
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 06 December 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.765943
This article is part of the Research Topic Searching for the Boundaries of Microbial Speciation in a Rapidly Evolving World View all 7 articles
 Nitin K. Singh1†
Nitin K. Singh1† Céline Lavire2†
Céline Lavire2† Joseph Nesme3†
Joseph Nesme3† Ludovic Vial2
Ludovic Vial2 Xavier Nesme2
Xavier Nesme2 Christopher E. Mason4
Christopher E. Mason4 Florent Lassalle5*
Florent Lassalle5* Kasthuri Venkateswaran1*
Kasthuri Venkateswaran1*Strains of Agrobacterium genomospecies 3 (i.e., genomovar G3 of the Agrobacterium tumefaciens species complex) have been previously isolated from diverse environments, including in association with plant roots, with algae, as part of a lignocellulose degrading community, from a hospital environment, as a human opportunistic pathogen, or as reported in this study, from a surface within the International Space Station. Polyphasic taxonomic methods revealed the relationship of Agrobacterium G3 strains to other Agrobacterium spp., which supports the description of a novel species. The G3 strains tested (n = 9) were phenotypically distinguishable among the strains from other genomospecies of the genus Agrobacterium. Phylogenetic analyses of the 16S rRNA gene, gyrB gene, multi-locus sequence analysis, and 1,089-gene core-genome gene concatenate concur that tested G3 strains belong to the Agrobacterium genus and they form a clade distinct from other validly described Agrobacterium species. The distinctiveness of this clade was confirmed by average nucleotide identity (ANI) and in silico digital DNA–DNA hybridization (dDDH) comparisons between the G3 tested strains and all known Agrobacterium species type strains, since obtained values were considerably below the 95% (ANI) and 70% (dDDH) thresholds used for the species delineation. According to the core-genome phylogeny and ANI comparisons, the closest relatives of G3 strains were Agrobacterium sp. strains UGM030330-04 and K599, members of a novel genomospecies we propose to call genomovar G21. Using this polyphasic approach, we characterized the phenotypic and genotypic synapomorphies of Agrobacterium G3, showing it is a bona fide bacterial species, well separated from previously named Agrobacterium species or other recognized genomic species. We thus propose the name Agrobacterium tomkonis for this species previously referred to as Agrobacterium genomospecies 3. The type strain of A. tomkonis is IIF1SW-B1T (= LMG 32164 = NRRL B-65602). Comparative genomic analysis show A. tomkonis strains have species-specific genes associated with secretion of secondary metabolites, including an exopolysaccharide and putative adhesins and resistance to copper. A. tomkonis specific gene functions notably relate to surface adhesion and could be involved to colonize nutrient-poor and harsh habitats. The A. tomkonis strains from the ISS showed presence of a 40-kbp plasmid and several other potential mobile genetic elements detected that could also be part of conjugative elements or integrated prophages.
Agrobacterium species are firstly known for their pathogenic potential on plant, causing either crown gall or hairy root disease, depending on the presence of a tumor-inducing (Ti) or root-inducing (Ri) megaplasmid in their genome; however, most strains lack such plasmids and are not pathogenic. Agrobacterium species are common members of soil communities, and are efficient colonizers of the rhizospheres of a wide variety of plant hosts (Macrae et al., 1988; Matthysse and McMahan, 1998), with which they have a commensal relationship (Sanguin et al., 2006; Dessaux and Faure, 2018). Agrobacterium is a polyphyletic group, which taxonomy, originally aimed at reflecting the pathogenicity status of strains (Smith and Townsend, 1907), was revised multiple times, in the light of growing phenotypic and genotypic evidence, notably showing that the pathogenicity status was not correlated to the diversity of agrobacteria. Based on biochemical testing and DNA-DNA hybridization (DDH), Agrobacterium species were initially classified into three biovars (Keane et al., 1970; Kersters et al., 1973; Ophel and Kerr, 1987). Agrobacterium species belonging to biovar 1, as well as closely related species such as Agrobacterium rubi and Agrobacterium larrymoorei can be recognized by the presence of a linear chromid as the second major molecule of the genome, a synapomorphic trait enabled by the landmark acquisition of the telA gene (Ramírez-Bahena et al., 2014). The strains that belong to biovar 2 and biovar 3 are only distantly related to Agrobacterium biovar 1, which later led to their reclassification into the genera Rhizobium and Allorhizobium, respectively (Young et al., 2001; Mousavi et al., 2015). Further refinement of the taxonomy of Agrobacterium biovar 1, also known as the Agrobacterium tumefaciens species complex, was conducted based on whole-genome information using DDH, leading to the classification of strains into species-level units called genomic species or genomovars (Popoff et al., 1984). This classification framework was later validated and enriched by studies using amplified fragment-length polymorphism (AFLP), marker gene phylogeny, multi-locus sequence analysis (MLSA), comparative genome hybridization (CGH) and core-genome phylogeny (Portier et al., 2006; Costechareyre et al., 2010; Lassalle et al., 2011, 2017; Mousavi et al., 2015), as recently reviewed by Flores-Felix et al. (2020). At the time of writing this communication, the Agrobacterium genus contained 15 named species, of which 11 had validly published names. Moreover, nine out of 15 described genomic species within the Agrobacterium biovar 1 are not yet formally named: G3 (this study), G5, G6, G7 (“Agrobacterium deltaense”), G8 (“Agrobacterium fabrum”), G13, G15 (“Agrobacterium viscosum”), G19 and G20 (Mafakheri et al., 2019).
Several hundred microbial strains were isolated and identified in an ongoing investigation to map microbial diversity of the International Space Station (ISS; Checinska Sielaff et al., 2019). The ISS is a hermetically sealed closed system and their modules are at least 20-year-old, depending on their time of addition. The source of microorganisms is mainly through the human traffic, cargo transport and also associated with other experimental components. The air of the ISS is recirculated via an advanced environmental control system, and surfaces are maintained by implementing periodic cleaning. Previous attempts to isolate microorganisms from ISS environmental surfaces revealed that they generally consist of predominantly benign and commensal microorganisms (Mhatre et al., 2020; Bijlani et al., 2021), but potentially pathogenic microorganisms were also sporadically isolated (Knox et al., 2016; Singh et al., 2018a). In 2015, several bacterial strains were collected from the observation deck (Cupola) of the ISS, and whole-genome sequences (WGS) were generated for these isolates (Bharadwaj et al., 2020a,b; Bijlani et al., 2020a,b; Daudu et al., 2020a,b; Solomon et al., 2020; Simpson et al., 2021). Based on preliminary genomic analyses, three ISS strains were identified as belonging to the Agrobacterium genomospecies 3 (also known as A. tumefaciens genomovar G3 or Agrobacterium G3).
The first objective of this study is to describe the phylogenomic novelty and characterize the taxonomic affiliation of the three strains isolated from the ISS environment. We, therefore, assembled a dataset of complete genomes of Agrobacterium G3, including previously released sequences from five strains isolated from various geographical regions and environments: an eosin flask from a hospital environment in France (CFBP 6623, sequenced twice), a cave in Lechuguilla, United States (LC34), algae in the United Kingdom (SUL3), Arabidopsis roots in Germany (Root651), and bioprospecting for lignocellulolytic microbes and enzymes from natural, highly evolved plant biomass-degrading systems in the United States (UGM030330-04). We added two novel sequences from Agrobacterium G3 strains from our own collection, including an opportunistic pathogen isolated from cerebrospinal fluid (CFBP 6624) and one isolated from a tobacco plant rhizosphere (RTP8). In addition to traditional phenotype testing, molecular taxonomy utilizing 16S rRNA gene, gyrB, MLSA (gyrB, parE, recA, and rpoB genes), and core-gene-based phylogenic analyses were carried out to describe Agrobacterium G3 strains.
Second objective of this study is to perform a comparative genomic analysis of Agrobacterium G3 strains together with representatives of other Agrobacterium species (n = 41 genomes) to elucidate genomic complexity. Subsequently, experimental characterization of the plasmid content was carried out on the set of Agrobacterium strains available to us in culture, i.e., G3 strains IIF1SW-B1T, IIF1SW-B3, IIF1SW-B4, CFBP 6623, CFBP 6624, and RTP8 and reference strains “A. fabrum C58,” Agrobacterium radiobacter B6T, and Agrobacterium pusense CFBP 5494. Thirdly, a pangenome analysis was implemented to identify core homologous gene clusters specific to ISS isolates and other Agrobacterium G3 strains. Finally, the metagenome reads of ISS environmental surfaces (Singh et al., 2018b) were mined for the presence of Agrobacterium G3 species to understand the prevalence of these novel species in ISS.
The three strains collected from ISS belonging to Agrobacterium G3 along with six other G3 strains were subjected to a polyphasic characterization to determine the variable, conserved or distinctive traits of this genomic species.
Table 1 summarizes assembly statistics of all five novel ISS strains sequenced in this study. As an example, for strain IIF1SW-B1T, the Illumina NovaSeq platform yielded 1.8 × 107 paired-end (PE) reads were reduced to 1.79 × 107 PE reads after performing trimming and quality filtering. The assembled draft genome of ISS strains consisted of 76–81 contigs with a genome size of 6.25–6.29 × 106 bp; the assembled genomes of CFBP 6624 and RTP8 had 35 and 38 contigs, for a size of 5.48 × 106 bp and 5.44 × 106 bp, respectively. The contig N50 size was 3.21–3.78 × 105-bp, with a mean coverage of 100x for ISS strains, and 18x and 19x for strains CFBP 6624 and RTP8, respectively. G + C% of the five genomes was 59.12–59.21.

Table 1. Summary of the draft whole-genome sequences of several Agrobacterium tomkonis strains.
A Maximum Likelihood (ML) tree of 16S ribosomal RNA (rRNA) gene sequences shows that this marker is not able to resolve and distinguish all Agrobacterium species (Figure 1A). All strains from genomospecies G3 and G4; G7 and G13; as well as G2 and G9 were found to have identical 16S rRNA gene sequences. In addition, the support for the topology of the 16S tree is generally low, with only branches leading to species A. rubi, Agrobacterium bohemicum and the clade formed by Agrobacterium genomospecies G6 and G8 being well supported. In addition, the type strains of non-biovar 1 species A. larrymoorei and “Agrobacterium albertimagni” are positioned within the clade containing otherwise only Agrobacterium biovar 1 strains, indicating that these 16S alleles are likely the result of horizontal gene transfer events. Similar results showing high similarities or identical 16S rDNA sequences for Agrobacterium rosae NCPPB-1650T, Agrobacterium skierniewicense Di1472 (99.7%), and A. rubi NBRC 13261T (99.5%) were reported (Kuzmanović et al., 2018). This altogether suggests that the 16S rRNA gene is not a good marker to identify Agrobacterium species or to study their relationships, as observed in other taxa (La Duc et al., 2004).
