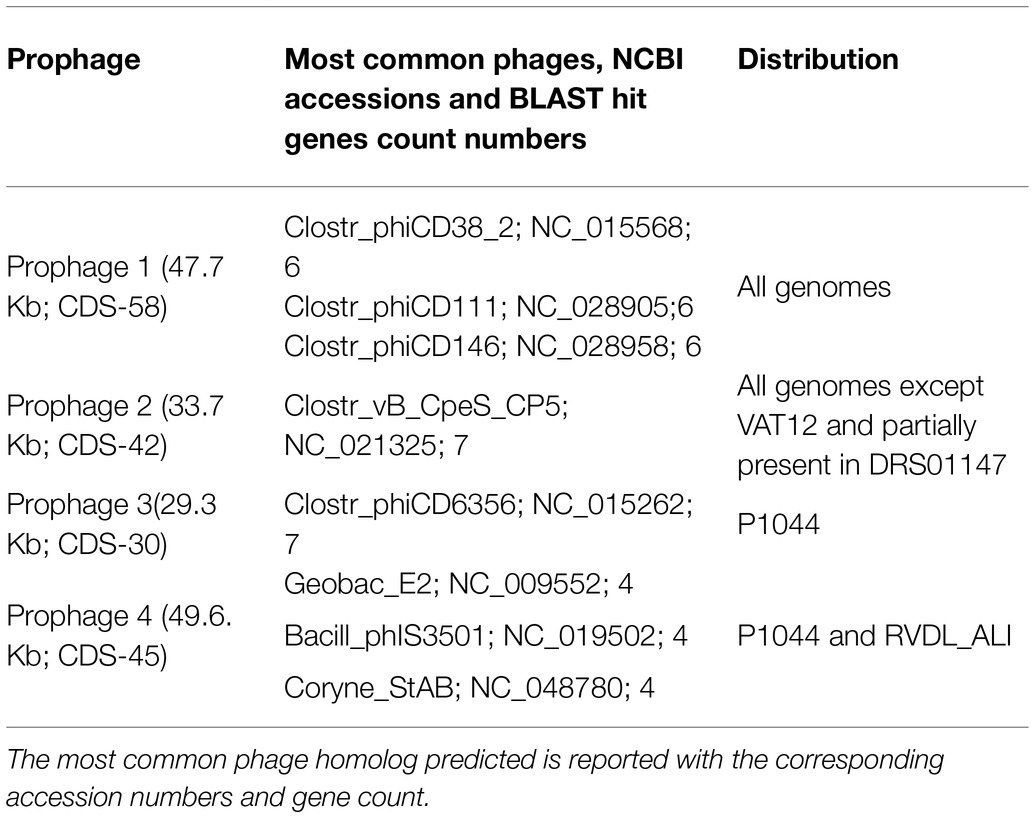
Table 3. Predicted prophages and their distribution across C. septicum genomes.
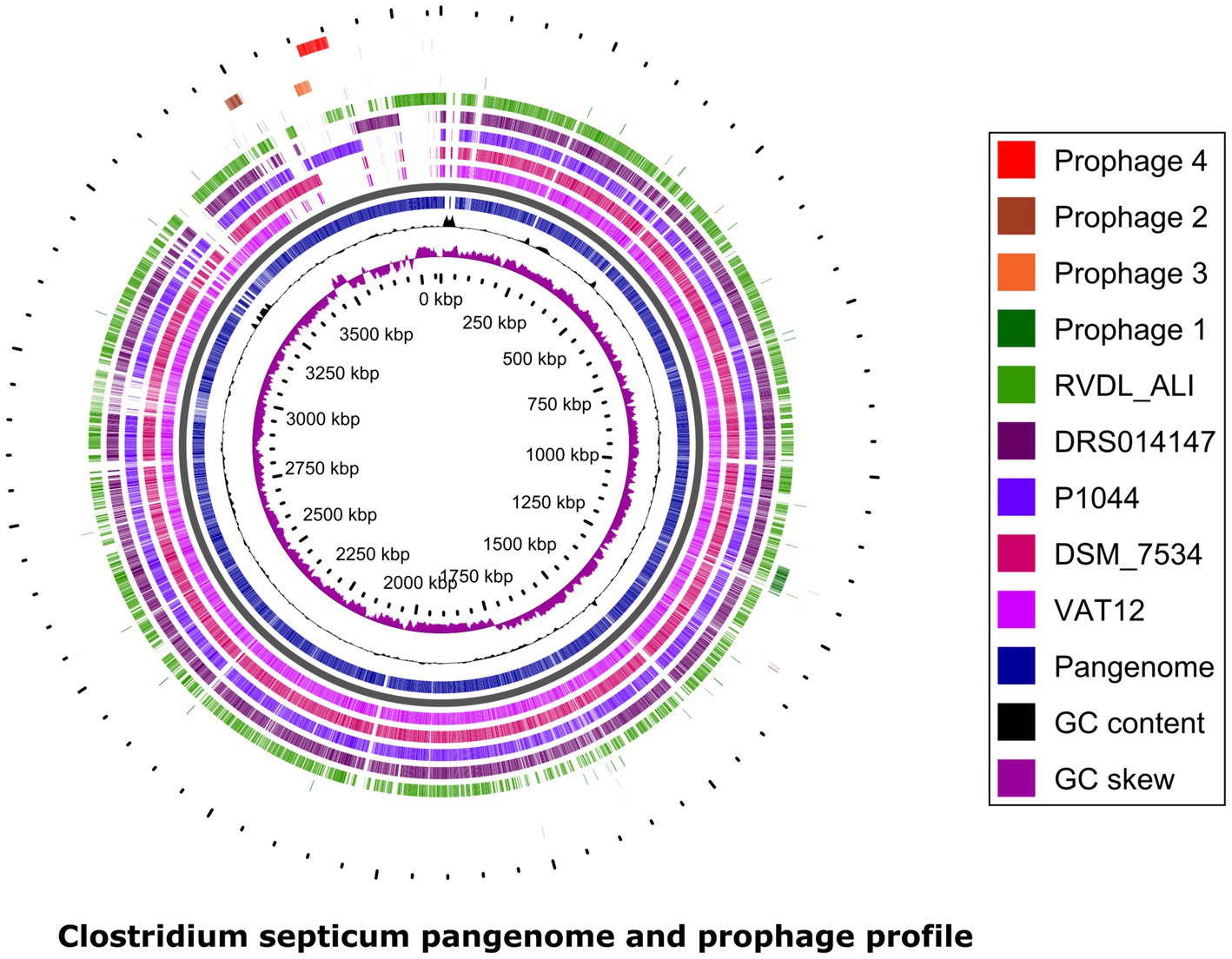
Figure 3. Clostridium septicum pangenome prophage profile. The pangenome plot depicts BLASTP relatedness among protein coding genes in five C. septicum genomes to species pangenome. The plot also represents the GC content and GCskew. Prophages (1–4) mapped to C. septicum pangenome shows the shared and unique presence of few phages in all strains or some strains, respectively. Pangenome prophage profile was created using GView Server.
Restriction-Modification (RM) Systems and Methylation Motifs
The complete genomes DSM 7534T and VAT 12 share putative type I, II, and IV RM systems whereas a putative type III system was additionally present in DSM 7534T strain. The type IV RM system encodes one restriction endonuclease (REase) as reported for the type IV RM systems for harboring only REases (Rusinov et al., 2015). The confirmed DNA methylations motifs of the identified RM systems were AAmGNNNNNRTmGAA and GTmATAmC for both genomes and AGAmGC for the C. septicum DSM 7534T genome (“Am” is the methylated base m6A; Table 4). The methylation motif summary analysis was carried out for the strain DSM 7534T (Table 5). The total number of modifications identified for the C. septicum DSM 7534T genome was 1,014,579 which included 66,811 4-methyl-cytosine (m4C), 14,780 6-methyl-adenine (m6A) and remaining were unidentified modifications types.
