 Dandan Xiao
Dandan Xiao Ke Zhou1,2,3,4
Ke Zhou1,2,3,4 Xiaoqian Yang
Xiaoqian Yang Yuzhang Yang
Yuzhang Yang Yanwei Wang
Yanwei Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 28 December 2021
Sec. Microbe and Virus Interactions with Plants
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.750089
This article is part of the Research Topic Systematics, Genomics, and Pathogenesis of Forest Pathogens View all 8 articles
DNA methylation plays crucial roles in responses to environmental stimuli. Modification of DNA methylation during development and abiotic stress responses has been confirmed in increasing numbers of plants, mainly annual plants. However, the epigenetic regulation mechanism underlying the immune response to pathogens remains largely unknown in plants, especially trees. To investigate whether DNA methylation is involved in the response to infection process or is related to the resistance differences among poplars, we performed comprehensive whole-genome bisulfite sequencing of the infected stem of the susceptible type Populus × euramerican ‘74/76’ and resistant type Populus tomentosa ‘henan’ upon Lonsdalea populi infection. The results revealed that DNA methylation changed dynamically in poplars during the infection process with a remarkable decrease seen in the DNA methylation ratio. Intriguingly, the resistant P. tomentosa ‘henan’ had a much lower basal DNA methylation ratio than the susceptible P. × euramerican ‘74/76’. Compared to mock-inoculation, both poplar types underwent post-inoculation CHH hypomethylation; however, significant decreases in mC and mCHH proportions were found in resistant poplar. In addition, most differentially CHH-hypomethylated regions were distributed in repeat and promoter regions. Based on comparison of DNA methylation modification with the expression profiles of genes, DNA methylation occurred in resistance genes, pathogenesis-related genes, and phytohormone genes in poplars during pathogen infection. Additionally, transcript levels of genes encoding methylation-related enzymes changed during pathogen infection. Interestingly, small-regulator miRNAs were subject to DNA methylation in poplars experiencing pathogen infection. This investigation highlights the critical role of DNA methylation in the poplar immune response to pathogen infection and provides new insights into epigenetic regulation in perennial plants in response to biotic stress.
Epigenetic mechanisms, especially DNA methylation, are conserved and associated with gene silencing, chromatin remodeling, histone modification, and epigenetic variations induced by the environment (Law and Jacobsen, 2010; Shipony et al., 2014; Tirnaz and Batley, 2019). Moreover, DNA methylation occurs throughout the whole life of the plant, playing a role in adaptation to stress conditions, genome management, and developmental regulation (Sherman and Talbert, 2002; Meng et al., 2003; Feng et al., 2014; Elhamamsy, 2016). In Arabidopsis, research results support a model whereby DNA methylation imparts persistent control over some defense genes under non-stressful conditions, but can shift dynamically to modulate gene expression in response to environmental stimuli (Dowen et al., 2012). In plants, all three types of cytosine DNA methylation (CG, CHG, and CHH) are catalyzed by DOMAINS REARRANGED METHYLTRA NSFERASE2 (DRM2), but with differing maintenance pathways. Symmetrical CG and CHG DNA methylation grouping is maintained by METHYLTRANSFER ASE1 (MET1) and CHROMOMETHYLASE3 (CMT3), respectively, while asymmetric CHH methylation is maintained by DRM2 (Law and Jacobsen, 2010; Zhang et al., 2018).
DNA methylation in plants changes in response to diverse abiotic stress conditions, including heat, cold, drought, high salinity, osmotic stress, ultraviolet radiation, soil nutrient deficiency, laser irradiation, metal stress, anoxia and re-oxygenation, pesticides, and climate change. These changes have been investigated thoroughly in a wide range of plants including Arabidopsis, cotton, winter wheat, rice, Brassica rapa, Isoetes sinensis Palmer, Camellia sinensis, and poplar (Sherman and Talbert, 2002; Song et al., 2016; Liu et al., 2017; Zheng et al., 2017; Zhang et al., 2018, 2019; Ding et al., 2019; Van Dooren et al., 2020; Zhu et al., 2020). Recently, reports of DNA methylation modification in plants in response to biotic stressors have increased. Biotic stressors include the effects of bacteria, fungi, and viruses that impact the normal growth and development of plants. DNA methylation has been reported to play a crucial role in plant resistance to biotic stress (Hewezi et al., 2017). For example, the level of DNA methylation in tobacco changed in response to external tobacco mosaic virus (TMV) infection, and this change was closely related to the activation of stress-responsive genes (Wada et al., 2004). Dynamic DNA methylation had been suggested to represent a regulatory layer in the complex mechanism of plant immunity, which may be exploited to improve disease resistance in common wheat (Geng et al., 2019). Additionally, DNA demethylation was presumed to be involved in the immune activity of Arabidopsis against microbial pathogens (Pavet et al., 2006). Together, these reports demonstrate that changes in DNA methylation in response to various biotic stressors are common in plants. However, these response patterns may differ among plant species, or in response to infection with different pathogens. In grape berries, melatonin treatment increased plant disease resistance and flavonoid biosynthesis by decreasing the methylation levels (MLs) of gene promoters (Gao et al., 2020). Stable methylation was observed in Populus simonii under cold, osmotic, heat, and salt stress, based on analysis of the methylome and gene expression (Song et al., 2016). Single-base-resolution methylomes of Populus trichocarpa showed a significant increase in cytosine methylation after drought treatment (Liang et al., 2014). These investigations demonstrated that DNA methylation plays important roles in perennial plant responses to abiotic stress. However, whether stable global DNA methylation is involved in the immune response to biotic stress in poplar, a model woody perennial plant, remains unknown.
Lonsdalea canker, caused by Lonsdalea populi, was first observed in poplars of Populus × euramericana ‘74/76’ and P. × euramericana ‘Zhonglin 46’ in both Henan and Shandong provinces of China in 2006 (Li et al., 2014). Large numbers of poplars in China and Hungary had been affected by Lonsdalea canker (Li and He, 2019). Several studies of this disease have been conducted on poplars and have identified the pathogen types, molecular basis of the pathogenesis of L. populi, tolerance/resistance of different poplar species, salicylic acid and jasmonic acid signal transduction pathways, and transcriptomic analysis of poplars infected with L. populi (Li et al., 2014; Liu Z. et al., 2015; Hou et al., 2016; Yang et al., 2018). Previous research has focused primarily on the bacterium itself, or on the plant immune response or poplar transcriptomes. Poplar species differing in survivability to L. populi infection have been investigated and validated, making it possible and necessary to explore the molecular mechanism of resistant and susceptible poplars showing different defense responses to L. populi. However, few reports have addressed whether the molecular mechanism of DNA methylation plays an important role in the immune responses of various poplar species.
Given increasing evidences for the involvement of DNA methylation in plant responses to biotic stress, as well as of a role of DNA methylation in regulating gene expression and genomic stability (Tirnaz and Batley, 2019), we hypothesized that DNA methylation changed in poplars during pathogen infection and may have important effects on the expression of biotic-stress-responsive genes. Here, the role of DNA methylation in the modification between two poplar species with different resistance levels and L. populi was comprehensively investigated at the genome-wide level. We found that DNA methylation changed dynamically during the inoculation process and showed similar change trends between the susceptible and resistant poplar types investigated, albeit with different DNA MLs. Additionally, the DNA methylation changes might be involved in differential expression of resistance (R) genes, pathogenesis-related (PR) genes, and phytohormone genes in poplar species. This investigation provides a new insight into the role of DNA methylation in the immune response upon infection of trees with bacterial pathogens, which could be potentially used in endowing resistance of perennial plants to pathogen infection.
