
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 09 November 2021
Sec. Food Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.752529
 Zhonghuan Tian1,2,3Yujie Du1,2,3Fan Yang1,2,3Juan Zhao1,2,3Shuqi Liu1,2,3Deyao Zhang1,2,3Chao-an Long1,2,3*
Zhonghuan Tian1,2,3Yujie Du1,2,3Fan Yang1,2,3Juan Zhao1,2,3Shuqi Liu1,2,3Deyao Zhang1,2,3Chao-an Long1,2,3*Biological control is an environmentally friendly, safe, and replaceable strategy for disease management. Genome sequences of a certain biocontrol agent could lay a solid foundation for the research of molecular biology, and the more refined the reference genome, the more information it provides. In the present study, a higher resolution genome of Kloeckera apiculata 34-9 was assembled using high-throughput chromosome conformation capture (Hi-C) technology. A total of 8.07 M sequences of K. apiculata 34-9 genome was anchored onto 7 pesudochromosomes, which accounting for about 99.51% of the whole assembled sequences, and 4,014 protein-coding genes were annotated. Meanwhile, the detailed gene expression changes of K. apiculata 34-9 were obtained under low temperature and co-incubation with Penicillium digitatum treatments, respectively. Totally 254 differentially expressed genes (DEGs) were detected with low temperature treatment, of which 184 and 70 genes were upregulated and downregulated, respectively. Some candidate genes were significantly enriched in ribosome biosynthesis in eukaryotes and ABC transporters. The expression of gene Kap003732 and Kap001595 remained upregulated and downregulated through the entire time-points, respectively, indicating that they might be core genes for positive and negative response to low temperature stress. When co-incubation with P. digitatum, a total of 2,364 DEGs were found, and there were 1,247 upregulated and 1,117 downregulated genes, respectively. Biosynthesis of lysine and arginine, and phenylalanine metabolism were the highest enrichment of the cluster and KEGG analyses of the co-DEGs, the results showed that they might be involved in the positive regulation of K. apiculata 34-9 response to P. digitatum. The completeness of K. apiculata 34-9 genome and the transcriptome data presented here are essential for providing a high-quality genomic resource and it might serve as valuable molecular properties for further studies on yeast genome, expression pattern of biocontrol system, and postharvest citrus storage and preservation.
Hanseniaspora species, including H. uvarum, H. vineae, and H. guilliermondii, are widely distributed in various environments with lemon-shaped cell morphology, and they are broadly known to be plentiful in wine fermentation and have a great influence on wine quality compared to other non-Saccharomyces yeast species (Langenberg et al., 2017; Guaragnella et al., 2020). Their abilities to provide enhanced levels of acetate esters, benzenoids, and other valuable compounds make them very promising fermentation strains (Tristezza et al., 2016; Martin et al., 2018). Moreover, enough studies had reported that H. uvarum (anamorph Kloeckera apiculata), one of the extensively known apiculata yeasts, could be frequently detected on mature fruits (especially on grapes), fermented beverages, soils, and plants (Valles et al., 2007; Zott et al., 2008). In addition, H. uvarum exhibited strong antagonistic properties against some phytopathogens which were responsible for citrus, strawberries, grapes, and plum fruit spoilage, thus, it was regarded as a potential biological control agent (Long et al., 2005; Cai et al., 2015; Apaliya et al., 2017; Zhang et al., 2017). In our previous studies, K. apiculata 34-9 (CCTCC M204025) showed biocontrol efficacy against Penicillium digitatum, P. italicum, and Botrytis cinerea (Long et al., 2005; Liu P. et al., 2014), and its biological control mechanisms had been studied including secretion of antifungal compounds (Liu P. et al., 2014), induction of citrus fruit resistance (Liu P. et al., 2016), and competition for nutrients with the fungi (Liu P. et al., 2013). Genome sequences of K. apiculata 34-9 were obtained using the Illumina HiSeq 2000 platform, a total of 8.1 M sequences were assembled into 41 scaffolds, and 3,786 protein-coding genes were defined after genome assembly and annotation (Chen et al., 2017). However, knowledge of the genome version at the chromosomal level is lacking, and the completeness of the genome is essential for providing high-quality genomic resources for further studies.
Abiotic and biotic stresses are the threatening environmental restrictions of all organisms (Suzuki et al., 2014; Bai et al., 2018; Sies and Jones, 2020; Roeber et al., 2021). They also play important roles in the inhibitory effect of antimicrobial agents. Heat stress treatment adversely affects biocontrol efficiency of Debaryomyces hansenii against kiwifruit gray mold, and it showed decreased expression of genes related to stimulus response and developmental process (Dai et al., 2021). The transcriptome profiles of S. cerevisiae and S. kudriavzevii showed diverse responses to cold shock of these two yeast strains, the latter strain exhibited increased translation rate in cold adaptation (Tronchoni et al., 2014). Low temperature may influence the energy absorption and cause metabolic imbalance in yeast cells, and it could decrease their biocontrol efficiency against phytopathogens. So, the relevant tolerance mechanisms of yeast exposed to low temperature are important for the food industry. However, the elucidation of cold resistance mechanisms of K. apiculata and the genes involved were still largely unknown.
Genome analyses have been conducted to study the genetics and molecular biology of different Hanseniaspora species recently. High similarity in size and genes annotation were revealed within several strains of H. vineae, and that it diverged from the Saccharomyces complex clade before the whole-genome duplication event (Giorello et al., 2019). Seixas et al. (2019) found that H. guiliermondii was destitute of the functional gluconeogenesis or glyoxylate cycle, and it was believed to be utilized in wine-making and other relevant biotechnological industrial processes. Accessibility of these sequenced genomic resources could therefore contribute to a better comprehension of these species on molecular and genetic aspects. This would affect genetic modification on fermentation traits and facilitate the application of these yeasts.
Here we established a chromosome-level assembly of the genome of K. apiculata, using a combination of next-generation sequencing and Hi-C (high-throughput chromosome conformation capture) scaffolding, which had been successfully utilized for the assembly of plant, animal, and fungal genomes (Dekker et al., 2013; Shao et al., 2018; Peng et al., 2019; Yang et al., 2020), to study key questions about K. apiculata genome biology that have been previously difficult to address due to the fragmentary genome assembly. Meanwhile, transcriptome analyses were performed to gain more knowledge on the growth of K. apiculata at low temperature, and to better understand the possible biocontrol mechanisms of K. apiculata against P. digitatum. These findings may reveal comprehensions into the molecular mechanisms of low temperature tolerance in K. apiculata and provide diverse transcriptome resources related to low temperature tolerance and biotic stress research.
The antagonist yeast K. apiculata 34-9 was isolated and preserved in a postharvest laboratory of Huazhong Agricultural University. It was cultured on YPDA (1% yeast extract, 2% peptone, 2% dextrose, 2% agar) plate for 2 days, or YPD medium (1% yeast extract, 2% peptone, 2% dextrose) in a rotary shaker coupled with 150 rpm shaking for 1 day at 25°C, respectively.
The plant pathogen P. digitatum N1 which was isolated and preserved in a postharvest lab of Huazhong Agricultural University was used in this study. An even mycelial layer was obtained according to a previous study (Tian et al., 2020). Briefly, 1 mL spore suspensions at a concentration of 1.0 × 106 CFU/mL of P. digitatum was transferred into 100 mL liquid PDA medium (below 50°C) and immediately divided into Petri dishes followed by absolutely mixed. The Petri dishes were air-dried, then incubated in a climate chamber for 4 days at 25°C.
The Hi-C sequencing approach was explored to obtain high-quality assembly and annotated genome. Fresh K. apiculata 34-9 cell samples grown on YPDA plates were harvested after incubation for 2 days at 25°C, then fixed in 1.0% formaldehyde at room temperature for crosslinking to maintain its cellular three-dimensional structure. The yeast cells were lysed, and the cross-linked DNA obtained was then digested with HindIII restriction enzyme. The digested DNA ends were filled and labeled with biotin, then ligated to form a circular chimeric molecule, and the DNA was purified and cut into 300–700 bp fragments. The density and insert size of the constructed library was determined using Qubit 2.0 (Thermo Fisher Scientific Inc., Waltham, United States) and Agilent 2100 Bioanalyzer system (Agilent Technologies Inc., Santa Clara, United States). Then the constructed library was processed for paired-end sequencing on the Illumina Hiseq X Ten platform. The Hi-C reads were filtered using Hi-C Pro to validate paired-end reads (Servant et al., 2015). In addition, the draft contigs obtained from Hi-C sequencing were grouped, ordered, and oriented for chromosomes assembly using the software Lachesis with default parameters (Burton et al., 2013).
