 Ana Durán-Viseras
Ana Durán-Viseras Cristina Sánchez-Porro
Cristina Sánchez-Porro Antonio Ventosa
Antonio Ventosa- Department of Microbiology and Parasitology, Faculty of Pharmacy, University of Sevilla, Sevilla, Spain
Metagenomic studies on prokaryotic diversity of hypersaline soils from the Odiel saltmarshes, South-west Spain, revealed a high proportion of genomic sequences not related to previously cultivated taxa, that might be related to haloarchaea with a high environmental and nutritional flexibility. In this study, we used a culturomics approach in order to isolate new haloarchaeal microorganisms from these hypersaline soils. Four haloarchaeal strains, designated strains F24AT, F28, F27T, and F13T, phylogenetically related to the genus Halomicroarcula, were isolated and characterized in detail. The phylogenomic tree based on the 100 orthologous single-copy genes present in the genomes of these four strains as well as those of the type strains of the species Halomicroarcula pellucida CECT 7537T, Halomicroarcula salina JCM 18369T and Halomicroarcula limicola JCM 18640T, that were determined in this study, revealed that these four new isolates clustered on three groups, with strains F24AT and F28 within a single cluster, and altogether with the species of Halomicroarcula. Additionally, Orthologous Average Nucleotide Identity (OrthoANI), digital DNA-DNA hybridization (dDDH) and Average Amino-acid Identity (AAI) values, likewise phenotypic characteristics, including their polar lipids profiles, permitted to determine that they represent three new species, for which we propose the names Halomicroarcula rubra sp. nov. (type strain F13T), Halomicroarcula nitratireducens sp. nov. (type strain F27T) and Halomicroarcula salinisoli sp. nov. (type strain F24AT). An in deep comparative genomic analysis of species of the genus Halomicroarcula, including their metabolism, their capability to biosynthesize secondary metabolites and their osmoregulatory adaptation mechanisms was carried out. Although they use a salt-in strategy, the identification of the complete pathways for the biosynthesis of the compatible solutes trehalose and glycine betaine, not identified before in any other haloarchaea, might suggest alternative osmoadaptation strategies for this group. This alternative osmoregulatory mechanism would allow this group of haloarchaea to be versatile and eco-physiologically successful in hypersaline environments and would justify the capability of the species of this genus to grow not only on environments with high salt concentrations [up to 30% (w/v) salts], but also under intermediate to low salinities.
Introduction
Hypersaline environments are extreme habitats that have permitted the isolation and characterization of many halophilic microorganisms that have been used as models for the study of basic and applied purposes (Ventosa, 2006; Ventosa et al., 2015). These habitats are characterized by high levels of salinity, frequently accompanied by other extreme conditions, such as high or low temperature or pH values, UV radiation, hydrostatic pressure or high concentrations of toxic compounds (Rodríguez-Valera, 1988; Ventosa, 2006). To withstand these high salt concentrations and the constant fluctuations in salinity levels, halophilic microorganisms have developed diverse physiological adaptations (Galinski and Trüper, 1994; Kempf and Bremer, 1998; Gunde-Cimerman et al., 2018). The best adapted organisms to these high salt concentrations are prokaryotes belonging to the extremely halophilic archaea, members of the class Halobacteria (also called haloarchaea) (Amoozegar et al., 2017; Oren and Ventosa, 2017a). Although haloarchaea have been traditionally considered as a coherent group limited to use a salt-in osmoadaptation strategy, i.e., the accumulation of K+, Na+, and Cl– ions, recent studies have brought to light their potential to employ additional alternative mechanisms (Anderson et al., 2011; Youssef et al., 2014).
Hypersaline environments are represented by a wide range of habitats, from aquatic or terrestrial to deep-sea, salt mines, salt-cured food or plants (Ventosa, 2006). However, hypersaline aquatic systems and more recently saline soils constitute the most extensively studied hypersaline environments (Ventosa et al., 2015). Traditionally, aquatic environments have been thoroughly studied, specially saline lakes and salterns (Ventosa et al., 2014, 2015; Oren, 2015), but the number of studies in hypersaline soils is much more reduced (Ventosa et al., 2008; Oren, 2011). Recent metagenomic studies carried out on hypersaline soils from the Odiel saltmarshes (South-west Spain) revealed a high proportion of genomic sequences not related to any cultivated organisms as well as a high environmental and nutritional flexibility of microorganism inhabiting these systems (Vera-Gargallo and Ventosa, 2018; Vera-Gargallo et al., 2019). With the purpose of isolating such of these metabolically diverse groups not isolated to date, we focused on these habitats by using culture-dependent methods. For that purpose, we used the current “culturomics” approach (Durán-Viseras et al., 2019a, 2021), in order to isolate new haloarchaeal groups taking advantage of the previous metagenomic studies, that showed an unexpected large proportion of haloarchaea in these hypersaline soils (Vera-Gargallo and Ventosa, 2018). As a result, four haloarchaeal strains phylogenetically related to the genus Halomicroarcula were isolated in pure culture.
The genus Halomicroarcula belongs to the family Haloarculaceae, within the order Halobacteriales, class Halobacteria and phylum Euryarchaeota (Echigo, 2016; Oren and Ventosa, 2017b). At the time of writing the genus Halomicroarcula comprises four species: Halomicroarcula pellucida (Echigo et al., 2013), which is the type species of the genus and was isolated from French marine salt, designated “Sel marin de Guérande,” Halomicroarcula limicola (Zhang and Cui, 2014) and Halomicroarcula salina (Zhang and Cui, 2015), both isolated from Yinggehai solar saltern, located in Hainan Province, China; and more recently, Halomicroarcula amylolytica (Chen et al., 2020), isolated from a salt mine in Yunnan Province, China. The genus Halomicroarcula includes Gram-stain-negative, motile and pleomorphic cells. They form non-pigmented and transparent or red-pigmented colonies. They are halophilic, neutrophilic and mesophilic. Other characteristics of this genus are their aerobic and heterotrophic metabolism (Echigo et al., 2013). Their major polar lipids are phosphatidylglycerol (PG), phosphatidylglycerolphosphate methyl ester (PGP-Me) and phosphatidylglycerol sulfate (PGS). Glycolipids, including sulfated mannosyl glucosyl diether (S-DGD-1) and mannosyl glucosyl diether (DGD-1) may be present in some species (Zhang and Cui, 2014; Echigo, 2016). The G + C content of strains of the genus Halomicroarcula range between 64.0 and 64.5 mol% (Echigo et al., 2013; Zhang and Cui, 2014; Echigo, 2016).
In this work, we have carried out an exhaustive taxogenomic study of the genus Halomicroarcula, and we have addressed the description of three new species of the genus Halomicroarcula. Besides, we have performed a comparative genomic analysis of species of this genus aimed at studying in depth their metabolism, their capacity to biosynthesize secondary metabolites and their osmoregulatory adaptation mechanisms that allow this group of microorganisms to be versatile and eco-physiologically successful in hypersaline environments.
Materials and Methods
Strains Isolation, Reference Strains and Culture Conditions
Strains F24AT, F28, F27T, and F13T were isolated from saline soil samples (conductivity 54.5 CE1:5 mS/cm and pH 8.9) collected from the Odiel saltmarshes (Huelva, South-west Spain) (37°12′26″N 6°57′58″O). Samples were diluted, plated under sterile conditions and incubated at 37°C up to two months. Strains F24AT, F28, F27T, and F13T were isolated and routinely grown in R2A medium (Difco) (pH adjusted to 7.5) supplemented with 25% (w/v) seawater salt solution prepared by dilution of SW 30% (w/v) stock solution containing (g/L): NaCl, 195; MgCl2⋅6H2O, 32.5; MgSO4⋅7H2O, 50.8; CaCl2, 0.83; KCl, 5.0; NaHCO3, 0.17; NaBr, 0.58. Purified agar (2%; Oxoid) was used as solidifying agent, when needed. For long term maintenance, cultures were preserved at –80°C in this medium containing 20% (v/v) glycerol (Durán-Viseras et al., 2021).
For taxonomic comparative purposes strains Halomicroarcula pellucida CECT 7537T, Halomicroarcula salina JCM 18369T and Halomicroarcula limicola JCM 18640T, obtained from culture collections, were used in this study. These strains were also grown in the same medium and culture conditions as described above.
DNA Extraction, Purification and Sequencing
The genomic DNA of strains F24AT, F28, F27T, F13T, Halomicroarcula pellucida CECT 7537T, Halomicroarcula salina JCM 18369T and Halomicroarcula limicola JCM 18640T was extracted and purified using the method described by Marmur (1961). DNA quality and concentration was checked by spectrophotometry (DeNovix DS-11 FX, DeNovix Technologies, Wilmington, Delaware, United States) and fluorometry (Qubit 3.0 Fluorometer, Thermofisher Scientific, United States). The 16S rRNA gene was amplified by PCR using the universal primers ArchF and ArchR (DeLong, 1992; Arahal et al., 1996), then cloned and sequenced as described previously (Durán-Viseras et al., 2019b). The rpoB′ gene was amplified by PCR using the primers rpoBF and rpoBR (Fullmer et al., 2014). The genome of strains F13T, F24AT, F27T, F28, Halomicroarcula pellucida CECT 7537T, Halomicroarcula limicola JCM 18640T and Halomicroarcula salina JCM 18369T were sequenced using the Illumina HiSeq 4000 platform at StabVida (Oeiras, Portugal) and Novogene (Cambridge United Kingdom).
Phylogenetic and Phylogenomic Analyses
Identification of phylogenetic neighbors and calculation of pairwaise 16S rRNA and rpoB′ gene sequences similarities were conducted by using the Ez-BioCloud server (Yoon et al., 2017) and BLAST (Altschul et al., 1990), respectively. Additional 16S rRNA and rpoB′ gene sequences, and genomes used for comparisons were retrieved from the GenBank/EMBL/DDBJ databases. Clustering of 16S rRNA and rpoB′ gene sequences were determined using the neighbor-joining (Saitou and Nei, 1987) and maximum-likelihood (Felsenstein, 1981) algorithms implemented in the MEGA-X software (Kumar et al., 2018), using the Jukes-Cantor method (Jukes and Cantor, 1969) for evolutionary distances calculation. For phylogenetic tree branch support estimation, a bootstrap analysis based on 1000 replications was calculated (Felsenstein, 1985).
For phylogenomic analyses, core orthologous genes were identified from all analyzed genomes by an all-versus-all BLAST as implemented in the Enveomics collection toolbox (Rodriguez-R and Konstantinidis, 2016). As a result, a set of 100 conserved genes were retrieved and aligned using MUSCLE (Edgar, 2004). Phylogenomic tree of concatenated sequences was reconstructed by using the software MEGA-X (Kumar et al., 2018) with the neighbor-joining and maximum-likelihood methodology and Jukes-Cantor correction.
Genome Assembly, Annotation and Comparative Genomics
The de novo assembly of the reads was performed using Spades 3.9.1 (Bankevich et al., 2012). Draft genome assembly was quality checked using Quast v2.3 (Gurevich et al., 2013). Genome completeness, contamination and strain heterogeneity was estimated using CheckM v1.0.5 (Parks et al., 2015).