Two-year-old P. × euramericana ‘74/76’ and Populus tomentosa ‘henan’ plantlets were grown under normal field conditions in the nursery of the Puyang Academy of Forestry (114° 87′ E, 35° 81′ N) in Henan Province, China. The plantlets were inoculated with L. populi strain N-5-1 (provided by Laboratory of Forest Pathology, Beijing Forestry University) or sterile water as a mock inoculation, as described previously (Hou et al., 2016).
Bark, including cambium, of the inoculated and mock inoculated poplar stems was sampled at 6 days post-inoculation (dpi) for both poplar types, which differed in resistance levels, and P. × euramericana trees were further sampled at 2 and 4 dpi for exploration of the dynamic response to infection of L. populi. Three biological replicates were conducted for each condition. Samples were immediately frozen in liquid nitrogen and stored at −80°C.
Genomic DNA was extracted using a combination of cetyltrimethylammonium bromide (CTAB) and sodium dodecyl sulfate (SDS; Zhao et al., 2016). DNA purity and concentration were checked using the Nano Photometer® spectrophotometer (Implen, Westlake Village, CA, United States). Genomic DNA degradation and contamination were monitored using 1% agarose gels.
The genomic and lambda DNA were fragmented via sonication to 200–300 bp with a Covaris S220 sonicator, followed by end repair and adenylation. Cytosine-methylated barcodes were ligated to the sonicated DNA. Then, these DNA fragments were treated with bisulfite using Accel-NGS® Methyl-Seq DNA Library Kit in accordance with the manufacturer’s instructions. Library concentration was quantified with a Qubit® 2.0 flurometer (Life Technologies, Carlsbad, CA, United States) and quantitative PCR, and the insert size was assessed with the Bioanalyzer 2100 system (Agilent, Santa Clara, CA, United States). The libraries were sequenced on a NovaSeq6000 sequencer (Novogene Co., Ltd., Beijing, China).
Bismark software (v.0.16.3) (Krueger and Andrews, 2011) was used for alignment of bisulfite-treated reads to poplar genome v3.01 with parameters set as –score_min L, 0, -0.2, -X 700 –dovetail. The reference genome was first transformed into a bisulfite-converted version (C-to-T and G-to-A) and then indexed using bowtie2 (v.2.2.5) (Langmead and Salzberg, 2012).
Sequence reads were transformed into fully bisulfite-converted versions before alignment to similarly converted versions of the genome in a directional manner. Sequence reads that produced a unique best hit from the two alignment processes (original top and bottom strand) were compared to the normal genomic sequence, and the methylation state of all cytosine positions in the read was inferred. Reads aligned to the same regions of the genome were regarded as duplicates. The sequencing depth and coverage were summarized using deduplicated reads.
To identify methylation sites, we summed the methylated cytokines (mC) as a binomial (Bin) random variable with a methylation rate (r), which was calculated as follows: mC ∼ Bln (mC + umC*r). The sum of methylated and unmethylated read counts in each window was calculated. Bisulphite conversion efficiency was calculated based on lambda DNA genome. The reliability of methylation site level data was evaluated with a binomial distribution test, and thresholds were set to precisely determine the methylation sites, as follows: the sequencing depth ≥5; Q-value ≤ 0.01; and conversion rate ≥99%. The ML for each window or C site indicated the fraction of methylated Cs, and was defined as: ML (C) = reads (mC)/reads (mC + umC). This was corrected based on the bisulfite non-conversion rate reported previously (Lister et al., 2013). The corrected ML was then estimated as follows: ML(correlated) = (ML – r)/(1 – r).
Differentially methylated regions (DMRs) were identified using DSS software (v.2.12.0) (Wu et al., 2015; Park and Wu, 2016) with the parameters including smoothing.span = 200, delta = 0, p.threshold = 1e-05, minlen = 50, minCG = 3, dis.merge = 100, pct.sig = 0.5. Based on the distribution of DMRs throughout the genome, genes were defined as DMR-related, if the genebody region (from the transcription start site, TSS, to the transcription end site, TES) or promoter region (2 kb upstream from the TSS) had overlaps with DMRs. GraphPad Prism software (ver. 8.0.1; GraphPad Software Inc., La Jolla, CA, United States) was used for the statistical analysis.
Gene Ontology (GO)2 enrichment analysis of genes related to DMRs was conducted using the GOseq R package to correct for gene length bias (Young et al., 2010). GO terms with corrected P-values < 0.05 were considered significantly enriched. Kyoto Encyclopedia of Genes and Genomes (KEGG)3 pathway enrichment analysis was conducted based on P-values < 0.05. KOBAS (v.3.0) software was used to test for enrichment of DMR-related genes in KEGG pathways (Mao et al., 2005; Kanehisa et al., 2008).
Joint analysis was conducted between DNA methylation and gene expression, comparing resistant and susceptible poplar samples infected with the pathogen (6 dpi) to samples subjected to mock inoculation. The types of genes encoding methylation-related enzymes were analyzed based on previous research (Song et al., 2016). The DNA MLs of R, PR, and phytohormone genes were identified, with overlapping differentially expressed genes (DEGs) and DMRs considered to be associated with each other.
miRNAs corresponding to target gene were predicted using psRNATarget4 and screened according to the following conditions: the gene appeared in combined analysis of DMRs and DEGs, and exhibited CHH hypomethylation in 5′ flanking region.
Lonsdalea populi strain N-5-1, a Gram-negative, rod-shaped bacterium, causes a lethal disease known as bark canker in some poplars. Based on our previous reports, 6 dpi was selected as the time point for investigating lesions (Li et al., 2014; Hou et al., 2016). In this investigation, mock-inoculated and inoculated susceptible P. × euramericana and resistant P. tomentosa poplars were observed at 6 dpi. In P. × euramericana, the bark had a canker with white, odorous, and exudates dripping from the inoculation point and expanding lesions relative to mock-inoculation group (Supplementary Figures 1A,B,E,F). These symptoms were typical of L. populi infection (Li et al., 2014) but did not appear in the resistant P. tomentosa (Supplementary Figures 1C,D,G,H).
To investigate whether DNA methylation is involved in the poplar response to L. populi infection, whole-genome bisulfite sequencing (WGBS) of poplar bark from the mock-inoculation and inoculation groups was performed. In total, 19–26 million unique mapping clean reads on average were obtained for further analysis (Supplementary Table 1A). The bisulfite conversation rates of all samples were over 99% (Supplementary Table 1B). After methylation site calling, 3,911,428 and 3,702,079 mC on average were identified in P. tomentosa libraries by mock inoculation and at 6 dpi, accounting for 2.74 and 2.59% of all reads, respectively. In total, 7,896,041, 8,707,359, 6,821,148, and 6,332,367 mC on average were identified in P. × euramericana libraries by mock-inoculation, and at 2, 4, and 6 dpi, respectively, accounting for 4.43–6.09% of all reads (Supplementary Table 1C). To ensure the repeatability of the analysis, the correlations of MLs between samples were determined through Pearson correlation analysis. After inoculation, DNA methylation similarity to the control was higher for CG and CHG than CHH, in both poplar types. In addition, the R2 values of all samples were greater than 0.90 (Supplementary Figure 2).