Tandem repeats were identified with the utilization of a Tandem Repeat Finder (TRF) in the genome of K. apiculata 34-9 (Benson, 1999). Transposon elements (TEs) of K. apiculata 34-9 genome were annotated with the combination of homology and de novo approaches. Briefly, RepeatMasker1 and RepeatProteinMask were used for the annotation of TEs of K. apiculata 34-9 genome based on the RepBase database2 and RepeatMasker package TE protein database, respectively. RepeatModeler and LTR_FINDER were applied for de novo repeat prediction (Xu and Wang, 2007), and RepeatMasker was utilized for the annotation of TEs with default parameters.
De novo gene prediction and homology-based strategy were employed to perform the gene prediction and functional annotation, and these results were integrated into a gene model using MAKER2 (Holt and Yandell, 2011). The completeness and accuracy of the assembly was assessed using CEGMA (Parra et al., 2007), and the final reliable gene set was obtained in a HiCESAP process. Protein sequences of H. guilliermondii (accession number: GCA_900119595.1), H. opuntiae (accession number: GCA_001749795.1), H. uvarum (accession number: GCA_001747055.1), H. valbyensis (accession number: GCA_001664025.1) genomes were aligned to the genome of K. apiculata 34-9 to perform homology-based gene prediction. All predicted protein-coding sequences were aligned to the SwissProt, non-redundant protein (NR), Gene Ontology (GO), InterPro, Kyoto Encyclopedia of Genes and Genomes (KEGG), and TrEMBL databases for further annotation using BLASTP with default parameters.
For annotation of non-coding RNA, tRNAscan-SE was used to screen the tRNA of K. apiculata 34-9 genome, and BLASTN was applied to identify rRNA based on sequence alignment with homologous species, micro-RNA (miRNA) and small nuclear RNA (snRNA) were annotated by INFERNAL from Rfam.3
The re-assembled genome of K. apiculata 34-9 was compared with H. uvarum (assembly GCA_001747055.1) and S. cerevisiae (assembly GCA_002057635.1). Collinearity among these three yeasts was performed using TBtools (Chen et al., 2020) with default parameters.
For low temperature (LT) treatment, yeast samples were collected after incubation for 1 day in YPD at 25°C, the cells were rapidly frozen in liquid nitrogen after centrifugation at 3,000 × g for 10 min, and marked as 0 h, then stored at −80°C. From that time, some YPD flasks were placed into a 4°C incubator for continuous incubation, and yeast samples were harvested after 5, 12, 24, and 48 h, respectively. Yeast grown at 25°C (normal temperature, Kap) was handled as the same procedure.
For the preparation of interaction between antagonist K. apiculata 34-9 and P. digitatum N1 (Pdi), yeast samples harvested after incubation for 1 day in YPD at 25°C were adjusted to a concentration of 1.0 × 108 CFU/mL, and a total of 1 mL K. apiculata cell suspension was inoculated to each P. digitatum plate and continuously cultured at 25°C, from that time (marked 0 h), samples were harvested as the above time course. There were three biological replicates of each sample. All samples were rapidly frozen in liquid nitrogen and immediately stored at −80°C.
Total RNA from collected yeast samples was isolated using TRIzon Reagent (Cwbiotech, China) following the manufacturer’s instruction. RNA concentration and integrity were measured using NanoDrop 2000 (Thermo Fisher Scientific Inc., Waltham, United States) and Agilent 2100 Bioanalyzer system (Agilent Technologies Inc., Santa Clara, United States), respectively. RNA-sequencing (RNA-seq) libraries were prepared using a NEBNext Ultra RNA Library Prep Kit for Illumina (New England BioLabs® Inc.) according to the manufacturer’s guide, and the libraries were sequenced on an Illumina HiSeq X 10 platform (Illumina Inc., San Diego, CA). The generated reads were trimmed to eliminate adaptors and enhance quality. The filtrated clean reads were mapped to K. apiculata 34-9 genome using Hisat2 (version 2.1.0) and aligned to the generated transcriptome assembly using Bowtie2 (version 2.2.2), respectively. The output results were estimated using RSEM (version 1.3.0) to obtain normalized counts of gene expression level. The values of expression genes were calculated as fragments per kilobase per million mapped reads (FPKM).
The analyses of differentially expressed genes (DEGs) were conducted using R package DEseq2, and the DEGs were selected if their false discovery rate (FDR) < 0.05 and a log2 fold change > 1, then the DEGs were annotated to GO terms (Ye et al., 2018) and KEGG enrichments (Xie et al., 2011) for functional analyses.
Quantitative real-time PCR (qRT-PCR) assays were conducted to confirm RNA-seq results independently. Total RNA was extracted with Fungal RNA Kit (OMEGA Biotech, Guangzhou, China) followed by the operating instructions. The first-strand cDNA synthesis was conducted using a PrimeScriptTM RT reagent Kit with gDNA Eraser (TaKaRa Biotechnology, Dalian, China). The PowerUPTM SYBRTM Green Master Mix (Life Technologies, United States) was used for the detection of the relative expression profiles according to the recommended procedures. qRT-PCR was performed on a Q6 Flex Real-Time PCR system (Life Technologies, United States). The relative gene expression level was analyzed by the 2–ΔΔCt method (Livak and Schmittgen, 2001). The β-actin was used as a reference gene for normalization, all qRT-PCR primers were designed using Primer-BLAST4 and shown in Supplementary Table 1.
The Hi-C sequencing and RNA-seq data generated in this study were released in NCBI Sequence Read Archive database5 under accession numbers PRJNA667993 and PRJNA668195.
The next-generation sequencing (previous data) and Hi-C scaffolding were combined to improve K. apiculata yeast genome assembly and chromosome anchoring accuracy. A total of 3.47 Gb (428.40 × coverage) clean data were generated after quality control. K. apiculata genome was assembled into 8.1 Mb of scaffolds sequences and the assembly contained 15 scaffolds (more than 1 kb per scaffold), with a scaffold N50 of 1,349,021 bp (about 4.8-fold larger than that of the previous version), and the maximum scaffold length was 1,727,301 bp (Table 1). Meanwhile, the assembly comprised 155 contigs (more than 1 kb per contig), with a contig N50 of 104,210 bp, and the maximum contig length was 553,666 bp. The GC content was 31.98%, and there were 4,014 protein-coding genes of the new genome version, which was 228 more than the previous annotation version. In addition, 99.51% (8.06 Mb) of the genome sequences had been divided into seven groups (Supplementary Table 2). As many as 75.55% of the reads were uniquely mapped to the genome of K. apiculata 34-9, which were good enough for further analyses. A Hi-C contact heatmap was visualized to confirm the accuracy of Hi-C assembly at 20 kb resolution, all bins could be clearly anchored onto seven pseudochromosomes, the intensity of red color represents the number of Hi-C links on the pseudochromosomes of final assembly (Figures 1A,B). In addition, synteny blocks among K. apiculata 34-9, H uvarum and S. cerevisiae were visualized (Figure 1C).
