
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 20 January 2022
Sec. Microbial Physiology and Metabolism
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.768562
This article is part of the Research TopicMicroorganisms for Consolidated 2nd Generation BiorefiningView all 5 articles
 Nicoletta Gronchi1
Nicoletta Gronchi1 Nicola De Bernardini2
Nicola De Bernardini2 Rosemary A. Cripwell3
Rosemary A. Cripwell3 Laura Treu2
Laura Treu2 Stefano Campanaro2
Stefano Campanaro2 Marina Basaglia1
Marina Basaglia1 Maria R. Foulquié-Moreno4Johan M. Thevelein4,5
Maria R. Foulquié-Moreno4Johan M. Thevelein4,5 Willem H. Van Zyl3
Willem H. Van Zyl3 Lorenzo Favaro1*†
Lorenzo Favaro1*† Sergio Casella1†
Sergio Casella1†
Natural yeast with superior fermentative traits can serve as a platform for the development of recombinant strains that can be used to improve the sustainability of bioethanol production from starch. This process will benefit from a consolidated bioprocessing (CBP) approach where an engineered strain producing amylases directly converts starch into ethanol. The yeast Saccharomyces cerevisiae L20, previously selected as outperforming the benchmark yeast Ethanol Red, was here subjected to a comparative genomic investigation using a dataset of industrial S. cerevisiae strains. Along with Ethanol Red, strain L20 was then engineered for the expression of α-amylase amyA and glucoamylase glaA genes from Aspergillus tubingensis by employing two different approaches (delta integration and CRISPR/Cas9). A correlation between the number of integrated copies and the hydrolytic abilities of the recombinants was investigated. L20 demonstrated important traits for the construction of a proficient CBP yeast. Despite showing a close relatedness to commercial wine yeast and the benchmark Ethanol Red, a unique profile of gene copy number variations (CNVs) was found in L20, mainly encoding membrane transporters and secretion pathway proteins but also the fermentative metabolism. Moreover, the genome annotation disclosed seven open reading frames (ORFs) in L20 that are absent in the reference S288C genome. Genome engineering was successfully implemented for amylase production. However, with equal amylase gene copies, L20 proved its proficiency as a good enzyme secretor by exhibiting a markedly higher amylolytic activity than Ethanol Red, in compliance to the findings of the genomic exploration. The recombinant L20 dT8 exhibited the highest amylolytic activity and produced more than 4 g/L of ethanol from 2% starch in a CBP setting without the addition of supplementary enzymes. Based on the performance of this strain, an amylase/glucoamylase ratio of 1:2.5 was suggested as baseline for further improvement of the CBP ability. Overall, L20 showed important traits for the future construction of a proficient CBP yeast. As such, this work shows that natural S. cerevisiae strains can be used for the expression of foreign secreted enzymes, paving the way to strain improvement for the starch-to-bioethanol route.
The increasing global fuel demand claims for the develop of a sustainable and cost-effective technology to convert polysaccharides into bioethanol. Nowadays, the production of ethanol from starch is particularly significant in the United States and Europe, being the leading producers of first-generation bioethanol from corn and wheat, respectively. Being more readily degradable than lignocellulose, starch is the preferred raw material for conversion into ethanol. Therefore, it is not surprising that one-third of the current global corn production is dedicated to the biofuel industry (Mohanty and Swain, 2019). However, despite the benefits from the reduced petroleum reliance, the use of corn contributes to the price increase in food and feed commodities, as well as the depletion of water resources and soil degradation. Alternatively, the use of starchy residual biomass from forestry, agricultural and industrial activities has been proposed as second-generation feedstock to preserve the food supply chain and to reduce the environmental threat (Lin and Tanaka, 2006; Castro et al., 2011; Vohra et al., 2014; Gupta and Verma, 2015; Aditiya et al., 2016; Zabed et al., 2017; Robak and Balcerek, 2018; Myburgh et al., 2019).
The starch-to-ethanol conversion is a well-established and technically mature technology. The process involves significant heat-intensive steps for starch liquefaction, as well as the use of commercial thermostable hydrolase mixtures (α-amylase and glucoamylase) for the complete saccharification of the substrate (Ishizaki and Hasumi, 2014; Vohra et al., 2014; Cinelli et al., 2015; Zabed et al., 2017; Cripwell et al., 2020). With the aim of limiting the operational costs as well as the capital by employing waste biomass, the integration of all steps into a single fermentative unit simplifies the industrial process and is expected to save up to 10–50% of the cost (Lynd et al., 2008; Brown et al., 2020; Cunha et al., 2020). In this scenario the employment of a consolidated bioprocessing (CBP) yeast, able to simultaneously hydrolyze starchy biomass and directly ferment the resulting glucose at fermentation temperatures would represent a cost-savings approach. However, to date, no natural yeast isolate has been described to perform CBP for sustainable bioethanol production (Favaro et al., 2013, 2015; Kricka et al., 2014; Cripwell et al., 2019a; Adegboye et al., 2021).
The ethanologenic yeast Saccharomyces cerevisiae represents the ultimate candidate for bioethanol production due to the ease of cultivation and the generally recognized as safe (GRAS) and qualified presumption of safety (QPS) status (Sharma et al., 2018; Favaro et al., 2019b). Moreover, robust industrial strains have been adapted to stressful conditions and present favorable traits such as high fermentation rate, general robustness, tolerance to low pH and osmotic stress. The major limitation, however, is their inability to produce amylases (Görgens et al., 2015; Cripwell et al., 2020).
Despite the large employment of S. cerevisiae in biotechnological research, only CBP yeasts with limited amylolytic activity are currently employed on an industrial scale. The genome engineering for amylase expression in industrial S. cerevisiae strains has already been reported by integration of heterologous genes at delta sequences of the Ty retrotransposon (Cho et al., 1999; Kang et al., 2003; Favaro et al., 2010, 2015; Cripwell et al., 2019a) or ribosomal DNA (Lopes et al., 1989, 1996; Nieto et al., 1999; Choi et al., 2002; Liao et al., 2012). Although these strategies are known as very efficient in S. cerevisiae because of the native homologous recombination machinery, the inserts often result in long tandem repeats at one location leading to genome instability and unstable phenotypes. Likewise, multiple chromosome integrations can be hampered by the limited availability of selective markers.
Genetic modification of complex industrial yeast has advanced rapidly with the use of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR associated (Cas) protein system, which is by now widely considered as the technology of choice for metabolic engineering (Jansen et al., 2017; Zhang et al., 2020; Adegboye et al., 2021; Riley and Guss, 2021). Compared to other endonuclease-based and in vivo recombineering methods, it has proven to be a fast, marker-free, versatile, and most importantly site-directed genome-editing technique (Jakočiūnas et al., 2016; Roggenkamp et al., 2017).
From an industrial perspective, complete starch hydrolysis without liquefaction can only be achieved by a CBP yeast co-producing raw starch α-amylase and glucoamylase genes at high titers and able to ferment raw starch at high substrate loading (Cripwell et al., 2020; den Haan et al., 2021). Few groups have reported successful bioethanol production from raw corn starch using recombinant S. cerevisiae strains, mostly developed from laboratory backgrounds (Murai et al., 1998; Khaw et al., 2006; Yamada et al., 2009; Favaro et al., 2015; Cripwell et al., 2019a,b). Moreover, the exploration of the immense, and still largely unknown, potential of natural yeast strains could be of great relevance for the improvement of the starch-to-ethanol process (Favaro et al., 2019b). As reported in literature, natural yeast isolates have been screened for lignocellulosic bioethanol production (Basso et al., 2008; Jansen et al., 2017) but only a little is known for the starch process (Mohd Azhar et al., 2017; Favaro et al., 2019b; Cripwell et al., 2020). For instance, Gronchi et al. (2019) evaluated a cluster of natural S. cerevisiae isolates in simultaneous saccharification and fermentation (SSF) of raw starch and identified S. cerevisiae L20 as outperforming the industrial benchmark S. cerevisiae Ethanol Red (Lesaffre, France), which is one of the most widely used yeast strains for first-generation bioethanol production.
In this study, the L20 strain was examined at a genome level to highlight possible traits that could elucidate its superior fermentative abilities. The genome was assembled de novo by a hybrid Illumina/Nanopore approach to increase the genome completeness, and then subjected to a comparative analysis with a dataset of other S. cerevisiae strains. The dataset was constructed with the deposited genomic sequences of S. cerevisiae strains that are involved in alcoholic beverages and bioethanol production. The genome of the strain Ethanol Red was also sequenced and included in the dataset as a benchmark. Strain L20, which showed a unique amplification profile of genes, was then selected as suitable candidate for genome engineering in order to develop an efficient CBP strain. The α-amylase (amyA) and glucoamylase (glaA) genes from Aspergillus tubingensis, previously employed by Viktor et al. (2013) on a multicopy plasmid, were cloned and stably expressed in both strains L20 and Ethanol Red by adopting delta integration and CRISPR/Cas9 strategies. The recombinant strains were investigated with regards to their enzymatic and fermenting abilities on corn starch, giving particular attention to the correlation between the gene copy number and hydrolytic activity.
The results demonstrated that S. cerevisiae L20 exhibited a great potential for the application in the bioethanol industry. At the same time, the strain was successfully employed as microbial platform for the development of a starch-CBP yeast. For the first time, a natural yeast strain has been engineered by the CRISPR/Cas9 technology for fungal amylase production, representing the earliest example of a drop-in yeast for the starch-to-ethanol industry.
All chemicals were of analytical grade and were obtained from Sigma-Aldrich, unless stated otherwise. Escherichia coli cells were grown at 37°C in Luria Bertani broth (LB; g/L: yeast extract, 5; tryptone, 10; NaCl, 10) supplemented with ampicillin (100 mg/L). S. cerevisiae strains were maintained in yeast extract peptone dextrose (YPD; g/L: yeast extract, 10; peptone, 20; glucose, 20) or on selective YPD plates supplemented with agar (15 g/L) and antibiotics: geneticin sulfate (200 mg/L, Sigma-Aldrich), hygromycin B (300 mg/L, Invivogen), or nourseothricin (100 mg/L, Jena Bioscience). Ampicillin and streptomycin (75 mg/L, Sigma-Aldrich) were added to prevent bacterial contamination during fermentation. Yeast nitrogen base plates (YNB; g/L: yeast nitrogen base, 6.7; agar, 15) containing 0.2% soluble corn starch were used for amylase plate assays.
The strains and plasmids used in this study are summarized in Table 1.
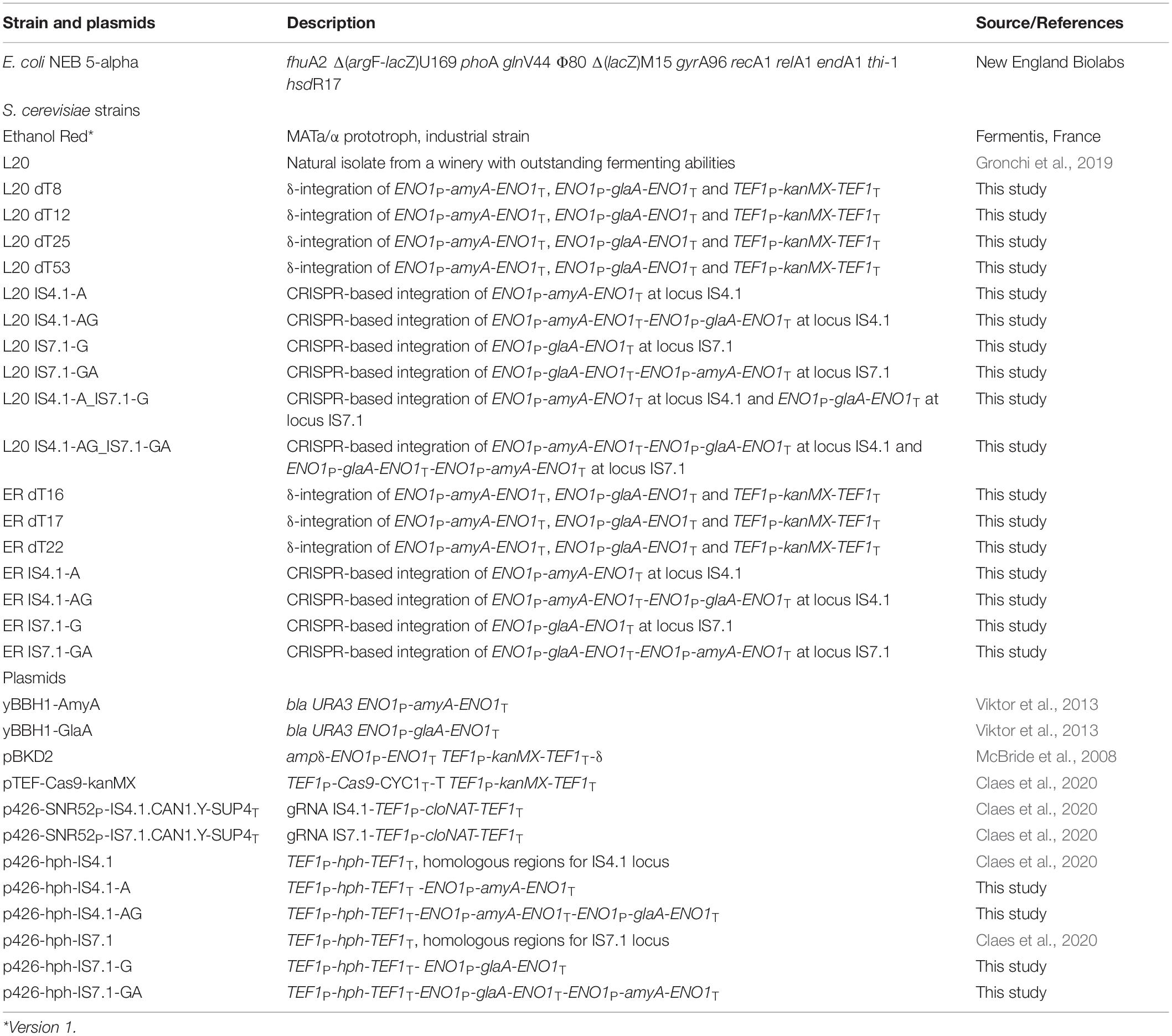
Table 1. Strains and plasmids used in this study.
