
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 29 November 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.766464
This article is part of the Research Topic The CRISPR/Cas System in Pathogen Resistance, Virulence, Diagnosis and Typing View all 8 articles
 Hailong Lv1,2
Hailong Lv1,2 Jian Wang1
Jian Wang1 Jian Zhang1
Jian Zhang1 Yijian Chen3,4Lei Yin5Dian Jin6Dayong Gu1*Huailong Zhao7*Yong Xu1*
Yijian Chen3,4Lei Yin5Dian Jin6Dayong Gu1*Huailong Zhao7*Yong Xu1* Jin Wang1,8*
Jin Wang1,8*The CRISPR diagnostic (CRISPR-Dx) technology that employs the trans-cleavage activities has shown great potential in diagnostic sensitivity, specificity, convenience, and portability, and has been recognized as the next-generation diagnostic methods. However, due to the lack of standardized definition of Cas trans-cleavage enzymatic units, it is difficult to standardize the present CRISPR-Dx systems, which have undoubtedly impeded the development of the CRISPR-Dx industry. To solve the problem, we here first systematically optimized the reaction systems for Cas12a, and then defined its trans-cleavage units (transU), which we believe will be of great importance and interest to researchers in both molecular diagnostic industry and basic research. Moreover, a simple protocol was provided to facilitate a step-by-step measurement of the Cas12a transU, which can also act as a reference for the definition of the transU for other Cas proteins.
The recently characterized trans-cleavage activities of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) Cas enzymes such as Cas12 and Cas13 has undoubtedly sparked the interests in developing CRISPR diagnostic (CRISPR-Dx) systems (Gootenberg et al., 2017, 2018; Chen et al., 2018; Li et al., 2018b; Li Y. et al., 2019). The trans-cleavage activities of Cas12a were first discovered during the characterization of its cleavage behaviors against target single-stranded DNA (ssDNA) (Chen et al., 2018; Li et al., 2018a; Li Y. et al., 2019). Under the guidance of CRISPR RNA (crRNA), Cas12a recognizes target DNA, forms a ternary complex of Cas12a, crRNA, and target DNA, and then trans-cleaves non-specific ssDNA in the system, which activity is designated as the trans-cleavage activity (Chen et al., 2018; Li et al., 2018a). Similar ssDNA trans-cleavage activities have been found in other Cas12 subtypes such as the thermophilic Cas12b and the Cas12f (previously known as Cas14) that specifically recognizes ssDNA targets (Leung et al., 2021). With the employment of these Cas12 trans-cleavage activities, dozens of CRISPR-Dx systems have been successfully created, among which HOLMES (one-hour low-cost multipurpose highly efficient system), HOLMESv2 and DETECTR (DNA Endonuclease Targeted CRISPR Trans Reporter) are the representatives (Chen et al., 2018; Li et al., 2018b; Li L. et al., 2019).
HOLMES first uses the polymerase chain reaction (PCR) technology to amplify the target nucleic acids and then detects the amplicons with the Cas12a trans-cleavage activities, illuminating the fluorescent signals. HOLMES detects target nucleic acid sequences with the attomolar sensitivity and distinguishes single-base mismatches (Li et al., 2018b). However, the PCR amplification and Cas12a trans-cleavage processes are separated in HOLMES and the transfer of amplicons may result in aerosol contamination. To solve the problem, the physically separated amplification and Cas12a trans-cleavage steps may either be designed in a closed microfluidic system or operated in a standard PCR laboratory that contains separate rooms for distinct purposes; however, it may cause inconvenience in use of the HOLMES technology. Alternatively, HOLMESv2 integrates the thermophilic Cas12b with the Loop-mediated isothermal amplification (LAMP) and supports one-pot diagnosis of target nucleic acids (Li L. et al., 2019). Unlike Cas12, Cas13 targets RNA and is triggered to trans-cleave collateral single-stranded RNA reporter sequences in the system, with which diagnostic systems such as SHERLOCK (Specific High Sensitivity Enzymatic Reporter UnLOCKing) (Gootenberg et al., 2017) were developed. Then, with the employment of Cas12 and Cas13, SHERLOCKv2 was created to simultaneously detect multiple target nucleic acids in one diagnostic system (Gootenberg et al., 2018). Recently, with the use of either tandem crRNAs or remarkably reduced reaction volumes for target nucleic acid detection, diagnostic sensitivities can be greatly improved, and several amplification-free systems have been successfully developed (Fozouni et al., 2021; Liu et al., 2021; Shinoda et al., 2021; Tian et al., 2021; Yue et al., 2021).
In comparison with traditional molecular diagnostic methods such as polymerase chain reaction (PCR), CRISPR-Dx methods have shown great advantages in sensitivity, specificity, simplicity and portability and have been well recognized as the next-generation diagnostic methods (Chertow, 2018). Therefore, soon after the outbreak of the COVID-19 pandemic, a large number of diagnostic methods were successfully developed, most of which employed the immunochromatography and the real-time PCR technologies and have played an important role in the epidemic prevention and control. Meanwhile, dozens of CRISPR-Dx methods were also developed for COVID-19 diagnosis, including the one-pot systems that combine either recombinase polymerase amplification (RPA) with Cas12a (Aman et al., 2020; Ding et al., 2020) or LAMP with Cas12b (Joung et al., 2020), all showing advantages in diagnostic sensitivity, specificity, and convenience (Han et al., 2021). Noticeably, within 3 years since the first report of the CRISPR-Dx technology in 2017 (Gootenberg et al., 2017), the United States Food and Drug Administration (FDA) granted the Emergency Use Authorization (EUA) to Sherlock biosciences for the CRISPR SARS-CoV-2 rapid diagnostic kit (Martin et al., 2021), which once again confirmed the great potential of CRISPR technologies in pathogen diagnosis.