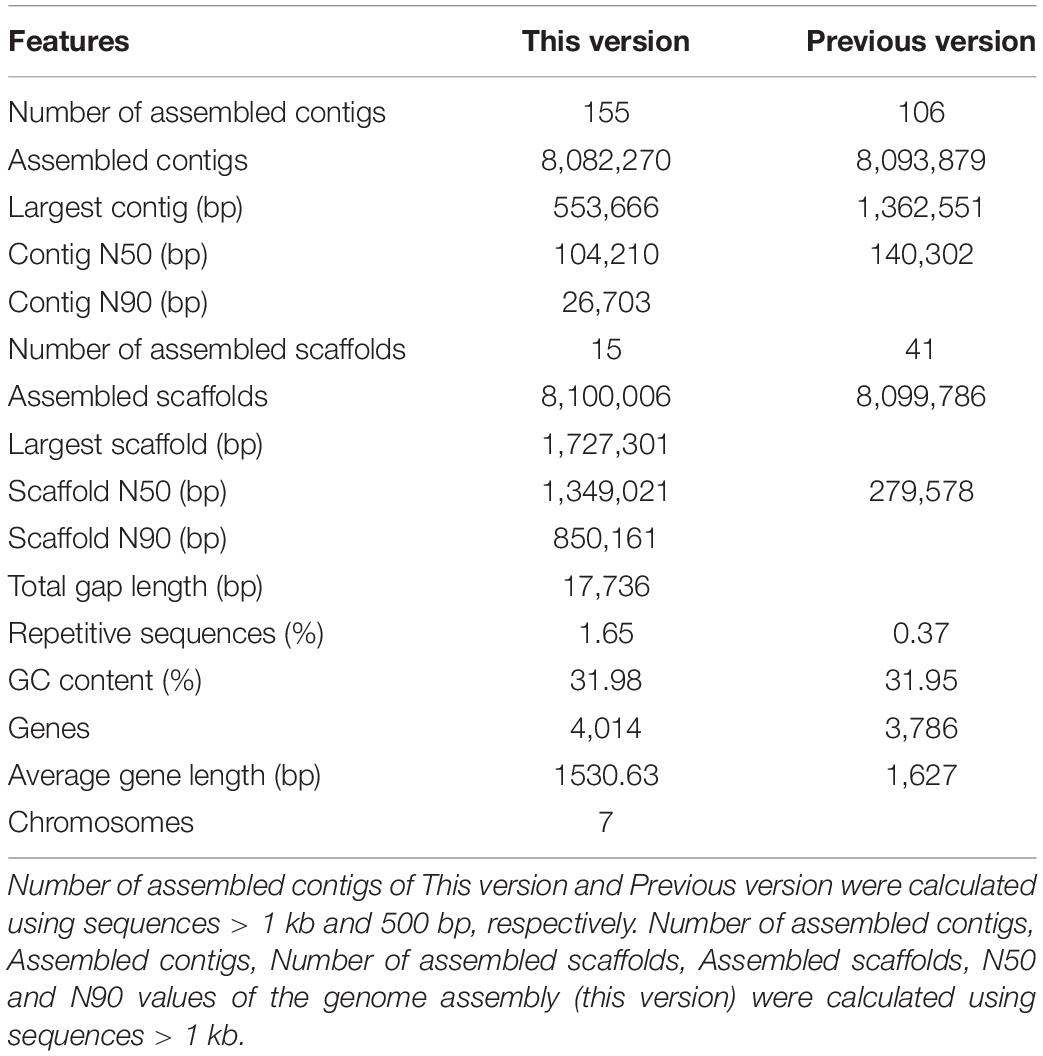
Table 1. Statistics of the Kloeckera apiculata draft genome assembly assisted by Hi-C sequencing.
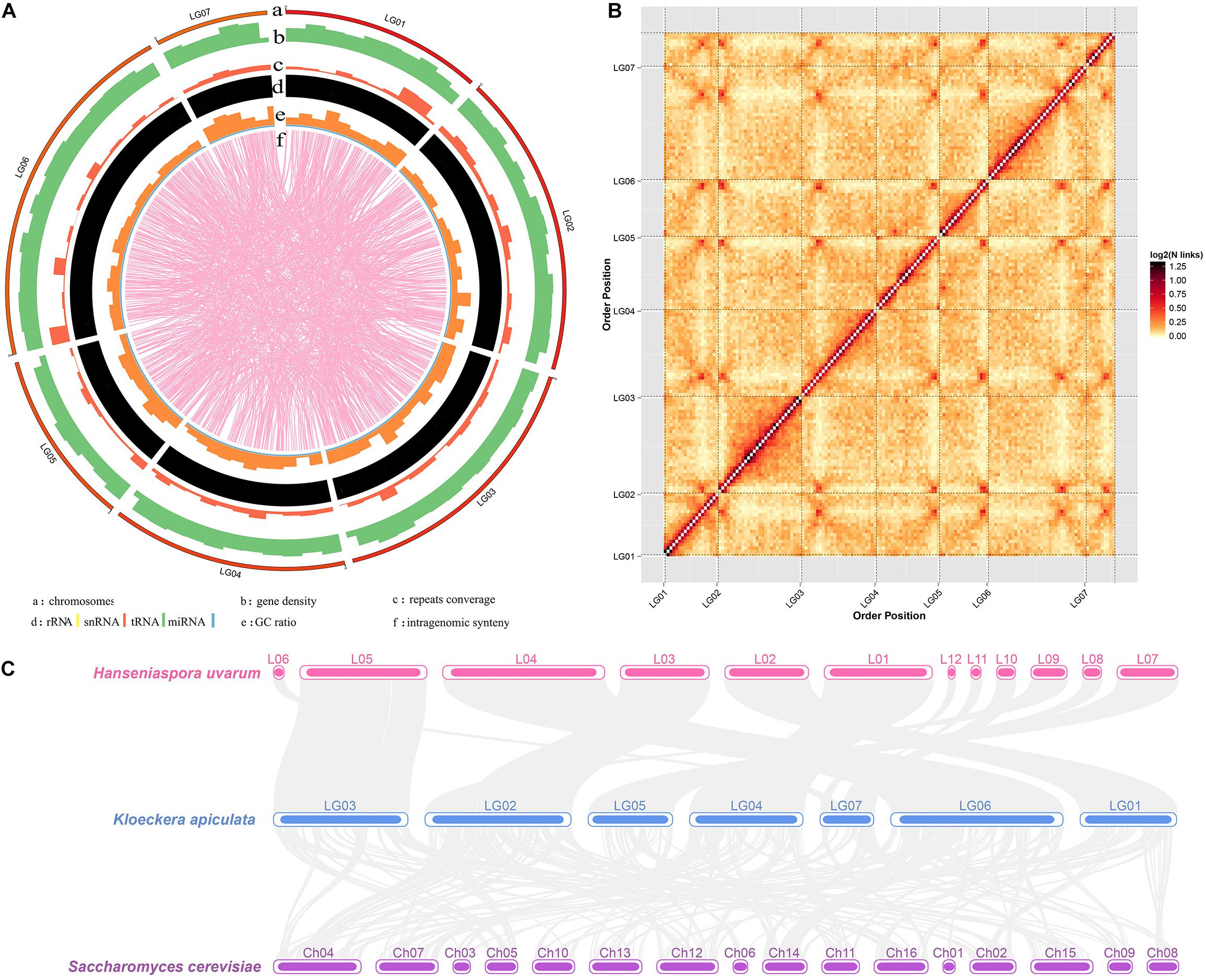
Figure 1. Chromosome resolution genome of K. apiculata. Genome synteny of Hanseniaspora uvarum, K. apiculate, and Saccharomyces cerevisiae are obtained from scaffolds of H. uvarum, and chromosomes of K. apiculata 34-9 and S. cerevisiae.
Repeat elements are important parts of the genome. A total of 133,569 bp (1.65%) repeat sequences were identified in the genome of K. apiculata 34-9. Among them, the proportion of long terminal repeats (LTRs), long interspersed repeats, short interspersed repeats, and DNA transposon was 0.58, 0.16, 0.01, and 0.36% of the genome, respectively (Supplementary Table 3).
In the present study, we aimed to uncover the mystery of K. apiculata responses to cold shock and P. digitatum stress, and comparative transcriptomics studies were conducted to gain more insights into the underlying molecular basis. A total of 304.08 Gb clean data were generated from the sequenced 39 yeast samples (at least 21,559,868 reads pairs of each sample), the Q30 was more than 92.54 and 80.54–85.50% of the reads were uniquely mapped to the genome of K. apiculata 34-9 (Supplementary Table 4). Since the trials were performed within different time courses, yeast samples obtained at the initial time (incubation for 1 day at 25°C) were marked as 0 h, and their transcriptome data were filtered out, then samples from the rest of the four time periods were conducted for transcription profiles analyses.
The detailed number of DEGs are illustrated in Table 2, and as many as 254 (184 up-regulated, 70 down-regulated) and 2,364 (1,247 up-regulated, 1,117 down-regulated) DEGs were detected in LT and Pdi, respectively. As it can be seen in Table 3, the largest proportion of genes in LT are annotated in cellular component organization or biogenesis, and the maximal percentage of genes in Pdi are annotated in catalytic activity. The majority of DEGs were up-regulated in both treatments (Figures 2A,B), with nearly 72.44 and 52.75% percentage of all the DEGs, respectively. In addition, the largest number of DEGs in LT was found at 48 h, and Pdi was at 24 h. Genes differentially expressed at two or more time points were defined as CEGs (co-differentially expressed genes), a total of six CEGs (three up-regulated genes and three down-regulated genes) appeared in both LT and Pdi at the four time points (Figure 2C), and five CEGs were annotated as encoding hypothetical protein, with only one gene (Kap003732) annotated as encoding geranylgeranyl transferase type-2 subunit beta. Here, we found the three up-regulated genes and the three down-regulated genes might be involved in biotic and abiotic stresses responses of K. apiculata.