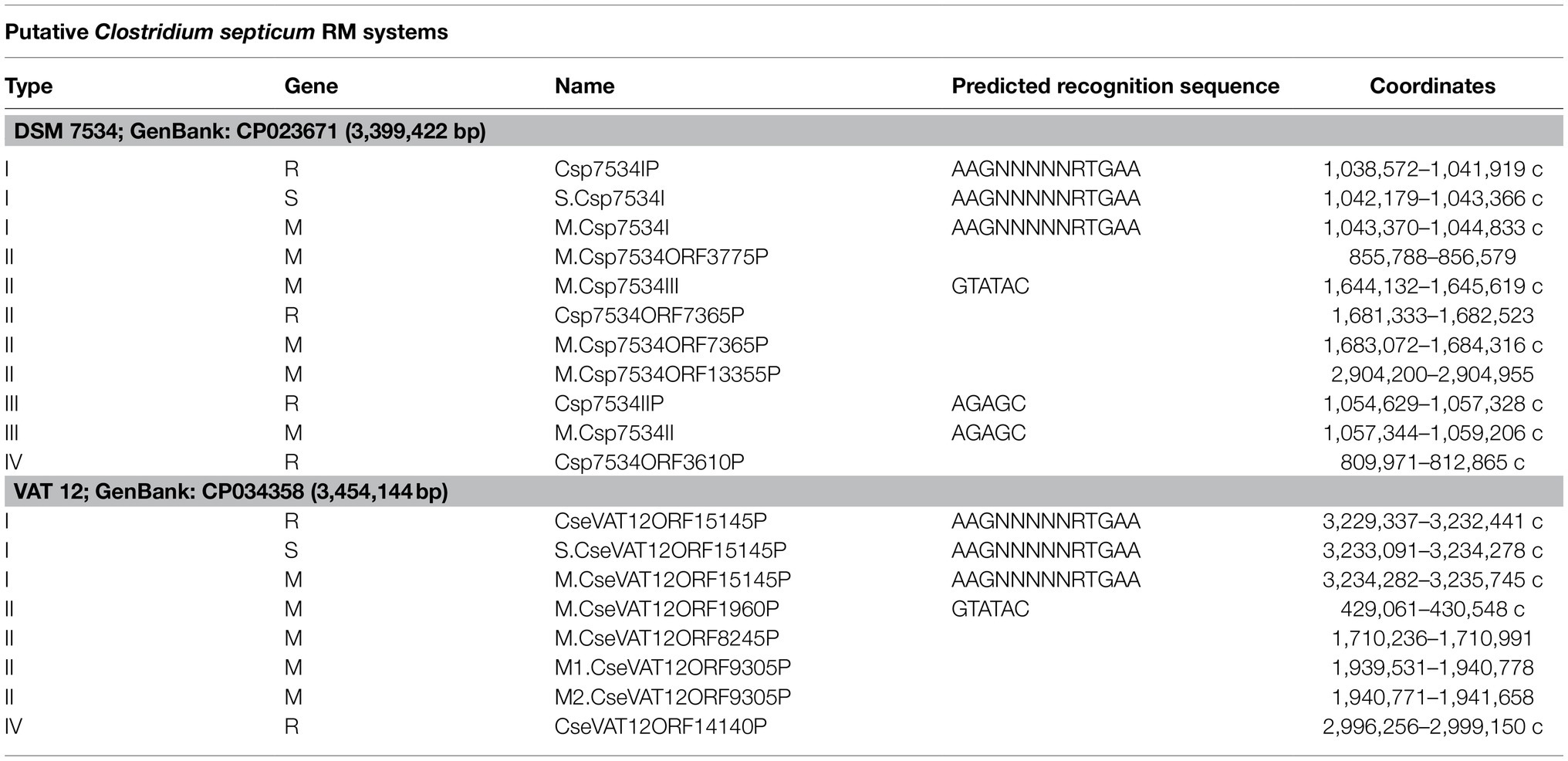
Table 4. Restriction-modification (RM) systems identified in C. septicum genomes RM systems involved, associated rrestriction endonucleases (REases) and methyltransferases (MTases), type/subtype information and recognition sequence they methylate, respectively, for DSM 7534T and VAT 12 are indicated.
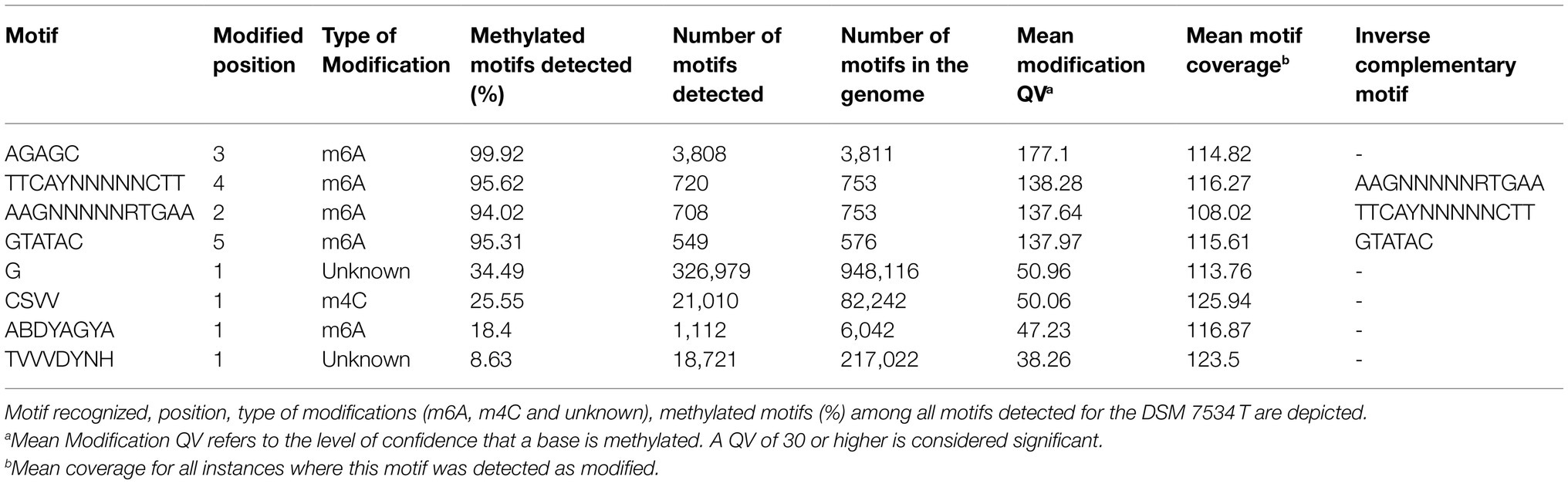
Table 5. Methylated motifs detected in C. septicum DSM 7534T genome.
Comparative Genomics and Phylogeny
Genome alignment of the five C. septicum genomes revealed 15–17 conserved local collinear blocks (LCBs; Figure 4A). In addition, genome alignment of C. septicum and C. chauvoei revealed conserved LCBs (Figure 4B). These LCBs indicate regions of conservation among genomes and unshared regions indicate unique genetic elements.
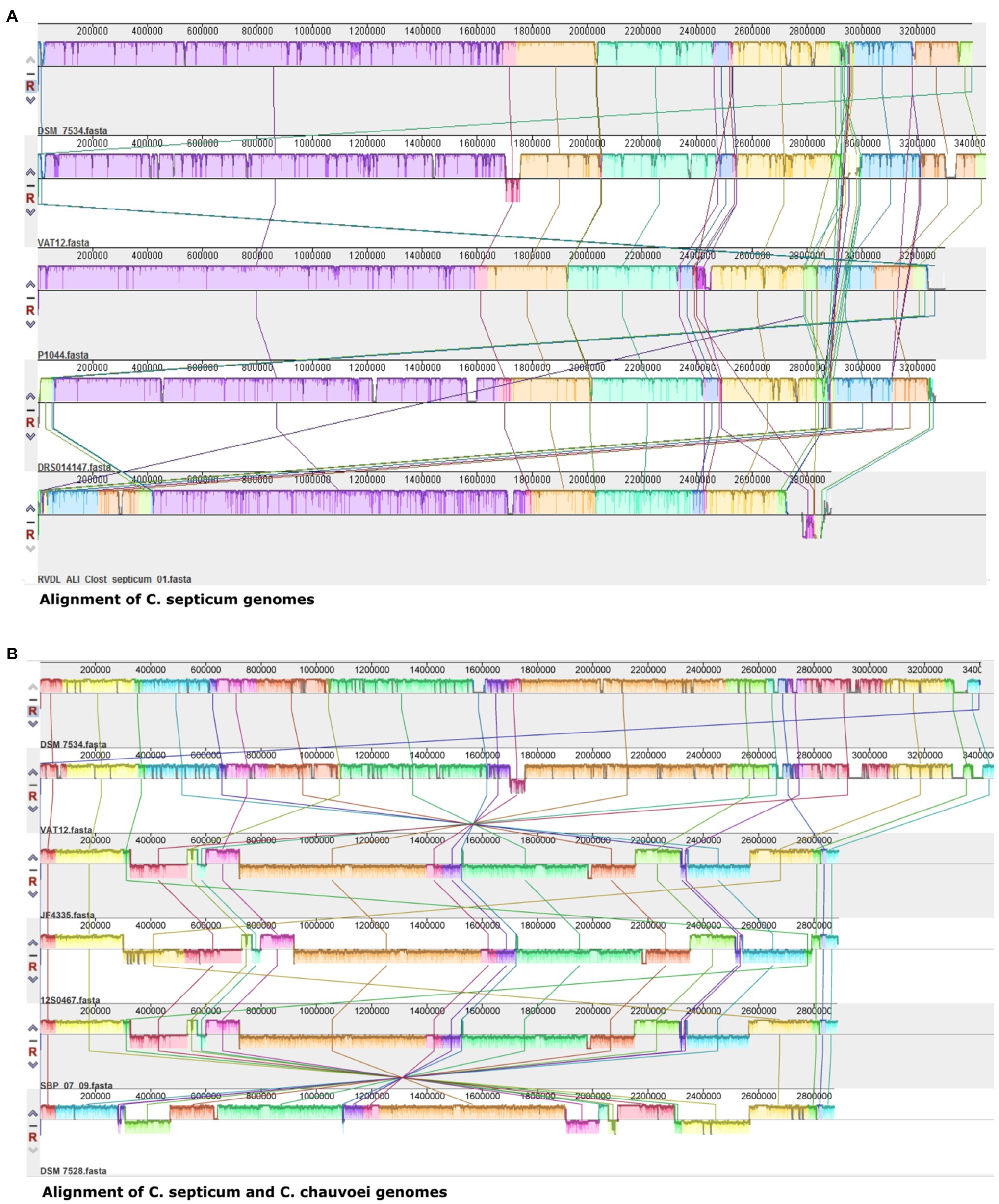
Figure 4. Multiple genome alignment with progressiveMauve shows the presence of locally collinear blocks (LCBs) among the genomes. (A) Alignment of all C. septium genomes. (B) Alignment of complete C. septium (DSM 7534T and VAT 12) and C. chauvoei (DSM 7528T, JF4335, 12S0467, and SBP 07/09) genomes. The sharing of LCBs indicates regions of conservation among genomes.