However, due to the lack of unified trans-cleavage units (transU), the Cas enzymes used in present systems are usually quantitated in concentration instead of enzymatic units. As the Cas specific trans-cleavage activities may vary from different commercial providers, distinct diagnostic performances can be obtained for a defined CRISPR-Dx system if the Cas enzyme is definitized in concentration, which will undoubtedly limit large-scale industrial applications of CRISPR-Dx technologies. Besides, it is well known that enzymatic cleavage activities can be affected by many reaction factors, including ions, salt concentration, pH values and temperature. Therefore, we here systematically studied the factors affecting Cas12a trans-cleavage activities, optimized the reaction systems and finally defined its transU. Moreover, a protocol was provided to simplify the transU definition procedures.
To precisely calculate the Cas12a trans-cleavage activity, the ssDNA reporter used was dual labeled with fluorophore and quencher (i.e., FQ-reporter), and the reaction was monitored by a fluorescence reader. To simplify the calculation, the trans-cleavage activity was calculated on the basis of the initial fluorescence growth rate vg (vg = ΔF/Δt), following the recently published Michaelis-Menten model (Ramachandran and Santiago, 2021).
Non-paired ssDNA FQ-reporters are the substrates for Cas12a trans-cleavage reactions, and the sequences may affect the cleavage efficiencies (Li et al., 2018a). We first tested four homopolymers of 6 nucleotides in length, including poly-adenine (A), poly-cytosine (C), poly-guanine (G), and poly-thymine (T), respectively, and found Cas12a trans-cleaved the poly-C reporter with the highest efficiency but failed to cleave the poly-G reporter (Figures 1A,B), which was consistent with the previous findings (Yue et al., 2021). Along with the elongation of the ssDNA reporter sequence, the vg gradually increased and reached the peak when the sequence was longer than 8 nucleotides (Figures 1C,D). Considering the fact that longer ssDNA FQ-reporter has higher background fluorescence and is more expensive to synthesize, the 8C FQ-reporter was chosen for the following assays.
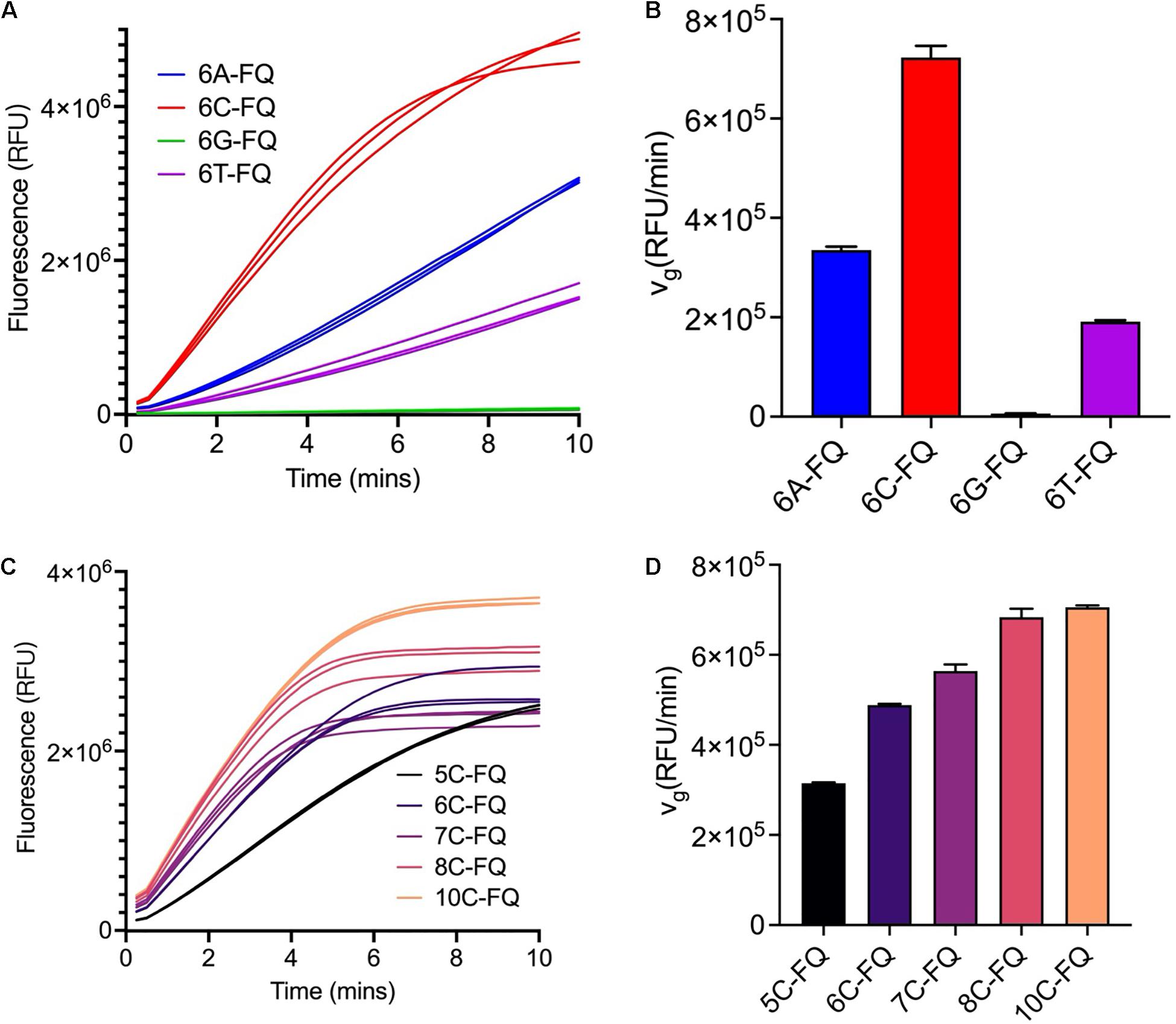
Figure 1. Optimization of the ssDNA reporter sequences. (A) Fluorescence curves of the Cas12a trans-cleavage reactions using different ssDNA reporter sequences. The reaction systems were comprised of 250 nM crRNA, 250 nM Cas12a, 3 nM target dsDNA, 1000 nM reporter and 1× NEBuffer 3.1. (B) Initial fluorescence growth rate of the reporters used in panel (A). (C) Fluorescence curves of the Cas12a trans-cleavage reactions using poly-C ssDNA reporters of different lengths. The reaction systems were comprised of 250 nM crRNA, 250 nM Cas12a, 3 nM target dsDNA, 800 nM reporter, and 1× NEBuffer 3.1. (D) Initial fluorescence growth rate of the reporters used in panel (C).