Genome sequences were annotated with Prokka (Seemann, 2014). BlastKOALA (Kanehisa et al., 2016) was used to assign KO identifiers (K numbers) to orthologous genes present in the genomes and mapped to the KEGG pathways and KEGG modules to perform the metabolic pathways reconstructions.
CRISPR/Cas systems, prophage sequences and secondary metabolites were identified by the tools CRISPRCasFinder (Couvin et al., 2018), PHASTER (Zhou et al., 2011; Arndt et al., 2016) and antiSMASH 5.0 (Blin et al., 2019), respectively. Isoelectric points of predicted proteins were computed using the iep program from EMBOSS package (Rice et al., 2000). The Orthologous Average Nucleotide Identity (ANI), Average Amino-acid Identity (AAI), and digital DNA-DNA hybridization (dDDH) were calculated using the tools OAT v0.93.1 (Lee et al., 2016), AAI-Matrix (Rodriguez-R and Konstantinidis, 2016) and Genome-to-Genome Distance Calculator (GGDC) (Meier-Kolthoff et al., 2013), respectively.
Phenotypic and Chemotaxonomic Characterization
Phenotypic features of strains F24AT, F28, F27T, and F13T were performed according to the minimal standards established for the taxonomic description of novel taxa of the class Halobacteria (Oren et al., 1997) and following the methodology previously described by Durán-Viseras et al., 2020. Halomicroarcula pellucida CECT 7537T, Halomicroarcula salina JCM 18369T and Halomicroarcula limicola JCM 18640T were used as reference strains for taxonomic comparisons.
Comparative polar lipids analysis of strains F24AT, F28, F27T, and F13T were determined by High-Performance Thin Layer Chromatography (HPTLC) as described elsewhere (Corral et al., 2015, 2016), using as spray reagents 5% H2SO4 (in water) or molybdenum blue. In this case, strains Halomicroarcula pellucida CECT 7537T, Halomicroarcula salina JCM 18369T, Halomicroarcula limicola JCM 18640T, Halobacterium salinarum DSM 3754T and Halorubrum saccharovorum DSM 1137T were used as reference species for polar lipids characterization. The polar lipids of strains F24AT, F28, F27T, F13T, Halomicroarcula pellucida CECT 7537T, Halomicroarcula salina JCM 18369T and Halomicroarcula limicola JCM 18640T were obtained from biomass cultured in R2A medium supplemented with 25% (w/v) seawater salt solution and pH adjusted to 7.5.
Results and Discussion
Phylogenetic and Phenotypic Analyses
Previous metagenomic studies on prokaryotic diversity of hypersaline soils showed a large proportion of haloarchaea, with a high percentage not closely related to any previously described taxa. For that reason, we isolated a large collection of strains from the Odiel saltmarsh hypersaline soils located in Huelva, South-west Spain, using different complex oligotrophic media and culture conditions, based on culturomics techniques as previously detailed (Durán-Viseras et al., 2021). Partial sequencing of the 16S rRNA gene permitted us to preliminary delineate the taxonomic position of the isolates. For this study we selected four new isolates, designated as strains F24AT, F28, F27T, and F13T, that were identified as members of the genus Halomicroarcula.
According to current practice, we determined the almost complete 16S rRNA gene sequences of strains F24AT, F28, F27T, and F13T. Similarly to other species of the genus Halomicroarcula, the four new strains showed two different copies of the 16S rRNA gene (designated as rrnA and rrnB), with sizes of 1441 bp and 1441 bp (for strain F13T), 1441 bp and 1442 bp (for strain F27T), 1446 bp and 1446 bp (for strain F24AT), and 1447 bp and 1446 bp (for strain F28), respectively. Their percentages of similarity with the type strains of species of Halomicroarcula (detailed in Supplementary Table 1), were in all cases equal or lower than 99.2% with Halomicroarcula limicola YGHS32T, 96.5% with Halomicroarcula pellucida BNERC31T and 96.0% with Halomicroarcula salina YGHS18T. The phylogenetic tree generated on the basis of the 16S rRNA gene showed that they clustered within the Halomicroarcula branch (Supplementary Figure 1A). However, since this gene has been proved to be not very useful as a phylogenetic marker for haloarchaea, we also sequenced the rpoB′ gene, which has been recommended as an alternative for single-gene phylogenetic analyses (Enache et al., 2007; Minegishi et al., 2010). The phylogenetic tree obtained by the neighbor-joining method (Supplementary Figure 1B) showed a similar topology with the 16S rRNA gene tree, in which the four new strains were placed on the Halomicroarcula cluster, with strains F24AT and F28 clustering together.
In order to determine in more detail the phylogenomic relationships of these four new isolates with respect to the species of Halomicroarcula and according to the Minimal Standards recommendations (Chun et al., 2018), we sequenced their genomes as well as those of the type strains of the species Halomicroarcula pellucida CECT 7537T, Halomicroarcula salina JCM 18369T and Halomicroarcula limicola JCM 18640T. Additionally, the genome of Halomicroarcula amylolytica LR21T, previously sequenced, was also included in this study. The main characteristics of these genomes are detailed in Table 1. All these genomic features are in accordance with the Minimal Standards established for the use of genomic data in prokaryotic taxonomy (Chun et al., 2018). The phylogenomic tree based on 100 orthologous single-copy genes present in all the genomes under study is shown in Figure 1. Bootstrap values of 100% in all branches related to Halomicroarcula supported this tree, generated by the neighbor-joining algorithm. This tree shows that the four new strains cluster within the Halomicroarcula branch, with strains F27T and F13T most closely related to Halomicroarcula limicola JCM 18640T and Halomicroarcula pellucida CECT 7537T, respectively, while strains F24AT and F28 clustered very close each other, suggesting that they may be members of the same species. Besides, this tree shows that the type strain of the species Halomicroarcula salina JCM 18639T clustered with the genus Haloarcula, suggesting its taxonomic position should be revised.

Table 1. Main features of the sequenced genomes of strains F13T, F24AT, F28, F27T, and the type strains of species of Halomicroarcula used in this study.

Figure 1. Neighbor-joining phylogenomic tree based on the concatenation of 100 orthologous single-copy genes shared by strains F13T, F24AT, F27T, F28, members of the genus Halomicroarcula and other related genera. Filled circles indicate branches that were also supported by the maximum-likelihood algorithm. Sequence accession number are shown in parentheses. Bootstrap values ≥70% are shown at branch points. Bar, 0.05 changes per nucleotide position.
Currently, several Overal Genome Relatedness Indexes (OGRI) are used in order to measure similarities between genome sequences (Chun and Rainey, 2014). Many algorithms have been proposed for this purpose, but the three most widely used for taxonomic purposes are Orthologous Average Nucleotide Identity (OrthoANI) (Lee et al., 2016), and digital DNA-DNA hybridization (dDDH) (Meier-Kolthoff et al., 2013), especially useful for delineation at the species level, and Average Amino-acid Identity (AAI) (Rodriguez-R and Konstantinidis, 2016), for genus delineation. It is widely accepted that for delineation of prokaryotic species the threshold values for OrthoANI and dDDH are 95% and 70%, respectively (Konstantinidis and Tiedje, 2005; Auch et al., 2010; Chun and Rainey, 2014). However, for the delineation at the genus level there is no clear universal AAI cutoff value, with approximately 65% as a reference percentage (Konstantinidis et al., 2017). Figure 2 shows the OrthoANI and dDDH percentages calculated for all pairs of strains F24AT, F28, F27T, F13T, Halomicroarcula pellucida CECT 7537T, Halomicroarcula salina JCM 18369T, Halomicroarcula limicola JCM 18640T and Halomicroarcula amylolytica LR21T, as well as for the type strains of other related haloarchaea. OrthoANI and dDDH values between strains F24AT and F28 were 99.2% and 94.5%, respectively, confirming unequivocally that these two strains are members of the same species. On the other hand, their percentages with respect to the strains F27T and F13T and the type strains of species of Halomicroarcula and to other haloarchaea are equal or lower than 80.8% and 29.5%, respectively (Figure 2), which clearly indicates that they constitute a separated species of the genus Halomicroarcula. With respect to strains F27T and F13T, their percentages of OrthoANI and dDDH with the most closely related species of Halomicroarcula are equal or lower than 92.3% and 52.6%, respectively (for strain F27T) and 88.7% and 41.3%, respectively (for strain F13T) (Figure 2), all of them values lower than those defined for the delineation of species, and thus supporting the new species status for both new isolates.
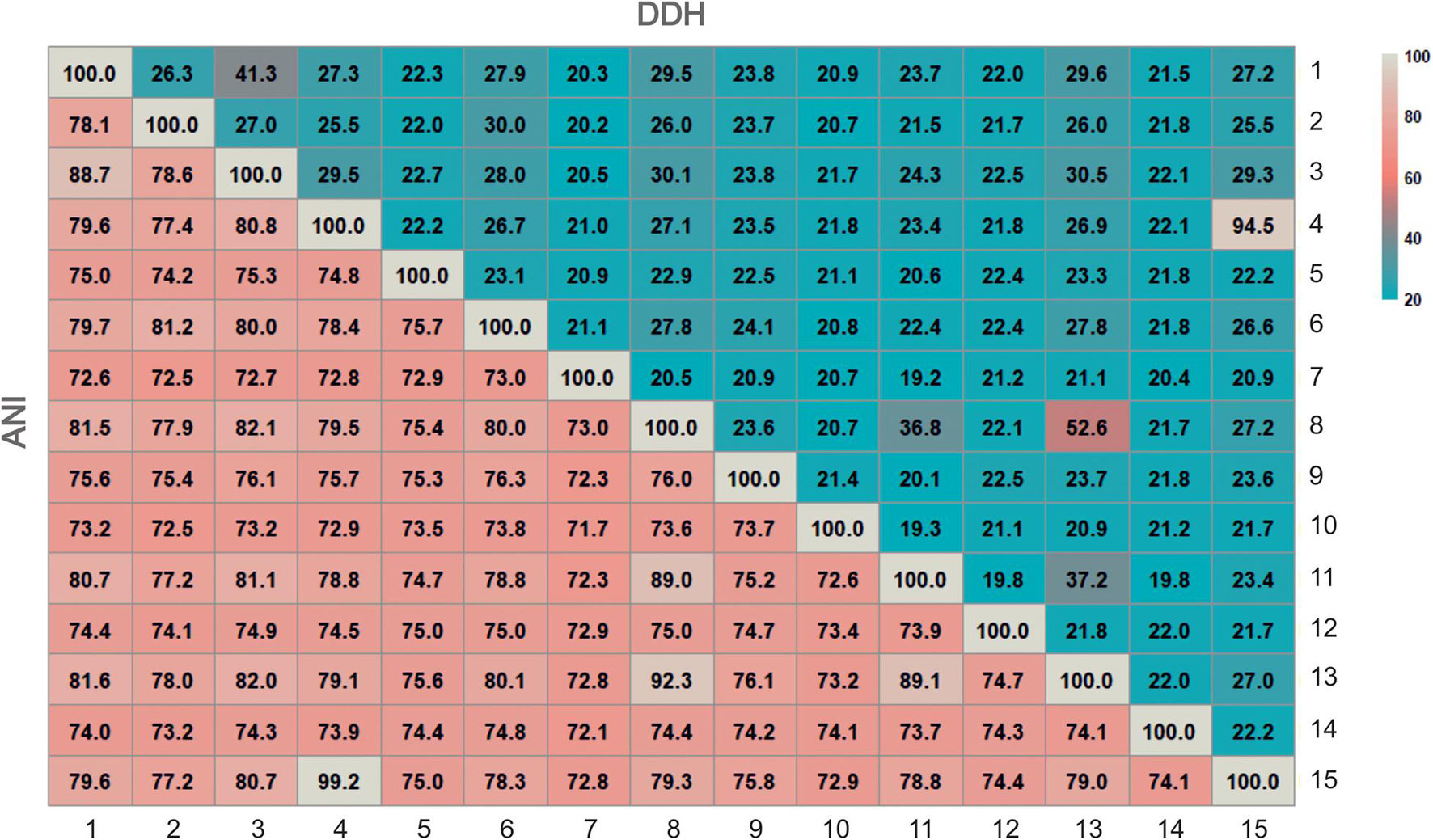
Figure 2. Heatmap of genome relatness among strains F13T, F24AT, F27T, F28, members of the genus Halomicroarcula and other related genera by means of OrthoANI and digital DDH. Strains: 1. Halomicroarcula pellucida CECT 7537T, 2. Haloarcula vallismortis ATCC 29715T, 3. Strain F13T, 4. Strain F24AT, 5. Halosimplex carlsbadense 2-9-1T, 6. Halomicroarcula salina JCM 18369T, 7. Natronomonas pharaonis DSM 2160T, 8. Halomicroarcula limicola JCM 18640T, 9. Halomicrobium mukohatei DSM 12286T, 10.Halorhabdus utahensis DSM 12940T, 11. Halomicroarcula amylolytica LR21T, 12. Halorientalis regularis IBRC-M 10760T, 13. Strain F27T, 14.Halapricum salinum CBA1105T, 15. Strain F28T. Genome accession numbers are indicated in Figure 1.