Orthologous gene clustering with Panaroo (Tonkin-Hill et al., 2020) predicted 3,740 genes, comprising the pangenome of the five genomes. Of these, 2,311 (core genome) were present in all genome datasets while 1,429 represented the accessory genome. The COG annotation indicated that most of the accessory genes belonged to the COG category representing function unknown (S; 215 genes) followed by replication, recombination and repair (L; 182 genes; Supplementary Figure S3). Some of the accessory genetic elements were also contributed by prophages (Figure 3). Of the core genome, 1,400 (61.1%) genes were assigned to a specific functional category in the KEGG database, of which 371 were associated with metabolic pathways. Further 197 proteins represent the processing of genetic information, followed by 172 proteins for carbohydrate metabolism. The list of 32 metabolic pathway modules (complete) representing different metabolic processes identified in the C. septicum core genome is shown in Supplementary Table S4. Of the accessory genes, only 317 genes (24.5%) were annotated using the KEGG database, including 61 proteins for signaling and cellular processes, 52 for genetic information processing, and 33 for environmental information processing. Fifty-three accessory genes were predicted to encode for metabolic pathways. Only one complete pathway module for central carbohydrate metabolism was predicted for accessory genome.
The pan-genome trajectory pattern showed a large expansion of the pangenome and a decrease in the core genomes. The fit for the pangenome profile curve using the power-law regression model resulted in B = 0.01 (r2 = 0.999), confirming the openness of the pangenome (Figure 5A). Core genome phylogeny based on 2,311 genes (2,234,611-bp) revealed 7,870 core variable positions in the alignment with 2,976 of them were parsimony informative. The genomes of strains DSM 7534T and VAT12 were also phylogenetically related with 100% bootstrap values, while the genomes of strains RVDL_ALI and DRS014147 were observed as singletons (Figure 5B). Pairwise SNP variations between the genomes ranged from 1,026 (DSM 7534T and VAT12) to 4,886 SNPs (DSM 7534T and RVDL_ALI; Figure 5C).
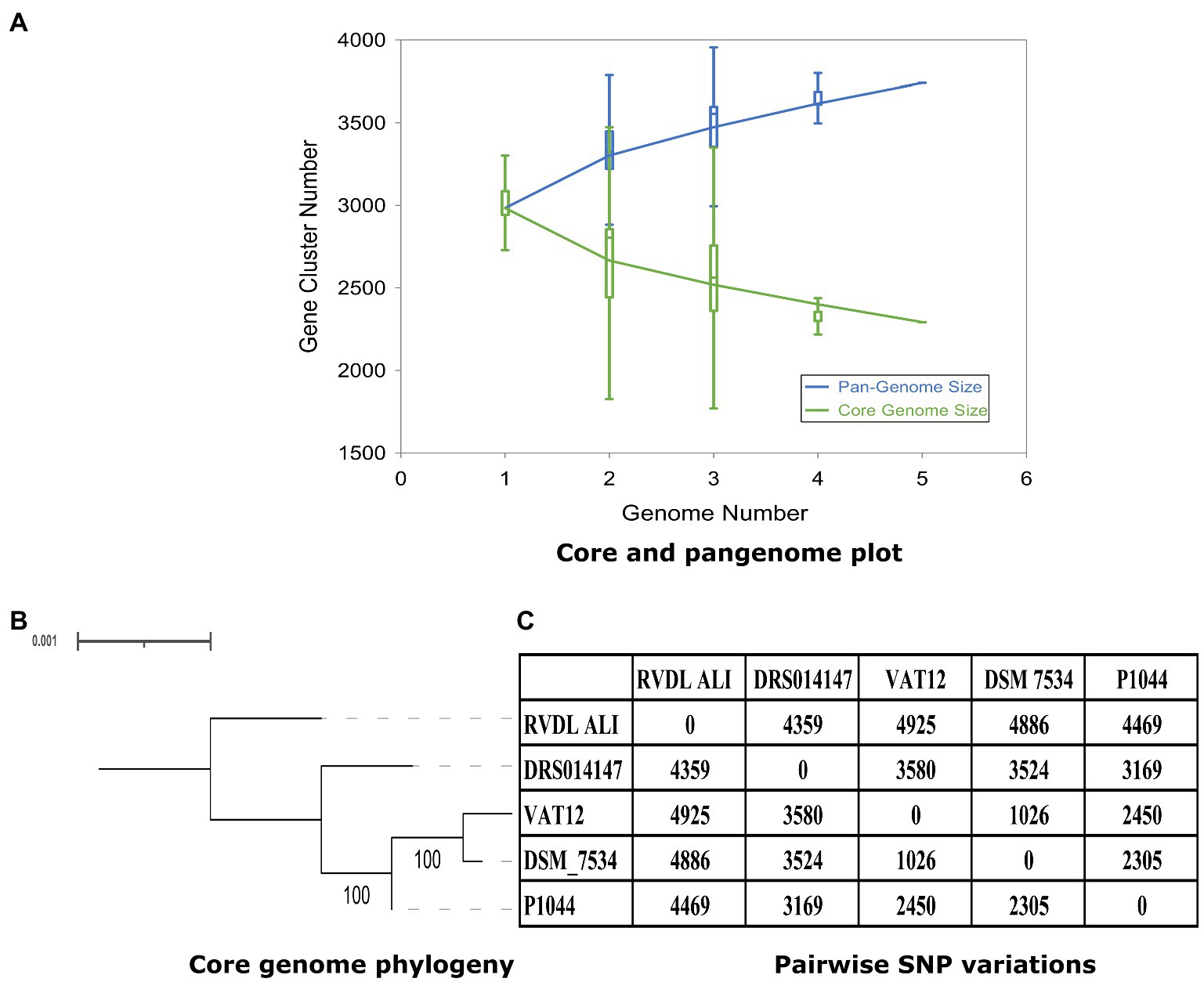
Figure 5. Pangenome, core genome plots and core genome phylogeny. (A) The pan-genome and core genome plot. Pangenome analysis indicated 2,311 and 1,429 core and accessory genes, respectively, and the pangenome profile indicated and open pangenome for C. septicum. (B) Core genome phylogeny based on 2,311 genes. (C) Pairwise SNP variations with in the core genome.
Virulence Genes
Clostridium septicum α-Toxin
The C. septicum α-toxin gene (csa) was present in all genomes and showed pairwise sequence identity of over 99.9%, with only a single synonymous SNP (T/C) observed at position 279. The genome RVDL_ALI had however four additional SNPs, two of which were nonsynonymous (Supplementary Figure S5A). To further investigate the sequence conservation of the csa-gene on a larger data set, we downloaded 29 csa sequences with complete CDS from NCBI. Sequence comparison revealed a sequence identity of more than 99% except for three strains with 97 to 98%. Phylogenetic analysis of the csa genes showed clustering of strain RVDL_ALI with two strains also isolated from cattle (Alhassa1 and Yamaguchi 6335). Strains DSM 7534T and VAT12 clustered with a strain from Japan (Tokachi). Strains DRS014147 and P1044 clustered with strains from Japan (Fukushima 5) and China (AY829447.1; Supplementary Figure S4).
Clostridium septicum Novel Toxin Homolog
A gene encoding a novel toxin homolog was identified in all C. septicum genomes. The predicted protein contains a leukocidin/hemolysin domain similar to beta-channel forming cytolysin reported for other Clostridium species. In particular, it shares 71.10% aa identity with C. chauvoei toxin A (CctA; Table 6) that is a potent cytolysin involved in C. chauvoei virulence in blackleg disease in cattle and sheep (Frey and Falquet, 2015). The predicted protein also has the exact same size as CctA (317 aa). In addition, phylogenetic analysis with several pore-forming toxins revealed a closer relationship to CctA, with 100% bootstrap support, than to other Clostridium pore-forming toxins, including C. perfringens beta, NetB, NetE, NetG, NetF, and delta toxins (Figure 6B). We designated the identified homolog, Clostridium septicum toxin A (CstA). Sequence analysis of CstA-encoding gene (cstA) revealed 100% sequence identity in all C. septicum genomes.
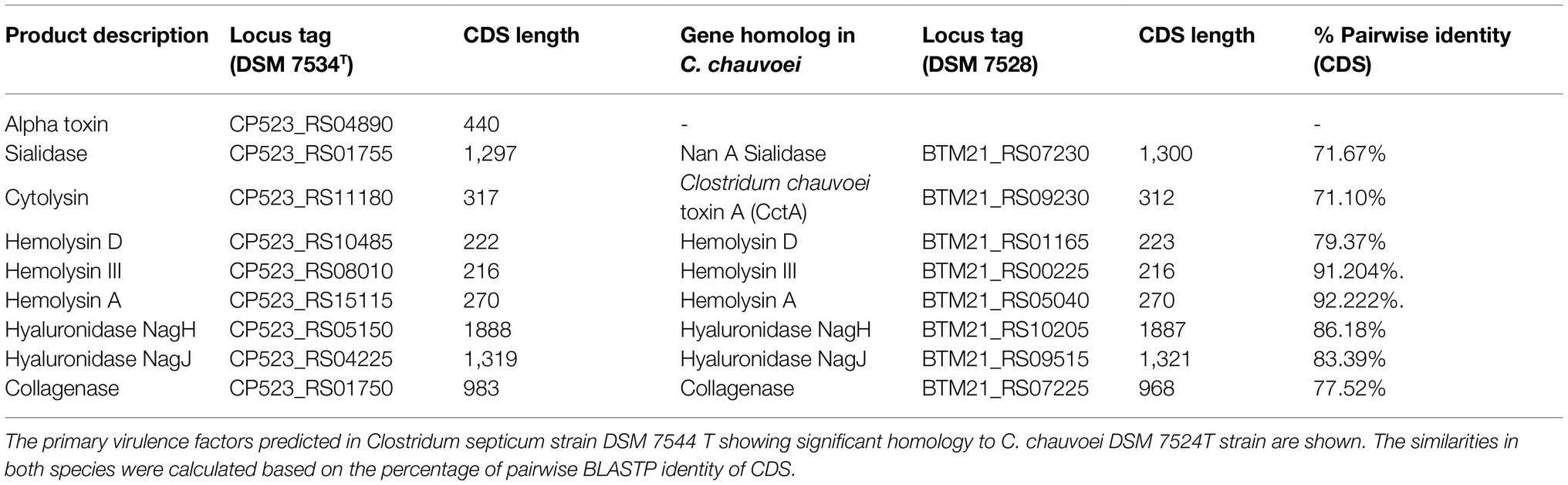
Table 6. Virulence factors predicted for Clostridium septicum strains.