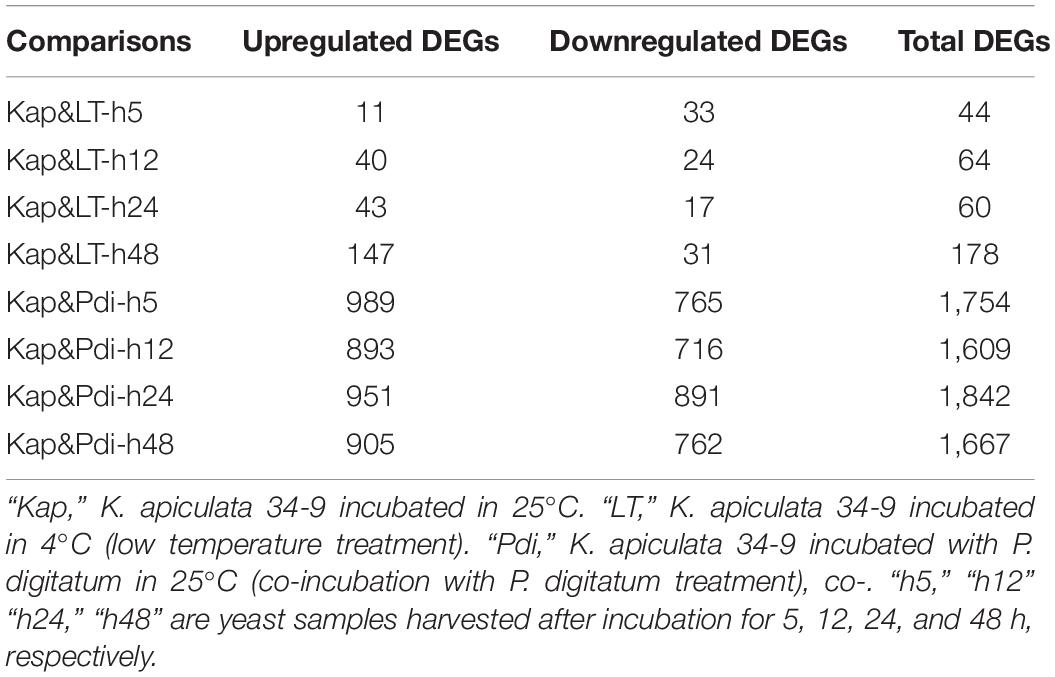
Table 2. Number of differentially expressed genes of RNA-seq samples.
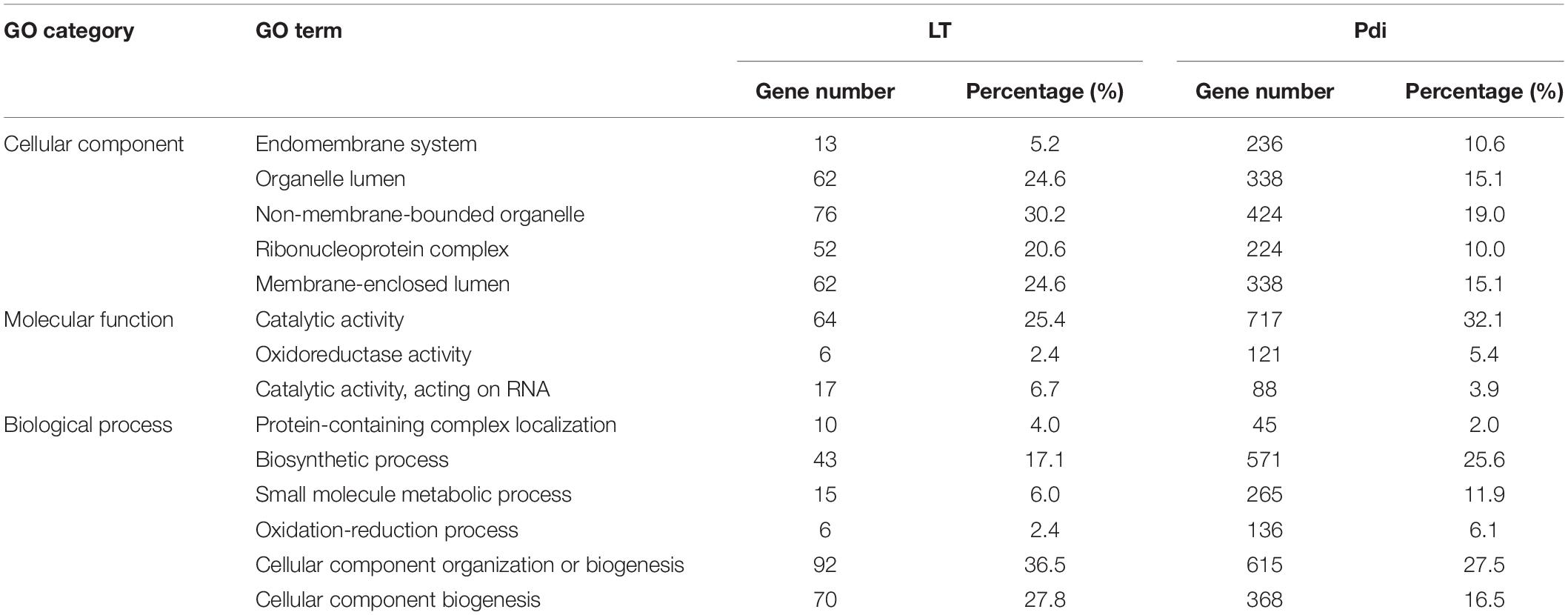
Table 3. Number and proportion of genes in each GO category between LT and Pdi.
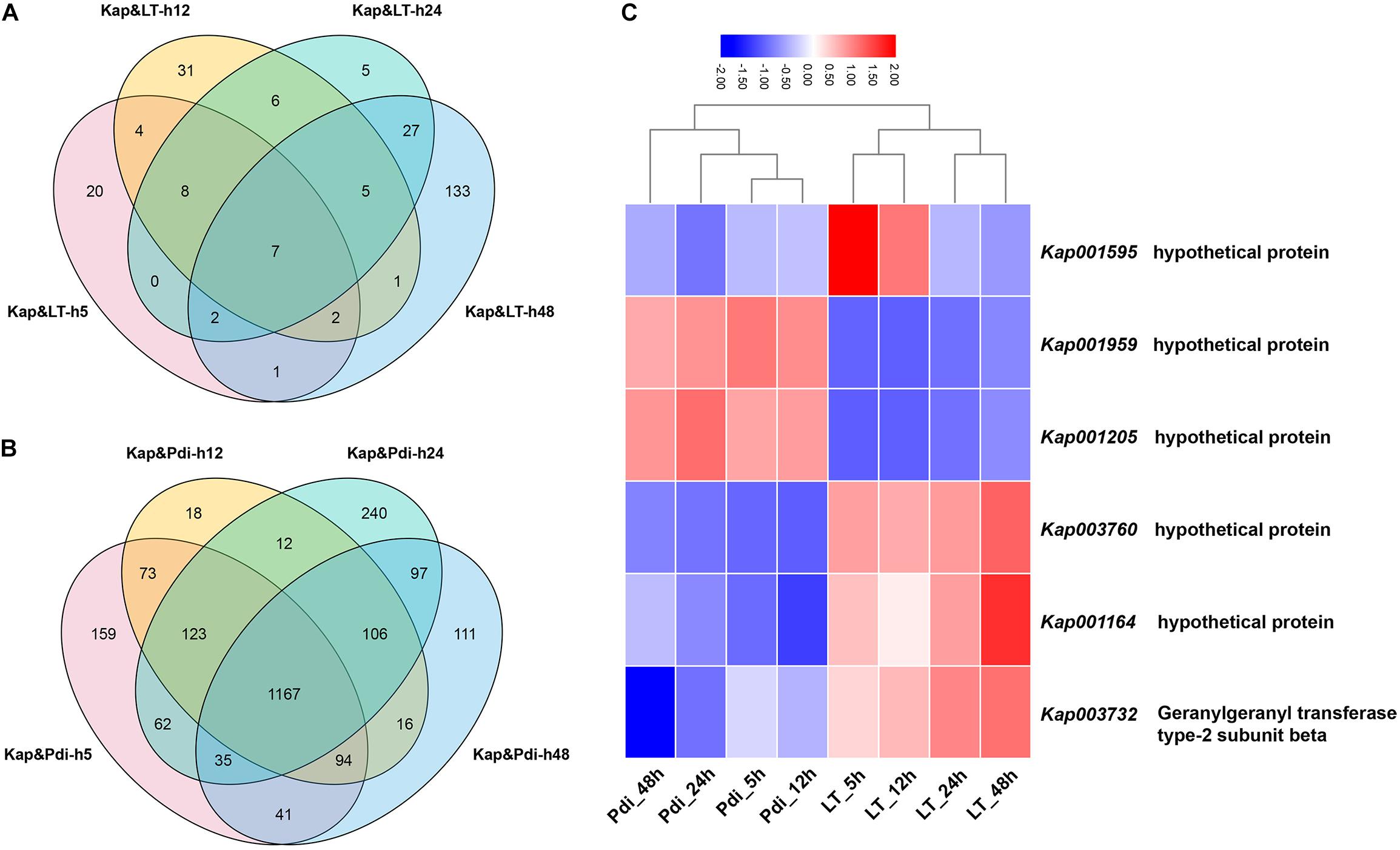
Figure 2. Veen diagram of differentially expressed genes. (A) Differentially expressed genes (DEGs) after low temperature treatment. (B) DEGs after co-incubation with Penicillium digitatum. (C) Heat map of co-DEGs (CEGs) between LT and Pdi at the four time points. “Kap,” “LT,” and “Pdi” are K. apiculata 34-9 incubated in 25°C, 4°C and co-incubation with P. digitatum in 25°C, respectively. “h5,” “h12,” “h24,” and “h48” are yeast samples harvested after incubation for 5, 12, 24, and 48 h, respectively.