Four successive disease development stages have been described for L. populi infection: the contact, penetration, incubation, and symptom appearance periods (Liu M. et al., 2015). The incubation period lasted approximately 3 days after the bacteria were inoculated onto the poplar stems (Shang et al., 2014). To explore the changes in DNA methylation among periods of L. populi infection, detection of DNA methylation variations in susceptible P. × euramericana was performed. After inoculation with L. populi, the methylation ratio of total cytosine was 6.09, 4.77, and 4.73% at 2, 4, and 6 dpi in P. × euramericana, respectively. Compared to the cytosine methylation ratio of the mock-inoculated group (5.53%), DNA methylation increased slightly at 2 dpi, while continuously decreased from 2 to 6 dpi. The result demonstrated that the proportion of total cytosine methylation differed among infection periods, indicating that gene expression in poplars was modulated in response to L. populi infection. This finding was consistent with previous reports that DNA methylation regulated plant gene expression in response to pathogen infection (Hewezi et al., 2017). The CG, CHG, and CHH methylation ratios increased slightly at 2 dpi and then decreased continuously, similar to the change trend of total cytosines and previous reports that DNA hypomethylation affected plant defense against nematodes (Figure 1; Atighi et al., 2020). In addition, the variation of CHH methylation was similar to that of total cytosines, with a sharp decline from 2 to 4 dpi and gradual decrease from 4 to 6 dpi. Meanwhile, the variation of CG and CHG methylation gradually decreased from 2 to 6 dpi. More importantly, compared to the mock-inoculation group, the decrease in methylation ratio was greatest for CHH. Overall, the level of DNA methylation initially increased, and then decreased, during the infection process in P. × euramericana.
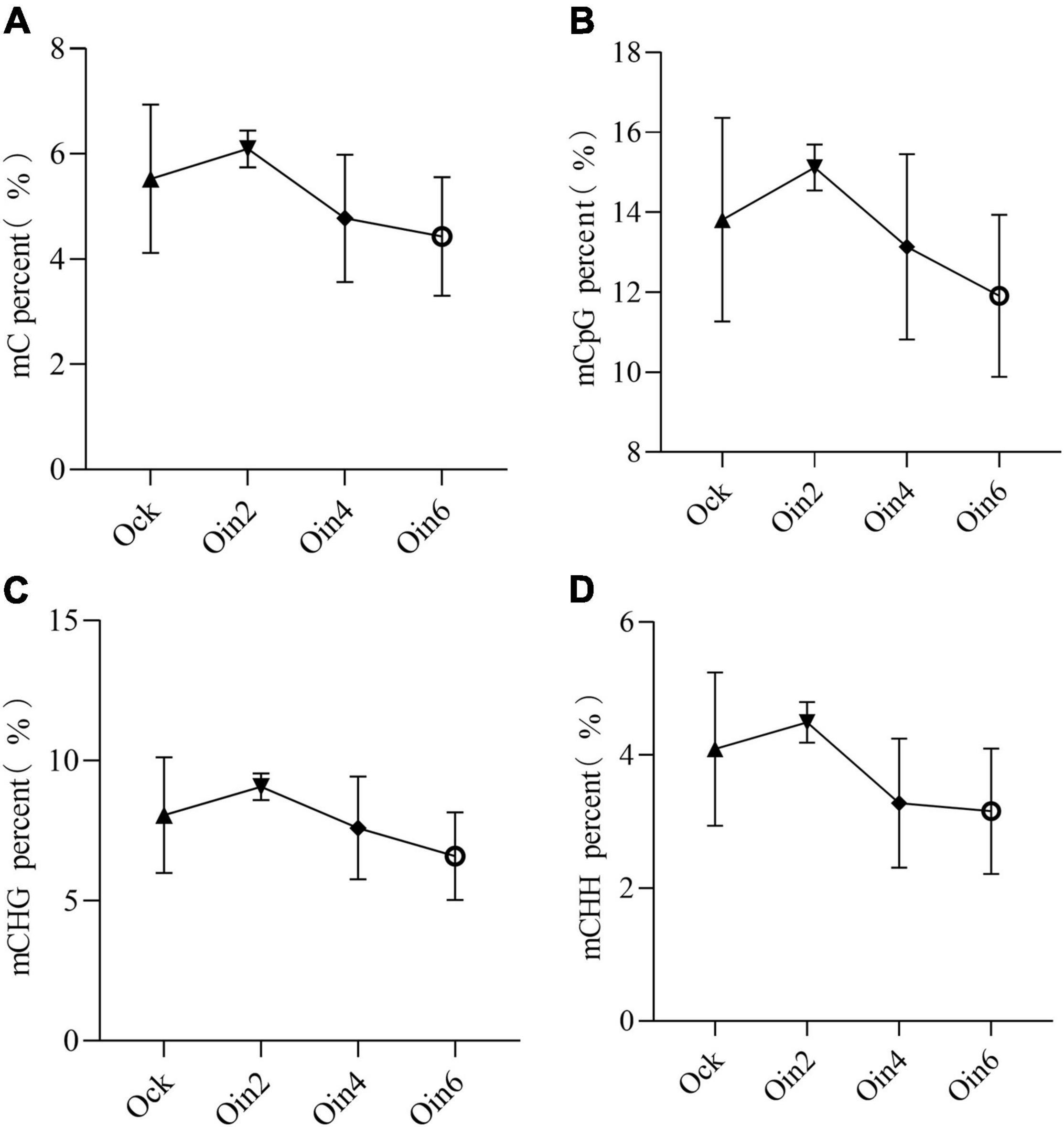
Figure 1. DNA methylation dynamic change tendency of Populus × euramericana “74/76” in response to Lonsdalea populi infection at different contexts. The mC percent (A), mCpG percent (B), mCHG percent (C), and mCHH percent (D) of P. × euramericana in response to L. populi infection at different infection time points. The data were plotted as the mean ± SEM. Regular triangle, inverted triangle, diamond, and circle ring indicate the mean value of different infection time points. Oin/Ock means inoculation/mock-inoculation in P. × euramericana, and the Arabic numerals indicate the specific inoculation days.
To elucidate how CHH methylation changed with the extension of infection course, we further analyzed CHH ML in genebody regions in P. × euramericana. Through pairwise analysis (Oin2_vs_Ock, Oin4_vs_Oin2 and Oin6_vs_Oin4), CHH methylation was compared among the different stages of disease development. DNA methylation region occurred mainly in the repeat region followed by the promoter region. Methylation was low in the intron, exon, 5′ untranslated region (UTR), and 3′ UTR (Figures 2A–C). This pattern was seen when comparing all infection conditions against the mock treatment (Figure 2A and Supplementary Figures 3A,B). Although the level of CHH methylation was lowest at 4 dpi, it still occurred mainly in the repeat and promoter regions (Figures 2B,C). Compared to mock inoculation, hyper and hypo CHH DMR genes had a roughly equal number at 2 dpi (Figure 2D). Interestingly, a large number of DMR genes exhibited CHH hypomethylation from 2 to 4 dpi (Figure 2E). In contrast, numerous DMR genes exhibited CHH hypermethylation from 4 to 6 dpi (Figure 2F). Relative to the control, the difference between hypo- and hypermethylated CHH DMR genes was most apparent at 4 dpi (Figures 2D, 5B, and Supplementary Figure 3C). Together, these results indicated that DNA methylation was lowest at 4 dpi, and that CHH DMRs were mainly in repeat and promoter regions in P. × euramericana. Additionally, we randomly chosen 200 random regions (RRs) from each of 19 chromosomes of poplar with each RR 150 bp in length to explore DMRs in RRs. The RRs were defined and chosen as candidates if they had overlapping hits with identified DMRs. Based on these 3,800 RRs results (Supplementary Table 2A), the number of DMRs for expectation in different categories of Figures 2D–F was further in silico predicted. It showed that the number of DMRs for observation differed from that for expectation identified based on the RRs in genome, especially DMRs in repeat regions were more, compared to the expectation values (Supplementary Table 2B).