Concerning the AAI values between the four new strains and members of the genus Halomicroarcula and other related genera, they clearly confirm that these four new strains are members of the genus Halomicroarcula. Percentages of AAI of strains F24AT, F28, F27T, and F13T among themselves and the species of Halomicroarcula are equal or higher than 71.3%, 71.5%, 72.7%, and 74.3%, respectively, values higher than the 65% generally accepted for the delineation at the genus level. It is noticeable that the AAI percentages for the new isolates and current species of the genus Halomicroarcula with Haloarcula vallismortis (type species of the genus Haloarcula) are 71.0%, 71.0%, 71.5%, and 73.1% for strains F24AT, F28, F27T, and F13T, respectively, and 73.9%, 79.6%, 72.0%, and 71.6%, respectively, for the species Halomicroarcula pellucida CECT 7537T, Halomicroarcula salina JCM 18369T, Halomicroarcula limicola JCM 18640T and Halomicroarcula amylolytica LR21T. In fact, Halomicroarcula salina JCM 18369T shows the higher percentage of AAI (79.6%) with Haloarcula vallismortis ATCC 29715T, and lower values with the remaining members of the genus Halomicroarcula: 71.3%, 71.5%, 73.5%, 74.3%, 74.5%, 74.2%, and 72.8% with strains F24AT, F28, F27T, and F13T and the species Halomicroarcula pellucida CECT 7537T, Halomicroarcula limicola JCM 18640T and Halomicroarcula amylolytica LR21T, respectively. Although the cutoff value of 65% for AAI delineation at the genus level is not universally accepted, considering these percentages of similarity, the species Halomicroarcula salina JCM 18369T should be considered as a member of the genus Haloarcula. However, it must be noted that the AAI values for the remaining species of Halomicroarcula with respect to Haloarcula vallismortis ATCC 29715T are also higher than 65% (71.6% for Halomicroarcula amylolytica LR21T, 72.0% for Halomicroarcula limicola JCM 18640T and 73.9% for the type species of the genus, Halomicroarcula pellucida CECT 7537T). According to these results the species of the genera Halomicroarcula and Haloarcula should be merged into a single genus, as Haloarcula. A more exhaustive study, including a larger set of strains and based on additional genomic analysis reflecting their evolutionary relationships as well as their phenotypic features should be necessary in order to dilucidate the taxonomic status of the species of Halomicroarcula and Haloarcula.
We carried out a detailed phenotypic characterization of the four new isolates with respect to the type strains of species of Halomicroarcula, following the recommended minimal standards for describing new taxa of the class Halobacteria (Oren et al., 1997). These features included morphological, physiological, biochemical and nutritional characteristics, as well as the determination of the membrane polar lipids, which has been proved to be an important feature for the characterization of haloarchaeal genera (Oren et al., 1997, 2009). The results of the phenotypic features of the new isolates are shown in Supplementary Table 2 and in the new species descriptions included at the conclusion section. These data confirm that there are differential features supporting the proposal of the three new species of the genus Halomicroarcula (Supplementary Table 2). Concerning the polar lipids composition, they were analyzed by High-Performance Thin Layer Chromatography (HPTLC) and the results are shown in Supplementary Figure 2. The four new isolates, strains F24AT, F28, F27T, and F13T showed the typical polar lipid profile of species of the genus Halomicroarcula, composed of phosphatidylglycerol (PG), phosphatidylglycerol phosphate methyl ester (PGP-Me), phosphatidylglycerol sulfate (PGS) and sulfated diglycosil diether (S-DGD-1) (Echigo et al., 2013; Zhang and Cui, 2014). Overall, all these phenotypic features are in agreement with the previous genomic results and support the placement of the new isolates into three new species of the genus Halomicroarcula.
Genomic Features
In order to gain insights into the genome diversity of the genus Halomicroarcula we sequenced and analyzed the genome of the four strains isolated in this study and of the four already described species of the genus Halomicroarcula. The size of the genomes among the representatives of the genus Halomicroarcula ranged from 3.8 to 5.2 Mb, the DNA G + C content from 62.0 to 66.7 mol% and the total number of genes from 3827 to 5281. Most genomes of these strains ranged from 3.8 to 4.0 Mb, with the exception of three genomes that showed higher values (Table 1), even considering that the quality of these genomes are within the standards already described for prokaryotes (Chun et al., 2018).
On the other hand, several genetic elements detailed in Table 2 were also detected in those genomes. Members of the genus Halomicroarcula presented a large number of integrases and transposases, especially abundant in the isolated strains in comparison with the type strains of the previously described species of Halomicroarcula. Moreover, several CRISPR loci were also found, their number ranged from 2 in Halomicroarcula salina JCM 18369T to 8 in strain F27T (Table 2). While strains F24AT, F13T, F27T and F28 presented cas cluster type IB and 94, 86, 84 and 74 spacers, respectively, not detectable cas genes were found in Halomicroarcula limicola JCM 18640T, Halomicroarcula pellucida CECT 7537T, Halomicroarcula salina JCM 18369T and Halomicroarcula amylolytica LR21T genomes, suggesting the lack of functionality of these systems in those strains. These data correlate with the high number of prophage sequences detected in Halomicroarcula genomes (Table 2), with a size between 5.4 and 33.1 kb. However, most of these prophage sequences were not complete.

Table 2. CRISPR loci and mobile genetic elements determined in the genomes of strains F13T, F24AT, F27T, F28 and the type strains of species of Halomicroarcula.
The presence of these elements in high copy numbers in the genomes of members of the genus Halomicroarcula reflect a high genomic plasticity of these strains (including genetic rearrangements or horizontal gene transfer events), particularly in those isolated in this study from hypersaline soils. This fact could suggest a great adaptation of these taxa to different ecological niches and their success in nature.
Metabolism
Based on the genome annotation of members of the genus Halomicroarcula the major metabolic pathways of those strains could be reconstructed. For the carbohydrates uptake a large number of transporters were enconded in Halomicroarcula genomes reflecting their heterotrophic capabilities. Complete pathways involved in central carbohydrate metabolism (tricarboxylic acid cycle, oxidative pentose phosphate, Entner-Doudoroff or gluconeogenesis pathways) were present. However, in accordance with previous metabolic studies in haloarchaea (Falb et al., 2008; Anderson et al., 2011; Durán-Viseras et al., 2019a, 2021) the glycolysis pathway (Embden-Meyerhoff-Parnas) was truncated, therefore suggesting that alternative pathways like Entner-Doudoroff or the oxidative pentose phosphate could be used instead (Verhees et al., 2003). For the oxidation of the generated pyruvate to acetyl-CoA, both aerobic and anaerobic routes via pyruvate dehydrogenase and pyruvate ferredoxin oxidoreductase, respectively, were identified. Other carbohydrate metabolic pathways like the methylaspartate cycle, an anaplerotic acetate assimilation pathway, was also dilucidated in all Halomicroarcula genomes. This cycle has been previously identified in many other haloarchaea, such as the phylogenetically closest neighbor of the genus Halomicroarcula, the genus Haloarcula (Borjian et al., 2016). Its presence in haloarchaea has been frequently associated with the possession of genes for polyhydroxyalkanoate biosynthesis (Han et al., 2010; Borjian et al., 2016), which were also identified in Halomicroarcula genomes. Therefore, the presence of both pathways in Halomicroarcula genomes suggest the capability of members of this genus to biosynthetize polyhydroxialcanoates, as well as their adaptation for the assimilation of acetyl-CoA, produced from the internal carbon storage, during carbon starvation periods. This is a crucial factor for haloarchaea living under frequent starvation periods, such as on hypersaline soils from which our four strains were isolated. The genome of some members of the genus Halomicroarcula also encode for additional pathways related to carbohydrate metabolism (D-glucuronate and D-galacturonate degradation or glycogen biosynthesis), and with the hydrolysis of complex polysaccharides (α-amylase, chitinase, and endoglucanase enzymes), thus reflecting the metabolic diversity of representatives of this genus.
On the other hand, Halomicroarcula genomes encode the whole set of genes responsible for ammonia assimilation as a part of their nitrogen metabolism, such as enzymes for nitrate and nitrite reduction, the high-affinity ammonium transporter and glutamine synthetase and glutamate synthase. Moreover, complete biosynthesis pathways of several amino acids (i.e., arginine, cysteine, histidine, isoleucine, lysine, proline, serine, threonine, tryptophan, and valine) and few polyamine biosynthesis were identified along the genomes. Other determined sources of potential nitrogen rich compounds are different kinds of transporters for amino acids, urea and spermidine/putrescine uptake or a complete urease gene cluster.
Furthermore, Halomicroarcula genomes also encode genes for the ABC transporters for phosphate (pstSCAB) uptake, and in the case of H. salina JCM 18369T and H. pellucida CECT 7537T for phosphonate (phnCDE), as well as several mfs transporters related with multridrug efflux pump systems. Genes enconding archaella were also found in all Halomicroarcula genomes, confirming their motility as phenotypic feature.
In addition, rhodopsin-like sequences were also identified in members of the genus Halomicroarcula, suggesting the phototrophic capabilities of this group of prokaryotic microorganisms. Strains F13T, F24AT, F28 and Halomicroarcula salina JCM 18369T presented sensory rhodopsins, haloarchaeal proton pumps and halorhodopsins, while Halomicroarcula pellucida CECT 7537T exhibited sensory rhodopsin and haloarchaeal proton pump, and strain F27T and Halomicroarcula amylolytica LR21T only sensory rhodopsins. No rhodopsin-like sequences were identified in Halomicroarcula limicola JCM 18640T. The presence of rhodopsin like-sequences in most Halomicroarcula genomes suggest a versatile metabolic flexibility in illuminated conditions for members of this genus. These results are in accordance with recent comparative genomic studies in other haloarchaeal genera (Durán-Viseras et al., 2019a, 2020, 2021) in which rhodopsins were also identified, and also with previous metagenomic analyses on hypersaline systems (Fernández et al., 2014; Vera-Gargallo and Ventosa, 2018) that showed the existence of a large number of rhodopsin coding genes, clearly indicating the wide use of light by haloarchaea in these extreme habitats.