To optimize the Cas12a reaction buffer, we first compared several commercially available reaction buffers and found that Cas12a in buffer F had the highest trans-cleavage efficiencies (Figure 2A and Supplementary Table 1). As the pH values may have influences on the Cas12a trans-cleavage activities, a group of commercially available standard buffers with pH values ranging from 3.5 to 9.6 in increments of 0.1 were further analyzed, through individually substituting the (hydroxymethyl)aminomethane (abbreviated as Tris) salt in buffer F. Among the tested conditions, Cas12a generally preferred buffers with high pH values and exhibited obviously higher trans-cleavage activities in the 1 M glycine buffer (pH 8.6) and the 1 M Tris–HCl buffer (pH 8.5) (Figure 2B). Considering the buffering capacity in maintaining a stable pH value of the reaction buffers, especially when complicated samples are detected, the Tris–HCl buffer (pH 8.5) was finally chosen. Meanwhile, the loss of Cas12a trans-cleavage activities in buffer C could be caused by either the relatively low pH value or the low concentration of Mg2+, which has been demonstrated to be important for the maintenance of Cas12a cis-cleavage activities in a previous study (Fonfara et al., 2016; Supplementary Table 1).
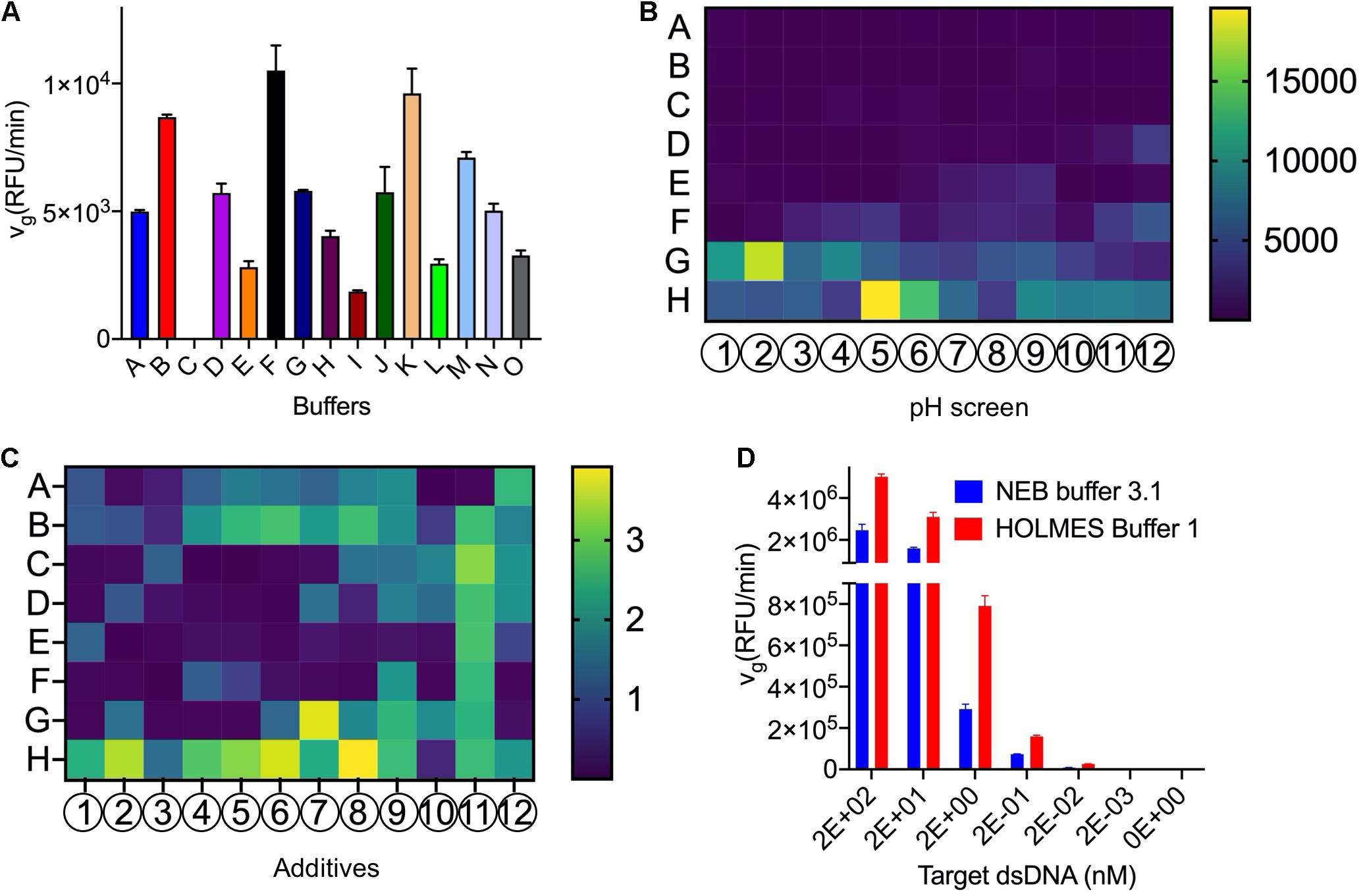
Figure 2. Optimization of the Cas12a trans-cleavage reaction buffer. (A) Screening of the commercially available buffers. Detailed compositions of the tested buffers were listed in Supplementary Table 1. (B) Screening of the pH values with the pH slice plate (Cat. No. #HR2-070) from Hampton Research. The A–H and 1–12 indicated the positions in the 96-well plate and detailed components in each well were shown in Supplementary Sheet 1. Specifically, Cas12a in conditions of H5 (pH 8.6 in 1 M glycine buffer) and G2 (pH 8.5 in 1 M Tris–HCl buffer) showed obviously higher trans-cleavage activities and the Tris buffer with pH 8.5 was finally chosen for further analysis. The trans-cleavage activities (RFU/min) were reflected by the colored histogram on the right side. (C) Screening of additives with the plate (Cat. No. #HR2-072) from Hampton Research. The A–H and 1–12 indicated the positions in the 96-well plate and detailed components in each well were shown in Supplementary Sheet 2. In comparison with the water control in A1, the Cas12a showed obviously higher trans-cleavage activities with several additives such as the G7 (glycerol), H5 (PEG 3,350), H6 (PEG 8,000), and H8 (PEG 20,000), and the most efficient H8 was employed for further analysis. The fold changes, relative to the activities in A1, were indicated by the colored histogram on the right side. (D) Comparison of the vg of Cas12a in HOLMES Buffer 1 and NEB buffer 3.1.