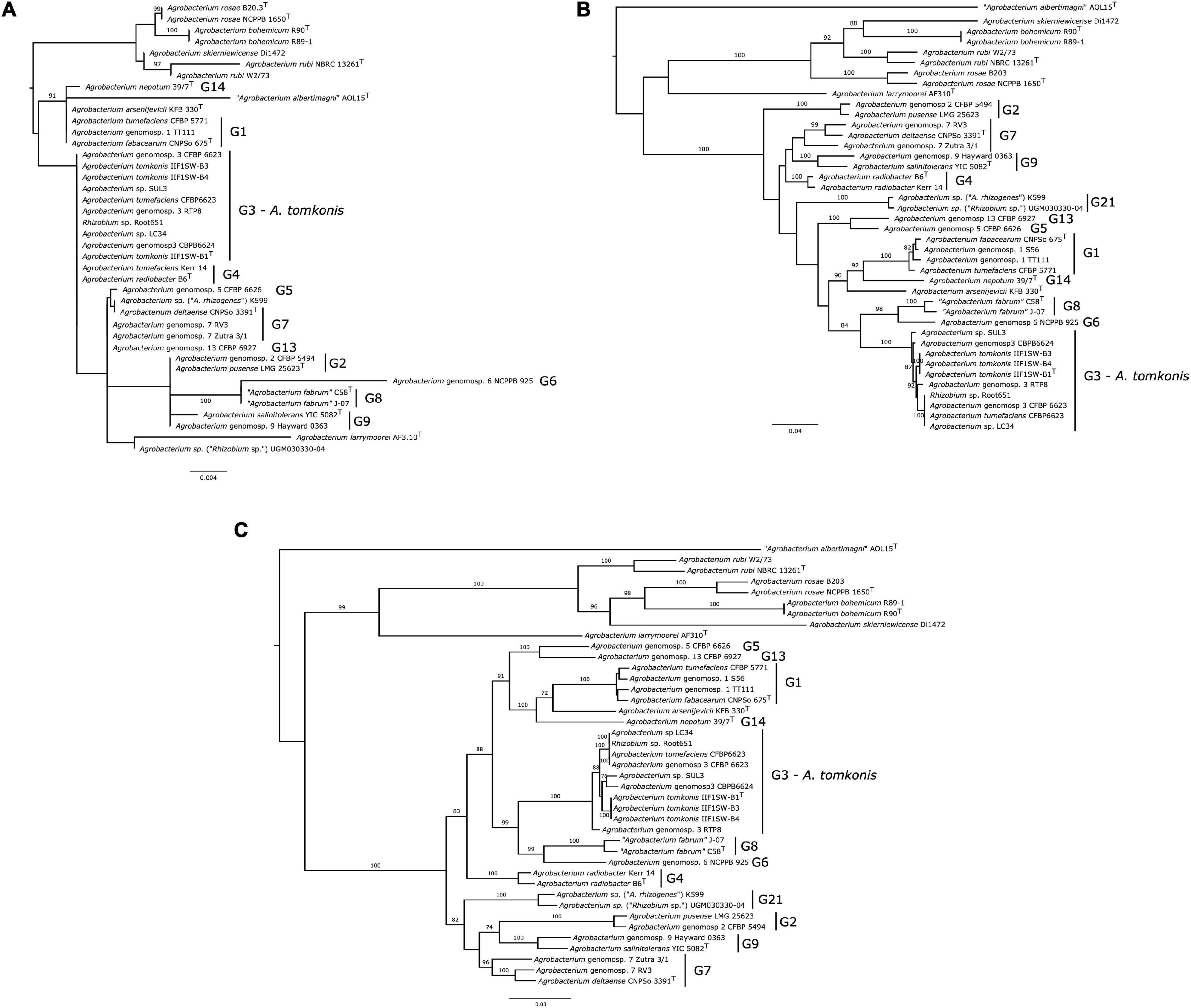
Figure 1. Phylogeny of 40 Agrobacterium spp. strains based on 16S rRNA gene, gyrB, and MLSA. Conserved marker gene phylogenies of 40 Agrobacterium distinct strains (41 when including the two published genome versions for strain CFBP 6623. (A) 16S rRNA gene phylogeny, based on 1,602 aligned positions), rooted with sequence from Rhizobium leguminosarum USDA 2370T; (B) gyrB gene phylogeny, based on 2,477 aligned positions; (C) multi-locus sequence analysis phylogeny, based on the concatenated alignments of genes parE, gyrB, recA, and rpoB, resulting into 9,968 aligned positions. All gene sequences were extracted from the 41 studied and then aligned with Clustal Omega (for coding genes, alignment was performed at the protein level and then reverse-translated into codons). Maximum-likelihood trees were inferred with RAxML-NG 1.0.0 under the model GTR + FO + G4m, taking the best of 20 independent inferences, started with 10 random trees and 10 parsimony-optimized tree. Branch supports were estimated with 200 Felsenstein bootstrap trees. Only branch support over 70% are displayed; full information on the trees are available at https://doi.org/10.6084/m9.figshare.14792148; https://doi.org/10.6084/m9.figshare.14792169; https://doi.org/10.6084/m9.figshare.14792100.
The gene gyrB provides a much better phylogenetic resolution of relationship among Agrobacterium species, with all except genomospecies G7 forming highly supported clades (Figure 1B). All nine strains belonging to G3 were grouped into one clade. The shallow grouping of species into clades is highly supported except G4, G7, and G9 strains, but deeper relationships within the Agrobacterium biovar 1 are not. This indicate that gyrB is a bona fide marker gene to identify and study the diversity of Agrobacterium species, even though its use as a marker for amplicon-based surveys has been shown to be impractical due to the presence of the paralogous gene parE in the genome rendering selective amplification difficult (Puławska and Kałużna, 2012).
The MLSA tree provides further resolution for Agrobacterium species, where strains belonging to various genomospecies were placed in a tight clade with high bootstrap values (Figure 1C). The relationships between species according to this MLSA tree are mostly in agreement with previous reports based on the concatenation of multiple loci or core-genome genes (Mousavi et al., 2014, 2015; Lassalle et al., 2017), indicating that this four-loci MLSA scheme (including gyrB, parE, recA, and rpoB genes) provides an efficient way to affiliate phylogenetic relationships among Agrobacterium species. All nine strains of Agrobacterium G3 formed a clade with a bootstrap value >88%.
Finally, a tree based on the concatenation of 1,089 core-genome genes is fully resolved, with almost all branches having the highest support (Figure 2, bootstrap supports indicated if not 100%), and thus constitutes the gold standard for depicting the phylogenetic relationships of Agrobacterium species.
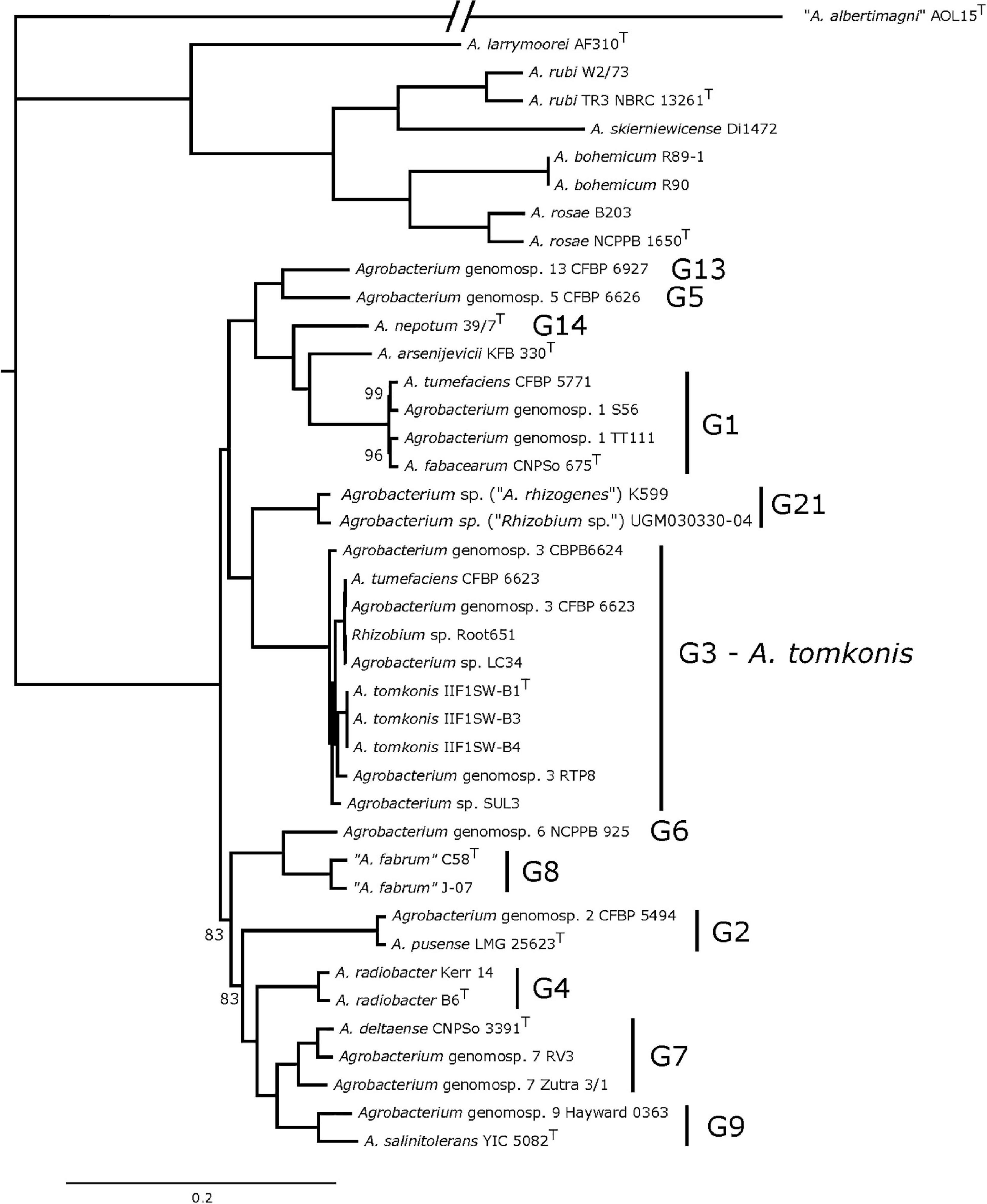
Figure 2. Phylogeny of 40 Agrobacterium spp. strains based on the concatenation of 1,089 core-genome genes. Maximum-likelihood phylogenetic tree of 40 Agrobacterium distinct strains (41 when including the two published genome versions for strain CFBP 6623) obtained using the bioinformatic pipeline Pantagruel (data available at https://doi.org/10.6084/m9.figshare.14792178). The tree was computed with RAxML 8.1.2 based on the concatenation of 1,089 core-genome gene alignments under the GTRCATX model, with 200 rapid bootstraps and rooted with RAxML tree-balance algorithm. All branch supports are 100%, unless displayed.