Regarding vitamins and cofactors metabolism the complete pathway for the vitamin B12 biosynthesis was determined in all members of the genus Halomicroarcula, suggesting the capability of this genus for its de novo synthesis. Remarkably, this pathway was recently identified by genomic studies in other haloarchaeal members of the genus Halonotius (Durán-Viseras et al., 2019a). An ABC transporter for biotin uptake was also encountered in Halomicroarcula genomes.
Besides, as a part of the metabolic analyses we also searched for metal resistance genes in Halomicroarcula genomes. While Halomicroarcula salina JCM 18369T and Halomicroarcula amylolytica LR21T encode the complete arsenite resistance gene cluster (ars), this cluster was truncated in the other Halomicroarcula members. However, strains isolated in this study (F13T, F24AT, F27T, and F28) and Halomicroarcula amylolytica LR21T encode the CzcD transporter, a member of the cation diffusion facilitator (CDF) protein family, which was absent in the other Halomicroarcula reference strains. This transporter not only reflects a heavy metal resistance against cobalt, zinc or cadmium, but has also been suggested as a biomarker of nickel and vanadium pollution in some Bacteria (Anton et al., 1999; Fierros-Romero et al., 2020). On the contrary, none of the genes involved in copper or mercury resistance were identified in any of the studied genomes. These results could be related to the different stress conditions affecting the diverse ecological niches that they inhabit. In addition, several ABC metal transporters for zinc, cobalt and nickel were identified, which display a less critical role in maintaining metal homeostasis than CDF family transporters (Kaur et al., 2006).
Osmoadaptative Capabilities
To cope with the high salt concentrations and salinity fluctuations of hypersaline environments, halophilic microorganisms have developed diverse mechanisms of adaptation (Gunde-Cimerman et al., 2018). In order to delucidate the osmorregulatory strategy employed by Halomicroarcula members and their closest phylogenetic neighbors (the genera Haloarcula and Halomicrobium), their proteome was analyzed and compared with those of salt-in microorganisms [i.e., Haloquadratum walsbyi C23T (Dyall-Smith et al., 2011) and Salinibacter ruber DSM 13855T (Mongodin et al., 2005)] and with that of a salt-out bacterium [Spiribacter salinus M19-40T (López-Pérez et al., 2013)] (Supplementary Figure 3). In accordance to Haloquadratum walsbyi C23T and Salinibacter ruber DSM 13855T and by contrast with Spiribacter salinus M19-40T, Halomicroarcula representatives exhibited an acidic proteome with a low isoelectric point peak around 4.0 (Supplementary Figure 3A). Hence, highlighting a typical salt-in strategy for all members of this genus. Similar results were obtained for Haloarcula and Halomicrobium representatives (Supplementary Figure 3B). Besides, during the indeep genomic analysis of members of the genus Halomicroarcula we investigated the presence of genes putatively involved in osmorregulation. In conformity with results mentioned above, several transporters for Na+ extrusion, K+ uptake and Cl– homeostasis were also identified, reinforcing its salt-in strategy.
According to previous suggestions (Youssef et al., 2014) and considering the frequent periods of low or fluctuenting salinity levels of the saline soils from which strains F13T, F24AT, F27T, and F28 were isolated, it is not rare that they could not exclusively employ a salt-in strategy. Noteworthy, genes encoding de novo synthesis of trehalose via trehalose-6-phosphate synthase (OtsA) and trehalose-6-phosphatase (OtsB) were encountered in the genomes of strains F24AT, F27T, F28, Halomicroarcula amylolytica LR21T and Halomicroarcula limicola JCM 18640T suggesting their capability for trehalose biosynthesis. Trehalose is a dissacharide which has been reported to be used as an osmolyte by different organisms (Shivanand and Mugeraya, 2011). On the other side, the otsAB pathway was not found complete for strains F13T, Halomicroarcula salina JCM 18369T and Halomicroarcula pellucida CECT 7537T, which lack the OtsB enzyme. However, the genome of strain F13T was the only one exhibiting the enzyme trehalose synthase (TreT) suggesting this is the pathway used by this strain to produce trehalose. Although the synthesis of the compatible solute trehalose has been previously reported for other members of the haloarchaea (Youssef et al., 2014), no evidence has been demonstrated for any Haloarcula representatives (Youssef et al., 2014), the phylogenetically closest neighbors of the genus Halomicroarcula. Additionally, the presence of genes encoding the different trehalose biosynthesis pathways were analyzed during the course of this study in all members of the genera Haloarcula and Halomicrobium with available genomes (Supplementary Table 3). The enzyme trehalose synthase (TreT) was not found in any Haloarcula or Halomicrobium genomes, whereas the OtsAB pathway was only found complete in Halomicrobium zhouii CGMCC 1.10457T (only OtsA was present in Haloarcula sebkhae JCM 19018T, H. salaria ZP1-2, H. quadrata DSM 11927T, H. marismortui ATCC 43049T, H. hispanica ATCC 33960T and H. argentinensis DSM 12282T genomes; while Halomicrobium katesii DSM 19301T only possessed OtsB). These results suggest that the ability to synthethize trehalose is not extended neither in the genera Haloarcula and Halomicrobium, in accordance with previous presumptions of Youssef et al. (2014) for the genus Haloarcula. The ability to synthethize trehalose has been previously described for members of the class Halobacteria dwelling environments with lower salinity or salinity fluctuations (Youssef et al., 2014), such as the hypersaline soil from which strains F13T, F24AT, F27T and F28 were isolated.
Surprinsingly, the complete pathway for the biosynthesis of the compatible solute glycine betaine from choline, was identified in the genomes of strain F13T and Halomicroarcula limicola JCM 18640T. No evidencies were identified in any of the genomes from species of the genera Haloarcula or Halomicrobium. Accordingly, the BCCT family transporters: OpuD and BetT, possibly for glycine betaine uptake or for other types of compatible solutes (Ziegler et al., 2010), were also present in the genomes of strains F13T, F27T, Halomicroarcula pellucida CECT 7537T, Halomicroarcula amylolytica LR21T and Halomicroarcula salina JCM 18369T (in the case of OpuD) and strains F13T, F27T, Halomicroarcula limicola JCM 18640T and Halomicroarcula pellucida CECT 7537T (in the case of BetT). In the same way, OpuD was also present in Haloarcula sebkhae JCM 19018T, H. argentinensis DSM 12282T and Halomicrobium zhouii CGMCC 1.10457T genomes, and BetT in the genomes of Haloarcula sebkhae JCM 19018T and H. argentinensis DSM 12282T. Moreover, Halomicroarcula salina JCM 18369T and Halomicroarcula amylolytica LR21T also showed the ABC transporter OpuA for betaine incorporation (Kempf and Bremer, 1995). Small conductance mechanosensitive channels from the MscS family were identified as well in Halomicroarcula genomes, and in the genomes of few members of the genera Haloarcula and Halomicrobium. These systems are ubiquitously used by microorganisms to manage the rapid transition from high salinity surroundings to environments with moderate salinities (Booth and Blount, 2012). The functionality of such safety valves has been demonstrated in some Archaea such as the marine thaumarchaeon Nitrosopumilus maritimus (Widderich et al., 2016). While BCCT family transporters are quite common in members of the class Halobacteria (Anderson et al., 2011; Youssef et al., 2014; Gunde-Cimerman et al., 2018), to the best of the authors knowledge, this is the first time that genes encoding glycine betaine synthesis are reported for any haloarchaea by far. This fact, could indicate an additional osmoadaptative mechanism for strains of the genus Halomicroarcula, which would corroborate the versatility of this group of microorganisms to adapt to environments with different salinities. The large amount of genes related with glycine betaine in the genomes of the genus Halomicroarcula raises the possibility that this organic solute could play an important role as osmoprotectant in this archaeal group that needs to be further investigated.
Biosynthesis of Secondary Metabolites
Secondary metabolites are a range of bioactive compounds of high interest for biotechnological, pharmaceutical or industrial applications (Charlesworth and Burns, 2015). The production of secondary metabolites give some environmental advantages to the microorganisms producing them (such as tolerance against environmental stress or interspecies defenses) (Osbourn, 2010; Piasecka et al., 2015; Wang and Lu, 2017; Wang et al., 2019). While secondary metabolites in the domains Bacteria and Eukarya have been deeply studied, analysis of these compounds in Archaea are more scarce (Charlesworth and Burns, 2015; Corral et al., 2020). As a part of our genomic analysis we also investigated the presence of genes involved on the biosynthesis of secondary metabolites in the genomes of members of the genus Halomicroarcula. The clusters that we have identified on these genomes are detailed in Table 3 and Figures 3–6. Two different terpene clusters were detected in all Halomicroarcula genomes, which shared similarities within the different analyzed strains (Figure 3). To the best of the authors knowledge the presence of terpenes in Archaea has only been reported in a previous study (Wang et al., 2019), which also indicated the presence of two terpene clusters in the Halobacteria genomes. Terpenes are metabolites widely distributed in plants, fungi and bacteria in which they play a protective role; these compounds have also been suggested to be a source for the discovery of natural products (Pichersky et al., 2006; Yamada et al., 2015). On the other hand, siderophores, iron-chelating molecules produced during stress conditions or iron deficiency (Srivastava and Kowshik, 2013), were also distributed in the seven analyzed genomes of members of the genus Halomicroarcula (Figure 4). Despite the ubiquity of iron in the environment its solubility is very low, leading to the siderophore strategy to overcome this scarcity. The production of siderophores has also been detected before in some other haloarchaea (Dave et al., 2006). Finally, we detected a thiopeptide cluster in the genomes of strain F24AT and Halomicroarcula pellucida CECT 7537T (Figure 5), and a lanthipeptide in the genome of Halomicroarcula salina JCM 18369T (Figure 6), both of them members of the ribosomally synthesized and post-translationally modified peptides (RiPP) family of natural products. No similarity was observed between the identified thiopeptide clusters (Figure 5). While thiopeptides constitute important components of the defense system in archaea, lanthipeptides display different biological activities, such as antimicrobial or antiallodynic (Repka et al., 2017; Wang and Lu, 2017). Thus, the biosynthesis of this wide variety of secondary metabolites by strains of the genus Halomicroarcula suggests the versatility of members of this genus, which have developed several ecological advantages such as a great adaptability to extreme conditions or defense mechanisms. In addition, our results brought to light their possible applications on the biotechnological field as a source of new natural compounds, also recently proposed for other novel haloarchaeal taxa (Durán-Viseras et al., 2021).

Table 3. Number of genetic clusters involved in the biosynthesis of secondary metabolites determined in the genomes of strains F13T, F24AT, F28, F27T, and the type strains of species of Halomicroarcula.