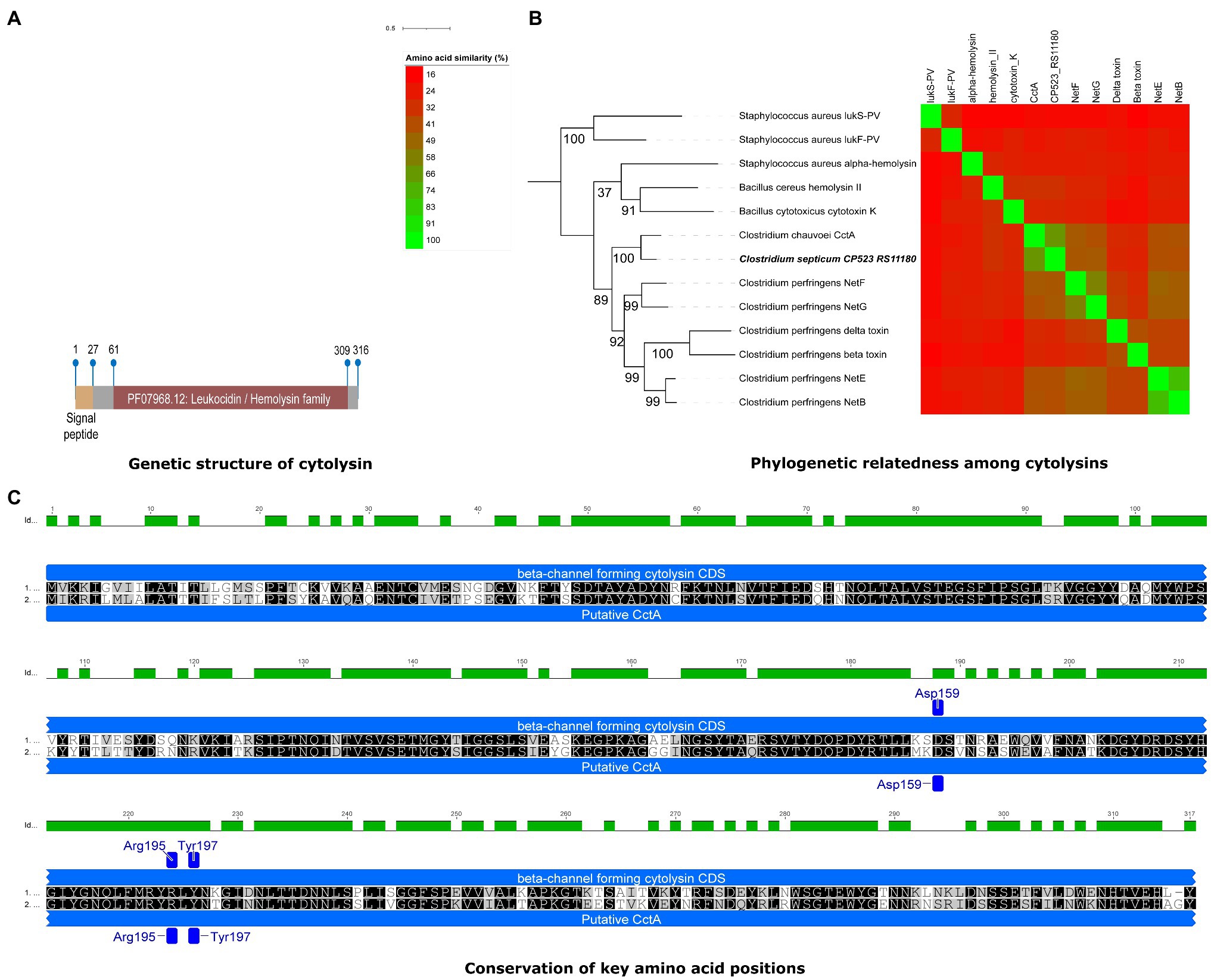
Figure 6. Novel cytolysin identified in C. septicum genomes. (A) Genetic structure of novel cytolysin identified in C. septicum with respect to genetic structure such as signal peptide and Leucocidin/Hemolysin toxin domain. (B) Phylogenetic relatedness of cytolysin to C. chauvoei CctA (100% boostrap support value) and other pore forming toxins in related species. (C) Conservation of key amino acid positions in C. septicum cytolysin (upper) with C. chauvoei CctA (lower) reported for virulence.
The CstA protein sequences include a signal peptide region with 27 aa residues (Sec/SPI) followed by a leucocidin/hemolysin domain (aa position 69 to 309). The cleavage site A of signal peptidase I (Tat/SPI) was between nucleotide positions 27 and 28 (ATA-TT; Figure 6A). Three conserved aa residues (Asp159, Arg195, and Tyr197) reported by Frey et al. (2012) in the mature protein of C. chauvoei CctA were also identified in CstA. These aa residues are important for membrane specificity and activity of the pore-forming toxins, C. perfringens NetB and beta toxin, and S. aureus alpha toxins (Song et al., 1996; Steinthorsdottir et al., 1998; Nagahama et al., 1999). The conserved aa positions for the predicted CstA might therefore indicate similar pore-forming activity (Figure 6B).
ABCpred server predicted 17 and 19 linear epitopes for the CctA and CstA, respectively. Considering the score of predicted epitopes (indicative of epitope potency), CstA was predicted with more epitopes with the highest scores (three epitopes with 0.89 score) as compared to CctA (1 epitope with 0.89 score). Also, the cumulative total score when all epitopes above cutoff value 0.75 are considered was higher for CstA (total score 15.53) as compared to CctA (total score 13.92; Supplementary Table S5).
Clostridium septicum Sialidases
Genes encoding sialidases were identified in the C. septicum strains (Table 6). The C. septicum sialidase gene encodes 1,296 aa (EC 3.2.1.18), exhibiting 71.6% aa identity to NanA sialidase of C. chauvoei. The predicted protein sequence of sialidase was conserved; three aa variations in the genome DRS014147, one aa variation for RVDL_ALI. The latter had also a premature stop codon, resulting in a shorter predicted protein (1,237 aa; Supplementary Figure S5B).
Clostridium septicum Hemolysin
Three genes (locus tags CP523_RS10485, CP523_RS08010, and CP523_RS15115) encoding hemolysin proteins were identified in the C. septicum genomes (Table 6). CP523_RS10485 was similar to the C. chauvoei hemolysin D (79.3% aa identity; Frey and Falquet, 2015), CP523_RS08010 was similar to C. chauvoei hemolysin III (91.2% aa identity), and CP523_RS15115 was similar to C. chauvoei hemolysin A (FtsJ RNA methyltransferase; 92.2% aa identity; Frey and Falquet, 2015). The C. septicum hemolysin genes were detected in all five genomes with 100% sequence identity, except for the hemolysin A gene in the RVDL_ALI genome, which showed one aa variation (Supplementary Figure S5C).
In addition, a gene (CP523_RS13325) with very limited homology (16% at aa level) to C. chauvoei hemolysin XhlA gene (Frey and Falquet, 2015) was identified in four C. septicum genomes showing 100% identity at aa level.
Clostridium septicum Hyaluronidases
Clostridium septicum genomes harbored homologs for hyaluronidases NagH and NagJ reported in C. chauvoei (Frey and Falquet, 2015). The hyaluronidases genes were identified in the five C. septicum genomes with a pairwise aa sequence identity of 99.6 and 99.7% for NagH and NagJ, respectively Supplementary Figures S5D,E.
Clostridium septicum Collagenase
A gene coding for collagenase (EC 3.4.24.3) was predicted in C. septicum genomes with identical aa sequences except for RVDL_ALI genome which had a short collagenase gene.
Clostridium septicum Flagellin
The genome sequence of C. septicum DSM 7534T and P1044 revealed four complete flagellin (fliC) genes, and a similar gene presence with reduced homology was also seen in the other strains. The four fliC genes showed a considerable homology of 95.4% pairwise identity at aa level and had a central variable region.
Discussion
Clostridial gas gangrene (myonecrosis) is a lethal infection that affects a wide range of hosts with the involvement of different pathogenic species, which may influence the symptoms and clinical course of the disease. Potent exotoxins released by these pathogens in the host tissues are thought to be a major contributor to the observed pathogenesis (Stevens et al., 2012; Carter et al., 2014; Oda et al., 2015; Yamamura et al., 2019). Clostridium perfringens and C. septicum are the most commonly isolated organisms in these infections. However, unlike C. perfringens, detailed genomic characterization of C. septicum has not been yet performed. The study reports the first complete sequence of the C. septicum type strain (DSM 7534T). In addition, a comparative analysis involving all available C. septicum genomes (n = 5) was performed in order to decipher genomic and virulence aspects that are possibly associated with the development of gas gangrene in humans and animals. The size of the circularized C. septicum genomes (3.3–3.4-Mb) was similar to C. perfringens (3–3.5-Mb) but slightly larger than the phylogenetically related species, C. chauvoei (2.8-Mb; Shimizu et al., 2002; Falquet et al., 2013; Rychener et al., 2017; Table 2). The DSM 7534T genome also harbored a small plasmid (5.2-kb) similar to that reported for C. chauvoei (4.1-kb; Falquet et al., 2013; Thomas et al., 2017). The plasmid also encoded a viroplasmin family protein and Teicoplanin resistance protein (vanZ; Figure 1). Previous studies have reported RNase H1/viroplasmin domain-containing protein associated with Caulimovirus (Volovitch et al., 1990), whereas the role of the similar protein in bacteria is not reported so far. The vanZ protein is associated with teicoplanin resistance and the gene orthologs have been reported from several bacterial genera (Vimberg et al., 2020). A previous study has shown clinical resistance to vancomycin in two strains recovered from clinical cases (Aldape et al., 2018). The vancomycin resistance cassette was also reported to be encoded on a 5-kb plasmid (Aldape et al., 2018). The present study also shows the existence of vanZ protein gene on a 5-kb plasmid similar to the report.