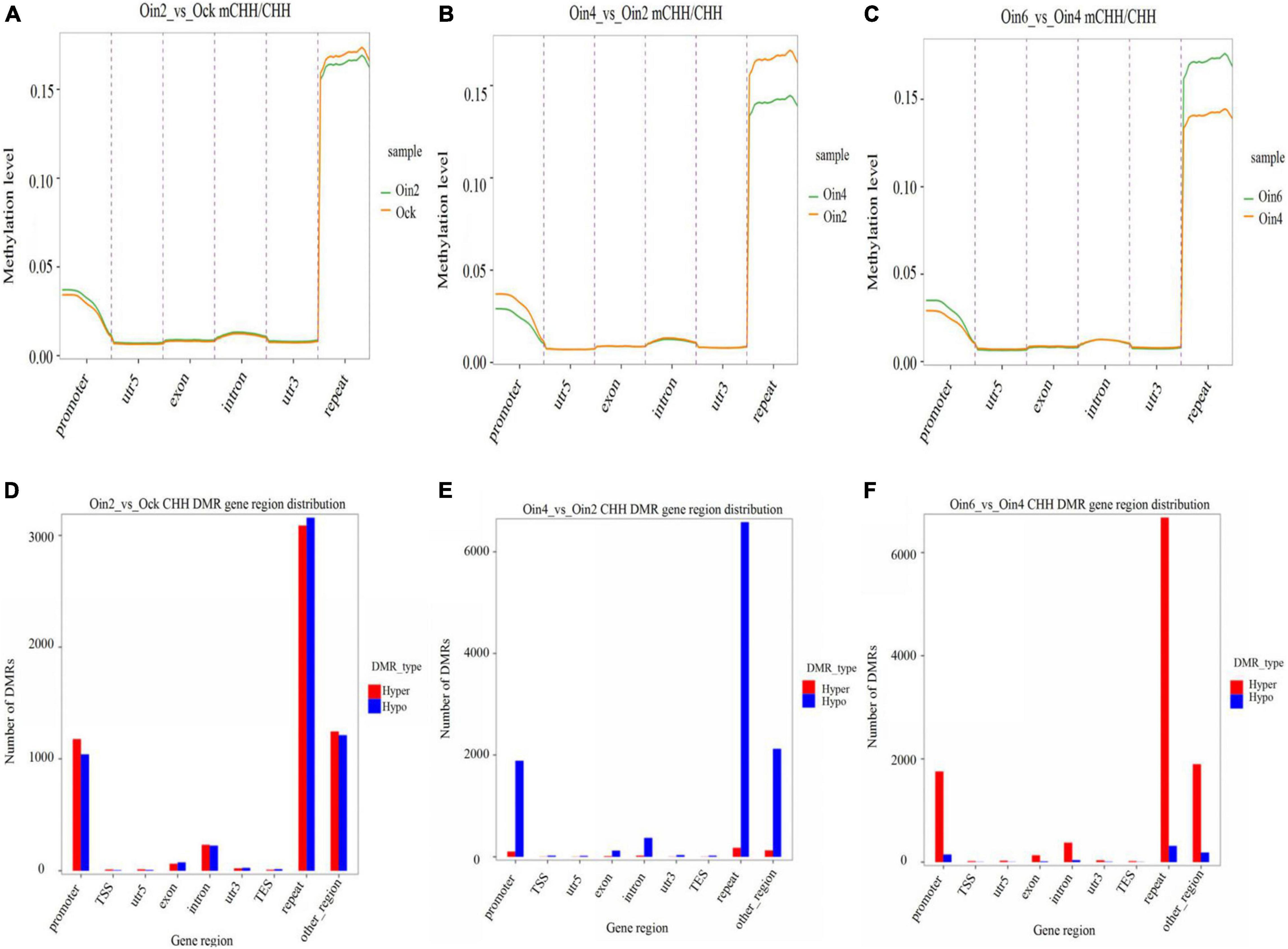
Figure 2. Changes in CHH methylation of Populus × euramericana ‘74/76’. Difference in CHH methylation level of P. × euramericana 2 dpi (Oin2) compared with mock-inoculation (Ock) (A), 4 dpi (Oin4) compared with 2 dpi (B), and 6 dpi (Oin6) compared with 4 dpi (C); distribution of differentially methylated genes in P. × euramericana 2 dpi compared with mock-inoculation (D), 4 dpi compared with 2 dpi (E), and 6 dpi compared with 4 dpi (F).
Furthermore, CHH DMRs were annotated within the genebody and promoter regions to explore conserved and specific DMRs annotated genes of P. × euramericana under different conditions. A total of 70 conserved DMR-annotated genes were found in the genebody region during different infection periods, while 153 in gene promoter region (Oin2_vs_Ock, Oin4_vs_Ock, and Oin6_vs_Ock, Supplementary Figures 4A,C). Additionally, 334, 186, and 243 specific genes were found in the genebody region in pairwise comparisons (Oin2_vs_Ock, Oin4_vs_Oin2, and Oin6_vs_Oin4), respectively (Supplementary Figure 4B). There were 1,598, 981, and 1,059 specific genes in gene promoter region in Oin2_vs_Ock, Oin4_vs_Oin2, and Oin6_vs_Oin4 comparisons, respectively (Supplementary Figure 4D). Interestingly, conserved and specific DMR-annotated genes were more abundant in gene promoter region than that in genebody region, which was similar to our finding of more CHH DMRs in gene promoter region than that in genebody region.
To further explore the role of DNA methylation involved in the different resistances of poplars, susceptible P. × euramericana and resistant P. tomentosa were inoculated with L. populi. For mock-inoculation, the DNA ML of resistant type was much lower than that of susceptible type, suggesting the basal DNA methylation difference might be related to differences in the immune response. Interestingly, a sharp decrease in methylation occurred in P. tomentosa infected with L. populi, and significant decreases were observed in the percentages of both mC and mCHH (Figure 3), but not in mCHG. DNA methylation also decreased, although not significantly, for mCHH, mCHG, and mCG in P. × euramericana at 6 dpi (Figure 3). DNA methylation percentages were lower for the resistant than susceptible type in all three sequences, and with both mock inoculation and inoculation. The result showed that DNA methylation decreased significantly in P. tomentosa infected with L. populi, but not in P. × euramericana. CHH methylation plays an important role in disease resistance against pathogens (Geng et al., 2019). To further identify the methylation characteristics of various sequences in the genebody, the percent of mCpG, mCHG, and mCHH were analyzed. The decreasing trend of CHH methylation was consistent with the general trend of DNA hypomethylation (Figure 3), suggesting that CHH methylation was an important component of DNA methylation in the poplar response to pathogen infection, as observed in other plants.