Even though the counts of DEGs generated from qRT-PCR were not the same as FPKMs generated from RNA-seq, they nearly all showed the same trends (Figure 3). The high correlation between qRT-PCR and RNA-seq data were a validation of the generated RNA-seq results.

Figure 3. qRT-PCR analyses of DEGs. (A) Low temperature treatment. (B) Co-incubation with P. digitatum treatment. “Kap,” “LT,” and “Pdi” are the qRT-PCR results (left Y-axis) of differentially expressed genes, respectively. “K,” “L,” and “P” are the transcription expression abundance (FPKM, right Y-axis) of the differentially expressed genes in the RNA-seq results, respectively.
A total of 451 GO terms were obtained in the GO functional category analyses, the number of GO terms of biological process, cellular component and molecular function were 159, 161, and 131, respectively (Figure 4A). For biological process annotation, cellular process (GO: 0009987) and metabolic process (GO: 0008152) were the most plentiful terms, and terms annotated as response to stimulus (GO: 0050896) were also abundant. Meanwhile, binding (GO: 0005488), catalytic activity (GO: 0003824), and molecular function regulator (GO: 0098772) were largely associated terms in the annotation of molecular function. Moreover, cell part (GO: 0044464), protein-containing complex (GO: 0032991), and membrane-enclosed lumen (GO: 0031974) were the most involved terms for cellular component.
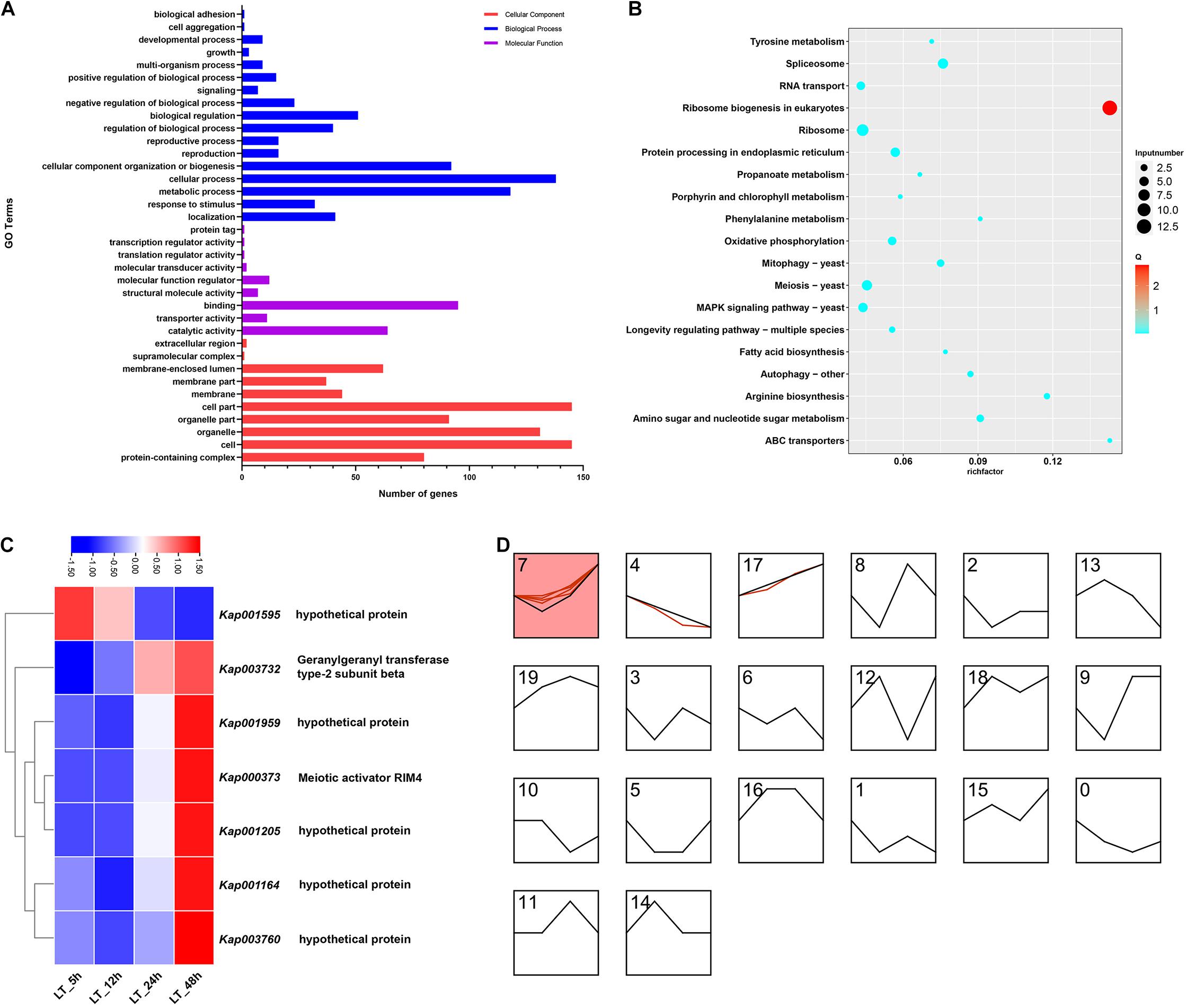
Figure 4. Bioinformatics analyses of DEGs of K. apiculata with low temperature treatment. (A) GO category of total DEGs. (B) KEGG enrichment pathway of total DEGs. (C) Heat map of common DEGs. (D) Expression patterns analysis of DEGs.
For KEGG enrichment analyses, ribosome biogenesis in eukaryotes (KEGG: sce03008), ABC transporters (KEGG: sce02010), and arginine biosynthesis (KEGG: sce00220) were the significantly enriched pathways, they all showed a higher enrichment factor (Figure 4B). In addition, phenylalanine metabolism (KEGG: sce00360) pathway was also enriched in the analyses.
There were 7 CEGs in the four time points in LT (Figure 2A), and the normalized expression counts of the 7 CEGs were visualized in a heatmap, and the expression patterns were clustered in STEM (Figures 4C,D), the 7 CEGs were grouped in 3 clusters (7, 4, 17), gene Kap003732 remained up-regulated throughout the entire treatments. Interestingly, we also observed one CEG (Kap003067) with annotation as heat shock protein 78 (HSP78) was downregulated at 5, 12, and 48 h, but it was not detected differentially expressed at 24 h.