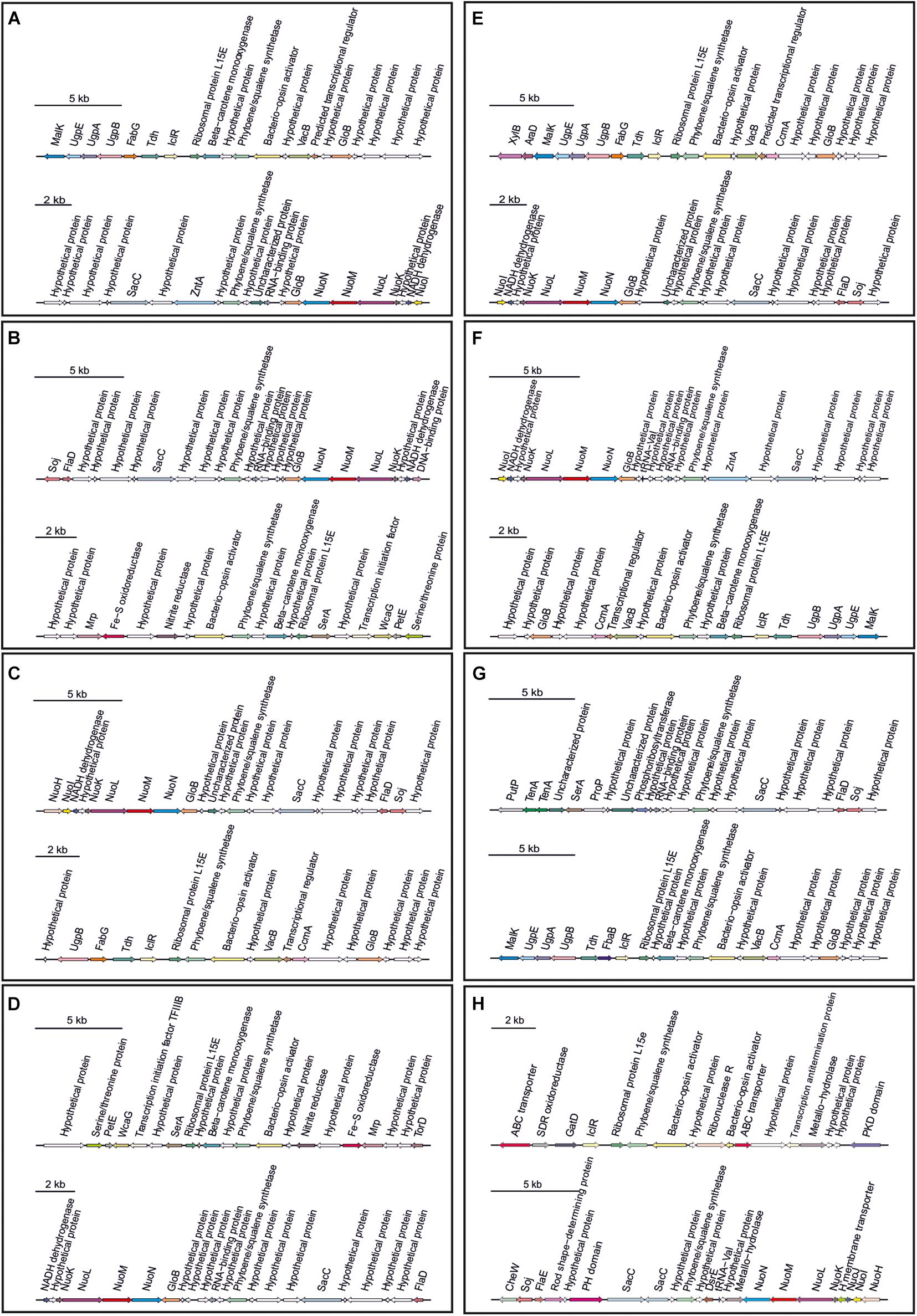
Figure 3. Terpene biosynthetic gene clusters identified in Halomicroarcula genomes: (A) Strain F13T, (B) Strain F24AT, (C) Strain F27T, (D) Strain F28, (E) Halomicroarcula limicola JCM 18640T, (F) Halomicroarcula pellucida CECT 7537T, (G) Halomicroarcula salina JCM 18369T, and (H) Halomicroarcula amylolytica LR21T.
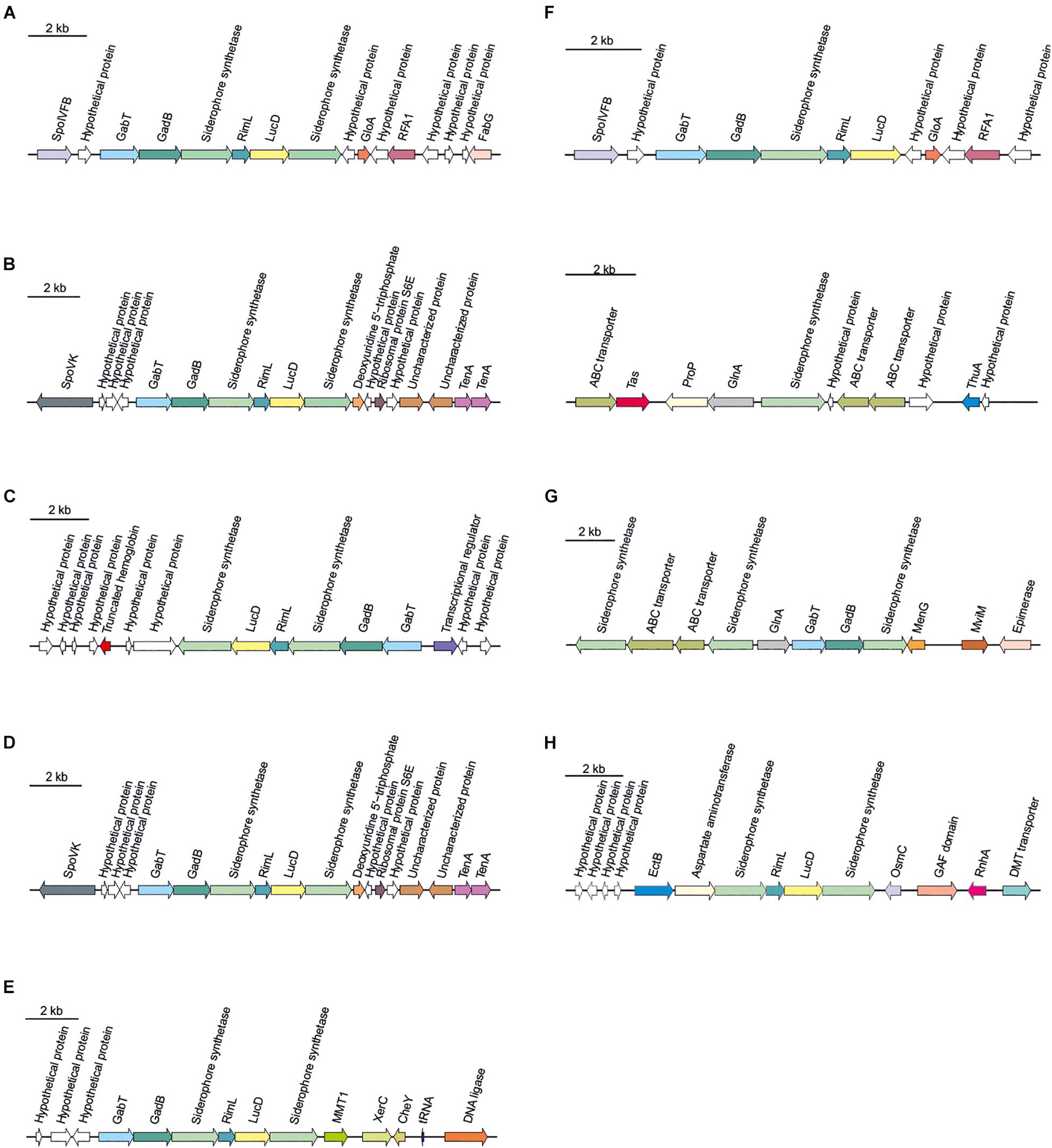
Figure 4. Siderophore biosynthetic gene clusters identified in Halomicroarcula genomes: (A) Strain F13T, (B) Strain F24AT, (C) Strain F27T, (D) Strain F28, (E) Halomicroarcula limicola JCM 18640T, (F) Halomicroarcula pellucida CECT 7537T, (G) Halomicroarcula salina JCM 18369T, and (H) Halomicroarcula amylolytica LR21T.
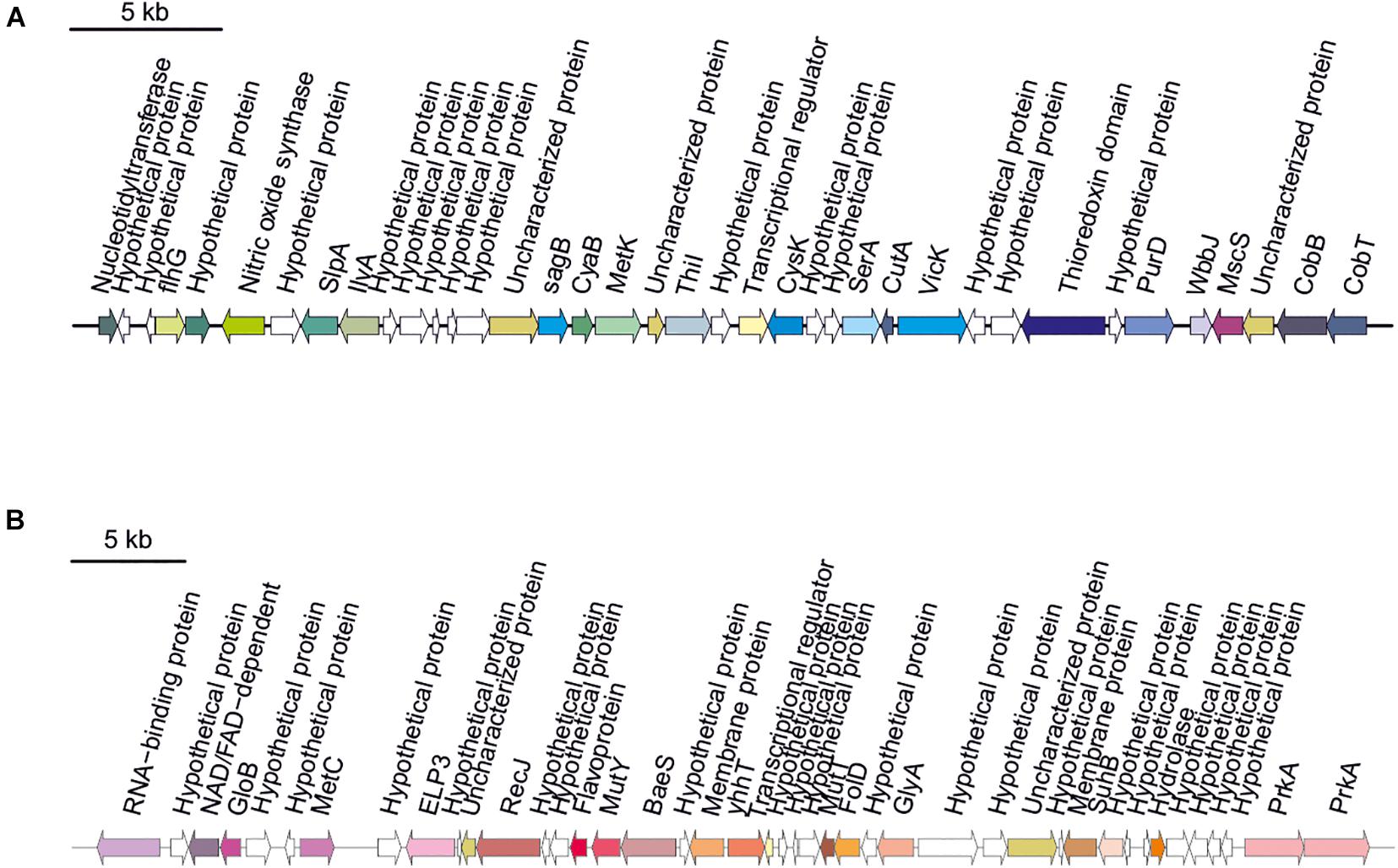
Figure 5. Thiopeptide biosynthetic gene clusters identified in Halomicroarcula genomes: (A) Strain F24AT and (B) Halomicroarcula pellucida CECT 7537T.