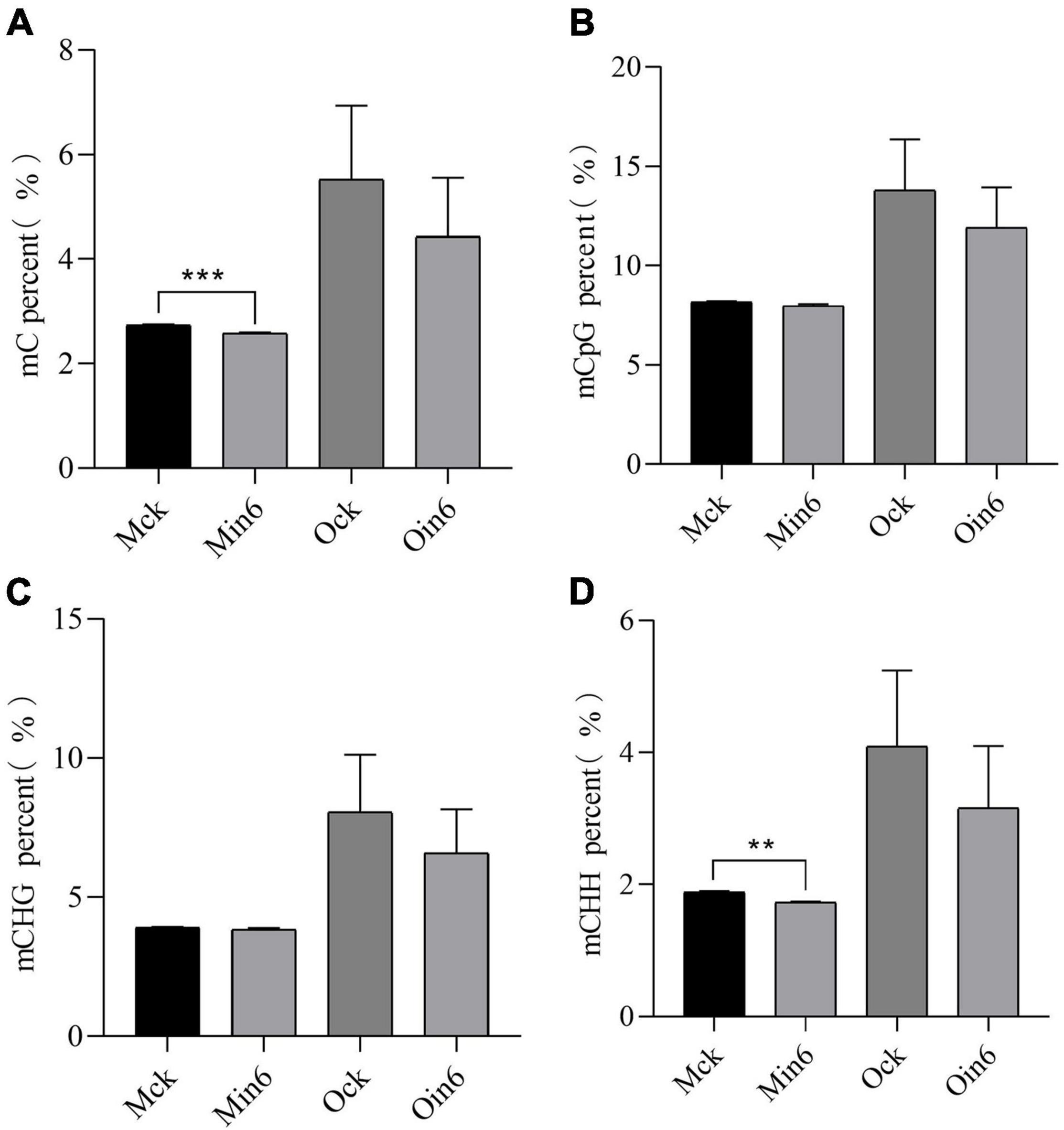
Figure 3. Lonsdalea populi infection induced methylation decline in different poplars. The mC percent (A), mCpG percent (B), mCHG percent (C), and mCHH percent (D) of P. tomentosa and P. × euramericana before and after pathogen infection. The data are plotted as mean ± SEM. For statistical analysis of differentially expressed genes, unpaired t-test was performed. ***P < 0.001, **P < 0.01. Min/Oin means inoculation of P. tomentosa or P. × euramericana, Mck/Ock means mock-inoculation of P. tomentosa or P. × euramericana, and the Arabic numerals indicated the specific inoculation days.
To identify DMRs, we analyzed differential DNA hypomethylation and hypermethylation regions in CG, CHG, and CHH in two poplar types at 6 dpi. The numbers of differential hypomethylation regions of CG, CHG, and CHH in P. tomentosa were 1,183, 1,243, and 5,899, respectively, while the differential hypomethylation regions in P. × euramericana numbered 284, 653, and 5,594, respectively. Meanwhile, the numbers of differential hypermethylation regions of CG, CHG, and CHH in P. tomentosa were 921, 1,211 and 1,829, respectively, and the corresponding counts in P. × euramericana were 227, 460, and 3,700. Although CG and CHG hypomethylation occurred at nearly equal levels, regions of CHH hypomethylation were much more common in both poplars than regions of CHH hypermethylation. Interestingly, the number of differential CHH hypermethylation regions in P. × euramericana was approximately double that in P. tomentosa (Figures 4A,B). These data demonstrated that the infection suppressed the methylation of CHH and suggested that CHH hypomethylation could play a role in genetic regulation in plants, including poplar in response to pathogen infection.
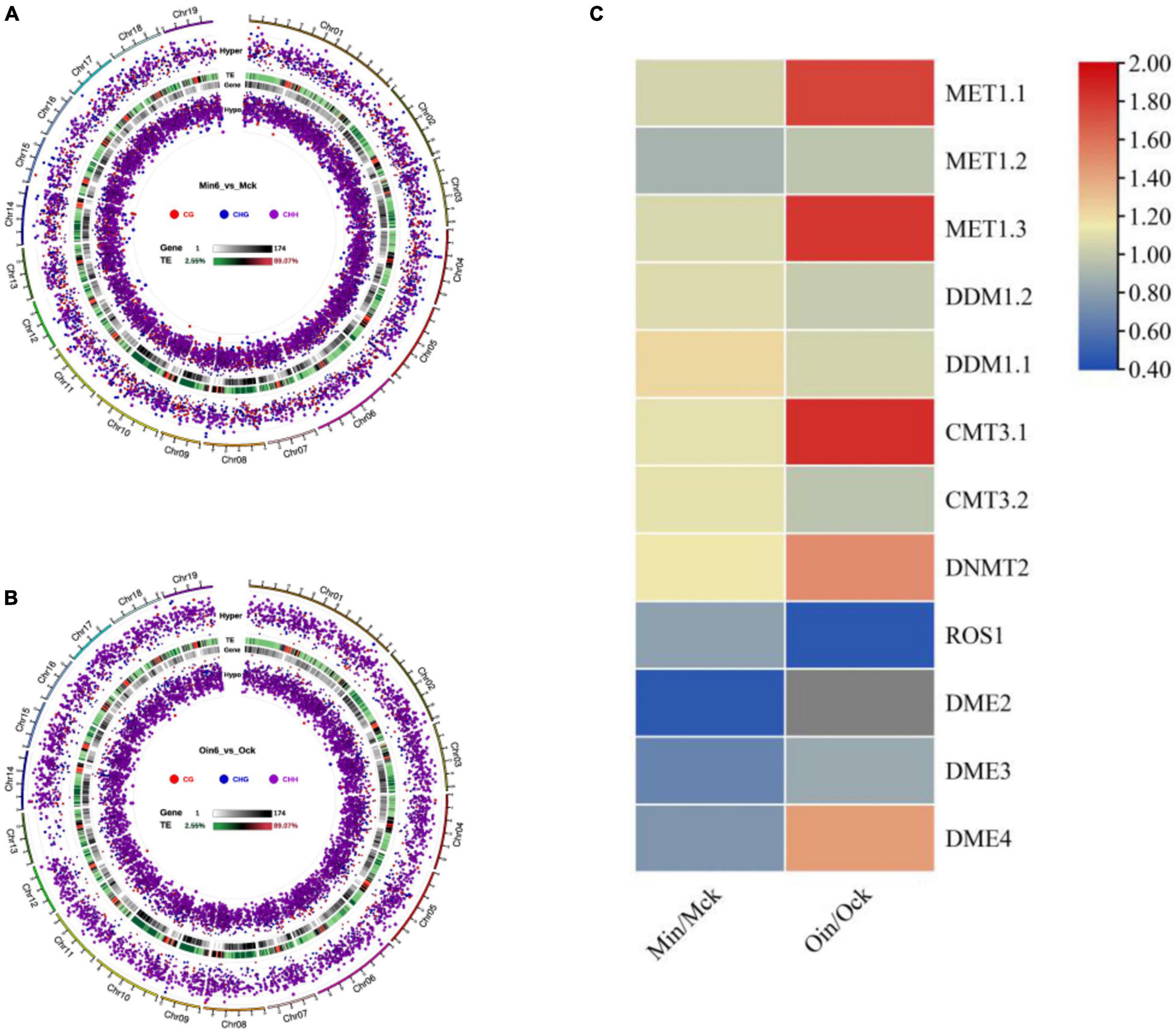
Figure 4. Distribution and significance of differential methylation regions (DMRs) in genome in three sequence contexts (CG/CHG/CHH) of two poplar types by Circos map. Display of three sequence contexts DMRs overall Circos of P. tomentosa (Min6_vs_Mck) (A), P. × euramericana (Oin6_vs_Ock) (B). Heatmap of hierarchical clustering for expression of DNA methylation- and demethylation-related genes in poplar under Lonsdalea populi infection (C). In panels (A,B), from the outside to the inside means: (a) Chromosomes. (b) Hyper differentially methylated regions (DMRs) statistical value log5 (| area stat|); the higher the outward dot, the more significant the position difference. (c) TE, the proportion of repeat elements. (d) Gene density. (e) Hypo DMR statistical value log5 (| area stat|); the higher the inward dot, the more significant the position difference.