Meanwhile, dozens of additives were individually added into the reaction buffer F to test their efficacy on the Cas12a trans-cleavage activities, and several components such as polyethylene glycol (PEG) and glycerol obviously enhanced the cleavage signals and were chosen for further analysis (Figure 2C). The chosen additives of different combinations and concentrations were compared and the condition of 0.4% PEG-20000 showed the most remarkable enhancement and was finally selected (Supplementary Figure 1). In addition, we found that non-ionic detergents such as Triton X-100 could also promoted the Cas12a trans activities.
After systematical analysis of the above selected components, an optimized reaction buffer namely HOLMES Buffer 1 showed better performance than buffer F in triggering the Cas12a trans-cleavage and was chosen for subsequent analysis (Supplementary Figure 2 and section “5.3.1” in Supplementary Protocol). Moreover, through comparison with the control buffer of NEB buffer 3.1, HOLMES Buffer 1 remarkably enhanced the Cas12a trans-cleavage activities as well as its detection sensitivities (Figure 2D).
Besides the reaction buffer, the ratio among Cas12a, crRNA, and target dsDNA could also be an important factor affecting the Cas12a trans-cleavage efficiencies, and different combinations were therefore tested in HOLMES Buffer 1. In theory, excessive amounts of crRNA and target dsDNA could promote the formation of the Cas12a ternary complex as well as triggering of the Cas12a trans-cleavage; however, we found that the Cas12a cleavage activity was slightly inhibited by high concentrations of crRNA and dsDNA with an unknown reason (Supplementary Figure 3). Therefore, a ratio of 1: 2: 2 was finally chosen to obtain the best kinetic parameters for Cas12a trans-cleavage in HOLMES Buffer 1.
At the presence of the target DNA, the Cas12a trans-cleaves the ssDNA FQ-reporter to illuminate fluorescent signals, which can be employed to measure the Cas12a enzymatic constants. However, as the fluorescence intensities may vary from different readers, standard curves should be first created to correlate the relative fluorescent units (RFUs) and the concentrations of the FQ-reporter, including both un-cleaved (i.e., the substrates) and the cleaved (i.e., the products) reporters (Figures 3A,B and Supplementary Figures 4a,b). Briefly, the ssDNA FQ-reporter should be serially diluted and both cleaved and un-cleaved fluorescence signals are continually measured till the fluorescence reaches the saturation point, and the highest values of each gradient are then used for calibration of the standard curves for both cleaved and un-cleaved reporters (Figures 3C,D and Supplementary Figures 4c,d).