According to the gyrB and MLSA trees, the closest species to Agrobacterium G3 strains is the group formed of “A. fabrum” (G8) and Agrobacterium G6 strains, and these results were in accordance with previous reports (Lassalle et al., 2017). However, the core-genome tree revealed that the closest relatives were strains Agrobacterium sp. (Rhizobium sp.) UGM030330-04 and Agrobacterium sp. (Agrobacterium rhizogenes) K599 whereas strains belonging to genomovars G8 and G6 formed a different and distant clade (Figure 2). We note that strains UGM030330-04 and K599 are misnamed given these phylogenies clearly show that they are members of the Agrobacterium genus and thus not part of Rhizobium, nor of A. rhizogenes, which is a synonym of R. rhizogenes; they are thus more correctly referred to as Agrobacterium sp.
All average nucleotide index (ANI) values among Agrobacterium G3 strains were over 97.5% whereas when comparing G3 strains to non-G3 strains, the ANI values were below 90.68% (Table 2 and Supplementary Table 1). The ANI values of Agrobacterium G3 strains and most closely related (as per core-genome) Agrobacterium sp. strains UGM030330-04 and K599 were ∼90.5% similarity. Similarly, the ANI values of Agrobacterium G3 strains with other closely related – according to gyrB and MLSA trees – Agrobacterium G6 (NCPPB 925) and G8 strains (J-07 and C58), exhibited ∼87.6% relatedness. Furthermore, average amino-acid identity (AAI) values among Agrobacterium G3 strains were over 97.6% while values between G3 and other agrobacteria were below 94%. Likewise, the digital DNA:DNA hybridization (dDDH) values among Agrobacterium G3 strains were over 83.6% while values between Agrobacterium G3 and other agrobacteria were 41.7% or below. More specifically the dDDH values were ∼41.7% for comparisons with strains UGM030330-04 and K599, 34.5% with G6 strains, and <33.9% with G8 strains. The ANI and dDDH values obtained for all the Agrobacterium G3 strains – including the ISS isolates – with other Agrobacterium species were below the threshold of 95% ANI (Yoon et al., 2017) and 70% dDDH values (Auch et al., 2010), which were established as standard for prokaryotic species delineation. We thus propose the name Agrobacterium tomkonis for the genomospecies 3 (= genomovar G3) of the Agrobacterium biovar 1 (i.e., A. tumefaciens species complex).
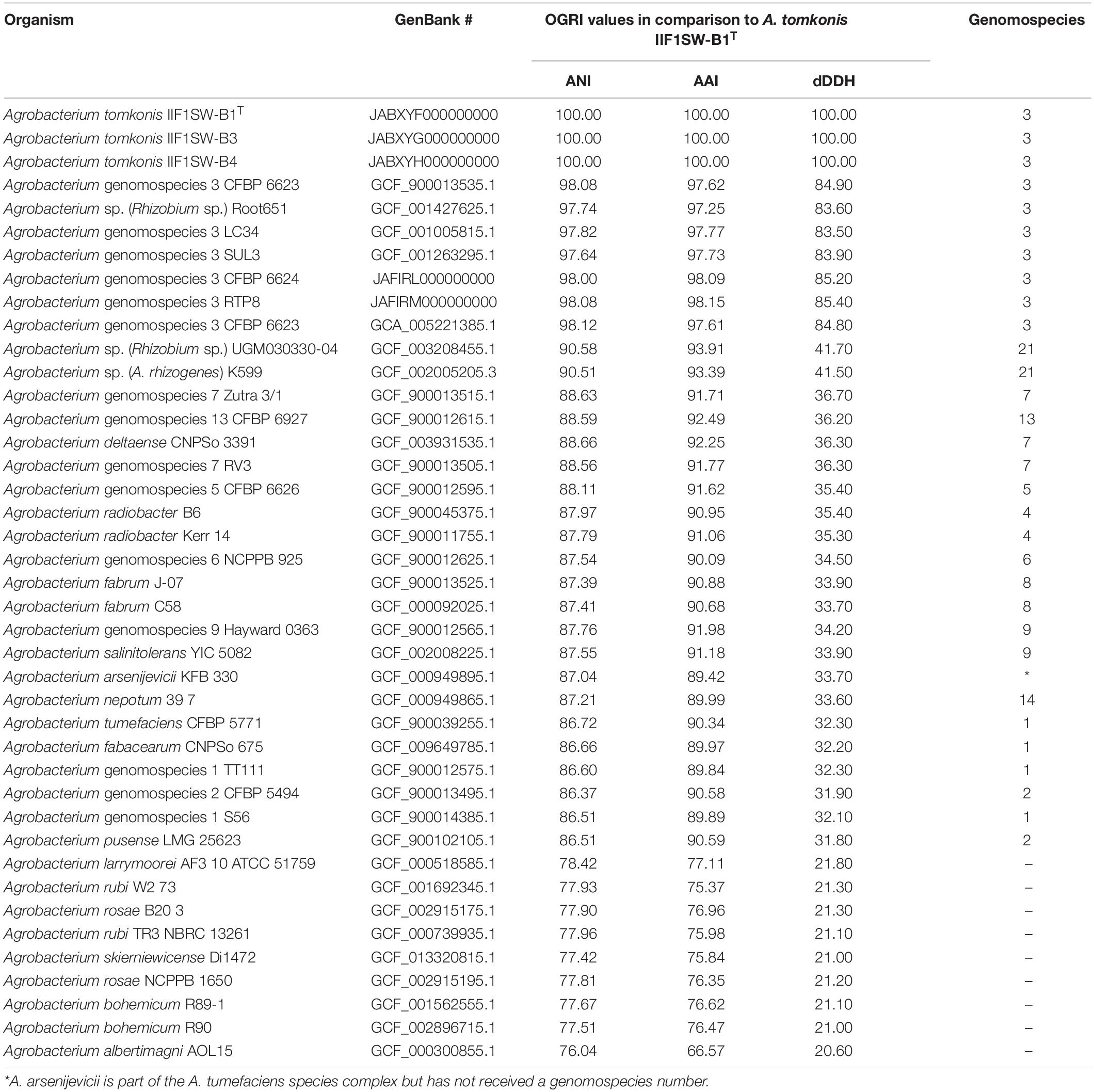
Table 2. List of Agrobacterium genomes used in this study and Average Nucleotide Identity (ANI), Average Amino acid Identity (AAI), and digital DNA-DNA Hybridization (dDDH) values of A. tomkonis IIF1SW-B1T compared with all other tested Agrobacterium strains.
Agrobacterium sp. strains UGM030330-04 and K599 consistently form a distinct group in the gyrB, MLSA and core-genome tree; these two strains share an ANI of 98.11% and have <90.68% ANI with any other Agrobacterium strain. We therefore propose to group them into a new genomospecies of the Agrobacterium biovar 1, to be named genomovar G21, following the recently described genomovars G19 and G20 (Mafakheri et al., 2019).
Differential phenotypic characteristics of A. tomkonis strains with six other Agrobacterium species are given in Table 3 and Supplementary Table 2. The biochemical tests were highly variable among Agrobacterium species tested, in accordance with previous reports (Kuzmanović et al., 2018). However, we found a distinctive trait in that all Agrobacterium biovar 1 strains except those belonging to the A. tomkonis assimilated D-galacturonic acid. In addition, A. tomkonis strains tested assimilated the following carbon sources (Supplementary Table 2). D-maltose, D-trehalose, D-cellobiose, gentiobiose, sucrose, D-turanose, stachyose, D-raffinose, α-D-lactose, D-melibiose, β-methyl-D-glucoside, D-salicin, N-acetyl-D-glucosamine, N-acetyl-D-galactosamine, α-D-glucose, D-mannose, D-fructose, D-galactose, D-fucose, L-fucose, L-rhamnose, D-sorbitol, D-mannitol, D-arabitol, myo-inositol, glycerol, D-glucose- 6-PO4, D-fructose- 6-PO4, L-alanine, L-arginine, L-aspartic acid, L-glutamic acid, L-histidine, L-pyroglutamic acid, L-serine, pectin, D-gluconic acid, D-Glucuronic acid, quinic acid, methyl pyruvate, L-malic acid, γ-amino-butyric acid, propionic acid, acetic acid. Moreover, L-lactic acid, D-malic acid, bromosuccinic acid, β-hydroxy-D, L-butyric acid and acetoacetic acid support at least a weak metabolic activity. However, no metabolic activity was observed for A. tomkonis strains on N-acetyl-β-D-mannosamine, N-acetyl neuraminic acid, 3-Methyl Glucose, fusidic acid, D-serine, D-aspartic acid, D-galacturonic acid, L-galactonic acid lactone, mucic acid, D-saccharic acid, p-hydroxyphenylacetic acid, and sodium butyrate.