To further examine the genetic changes that occurred in poplar during L. populi infection, the expression of genes encoding methylation-related enzymes was analyzed. In most plants, four classes of DNA methyltransferases participate in the establishment and maintenance of DNA methylation, and two classes of DNA glycosylases are responsible for demethylation. In this investigation, we analyzed the expression of 14 DNA methyltransferase- and glycosylase-related genes in resistant and susceptible poplars at 6 dpi under L. populi infection (Supplementary Table 3). The transcript levels of methylation-related genes increased, including METHYLTRANSFERASE 1.1 (MET1.1) and DECREASE IN DNA METHYLATION (DDM1), excepting that expression of MET1.2 slightly decreased. Demethylation-related genes including REPRESSOR OF SILENCING 1 (ROS1) and DEMETER (DME) were repressed in both poplars, although the expression of DME4 increased in P. × euramericana. Intriguingly, in P. tomentosa, genes related to the establishment and maintenance of DNA methylation were induced, including CMT3.1, CMT3.2, MET1.1, and DNMT2; the corresponding genes induced in P. × euramericana were CMT3.1, MET1.1, DNMT2, and DME4. Therefore, L. populi infection induced different patterns of methylation-related enzyme gene activation in P. × euramericana and P. tomentosa (Figure 4C). Differential expression of genes encoding methylation-related enzymes supported the occurrence of DNA methylation changes in poplar upon pathogen infection, which gave an impetus for poplar response to biotic stress.
In this investigation, GO annotation was used to classify the functions of CHH DMR genes. Based on WGBS data, genes showing DNA methylation were classified into three categories, “molecular function,” “biological process,” and “cellular component” (Supplementary Figure 5). In P. tomentosa (Min6_vs_Mck), 78 DMR genes were significantly enriched in the “biological process” category (Supplementary Table 4A). However, only 21 DMR genes in P. × euramericana (Oin6_vs_Ock) were significantly classified into “molecular function” group (Supplementary Table 4B), and no DMR genes were significantly enriched in “biological process.” These GO significant enrichment results indicate differing responses between resistant and susceptible poplar types.
To further explore the biological functions of poplar genes involved in the response to L. populi infection, the CHH DMRs were mapped to reference pathways in the KEGG database. In P. tomentosa (Min6_vs_Mck), 275 CHH DMRs were assigned to 79 pathways, including four genes significantly enriched in monoterpenoid biosynthesis pathway (Supplementary Table 5A). In P. × euramericana (Oin6_vs_Ock), 205 CHH DMRs were enriched in 71 pathways, including six genes significantly enriched in alpha-linolenic acid metabolism and 31 significantly enriched in biosynthesis of secondary metabolites (Supplementary Table 5B). Taken together, the significant enrichment of CHH DMR genes in monoterpenoid biosynthesis pathway supported the occurrence of systemic acquired resistance (SAR) in plants, suggesting that the resistant type might ward off pathogens via CHH DNA methylation modification of monoterpenoid biosynthesis genes (Riedlmeier et al., 2017).
Cytosine methylation mostly resided in repetitive sequences in plants, mammals, and fungi (Goll and Bestor, 2005). To further analyze the methylation distribution among regions of gene bodies, we annotated DNA methylation differences in diverse parts of genes. Considering genotype-specific differences in methylation, CHH DMR gene region distribution of mock-inoculated samples was compared in two types of poplars (Ock vs. Mck) (Supplementary Figure 6). In the repeat region, 1,832 hypo DMRs and 1,959 hyper DMRs were found. The promoter region contained 1,372 hypo DMRs and 993 hyper DMRs. Together, these results showed no apparent difference in repeat or promoter region between resistant and susceptible poplars. We found that the most abundant CHH hypomethylation region in P. tomentosa (Min6 vs. Mck) was the repeat region (3,905 DMRs), followed by promoter region (1,208 DMRs). The same pattern was observed in P. × euramericana (Oin6 vs. Ock), with 3,567 hypo DMRs in repeat region and 1,252 in promoter region (Figure 5). In contrast, CHH methylation in exon, intron, or other regions in both poplars was much lower than that of repeat or promoter regions. These data demonstrated that a large number of promoter and repeat gene regions were CHH hypomethylated in both poplars, implying that CHH hypomethylation might be involved in poplar response to pathogen. Additionally, 1,133 DMRs were found in the repeat region, and 302 in the promoter region, in the analysis of CHH hypermethylation in P. tomentosa. Meanwhile, CHH hypermethylation in the P. × euramericana repeat and promoter regions included 2,376 and 830 DMRs, respectively. Nevertheless, the level of CHH hypermethylation in the promoter and repeat regions of P. × euramericana was approximately double that of P. tomentosa, further suggesting that CHH hypermethylation might be related to the difference in resistance between these poplars other than CHH hypomethylation.