Genomic DNA from S. cerevisiae L20 and Ethanol Red strains was isolated from overnight cultures using zymolyase digestion and standard phenol-chloroform extraction (Treu et al., 2014). A combined sequencing approach was then applied using Illumina and Oxford Nanopore MinION single-molecule sequencing. The Illumina library was generated using the TruSeq DNA PCR-Free Library Prep Kit (Illumina Inc., San Diego, CA, United States) and Covaris S2 (Woburn, MA, United States) for a 550-bp average fragment size. The library was loaded onto the flow cell provided in the NextSeq 500 Reagent kit v2 (150 cycles, Illumina Inc., San Diego, CA, United States) and sequenced on a NextSeq 500 (Illumina Inc., San Diego, CA, United States) platform with a paired-end protocol and read lengths of 151 bp at the CRIBI Biotechnology Center (Padua, Italy). The Nanopore library was prepared according to the SQK-LSK109/SQK-RBK004 ligation sequencing kit and sequenced on a FLO-MIN106 R9/FLO-MIN106 D flowcell; a detailed procedure of DNA extraction/purification and library preparation was reported in Basile et al. (2021). The genome assembly was performed with a de novo approach with an in-house pipeline developed to combine Nanopore and Illumina sequences analysis. Briefly, the long reads were corrected and assembled with the Canu (Koren et al., 2017) software. The obtained contigs were polished with Pilon (Walker et al., 2014) software using the independent high-quality Illumina sequences. A whole-genome alignment was then obtained using Mauve software (Darling et al., 2010) to highlight genome completeness and structural variants in comparison to the reference S. cerevisiae S288C. For the CRISPR/Cas9 application, the non-coding loci IS4.1 and IS7.1 were selected from Claes et al. (2020) and confirmed as suitable targets for p426-SNR52P-IS4.1.CAN1.Y-SUP4T and p426-SNR52P-IS7.1.CAN1.Y-SUP4T guide RNA vectors in L20 and Ethanol Red strains.
The genomic DNA was isolated from the recombinant strains for Illumina sequencing to verify the copy number of integrated amylase cassettes. DNA was extracted from overnight cultures according to DNeasy PowerSoil Kit (Qiagen). An additional cleaning step with phenol:chloroform:isoamyl alcohol (PCI; 25:24:1, v/v) solution (Sigma-Aldrich) was performed before DNA isolation. Illumina library and assembly were performed as previously described for host strains. The sequences of integrated genes (amyA and glaA) and single-copy reference genes (ACT1, ALG9, PGK1, and TFC1) were used as queries for BLAST analyses. The integrated gene copy numbers were assessed based on the ratio between the average coverage of selected reference genes and the average coverage of the heterologous genes (Cripwell et al., 2019a).
A comparative genomic approach was used to characterize L20. Fifty-four S. cerevisiae strains, whose genome has been previously deposited in online repositories, were selected due to their commercial/industrial relevance. Wine and beer-producing strains were chosen because of the L20’s enological background, and bioethanol producing strains were included in view of the final application (CBP strain development). The S288C and Ethanol Red genomes were also included. The selected strains were grouped according to their industrial application (Table 2).
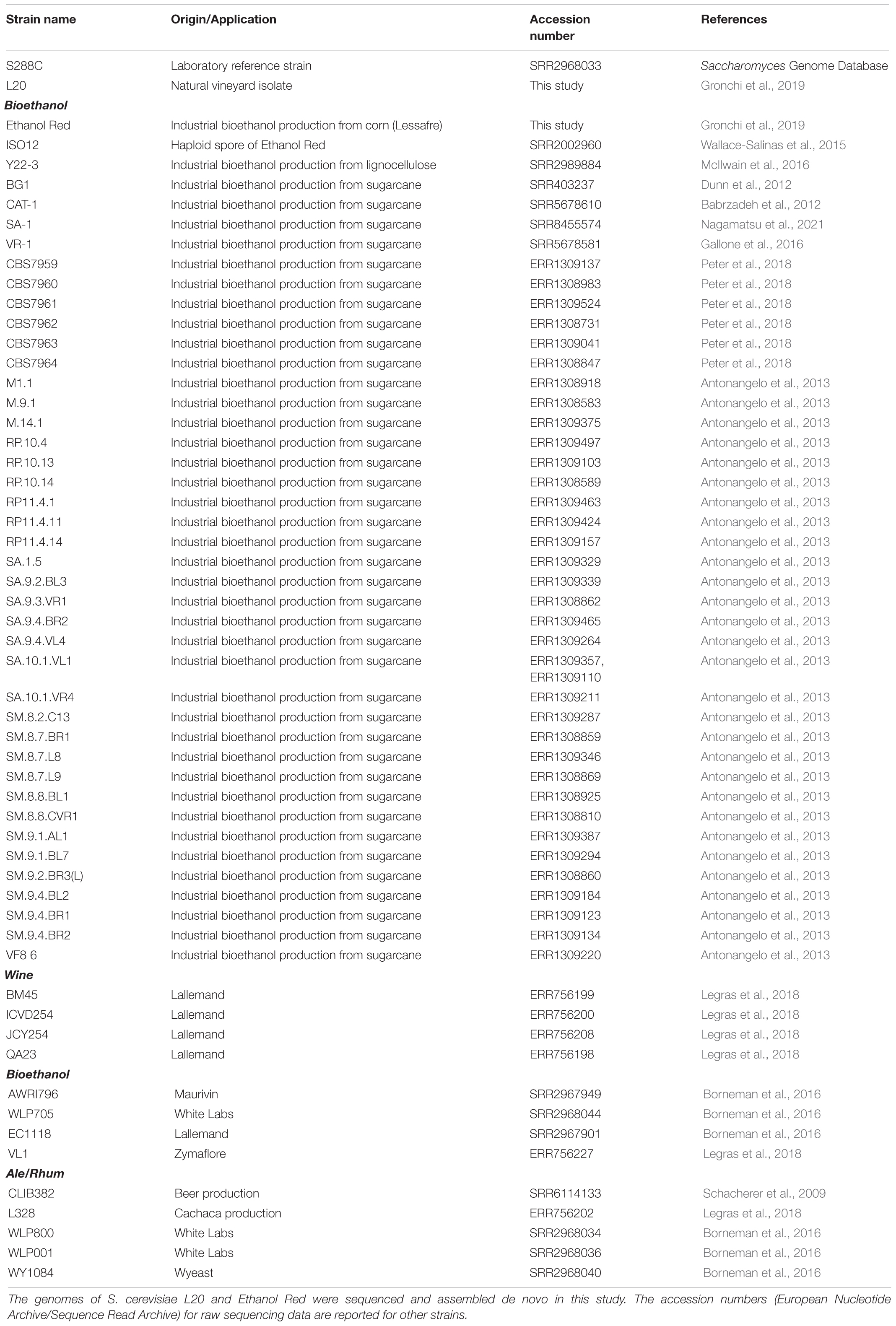
Table 2. Yeast strains used in the comparative genomic analysis.
The genetic variants present in open reading frames (ORFs) were investigated for their potential as a source of phenotypic variation. Variant calling analysis was performed following the Genome Analysis Toolkit (GATK4, v4.1.9.0) Best Practices as first discussed by DePristo et al. (2011). Through the comparison with a reference genome, this framework allows the discovery of small genetic variants, such as single nucleotide polymorphisms (SNPs) and insertion-deletions (INDELs), and the genotyping of multiple samples simultaneously. The strain selected for all the reference-based analyses was the S. cerevisiae S288C R64-1-1. The key phases of the pipeline were applied as previously described by Basile et al. (2021). Briefly, (I) filtered reads were aligned using bwa mem (v0.7.17); (II) base quality scores were recalibrated using a machine learning model implemented in “BQSR”; (III) variants were identified using the haplotype caller algorithm; (IV) detected variants were filtered using a variant quality score recalibration model trained on three different subsets of a previously published dataset (Peter et al., 2018). Functional effect prediction and genetic variants annotation were performed with the SnpEff software (v5.0) (Cingolani et al., 2012).
The genetic variants of the yeast dataset were identified and only the biallelic SNPs were retained using VCFtools (v0.1.16) for the phylogenetic analysis. Overall, the resulting subset contains 299,604 strain-specific variants which have been subsequently processed to generate a multiple sequence alignment (MSA) in FASTA format required as input by IQ-TREE v2.0.3 (Nguyen et al., 2015). The tools used for the conversion include “vcf-to-tab” from the VCFtools suite (Danecek et al., 2011), GNU Datamash (Free Software Foundation Inc., 2014) and a custom Perl (Wall et al., 2000) script. Finally, IQ-TREE was used to reconstruct a maximum likelihood (ML) phylogenetic tree. The substitution model (SYM + R3) adopted was selected with ModelFinder (Kalyaanamoorthy et al., 2017) and the robustness of the topology was further assessed using 500 ultrafast bootstrap iterations (Hoang et al., 2018). The Interactive Tree Of Life (iTOL)1 was finally used for the graphic representation of the phylogenetic tree (Letunic and Bork, 2019).
The similarity between L20 and S288C orthologous genes was estimated using two strategies: (1) annotated genes from the reference were mapped to the target assembly using Liftoff (v1.6.1) to predict which are common; differences among strains were determined (2) by predicting the total ORFs and extracting L20 specific genes and (3) by predicting ORFs in L20 accessory region with respect to the reference. To limit the gene finding process only to strain-specific regions of L20, a strategy previously reported by Basile et al. (2021) was implemented. It consisted in the identification of strain-specific regions of at least 500 bp with AGEnt (Ozer et al., 2014) followed by the prediction of protein-encoding genes within these regions. GeneMark-ES (v4.67) was used in both analyses, accounting for fungal-specific intron organization and assuming a maximum intron length of 500 for the prediction (Ter-Hovhannisyan et al., 2008). Afterward, the genes identified in L20 were translated into protein sequence and clustered with the reference proteome using cd-hit software (v4.8.1) (Fu et al., 2012). The thresholds used for clustering were minimum length of 100 nucleotides per sequence and minimum overlap with the cluster longest sequence of at least 10%. Different identity thresholds between clustered sequences have been tested, ranging from 80 to 95%, but only results derived from the most conservative ones (80%) were used for further analysis. The ORFs found in L20 but not in the reference S288C were annotated using RPS-BLAST (v2.6.0+).
The copy number variations (CNVs) were estimated based on whole genome sequencing data using CNVpytor (v1.0; Suvakov et al., 2021). The raw reads were polished using Trimmomatic (v.0.39), aligned to the reference genome with bwa (v.0.7.17) and eventually run with CNVpytor. The software estimated CNV values of entire genome regions based on read depth (RD) and allowed the extraction of predicted copy numbers. A deeper insight was dedicated to the CNV for the Gene Ontology (GO) terms “transmembrane transport” (GO:0090662; GO:0006899), “energy derivation by oxidation of organic compounds” (GO:0015980) and “response to stress” (GO:0006950) as annotated in AmiGO database for S. cerevisiae S288C and SGD database.
Standard protocols were followed for DNA manipulations and E. coli transformation (Sambrook and Russell, 2001). Restriction enzymes were supplied by New England Biolabs or Thermofisher and used as recommended by the supplier. DNA was eluted from 1% agarose gels using the Wizard SV Gel and PCR Clean-Up System (Promega). Plasmids were isolated using the NucleoSpin Plasmid Easy Pure kit (Macherey-Nagel). The Q5 High Fidelity (New England Biolabs) polymerase was used for PCR amplification.