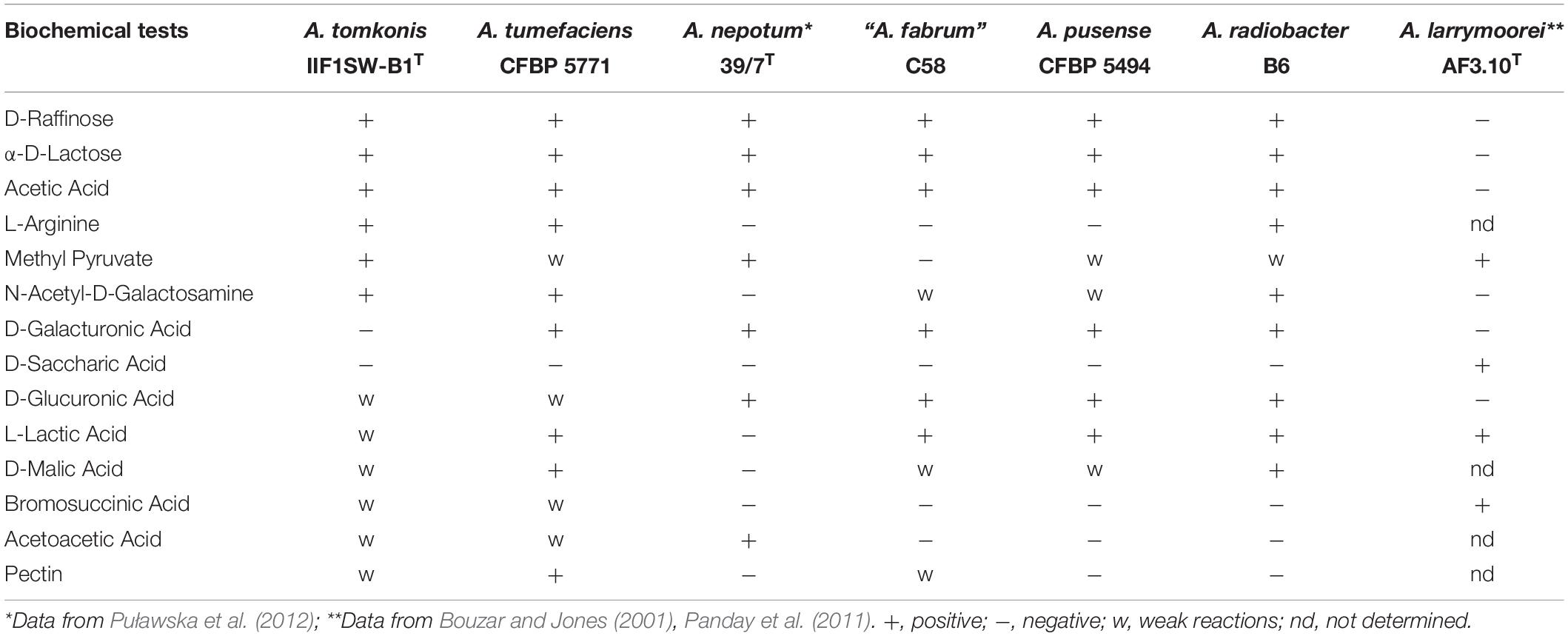
Table 3. Differential biochemical characteristics between A. tomkonis and closely related species.
Among various compounds tested, ISS A. tomkonis strains did not metabolize citric acid and Niaproof 4 (an anionic surfactant) whereas other A. tomkonis strains utilized them. Most of the compounds tested were metabolized unvaryingly by all ISS strains whereas strain IIF1SW-B3 was able to utilize lithium chloride as sole carbon source as that of other A. tomkonis strains tested. Similarly, strain IIF1SW-B3 was metabolizing Tween 40 (polysorbate, surfactant) but other A. tomkonis strains did not assimilate it. However, all six tested A. tomkonis strains are tolerant to troleandomycin, rifamycin SV, lincomycin, guanidine HCl, vancomycin, potassium tellurite, aztreonam, 1% sodium lactate, tetrazolium violet, and tetrazolium blue, but not to minocycline. Moreover, they all grew at pH 6 but not or only weakly at pH 5. All six tested A. tomkonis strains did not grow at 4% NaCl and beyond. The strains IIF1SW-B1T, IIF1SW-B4, and RTP8 tested on tomato produced no tumor (results not shown) and are therefore considered not pathogenic on tomato plant.
Forty-nine genes were specifically found in A. tomkonis genomes. These genes were mostly interspersed in the genome and coded a range of functions (Supplementary Table 3), but a fraction clustered into three genomic islands, two on the linear chromosome and one on the largest of the two megaplasmids present in strain CFBP 6623 (pCFBP6623a). The first cluster of seven A. tomkonis specific genes clearly encodes the biosynthesis of a surface polysaccharide; the second encodes mostly proteins of unknown function, with one potentially related to type 6 secretion systems; the last A. tomkonis specific gene cluster on the plasmid encodes resistance to copper. Other A. tomkonis specific genes are scattered in the genome, but have related functions. There are notably two genes encoding ankyrin-repeat domain-containing proteins, as well as a protein containing an extracellular HAF repeat domain (InterPro domain IPR014262) and an autotransporter domain, all three exported to the cell surface and likely mediating attachment to an extracellular substrate. Together with the biosynthesis of a likely extracellular polysaccharide, these species-specific genes present a convergent function related to surface attachment and potentially resistance to stress from the external medium. This enrichment in the specific core-genome of A. tomkonis suggests natural selection was at work and that surface attachment may have a strong role in A. tomkonis ecology. Indeed, the first reported A. tomkonis strains CFBP 6623 and CFBP 6624 were isolated from within an eosin flask and from a human clinical sample (likely as an opportunist taking advantage of another pathogen infection), respectively (Popoff et al., 1984), showing already unusual colonization abilities for agrobacteria. Moreover, the strains recovered from the ISS were isolated from the surface of the observation dome (Cupola), which panel is made of aluminum with a polyurethane topcoat (e.g., Aeroglaze A276 or BMS10-60), an ultra-clean and smooth surface that carries very little microbial life. The independent isolation of G3 strains in such locations suggest that attachment to inhospitable surfaces may be a way for A. tomkonis to colonize new habitats where it can evade competition with other microbes.
Similarly, the gene clustering analysis between the genomes of 40 different Agrobacterium strains allowed the identification of ISS strains-specific gene clusters (Figure 3 and Supplementary Table 4). We found 556 genes specifically present in the three ISS strains; we identified these genes based on their presence in all ISS strains and none of the 17 genomes in the background clade comprising the other A. tomkonis strains, all strains of Agrobacterium arsenijevicii, Agrobacterium nepotum, Agrobacterium genomospecies G1, G5, G13, and G2 (Supplementary Data online)1. Using a more relaxed criterion allowing presence of the genes in a maximum of 2 out the 17 genomes in the background clade, to allow the detection of genes specifically gained by the ancestor of the ISS strains but that may have independently introduced in close relatives via horizontal gene transfer, we found 838 genes (Supplementary Table 4), most of them located in large contiguous regions.
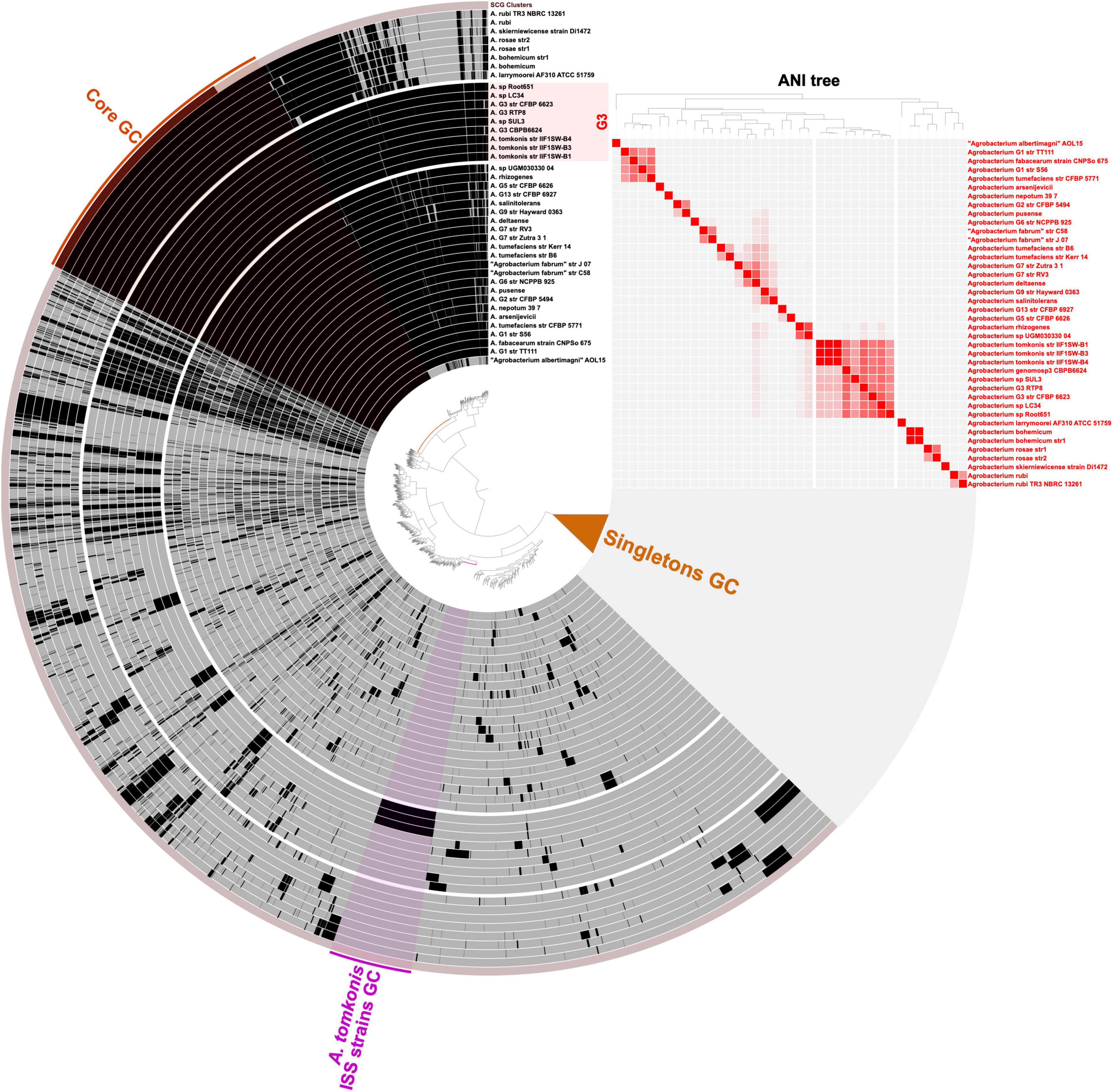
Figure 3. Visualization of the Agrobacterium pangenome. Genomes are ordered as layers using a tree based on average nucleotide identity (ANI) values matrix (Euclidean distance, Ward clustering). Color hue of ANI values are ranging from 70% ANI (white) to 100% ANI (red) and displayed as a heatmap on the right-side panel. Gene clusters are represented as vertical black bars indicating their presence in each genome layer. The central tree is ordering gene clusters based on their presence/absence in genomes using Euclidean distance and Ward clustering. The “Core GC” highlight indicates the gene clusters identified in all genomes. Gene clusters that are found in a single genome are collapsed as “Singletons GC.” The “A. tomkonis ISS strains GC” highlight indicates gene clusters found exclusively on the three strains isolated from the International Space Station. The figure was created in Anvi’o v6.