Because there were many DEGs at different time points, the CEGs from Pdi were utilized for GO category to investigate the possible functions analyses. Results from GO category analyses were on a same occasion to LT, the CEGs were primarily annotated in biological process and cell components (Figure 5A). To get a better understanding of the CEGs, the entire 1,167 CEGs at the four time points in Pdi were employed for further analyses. The normalized expression counts of the 1,167 CEGs were visualized in a heatmap and expression patterns were clustered in STEM (Figures 5B,D), and the 1,167 CEGs were clustered into 20 different groups, group 17 (79 CEGs) was observed up-regulated throughout the entire treatments (Figure 5D). We also performed KEGG enrichment analyses to detect the potential biological pathways, and the results showed that many DEGs were annotated to metabolic pathways (KEGG: sce01100) and biosynthesis of secondary metabolites (sce01110) (Figure 5C). Moreover, lysine biosynthesis (KEGG: sce00300) pathway was enriched with the highest factor, and steroid biosynthesis (KEGG: sce00100) followed. CEGs involved in lysine biosynthesis (KEGG: sce00300), steroid biosynthesis (KEGG: sce00100), phenylalanine metabolism (KEGG: sce00360), and riboflavin metabolism (KEGG: sce00740) KEGG pathways were selected and the FPKM were normalized and visualized in a heatmap (Figure 6).
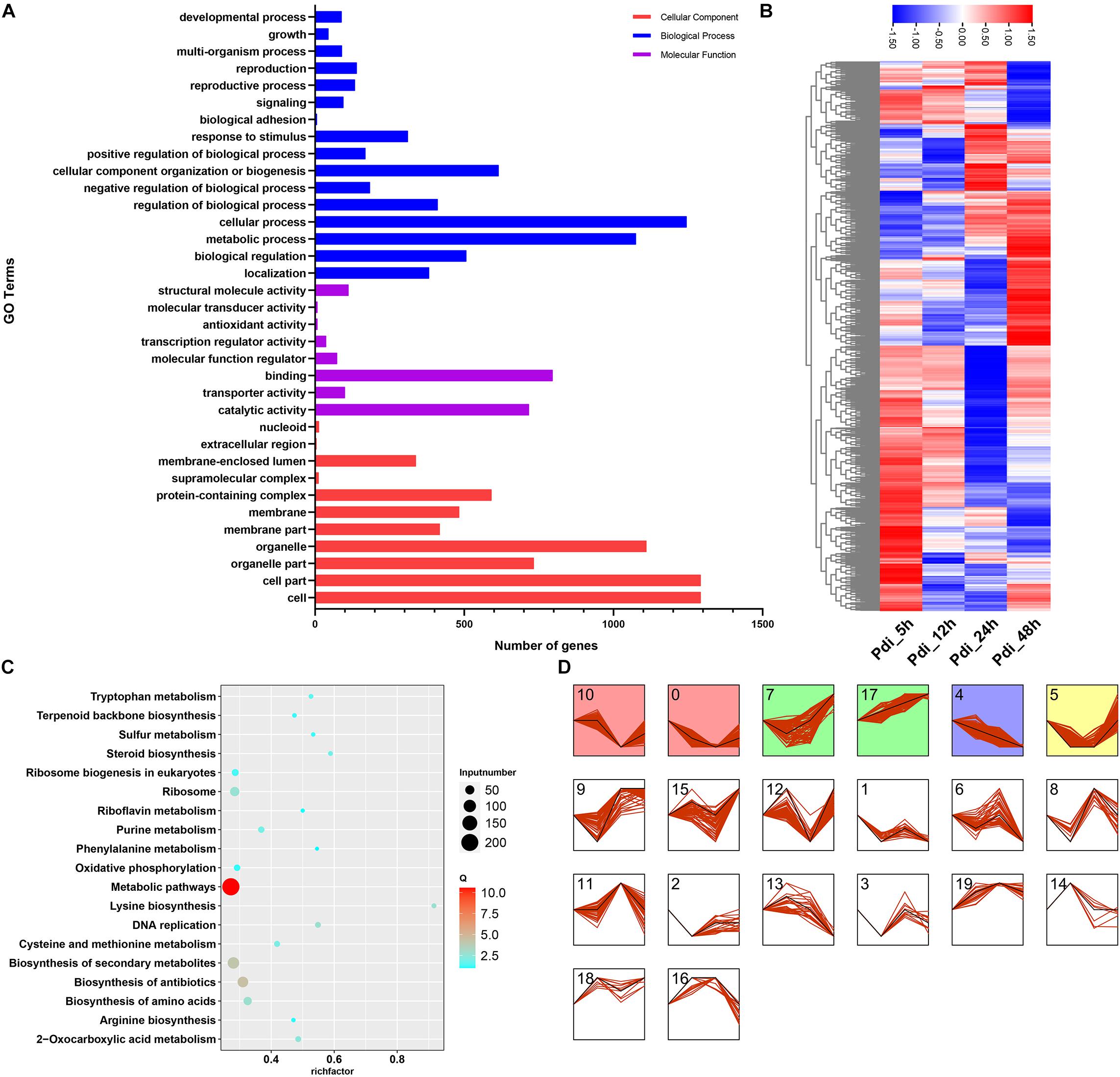
Figure 5. Bioinformatics analyses of DEGs of K. apiculata after co-incubation with P. digitatum. (A) GO category of total DEGs. (B) KEGG enrichment pathway of total DEGs. (C) Heat map of common DEGs. (D) Expression patterns analysis of DEGs.

Figure 6. The expression of lysine biosynthesis, phenylalanine metabolism, riboflavin metabolism, and steroid biosynthesis of K. apiculata co-incubation with P. digitatum treatment.
We also performed GO category and KEGG enrichment analyses of each individual time point. DEGs were associated with catalytic activity (GO: 0003824), hydrolase activity (GO: 0016787), and phosphatase activity (GO: 0016791) after co-incubation for 5, 12, and 24 h (Supplementary Figures 1–3). In addition, DEGs were involved in co-translational protein targeting to membrane (GO: 0006613), sodium ion transport (GO: 0006814), and others after co-incubation for 48 h (Supplementary Figure 4).
In the present study, two different kinds of treatments (or stresses) were carried out on K. apiculata yeast, and the expression levels were obtained by RNA-seq. A comparison was conducted using WEGO to extend a comprehensive understanding of these treatments, and the detailed GO terms were shown in Table 2. The number of DEGs annotated to catalytic activity (GO: 0003824) of LT and Pdi were 64 (25.4%) and 717 (32.1%), respectively. However, a higher percentage of DEGs annotated to catalytic activity (acting on RNA) (GO: 0140097) of LT than Pdi, with proportion of 6.6 and 3.9%, respectively. DEGs annotated to oxidoreductase activity (GO: 0016491) of LT and Pdi were 6 (2.4%) and 121 (5.4%), respectively. The percentage of DEGs annotated to the cellular component term of LT were mainly reduced compared to Pdi, excluding the endomembrane system.
Here, we present a chromosome level genome with combination of next-generation sequencing (previous data) and Hi-C sequencing to address the limited information of K. apiculata 34-9 genome. The newly re-assembled chromosome level genome of K. apiculata 34-9 exhibited appreciable improvements in completeness compared to the previous assembled draft genome (Chen et al., 2017), with 228 number of genes added, and more proportion of repeat sequences (1.65%) were annotated. The freshly annotated genes might be owing to the improvements of the continuity of the draft genome, and more useful sequences were obtained. Moreover, it was also because of the development of the algorithm of the software and the updated genome annotation database (Grünberger et al., 2019; Liu T. et al., 2021). The new genome was assembled in scaffolds N50 and contig N50 of 1,349,021 and 104,210 bp, respectively, which were more meticulously examined than those of the former version (scaffolds N50 and contig N50 were 279,578 and 140,302 bp, respectively). In addition, the gene number was near the average number of genes of available sequenced Hanseniaspora species (about 4,223 genes) inferred in a previous study (Guaragnella et al., 2020).
RNA-seq had been widely used to study the transcriptome profiles during different treatments (Fracasso et al., 2016; Deng et al., 2018). Many studies were usually performed to detect the transcriptome changes of pythopathogens in response to the presence of biocontrol agents (Gkarmiri et al., 2015; Liu D. et al., 2019). However, few studies have focused on the transcriptome of the biocontrol agents (especially yeast) response to phytopathogens, thus it posed a mystery of these organisms. Transcriptome profiling of Chaetomium golbosum in the presence of Bipolaris sorokiniana demonstrated that genes related to hydrolytic activity and some secondary metabolites might play a role in antagonistic mechanisms in the antagonist-pathogen interaction (Darshan et al., 2020). In this study, transcriptional profiles of K. apiculata co-incubation with P. digitatum under different time points were also performed to evaluate the relevant molecular responses; a total of 2,364 genes were found to be differentially expressed in the presence of P. digitatum. After co-incubation with P. digitatum at 5, 12, 24, and 48 h, a number of 989, 893, 951, and 905 DEGs were up-regulated and 765, 716, 891, and 762 DEGs were down-regulated, respectively. The temporal transcriptome analyses detected numerous candidate genes associated with the response to P. digitatum stress. Gene Kap002298 annotated as aromatic/aminoadipate aminotransferase 1 was involved in lysine biosynthesis and phenylalanine metabolism, and it remained up-regulated with the extension of co-incubation time. Phenylalanine metabolism was important for organisms to survive the stresses (Tonnessen et al., 2015; Solekha et al., 2020). Our results revealed that the significant changes of DEGs associated with vital metabolic pathways such as steroid biosynthesis and riboflavin metabolism among different time points, indicating that it must be a complex regulatory mechanism of K. apiculata response to P. digitatum stress. The transcriptome data present here might work as a valuable molecular property for future studies.