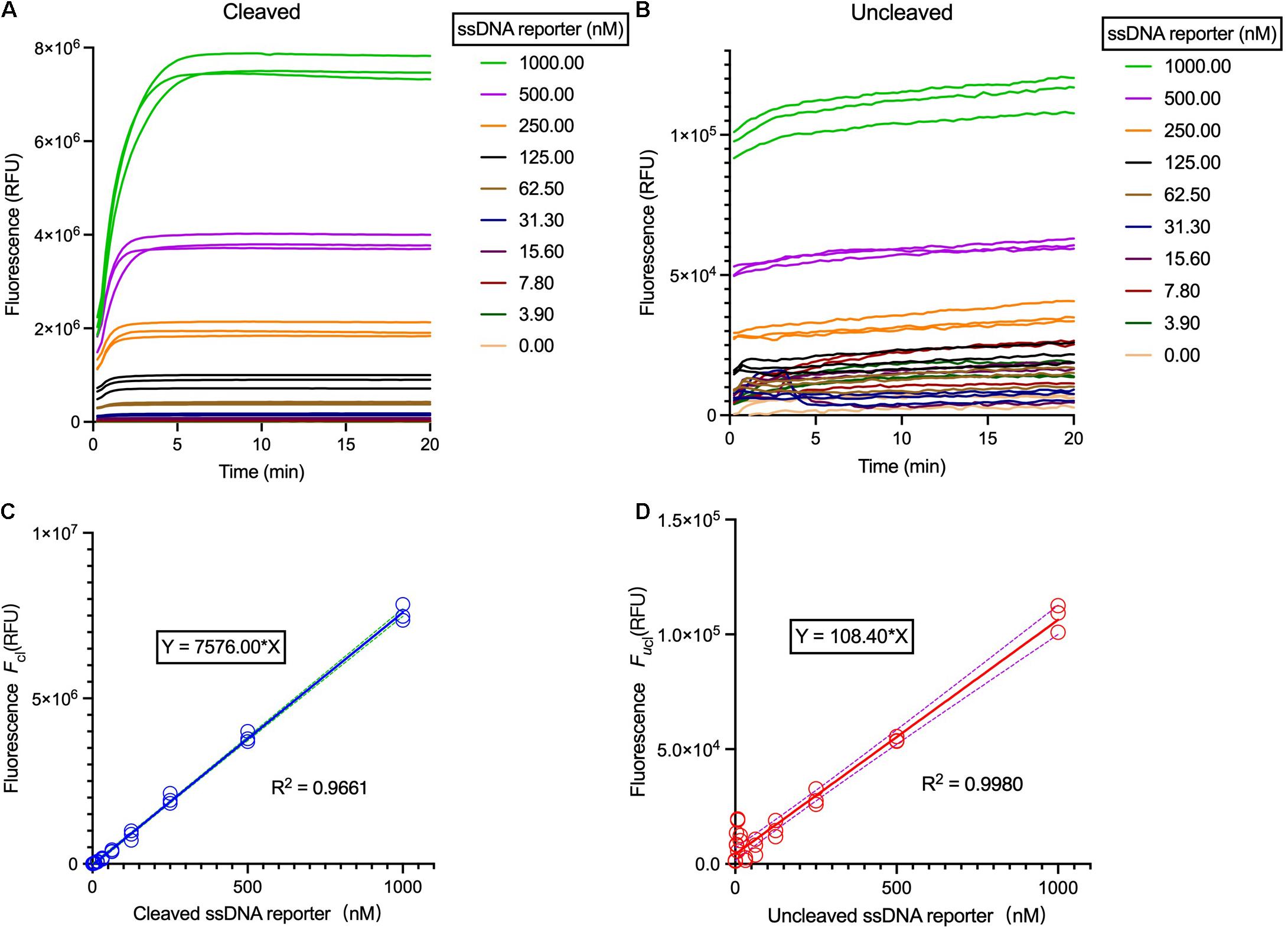
Figure 3. Correlation of the relative fluorescent units (RFUs) and the FQ-reporter concentration. (A) Fluorescence curves of the Cas12a trans-cleavage reactions using different ssDNA reporter concentration. (B) Fluorescence curves of the Cas12a trans-cleavage reactions using different ssDNA reporter concentration without target. The reaction systems were comprised of 40 nM crRNA, 20 nM Cas12a, 40 nM target dsDNA, and 1× HOLMES Buffer 1. (C) Background-subtracted fluorescence Fcl versus concentration of cleaved reporters. (D) Background-subtracted fluorescence Fucl versus concentration of un-cleaved reporters. Solid lines are linear regression fits to experimental data (in symbols). Three replicates were measured for each concentration. Both cleaved and un-cleaved reporters were detected by the Applied Biosystems QuantStudio 3 machine.
Then, with the employment of a limited concentration of Cas12a and crRNA and varied concentrations of ssDNA reporters, trans-cleavage enzymatic kinetics for Cas12a were determined, including the values of vmax, Km, and Kcat (Figure 4 and Supplementary Table 2). Noticeably, the constants of Km, Kcat as well as the Kcat/Km were miscalculated in a previous study (Chen et al., 2018), which were much higher than the data in this study and another work (Ramachandran and Santiago, 2021) and have been recently corrected (Supplementary Table 3).