Genome sequencing provides a better resolution for depicting phylogenetic and taxonomic relatedness of bacterial genomes compared to 16S rRNA sequence analysis. The taxonomical classification of C. septicum was congruent between genome-based approaches and previous reports using 16S rRNA sequences (Kuhnert et al., 1996). The results indicate a clear species delimitation of C. septicum and confirm its phylogenetic relationship with C. chauvoei, as previously reported (Kuhnert et al., 1996).
C. septicum had CRISPR elements present in all genomes. This was similar to C. chauvoei, but different to C. perfringens, where studies showed the absence of CRISPR regions in many strains (Long et al., 2019). The CRISPR repeat arrays were interrupted by two mobile elements in strain DSM 7534T, which was also found in C. chavuoei (Rychener et al., 2017). Interestingly, the C. septicum genomes also showed genetic variability for the CRISPR array and spacers. This heterogeneity of CRISPR spacer sequences may provide a basis for genotyping C. septicum strains, as previously shown for related pathogen such as C. chauvoei (Rychener et al., 2017) and C. difficile (Andersen et al., 2016). Indeed, the diversity of C. septicum CRISPR spacer arrays showed a good correlation with the core genome phylogeny (Supplementary Figure S2). Hence, a CRISPR spacer-based typing approach may be another option for strain typing of this species. However, to achieve this, sequence data will need to be generated from more strains obtained under different geographic, host, and disease conditions. These will provide further insight into genetic diversity and could facilitate the identification of specific host and disease associations in C. septicum.
C. septicum genomes were predicted to encode four prophages (Table 3). Prophage 1 and 3 matched to phiCD38-2/phiCD111/phiCD146 and phiCD6356 phages respectively, reported earlier from C. difficle (Fortier and Moineau, 2007; Sekulovic et al., 2014). On the other hand, prophages (Geobac_E2/Bacill_phIS3501/Coryne_StAB) identical to prophage 4 are also reported in the genus Clostridium (Smith et al., 2021) as well as in other species such as Shigella (Muthuirulandi Sethuvel et al., 2019) and Corynebacterium (NC_048780). Phage vB_CpeS-CP51 (prophage 2), has been initially recognized as a temperate bacteriophage of C. perfringens (Gervasi et al., 2013) and was later reported also in C. chauvoei (Frey and Falquet, 2015). The presence of relatively higher numbers of prophages compared to C. chauvoei (Thomas et al., 2017), as well as the presence of CRISPR elements in all C. septicum genomes analyzed, likely indicates limited functionality of CRISPR elements in phage inhibition. This is however similar to C. perfringens, in which no direct correlation was found between the prophage frequencies and the absence or presence of CRISPR elements (Abdel-Glil et al., 2021).
RM systems include a restriction endonuclease (REase) and a modification methyltransferase (MTase). The REase degrades DNA from any exogenous source, whereas the MTase methylates the host REase target sites in the genome and thus protects them from cleavage (Blow et al., 2016). The RM system detected for the two circularized genomes showed variations with respect to the RM system types they carried (Table 4). Highly variable RM systems were identified in 302 environmental and outbreak-associated Listeria monocytogenes strains, with type II followed by type I and other types (III and IV) predominating, indicating a wide diversity of RM systems even within the same species (Chen et al., 2017). The RM system of C. chauvoei was conserved among three strains and included type I, II, and IV systems (http://rebase.neb.com/rebase/rebase.html; assessed April 14, 2021). In contrast, RM systems in C. perfringens strains (n = 62 strains) were diverse, ranging from zero to all combinations of RM systems (Type I to IV systems).7 The characterized C. septicum strain (DSM 7534T) harbored methyltransferase for m6A methylation. In addition, an absence of m5C methylation was observed in C. septicum (Table 5), reflecting either the true absence or the lower sensitivity of SMRT sequencing to detect m5C methylation (Murray et al., 2012).
Pangenome analysis based on orthologous gene clustering for the five genomes revealed an open pangenome, with the accessory genome comprising 1,429 genes (Figure 5). Most of the accessory genes belonged to the replication, recombination and repair category or to the unknown function category in COG annotation (Supplementary Figure S3). This suggests a role of horizontal genetic transfers in shaping the composition of the accessory genome of C. septicum. Similar results were reported for C. perfringens, wherein 849 genes within the accessory genome in contrast to 79 genes within the core genome belonged to replication, recombination, and repair mechanism (Kiu et al., 2017). A comparative genomic study of 206 genomes of C. perfringens strains showed that the major phylogroups exhibit a collinear distribution pattern of accessory genes, indicating the possible relevance of specific accessory genes in bacterial specialization (Abdel-Glil et al., 2021). The accessory genetic elements in C. septicum are resembling in frequency and COG category those in the genomes of C. perfringens. This contrasts with the limited accessory genetic variability found in C. chauvoei, the species most closely phylogenetically related to C. septicum (Rychener et al., 2017; Thomas et al., 2017). All core metabolic pathways with respect to energy, carbohydrate, amino acids, nucleotides and cofactor, and vitamins were represented in the core genome (Supplementary Table S4). Similar presence of high number of genes involved in carbohydrate metabolism was represented in the core genome of Clostridium butyricum (Zou et al., 2021).
The Clostridium septicum alpha toxin (csa) gene was mostly conserved in the analyzed five genomes, but variations were observable with other published csa genes (Supplementary Figure S4). A previous study reported seven unique patterns in the deduced α-toxin aa sequences of 25 C. septicum strains. The same study also reported unique insertion, altered stop codon position, and deletion of 9-bp for the involved strains (Amimoto et al., 2006). In the present study, similarly, a higher genetic variation was found between strains for α-toxin.
Besides the α-toxin, a novel putative cytolysin gene, Clostridium septicum toxin A (CstA) present in all C. septicum genomes was identified. It showed 71.1% aa identity to the CctA gene of C. chauvoei, predicted to encode a similar signal peptide and Leucocidin/Hemolysin toxin domains (Figure 6). Strong conservation of the CctA gene between C. chauvoei strains has been reported (Rychener et al., 2017), and the genomes of the current study also showed similar conservation (100%) for the putative cytolysin gene in C. septicum. Previous studies have demonstrated CctA as one of the main protective antigens in vaccines against blackleg (Frey et al., 2012), and a recent study reported the production of neutralizing antibodies against CctA following the blackleg vaccination (Nicholson et al., 2019). The presence of analogous potent B-cell epitopes predicted for the C. septicum CstA (Supplementary Table S5) suggests that it is suitable as another potent vaccine target candidate for clostridial infections, in addition to known toxins such as α-toxin (Haghroosta et al., 2020). It must be noted that a toxin known as septicolysin (delta-toxin) belong to cholesterol-dependent cytolysin (CDC) family has been proposed to have a synergistic effect with α-toxin in the pathogenesis of C. septicum infection (Popoff, 2016). Septicolysin associated hemolytic activity was reported to be less than 5% of the total detectable hemolytic activity and the remaining was attributed to α-toxin in C. septicum (Ballard et al., 1992). Based on a published Clostridium septicum partial septicolysin (spl) gene sequence (AJ539084.1), we did a BLASTN query with all five C. septicum CDSs. There were no homologous counterparts identified within the strains with high identity. This spl gene had 78.7% nucleotide identity with another gene encoding for an amino acid permease present in all strains (Locus tag: CP523_RS09335 for DSM 7534T). Interestingly, the spl gene sequence is 53.1% similar to the identified CstA indicating that this novel cytolysin homolog (CstA) is divergent from the C. septicum streptolysin (delta toxin) previously reported.
Between the species C. chauvoei and C. septicum, both cytolysin and sialidase homologs showed reduced similarity (<75%), whereas the hyaluronidase genes (NagH and NagJ), hemolysins and collagenase showed higher similarity (Table 6). This suggests that speciation likely had a smaller effect on genetic divergence of other virulence genes than for cytolysin and sialidase genes. Among the virulence factors identified in the genomes of both species, the predominant classes were sialdiases, hyaluronidases, hemolysins, leucocidin, and aerolysin family toxins (CctA in the case of C. chauvoei and CstA and α-toxin in the case of C. septicum), as well as collagenase and internalin. With the known and predicted virulence factors, C. septicum was unique to harbor the α-toxin gene as compared to C. chauvoei. In summary, our analysis revealed a core set of putative virulence-related genes likely involved in C. septicum disease progression, as most of the identified genes encode enzymes that act extracellularly, and some of these homologs, particularly CctA, have been significantly associated with blackleg pathogenesis in cattle.
Conclusion
To conclude, this is the first comparative genomic study for the C. septicum species using five genomes. Although our analysis included all publicly-available genomes of C. septicum in NCBI, the small number of available genomes may have made it difficult to portrait the overall population diversity of the species, which may be considered a limitation of our analysis. Nevertheless, the analysis described here represents a starting point for understanding the genomic variation of C. septicum that is a relevant human and animal pathogen. The high fatality rate of diseases caused by this bacterium will motivate further studies to sequence its genome and more deeply investigate its genetic properties. The analysis presented here will be useful for future studies in several ways. First, the taxonomic classifications based on 16S rRNA and genome-wide approaches reconfirm the phylogenetic proximity to C. chauvoei. Second, unlike C. chauvoei, a less variable species, the genomes of C. septicum strains exhibited high genetic diversity in terms of prophages, CRISPR spacers, RM systems, accessory genomes, and core genome SNPs, possibly due to the diverse lifestyle and broad host range. Third, C. septicum encodes several virulence factors that are likely to be involved in the clinical course of C. septicum disease. These virulence factors were genetically most closely related to those of C. chauvoei. Most importantly, a novel toxin homolog CstA was identified that resembled the C. chauvoei key virulence factor CctA. The role of the virulence factors predicted in this study needs further investigation in in vitro and in vivo models to understand the disease and pathogenesis occurring during C. septicum infections in animals and humans.