For the delta integration approach, linear donor DNA fragments were constructed by providing 500 bp-homologous flanking regions to the amylase cassettes. Briefly, the yBBH1-AmyA and yBBH1-GlaA plasmids (Viktor et al., 2013) were used as templates to amplify the δ-ENO1P-amyA-ENO1T and δ-ENO1P-glaA-ENO1T amylase fragments, respectively, using the ENO1P Delta-L and ENO1T-R primers (Table 3). The geneticin marker was used for the selection: the TEF1P-kanMX-TEF1T-δ cassette was amplified from pBKD2 using ENO1T marker-L and Delta-R Primers (Table 3) and ligated in vivo at the 3′ of the amylase fragments to generate δ-ENO1P-amyA-ENO1T-kanMX-δ and δ-ENO1P-glaA-ENO1T-kanMX-δ, respectively.

Table 3. Primers used for amplification of amylase cassettes, with italicized oligos representing regions for homologous recombination.
Yeast cells were transformed according to Favaro et al. (2012) with δ-ENO1P-amyA-ENO1T, δ-ENO1P-glaA-ENO1T and TEF1P-kanMX-TEF1T-δ cassettes simultaneously. In 0.2 cm electroporation cuvettes, an electric pulse of 1.4 kV, 200 Ω and 25 μF was applied using a Bio-Rad system (GenePluserXcell, Bio-Rad, Hercules, CA, United States). Cells were immediately suspended in 1 mL of YPD containing 1 M sorbitol (YPDS) and incubated at 30°C for 3 h to allow recovery. Electroporated cells were then spread onto YPD plates supplemented with geneticin and incubated at 30°C for 48 h. The recombinants were named according to the transformation method (“d” for delta integration and then consecutively numbered).
A three plasmid-based approach was used for CRISPR/Cas9. The p426-hph vector was used as donor plasmid containing homologous regions for IS4.1 or IS7.1 loci and constructed to encode a single or a combination of amyA and glaA sequences (Table 1). The plasmid maps used in this study are reported in Figure 1. Briefly, the ENO1P-amyA-ENO1T and ENO1P-glaA-ENO1T cassettes were amplified from yBBH1 (Table 1) with primers reported in Table 3. The primers design and fragment assembly were performed according to the Gibson Assembly Cloning Kit (New England Biolabs) manufacturer’s recommendations. The plasmids were sent for Sanger sequencing (Mix2Seq; Eurofins Genomics, Germany).

Figure 1. Donor DNA plasmids for the CRISPR/Cas9 method used in this study: plasmids were constructed to carry single A. tubingensis amyA (A) or glaA (B) or double cassette for the simultaneous expression of the A. tubingensis amyA and glaA genes (C,D). IS4.1 and IS7.1 indicate the locus position for gene integration. The homologous regions (HR1 and HR2) are sequences flanking the designated genomic locus. All plasmids contained bacterial ori and amp genes for plasmid replication and ampicillin resistance, respectively.
The transformation was carried out using the LiAC/SS carrier DNA/PEG method (Gietz and Schiestl, 2007). Salmon sperm DNA (ssDNA 10 mg/mL) was purchased from Roche. The pTEF-Cas9-kanMX, p426-hph donor DNA and the gRNA plasmids were transformed separately in this order. Yeast cells were engineered for the expression of a single gene (donor vectors p426-hph-IS4.1-A for amyA in locus IS4.1; p426-hph-IS7.1-G for glaA in locus IS7.1), or double construct (p426-hph-IS4.1-AG and p426-hph-IS7.1-GA for amyA and glaA in locus IS4.1 and IS7.1, respectively; Table 1 and Figure 1). The L20 recombinants already transformed at IS4.1 and IS7.1 loci were subjected to a second round of transformation to target the alternative locus with a single or double cassette donor plasmid (Table 1 and Figure 1).
Cells were spread onto selective plates and incubated at 30°C for 48–72 h. The recombinants were named according to the donor plasmid/plasmids used (L20 IS4.1-A, IS4.1-AG, IS7.1-G, IS7.1-GA, IS4.1-A_IS7.1-G, IS4.1-AG_IS7.1-GA, Ethanol Red IS4.1-A, IS4.1-AG, IS7.1-G and IS7.1-GA).
Starch plate assays were used for qualitative analysis to verify the hydrolytic activity of the transformants. After 72 h growth in YPD, cultures were spotted onto YNB containing soluble corn starch and incubated for 48 h at 30°C. Strains expressing the amylase genes produced a clear surrounding halo after Lugol staining (Favaro et al., 2015). The yeast strains constructed using the CRISPR/CAS approach were subjected to sequential batch cultures using non-selective YPD broth for plasmid curing. The mitotic stability was verified according to Favaro et al. (2012). Single cell colonies were isolated on YPD plates by Singer Instruments MSM-400 micromanipulator.
Yeast colonies that produced clearing zones during plate assays were screened using the polymerase chain reaction (PCR) to confirm the presence of the integrated gene(s). Genomic DNA was extracted using the PCI solution with subsequent ethanol precipitation. PCR was performed with primers reported in Table 4. The genomic DNA of the parental strains was used as negative control.

Table 4. Primers used to confirm the integration of heterologous amyA and glaA genes in S. cerevisiae.
The supernatant from yeast cultures, grown for 24, 48, and 72 h, was denatured at 100°C for 3 min. The protein fractions were separated by SDS-PAGE using an 8% separation gel (Laemmli, 1970). Electrophoresis was carried out at 100 V for 90 min at room temperature and the proteins were visualized using the silver staining method (O’Connell and Stults, 1997). Supernatant from the parental strains was used as negative control. The broad-range PageRuler Prestained Protein Ladder (Fermentas) was used as a molecular mass marker.
Recombinant strains were cultured in 20 mL YPD in 125 mL Erlenmeyer flasks with agitation at 120 rpm, with an initial optical density of 0.2 (OD600). The supernatant, collected after 24, 48, and 72 h of cultivation, was used to assess the enzymatic activity as described by Viktor et al. (2013). The total amylase activity was colorimetrically determined by using the DNS (3,5-dinitro salicylic acid) method described by Miller (1959) at 50°C for 5 min. For glucoamylase activity, 50 μL supernatant was incubated for 15 min with 450 μL of a 0.2% soluble corn starch solution (50°C, pH 5). The resulting glucose concentration was determined with the D-Glucose HK Assay Kit (Megazyme, Ireland) (adapted from Viktor et al., 2013). Enzymatic activities were expressed as nanokatals per mL (nKat/mL), which is defined as the enzyme activity needed to release 1 nmol of glucose per second per mL of culture. All experiments were carried out in triplicate. The parental strains were used as negative controls.
Small-scale fermentations were performed on both soluble and raw corn starch in oxygen-limited conditions. Yeasts were cultured in 300 mL of YPD in 1-L Erlenmeyer flasks and incubated overnight at 30°C on a rotatory shaker at 120 rpm. Cells were collected by centrifugation for 5 min at 4000 rpm and inoculated at an OD600 value of 5 in 120-mL serum bottles containing 100 mL YP supplemented with 0.05% glucose, 2% soluble and raw starch and antibiotics (ampicillin and streptomycin) to prevent bacterial contamination. Serum bottles were sealed with rubber stoppers and provided with a needle for CO2 removal, then incubated at 30°C on a magnetic stirrer (Cimarec i Poly 15 Multipoint stirrer, Thermo Scientific) with agitation at 700 rpm. Samples were taken every 24 h, filtered through 0.22-μm for high-performance liquid chromatography (HPLC) analysis. The experiments were carried out in triplicate. Parental strains were used as negative control.
Samples were analyzed for glucose, glycerol and ethanol through liquid chromatography using a Shimadzu Nexera HPLC system, equipped with a RID-10A refractive index detector (Shimadzu, Kyoto, Japan). The chromatographic separations were performed using a Rezex ROA-Organic Acid H+ (8%) column (300 mm 7.8 mm, Phenomenex, Torrance, CA, United States). The column temperature was set at 60°C and the analysis was performed at a flow rate of 0.6 mL/min using isocratic elution, with 2.5 mM H2SO4 as a mobile phase (Cagnin et al., 2019).
From an industrial perspective, the implementation of a CBP yeast for complete starch utilization would require co-production of raw starch α-amylase and glucoamylase enzymes and fermentation at high substrate loadings. Considering the future development of an efficient CBP yeast, the host strain must, among other traits, yield superior ethanol levels. Bearing this in mind, S. cerevisiae L20, which was previously selected as a superior yeast strain under high gravity SSF conditions, was engineered to produce amylases. The ethanol yield of strain L20 was much greater than those exhibited by the industrial benchmark Ethanol Red. Genome sequencing data was used to unravel the basis of L20’s superior fermenting abilities and an engineering approach was pursued for the co-secretion of the A. tubingensis α-amylase amyA and glucoamylase glaA.
The whole-genome sequence of L20 was obtained using a novel strategy that integrates MinION and Illumina technologies: the first platform is expected to produce robust scaffolds against which the Illumina reads can be mapped to in order to increase the assembly quality. The number of paired-end reads (2 × 150 bp) was 1,022,547, resulting in a 25-fold genome coverage. The number of MinION sequences was 58,954 with an average length of 6,649 bp. The de novo assembly generated a genome of 11.9 Mb, composed of 18 contigs with a N50 of 788,913 and 14 chromosomes assembled in a single contig. The genome size is comparable to the average of other natural and industrial S. cerevisiae strains (Gallone et al., 2016; Duan et al., 2018).
The whole-genome sequencing of Ethanol Red resulted in 136-fold genome coverage, with a total number of paired-end reads (2 × 150 bp) of 5,302,549. The number of MinION sequences was 121,382 with an average length of 4,493 bp. The de novo assembly produced a genome of 12.1 Mb, composed of 29 contigs with a N50 of 779,629.
Genome assembly details for all strains considered in this study are reported in Supplementary Table 1. Raw reads of S. cerevisiae L20 and Ethanol Red were deposited at GenBank under the BioProject accession number PRJNA762028.
The analysis of variants among the S. cerevisiae L20 and selected industrially relevant S. cerevisiae strains revealed 363,159 single nucleotide variants (SNV) in 343,963 loci. Variants were equally distributed among the 16 chromosomes, with an average rate of 1 every 33 bases. When grouped by type, 317,682 were SNPs (87.5%), and 24,668 (6.8%) and 20,809 (5.8%) were classified as insertion or deletion, respectively. A total of about 2.5 million effects were predicted, out of which 91.6% were found in non-coding regions. The 8.5% (212,137) and 6% (148,602) of effects were found in exons and intergenic regions, respectively. However, the majority of effects was detected within 5 kb upstream (5′-) or downstream (3′-) regions (44 and 42%, respectively). The details are reported in Supplementary Tables 1, 2.
The phylogenetic analysis was inferred on the dataset of small biallelic variants (299,604) and a maximum-likelihood tree was constructed and is shown in Figure 2.
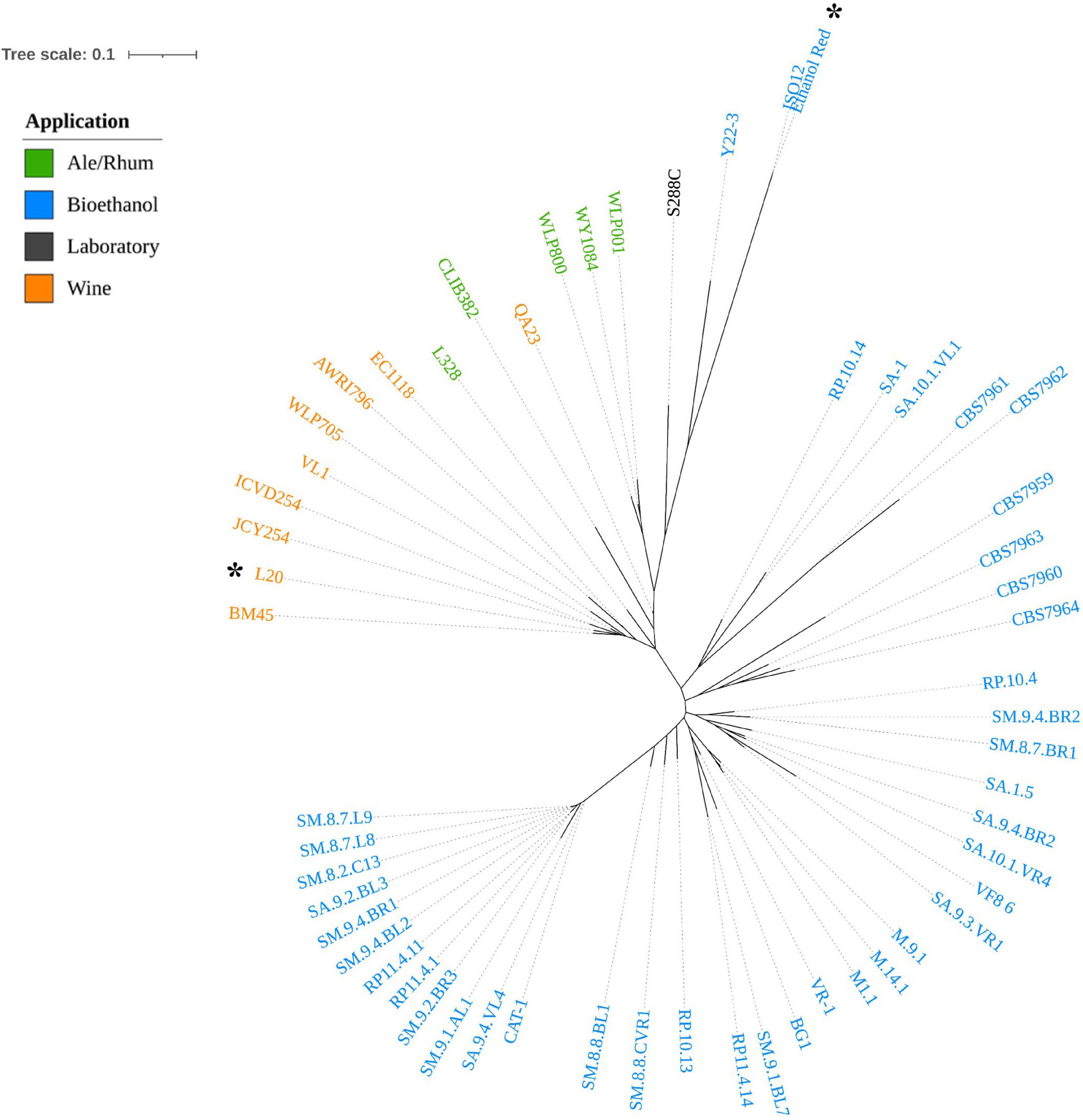
Figure 2. A maximum-likelihood phylogenetic tree based on SNP dataset representing the genetic distances among the 56 S. cerevisiae strains. The L20 and Ethanol Red strains sequenced in this study are marked with a black asterisk. The colors depict the industrial application of each strain (orange: Wine; green: Ale/Rhum; black: Laboratory; blue: Bioethanol).