The inhibitory effect of antimicrobial agents is usually affected by environmental circumstances (Liu J. et al., 2013). In addition, transcriptome analysis has been employed to study the responses of biocontrol agents to abiotic stresses (Spadaro and Droby, 2016; Dai et al., 2021). In this study, transcriptional analyses of biocontrol yeast K. apiculata 34-9 between different temperatures (4° and 25°C) were also conducted, and approximately 6.33% of the total genes with 254 DEGs were found after LT treatment at four different time points. Previous studies have shown that ABC transporters play vital roles in diverse biological processes, such as mitochondrial iron homoeostasis, fatty acid metabolism, and mRNA translation (Klein et al., 2011; Do et al., 2018). In the present study, we found that ABC transporters were significantly enriched in the KEGG analyses, they might be involved in the adaption of K. apiculata to low temperatures. Heat shock proteins (HSPs) are essential for many cellular processes and organisms to survive stress conditions (Mittal et al., 2011; Park and Seo, 2015). Von Janowsky et al. (2006) responded that HSP78 has a protective role and it could cooperate with the molecular chaperone Ssc1 for disaggregation activity of S. cerevisiae during heat stress. In our transcriptome analyses, a protein homologous to HSP78 was down-regulated after being exposed in LT treatment for 5, 12, and 48 h, respectively. It could be inferred that HSP78 of K. apiculata might function in the negative regulation of K. apiculata to LT stress. Different transcriptomic studies have shown that several genes were considered as cold stress markers, including NSL1 and HSP12, which were up-regulated after cold shock (Tai et al., 2007; Tronchoni et al., 2014). In this study, a maintained up-regulated gene (Kap003732) and a maintained down-regulated gene (Kap001595) were detected in LT responses, however, these two genes were not significantly enriched in either pathway exhibited in this part. It is possible that the expression changes of these two genes at different time points could be indicators of K. apiculata response to LT stress in a short-term adaptation. Low temperature-responsive transcriptome analyses in this study may provide a data-based model for elucidating the molecular mechanisms of K. apiculata low temperature tolerance.
In conclusion, a chromosome resolution genome of K. apiculata was obtained, and transcriptome expression profiles of K. apiculata 34-9 under low temperature and co-incubation with P. digitatum were obtained. These results may establish a foundation for the genome study of Hanseniaspora spp., and the regulatory mechanisms of low temperature adaptation. Meanwhile, the transcriptome data of K. apiculata co-incubation with P. digitatum may extend our knowledge on Kloeckera yeast as biocontrol agents against citrus green mold, and the involved mechanisms might be a study model on other phytopathogens. These findings might provide relevant information of biocontrol agents and postharvest citrus storage and preservation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioProjects accession numbers PRJNA667993 and PRJNA668195.
CL: conceptualization, writing—review and editing, supervision, and funding acquisition. ZT: conceptualization, methodology, data curation, and writing—original draft. YD, FY, JZ, SL, and DZ: methodology, validation, investigation, and data curation. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31972122 and 32172255), the Earmarked Fund for China Agriculture Research System (Grant No. CARS-26), and the Fundamental Research Funds for the Central Universities (Grant No. 2662020YLPY008) for financial support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.752529/full#supplementary-material
Apaliya, M. T., Zhang, H. Y., Yang, Q. Y., Zheng, X. F., Zhao, L. N., Kwaw, E., et al. (2017). Hanseniaspora uvarum enhanced with trehalose induced defense-related enzyme activities and relative genes expression levels against Aspergillus tubingensis in table grapes. Postharvest Biol. Technol. 132, 162–170.
Bai, Y., Kissoudis, C., Yan, Z., Visser, R. G. F., and van der Linden, G. (2018). Plant behaviour under combined stress: tomato responses to combined salinity and pathogen stress. Plant J. 93, 781–793.
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580.
Burton, J. N., Adey, A., Patwardhan, R. P., Qiu, R., Kitzman, J. O., and Shendure, J. (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125.
Cai, Z. K., Yang, R., Xiao, H. M., Qin, X. J., and Si, L. Y. (2015). Effect of preharvest application of Hanseniaspora uvarum on postharvest diseases in strawberries. Postharvest Biol. Technol. 100, 52–58.
Chen, C. J., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y. H., et al. (2020). TBtools: an Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 13, 1194–1202.
Chen, K., Yang, X. P., Zheng, F., and Long, C. A. (2017). Genome sequencing and analysis of Kloeckera apiculata strain 34-9, a biocontrol agent against postharvest pathogens in citrus. Genes Genom. 39, 87–99.
Dai, Y., Wang, Z., Leng, J., Wang, Q., and Liu, J. (2021). Heat stress alters the transcriptome of Debaryomyces hansenii and reduces its biocontrol activity against postharvest gray mold on kiwifruit. Postharvest Biol. Technol. 178:111541.
Darshan, K., Aggarwal, R., Bashyal, B. M., Singh, J., Shanmugam, V., Gurjar, M. S., et al. (2020). Transcriptome Profiling Provides Insights Into Potential Antagonistic Mechanisms Involved in Chaetomium globosum Against Bipolaris sorokiniana. Front. Microbiol. 11:578115. doi: 10.3389/fmicb.2020.578115
Dekker, J., Marti-Renom, M. A., and Mirny, L. A. (2013). Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet. 14, 390–403.
Deng, B., Wang, W., Deng, L., Yao, S., Ming, J., and Zeng, K. (2018). Comparative RNA-seq analysis of citrus fruit in response to infection with three major postharvest fungi. Postharvest Biol. Technol. 146, 134–146.
Do, T. H. T., Martinoia, E., and Lee, Y. (2018). Functions of ABC transporters in plant growth and development. Curr. Opin. Plant Biol. 41, 32–38.
Fracasso, A., Trindade, L. M., and Amaducci, S. (2016). Drought stress tolerance strategies revealed by RNA-Seq in two sorghum genotypes with contrasting WUE. BMC Plant Biol. 16:115. doi: 10.1186/s12870-016-0800-x
Giorello, F., Valera, M. J., Martin, V., Parada, A., Salzman, V., Camesasca, L., et al. (2019). Genomic and Transcriptomic Basis of Hanseniaspora vineae’s Impact on Flavor Diversity and Wine Quality. Appl. Environ. Microbiol. 85, e01959–18.
Gkarmiri, K., Finlay, R. D., Alström, S., Thomas, E., Cubeta, M. A., and Högberg, N. (2015). Transcriptomic changes in the plant pathogenic fungus Rhizoctonia solani AG-3 in response to the antagonistic bacteria Serratia proteamaculans and Serratia plymuthica. BMC Genomics 16:630. doi: 10.1186/s12864-015-1758-z
Grünberger, F., Reichelt, R., Bunk, B., Spröer, C., Overmann, J., Rachel, R., et al. (2019). Next Generation DNA-Seq and Differential RNA-Seq Allow Re-annotation of the Pyrococcus furiosus DSM 3638 Genome and Provide Insights Into Archaeal Antisense Transcription. Front. Microbiol. 10:1603. doi: 10.3389/fmicb.2019.01603
Guaragnella, N., Chiara, M., Capece, A., Romano, P., Pietrafesa, R., Siesto, G., et al. (2020). Genome Sequencing and Comparative Analysis of Three Hanseniaspora uvarum Indigenous Wine Strains Reveal Remarkable Biotechnological Potential. Front. Microbiol. 10:3133. doi: 10.3389/fmicb.2019.03133
Holt, C., and Yandell, M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12:491. doi: 10.1186/1471-2105-12-491
Klein, C., Kuchler, K., and Valachovic, M. (2011). ABC proteins in yeast and fungal pathogens. Essays Biochem. 50, 101–119.