Data Availability Statement
The original contributions presented in the study are publicly available. The sequence data generated for this study can be found in the National Center for Biotechnology Information Reference Sequences (RefSeq) using accession numbers, NZ_CP023671.1 (chromosome) and NZ_CP023672.1 (plasmid; https://www.ncbi.nlm.nih.gov/bioproject/412368).
Author Contributions
PT and MA-G conceptualized and designed the study, performed the bioinformatic analysis, and wrote the manuscript. AS and AB provided support for the bioinformatic analysis. IE performed the next-generation sequencing. LW, HN, and MP supervised the study. CS conceptualized and supervised the study and critically revised and improved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
AB was supported by the Deutsche Forschungsgemeinschaft (German Research Foundation) under Germany’s Excellence Strategy (EXC 2051, project 390713860).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Sandra Pfeifer, Jutta Carmon, and Renate Danner for their excellent technical assistance. The international fellowship from Indian Council of Agricultural Research (ICAR), New Delhi, India, for Prasad Thomas is gratefully acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.771945/full#supplementary-material
Supplementary Figure S1 | Taxonomic classification of C. septicum based on 16srRNA gene. The 16S rRNA gene based phylogenetic analysis of C. septicum indicates close relatedness with C. chauvoei within Clostridium genus cluster 1 (Clostridium sensu stricto).
Supplementary Figure S2 | Diversity and structure of CRISPR regions and CRISPR spacers. CRISPR spacers present among strains are represented in the form of an array. (A) Every unique spacer present within any strain is represented with different color codes. (B). Spacers that are shared among strains are represented with same color codes and unshared spacers are represented in gray. The three CRISPR regions in the type strain DSM 7534T intervened by IS256 family transposons are indicated as white space.
Supplementary Figure S3 | Clusters of Orthologous Groups (COGs) categorization of core and accessory genes. Functional annotation of the core and accessory genes were carried out for COGs. Most of the accessory were belonging to the category of representing function unknown (S) and replication, recombination and repair (L).
Supplementary Figure S4 | Phylogenetic tree involving C. septicum alpha toxin cds at nucleotide level. Phylogenetic relatedness of alpha toxin gene (full CDS) among C. speticum strains from diverse host/source and geographical sources was inferred.
Supplementary Figure S5 | Protein alignment of Clostridium septicum virulence factors. Alignment of primary virulence factors showing amino acid variations are shown in Figure S5. (A) Alpha toxin, (B) Sialidase, (C) Hemolysin A, (D), Hyaluronidase NagJ and (E) Hyaluronidase NagH.
Supplementary Table S1 | Metadata summary of sequences retrieved from NCBI. Details with respect to species, strain name, and NCBI accession numbers of species of Clostridium sensu stricto used for 16S rRNA phylogenetic analysis are shown.
Supplementary Table S2 | Taxonomic classification of C. septicum based on in silico DNA-DNA hybridization.
Supplementary Table S3 | Prophages identified in C. septicum genomes. The prophages identified in five C. septicum strains indicating the size in bases, score predictions based on PHASTER, protein coding genes and BLAST identity are given.
Supplementary Table S4 | KEGG pathway modules identified in the core genome of C. septicum.
Supplementary Table S5 | Predicted B-cell epitopes in C. chauvoei toxin A (CctA) and in the identified homolog, C. septicum toxin A (CstA) identified by ABCpred server.
Footnotes
1. ^https://github.com/tseemann/shovill
2. ^https://github.com/tseemann/barrnap
5. ^https://github.com/tseemann/abricate
6. ^https://github.com/tseemann/snp-dists
7. ^http://rebase.neb.com/rebase/rebase.html (Assessed April 14, 2021).
References
Abdel-Glil, M. Y., Thomas, P., Linde, J., Busch, A., Wieler, L. H., Neubauer, H., et al. (2021). Comparative in silico genome analysis of Clostridium perfringens unravels stable phylogroups with different genome characteristics and pathogenic potential. Sci. Rep. 11, 6756. doi: 10.1038/s41598-021-86148-8
Afgan, E., Baker, D., Batut, B., Van Den Beek, M., Bouvier, D., Cech, M., et al. (2018). The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. doi: 10.1093/nar/gky379
Aldape, M. J., Bayer, C. R., Rice, S. N., Bryant, A. E., and Stevens, D. L. (2018). Comparative efficacy of antibiotics in treating experimental Clostridium septicum infection. Int. J. Antimicrob. Agents 52, 469–473. doi: 10.1016/j.ijantimicag.2018.07.009
Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/s41587-019-0036-z
Almeida E Macêdo, J. T. S., Pires, P. S., Pinheiro, E. E. G., Oliveira, R. S. D., Silva, R. O. S., Lobato, F. C. F., et al. (2013). Malignant edema caused by Clostridium chauvoei in a horse. Acta Sci. Vet. 41:24
Almeida, A., Nayfach, S., Boland, M., Strozzi, F., Beracochea, M., Shi, Z. J., et al. (2021). A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114. doi: 10.1038/s41587-020-0603-3
Amimoto, K., Sasaki, Y., Fukuyama, S., and Tamura, Y. (2006). Genetic variation and cross-reactivity of Clostridium septicum alpha-toxin. Vet Microbiol 114, 51–59. doi: 10.1016/j.vetmic.2005.10.039
Andersen, J. M., Shoup, M., Robinson, C., Britton, R., Olsen, K. E., and Barrangou, R. (2016). CRISPR Diversity and Microevolution in Clostridium difficile. Genome Biol. Evol. 8, 2841–2855. doi: 10.1093/gbe/evw203
Annapureddy, N., Agarwal, S. K., Kanakadandi, V., Sabharwal, M. S., Ammakkanavar, N., Simoes, P., et al. (2012). Clostridium septicum aortitis in a patient with extensive atheromatous disease of the aorta. J. Infect. Chemother. 18, 948–950. doi: 10.1007/s10156-012-0394-7
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Auch, A. F., von Jan, M., Klenk, H.-P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Ballard, J., Bryant, A., Stevens, D., and Tweten, R. K. (1992). Purification and characterization of the lethal toxin (alpha-toxin) of Clostridium septicum. Infect. Immun. 60, 784–790. doi: 10.1128/iai.60.3.784-790.1992
Barnes, C., Gerstle, J. T., Freedman, M. H., and Carcao, M. D. (2004). Clostridium septicum myonecrosis in congenital neutropenia. Pediatrics 114, e757–e760. doi: 10.1542/peds.2004-0124
Benamar, S., Cassir, N., Caputo, A., Cadoret, F., and La Scola, B. (2016). Complete genome sequence of Clostridium septicum strain CSUR P1044, isolated from the human gut microbiota. Genome Announc 4, e00922–e00916. doi: 10.1128/genomeA.00922-16
Bernheimer, A. W. (1944). Parallelism in the lethal and hemolytic activity of the toxin of Clostridium septicum. J. Exp. Med. 80, 309–320. doi: 10.1084/jem.80.4.309
Biswas, A., Staals, R. H., Morales, S. E., Fineran, P. C., and Brown, C. M. (2016). CRISPRDetect: A flexible algorithm to define CRISPR arrays. BMC Genomics 17:356. doi: 10.1186/s12864-016-2627-0
Blow, M. J., Clark, T. A., Daum, C. G., Deutschbauer, A. M., Fomenkov, A., Fries, R., et al. (2016). The Epigenomic landscape of prokaryotes. PLoS Genet. 12:e1005854. doi: 10.1371/journal.pgen.1005854
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. doi: 10.1186/1471-2105-10-421
Carter, G. P., Cheung, J. K., Larcombe, S., and Lyras, D. (2014). Regulation of toxin production in the pathogenic clostridia. Mol. Microbiol. 91, 221–231. doi: 10.1111/mmi.12469
Chakravorty, A., Awad, M. M., Cheung, J. K., Hiscox, T. J., Lyras, D., and Rood, J. I. (2015). The pore-forming α-toxin from Clostridium septicum activates the MAPK pathway in a Ras-c-Raf-dependent and independent manner. Toxins 7, 516–534. doi: 10.3390/toxins7020516
Chen, P., Den Bakker, H. C., Korlach, J., Kong, N., Storey, D. B., Paxinos, E. E., et al. (2017). Comparative genomics reveals the diversity of restriction-modification systems and DNA methylation sites in Listeria monocytogenes. Appl. Environ. Microbiol. 83, e02091–e02016. doi: 10.1128/AEM.02091-16
Clark, T. A., Murray, I. A., Morgan, R. D., Kislyuk, A. O., Spittle, K. E., Boitano, M., et al. (2012). Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 40:e29. doi: 10.1093/nar/gkr1146
Curtis-Martínez, C., and Sánchez-Guillén, L. (2019). Emphysematous Aortitis by Clostridium septicum: A rare and lethal complication of right colon cancer. Eur. J. Vasc. Endovasc. Surg. 57:509. doi: 10.1016/j.ejvs.2018.12.022
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Dion, M. B., Labrie, S. J., Shah, S. A., and Moineau, S. (2018). CRISPRStudio: a user-friendly software for rapid CRISPR Array visualization. Viruses 10:602. doi: 10.3390/v10110602
Eplinius, F., and Hädrich, C. (2014). Acute aortic dissection caused by Clostridium septicum aortitis. Forensic Sci. Int. 244, e38–e41. doi: 10.1016/j.forsciint.2014.08.032
Falquet, L., Calderon-Copete, S. P., and Frey, J. (2013). Draft genome sequence of the virulent Clostridium chauvoei reference strain JF4335. Genome Announc. 1, e00593–e00513. doi: 10.1128/genomeA.00593-13
Fortier, L.-C., and Moineau, S. (2007). Morphological and genetic diversity of temperate phages in Clostridium difficile. Appl. Environ. Microbiol. 73, 7358–7366. doi: 10.1128/AEM.00582-07
Frey, J., and Falquet, L. (2015). Patho-genetics of Clostridium chauvoei. Res. Microbiol. 166, 384–392. doi: 10.1016/j.resmic.2014.10.013
Frey, J., Johansson, A., Burki, S., Vilei, E. M., and Redhead, K. (2012). Cytotoxin CctA, a major virulence factor of Clostridium chauvoei conferring protective immunity against myonecrosis. Vaccine 30, 5500–5505. doi: 10.1016/j.vaccine.2012.06.050
Gazioglu, A., Karagülle, B., Yüksel, H., Nuri Açık, M., Keçeci, H., Dörtbudak, M. B., et al. (2018). Sudden death due to gas gangrene caused by Clostridium septicum in goats. BMC Vet. Res. 14:406. doi: 10.1186/s12917-018-1747-y
Gervasi, T., Curto, R. L., Narbad, A., and Mayer, M. J. (2013). Complete genome sequence of ΦCP51, a temperate bacteriophage of Clostridium perfringens. Arch. Virol. 158, 2015–2017. doi: 10.1007/s00705-013-1647-1
Glenn Songer, J. (2009). “Chapter 18: Clostridium novyi (Myonecrosis, Black Disease, and Bacillary Hemoglobinuria) and Clostridium septicum (Braxy) Infections,” in Food Animal Practice. eds. D. E. Anderson and D. M. Rings. 5th Edn. (Saint Louis: W.B. Saunders), 58–61.