The tree showed a clear separation of strains into two clusters: the first, represented by spirits-producing strains (Ale/Rhum and Wine), and the second including the fuel ethanol producers (Bioethanol). L20, which appeared to be clearly distinguished from other enological strains, was predicted as functionally related to commercial wine producers, in particular those isolated in Italy (BM45) and in France (JCY254 and ICVD254).
Interestingly, Ethanol Red, the benchmark yeast for first-generation bioethanol production, was phylogenetically assigned to the first cluster and closely related to the reference S288C. Ethanol Red and Y22-3 are the only bioethanol strains in this cluster. Y22-3 is a monospore engineered derivative of the stress-tolerant NRRL YB-210, which is a natural isolate from Costa Rican bananas and a progenitor of S288C (Mortimer and Johnston, 1986). However, the close relatedness to wine strains shows that Ethanol Red could share more genetic traits with domesticated strains rather than with the second cluster. These results are consistent with what was reported by Nagamatsu et al. (2021). However, Ethanol Red is still the most closely related strain to the sister branch of sugar-cane bioethanol strains, possibly representing a link between the two clusters. With the aim of selecting an alternative S. cerevisiae strain for the sustainable production of bioethanol from starch, these findings may indicate that enological yeast could be employed as promising host in genome engineering for the construction of a CBP starch-fermenting yeast.
The genome of L20 was assembled resulting in high-quality telomere-to-telomere reconstruction. The hybrid genomes obtained were used for the structural analysis by whole-genome alignment. The reference S. cerevisiae S288C R64-1-1 strain and one of the most studied industrial wine strains, EC1118 strain, were included (Figure 3).
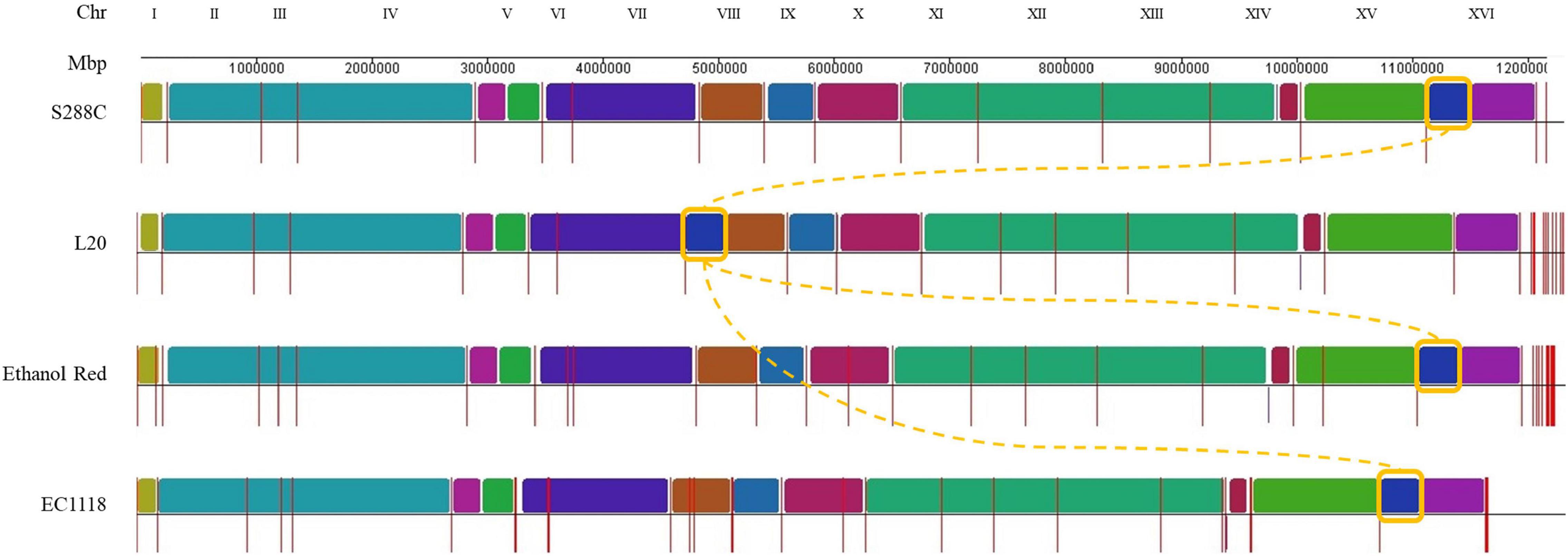
Figure 3. Multiple genome alignment of selected S. cerevisiae strains. The newly sequenced L20 and Ethanol Red genomes are compared to the reference S288C and with EC1118 strains. Chromosomes are ordered according to the S288C strain (first row) and syntenic regions are represented using different colors. Contiguous regions (chromosomes or scaffolds) are separated by red vertical bars. The translocation identified in L20 is highlighted using a yellow box.
Most of the L20 chromosomes assembled entirely in one contig except for chromosomes XII and XIV, which assembled in two fragments. The alignments in Figure 3 highlights a significant translocation between chromosomes VIII and XVI, which is a widespread translocation among enological yeast strains, although it has been identified in non-wine strains in the past as well (Perez-Ortin et al., 2002; Hou et al., 2014; Treu et al., 2014; García-Ríos and Guillamón, 2019; Crosato et al., 2020; Basile et al., 2021). It was correlated to an increased sulfur dioxide resistance, which is a critical parameter in winemaking. Since L20 was isolated from grape marcs (Gronchi et al., 2019), it can be assumed that such ecological background selected for this modification in L20 as well. This was further supported by the phylogenetic analysis (Figure 2) showing L20’ closest relatedness to commercial wine strains.
Yeast employed in industrial bioethanol fermentations are exposed to multiple stresses such as high sugar and ethanol concentrations, but also the presence of salts, sulfites, low pH, and bacterial contamination (Bauer and Pretorius, 2000; Favaro et al., 2019b; Brown et al., 2020). The variations of the gene copy number is usually associated with the adaptation to such specific conditions. Nagamatsu et al. (2021) examined the gene families having a positive selection in bioethanol yeast: due to the high metal concentration in sugarcane hydrolyzates, genes related to the metal homeostasis and detoxification were positively amplified in Brazilian strains. Strains producing bioethanol from corn, on the other hand, must cope with high ethanol concentration and high osmotic pressure, thus gene families related to membrane maintenance were often amplified.
All the strains in the dataset were investigated for CNV by considering S288C as reference. The full list of ORFs showing a CNV for at least one of the strains is reported in Supplementary Table 3. The analysis of over-represented genes was performed for three selected GO terms, namely those of utmost importance for bioethanol production (stress response, energy metabolism and transmembrane transport). The copy number was represented on a heatmap by color scale to better understand the relative abundance among the clusters of strains (Supplementary Table 4). The gene identifiers were pooled together and ordered by the location on the chromosomes, while strains were grouped according to their current industrial application.
Out of 199 genes considered in the heatmap, 130 were related to the transport mechanism. These included transmembrane transporters (73) but also the vesicle trafficking (53) from the endoplasmic reticulum and the Golgi apparatus, then moving to the secretory pathway (toward the plasma membrane) or the vacuole. L20 showed a great number of amplified genes related to the early vesicle-mediated transport (YPT1, SEC23, ERP1, ERP2, ERV46, EMP47, BST1, GYP8, RET2, MST27, and VPS8) but also exocytosis/secretion (SYN8, SNC1, SRO77, SEC4, and SED4). Transportation across the cell membrane is ensured by membrane transporters. Such over-represented genes in L20 belong to amino acid (AVT5, AGP1, DIP5, AGP3, GAP1, VBA3, VBA5, and RHB1), oligopeptide (OPT2), acetate (ADY2), allantoate (DAL5, DAL4), carboxylic acid (JEN1, BIO3, BIO4, and BIO5), glycerol (AQY3), glycerol-3-phosphate (GIT1), zinc ion (ZRT1), metal (ALR2, ENB1, FET5, and NFT1), organic hydroxy compound (HOL1), water (AQY1), nucleoside (FUN26) transporters. The monocarboxylic acids transporter MCH2 was not amplified in L20 and Ethanol Red but duplicated in other wine and bioethanol strains (CAT-1; Babrzadeh et al., 2012). The allantoate transporter SEO1 was absent in L20 but duplicated in Ethanol Red and many other strains (CAT-1; Babrzadeh et al., 2012).
In S. cerevisiae, sugar transporters play a critical role in biomass utilization by linking the extra- to the intra-cellular compartment. The number of genes involved in monosaccharide transmembrane transport was 12. MALx1 are low-affinity sucrose-H + symporters involved in maltose fermentation, which accounts for 50 to 60% of the total fermentable sugars in wort. The CNV analysis reported that Ethanol Red had a large amplification of MAL31 on chromosome II (5–6 copies), in agreement with the findings from Nagamatsu et al. (2021). With a few exceptions, Bioethanol strains showed at least a duplication of MAL31, while L20 was the only wine strain showing a higher copy number (2.5 copies). With regards to sugar metabolism, the L20 strain showed duplication for glucokinase (GLK1) and hexokinases (HXK1 and HXK2) involved in the glycolytic process, as well as the alcohol dehydrogenase (ADH4) involved in fermentation.
The extracellular environment is continuously changing during fermentation, thus yeast cells are adapting their metabolic response to different conditions (Bauer and Pretorius, 2000). Of the 199 genes, 69 were attributed to the GO term of stress response. Many genes related to the oxidative (NTG1, FRM2, HBN1, MXR2, GRX1, HSP30, HCM1, TRX3 CMK1, CUP1-1, CUP1-2, HYR1, MDL2, GEX1, and GEX2) and osmotic (HSP30 and YPD1) stress were amplified in the L20 strain. The HSP30 and HSP12 genes play a critical role in ethanol-induced stress, protecting the plasma membrane integrity. It is noteworthy to mention that among all the strains considered, L20 was the only strain that had an over-representation of both oxidative and stress genes.
This analysis revealed the genomic peculiarities of the L20 strain when compared to other relevant industrial strains. Moreover, the occurrence of higher copy numbers of genes linked to sugar transport (i.e., GLK1, HXT6, HXT7, MAL31, and MCH2) and ethanol tolerance (i.e., HSP12) support the higher ethanol production performance of strain L20 when compared to Ethanol Red under high-gravity SSF of broken rice (Gronchi et al., 2019). Furthermore, in strain L20 genes related to secretion had higher CNV compared to Ethanol Red (i.e., BOI1, SEC4, SNC1, SRO77, SWH1, and SYN8).
Overall, an important fraction of CNVs is localized in L20 on the chromosomes I, III, and VI, and a distinguishable CNV pattern can be observed for the bioethanol producers. The latter strains share a considerable number of deletions (red boxes in Supplementary Table 4) that are not common in wine and ale/rhum strains, confirming the evolutionary distance reported in Figure 2. Rather than a higher number of gene copies, strain L20 showed an amplification (mostly duplication) of a high number of genes correlated to the selected GO terms. EC1118, CBS7959, CBS7963, SA.9.2.BL3, and RP11.4.14 showed amplification for chromosomes I, III, and VI but none of them showed a similar CNV as was observed for L20.
A total of 5,626 ORFs were predicted for the nuclear genome of L20, out of which 4,903 were shared with S288C. Up to 43 Ty elements were identified in L20 (27 Ty1, 13 Ty2, 2 Ty3, 1 Ty4, and 0 Ty5). The number of delta sequences in L20 was higher than in Ethanol Red (263 versus 237, respectively), whereas 298 are annotated in S288C.