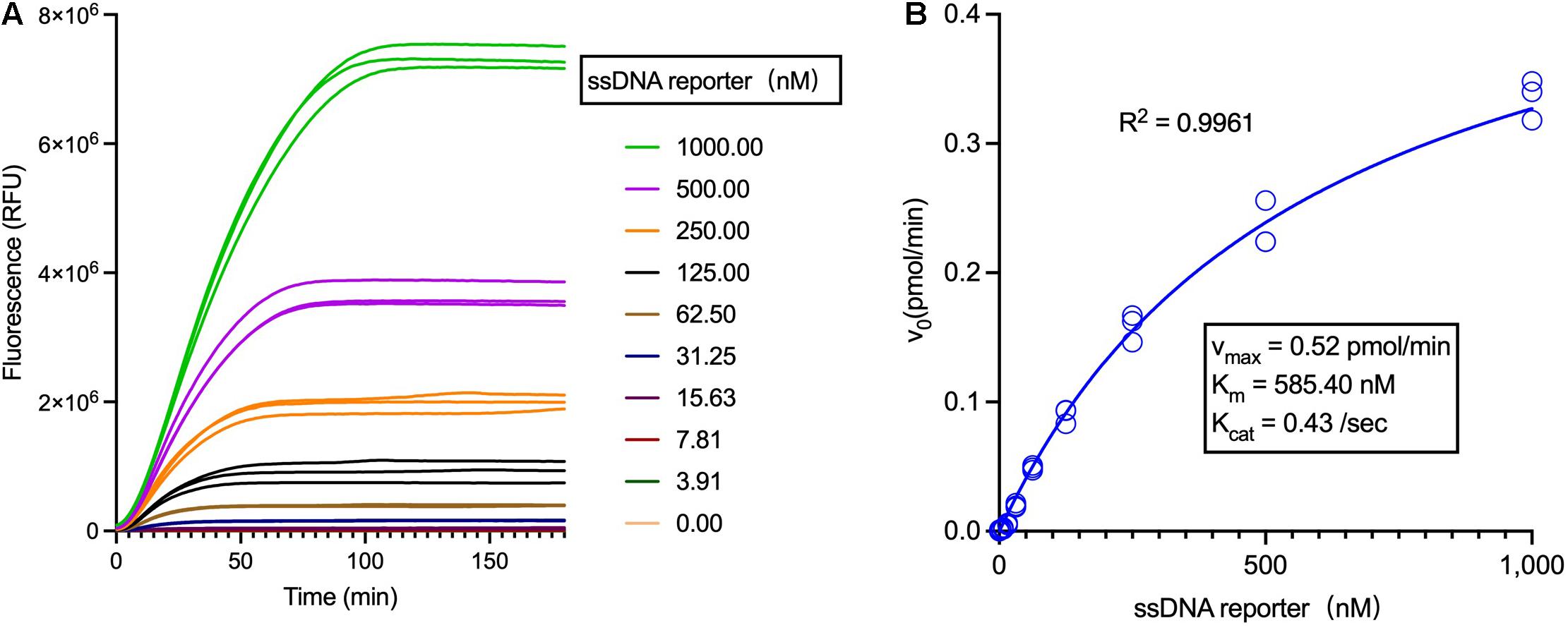
Figure 4. Analysis of the LbCas12a enzymatic kinetics. (A) Fluorescent curves of the Cas12a trans-cleavage reactions using different concentrations of the ssDNA reporter. (B) Analysis of the Michaelis-Menten constants of LbCas12a (Tolo Biotech.), employing 1 nM Cas12a and varied concentrations of ssDNA reporters.
In the above-mentioned optimized reaction condition, the Cas12a trans-unit (transU) was defined as the amount of Cas12a that trans-cleaves 1 pmol 8C FQ-reporter in 1 min at 37°C in a total reaction volume of 20 μL in HOLMES Buffer 1. To simplify the transU measurement, a step-by-step protocol was provided in the Supplementary Protocol, which can be run on different fluorescence readers. Noticeably, as the Cas12a trans-cleavage efficiencies can be remarkably affected by the crRNA and target dsDNA sequences as well as their concentrations, the transU should be determined using the same sequences and concentrations as provided in this study to unify the measurement (ref to sections “5 and 6.2” in Supplementary Protocol).
Briefly, a standard curve correlating the RFUs and the reporter concentrations should be drawn first, and then the trans-cleavage reaction is performed with serially diluted Cas12a and excess ssDNA reporters. The obtained data in the linear region are then used for calculation of the Cas12a transU. Following this protocol, a commercially available LbCas12a was then analyzed, and the trans-cleavage activities were determined to be 51.65 transUs/pmol (i.e., 19.36 fmol per transU) (Figure 5).
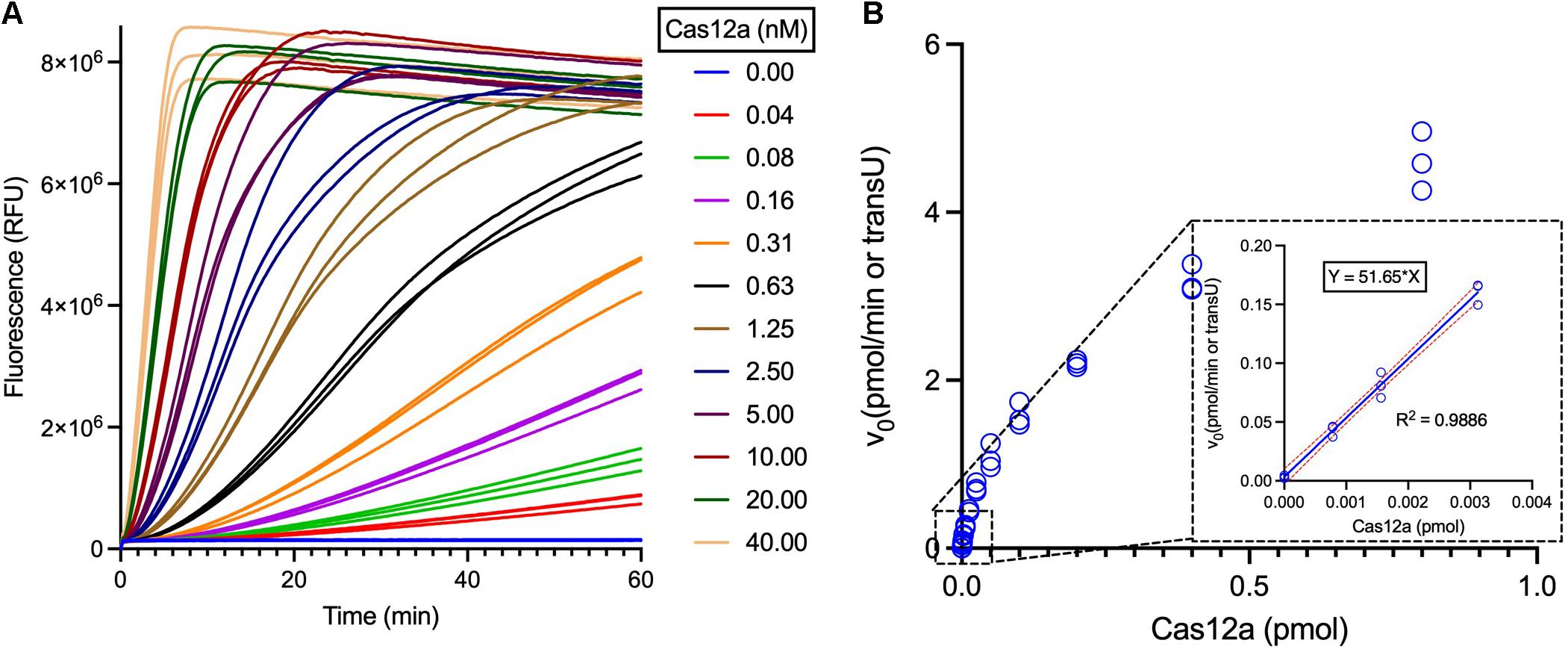
Figure 5. Measurement of the LbCas12a specific trans-cleavage activities. (A) Fluorescent curves of the Cas12a trans-cleavage reactions with different concentrations of LbCas12a. LbCas12a was twofold serially diluted for the transU measurement, using 1 μM ssDNA reporter as the substrate and following the Supplementary Protocol. (B) Curve plotting of calculated initial reaction rate versus the LbCas12a concentrations. The linear region of the reactions was boxed and enlarged, and the data were further employed for transU calculation.
Taken together, we here systematically optimized the Cas12a trans-cleavage reaction conditions, measured the Cas12a enzymatic constants and defined its transU, which will undoubtedly facilitate the development and application of robust Cas12a-based CRISPR-Dx systems in the future. Moreover, following a similar approach, the transU of other Cas effectors such as Cas12b, Cas12f, and Cas13 can be defined and their measurement protocols can be easily developed. On the other hand, although Cas12a exhibits much better performance in HOLMES Buffer 1 (e.g., with a higher Kcat value), one should be aware that there is still much work to do to further improve the Cas12a trans-cleavage activities, which may include the optimization of not only the reaction buffers but also the crRNA sequences in the future.