Glenn Songer, J., and Miskimins, D. W. (2005). Clostridial abomasitis in calves: case report and review of the literature. Anaerobe 11, 290–294. doi: 10.1016/j.anaerobe.2004.12.004
Gornatti-Churria, C. D., Crispo, M., Shivaprasad, H. L., and Uzal, F. A. (2018). Gangrenous dermatitis in chickens and turkeys. J. Vet. Diagn. Investig. 30, 188–196. doi: 10.1177/1040638717742435
Grant, J. R., and Stothard, P. (2008). The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36, W181–W184. doi: 10.1093/nar/gkn179
Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., et al. (2014). ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220. doi: 10.1128/AAC.01310-13
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haghroosta, A., Goudarzi, H., Faghihloo, E., Ghalavand, Z., Ranjbar, M. M., and Langroudi, R. P. (2020). In silico analysis of a chimeric fusion protein as a new vaccine candidate against Clostridium perfringens type A and Clostridium septicum alpha toxins. Comp. Clin. Path., 1–9. doi: 10.1007/s00580-020-03136-6 [Epub ahead of print].
Hickey, M. J., Kwan, R. Y. Q., Awad, M. M., Kennedy, C. L., Young, L. F., Hall, P., et al. (2008). Molecular and cellular basis of microvascular perfusion deficits induced by Clostridium perfringens and Clostridium septicum. PLoS Pathog. 4:e1000045. doi: 10.1371/journal.ppat.1000045
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., Von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–d314. doi: 10.1093/nar/gky1085
Hunt, M., Silva, N. D., Otto, T. D., Parkhill, J., Keane, J. A., and Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16, 1–10. doi: 10.1186/s13059-015-0849-0
Ito, F., Inokuchi, R., Matsumoto, A., Kumada, Y., Yokoyama, H., Ishida, T., et al. (2017). Presence of periaortic gas in Clostridium septicum-infected aortic aneurysm aids in early diagnosis: a case report and systematic review of the literature. J. Med. Case Rep. 11:268. doi: 10.1186/s13256-017-1422-0
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Junior, C. A. O., Silva, R. O. S., Lobato, F. C. F., Navarro, M. A., and Uzal, F. A. (2020). Gas gangrene in mammals: a review. J. Vet. Diagn. Investig. 32, 175–183. doi: 10.1177/1040638720905830
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and Metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Katoh, K., Misawa, K., Kuma, K. I., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kennedy, C. L., Krejany, E. O., Young, L. F., O'connor, J. R., Awad, M. M., Boyd, R. L., et al. (2005). The alpha-toxin of Clostridium septicum is essential for virulence. Mol. Microbiol. 57, 1357–1366. doi: 10.1111/j.1365-2958.2005.04774.x
Kennedy, C. L., Lyras, D., Cordner, L. M., Melton-Witt, J., Emmins, J. J., Tweten, R. K., et al. (2009). Pore-forming activity of alpha-toxin is essential for Clostridium septicum-mediated myonecrosis. Infect. Immun. 77, 943–951. doi: 10.1128/IAI.01267-08
Kiu, R., Caim, S., Alexander, S., Pachori, P., and Hall, L. J. (2017). Probing genomic aspects of the multi-host pathogen Clostridium perfringens reveals significant pangenome diversity, and a diverse array of virulence factors. Front. Microbiol. 8:2485. doi: 10.3389/fmicb.2017.02485
Kornbluth, A. A., Danzig, J. B., and Bernstein, L. H. (1989). Clostridium septicum infection and associated malignancy. Report of 2 cases and review of the literature. Medicine 68, 30–37. doi: 10.1097/00005792-198901000-00002
Kousa, O., Essa, A., Ramadan, B., Aly, A., Awad, D., Zhao, X., et al. (2020). Multiorgan fatal gas gangrene in the setting of Clostridium septicum bacteremia: a case report. J. Emerg. Crit. Care Med. 4, 1–7. doi: 10.21037/jeccm.2019.12.04
Kuhnert, P., Capaul, S. E., Nicolet, J., and Frey, J. (1996). Phylogenetic positions of Clostridium chauvoei and Clostridium septicum based on 16S rRNA gene sequences. Int. J. Syst. Bacteriol. 46, 1174–1176. doi: 10.1099/00207713-46-4-1174
Letunic, I., and Bork, P. (2019). Interactive tree Of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Long, J., Xu, Y., Ou, L., Yang, H., Xi, Y., Chen, S., et al. (2019). Diversity of CRISPR/Cas system in Clostridium perfringens. Mol. Gen. Genomics. 294, 1263–1275. doi: 10.1007/s00438-019-01579-3
Maclennan, J. D. (1962). The histotoxic clostridial infections of man. Bacteriol. Rev. 26, 177–274. doi: 10.1128/br.26.2_pt_1-2.177-274.1962
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J. J., Charpentier, E., Horvath, P., et al. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477. doi: 10.1038/nrmicro2577
Mcarthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14, 60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., Klenk, H.-P., and Göker, M. (2014). Taxonomic use of DNA G+C content and DNA–DNA hybridization in the genomic age. Int. J. Syst. Bacteriol. 64, 352–356. doi: 10.1099/ijs.0.056994-0
Minh, B. Q., Nguyen, M. A. T., and Von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Gustavo, A. S., Sonnhammer, E. L. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi: 10.1093/nar/gkaa913
Morris, W. E., Uzal, F. A., Fattorini, F. R., and Terzolo, H. (2002). Malignant oedema associated with blood-sampling in sheep. Aust. Vet. J. 80, 280–281. doi: 10.1111/j.1751-0813.2002.tb10839.x
Moussa, R. S. (1958). Complexity of toxins from Clostridium septicum and Clostridium chauvoei. J. Bacteriol. 76, 538–545. doi: 10.1128/jb.76.5.538-545.1958
Murray, I. A., Clark, T. A., Morgan, R. D., Boitano, M., Anton, B. P., Luong, K., et al. (2012). The methylomes of six bacteria. Nucleic Acids Res. 40, 11450–11462. doi: 10.1093/nar/gks891
Muthuirulandi Sethuvel, D. P., Veeraraghavan, B., Vasudevan, K., Devanga Ragupathi, N. K., Murugan, D., Walia, K., et al. (2019). Complete genome analysis of clinical Shigella strains reveals plasmid pSS1653 with resistance determinants: a triumph of hybrid approach. Gut Pathog. 11, 55–55. doi: 10.1186/s13099-019-0334-5
Nagahama, M., Kihara, A., Miyawaki, T., Mukai, M., Sakaguchi, Y., Ochi, S., et al. (1999). Clostridium perfringens β-toxin is sensitive to thiol-group modification but does not require a thiol group for lethal activity. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 1454, 97–105. doi: 10.1016/S0925-4439(99)00026-5
Nanjappa, S., Shah, S., and Pabbathi, S. (2015). Clostridium septicum gas gangrene in colon cancer: importance of early diagnosis. Case Rep. Infect. Dis. 2015:694247. doi: 10.1155/2015/694247
Neumann, A. P., and Rehberger, T. G. (2009). MLST analysis reveals a highly conserved core genome among poultry isolates of Clostridium septicum. Anaerobe 15, 99–106. doi: 10.1016/j.anaerobe.2009.01.005
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nicholson, P., Furrer, J., Hässig, M., Strauss, C., Heller, M., Braga-Lagache, S., et al. (2019). Production of neutralizing antibodies against the secreted Clostridium chauvoei toxin A (CctA) upon blackleg vaccination. Anaerobe 56, 78–87. doi: 10.1016/j.anaerobe.2019.02.011
Oda, M., Terao, Y., Sakurai, J., and Nagahama, M. (2015). Membrane-binding mechanism of Clostridium perfringens alpha-toxin. Toxins 7, 5268–5275. doi: 10.3390/toxins7124880
Odani, J. S., Blanchard, P. C., Adaska, J. M., Moeller, R. B., and Uzal, F. A. (2009). Malignant edema in postpartum dairy cattle. J. Vet. Diagn. Investig. 21, 920–924. doi: 10.1177/104063870902100631
Popoff, M.R. (2016). “Toxins of histotoxic clostridia,” in Clostridial Diseases of Animals eds. F. A. Uzal and J. G. Songer, Prescott, J. F., and Popoff, M. R.. (Ames, Iowa: Wiley Blackwell), 21–43.