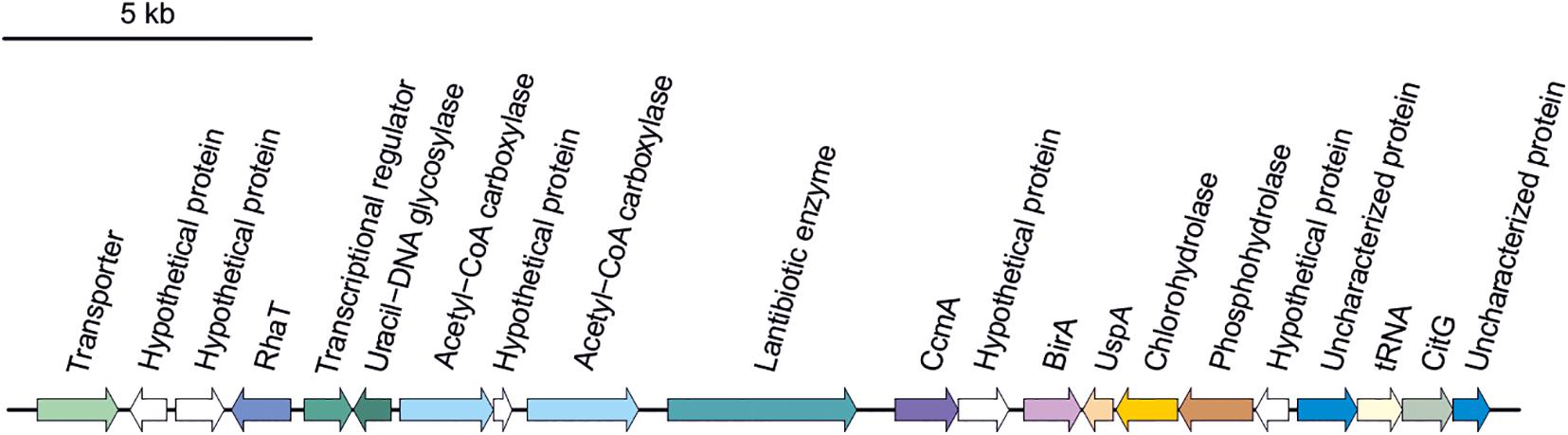
Figure 6. Lanthipeptide biosynthetic gene cluster identified in Halomicroarcula salina JCM 18369T.
Conclusion
The indeep comparative genomic analysis of the genus Halomicroarcula brought to light the presence of integrases, transposases and other gene transfer systems in high copy in the studied genomes, suggesting a vast plasticity for genes adquisition in members of the genus Halomicroarcula. This genomic plasticity leads to the versatile metabolism observed in the studied strains, such as the presence of metal resistance genes and genes coding for diverse carbohydrates pathways, some of them (i.e., methylaspartate cycle and polyhydroxyalkanoate biosynthesis) advantageous during carbon starvation periods. Besides, the capacity to biosynthethize diverse secondary metabolites (i.e., terpenes, siderophores, lanthipeptides or thiopeptides), could provide an ecological benefit for these microorganisms such as defense systems or adjustability to limiting conditions, and could also be source of novel compounds for biotechnological applications. The analysis of the proteome of members of the genus Halomicroarcula indicate they use a salt-in strategy. Nevertheless, complete pathways for the biosynthesis of compatible solutes (i.e., trehalose and glycine betaine), identified for the first time in haloarchaea during the detailed genomic analysis carried out in this study, suggests that alternative osmoadaptation strategies could be additionally employed. All these facts could give an ecological advantage for these microorganisms, which provide the genus Halomicroarcula the basis for the adaptation to a wide range of ecological niches and hence, playing a crucial role in the ecophysiological success of this taxa in the nature. Besides, this fact might justify the ability of these haloarchaea to grow at intermediate to low salinity environments.
On the other side, the exhaustive taxogenomic and phenotypic study carried out in this work, has permitted the characterization and description of three new species within the genus Halomicroarcula, for which we propose the new names Halomicroarcula rubra sp. nov., Halomicroarcula nitratireducens sp. nov. and Halomicroarcula salinisoli sp. nov.; and whose descriptions are detailed below.
Halomicroarcula rubra sp. nov.
Halomicroarcula rubra (ru′bra. L. fem. adj. rubra, red).
Cells are Gram-stain-negative, motile rods with 1 × 1.2-2.5 μm. Does not grow anaerobically with L-arginine, dimethyl sulfoxide (DMSO) or potassium nitrate. Colonies are circular, entire, red pigmented with 0.2–0.3 mm in diameter on R2A25 medium after 14 days of incubation at 37°C. Extremely halophilic, able to grow in media with 10–30% (w/v) salts, with optimal growth at 25–30% (w/v) salts. No growth occurs in the absence of NaCl. Mg2+ is not required for growth. Able to grow in the pH range of 6.0–9.0 and from 25 to 50°C, with optimal growth at pH 7.5–8.0 and at 37°C. Chemoorganotrophic and aerobic. Catalase positive and oxidase negative. Gelatin is hydrolyzed but starch, Tween 80 and aesculin are not. Nitrate and nitrite are reduced, without gas production. H2S is produced but indole and urease are not. Methyl red test is positive. Voges-Proskauer is negative. Acid is produced from D-arabinose, arbutin, L-citrulline, D-fructose, glycerol, D-glucose, D-ribose, L-xylitol and D-xylose but not from D-amygdalin, D-cellobiose, dulcitol, D-galactose, lactose, D-maltose, D-mannitol, D-mannose, D-melezitose, D-raffinose, D-sucrose, sorbitol or D-trehalose. The following compounds are used as carbon and energy source: D-melibiose, L-arginine, and L-methionine. The following compounds are not used as sole carbon and energy source: D-arabinose, D-cellobiose, fructose, D-galactose, D-glucose, lactose, maltose, D-mannose, L-raffinose, ribose, sucrose, D-trehalose, D-xylose, D-melezitose, salicin, butanol, dulcitol, ethanol, glycerol, D-mannitol, D-sorbitol, xylitol, methanol, benzoate, citrate, formate, fumarate, propionate, valerate, hippurate, malate, pyruvate, tartrate, L-alanine, L-cysteine, glutamine, L-glycine, L-lysine, isoleucine or valine. The major polar lipids are phosphatidylglycerol (PG), phosphatidylglycerol phosphate methyl ester (PGP-Me), phosphatidylglycerol sulfate (PGS) and sulfated mannosyl glucosyl diether (S-DGD-1). The DNA G + C content is 64.4 mol% (genome).
The type strain is F13T (= CCM 8888T = CECT 9686T = IBRC-M 11249T = JCM 33313T), isolated from a hypersaline soil located in Odiel saltmarshes, Huelva, Spain.
The GenBank/EMBL/DDBJ accession number for the 16S rRNA and rpoB′ gene sequences of Halomicroarcula rubra F13T are MH447277 (rrnA gene), MH447279 (rrnB gene) and MH454085, respectively, and that of the complete genome is RKLR00000000.
Halomicroarcula nitratireducens sp. nov.
Halomicroarcula nitratireducens (ni.tra.ti.re.du’cens. N.L. masc. n. nitras (gen. nitratis), nitrate; L. pres. part. reducens, converting to a different state, reducing; N.L. part. adj. nitratireducens, reducing nitrate).
Cells are Gram-stain-negative, motile rods with 1 × 1.2–2.5 μm. Does not grow anaerobically with L-arginine, DMSO or potassium nitrate. Colonies are circular, entire, red to orange pigmented with 0.2–0.3 mm in diameter on R2A25 medium after 14 days of incubation at 37°C. Extremely halophilic, able to grow in media with 10–30% (w/v) salts, with optimal growth at 25–30% (w/v) salts. No growth occurs in the absence of NaCl. Mg2+ is not required for growth. Able to grow in the pH range of 6.0–9.0 and from 25 to 50°C, with optimal growth at pH 7.5 and at 37°C. Chemoorganotrophic and aerobic. Catalase and oxidase negative. Starch and aesculin are hydrolyzed but gelatin and Tween 80 are not. Nitrate and nitrite are reduced, without gas production. H2S, indole and urease are not produced. Methyl red test is positive. Voges-Proskauer is negative. Acid is produced from D-arabinose, arbutin, L-citrulline, D-fructose, D-glucose, D-ribose and D-xylose but not from D-amygdalin, D-cellobiose, dulcitol, D-galactose, glycerol, lactose, D-maltose, D-mannitol, D-mannose, D-melezitose, D-raffinose, D-sucrose, sorbitol, D-trehalose or L-xylitol. The following compounds are used as carbon and energy source: fructose, D-galactose, D-glucose, maltose, ribose, sucrose, salicin, glycerol, D-mannitol, D-sorbitol, methanol, fumarate, or L-lysine. The following compounds are not used as sole carbon and energy source: D-arabinose, D-cellobiose, lactose, D-xylose, butanol, dulcitol, ethanol, xylitol, benzoate, citrate, formate, propionate, valerate, hippurate, malate, pyruvate, tartrate, L-alanine, L-arginine, L-cysteine, glutamine, L-methionine, L-glycine, isoleucine or valine. The major polar lipids are phosphatidylglycerol (PG), phosphatidylglycerol phosphate methyl ester (PGP-Me), phosphatidylglycerol sulfate (PGS) and sulfated mannosyl glucosyl diether (S-DGD-1). The DNA G + C content is 63.2 mol% (genome).
The type strain is F27T (= CCM 8887T = CECT 9636T = IBRC-M 11233T = JCM 33314T), isolated from a hypersaline soil located in Odiel saltmarshes, Huelva, Spain.
The GenBank/EMBL/DDBJ accession number for the 16S rRNA and rpoB′ gene sequences of Halomicroarcula nitratireducens F27T are MH447286 (rrnA gene), MH447284 (rrnB gene) and MH454093, respectively, and that of the complete genome is RKLT00000000.
Halomicroarcula salinisoli sp. nov.
Halomicroarcula salinisoli (sa.li.ni.so’li. N.L. masc. adj. salinus, salty; L. neut. n. solum, soil; N.L. gen. n. salinisoli, of salty soil).