Among this wider set of ISS clone-specific genes, some were found to cover 10 entire contigs of the genome assembly of strain IIF1SW-B1T (Supplementary Table 5), totaling to 347 genes over 613 kb of ISS clone-specific contigs, which possibly represent (parts of) extrachromosomal elements. Indeed, four of these ISS clone-specific contigs carried genes with functions associated to type 4 secretion systems (T4SS), with two seemingly complete clusters, indicating they may constitute conjugative elements such as plasmids or integrative and conjugative elements (ICE). Most of the other ISS clone-specific contigs carried transposase or genetic mobility-associated genes, suggesting they also constitute mobile genetic elements (MGEs). In addition, large contiguous portions of other contigs that were specific to the ISS strains carried transposase genes – often located at the extremity of the ISS clone-specific region – suggesting they may represent inserted MGEs such as transposons or genomic islands (Supplementary Tables 4, 5). In particular, the ISS clone-specific region of contigs #27 of strain IIF1SW-B1T genome assembly clearly corresponds to a prophage.
Beyond genetic mobility, the ISS clone-specific genes displayed a range of other functions, which were enriched for a range of functions, some forming coherent cellular processes (Supplementary Table 6). Among the top enriched functions, we found a multi-copper oxidase, accompanied by a chaperone for its maturation and transporters that might allow the assimilation of copper ions from the environment; these functions may form a pathway for biosynthesis of a redox enzyme that could be involved in a respiratory chain. Another set of coherent functions were found to be involved in scavenging and uptake of a ferric ion-siderophore complex, as well as a non-ribosomal peptide synthases-polyketide synthases (NRPS/PKS) biosynthetic cluster, which might be involved in the biosynthesis of the siderophore.
Based on migration of genomic DNA on Eckhardt gels (Vaudequin-Dransart et al., 1995), all A. tomkonis strains were evidenced to carry plasmids, although differences in size and number were observed between strains (Supplementary Figure 1). Indeed, strain CFBP 6624 presented a single plasmid of approximately 500 kb, whereas CFBP 6623, RTP8 and the three ISS strains showed one to three plasmids, with a common largest band around 260 kb. RTP8 and the three ISS strains have in common a 130-kb plasmid, and a 40-kb plasmid is present exclusively in the three ISS strains. The ISS strains do not harbor a pTi plasmid, as no classical vir genes were detected in their genomes (data not shown). Accordingly, no symptoms of crown-gall diseases were observed on injured tomato plants inoculated with any of the ISS strains (data not shown).
In order to evaluate the relative abundance of A. tomkonis onboard the ISS, we performed metagenomic read recruitment obtained from ISS environmental DNA shotgun datasets (Singh et al., 2018b) with strain IIF1SW-B1T genome as a reference. Visual inspection of the mapped read profiles showed that most recruited reads were located on conserved bacterial genes encoding products such as ribosomal RNA operons, ribosomal proteins, DNA-directed RNA polymerase rpoB or elongation factor Tu. Considering the high degree of conservation of these genes across distant bacterial lineages and the non-uniform read coverage of G3 contigs we conclude that despite being isolated from the same environment, A. tomkonis is not present in any detectable abundance in the ISS metagenomes.
In summary, A. tomkonis genomes are diverse, and notably carry a variety of plasmids, even though none of the G3 strain in our dataset carried a tumorigenic plasmid. As previously shown for other Agrobacterium genomic species (Lassalle et al., 2017), A. tomkonis strain genomes share a number of species-specific genes. A. tomkonis specific gene functions notably relate to surface adhesion and could be involved in the ecological specificity of A. tomkonis suggested by their varied source of isolation (Popoff et al., 1984). Indeed, while most species of the Agrobacterium biovar 1 are usually isolated from soils, plant rhizospheres or as plant pathogens inducing tumors (Barton et al., 2018; Dodueva et al., 2020; Weisberg et al., 2020), A. tomkonis strains never have been isolated as a plant pathogen, nor a tumor-inducing plasmid was observed in their genome, which we confirmed by negative infection tests on tomato plants. Even though the isolation of A. tomkonis strains RTP8 from a tobacco plant rhizosphere and Root651 from Arabidopsis roots prove that A. tomkonis strains are able to survive in plant rhizospheres, A. tomkonis strains were otherwise all isolated from a variety of unhospitable environments, including from an antiseptic flask, as an opportunistic pathogen of the human central nervous system, from a cave wall, and lastly from the inert surface of the ISS cupola. The ability to colonize these substrates, unusual for most agrobacteria, resonate with the finding that A. tomkonis genomes specifically carry genes involved in mediating attachment to surfaces, including production of putative adhesins and biofilm. This suggests a particular ability of A. tomkonis to colonize a different kind of habitats, that are even poorer in nutrients and harsher than soil, thus possibly escaping competition with other agrobacteria that are better at growing in richer environments such as plant rhizospheres.
The genomes of A. tomkonis strains isolated from the ISS, appear to be highly similar, suggesting they are clonally related. As such, they carry further specific genomic traits, including the presence of a 40-kbp plasmid observed via electrophoresis, as well as several other potential MGEs detected in their specific genomic content, with several ISS clone-specific contigs carrying conjugation genes (cumulated length 346 kbp) and more contigs that could also be part of conjugative elements or other types of MGEs such as integrated prophages. These ISS clone-specific genes also include many potentially adaptive genes, including putative pathways for respiratory chain biosynthesis and iron scavenging, but it is unclear what would be the selective advantage those genes could bring to the ISS strains in the context they were isolated from.
Using a polyphasic approach, we characterized the phenotypic and genotypic synapomorphies of Agrobacterium G3 (Agrobacterium genomospecies 3), showing it is a bona fide bacterial species, well separated from previously named Agrobacterium species or other recognized genomic species. Based on this evidence, we propose to name this species Agrobacterium tomkonis (formal description below). In addition, we performed an in-depth investigation of the pangenome, notably identifying species-specific genes, allowing us to relate the description of this new species to hypotheses on its ecology. Within this novel species, we further characterized the clonal group of strain isolated from the ISS, showing it possesses its own set of specific genes and MGEs that could carry adaptive traits linked to its survival in the ISS environment.
Agrobacterium tomkonis (tom. ko’ni.s. N.L gen. n. tomkonis referring to David Tomko, a well-known NASA Space Biology scientist who advanced space research in the United States). Cells are Gram-stain-negative, non-spore-forming, aerobic rods, 0.5–0.6 μm in width and 0.8–1.6 μm in length. Colonies are translucent and cream to white, with a diameter of 2–3 mm on R2A medium after incubation for 3 days at 25°C. Growth occurs at 20°C through 35°C, with an optimum temperature of 25°C. A. tomkonis strains grew at pH 6 but not or only weakly at pH 5 with a pH optimum at 7.5. All tested A. tomkonis strains failed to grow at 4% NaCl and beyond.
A. tomkonis strains metabolize D-maltose, D-trehalose, D-cellobiose, gentiobiose, sucrose, D-turanose, stachyose, D-raffinose, α-D-lactose, D-melibiose, β-methyl-D-glucoside, D-salicin, N-acetyl-D-glucosamine, N-acetyl-D-galactosamine, α-D-glucose, D-mannose, D-fructose, D-galactose, D-fucose, L-fucose, L-rhamnose, D-sorbitol, D-mannitol, D-arabitol, myo-inositol, glycerol, D-glucose- 6-PO4, D-fructose- 6-PO4, L-alanine, L-arginine, L-aspartic acid, L-glutamic acid, L-histidine, L-pyroglutamic acid, L-serine, pectin, D-gluconic acid, D-Glucuronic acid, quinic acid, methyl pyruvate, L-malic acid, γ-amino-butyric acid, propionic acid, acetic acid. However, did not utilize N-acetyl-β-D-mannosamine, N-acetyl neuraminic acid, 3-Methyl Glucose, fusidic acid, D-serine, D-aspartic acid, D-galacturonic acid, L-galactonic acid lactone, mucic acid, D-saccharic acid, p-hydroxyphenylacetic acid, and sodium butyrate.
A. tomkonis strains tested are tolerant to troleandomycin, rifamycin SV, lincomycin, guanidine HCl, vancomycin, potassium tellurite, aztreonam, 1% sodium lactate, tetrazolium violet, and tetrazolium blue, but not to minocycline.
The delineation of A. tomkonis from other Agrobacterium species was based on overall genome relatedness indexes (all A. tomkonis share ANI > 98% and dDDH > 84%), as well as based on well-supported clades in gyrB gene, MLSA (with gyrB, parE, recA, and rpoB genes) or core-genome phylogenies. The type strain, IIF1SW-B1T (= LMG 32164 = NRRL B-65602), was isolated from the ISS Port panel of the Cupola, which is the observation deck for the crew. The type strain DNA G + C content is 59.18 mol% and its genome sequence is available from GenBank under WGS accession JABXYF000000000.
The sampling of ISS surfaces performed for this study took place within the United States on-orbit segments. Samples collected during this study were: Node 3 (Locations #1, #2, and #3), Node 1 (Locations #4 and #5), Permanent Multipurpose Module (Location #6), U.S. Laboratory (Location #7), and Node 2 (Locations #8 and control). A detailed description of the various locations sampled was published elsewhere (Singh et al., 2018b). Sample collection from ISS environmental surfaces, processing, and cultivation of bacteria have been previously reported (Checinska Sielaff et al., 2019). Three strains isolated during the second flight from Location #1 (Cupola) surfaces, were later identified as belonging to the genus Agrobacterium via 16S rRNA gene sequencing of the cultivated strains; these were designated: IIF1SW-B1T, IIF1SW-B3, and IIF1SW-B4.
Strains IIF1SW-B1T, IIF1SW-B3, IIF1SW-B4, CFBP 6623, CFBP 6624, and RTP8 strains were subjected for the phenotypic characterization. All strains are grown on yeast extract-peptone-glucose (YPG) medium and collected from the surface of the agar plates (Scortichini, 2012) before inoculation and screened for various organic substrate utilization using Biolog GEN III system (Biolog, Inc., Hayward, CA, United States) following manufacturer’s recommendations. Concurrently, two replicates of Biolog tests were carried out, and Biolog plates were incubated at 28°C for 48 h. After incubation, the OmniLogTM system was used to measure carbon substrate utilization.