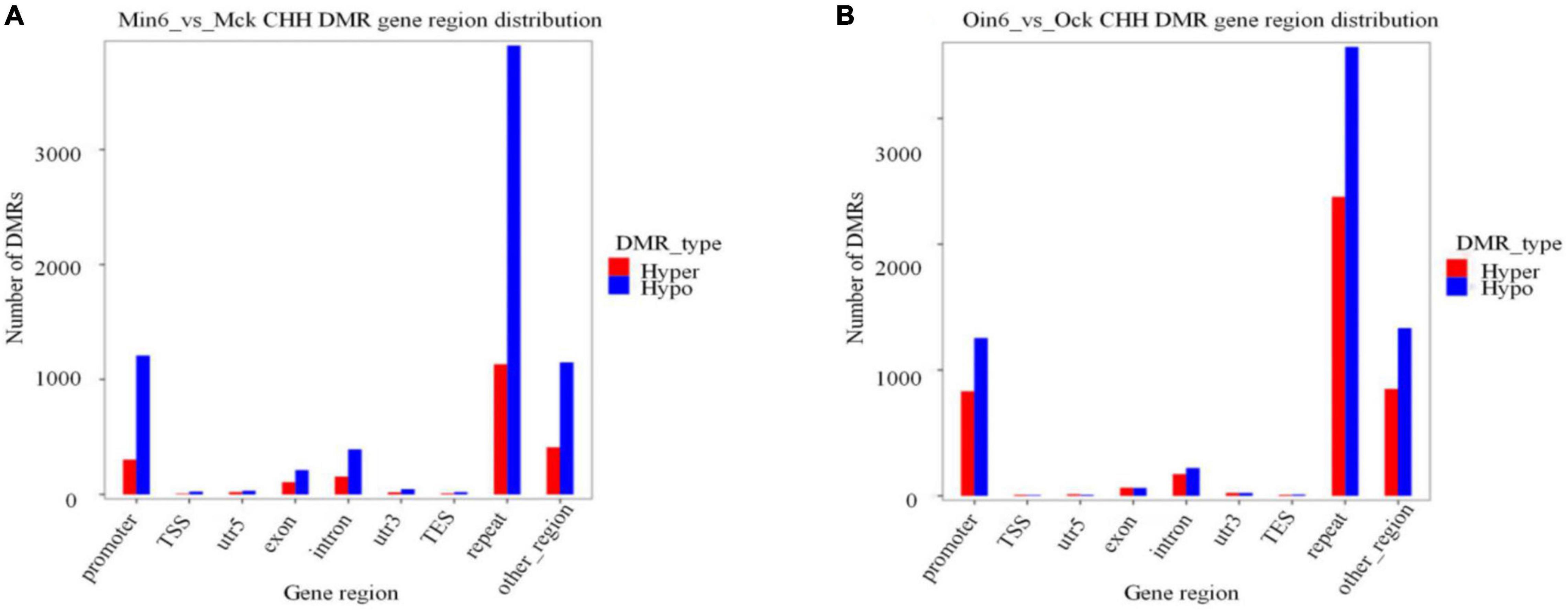
Figure 5. Number of CHH DMRs distributed in gene regions. Distribution of differentially methylated genes in P. tomentosa (A) and P. × euramericana (B) at 6 dpi compared with mock-inoculation. The abscissa represents the category of each area, and the ordinate represents the number of DMRs of Hyper/Hypo DMR in each region.
DNA methylation plays an important role in regulating the expression of genes (Wang et al., 2015; Gao et al., 2020). To assess whether DNA methylation is associated with gene activity, the correlations between DNA methylation and gene expression were analyzed. The relationship between the ML of DMR regions and the levels of associated DEGs was visualized as a scatter diagram (Supplementary Figures 7A,B). Intergroup comparison showed more genes of P. × euramericana in each quadrant than that of P. tomentosa. Furthermore, most of the interconnected genes showed hypomethylation compared to the control, in both poplars. To explore the CHH methylation distribution in more depth, we analyzed promoter DNA hypomethylation. The relationship between methylation and DMR-related gene expression was illustrated using scatter and box plots (Supplementary Figures 7C,D). In resistant type poplar, CHH hypomethylation level in the promoter region was negatively correlated with the expression level of DMR-related genes (Min6 vs. Mck). This correlation was relatively weak in the susceptible poplar.
Although promoter DNA methylation promoted gene transcription in some cases, it usually negatively regulated gene transcription (Zemach et al., 2010; Wang et al., 2015; Zhang et al., 2018). Promoter methylation has a negative correlation with gene expression levels (Xin et al., 2021). Therefore, we conducted combined analysis of DNA CHH hypo-DMRs in the promoter region (Min6 vs. Mck) and upregulated DEG transcriptome profiles in poplars generated under the same inoculation conditions. Five genes overlapping between the presence of DMRs and DEGs were found in P. tomentosa (Table 1). Through analysis of those five genes using the KEGG database, we found that two genes were enriched in the plant–pathogen interaction pathway (Supplementary Figure 8) and Vitamin B6 (VitB6) metabolism pathway (Supplementary Figure 9), respectively (Figure 6A). Functional annotations of Potri.007G127000 and Potri.001G182100 were obtained from Phytozome, indicating that these genes encode CALCIUM-DEPENDENT PROTEIN KINASE 24 (CDPK24) and pyridoxin biosynthesis PDX1-like protein 2, respectively. The correlation between DMRs and DEGs indicated that CDPK24, a protein kinase gene regulated by calcium ions in the plant pathogen interaction pathway, and PDX1.2, a VitB6 metabolism related gene, might enhance plant disease resistance.

Table 1. Transcriptional up-regulated expression genes with downregulated CHH methylation in the promoter region of Populus tomentosa ‘henan’.
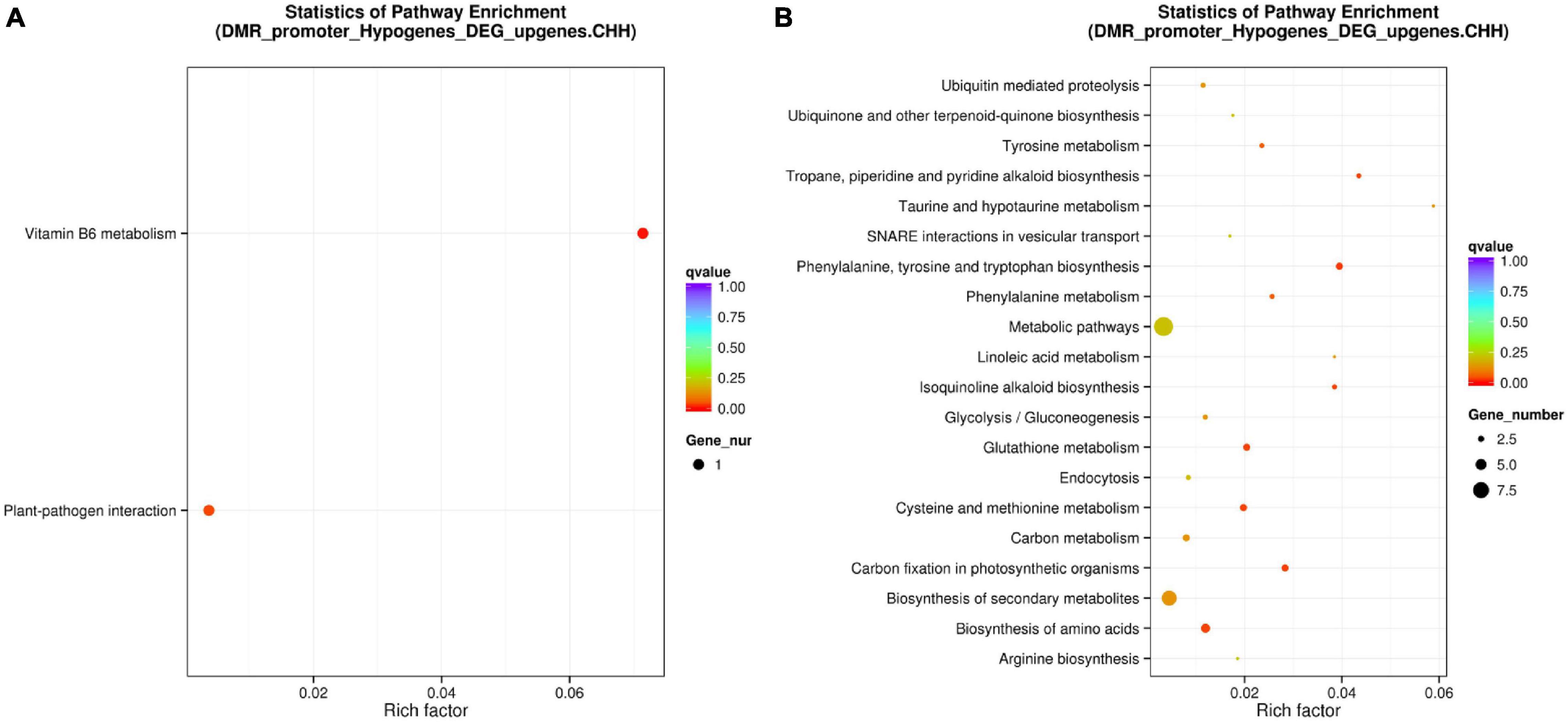
Figure 6. Combined analysis of promoter region CHH hypomethylation and differential gene expression of Populus tomentosa ‘henan’ and Populus × euramericana ‘74/76’. Scatter plot of enriched KEGG metabolic pathway of the intersection of DNA CHH hypo-DMRs for Min6 vs. Mck (A) and Oin6 vs. Ock (B) in promoter region and up regulated DEGs. For panels (A,B), the vertical axis represented the pathway name, the horizontal axis represented the richness factor, the size of the dot represented the number of DMR-related genes in this pathway, and the color of the dot corresponded to different Q value ranges.