Cells are Gram-stain-negative, motile, pleomorphic rods with 1 × 1.2–2.5 μm. Does not grow anaerobically with L-arginine, DMSO or potassium nitrate. Colonies are circular, entire, pink pigmented with 0.2–0.3 mm in diameter on R2A25 medium after 14 days of incubation at 37°C. Extremely halophilic, able to grow in media with 15–30% (w/v) salts, with optimal growth at 25% (w/v) salts. No growth occurs in the absence of NaCl. Mg2+ is not required for growth. Able to grow in the pH range of 6.0–8.5 and from 25 to 50°C, with optimal growth at pH 7–7.5 and at 37°C. Chemoorganotrophic and aerobic. Catalase positive and oxidase negative. Gelatin, aesculin and Tween 80 are hydrolyzed but starch is not. Nitrate and nitrite are reduced, without gas production. H2S production is variable, indole and urease are not produced. Methyl red test is positive. Voges-Proskauer is negative. Acid is produced from D-arabinose, arbutin, D-cellobiose, L-citrulline, D-fructose, D-glucose, D-ribose and D-xylose but not from dulcitol, D-galactose, lactose, D-maltose, D-mannitol, D-mannose, D-melezitose, D-raffinose, D-sucrose, sorbitol, D-trehalose or L-xylitol. The following compounds are used as carbon and energy source: D-cellobiose, D-glucose, maltose, sucrose, D-sorbitol, citrate, fumarate or tartrate. The following compounds are not used as sole carbon and energy source: D-arabinose, D-galactose, lactose, ribose, D-xylose, salicin, glycerol, xylitol, benzoate, propionate, valerate, hippurate, pyruvate, L-arginine, L-cysteine, L-methionine, isoleucine or valine. The major polar lipids are phosphatidylglycerol (PG), phosphatidylglycerol phosphate methyl ester (PGP-Me), phosphatidylglycerol sulfate (PGS) and sulfated mannosyl glucosyl diether (S-DGD-1). The DNA G + C content is 63.9–64.1 mol% (genome).
The type strain is F24AT (= CCM 8955T = CECT 9687T), isolated from a hypersaline soil located in Odiel saltmarshes, Huelva, Spain. The DNA G + C content of the type strain is 64.1 mol% (genome).
The GenBank/EMBL/DDBJ accession number for the 16S rRNA and rpoB′ gene sequences of Halomicroarcula salinisoli F24AT are MH447282 (rrnA gene), MH447281 (rrnB gene) and MH454092, respectively, and that of the complete genome is RKLQ00000000.
An additional strain of this species is strain F28. The DNA G + C content of this strain is 63.9 mol% (genome). The GenBank/EMBL/DDBJ accession number for the 16S rRNA and rpoB′ gene sequences of this strain are MH450228 (rrnA gene), MH447330 (rrnB gene) and MH454094, respectively, and that of the complete genome is RKLS00000000.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, the 16S rRNA and rpoB′ genes and the genome sequences generated for this study can be found in the GenBank/EMBL/DDBJ database under the accession numbers MH447277, MH447279, MH447282, MH447281, MH450228, MH447330, MH447286, MH447284, MH454085, MH454092, MH454094, MH454093, RKLR00000000, RKLQ00000000, RKLS00000000, RKLT00000000, JAHQXF000000000, RKLW00000000, and JAHQXE000000000.
Author Contributions
AD-V, CS-P, and AV did the conceptualization. AD-V did the strains isolation, the taxogenomic characterization, and the comparative genomic analyses. AD-V and AV wrote the manuscript. AD-V prepared the tables and figures. AV and CS-P did the funding acquisition. All authors read and approved the final version of the manuscript.
Funding
This research was funded by Junta de Andalucía, Spain (grants US-1263771 [US/JUNTA/FEDER/UE], P20_01066 and BIO-213, which included FEDER funds), and FEDER/Spanish Ministry of Science and Innovation-State Research Agency (projects CGL2017-83385-P and PID2020-118136GB-I00).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank A. Oren for his help on the nomenclature of the three new species.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.751746/full#supplementary-material
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Amoozegar, M. A., Siroosi, M., Atashgahi, S., Smidt, H., and Ventosa, A. (2017). Systematics of haloarchaea and biotechnological potential of their hydrolytic enzymes. Microbiology 163, 623–645. doi: 10.1099/mic.0.000463
Anderson, I., Scheuner, C., Göker, M., Mavromatis, K., Hooper, S. D., Porat, I., et al. (2011). Novel insights into the diversity of catabolic metabolism from ten haloarchaeal genomes. PLoS One 6:e20237. doi: 10.1371/journal.pone.0020237
Anton, A., Große, C., Reißmann, J., Pribyl, T., and Nies, D. H. (1999). CzcD is a heavy metal ion transporter involved in regulation of heavy metal resistance in Ralstonia sp. strain CH34. J. Bacteriol. 181, 6876–6881. doi: 10.1128/jb.181.22.6876-6881.1999
Arahal, D. R., Dewhirst, F. E., Paster, B. J., Volcani, B. E., and Ventosa, A. (1996). Phylogenetic analyses of some extremely halophilic archaea isolated from dead sea water, determined on the basis of their 16S rRNA sequences. Appl. Environ. Microbiol. 62, 3779–3786.
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Auch, A. F., von Jan, M., Klenk, H.-P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). AntiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Booth, I. R., and Blount, P. (2012). The MscS and MscL families of mechanosensitive channels act as microbial emergency release valves. J. Bacteriol. 194, 4802–4809. doi: 10.1128/JB.00576-12
Borjian, F., Han, J., Hou, J., Xiang, H., and Berg, I. A. (2016). The methylaspartate cycle in haloarchaea and its possible role in carbon metabolism. ISME J. 10, 546–557. doi: 10.1038/ismej.2015.132
Charlesworth, J. C., and Burns, B. P. (2015). Untapped resources: biotechnological potential of peptides and secondary metabolites in Archaea. Archaea 2015:282035. doi: 10.1155/2015/282035
Chen, F., Xu, Y., Sun, S., Shi, X., Liu, A., and Chen, S. (2020). Halomicroarcula amylolytica sp. nov., a novel halophilic archaeon isolated from a salt mine. Int. J. Syst. Evol. Microbiol. 70, 4978–4985. doi: 10.1099/ijsem.0.004368
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Chun, J., and Rainey, F. A. (2014). Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int. J. Syst. Evol. Microbiol. 64, 316–324. doi: 10.1099/ijs.0.054171-0
Corral, P., Amoozegar, M. A., and Ventosa, A. (2020). Halophiles and their biomolecules: recent advances and future applications in biomedicine. Mar. Drugs 18:33. doi: 10.3390/md18010033
Corral, P., Corcelli, A., and Ventosa, A. (2015). Halostagnicola bangensis sp. nov., an alkaliphilic haloarchaeon from a soda lake. Int. J. Syst. Evol. Microbiol. 65, 754–759. doi: 10.1099/ijs.0.000006
Corral, P., de la Haba, R. R., Sánchez-Porro, C., Amoozegar, M. A., Papke, R. T., and Ventosa, A. (2016). Halorubrum halodurans sp. nov., an extremely halophilicarchaeon isolated from a hypersaline lake. Int. J. Syst. Evol. Microbiol. 66, 435–444. doi: 10.1099/ijsem.0.000738
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/nar/gky425
Dave, B. P., Anshuman, K., and Hajela, P. (2006). Siderophores of halophilic archaea and their chemical characterization. Indian J. Exp. Biol. 44, 340–344.
DeLong, E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U. S. A. 89, 5685–5689. doi: 10.1073/PNAS.89.12.5685
Durán-Viseras, A., Andrei, A. Ş, Ghai, R., Sánchez-Porro, C., and Ventosa, A. (2019a). New Halonotius species provide genomics-based insights into cobalamin synthesis in haloarchaea. Front. Microbiol. 10:1928. doi: 10.3389/fmicb.2019.01928
Durán-Viseras, A., Sánchez-Porro, C., and Ventosa, A. (2019b). Halorientalis pallida sp. nov., an extremely halophilic archaeon isolated from a marine saltern. Int. J. Syst. Evol. Microbiol. 69, 3636–3643. doi: 10.1099/ijsem.0.003675
Durán-Viseras, A., Andrei, A. Ş, Vera-Gargallo, B., Ghai, R., Sánchez-Porro, C., and Ventosa, A. (2021). Culturomics-based genomics sheds light on the ecology of the new haloarchaeal genus Halosegnis. Environ. Microbiol. 23, 3418–3434. doi: 10.1111/1462-2920.15082
Durán-Viseras, A., Sánchez-Porro, C., and Ventosa, A. (2020). Natronomonas salsuginis sp. nov., a new inhabitant of a marine solar saltern. Microorganisms 8:605. doi: 10.3390/microorganisms8040605
Dyall-Smith, M. L., Pfeiffer, F., Klee, K., Palm, P., Gross, K., Schuster, S. C., et al. (2011). Haloquadratum walsbyi: limited diversity in a global pond. PLoS One 6:e20968. doi: 10.1371/journal.pone.0020968
Echigo, A. (2016). “Halomicroarcula,” in Bergey’s Manual of Systematics of Archaea and Bacteria, eds W. D. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. de Vos, et al. (Hoboken: John Wiley & Sons, Inc.), 1–9.
Echigo, A., Minegishi, H., Shimane, Y., Kamekura, M., Itoh, T., and Usami, R. (2013). Halomicroarcula pellucida gen. nov., sp. nov., a non-pigmented, transparent-colony-forming, halophilic archaeon isolated from solar salt. Int. J. Syst. Evol. Microbiol. 63, 3556–3562. doi: 10.1099/ijs.0.049965-0
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Enache, M., Itoh, T., Fukushima, T., Usami, R., Dumitru, L., and Kamekura, M. (2007). Phylogenetic relationships within the family Halobacteriaceae inferred from rpoB’ gene and protein sequences. Int. J. Syst. Evol. Microbiol. 57, 2289–2295. doi: 10.1099/ijs.0.65190-0
Falb, M., Müller, K., Königsmaier, L., Oberwinkler, T., Horn, P., von Gronau, S., et al. (2008). Metabolism of halophilic archaea. Extremophiles 12, 177–196. doi: 10.1007/s00792-008-0138-x
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution (N. Y) 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Fernández, A. B., Vera-Gargallo, B., Sánchez-Porro, C., Ghai, R., Papke, R. T., Rodríguez-Valera, F., et al. (2014). Comparison of prokaryotic community structure from Mediterranean and Atlantic saltern concentrator ponds by a metagenomic approach. Front. Microbiol. 5:196. doi: 10.3389/fmicb.2014.00196
Fierros-Romero, G., Gómez-Ramírez, M., Sharma, A., Pless, R. C., and Rojas-Avelizapa, N. G. (2020). czcD gene from Bacillus megaterium and Microbacterium liquefaciens as a potential nickel–vanadium soil pollution biomarker. J. Basic Microbiol. 60, 22–26. doi: 10.1002/jobm.201900323
Fullmer, M. S., Soucy, S. M., Swithers, K. S., Makkay, A. M., Wheeler, R., Ventosa, A., et al. (2014). Population and genomic analysis of the genus Halorubrum. Front. Microbiol. 5:140. doi: 10.3389/fmicb.2014.00140
Galinski, E. A., and Trüper, H. G. (1994). Microbial behaviour in salt-stressed ecosystems. FEMS Microbiol. Rev. 15, 95–108. doi: 10.1111/j.1574-6976.1994.tb00128.x
Gunde-Cimerman, N., Oren, A., and Plemenitaš, A. (2018). Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol. Rev. 42, 353–375. doi: 10.1093/femsre/fuy009/4909803
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Han, J., Hou, J., Liu, H., Cai, S., Feng, B., Zhou, J., et al. (2010). Wide distribution among halophilic archaea of a novel polyhydroxyalkanoate synthase subtype with homology to bacterial type III synthases. Appl. Environ. Microbiol. 76, 7811–7819. doi: 10.1128/AEM.01117-10
Jukes, T. H., and Cantor, C. R. (1969). “Evolution of protein molecules,” in Mammalian Protein Metabolism, ed. H. N. Munro (London: Elsevier), 21–132.