With reference to the genome of S288C, seven specific ORFs were detected in L20 (Supplementary Table 5). The RPS-BLAST annotation showed that such sequences codify for proteins belonging to the amino acid permease (SdaC), mannitol dehydrogenase (Mannitol_dh_C), acetate uptake transporter (Grp1_Fun34_YaaH), and superoxide dismutase (SodA) superfamilies.
The L20 and Ethanol Red strains were used as hosts for the expression of A. tubingensis amyA and glaA genes using delta integration and CRISPR/Cas9 strategies. For delta integration, linear amylase cassettes were constructed to randomly integrate at delta sites in combination with a kanMX cassette. The non-site-directed and random nature of delta integration resulted in large phenotypic variability in amylase secretion among isolates. This was evident when recombinants were cultivated on starch-containing plates and evaluated after Lugol staining (data not shown). Those displaying the largest halos were considered as the most efficient amylases secretors and designated as L20 dT8, dT12, dT25, and dT53 (S. cerevisiae L20 derivatives), as well as ER dT16, dT17, and dT22 (S. cerevisiae Ethanol Red derivatives), and were selected for further strain characterization. For the CRISPR/Cas9 strategy, a three-plasmid system was used to integrate the amylase cassettes into specific target sites in a controlled approach (IS4.1 and/or IS7.1; Claes et al., 2020), and was successfully implemented for both parental strains. Recombinants were designated according to the locus of integration and the amylase sequence.
The mitotic stability of delta or CRISPR/Cas9 recombinants was demonstrated by the preservation of antibiotic resistance and/or hydrolytic activity after 80 generations. PCR was performed to confirm gene integration (data not shown).
Recombinant L20 strains were cultured in YPD for 72 h and the supernatant used for SDS-PAGE analysis to confirm the secretion of heterologous amylases (Figure 4).
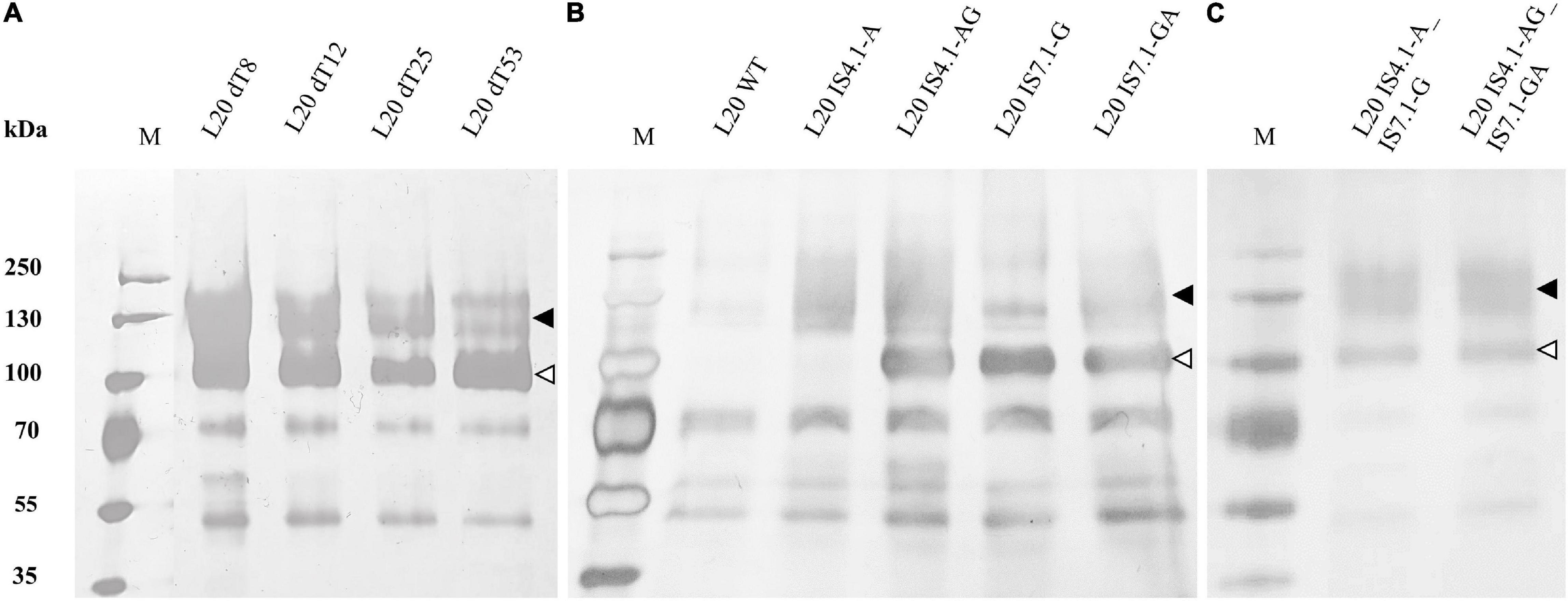
Figure 4. Supernatant from 72-h cultures of S. cerevisiae L20 strains was subjected to SDS-PAGE followed by silver staining. Arrows indicate the presence of recombinant protein species (▲) AmyA and (△) GlaA in the supernatant: (A) delta integrated, (B,C) CRISPR/Cas9 recombinants. WT indicates the parental strain. The PageRuler Prestained Protein Ladder (Fermentas) was used as protein size marker (M).
The SDS-PAGE analysis showed that AmyA and GlaA proteins were produced as differentially glycosylated species, with an average molecular size of 120 and 100 kDa, respectively. Similar results were found for Ethanol Red variants (Supplementary Figure 1). This is in agreement with previous studies (Viktor et al., 2013; Cripwell et al., 2017, 2019a,b).
The extracellular amylase activity was evaluated using liquid assays at 50°C on soluble starch (Figure 5).
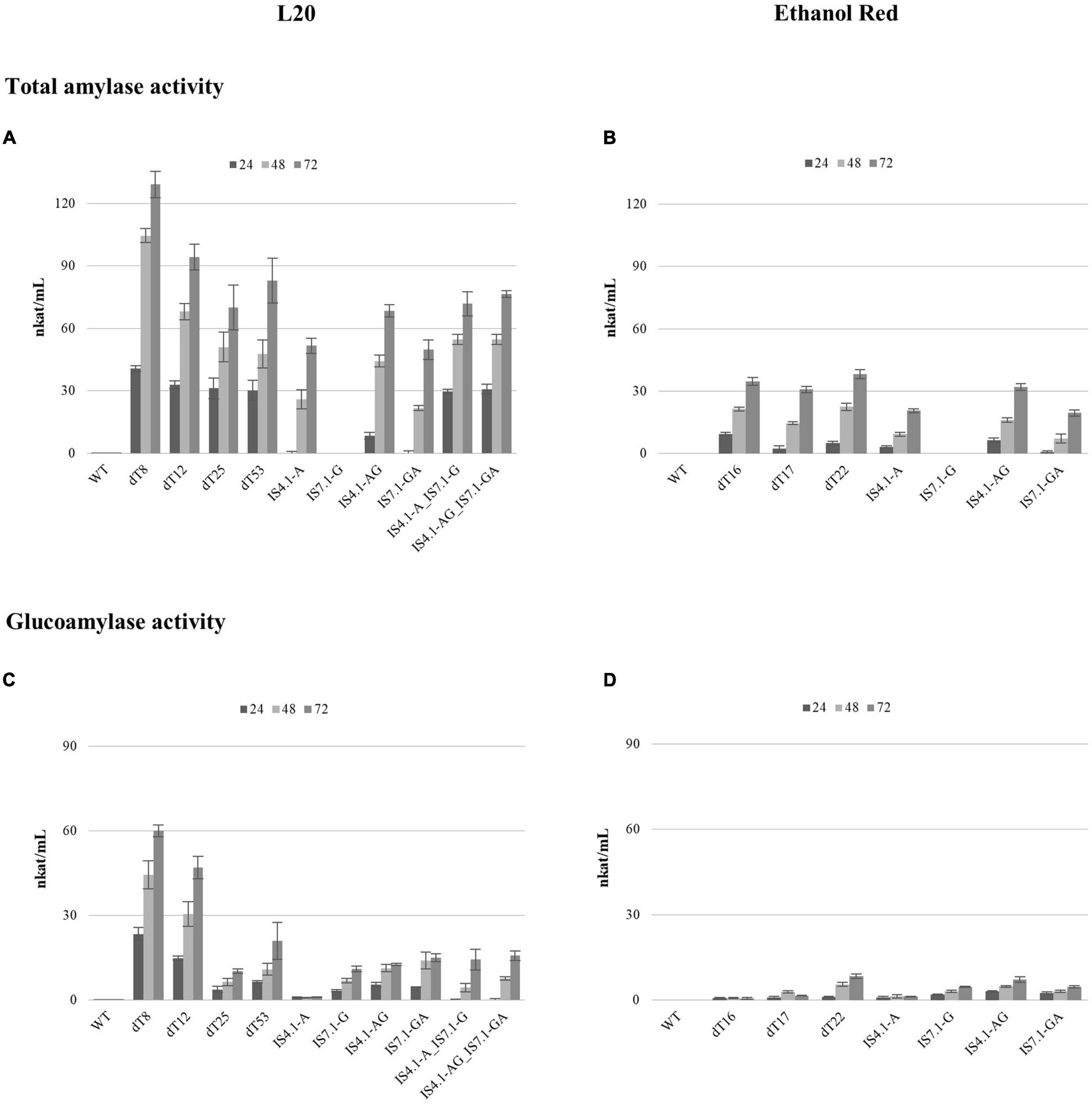
Figure 5. The total amylase (A,B) and glucoamylase (C,D) activity displayed by the S. cerevisiae L20 and Ethanol Red strains expressing amyA and/or glaA genes from A. tubingensis. WT indicates the parental strain. Enzymatic activity was determined using cell-free supernatant from cultures after 24, 48, and 72 h of incubation in YPD broth. Error bars represent the standard deviation from the mean of three replicates.
The DNS assay for secreted amylases revealed that enzymatic activity increased steadily for all strains over time (Figures 5A,B). However, the delta integrated strains showed a considerably higher hydrolytic activity, compared to those constructed using CRISPR/Cas9. L20 delta recombinants showed an average activity of 94 nkat/mL after 72 h of cultivation, which is 1.5-fold higher than the average activity obtained from the CRISPR/Cas9 recombinants (64 nkat/mL) (Figure 5A). The best performing L20 transformant was dT8, which exhibited 129 nkat/mL after 72 h growth. This varying degree of activity could be explained by the number/location of gene copies that were integrated.
Unexpectedly, such a large activity discrepancy among recombinants was not reported for Ethanol Red. The average activity displayed by delta integrated strains (35 nkat/mL) was 0.25-fold higher than the CRISPR/Cas9 strains (28 nkat/mL). The strain showing the highest activity was ER dT22 (38 nkat/mL at 72 h), which displayed a 1.36-fold higher activity than the CRISPR/Cas9 derivatives.
The activity of the CRISPR/Cas9 recombinant strains was significantly lower compared to those constructed using delta integration, and the results provide some interesting discussion points. Despite the locus, integration of a single, as well as both gene cassettes (simultaneously) resulted in much higher enzymatic activities in the case of L20 variants. By cloning a single amyA copy in locus IS4.1 (indicated by IS4.1-A), the L20 recombinant strain reached 51 nkat/mL after 72 h, whereas the maximum activity for an Ethanol Red transformants reached only 21 nkat/mL. When a double cassette amyA-glaA was inserted in the same locus (indicated by IS4.1-AG), in Ethanol Red, the activity increased by 0.52-fold (32 nkat/mL), while in strain L20 it only improved by 0.33-fold (68 nkat/mL). On average, a 2.1-fold higher activity was displayed for L20 compared to Ethanol Red strains. Moreover, when the same combination was integrated into the IS7.1 locus (strains IS7.1-GA) the hydrolytic activity was only 50 nkat/mL for L20 and 19.5 for Ethanol Red strains. Thus, the L20 strain showed a consistently higher activity over the Ethanol Red strain (2.56-fold).
Noteworthy, the L20 strain with the amyA and glaA cassettes integrated singularly (L20 IS4.1-A_IS7.1-G) and the transformant with two genes at both loci (L20 IS4.1-AG_IS7.1-GA) showed an activity of 71 and 76 nkat/mL, respectively, whereas a theoretical 2-fold improvement was expected.
This may be explained by the position of the integration event and the possible alteration of the chromosome structure, transcriptome, and epigenome (Flagfeldt et al., 2009; Wu et al., 2017; Gui et al., 2021). Wu et al. (2017) determined in a transcriptomic study the expression of an integrated fluorescent protein gene into different codifying genomic loci in S. cerevisiae, which revealed a genomic landscape of position effects besides the telomere and centromere regions. By observing their results, the closest codifying loci to our gRNA targets were considered as moderate (IS4.1) and high expression (IS7.1) levels. In this study, however, the heterologous cassette was inserted in intergenic regions, which are differently regulated and can result in modulated expression (Flagfeldt et al., 2009).