LbCas12a (LbCpf1) (Cat. No. #32108-03) was ordered from Tolo Biotech. (Shanghai, China). crRNAs were chemically synthesized by Biolino Acid Technology (Tianjin, China). FQ-labeled ssDNA reporters were ordered from Sangon Biotech. (Shanghai, China). The additive screening plates (Cat. No. #HR2-072) and the pH slice plate (Cat. No. #HR2-070) were ordered from Hampton Research (Aliso Viejo, CA).
For the short dsDNA target, two complementary oligonucleotides were synthesized by commercial companies and then annealed to form dsDNA. Specifically, a pair of oligonucleotides (4 μM each) were mixed in 1× PCR buffer and then annealed in the following procedure with a thermal cycler: 95°C for 5 min and then reduced from 95 to 20°C at a rate of 1°C per minute. The annealed dsDNA target was diluted to 1 μM and stored at −20°C before use.
The Cas12a trans-cleavage reaction was performed in a 20-μL volume, following the conditions as described before (Li et al., 2018b). In detail, 50 nM crRNA, 5 nM Cas12a nuclease, 4 nM dsDNA target, and 200 nM 8C FQ-reporter were pre-mixed in 1× buffer F and then 16 μL of the mixture was aliquoted into the 96-well PCR plate, which should be operated on ice. Then, the reaction was initiated by adding 4-μL additives into each well and the fluorescence signals were measured by the qPCR machine (Applied Biosystems QuantStudio 3). The program was set at 37°C and signals (λex: 488 nm; λem: 535 nm) were collected every 15 s. The initial fluorescence growth rate (vg) was calculated according to the maximum slope of the fluorescence curve using the ICEKAT software1.
The Michaelis-Menten was analyzed according to following equation: v0 = vmax [S]/(Km + [S]), where [S] is the substrate concentration, vmax is the maximum reaction rate, and Km is the Michaelis constant. The turnover number (Kcat) was determined by the equation: Kcat = vmax/[Et], where Et was the effective complex. The concentration of the activated Cas12a enzymatic complex was always kept at 1 nM in 1× reaction buffer. The Cas12a trans-cleavage reaction was initiated by the addition of the ssDNA FQ-reporter at the final concentrations of 0 nM, 3.9 nM, 7.8 nM, 15.6 nM, 31.2 nM, 62.5 nM, 125 nM, 250 nM, 500 nM, and 1 μM, respectively. Reactions were carried out in three replicates at 37°C and fluorescence readouts were taken every 15 s. Background-subtracted fluorescence signals were obtained by subtracting the signal from a negative control. The v0 data were converted to nM/s from Relative Fluorescent Units using the calibrated standard curve (ref to section “6.1” in Supplementary Protocol), and the Michaelis-Menten constants were determined by plotting v0 against the substrate concentration and then fitting into the Michaelis-Menten curve (Graphpad Software).