Potter, S. C., Luciani, A., Eddy, S. R., Park, Y., Lopez, R., and Finn, R. D. (2018). HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204. doi: 10.1093/nar/gky448
Powell, M. J., Sasapu, K. K., and Macklin, C. (2008). Metastatic gas gangrene and colonic perforation: a case report. World J. Emerg. Surg. 3, 15. doi: 10.1186/1749-7922-3-15
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Rahman, H., Chakraborty, A., Rahman, T., Sharma, R., Shome, B. R., and Shakuntala, I. (2009). Clostridial myonecrosis clinically resembling black quarter in an Indian elephant (Elephas maximus). Rev. Sci. Tech. 28, 1069–1075. doi: 10.20506/rst.28.3.1951
Rusinov, I., Ershova, A., Karyagina, A., Spirin, S., and Alexeevski, A. (2015). Lifespan of restriction-modification systems critically affects avoidance of their recognition sites in host genomes. BMC Genomics 16:1084. doi: 10.1186/s12864-015-2288-4
Rychener, L., Inalbon, S., Djordjevic, S. P., Chowdhury, P. R., Ziech, R. E., De Vargas, A. C., et al. (2017). Clostridium chauvoei, an evolutionary dead-end pathogen. Front. Microbiol. 8:1054. doi: 10.3389/fmicb.2017.01054
Saha, S., and Raghava, G. P. (2006). Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65, 40–48. doi: 10.1002/prot.21078
Sasaki, Y., Yamamoto, K., Kojima, A., Tetsuka, Y., Norimatsu, M., and Tamura, Y. (2000). Rapid and direct detection of Clostridium chauvoei by PCR of the 16S-23S rDNA spacer region and partial 23S rDNA sequences. J. Vet. Med. Sci. 62, 1275–1281. doi: 10.1292/jvms.62.1275
Sasaki, Y., Yamamoto, K., Tamura, Y., and Takahashi, T. (2001). Tetracycline-resistance genes of Clostridium perfringens, Clostridium septicum and Clostridium sordellii isolated from cattle affected with malignant edema. Vet. Microbiol. 83, 61–69. doi: 10.1016/S0378-1135(01)00402-3
Schadt, E. E., Banerjee, O., Fang, G., Feng, Z., Wong, W. H., Zhang, X., et al. (2013). Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases. Genome Res. 23, 129–141. doi: 10.1101/gr.136739.111
Schamber, G. J., Berg, I. E., and Molesworth, J. R. (1986). Braxy or Bradsot-like Abomastitis Caused by Clostridium septicum in a Calf. Can. Vet. J. 27, 194–194.
Seder, C. W., Kramer, M., Long, G., Uzieblo, M. R., Shanley, C. J., and Bove, P. (2009). Clostridium septicum aortitis: report of two cases and review of the literature. J. Vasc. Surg. 49, 1304–1309. doi: 10.1016/j.jvs.2008.11.058
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Segata, N., Börnigen, D., Morgan, X. C., and Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4, 2304. doi: 10.1038/ncomms3304
Sekulovic, O., Garneau, J. R., Néron, A., and Fortier, L.-C. (2014). Characterization of temperate phages infecting Clostridium difficile isolates of human and animal origins. Appl. Environ. Microbiol. 80, 2555–2563. doi: 10.1128/AEM.00237-14
Shimizu, T., Ohtani, K., Hirakawa, H., Ohshima, K., Yamashita, A., Shiba, T., et al. (2002). Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. PNAS 99, 996–1001. doi: 10.1073/pnas.022493799
Shivaprasad, H. L. (2016). “Gangrenous dermatitis in poultry” in Clostridial Diseases of Animals. eds. F. A. Uzal, J. G. Songer, J. F. Prescott, and M. R. Popoff (Ames, Iowa: Wiley Blackwell), 255–264.
Silva, R. O. S., Uzal, F. A., Oliveira, C. A., Lobato, F. C. F., Uzal, F. A., Songer, J. G., et al. (2016). “Gas Gangrene (Malignant Edema),” in Clostridial Diseases of Animals (Ames, Iowa, United States: John Wiley & Sons, Inc), 243–254.
Smith, T. J., Williamson, C. H. D., Hill, K. K., Johnson, S. L., Xie, G., Anniballi, F., et al. (2021). The distinctive evolution of orfX Clostridium parabotulinum strains and their Botulinum neurotoxin type A and F gene clusters is influenced by environmental factors and gene interactions via mobile genetic elements. Front. Microbiol. 12, 566908–566908. doi: 10.3389/fmicb.2021.566908
Smith-Slatas, C. L., Bourque, M., and Salazar, J. C. (2006). Clostridium septicum infections in children: a case report and review of the literature. Pediatrics 117, e796–e805. doi: 10.1542/peds.2005-1074
Song, L., Hobaugh, M. R., Shustak, C., Cheley, S., Bayley, H., and Gouaux, J. E. (1996). Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 274, 1859–1865. doi: 10.1126/science.274.5294.1859
Srivastava, I., Aldape, M. J., Bryant, A. E., and Stevens, D. L. (2017). Spontaneous C. septicum gas gangrene: a literature review. Anaerobe 48, 165–171. doi: 10.1016/j.anaerobe.2017.07.008
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Steinthorsdottir, V., Fridriksdottir, V., Gunnarsson, E., and Andrésson, O. S. (1998). Site-directed mutagenesis of Clostridium perfringens beta-toxin: expression of wild-type and mutant toxins in Bacillus subtilis. FEMS Microbiol. Lett. 158, 17–23. doi: 10.1111/j.1574-6968.1998.tb12794.x
Stevens, D. L., Aldape, M. J., and Bryant, A. E. (2012). Life-threatening clostridial infections. Anaerobe 18, 254–259. doi: 10.1016/j.anaerobe.2011.11.001
Stothard, P., Grant, J. R., and Van Domselaar, G. (2019). Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 20, 1576–1582. doi: 10.1093/bib/bbx081
Thomas, P., Semmler, T., Eichhorn, I., Lübke-Becker, A., Werckenthin, C., Abdel-Glil, M. Y., et al. (2017). First report of two complete Clostridium chauvoei genome sequences and detailed in silico genome analysis. Infect. Genet. Evol. 54, 287–298. doi: 10.1016/j.meegid.2017.07.018
Tonkin-Hill, G., Macalasdair, N., Ruis, C., Weimann, A., Horesh, G., Lees, J. A., et al. (2020). Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 21:180. doi: 10.1186/s13059-020-02090-4
Vimberg, V., Zieglerová, L., Buriánková, K., Branny, P., and Balíková Novotná, G. (2020). VanZ reduces the binding of lipoglycopeptide antibiotics to Staphylococcus aureus and Streptococcus pneumoniae cells. Front. Microbiol. 11:566. doi: 10.3389/fmicb.2020.00566
Volovitch, M., Modjtahedi, N., Chouikh, Y., and Yot, P. (1990). DNA sequence of gene VI of cauliflower mosaic virus strain PV147. Nucleic Acids Res. 18, 5297. doi: 10.1093/nar/18.17.5297
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Willoughby, D. H., Bickford, A. A., Cooper, G. L., and Charlton, B. R. (1996). Periodic recurrence of gangrenous dermatitis associated with Clostridium septicum in a broiler chicken operation. J. Vet. Diagn. Investig. 8, 259–261. doi: 10.1177/104063879600800222
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20:257. doi: 10.1186/s13059-019-1891-0
Yamamura, K., Ashida, H., Okano, T., Kinoshita-Daitoku, R., Suzuki, S., Ohtani, K., et al. (2019). Inflammasome activation induced by Perfringolysin O of Clostridium perfringens and its involvement in the progression of gas gangrene. Front. Microbiol. 10:2406. doi: 10.3389/fmicb.2019.02406
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zhao, Y., Jia, X., Yang, J., Ling, Y., Zhang, Z., Yu, J., et al. (2014). PanGP: a tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 30, 1297–1299. doi: 10.1093/bioinformatics/btu017
Keywords: Clostridium septicum, comparative analysis, genome, gas gangrene, DSM 7534T, virulence factors
Citation: Thomas P, Abdel-Glil MY, Subbaiyan A, Busch A, Eichhorn I, Wieler LH, Neubauer H, Pletz M and Seyboldt C (2021) First Comparative Analysis of Clostridium septicum Genomes Provides Insights Into the Taxonomy, Species Genetic Diversity, and Virulence Related to Gas Gangrene. Front. Microbiol. 12:771945. doi: 10.3389/fmicb.2021.771945
Edited by:
Frank T. Robb, University of Maryland, Baltimore, United StatesReviewed by:
Henrique César Pereira Figueiredo, Federal University of Minas Gerais, BrazilDieter Jahn, Technische Universitat Braunschweig, Germany
Rodrigo Otávio Silveira Silva, Federal University of Minas Gerais, Brazil
Copyright © 2021 Thomas, Abdel-Glil, Subbaiyan, Busch, Eichhorn, Wieler, Neubauer, Pletz and Seyboldt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mostafa Y. Abdel-Glil, bW9zdGFmYS5hYmRlbC1nbGlsQGZsaS5kZQ==
†These authors share first authorship