Langenberg, A. K., Bink, F. J., Wolff, L., Walter, S., von Wallbrunn, C., Grossmann, M., et al. (2017). Glycolytic Functions Are Conserved in the Genome of the Wine Yeast Hanseniaspora uvarum, and Pyruvate Kinase Limits Its Capacity for Alcoholic Fermentation. Appl. Environ. Microbiol. 83, e01580–17.
Liu, D., Li, K., Hu, J., Wang, W., Liu, X., and Gao, Z. (2019). Biocontrol and Action Mechanism of Bacillus amyloliquefaciens and Bacillus subtilis in Soybean Phytophthora Blight. Int. J. Mol. Sci. 20:2908.
Liu, J., Sui, Y., Wisniewski, M., Droby, S., and Liu, Y. (2013). Review: utilization of antagonistic yeasts to manage postharvest fungal diseases of fruit. Int. J. Food Microbiol. 167, 153–160.
Liu, P., Luo, L., and Long, C.-A. (2013). Characterization of competition for nutrients in the biocontrol of Penicillium italicum by Kloeckera apiculata. Biol. Control 67, 157–162.
Liu, P., Chen, K., Li, G., Yang, X., and Long, C.-A. (2016). Comparative transcriptional profiling of orange fruit in response to the biocontrol yeast Kloeckera apiculata and its active compounds. BMC Genomics 17:17. doi: 10.1186/s12864-015-2333-3
Liu, P., Cheng, Y. J., Yang, M., Liu, Y. J., Chen, K., Long, C. A., et al. (2014). Mechanisms of action for 2-phenylethanol isolated from Kloeckera apiculata in control of Penicillium molds of citrus fruits. BMC Microbiol. 14:242. doi: 10.1186/s12866-014-0242-2
Liu, T., Li, M., Liu, Z., Ai, X., and Li, Y. (2021). Reannotation of the cultivated strawberry genome and establishment of a strawberry genome database. Hortic. Res. 8:41.
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Long, C. A., Wu, Z., and Deng, B. X. (2005). Biological control of Penicillium italicum of citrus and Botrytis cinerea of grape by strain 34-9 of Kloeckera apiculata. Eur. Food Res. Technol. 221, 197–201.
Martin, V., Jose Valera, M., Medina, K., Boido, E., and Carrau, F. (2018). Oenological Impact of the Hanseniaspora/Kloeckera Yeast Genus on Wines-A Review. Fermentation 4:76.
Mittal, D., Enoki, Y., Lavania, D., Singh, A., Sakurai, H., and Grover, A. (2011). Binding affinities and interactions among different heat shock element types and heat shock factors in rice (Oryza sativa L.). FEBS J. 278, 3076–3085.
Park, C.-J., and Seo, Y.-S. (2015). Heat Shock Proteins: a review of the molecular chaperones for plant immunity. Plant Pathol. J. 31, 323–333.
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067.
Peng, X., Liu, H., Chen, P., Tang, F., Hu, Y., Wang, F., et al. (2019). A Chromosome-Scale Genome Assembly of Paper Mulberry (Broussonetia papyrifera) Provides New Insights into Its Forage and Papermaking Usage. Mol. Plant 12, 661–677.
Roeber, V. M., Bajaj, I., Rohde, M., Schmülling, T., and Cortleven, A. (2021). Light acts as a stressor and influences abiotic and biotic stress responses in plants. Plant Cell Environ. 44, 645–664.
Seixas, I., Barbosa, C., Mendes-Faia, A., Guldener, U., Tenreiro, R., Mendes-Ferreira, A., et al. (2019). Genome sequence of the non-conventional wine yeast Hanseniaspora guilliermondii UTAD222 unveils relevant traits of this species and of the Hanseniaspora genus in the context of wine fermentation. DNA Res. 26, 67–83.
Servant, N., Varoquaux, N., Lajoie, B. R., Viara, E., Chen, C.-J., Vert, J.-P., et al. (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16:259.
Shao, Y., Lu, N., Wu, Z., Cai, C., Wang, S., Zhang, L.-L., et al. (2018). Creating a functional single-chromosome yeast. Nature 560, 331–335.
Sies, H., and Jones, D. P. (2020). Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21, 363–383.
Solekha, R., Susanto, F. A., Joko, T., Nuringtyas, T. R., and Purwestri, Y. A. (2020). Phenylalanine ammonia lyase (PAL) contributes to the resistance of black rice against Xanthomonas oryzae pv. oryzae. J. Plant Pathol. 102, 359–365.
Spadaro, D., and Droby, S. (2016). Development of biocontrol products for postharvest diseases of fruit: the importance of elucidating the mechanisms of action of yeast antagonists. Trends Food Sci. Technol. 47, 39–49.
Suzuki, N., Rivero, R. M., Shulaev, V., Blumwald, E., and Mittler, R. (2014). Abiotic and biotic stress combinations. New Phytol. 203, 32–43.
Tai, S. L., Daran-Lapujade, P., Walsh, M. C., Pronk, J. T., and Daran, J.-M. (2007). Acclimation of Saccharomyces cerevisiae to low temperature: a chemostat-based transcriptome analysis. Mol. Biol. Cell 18, 5100–5112.
Tian, Z., Chen, C., Chen, K., Liu, P., Fan, Q., Zhao, J., et al. (2020). Biocontrol and the mechanisms of Bacillus sp. w176 against postharvest green mold in citrus. Postharvest Biol. Technol. 159:111022.
Tonnessen, B. W., Manosalva, P., Lang, J. M., Baraoidan, M., Bordeos, A., Mauleon, R., et al. (2015). Rice phenylalanine ammonia-lyase gene OsPAL4 is associated with broad spectrum disease resistance. Plant Mol. Biol. 87, 273–286.
Tristezza, M., Tufariello, M., Capozzi, V., Spano, G., Mita, G., and Grieco, F. (2016). The Oenological Potential of Hanseniaspora uvarum in Simultaneous and Sequential Co-fermentation with Saccharomyces cerevisiae for Industrial Wine Production. Front. Microbiol. 7:670. doi: 10.3389/fmicb.2016.00670
Tronchoni, J., Medina, V., Guillamón, J. M., Querol, A., and Pérez-Torrado, R. (2014). Transcriptomics of cryophilic Saccharomyces kudriavzevii reveals the key role of gene translation efficiency in cold stress adaptations. BMC Genomics 15:432. doi: 10.1186/1471-2164-15-432
Valles, B. S., Bedriñana, R. P., Tascón, N. F., Simón, A. Q., and Madrera, R. R. (2007). Yeast species associated with the spontaneous fermentation of cider. Food Microbiol. 24, 25–31.
Von Janowsky, B., Major, T., Knapp, K., and Voos, W. (2006). The disaggregation activity of the mitochondrial ClpB homolog Hsp78 maintains Hsp70 function during heat stress. J. Mol. Biol. 357, 793–807.
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322.
Xu, Z., and Wang, H. (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268.
Yang, H., Luan, Y., Liu, T., Lee, H. J., Fang, L., Wang, Y., et al. (2020). A map of cis-regulatory elements and 3D genome structures in zebrafish. Nature 588, 337–343.
Ye, J., Zhang, Y., Cui, H. H., Liu, J. W., Wu, Y. Q., Cheng, Y., et al. (2018). WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 46, W71–W75.
Zhang, J., Xie, J., Zhou, Y. H., Deng, L. L., Yao, S. X., and Zeng, K. F. (2017). Inhibitory effect of Pichia membranaefaciens and Kloeckera apiculata against Monilinia fructicola and their biocontrol ability of brown rot in postharvest plum. Biol. Control 114, 51–58.
Keywords: Kloeckera apiculata, genome, transcriptome, biocontrol, stress
Citation: Tian Z, Du Y, Yang F, Zhao J, Liu S, Zhang D and Long C (2021) Chromosome Genome Sequencing and Comparative Transcriptome-Based Analyses of Kloeckera apiculata 34-9 Unveil the Potential Biocontrol Mechanisms Against Citrus Green Mold. Front. Microbiol. 12:752529. doi: 10.3389/fmicb.2021.752529
Received: 03 August 2021; Accepted: 15 October 2021;
Published: 09 November 2021.
Edited by:
Javier Carballo, University of Vigo, SpainReviewed by:
Chuan Xu, Shanghai Jiao Tong University, ChinaCopyright © 2021 Tian, Du, Yang, Zhao, Liu, Zhang and Long. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chao-an Long, postharvest@mail.hzau.edu.cn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.