A loopful of purified microbial culture (only ISS strains) was subjected to DNA extraction with the UltraClean DNA kit (MO BIO, Carlsbad, CA, United States) or Maxwell Automated System (Promega, Madison, WI, United States) as per manufacturer instructions. The extracted DNA was eluted in 50-μL of molecular grade water and stored at −20°C until further analysis. The 16S rRNA gene was amplified using the forward primer, 27F (5′-AGA GTT TGA TCC TGG CTC AG-3′) and the reverse primer, 1492R (5′-GGT TAC CTT GTT ACG ACT T-3′) (Checinska Sielaff et al., 2019) and PCR was performed with the following conditions: denaturation at 95°C for 5 min, followed by 35 cycles consisting of denaturation at 95°C for 50 s, annealing at 55°C for 50 s, and extension at 72°C for 1.5 min, and finalized by extension at 72°C for 10 min. The amplified products were treated with Antarctic phosphatase and exonuclease (New England Biolabs, Ipswich, MA, United States) to remove 5′- and 3′- phosphates from unused dNTPs before sequencing. Sequencing was performed by Macrogen (Rockville, MD, United States) using 27F and 1492R primers for Bacteria. The resulting sequences were assembled using SeqMan Pro from the DNASTAR Lasergene package (DNASTAR Inc., Madison, WI, United States). Bacterial sequences were searched against the EzTaxon-e database (Kim et al., 2012) and identified based on the closest percentage similarity (>98.6%) to previously identified microbial-type strains.
Plasmid profiles were determined by a modified Eckhardt agarose gel electrophoresis technique as previously described (Vial et al., 2006). Isolates were grown overnight in YPG medium at 28°C until the optical density at 600 nm reached 0.4. Electrophoresis was carried out at 5 V for 30 min and 90 V for 5 h at 4°C on a 0.75% agarose gel containing 1% sodium dodecyl sulfate. Plasmids sizes were estimated by comparison with those of “A. fabrum” C58 and Allorhizobium vitis S4 (Wood et al., 2001; Slater et al., 2009).
To test if the ISS G3 strains were plant-pathogen strains, 3-week-old tomato plants, cultivated with a photoperiod of 16 h/8 h light/dark in a greenhouse were stem-injured and inoculated with 10 μL of bacterial overnight culture (108 CFU/ml). The plants were then incubated for 21 days, and the presence or absence of crown gall symptoms was established by visual inspection. “A. fabrum” C58 strain was used as the positive control.
The WGS sequencing of the three ISS G3 strains was carried out as per established procedures (Singh et al., 2018a). Shotgun libraries were prepared using the Illumina Nextera Flex protocol (Singh et al., 2018b) and sequenced using NovaSeq 6000 S4 flow cell 2 × 150 PE sequencing kit. Verification of the quality of the raw sequencing data was carried out using FastQC v0.11.7 using default parameters2. Quality control for adapter trimming and quality filtering were performed using fastp v0.20.0 (Chen et al., 2018), and then SPAdes v3.11.1 (Bankevich et al., 2012) was used to assemble all the cleaned sequences. Fastp quality control was based on the following three parameters: (i) correction of mismatches in overlapped regions of paired-end reads, (ii) trimming of autodetected adapter sequences, and (iii) quality trimming at the 59 and 39 ends. To determine the quality of the assembled sequences, the number of contigs, the N50 value, and the total length were calculated using QUAST v5.0.2 (Gurevich et al., 2013). Default parameters were used for all software. The WGS sequencing of strains RTP8 and CFBP 6624 was carried out by Beckman Coulter Genomics (Takeley, Essex, United Kingdom). SPAdes v3.8.1 (Bankevich et al., 2012) was used to assemble all the cleaned sequences. All five new genomes were annotated using Prokka v1.14.5 (Seemann, 2014) with the following options: “–force –addgenes –compliant –usegenus.” Protein function was annotated by performing a blastp similarity search against a reference proteome database made of 405 high-quality genome assemblies obtained from the NCBI RefSeq, selecting all genomes belonging to the Rhizobiaceae family available as on 14th August 2018 that had a contig N50 greater than 98 kbp (Supplementary Table 7).
The ANI (Yoon et al., 2017) between each pair of the 40 Agrobacterium genomes (41 genomes including the two published versions of strain CFBP 6623 genome) was calculated using pyANI (Pritchard et al., 2016). The dDDH analysis was performed using the Genome-to-Genome Distance Calculator 2.0 (GGDC 2.0) (Auch et al., 2010).
We applied two separate pangenome analysis pipelines to obtain robust estimates of the sets of genes present and absent in each genome and to compute core and clade-specific gene sets for the various taxonomic groups represented in our dataset, including Agrobacterium biovar 1, A. tomkonis strains including ISS isolates.
(1) On one hand, a pangenomic Snakemake workflow (Köster and Rahmann, 2012) was run in anvi’o v6.2 (Eren et al., 2015). Briefly, gene clusters (GC) were defined by clustering all-versus-all open-reading frames BLAST similarity scores using the Markov Clustering (MCL) algorithm (Enright et al., 2002) with an inflation score of 8. Open reading frames were identified with Prodigal 2.6.3 in single mode (Hyatt et al., 2010). Open reading frames were annotated using COG and KOfam databases (Eren et al., 2015; Delmont and Eren, 2018). Results were displayed using anvi-display-pan function (Delmont and Eren, 2018). Functional enrichment in the ISS and G3 strain sets was obtained using anvi-compute-functional-enrichment function, as described in Shaiber et al. (2020), based on the COG annotation of open reading frames. Only enrichment for which FDR adjusted p-value < 0.05 are reported in Supplementary Table 5.
(2) On the other hand, a phylogenomic database was built with the Pantagruel pipeline (Lassalle et al., 2019), using the ‘‘usingGeneRax’’ branch of the code3 version “df79fee577b72389926a0138353fa657c11f3368.” The following pipeline tasks were performed: init, 00-03 and 05, leading to the clustering of homologous protein families with MMseqs2 (Steinegger and Söding, 2017), the alignment of their reverse-translated coding sequences with Clustal Omega (Sievers et al., 2011) and Python scripts using Biopython (Weisberg et al., 2020), and the computation of a maximum-likelihood (ML) phylogenetic tree with RAxML 8.1.2 (Stamatakis et al., 2005) based on the concatenation of 1,089 core-genome gene alignments, as previously described (Lassalle et al., 2021) (Supplementary Data File doi: 10.6084/m9.figshare.14792178). In addition, clade-specific sets of homologous gene clusters were determined using the “get_clade_specific_genes.r” script from Pantagruel package using the homologous gene family presence/absence matrix as input.
Metagenomic datasets (n = 42) previously obtained from environmental swabbing of the ISS (Singh et al., 2018b) were downloaded from NCBI-SRA BioProject PRJNA438545, quality filtered and mapped using BWA-MEM (Li and Durbin, 2009) against the genome assembly of strain IIF1SW-B1T. These steps were performed using anvi’o v6.2 (Eren et al., 2015) using anvi’o metagenomic workflow in reference mode and default parameters. Visual inspection of read mapping alignment was carried out using Artemis BamView (Carver et al., 2012).
For the 16S rRNA gene phylogenetic analysis, we extracted the 16S rRNA sequences from the genomes and added the reference 16S gene sequence from Rhizobium leguminosarum type strain USDA 2370T (GenBank accession U29386.1) and then aligned all sequences with Clustal Omega (Sievers et al., 2011) (Supplementary Data File doi: 10.6084/m9.figshare.14792148). For the gyrB single-gene phylogenetic analyses, as well as multi-locus sequence analysis (MLSA), we used the coding sequence (CDS) alignments for the marker genes parE, gyrB, recA and rpoB from the Pantagruel database described above (see Supplementary Data Files available at doi: 10.6084/m9.figshare.14792169 and doi: 10.6084/m9.figshare.14792100). For the MLSA, we concatenated the CDS alignments of parE, gyrB, recA, and rpoB into a multi-locus alignment, totaling 9,968 positions. ML Phylogenies were then inferred based on these alignments with RAxML-NG 1.0.0 (Stamatakis et al., 2005) with the options “–model GTR + G4 –all,” implying the use of the model GTR + FO + G4m, and that 20 independent inferences were run, starting with 10 random trees and 10 parsimony-optimized tree, and retaining the best ML tree from these 20 inferences. Branch supports were estimated with 200 Felsenstein bootstrap trees. The 16S tree was rooted using R. leguminosarum USDA 2370T sequence, whereas the gyrB and MLSA trees were rooted using the sequence from “A. albertimagni” AOL15 – a distant relative incorrectly classified in the Agrobacterium genus for which a classification in the newly proposed genus “Peteryoungia” was proposed (Kuzmanović et al., 2021).
The draft genome sequences of three ISS strains including type strainIIF1SW-B1T, as well as strains RTP8 and CFBP 6624, were deposited in NCBI. The genome sequences of isolates A. tomknonis IIF1SW-B1T, RTP8, and CFBP 6624 have been deposited to GenBank under the BioSample accessions SAMN15333190, SAMN17917486, and SAMN17918142, respectively; the WGS project accession numbers are also given in Table 1. The genome versions described in this paper are the first versions. Analysis intermediary data and result tables are available under the FigShare project available at https://figshare.com/account/home#/projects/115866 with individual items available at the following DOIs: 10.6084/m9.figshare.14792100, 10.6084/m9.figshare.14792148, 10.6084/m9.figshare.14792169, 10.6084/m9.figshare.14792178, 10.6084/m9.figshare.14883546, 10.6084/m9.figshare.14883516, 10.6084/m9.figshare.14798289, and 10.6084/m9.figshare.16782964.
CL, LV, XN, KV, and NS conceived and designed the microbiological experiments. NS, CL, and LV performed the microbiological experiments. CL and LV carried out the phenotypic assays Biolog based biochemical characterization. CL performed the plant pathogenicity experiments. LV performed plasmid content analyses. XN managed the Agrobacterium strain collection and the non-ISS strain genome sequencing project. NS and KV managed the ISS strain collection and the ISS strain genome sequencing project. FL, JN, and NS analyzed the data. NS analyzed the de novo assemblies, including contig alignment, and annotation checks. FL performed phylogenetic and phylogenomic analyses. JN performed metagenomic analysis. FL and JN performed pangenome analyses. FL and KV wrote the manuscript. All authors read and approved the final manuscript.