The glucoamylase activity assay revealed the same discrepancy between delta and CRISPR/Cas9 recombinants (Figures 5C,D), in agreement to those reported from the DNS assay (Figures 5A,B). L20 dT8 displayed a glucoamylase activity of 60 nkat/mL after 72 h and the average activity among L20 delta strains was 34.5 nkat/mL (Figure 5C). On the other hand, in L20 recombinants obtained by CRISPR/Cas9 the average was 14 nkat/mL. This could once again be due to the higher gene copy number integrated into the delta recombinants compared to the CRISPR/Cas9 strains. Strains containing a single glaA in IS7.1 (namely L20 IS7.1-G, L20 IS7.1-GA and L20 IS4.1-A_IS7.1-G) or the IS4.1 locus (L20 IS4.1-AG) displayed similar activity levels. Unexpectedly, the glucoamylase activity of L20 IS4.1-AG_IS7.1-GA, which contained two glaA copies was only slightly higher than L20 IS4.1-AG, which had a single glaA integration (16 and 13 nkat/mL, respectively).
Glucoamylase activity is well known to be limited to the availability of starch non-reducing ends (Görgens et al., 2015) produced by α-amylases, and this indicates that an α-amylase:glucoamylase ratio of 1:1 is not optimal for efficient starch hydrolysis. Therefore, higher α-amylase titers are required.
A possible explanation for the comparatively lower enzymatic activity for Ethanol Red derivatives could be a constitutive resilience of Ethanol Red to genome editing (as reported by Zhang et al., 2014 in industrial strain S. cerevisiae ATCC 4124), or a lower number of delta sequences compared to strain L20. Furthermore, in the case of the CRISPR/Cas9 engineered strains, Ethanol Red derivatives demonstrated a lower enzymatic activity compared to the respective L20 strains, suggesting that the intraspecific genomic variability plays a fundamental role in gene expression and, therefore, in construction of strains with high performance.
To our knowledge, this is one of the first reports demonstrating this in S. cerevisiae engineering for amylase production and will be of great importance to support the future development of efficient amylolytic CBP strains.
The use of delta sequences as target for genomic integration allows the simultaneous construction of strains with a varying number of gene copies and, therefore, different ratios of amylase:glucoamylase genes (Table 5).

Table 5. Results of Illumina sequencing of recombinant S. cerevisiae L20 and Ethanol Red strains obtained in this study.
The copy numbers of amyA and glaA in recombinant strains were consistent with the methodology used. The highest number of gene copies (1.02 for amyA and 2.51 for glaA) was found for L20 dT8, in accordance with the results of the enzymatic assays where L20 dT8 showed the highest activity (Figures 5A,C). Considering L20 dT8 as the best amylase producer, it can be assumed that the α-amylase:glucoamylase ratio of 1:2.5 (1.02:2.51) represent the baseline for further increase in gene copies and enzymatic activity. However, the dissimilar activity levels displayed by those having a single gene copy (CRISPR/Cas9 approach), suggested that external factors might affect the gene expression, resulting in lower enzymatic activities. As previously mentioned, the integration events could induce chromosome alterations and alter the transcriptome (Flagfeldt et al., 2009; Wu et al., 2017; Gui et al., 2021).
The recombinant strains demonstrating the highest enzymatic activity were further examined for their ability to convert soluble and raw starch (2% w/v) to ethanol under CBP conditions and high cell loading (OD600 5; Figure 6).
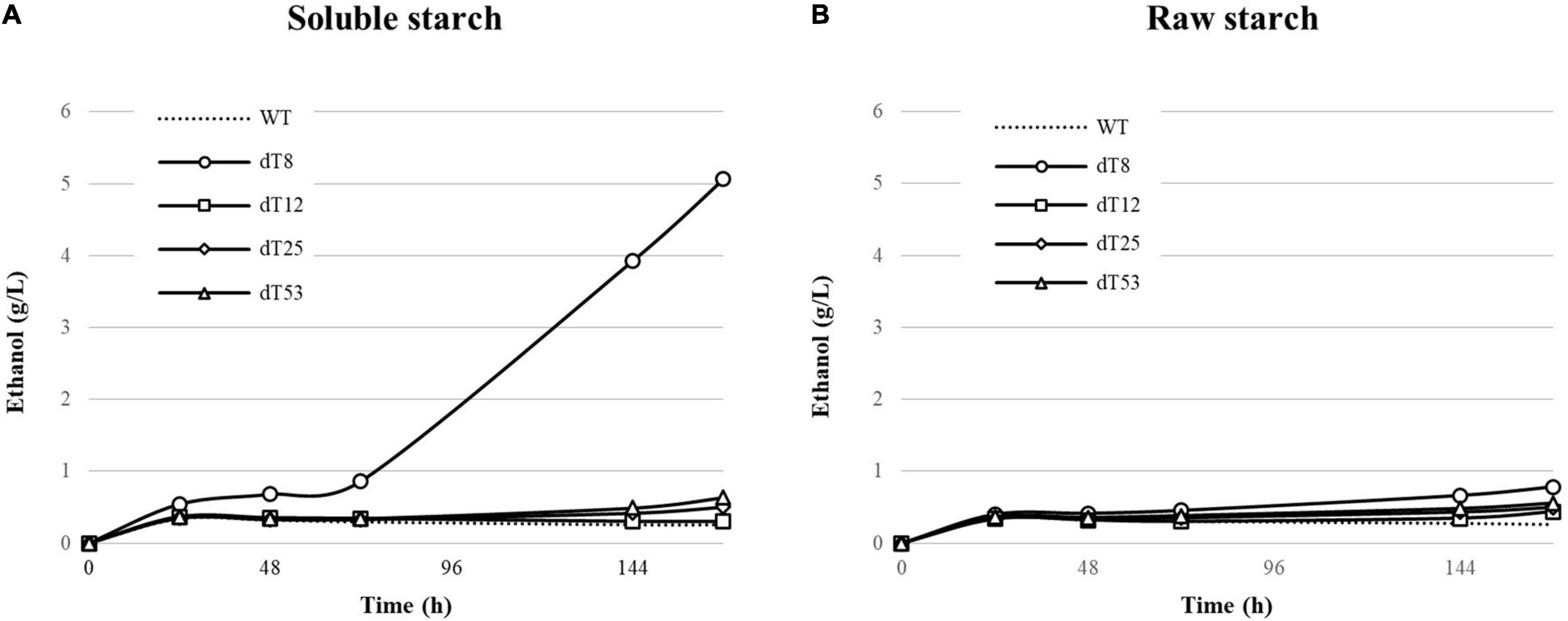
Figure 6. Ethanol production from 2% (w/v) soluble (A) and raw starch (B) by delta integrated S. cerevisiae L20 recombinants. WT indicates the parental strain. Strains were cultivated in YP medium with 0.05% glucose supplementation in oxygen-limited conditions. Values represent the mean of three replicates. The parental strain was used as reference.
All strains produced around 0.25 g/L ethanol (Figure 6A) from soluble starch within the first 24 h, corresponding to the theoretical conversion of the glucose supplementation. No further alcohol production was observed after this time point, indicating insufficient starch hydrolysis, except for L20 dT8 which produced up to 4 g/L ethanol (35% of theoretical yield) after 144 h, and this is in accordance with the corresponding enzyme activity (Figure 5A). By contrast, the other L20 recombinants (L20 dT25 and dT53) showed a modest ethanol production after 120 h. In particular, the strain dT12, although displaying good promise in terms of enzymatic activities at 50°C (Figure 5) produced ethanol levels slightly higher than those of the parental. This finding can be explained considering that at 30°C, temperature adopted for the CBP setting, both enzymes sharply decreased their activity (Viktor et al., 2013), thus releasing limited amount of glucose to support yeast cell growth. Moreover, the use of delta integration results in transformants with varying degrees of activity (Romanos et al., 1992; Cho et al., 1999; Favaro et al., 2012, 2015), which might not necessarily correlate to their fermentative abilities. Gene integrations can have caused a metabolic burden on S. cerevisiae L20 dT12 which in turn affects the strain’s ability to grow and ferment in a CBP context. This hypothesis is under investigation to further expand the scientific knowledge about metabolic burden in S. cerevisiae strains engineered for the expression of heterologous genes (Wu et al., 2016; Favaro et al., 2019a; Zahrl et al., 2019). The ethanol production from soluble starch was consistent with Nakamura et al. (1997) where the glucoamylase producing strain S. cerevisiae SR93 reached 3.3 g/L of ethanol after 48 h. Similarly, in Favaro et al. (2012) S. cerevisiae F2 and F3 produced 5.4 and 4.8 g/L of ethanol after 48 h, respectively.
On raw corn starch, the average ethanol production after 144 h was 0.48 g/L. As expected, L20 dT8 produced the highest ethanol titers 0.67 g/L (Figure 6B; 6% of theoretical yield). Despite the promising preliminary results, the hydrolytic activity was not sufficient to support the starch-to-ethanol route.
The amyA and glaA from A. tubingensis have previously been expressed using a multi-copy plasmid platform to engineer the S. cerevisiae Mnuα strain (Viktor et al., 2013). The recombinant strain was able to reach 80% of theoretical ethanol yield on 2% raw corn starch, demonstrating the hydrolytic ability of the amylases. Therefore, it is hypothesized that an increase in integrated copy number would improve the overall conversion of starch for the Ethanol Red and L20 derivatives. To enhance amylase secretion, further analysis has to be performed to identify the most favorable number of heterologous gene copies, as well as, the best amylase:glucoamylase ratio, while at the same time avoiding phenotypic alteration of the recombinant yeast strains. However, the lack of linearity between the number of integrated gene copies and the enzymatic activity suggests that the expression may be influenced by other, possibly strain-specific factors.
Overall, two different techniques were successfully employed for the development of amylase-producing yeast. They differ in terms of specificity of the target, number of gene copies and outputs. The delta integration approach resulted in recombinant strains displaying variable degrees of activity. The screening of numerous colonies could be time-consuming and difficult to handle. On the other hand, the CRISPR/Cas9 approach allows for a fine selection of target sites and modulation of gene copy numbers.
However, the combination of both approaches may lead to important advancements in CBP strain construction. For evaluation of a large number of recombinants concurrently, delta integration can ensure a rapid sorting of the most efficient in terms of saccharification and ethanol yields. After genome sequencing, the optimal ratio can be customizable and fine-tuned using CRISPR/Cas9.
Although the CRISPR/Cas9 approach was successful, one round of transformation was not sufficient for effective starch hydrolysis. Consistent improvements are expected to be achieved by identifying suitable genomic loci to integrate additional amylase copies (Jessop-Fabre et al., 2016).
In this work, a natural S. cerevisiae strain was described as a promising alternative for the development of a future CBP yeast. Genomic insight into L20’s genome revealed a distinctive profile for cellular transport systems, not only in terms of fermentative abilities but also for vesicle trafficking and secretion. This makes S. cerevisiae L20 an ideal candidate for the expression of heterologous hydrolase genes, which is fundamental for a CBP configuration. Future studies will investigate the fine tuning of amylase copy number for the efficient saccharification of starch, using L20 (or one of its derivatives).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: PRJNA762028.
NG performed the genome engineering, enzymatic activities, and fermentations experiments, participated in the experimental design, performed data analysis and data interpretation, and drafted the original manuscript. ND and LT performed genome assembly and whole genome analysis. RC participated in CBP strain construction, commented on the manuscript, and funding acquisition. MF-M participated in CBP strain construction. StC, JT, and WV commented and revised the manuscript. MB funding acquisition and commenting the revised manuscript. LF conceptualized the study and the experimental design, supervised the investigation, data interpretation, funding acquisition, editing and revision of the manuscript. SeC funding acquisition and commented the revised manuscript. All authors read and approved the final manuscript.
This work was supported by University of Padua [grants DOR1824847/18, DOR1827441/18, DOR1931153/19, DOR1928058/19, DOR2087054/20, DOR2084579/20, DOR2027838/20, and BIRD210708/21] and the National Research Foundation (NRF) of South Africa for financial support to grant holders and through the bilateral joint research project between Italy and South Africa [grants 113134 and ZA18MO04, respectively].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Authors are grateful to Valentino Pizzocchero (University of Padua, Italy) for HPLC analysis. Shaunita H. Rose (Stellenbosch University, South Africa) and Arne Claes (VIB, KU Leuven) are gratefully acknowledged for providing amylases sequences and CRISPR/Cas9 plasmid system.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.768562/full#supplementary-material
Supplementary Figure 1 | SDS-PAGE analysis of 72-h culture of recombinant S. cerevisiae Ethanol Red strains followed by silver staining. Arrows indicate the presence of recombinant protein species (▲) AmyA and (△) GlaA in the supernatant. WT indicates the parental strain. The PageRuler Prestained Protein Ladder (Fermentas) was used as protein size marker (M).