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kaur, A., Pan, M., Meislin, M., Facciotti, M. T., El-Gewely, R., and Baliga, N. S. (2006). A systems view of haloarchaeal strategies to withstand stress from transition metals. Genome Res. 16, 841–854. doi: 10.1101/gr.5189606
Kempf, B., and Bremer, E. (1995). OpuA, an osmotically regulated binding protein-dependent transport system for the osmoprotectant glycine betaine in Bacillus subtilis. J. Biol. Chem. 270, 16701–16713. doi: 10.1074/jbc.270.28.16701
Kempf, B., and Bremer, E. (1998). Uptake and synthesis of compatible solutes as microbial stress responses to high-osmolality environments. Arch. Microbiol. 170, 319–330. doi: 10.1007/s002030050649
Konstantinidis, K. T., Rosselló-Móra, R., and Amann, R. (2017). Uncultivated microbes in need of their own taxonomy. ISME J. 11, 2399–2406. doi: 10.1038/ismej.2017.113
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U. S. A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lee, I., Ouk Kim, Y., Park, S.-C., and Chun, J. (2016). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
López-Pérez, M., Ghai, R., León, M. J., Rodríguez-Olmos, A., Copa-Patiño, J. L., Soliveri, J., et al. (2013). Genomes of “Spiribacter”, a streamlined, successful halophilic bacterium. BMC Genomics 14:787. doi: 10.1186/1471-2164-14-787
Marmur, J. (1961). A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3, 208–218. doi: 10.1016/S0022-2836(61)80047-8
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Minegishi, H., Kamekura, M., Itoh, T., Echigo, A., Usami, R., and Hashimoto, T. (2010). Further refinement of the phylogeny of the Halobacteriaceae based on the full-length RNA polymerase subunit B’ (rpoB’) gene. Int. J. Syst. Evol. Microbiol. 60, 2398–2408. doi: 10.1099/ijs.0.017160-0
Mongodin, E. F., Nelson, K. E., Daugherty, S., DeBoy, R. T., Wister, J., Khouri, H., et al. (2005). The genome of Salinibacter ruber: convergence and gene exchange among hyperhalophilic bacteria and archaea. Proc. Natl. Acad. Sci. U.S.A. 102, 18147–18152. doi: 10.1073/pnas.0509073102
Oren, A. (2011). “Diversity of halophiles,” in Extremophiles Handbook, ed. K. Horikoshi (Tokyo: Springer), 309–325.
Oren, A. (2015). Halophilic microbial communities and their environments. Curr. Opin. Biotechnol. 33, 119–124. doi: 10.1016/j.copbio.2015.02.005
Oren, A., Arahal, D. R., and Ventosa, A. (2009). Emended descriptions of genera of the family Halobacteriaceae. Int. J. Syst. Evol. Microbiol. 59, 637–642. doi: 10.1099/ijs.0.008904-0
Oren, A., and Ventosa, A. (2017a). “Halobacteria,” in Bergey’s Manual of Systematics of Archaea and Bacteria, eds W. D. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. de Vos, et al. (Hoboken: John Wiley & Sons, Inc.), 1–5.
Oren, A., and Ventosa, A. (2017b). “Haloarculaceae,” in Bergey’s Manual of Systematics of Archaea and Bacteria, eds W. D. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. de Vos, et al. (Hoboken: John Wiley & Sons, Inc.), 1–5.
Oren, A., Ventosa, A., and Grant, W. D. (1997). Proposed minimal standards for description of new taxa in the order Halobacteriales. Int. J. Syst. Bacteriol. 47, 233–238. doi: 10.1099/00207713-47-1-233
Osbourn, A. (2010). Secondary metabolic gene clusters: evolutionary toolkits for chemical innovation. Trends Genet. 26, 449–457. doi: 10.1016/j.tig.2010.07.001
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Piasecka, A., Jedrzejczak-Rey, N., and Bednarek, P. (2015). Secondary metabolites in plant innate immunity: conserved function of divergent chemicals. New Phytol. 206, 948–964. doi: 10.1111/nph.13325
Pichersky, E., Noel, J. P., and Dudareva, N. (2006). Biosynthesis of plant volatiles: nature’s diversity and ingenuity. Science 311, 808–811. doi: 10.1126/science.1118510
Repka, L. M., Chekan, J. R., Nair, S. K., and Van Der Donk, W. A. (2017). Mechanistic understanding of lanthipeptide biosynthetic enzymes. Chem. Rev. 117, 5457–5520. doi: 10.1021/acs.chemrev.6b00591
Rice, P., Longden, L., and Bleasby, A. (2000). EMBOSS: the european molecular biology open software suite. Trends Genet. 16, 276–277. doi: 10.1016/S0168-9525(00)02024-2
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 4:e1900v1. doi: 10.7287/peerj.preprints.1900v1
Rodríguez-Valera, F. (1988). “Characteristics and microbial ecology of hypersaline environments,” in Halophilic Bacteria, ed. F. Rodríguez-Valera (Boca Raton: CRC Press), 3–30.
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shivanand, P., and Mugeraya, G. (2011). Halophilic bacteria and their compatible solutes -osmoregulation and potential applications. Curr. Sci. 100, 1516–1521.
Srivastava, P., and Kowshik, M. (2013). Mechanisms of metal resistance and homeostasis in haloarchaea. Archaea 2013:732864. doi: 10.1155/2013/732864
Ventosa, A. (2006). “Unusual micro-organisms from unusual habitats: hypersaline environments,” in Prokaryotic Diversity: Mechanisms and Significance, eds N. A. Logan, H. M. Lappin-Scrott, and P. C. F. Oyston (Cambridge: Cambridge University Press), 223–254.
Ventosa, A., de la Haba, R. R., Sánchez-Porro, C., and Papke, R. T. (2015). Microbial diversity of hypersaline environments: a metagenomic approach. Curr. Opin. Microbiol. 25, 80–87. doi: 10.1016/J.MIB.2015.05.002
Ventosa, A., Fernández, A. B., León, M. J., Sánchez-Porro, C., and Rodriguez-Valera, F. (2014). The Santa Pola saltern as a model for studying the microbiota of hypersaline environments. Extremophiles 18, 811–824. doi: 10.1007/s00792-014-0681-6
Ventosa, A., Mellado, E., Sánchez-Porro, C., and Márquez, M. C. (2008). “Halophilic and halotolerant micro-organisms from soils,” in Microbiology of Extreme Soils, eds P. Dion and C. S. Nautiyal (Berlin: Springer), 87–115. doi: 10.1007/978-3-540-74231-9_5
Vera-Gargallo, B., Chowdhury, T. R., Brown, J., Fansler, S. J., Durán-Viseras, A., Sánchez-Porro, C., et al. (2019). Spatial distribution of prokaryotic communities in hypersaline soils. Sci. Rep. 9:1769. doi: 10.1038/s41598-018-38339-z
Vera-Gargallo, B., and Ventosa, A. (2018). Metagenomic insights into the phylogenetic and metabolic diversity of the prokaryotic community dwelling in hypersaline soils from the Odiel Saltmarshes (SW Spain). Genes 9:152. doi: 10.3390/genes9030152
Verhees, C. H., Kengen, S. W. M., Tuininga, J. E., Schut, G. J., Adams, M. W. W., de Vos, W. M., et al. (2003). The unique features of glycolytic pathways in Archaea. Biochem. J. 375, 231–246. doi: 10.1042/bj20021472
Wang, S., and Lu, Z. (2017). “Secondary metabolites in archaea and extreme environments,” in Biocommunication of Archaea, ed. G. Witzany (Berlin: Springer), 235–239.
Wang, S., Zheng, Z., Zou, H., Li, N., and Wu, M. (2019). Characterization of the secondary metabolite biosynthetic gene clusters in archaea. Comput. Biol. Chem. 78, 165–169. doi: 10.1016/j.compbiolchem.2018.11.019
Widderich, N., Czech, L., Elling, F. J., Könneke, M., Stöveken, N., Pittelkow, M., et al. (2016). Strangers in the archaeal world: osmostress-responsive biosynthesis of ectoine and hydroxyectoine by the marine thaumarchaeon Nitrosopumilus maritimus. Environ. Microbiol. 18, 1227–1248. doi: 10.1111/1462-2920.13156
Yamada, Y., Kuzuyama, T., Komatsu, M., Shin-ya, K., Omura, S., Cane, D. E., et al. (2015). Terpene synthases are widely distributed in bacteria. Proc. Natl. Acad. Sci. U. S. A. 112, 857–862. doi: 10.1073/pnas.1422108112
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Youssef, N. H., Savage-Ashlock, K. N., McCully, A. L., Luedtke, B., Shaw, E. I., Hoff, W. D., et al. (2014). Trehalose/2-sulfotrehalose biosynthesis and glycine-betaine uptake are widely spread mechanisms for osmoadaptation in the Halobacteriales. ISME J. 8, 636–649. doi: 10.1038/ismej.2013.165
Zhang, W., and Cui, H. (2014). Halomicroarcula limicola sp. nov., isolated from a marine solar saltern, and emended description of the genus Halomicroarcula. Int. J. Syst. Evol. Microbiol. 64, 1747–1751. doi: 10.1099/ijs.0.062455-0
Zhang, W., and Cui, H. (2015). Halomicroarcula salina sp. nov., isolated from a marine solar saltern. Int. J. Syst. Evol. Microbiol. 65, 1628–1633. doi: 10.1099/ijs.0.000150
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J., and Wishart, D. S. (2011). PHAST: a fast phage search tool. Nucleic Acids Res. 39, W347–W352. doi: 10.1093/nar/gkr485
Keywords: Halomicroarcula, haloarchaea, comparative genomic analysis, compatible solutes, Halomicroarcula rubra sp. nov., Halomicroarcula nitratireducens sp. nov., Halomicroarcula salinisoli sp. nov.
Citation: Durán-Viseras A, Sánchez-Porro C and Ventosa A (2021) Genomic Insights Into New Species of the Genus Halomicroarcula Reveals Potential for New Osmoadaptative Strategies in Halophilic Archaea. Front. Microbiol. 12:751746. doi: 10.3389/fmicb.2021.751746
Received: 01 August 2021; Accepted: 11 October 2021;
Published: 04 November 2021.
Edited by:
André Antunes, Macau University of Science and Technology, ChinaCopyright © 2021 Durán-Viseras, Sánchez-Porro and Ventosa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Durán-Viseras, YW5hZHVyYW5AdXMuZXM=; Antonio Ventosa, dmVudG9zYUB1cy5lcw==