Part of the research described in this publication was carried out at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with National Aeronautics and Space Administration. This research was funded by a 2012 Space Biology NNH12ZTT001N grant no. 19-12829-26 under Task Order NNN13D111T award to KV, which also funded the post-doctoral fellowships for NS. This research was funded in part to FL by the Wellcome Trust Grant (206194). For the purpose of Open Access, the author has applied a CC-BY public copyright license to any author accepted manuscript version arising from this submission.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Aleksandra Checinska-Sielaff for isolating the strain and Achintya Bharathwaj for initial phylogenetic analyses, astronauts Captain Terry Virts for collecting samples aboard the ISS and the Implementation Team at NASA Ames Research Center (Fathi Karouia) for coordinating this effort, and Ryan Kemp (Zymo Research Corp.) for extracting DNA and Dan Butler (Weill Cornell Medicine) for performing shotgun sequencing using the NovaSeq platform. We acknowledged the Jet Propulsion Laboratory supercomputing facility staff, notably Narendra J. Patel (Jimmy) and Edward Villanueva, for their continuous support in providing the best possible infrastructure for BIGDATA analysis. © 2021 California Institute of Technology, Government sponsorship acknowledged. We would also like to thank Elise Lacroix for the plant cultures at the “Serre et Chambres Climatiques” platform of the FR BioEEnViS (UCBL) and L. Loiseau of PARMIC/CRB-EML platform of Ecologie Microbienne UMR CNRS 5557, UMR INRAE 1418.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.765943/full#supplementary-material
Supplementary Figure 1 | Plasmid profiles of A. tomkonis strains. Strains IIF1SW-B1, IIF1SW-B3 and IIF1SW-B4 showed the same plasmidic profil. Plasmids sizes were estimated by comparison with those of A. fabrum C58 and A. vitis S4 (data not shown). LC indicates the linear chromosome.
Supplementary Table 1 | ANI matrix of all tested Agrobacterium strains.
Supplementary Table 2 | Differential biochemical characteristics among A. tomkonis strains.
Supplementary Table 3 | COG function enriched in 9 Agrobacterium tomkonis (G3) strains, compared to the 31 other Agrobacterium genomes in the dataset.
Supplementary Table 4 | List of Agrobacterium clade-specific homologous gene families. Clade-specific genes are listed per clade; with each list starting by a hashtag and the clade id: “# cladeXX”. A. tumefaciens species complex/Agrobacterium biovar 1 is clade1; A. tomkonis is clade20; the clonal group of ISS strains is clade27. Genomes are listed using short codes, with the correspondence to strain names provided at the top.
Supplementary Table 5 | List of contigs specific to the clonal group of A. tomkonis strains isolated from the ISS and their characteristics. The count of genes with genetic mobility-related functions is based on the Prokka annotation of proteins.
Supplementary Table 6 | COG function enriched in 3 Agrobacterium tomkonis ISS strains, compared to the 37 other Agrobacterium genomes in the dataset.
Supplementary Table 7 | List of genome assemblies used as reference to annotate the new Agrobacterium genomes with Prokka.
Auch, A. F., Von Jan, M., Klenk, H.-P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barton, I. S., Fuqua, C., and Platt, T. G. (2018). Ecological and evolutionary dynamics of a model facultative pathogen: Agrobacterium and crown gall disease of plants. Environ. Microbiol. 20, 16–29. doi: 10.1111/1462-2920.13976
Bharadwaj, A. R., Daudu, R., Singh, N. K., Wood, J. M., Debieu, M., O’hara, N. B., et al. (2020a). Draft Genome Sequences of Enterobacteriales Strains Isolated from the International Space Station. Microbiol. Resour. Announc. 9, e817–e820.
Bharadwaj, A. R., Singh, N. K., Wood, J. M., Debieu, M., O’hara, N. B., Karouia, F., et al. (2020b). Draft Genome Sequences of Lactobacillales Isolated from the International Space Station. Microbiol. Resour. Announc. 9, e942–20.
Bijlani, S., Singh, N. K., Eedara, V. V. R., Podile, A. R., Mason, C. E., Wang, C. C. C., et al. (2021). Methylobacterium ajmalii sp. nov., Isolated From the International Space Station. Front. Microbiol. 12:639396. doi: 10.3389/fmicb.2021.639396
Bijlani, S., Singh, N. K., Mason, C. E., Wang, C. C. C., and Venkateswaran, K. (2020a). Draft Genome Sequences of Sphingomonas Species Associated with the International Space Station. Microbiol. Resour. Announc. 9, e00578–20.
Bijlani, S., Singh, N. K., Mason, C. E., Wang, C. C. C., and Venkateswaran, K. (2020b). Draft Genome Sequences of Tremellomycetes Strains Isolated from the International Space Station. Microbiol. Resour. Announc. 9, e00504–20.
Bouzar, H., and Jones, J. B. (2001). Agrobacterium larrymoorei sp. nov., a pathogen isolated from aerial tumours of Ficus benjamina. Int. J. Syst. Evol. Microbiol. 51, 1023–1026. doi: 10.1099/00207713-51-3-1023
Carver, T., Harris, S. R., Otto, T. D., Berriman, M., Parkhill, J., and Mcquillan, J. A. (2012). BamView: visualizing and interpretation of next-generation sequencing read alignments. Brief. Bioinform. 14, 203–212. doi: 10.1093/bib/bbr073
Checinska Sielaff, A., Urbaniak, C., Mohan, G. B. M., Stepanov, V. G., Tran, Q., Wood, J. M., et al. (2019). Characterization of the total and viable bacterial and fungal communities associated with the International Space Station surfaces. Microbiome 7:50. doi: 10.1186/s40168-019-0666-x
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Costechareyre, D., Rhouma, A., Lavire, C., Portier, P., Chapulliot, D., Bertolla, F., et al. (2010). Rapid and efficient identification of Agrobacterium species by recA allele analysis: Agrobacterium recA diversity. Microb. Ecol. 60, 862–872. doi: 10.1007/s00248-010-9685-7
Daudu, R., Parker, C. W., Singh, N. K., Wood, J. M., Debieu, M., O’hara, N. B., et al. (2020a). Draft Genome Sequences of Rhodotorula mucilaginosa Strains Isolated from the International Space Station. Microbiol. Resour. Announc. 9, e00570–20.
Daudu, R., Singh, N. K., Wood, J. M., Debieu, M., O’hara, N. B., Mason, C. E., et al. (2020b). Draft Genome Sequences of Bacillaceae Strains Isolated from the International Space Station. Microbiol. Resour. Announc. 9, e00701–20. doi: 10.1128/MRA.00701-20
Delmont, T. O., and Eren, A. M. (2018). Linking pangenomes and metagenomes: the Prochlorococcus metapangenome. PeerJ 6:e4320. doi: 10.7717/peerj.4320
Dessaux, Y., and Faure, D. (2018). “Niche Construction and Exploitation by Agrobacterium: How to Survive and Face Competition in Soil and Plant Habitats,” in Agrobacterium Biology: From Basic Science to Biotechnology, ed. S. B. Gelvin (Cham: Springer International Publishing), 55–86. doi: 10.1007/82_2018_83
Dodueva, I. E., Lebedeva, M. A., Kuznetsova, K. A., Gancheva, M. S., Paponova, S. S., and Lutova, L. L. (2020). Plant tumors: a hundred years of study. Planta 251:82. doi: 10.1007/s00425-020-03375-5
Enright, A. J., Van Dongen, S., and Ouzounis, C. A. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584. doi: 10.1093/nar/30.7.1575
Eren, A. M., Esen, O. C., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L., et al. (2015). Anvi’o: an advanced analysis and visualization platform for ’omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319
Flores-Felix, J. D., Menendez, E., Peix, A., Garcia-Fraile, P., and Velazquez, E. (2020). History and current taxonomic status of genus Agrobacterium. Syst. Appl. Microbiol. 43:126046. doi: 10.1016/j.syapm.2019.126046
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075.
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Keane, P., Kerr, A., and New, P. (1970). Crown Gall of Stone Fruit II. Identification and Nomenclature of Agrobacterium Isolates. Aust. J. Biol. Sci. 23, 585–596.
Kersters, K., De Ley, J., Sneath, P. H. A., and Sackin, M. (1973). Numerical Taxonomic Analysis of Agrobacterium. Microbiology 78, 227–239. doi: 10.1099/00221287-78-2-227
Kim, O. S., Cho, Y. J., Lee, K., Yoon, S. H., Kim, M., Na, H., et al. (2012). Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int. J. Syst. Evol. Microbiol. 62, 716–721. doi: 10.1099/ijs.0.038075-0
Knox, B. P., Blachowicz, A., Palmer, J. M., Romsdahl, J., Huttenlocher, A., Wang, C. C., et al. (2016). Characterization of Aspergillus fumigatus Isolates from Air and Surfaces of the International Space Station. mSphere 1, e00227–16.