Adegboye, M. F., Ojuederie, O. B., Talia, P. M., and Babalola, O. O. (2021). Bioprospecting of microbial strains for biofuel production: metabolic engineering, applications, and challenges. Biotechnol. Biofuels 14:5. doi: 10.1186/s13068-020-01853-2
Aditiya, H. B., Mahlia, T. M. I, Chong, W. T., Nur, H., and Sebayang, A. H. (2016). Second generation bioethanol production: a critical review. Renew. Sustain. Energy Rev. 66, 631–653. doi: 10.1016/j.rser.2016.07.015
Antonangelo, A. T. B. F., Alonso, D. P., Ribolla, P. E. M., and Colombi, D. (2013). Microsatellite marker-based assessment of the biodiversity of native bioethanol yeast strains. Yeast 30, 307–317. doi: 10.1002/yea.2964
Babrzadeh, F., Jalili, R., Wang, C., Shokralla, S., Pierce, S., Robinson-Mosher, A., et al. (2012). Whole-genome sequencing of the efficient industrial fuel-ethanol fermentative Saccharomyces cerevisiae strain CAT-1. Mol. Genet. Genomics 287, 485–494. doi: 10.1007/s00438-012-0695-7
Basile, A., De Pascale, F., Bianca, F., Rossi, A., Frizzarin, M., De Bernardini, N., et al. (2021). Large-scale sequencing and comparative analysis of oenological Saccharomyces cerevisiae strains supported by nanopore refinement of key genomes. Food Microbiol. 97:103753. doi: 10.1016/j.fm.2021.103753
Basso, L. C., De Amorim, H. V., De Oliveira, A. J., and Lopes, M. L. (2008). Yeast selection for fuel ethanol production in Brazil. FEMS Yeast Res. 8, 1155–1163. doi: 10.1111/j.1567-1364.2008.00428.x
Bauer, F. F., and Pretorius, I. S. (2000). Yeast stress response and fermentation efficiency: how to survive the making of wine. S. Afr. J. Enol. Vitic. 21, 27–51. doi: 10.21548/21-1-3557
Borneman, A. R., Forgan, A. H., Kolouchova, R., Fraser, J. A., and Schmidt, S. A. (2016). Whole genome comparison reveals high levels of inbreeding and strain redundancy across the spectrum of commercial wine strains of Saccharomyces cerevisiae. G3 Genes Genomes Genet. 6, 957–971. doi: 10.1534/g3.115.025692
Brown, A., Waldheim, L., Landälv, I., Saddler, J., Ebadian, M., McMillan, J. D., et al. (2020). Advanced Biofuels – Potential for Cost Reduction. Paris, France: IEA Bioenergy.
Cagnin, L., Favaro, L., Gronchi, N., Rose, S. H., Basaglia, M., van Zyl, W. H., et al. (2019). Comparing laboratory and industrial yeast platforms for the direct conversion of cellobiose into ethanol under simulated industrial conditions. FEMS Yeast Res. 19, 1–13. doi: 10.1093/femsyr/foz018
Castro, A. M., Castilho, L. R., and Freire, D. M. G. (2011). An overview on advances of amylases production and their use in the production of bioethanol by conventional and non-conventional processes. Biomass Convers. Biorefin. 1, 245–255. doi: 10.1007/s13399-011-0023-1
Cho, K. M., Yoo, Y. J., and Kang, H. S. (1999). δ-Integration of endo/exo-glucanase and β-glucosidase genes into the yeast chromosomes for direct conversion of cellulose to ethanol. Enzyme Microb. Technol. 25, 23–30. doi: 10.1016/S0141-0229(99)00011-3
Choi, E. Y., Park, J. N., Kim, H. O., Shin, D. J., Chun, Y. H., Im, S. Y., et al. (2002). Construction of an industrial polyploid strain of Saccharomyces cerevisiae containing Saprolegnia ferax β-amylase gene and secreting β-amylase. Biotechnol. Lett. 24, 1785–1790. doi: 10.1023/A:1020613306127
Cinelli, B. A., Castilho, L. R., Freire, D. M. G., and Castro, A. M. (2015). A brief review on the emerging technology of ethanol production by cold hydrolysis of raw starch. Fuel 150, 721–729. doi: 10.1016/j.fuel.2015.02.063
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92. doi: 10.4161/fly.19695
Claes, A., Deparis, Q., Foulquié-Moreno, M. R., and Thevelein, J. M. (2020). Simultaneous secretion of seven lignocellulolytic enzymes by an industrial second-generation yeast strain enables efficient ethanol production from multiple polymeric substrates. Metab. Eng. 59, 131–141. doi: 10.1016/j.ymben.2020.02.004
Cripwell, R. A., Favaro, L., Viljoen-Bloom, M., and Van Zyl, W. H. (2020). Consolidated bioprocessing of raw starch to ethanol by Saccharomyces cerevisiae: achievements and challenges. Biotechnol. Adv. 42:107579. doi: 10.1016/j.biotechadv.2020.107579
Cripwell, R. A., Rose, S. H., Favaro, L., and Van Zyl, W. H. (2019a). Construction of industrial Saccharomyces cerevisiae strains for the efficient consolidated bioprocessing of raw starch. Biotechnol. Biofuels 12, 1–16. doi: 10.1186/s13068-019-1541-5
Cripwell, R. A., Rose, S. H., Viljoen-Bloom, M., and Van Zyl, W. H. (2019b). Improved raw starch amylase production by Saccharomyces cerevisiae using codon optimisation strategies. FEMS Yeast Res. 19, 1–14. doi: 10.1093/femsyr/foy127
Cripwell, R. A., Rose, S. H., and Van Zyl, W. H. (2017). Expression and comparison of codon optimised Aspergillus tubingensis amylase variants in Saccharomyces cerevisiae. FEMS Yeast Res. 17, 1–12. doi: 10.1093/femsyr/fox040
Crosato, G., Nadai, C., Carlot, M., Garavaglia, J., Righetto Ziegler, D., Rossi, R. C., et al. (2020). The impact of CUP1 gene copy-number and XVI-VIII/XV-XVI translocations on copper and sulfite tolerance in vineyard Saccharomyces cerevisiae strain populations. FEMS Yeast Res. 20:foaa028. doi: 10.1093/femsyr/foaa028
Cunha, J. T., Romaní, A., Inokuma, K., Johansson, B., Hasunuma, T., Kondo, A., et al. (2020). Consolidated bioprocessing of corn cob-derived hemicellulose: engineered industrial Saccharomyces cerevisiae as efficient whole cell biocatalysts. Biotechnol. Biofuels 13:138. doi: 10.1186/s13068-020-01780-2
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Darling, A. E., Mau, B., and Perna, N. T. (2010). ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
den Haan, R., Rose, S. H., Cripwell, R. A., Trollope, K. M., Myburgh, M. W., Viljoen-Bloom, M., et al. (2021). Heterologous production of cellulose-and starch-degrading hydrolases to expand Saccharomyces cerevisiae substrate utilization: lessons learnt. Biotechnol. Adv. 53:107859. doi: 10.1016/j.biotechadv.2021.107859
Duan, S. F., Han, P. J., Wang, Q. M., Liu, W. Q., Shi, J. Y., Li, K., et al. (2018). The origin and adaptive evolution of domesticated populations of yeast from Far East Asia. Nat. Commun. 9:2690. doi: 10.1038/s41467-018-05106-7
Dunn, B., Richter, C., Kvitek, D. J., Pugh, T., and Sherlock, G. (2012). Analysis of the Saccharomyces cerevisiae pan-genome reveals a pool of copy number variants distributed in diverse yeast strains from differing industrial environments. Genome Res. 22, 908–924. doi: 10.1101/gr.130310.111
Favaro, L., Basaglia, M., Saayman, M., Rose, S. H., van Zyl, W. H., and Casella, S. (2010). Engineering amylolytic yeasts for industrial bioethanol production. Chem. Eng. Trans. 20, 97–102. doi: 10.3303/CET1020017
Favaro, L., Cagnin, L., Corte, L., Roscini, L., De Pascale, F., Treu, L., et al. (2019a). Metabolomic alterations do not induce metabolic burden in the industrial yeast M2n[pBKD2-Pccbgl1]-C1 engineered by multiple δ-integration of a fungal β-glucosidase gene. Front. Bioeng. Biotechnol. 7:376. doi: 10.3389/fbioe.2019.00376
Favaro, L., Jansen, T., and Van Zyl, W. H. (2019b). Exploring industrial and natural Saccharomyces cerevisiae strains for the bio-based economy from biomass: the case of bioethanol. Crit. Rev. Biotechnol. 39, 800–816. doi: 10.1080/07388551.2019.1619157
Favaro, L., Jooste, T., Basaglia, M., Rose, S. H., Saayman, M., Görgens, J. F., et al. (2012). Codon-optimized glucoamylase sGAI of Aspergillus awamori improves starch utilization in an industrial yeast. Appl. Microbiol. Biotechnol. 95, 957–968. doi: 10.1007/s00253-012-4001-8
Favaro, L., Jooste, T., Basaglia, M., Rose, S. H., Saayman, M., Görgens, J. F., et al. (2013). Designing industrial yeasts for the consolidated bioprocessing of starchy biomass to ethanol. Bioengineered 4, 97–102. doi: 10.4161/bioe.22268
Favaro, L., Viktor, M. J., Rose, S. H., Viljoen-Bloom, M., Van Zyl, W. H., Basaglia, M., et al. (2015). Consolidated bioprocessing of starchy substrates into ethanol by industrial Saccharomyces cerevisiae strains secreting fungal amylases. Biotechnol. Bioeng. 112, 1751–1760. doi: 10.1002/bit.25591
Flagfeldt, B. D., Siewers, V., Huang, L., and Nielsen, J. (2009). Characterization of chromosomal integration sites for heterologous gene expression in Saccharomyces cerevisiae. Yeast 26, 545–551. doi: 10.1002/yea.1705
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gallone, B., Steensels, J., Prahl, T., Soriaga, L., Saels, V., Herrera-Malaver, B., et al. (2016). Domestication and divergence of Saccharomyces cerevisiae beer yeasts. Cell 166, 1397–1410.e16. doi: 10.1016/j.cell.2016.08.020
García-Ríos, E., and Guillamón, J. M. (2019). “Mechanisms of yeast adaptation to wine fermentations,” in Yeasts in Biotechnology and Human Health. Progress in Molecular and Subcellular Biology, ed. I. Sá-Correia (Cham: Springer), doi: 10.1007/978-3-030-13035-0_2
Gietz, R. D., and Schiestl, R. H. (2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34. doi: 10.1038/nprot.2007.13
Görgens, J. F., Bressler, D. C., and Van Rensburg, E. (2015). Engineering Saccharomyces cerevisiae for direct conversion of raw, uncooked or granular starch to ethanol. Crit. Rev. Biotechnol. 35, 369–391. doi: 10.3109/07388551.2014.888048
Gronchi, N., Favaro, L., Cagnin, L., Brojanigo, S., Pizzocchero, V., Basaglia, M., et al. (2019). Novel yeast strains for the efficient saccharification and fermentation of starchy by-products to bioethanol. Energies 12:714. doi: 10.3390/en12040714
Gui, Q., Deng, S., Zhou, Z. Z., Cao, W., Zhang, X., Shi, W., et al. (2021). Transcriptome analysis in yeast reveals the externality of position effects. Mol. Biol. Evol. 38, 3294–3307. doi: 10.1093/molbev/msab104
Gupta, A., and Verma, J. P. (2015). Sustainable bio-ethanol production from agro-residues: a review. Renew. Sustain. Energy Rev. 41, 550–567. doi: 10.1016/j.rser.2014.08.032
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2018). UFBoot2: improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Hou, J., Friedrich, A., de Montigny, J., and Schacherer, J. (2014). Chromosomal rearrangements as a major mechanism in the onset of reproductive isolation in Saccharomyces cerevisiae. Curr. Biol. 24, 1153–1159. doi: 10.1016/j.cub.2014.03.063
Ishizaki, H., and Hasumi, K. (2014). “Ethanol production from biomass,” in Research Approaches to Sustainable Biomass Systems, eds S. Tojo and T. Hirasawa (Cambridge, MA, USA: Academic Press), 243–258. doi: 10.1016/B978-0-12-404609-2.00010-6
Jakočiūnas, T., Jensen, M. K., and Keasling, J. D. (2016). CRISPR/Cas9 advances engineering of microbial cell factories. Metab. Eng. 34, 44–59. doi: 10.1016/j.ymben.2015.12.003
Jansen, M. L. A., Bracher, J. M., Papapetridis, I., Verhoeven, M. D., de Bruijn, H., de Waal, P. P., et al. (2017). Saccharomyces cerevisiae strains for second-generation ethanol production: from academic exploration to industrial implementation. FEMS Yeast Res. 17, 1–20. doi: 10.1093/femsyr/fox044
Jessop-Fabre, M. M., Jakočiūnas, T., Stovicek, V., Dai, Z., Jensen, M. K., Keasling, J. D., et al. (2016). EasyClone-MarkerFree: a vector toolkit for marker-less integration of genes into Saccharomyces cerevisiae via CRISPR-Cas9. Biotechnol. J. 11, 1110–1117. doi: 10.1002/biot.201600147
Kalyaanamoorthy, S., Minh, B., Wong, T., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kang, N. Y., Park, J. N., Chin, J. E., Lee, H. B., Im, S. Y., and Bai, S. (2003). Construction of an amylolytic industrial strain of Saccharomyces cerevisiae containing the Schwanniomyces occidentalis α-amylase gene. Biotechnol. Lett. 25, 1847–1851. doi: 10.1023/A:1026281627466
Khaw, T. S., Katakura, Y., Koh, J., Kondo, A., Ueda, M., and Shioya, S. (2006). Evaluation of performance of different surface-engineered yeast strains for direct ethanol production from raw starch. Appl. Microbiol. Biotechnol. 70, 573–579. doi: 10.1007/s00253-005-0101-z
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kricka, W., Fitzpatrick, J., and Bond, U. (2014). Metabolic engineering of yeasts by heterologous enzyme production for degradation of cellulose and hemicellulose from biomass: a perspective. Front. Microbiol. 5:174. doi: 10.3389/fmicb.2014.00174
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. doi: 10.1038/227680a0
Legras, J. L., Galeote, V., Bigey, F., Camarasa, C., Marsit, S., Nidelet, T., et al. (2018). Adaptation of S. cerevisiae to fermented food environments reveals remarkable genome plasticity and the footprints of domestication. Mol. Biol. Evol. 35, 1712–1727. doi: 10.1093/molbev/msy066
Letunic, I., and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Liao, B., Hill, G. A., and Roesler, W. J. (2012). Stable expression of barley α-amylase in S. cerevisiae for conversion of starch into bioethanol. Biochem. Eng. J. 64, 8–16. doi: 10.1016/j.bej.2012.02.004
Lin, Y., and Tanaka, S. (2006). Ethanol fermentation from biomass resources: current state and prospects. Appl. Microbiol. Biotechnol. 69, 627–642. doi: 10.1007/s00253-005-0229-x
Lopes, T. S., De Wijs, I. J., Steenhauer, S. I, Verbakel, J., and Planta, R. J. (1996). Factors affecting the mitotic stability of high-copy-number integration into the ribosomal DNA of Saccharomyces cerevisiae. Yeast 12, 467–477. doi: 10.1002/(SICI)1097-0061(199604)12:5<467::AID-YEA933<3.0.CO;2-3
Lopes, T. S., Klootwijk, J., Veenstra, A. E., van der Aar, P. C., van Heerikhuizen, H., Raué, H. A., et al. (1989). High-copy-number integration into the ribosomal DNA of Saccharomyces cerevisiae: a new vector for high-level expression. Gene 79, 199–206. doi: 10.1016/0378-1119(89)90202-3
Lynd, L. R., Laser, M. S., Bransby, D., Dale, B. E., Davison, B., Hamilton, R., et al. (2008). How biotech can transform biofuels. Nat. Biotechnol. 26, 169–172. doi: 10.1038/nbt0208-169
McBride, J. E. E., Deleault, K. M., Lynd, L. R., and Pronk, J. T. (2008). Recombinant Yeast Strains Expressing Tethered Cellulase Enzymes. U.S. Patent Application No. 12/516, 175. Hanover, NH: The Trustees of Dartmouth College.