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
HL, JianW, and JZ performed most of the experiments. YC, LY, and DJ provided assistances in defining the Cas12a trans-cleavage units. HL and JinW drafted the manuscript. DG, HZ, YX, and JinW analyzed the data, revised the manuscript, and supervised the study. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (31922046 and 31770057), Sanming Project of Medicine in Shenzhen (SZSM202011017), the National Key Research and Development Program of China (2018YFA0903700 and 2018YFC0809200), Guangdong Science and Technology Foundation (2020B1111160001, B2019228, and 2020A1515110744), and Shenzhen Science and Technology Foundation (SGLH20180625171602058, 201906133000069, GJHZ20200731095604013, and JCYJ20210324102803009). The sponsors have no involvement in the study design; collection, analysis, and interpretation of data; the writing of the manuscript, and the decision to submit the manuscript for publication.
LY is employed by Tolo Biotechnology Company Limited, Shanghai, China.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.766464/full#supplementary-material
Aman, R., Mahas, A., Marsic, T., Hassan, N., and Mahfouz, M. M. (2020). Efficient, rapid, and sensitive detection of plant RNA viruses with one-pot RT-RPA-CRISPR/Cas12a assay. Front. Microbiol. 11:610872. doi: 10.3389/fmicb.2020.610872
Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Palefsky, J. M., et al. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439.
Ding, X., Yin, K., Li, Z., Lalla, R. V., Ballesteros, E., Sfeir, M. M., et al. (2020). Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nat. Commun. 11:4711.
Fonfara, I., Richter, H., Bratovic, M., Le Rhun, A., and Charpentier, E. (2016). The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521. doi: 10.1038/nature17945
Fozouni, P., Son, S., Diaz De Leon Derby, M., Knott, G. J., Gray, C. N., D’ambrosio, M. V., et al. (2021). Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 184, 323.e329–333.e329. doi: 10.1016/j.cell.2020.12.001
Gootenberg, J. S., Abudayyeh, O. O., Kellner, M. J., Joung, J., Collins, J. J., and Zhang, F. (2018). Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444. doi: 10.1126/science.aaq0179
Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J., Joung, J., et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. doi: 10.1126/science.aam9321
Han, T., Cong, H., Shen, Y., and Yu, B. (2021). Recent advances in detection technologies for COVID-19. Talanta 233:122609.
Joung, J., Ladha, A., Saito, M., Kim, N. G., Woolley, A. E., Segel, M., et al. (2020). Detection of SARS-CoV-2 with SHERLOCK One-Pot testing. N. Engl. J. Med. 383, 1492–1494.
Leung, R. K., Cheng, Q. X., Wu, Z. L., Khan, G., Liu, Y., Xia, H. Y., et al. (2021). CRISPR-Cas12-based nucleic acids detection systems. Methods [Online ahead of print] doi: 10.1016/j.ymeth.2021.02.018
Li, L., Li, S., Wu, N., Wu, J., Wang, G., Zhao, G., et al. (2019). HOLMESv2: a CRISPR-Cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synth. Biol. 8, 2228–2237. doi: 10.1021/acssynbio.9b00209
Li, Y., Li, S., Wang, J., and Liu, G. (2019). CRISPR/Cas systems towards next-generation biosensing. Trends Biotechnol. 37, 730–743.
Li, S. Y., Cheng, Q. X., Wang, J. M., Li, X. Y., Zhang, Z. L., Gao, S., et al. (2018b). CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 4:20.
Li, S. Y., Cheng, Q. X., Liu, J. K., Nie, X. Q., Zhao, G. P., and Wang, J. (2018a). CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 28, 491–493. doi: 10.1038/s41422-018-0022-x
Liu, P. F., Zhao, K. R., Liu, Z. J., Wang, L., Ye, S. Y., and Liang, G. X. (2021). Cas12a-based electrochemiluminescence biosensor for target amplification-free DNA detection. Biosens Bioelectron. 176:112954. doi: 10.1016/j.bios.2020.112954
Martin, J., Tena, N., and Asuero, A. G. (2021). Current state of diagnostic, screening and surveillance testing methods for COVID-19 from an analytical chemistry point of view. Microchem. J. 167:106305. doi: 10.1016/j.microc.2021.106305
Ramachandran, A., and Santiago, J. G. (2021). CRISPR enzyme kinetics for molecular diagnostics. Anal. Chem. 93, 7456–7464.
Shinoda, H., Taguchi, Y., Nakagawa, R., Makino, A., Okazaki, S., Nakano, M., et al. (2021). Amplification-free RNA detection with CRISPR-Cas13. Commun. Biol. 4:476.
Tian, T., Shu, B., Jiang, Y., Ye, M., Liu, L., Guo, Z., et al. (2021). An ultralocalized Cas13a assay enables universal and nucleic acid amplification-free single-molecule RNA diagnostics. ACS Nano 15, 1167–1178. doi: 10.1021/acsnano.0c08165
Keywords: CRISPR diagnostics, trans-cleavage, enzymatic units, Cas12, transU
Citation: Lv H, Wang J, Zhang J, Chen Y, Yin L, Jin D, Gu D, Zhao H, Xu Y and Wang J (2021) Definition of CRISPR Cas12a Trans-Cleavage Units to Facilitate CRISPR Diagnostics. Front. Microbiol. 12:766464. doi: 10.3389/fmicb.2021.766464
Received: 29 August 2021; Accepted: 08 November 2021;
Published: 29 November 2021.
Edited by:
Guangcai Duan, Zhengzhou University, ChinaReviewed by:
Tao Liu, Huazhong Agricultural University, ChinaCopyright © 2021 Lv, Wang, Zhang, Chen, Yin, Jin, Gu, Zhao, Xu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dayong Gu, d2FuaG9vZEAxNjMuY29t; Huailong Zhao, c2RqbG11QDE2My5jbQ==; Yong Xu, eHV5b25nXzIwMDBAdG9tLmNvbQ==; Jin Wang, d2FuZ2owMUBob3RtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.