Köster, J., and Rahmann, S. (2012). Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522. doi: 10.1093/bioinformatics/bts480
Kuzmanović, N., Fagorzi, C., Mengoni, A., Lassalle, F., and Dicenzo, G. C. (2021). Taxonomy of Rhizobiaceae revisited: proposal of a new framework for genus delimitation. bioRxiv [Preprint]. doi: 10.1101/2021.08.02.454807
Kuzmanović, N., Puławska, J., Smalla, K., and Nesme, X. (2018). Agrobacterium rosae sp. nov., isolated from galls on different agricultural crops. Syst. Appl. Microbiol. 41, 191–197. doi: 10.1016/j.syapm.2018.01.004
La Duc, M. T., Satomi, M., Agata, N., and Venkateswaran, K. (2004). gyrB as a phylogenetic discriminator for members of the Bacillus anthracis-cereus-thuringiensis group. J. Microbiol. Methods 56, 383–394. doi: 10.1016/j.mimet.2003.11.004
Lassalle, F., Campillo, T., Vial, L., Baude, J., Costechareyre, D., Chapulliot, D., et al. (2011). Genomic species are ecological species as revealed by comparative genomics in Agrobacterium tumefaciens. Genome Biol. Evol. 3, 762–781. doi: 10.1093/gbe/evr070
Lassalle, F., Dastgheib, S. M. M., Zhao, F.-J., Zhang, J., Verbarg, S., Frühling, A., et al. (2021). Phylogenomics reveals the basis of adaptation of Pseudorhizobium species to extreme environments and supports a taxonomic revision of the genus. Syst. Appl. Microbiol. 44:126165. doi: 10.1016/j.syapm.2020.126165
Lassalle, F., Planel, R., Penel, S., Chapulliot, D., Barbe, V., Dubost, A., et al. (2017). Ancestral Genome Estimation Reveals the History of Ecological Diversification in Agrobacterium. Genome Biol. Evol. 9, 3413–3431. doi: 10.1093/gbe/evx255
Lassalle, F., Veber, P., Jauneikaite, E., and Didelot, X. (2019). Automated reconstruction of all gene histories in large bacterial pangenome datasets and search for co-evolved gene modules with Pantagruel. bioRxiv [Preprint]. doi: 10.1101/586495
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Macrae, S., Thomson, J. A., and Staden, J. V. (1988). Colonization of Tomato Plants by Two Agrocin-Producing Strains of Agrobacterium tumefaciens. Appl. Environ. Microbiol. 54, 3133–3137. doi: 10.1128/aem.54.12.3133-3137.1988
Mafakheri, H., Taghavi, S. M., Puławska, J., De Lajudie, P., Lassalle, F., and Osdaghi, E. (2019). Two Novel Genomospecies in the Agrobacterium tumefaciens Species Complex Associated with Rose Crown Gall. Phytopathology 109, 1859–1868. doi: 10.1094/PHYTO-05-19-0178-R
Matthysse, A. G., and McMahan, S. (1998). Root Colonization by Agrobacterium tumefaciens Is Reduced in cel, attB, attD, and attR Mutants. Appl. Environ. Microbiol. 64, 2341–2345. doi: 10.1128/AEM.64.7.2341-2345.1998
Mhatre, S., Singh, N. K., Wood, J. M., Parker, C. W., Pukall, R., Verbarg, S., et al. (2020). Description of Chloramphenicol Resistant Kineococcus rubinsiae sp. nov. Isolated from a Spacecraft Assembly Facility. Front. Microbiol. 11:1957. doi: 10.3389/fmicb.2020.01957
Mousavi, S. A., Österman, J., Wahlberg, N., Nesme, X., Lavire, C., Vial, L., et al. (2014). Phylogeny of the Rhizobium–Allorhizobium–Agrobacterium clade supports the delineation of Neorhizobium gen. nov. Syst. Appl. Microbiol. 37, 208–215. doi: 10.1016/j.syapm.2013.12.007
Mousavi, S. A., Willems, A., Nesme, X., De Lajudie, P., and Lindström, K. (2015). Revised phylogeny of Rhizobiaceae: proposal of the delineation of Pararhizobium gen. nov., and 13 new species combinations. Syst. Appl. Microbiol. 38, 84–90. doi: 10.1016/j.syapm.2014.12.003
Ophel, K., and Kerr, A. (1987). “Ecology of Agrobacterium: Plasmids and Biovars,” in Molecular genetics of plant-microbe interactions: Proceedings of the Third International Symposium on the Molecular Genetics of Plant-Microbe Associations, Montréal, Québec, Canada, July 27–31, 1986, eds D. P. S. Verma and N. Brisson (Dordrecht: Springer), 3–5. doi: 10.1007/978-94-009-4482-4_1
Panday, D., Schumann, P., and Das, S. K. (2011). Rhizobium pusense sp. nov., isolated from the rhizosphere of chickpea (Cicer arietinum L.). Int. J. Syst. Evol. Microbiol. 61, 2632–2639. doi: 10.1099/ijs.0.028407-0
Popoff, M. Y., Kersters, K., Kiredjian, M., Miras, I., and Coynault, C. (1984). [Taxonomic position of Agrobacterium strains of hospital origin]. Ann. Microbiol. 135A, 427–442.
Portier, P., Fischer-Le Saux, M., Mougel, C., Lerondelle, C., Chapulliot, D., Thioulouse, J., et al. (2006). Identification of genomic species in Agrobacterium biovar 1 by AFLP genomic markers. Appl. Environ. Microbiol. 72, 7123–7131. doi: 10.1128/AEM.00018-06
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/c5ay02550h
Puławska, J., and Kałużna, M. (2012). Phylogenetic relationship and genetic diversity of Agrobacterium spp. isolated in Poland based on gyrB gene sequence analysis and RAPD. Eur. J. Plant Pathol. 133, 379–390. doi: 10.1007/s10658-011-9911-2
Puławska, J., Willems, A., De Meyer, S. E., and Süle, S. (2012). Rhizobium nepotum sp. nov. isolated from tumors on different plant species. Syst. Appl. Microbiol. 35, 215–220. doi: 10.1016/j.syapm.2012.03.001
Ramírez-Bahena, M. H., Vial, L., Lassalle, F., Diel, B., Chapulliot, D., Daubin, V., et al. (2014). Single acquisition of protelomerase gave rise to speciation of a large and diverse clade within the Agrobacterium/Rhizobium supercluster characterized by the presence of a linear chromid. Mol. Phylogenet. Evol. 73, 202–207. doi: 10.1016/j.ympev.2014.01.005
Sanguin, H., Remenant, B., Dechesne, A., Thioulouse, J., Vogel, T. M., Nesme, X., et al. (2006). Potential of a 16S rRNA-Based Taxonomic Microarray for Analyzing the Rhizosphere Effects of Maize on Agrobacterium spp. and Bacterial Communities. Appl. Environ. Microbiol. 72, 4302–4312. doi: 10.1128/AEM.02686-05
Scortichini, M. (2012). Detection and identification methods and new tests as developed and used in the framework of cost 873 for bacteria pathogenic to stone fruits and nuts Pseudomonas avellanae. J. Plant Pathol. 94, 115–16.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shaiber, A., Willis, A. D., Delmont, T. O., Roux, S., Chen, L.-X., Schmid, A. C., et al. (2020). Functional and genetic markers of niche partitioning among enigmatic members of the human oral microbiome. Genome Biol. 21:292. doi: 10.1186/s13059-020-02195-w
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Simpson, A. C., Urbaniak, C., Singh, N. K., Wood, J. M., Debieu, M., O’hara, N. B., et al. (2021). Draft Genome Sequences of Various Bacterial Phyla Isolated from the International Space Station. Microbiol. Resour. Announc. 10, e00214–e00221. doi: 10.1128/MRA.00214-21
Singh, N. K., Bezdan, D., Checinska Sielaff, A., Wheeler, K., Mason, C. E., and Venkateswaran, K. (2018a). Multi-drug resistant Enterobacter bugandensis species isolated from the International Space Station and comparative genomic analyses with human pathogenic strains. BMC Microbiol. 18:175. doi: 10.1186/s12866-018-1325-2
Singh, N. K., Wood, J. M., Karouia, F., and Venkateswaran, K. (2018b). Succession and persistence of microbial communities and antimicrobial resistance genes associated with International Space Station environmental surfaces. Microbiome 6:214.
Slater, S. C., Goldman, B. S., Goodner, B., Setubal, J. C., Farrand, S. K., Nester, E. W., et al. (2009). Genome sequences of three agrobacterium biovars help elucidate the evolution of multichromosome genomes in bacteria. J. Bacteriol. 191, 2501–2511. doi: 10.1128/JB.01779-08
Smith, E. F., and Townsend, C. O. (1907). A Plant-Tumor of Bacterial Origin. Science 25, 671–673. doi: 10.1126/science.25.643.671
Solomon, S. A., Bharadwaj, A. R., Singh, N. K., Wood, J. M., Debieu, M., O’hara, N. B., et al. (2020). Draft Genome Sequences of Klebsiella Species Isolated from the International Space Station. Microbiol. Resour. Announc. 9, e00923–20.
Stamatakis, A., Ludwig, T., and Meier, H. (2005). RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463. doi: 10.1093/bioinformatics/bti191
Steinegger, M., and Söding, J. (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028. doi: 10.1038/nbt.3988
Vaudequin-Dransart, V., Petit, A., Poncet, C., Ponsonnet, C., Nesme, X., Jones, J. B., et al. (1995). Novel Ti plasmids in Agrobacterium strains isolated from fig tree and chrysanthemum tumors and their opinelike molecules. Mol. Plant Microbe Interact. 8, 311–321. doi: 10.1094/mpmi-8-0311
Vial, L., Cuny, C., Gluchoff-Fiasson, K., Comte, G., Oger, P. M., Faure, D., et al. (2006). N-acyl-homoserine lactone-mediated quorum-sensing in Azospirillum: an exception rather than a rule. FEMS Microbiol. Ecol. 58, 155–168. doi: 10.1111/j.1574-6941.2006.00153.x
Weisberg, A. J., Davis, E. W., Tabima, J., Belcher, M. S., Miller, M., Kuo, C.-H., et al. (2020). Unexpected conservation and global transmission of agrobacterial virulence plasmids. Science 368:eaba5256. doi: 10.1126/science.aba5256
Wood, D. W., Setubal, J. C., Kaul, R., Monks, D. E., Kitajima, J. P., Okura, V. K., et al. (2001). The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science 294, 2317–2323. doi: 10.1126/science.1066804
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S., and Chun, J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286.
Young, J. M., Kuykendall, L. D., Martínez-Romero, E., Kerr, A., and Sawada, H. (2001). A revision of Rhizobium Frank 1889, with an emended description of the genus, and the inclusion of all species of Agrobacterium Conn 1942 and Allorhizobium undicola de Lajudie et al. 1998 as new combinations: Rhizobium radiobacter, R. rhizogenes, R. rubi, R. undicola and R. vitis. Int. J. Syst. Evol. Microbiol. 51, 89–103. doi: 10.1099/00207713-51-1-89
Keywords: Agrobacterium tomkonis, genomovar G3, ISS, metagenomics, phylogenomics, MLSA
Citation: Singh NK, Lavire C, Nesme J, Vial L, Nesme X, Mason CE, Lassalle F and Venkateswaran K (2021) Comparative Genomics of Novel Agrobacterium G3 Strains Isolated From the International Space Station and Description of Agrobacterium tomkonis sp. nov.. Front. Microbiol. 12:765943. doi: 10.3389/fmicb.2021.765943
Received: 27 August 2021; Accepted: 12 October 2021;
Published: 06 December 2021.
Edited by:
Eric Daniel Becraft, University of North Alabama, United StatesReviewed by:
Tatiana Matveeva, Saint Petersburg State University, RussiaCopyright © 2021 Singh, Lavire, Nesme, Vial, Nesme, Mason, Lassalle and Venkateswaran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Florent Lassalle, Zmw0QHNhbmdlci5hYy51aw==; Kasthuri Venkateswaran, a2p2ZW5rYXRAanBsLm5hc2EuZ292
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.