McIlwain, S. J., Peris, D., Sardi, M., Moskvin, O. V., Zhan, F., Myers, K. S., et al. (2016). Genome sequence and analysis of a stress-tolerant, wild-derived strain of Saccharomyces cerevisiae used in biofuels research. G3 Genes Genomes Genet. 6, 1757–1766. doi: 10.1534/g3.116.029389
Miller, G. L. (1959). Use of Dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31, 426–428. doi: 10.1021/ac60147a030
Mohanty, S. K., and Swain, M. R. (2019). “Bioethanol production from corn and wheat: food, fuel, and future,” in Ramachandran, Bioethanol Production from Food Crops, Chap. 3, eds C. Ramesh and S. Ray (Cambridge, Massachusetts: Academic Press), 45–59. doi: 10.1016/B978-0-12-813766-6.00003-5
Mohd Azhar, S. H., Abdulla, R., Jambo, S. A., Marbawi, H., Gansau, J. A., Mohd Faik, A. A., et al. (2017). Yeasts in sustainable bioethanol production: a review. Biochem. Biophys. Rep. 10, 52–61. doi: 10.1016/j.bbrep.2017.03.003
Mortimer, R. K., and Johnston, J. R. (1986). Genealogy of principal strains of the yeast genetic stock center. Genetics 113, 35–43. doi: 10.1093/genetics/113.1.35
Murai, T., Yoshino, T., Ueda, M., Haranoya, I., Ashikari, T., Yoshizumi, H., et al. (1998). Evaluation of the function of arming yeast displaying glucoamylase on its cell surface by direct fermentation of corn to ethanol. J. Ferment. Bioeng. 86, 569–572. doi: 10.1016/S0922-338X(99)80008-7
Myburgh, M. W., Cripwell, R. A., Favaro, L., and van Zyl, W. H. (2019). Application of industrial amylolytic yeast strains for the production of bioethanol from broken rice. Bioresour. Technol. 294:122222. doi: 10.1016/j.biortech.2019.122222
Nagamatsu, S. T., Coutouné, N., José, J., Fiamenghi, M. B., Pereira, G. A. G., Oliveira, J. V. D. C., et al. (2021). Ethanol production process driving changes on industrial strains. FEMS Yeast Res. 21, 1–10. doi: 10.1093/femsyr/foaa071
Nakamura, Y., Kobayashi, F., Ohnaga, M., and Sawada, T. (1997). Alcohol fermentation of starch by a genetic recombinant yeast having glucoamylase activity. Biotechnol. Bioeng. 53, 21–25. doi: 10.1002/(SICI)1097-0290(19970105)53:1<21::AID-BIT4<3.0.CO;2-0
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nieto, A., Prieto, J. A., and Sanz, P. (1999). Stable high-copy-number integration of Aspergillus oryzae α-amylase cDNA in an industrial Baker’s yeast strain. Biotechnol. Prog. 15, 459–466. doi: 10.1021/bp9900256
O’Connell, K. L., and Stults, J. T. (1997). Identification of mousse liver proteins on 2D electrophoresis gels by MALDI-MS of in situs enzymatic digests. Electrophoresis 18, 349–359.
Ozer, E. A., Allen, J. P., and Hauser, A. R. (2014). Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genomics 15:737. doi: 10.1186/1471-2164-15-737
Perez-Ortin, J. E., Querol, A., Puig, S., and Barrio, E. (2002). Molecular characterization of a chromosomal rearrangement involved in the adaptive evolution of yeast strains. Genome Res. 12, 1533–1539. doi: 10.1101/gr.436602
Peter, J., De Chiara, M., Friedrich, A., Yue, J. X., Pflieger, D., Bergström, A., et al. (2018). Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556, 339–344. doi: 10.1038/s41586-018-0030-5
Riley, L. A., and Guss, A. M. (2021). Approaches to genetic tool development for rapid domestication of non-model microorganisms. Biotechnol. Biofuels 14:30. doi: 10.1186/s13068-020-01872-z
Robak, K., and Balcerek, M. (2018). Review of second generation bioethanol production from residual biomass. Food Technol. Biotechnol. 56, 174–187. doi: 10.17113/ftb.56.02.18.5428
Roggenkamp, E., Giersch, R. M., Wedeman, E., Eaton, M., Turnquist, E., Schrock, M. N., et al. (2017). CRISPR-UnLOCK: multipurpose Cas9-based strategies for conversion of yeast libraries and strains. Front. Microbiol. 8:1773. doi: 10.3389/fmicb.2017.01773
Romanos, M. A., Scorer, C. A., and Clare, J. J. (1992). Foreign gene expression in yeast: a review. Yeast 8, 423–488. doi: 10.1002/yea.320080602
Sambrook, J., and Russell, D. W. (2001). Molecular Cloning: a Laboratory Manual, 3rd Edn. Cold Spring Harb. NY: Cold Spring Harbor Laboratory Press.
Schacherer, J., Shapiro, J., Ruderfer, D., and Kruglyak, L. (2009). Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature 458, 342–345. doi: 10.1038/nature07670
Sharma, S., Varghese, E., Arora, A., Singh, K. N., Singh, S., Nain, L., et al. (2018). Augmenting pentose utilization and ethanol production of native Saccharomyces cerevisiae LN using medium engineering and response surface methodology. Front. Bioeng. Biotechnol. 6:132. doi: 10.3389/fbioe.2018.00132
Suvakov, M., Panda, A., Diesh, C., Holmes, I., and Abyzov, A. (2021). CNVpytor: a tool for CNV/CNA detection and analysis from read depth and allele imbalance in whole genome sequencing. bioRxiv [Preprint]. doi: 10.1101/2021.01.27.428472
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O., and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990. doi: 10.1101/gr.081612.108
Treu, L., Toniolo, C., Nadai, C., Sardu, A., Giacomini, A., Corich, V., et al. (2014). The impact of genomic variability on gene expression in environmental Saccharomyces cerevisiae strains. Environ. Microbiol. 16, 1378–1397. doi: 10.1111/1462-2920.12327
Viktor, M. J., Rose, S. H., Van Zyl, W. H., and Viljoen-Bloom, M. (2013). Raw starch conversion by Saccharomyces cerevisiae expressing Aspergillus tubingensis amylases. Biotechnol. Biofuels 6:167. doi: 10.1186/1754-6834-6-167
Vohra, M., Manwar, J., Manmode, R., Padgilwar, S., and Patil, S. (2014). Bioethanol production: feedstock and current technologies. J. Environ. Chem. Eng. 2, 573–584. doi: 10.1016/j.jece.2013.10.013
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wall, L., Christiansen, T., and Orwant, J. (2000). Programming perl. Sebastopol: O’Reilly media, Inc.
Wallace-Salinas, V., Brink, D. P., Ahrén, D., and Gorwa-Grauslund, M. F. (2015). Cell periphery-related proteins as major genomic targets behind the adaptive evolution of an industrial Saccharomyces cerevisiae strain to combined heat and hydrolysate stress. BMC Genomics 16:514. doi: 10.1186/s12864-015-1737-4
Wu, G., Yan, Q., Jones, J. A., Tang, Y. J., Fong, S. S., and Koffas, M. A. G. (2016). Metabolic burden: cornerstones in synthetic biology and metabolic engineering applications. Trends Biotechnol. 34, 652–664. doi: 10.1016/j.tibtech.2016.02.010
Wu, X. L., Li, B. Z., Zhang, W. Z., Song, K., Qi, H., Dai, J., et al. (2017). Genome-wide landscape of position effects on heterogeneous gene expression in Saccharomyces cerevisiae. Biotechnol. Biofuels 10:189. doi: 10.1186/s13068-017-0872-3
Yamada, R., Bito, Y., Adachi, T., Tanaka, T., Ogino, C., Fukuda, H., et al. (2009). Efficient production of ethanol from raw starch by a mated diploid Saccharomyces cerevisiae with integrated α-amylase and glucoamylase genes. Enzyme Microb. Technol. 44, 344–349. doi: 10.1016/j.enzmictec.2009.01.001
Zabed, H., Sahu, J. N., Suely, A., Boyce, A. N., and Faruq, G. (2017). Bioethanol production from renewable sources: current perspectives and technological progress. Renew. Sustain. Energy Rev. 71, 475–501. doi: 10.1016/j.rser.2016.12.076
Zahrl, R. J., Gasser, B., Mattanovich, D., and Ferrer, P. (2019). Detection and elimination of cellular bottlenecks in protein-producing yeasts. Methods Mol. Biol. 1923, 75–95. doi: 10.1007/978-1-4939-9024-5_2
Zhang, G. C., Kong, I. I., Kim, H., Liu, J. J., Cate, J. H. D., and Jin, Y. S. (2014). Construction of a quadruple auxotrophic mutant of an industrial polyploid Saccharomyces cerevisiae strain by using RNA-guided Cas9 nuclease. Appl. Environ. Microbiol. 80, 7694–7701. doi: 10.1128/AEM.02310-14
Keywords: Saccharomyces cerevisiae, delta integration, CRISPR/Cas9, starch, Ethanol Red, consolidated bioprocessing, amylases, bioethanol
Citation: Gronchi N, De Bernardini N, Cripwell RA, Treu L, Campanaro S, Basaglia M, Foulquié-Moreno MR, Thevelein JM, Van Zyl WH, Favaro L and Casella S (2022) Natural Saccharomyces cerevisiae Strain Reveals Peculiar Genomic Traits for Starch-to-Bioethanol Production: the Design of an Amylolytic Consolidated Bioprocessing Yeast. Front. Microbiol. 12:768562. doi: 10.3389/fmicb.2021.768562
Received: 31 August 2021; Accepted: 17 December 2021;
Published: 20 January 2022.
Edited by:
Soo Rin Kim, Kyungpook National University, South KoreaReviewed by:
Nuno Pereira Mira, University of Lisbon, PortugalCopyright © 2022 Gronchi, De Bernardini, Cripwell, Treu, Campanaro, Basaglia, Foulquié-Moreno, Thevelein, Van Zyl, Favaro and Casella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorenzo Favaro, bG9yZW56by5mYXZhcm9AdW5pcGQuaXQ=
†These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.