In P. × euramericana (Oin6 vs. Ock), we found that 93 genes with DNA CHH hypo-DMRs in the promoter region were upregulated. Through comparison of these 93 genes against the KEGG enrichment database (Supplementary Table 6), we found that the two most abundant pathways were biosynthesis of secondary metabolites and metabolic pathways (Figure 6B). These genes and pathways in P. × euramericana differed markedly from those identified in P. tomentosa, which might be due to the abnormal metabolism of P. × euramericana under the influence of pathogen infection at 6 dpi (Figure 6).
Phytohormone genes, R genes, and PR genes are critical to plant defenses against pathogen infections (Derksen et al., 2013; Verma et al., 2016; Wang et al., 2018; Zou et al., 2018; Yang et al., 2021). To explore the DNA methylation characteristics of these genes, combined analysis of DMRs and DEGs was performed. In this analysis, 15 phytohormone-related DEGs, including one R gene in P. tomentosa, were found to be associated with differential DNA methylation, and the majority of these DEGs exhibited DNA hypomethylation, suggesting that they contributed to poplar defense against the pathogen infection (Figure 7A). In P. × euramericana, 214 phytohormone-related DEGs (Supplementary Table 7) including one R and two PR genes (Figure 7B) were involved in poplar response to biotic stress. Of these DEGs, 121 exhibited DNA hypomethylation and upregulated expression, while the others showed DNA hypermethylation and downregulated expression. These results implied that differentially expressed R, PR, and phytohormone genes might be modified by DNA methylation.
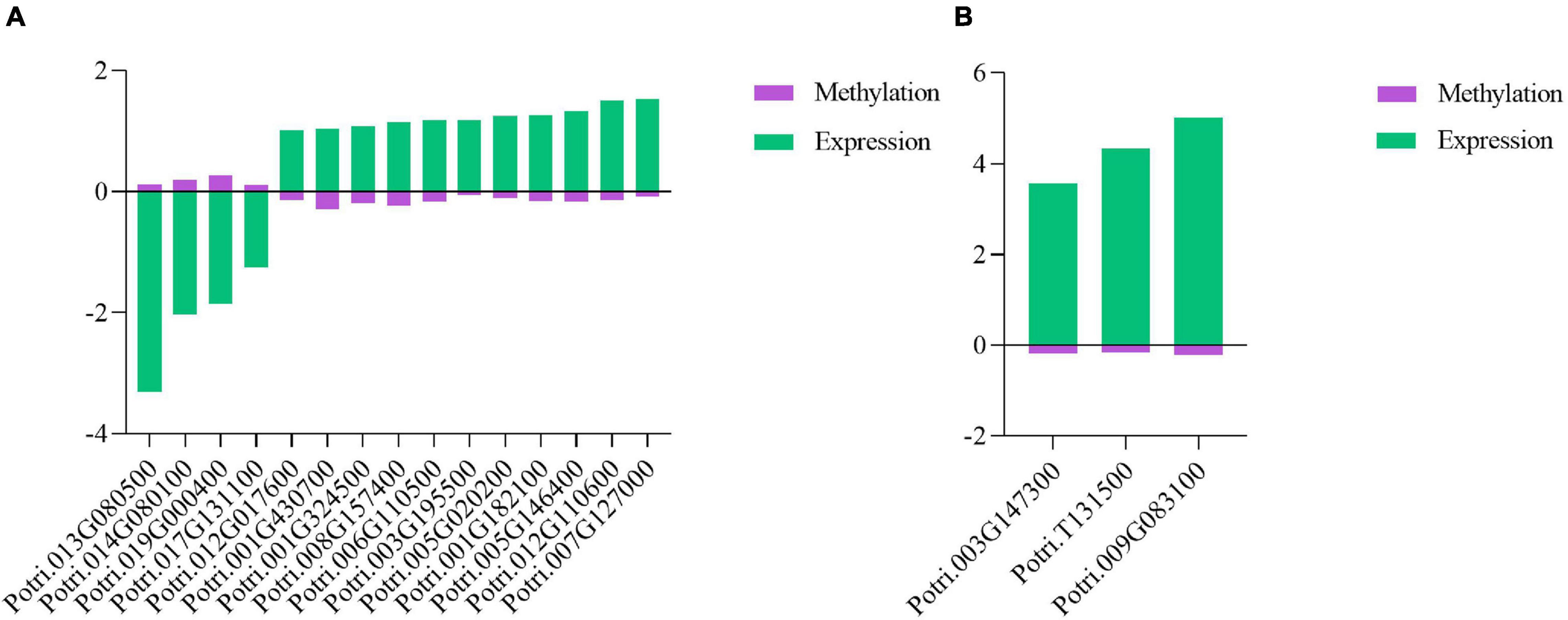
Figure 7. Co-relation between differentially expressed genes and differentially methylated regions in R/PR genes and phytohormone genes in poplar under pathogen infection. Co-relation between phytohormone DEGs and DMRs in P. tomentosa (A). Co-relation between R/PR DEGs and DMRs in P. × euramericana (B).
Considering the important roles of miRNAs in biotic stress responses, the 5′ flanking region, genebody, and 3′ flanking region of miRNA were analyzed to identify whether there were regulation of DNA methylation on miRNAs in poplar. We analyzed miRNA methylation in different regions under CG, CHG, and CHH contexts in both P. tomentosa (Mck, Min6) and P. × euramericana (Ock, Oin6). Intriguingly, the results showed that the pathogen infection induced miRNA hypomethylation of all regions in P. × euramericana. However, apparent miRNA CHH hypomethylation was detected only in the 5′ flanking regions in P. tomentosa, and no changes were apparent in other regions under different contexts (Figure 8). Overall, pathogen infection reduced the number of CHH-methylated miRNAs in the 5′ flanking region of P. tomentosa, and the number of methylated miRNAs in P. × euramericana, indicating that miRNA methylation might be involved in regulating the response to L. populi infection in poplars.
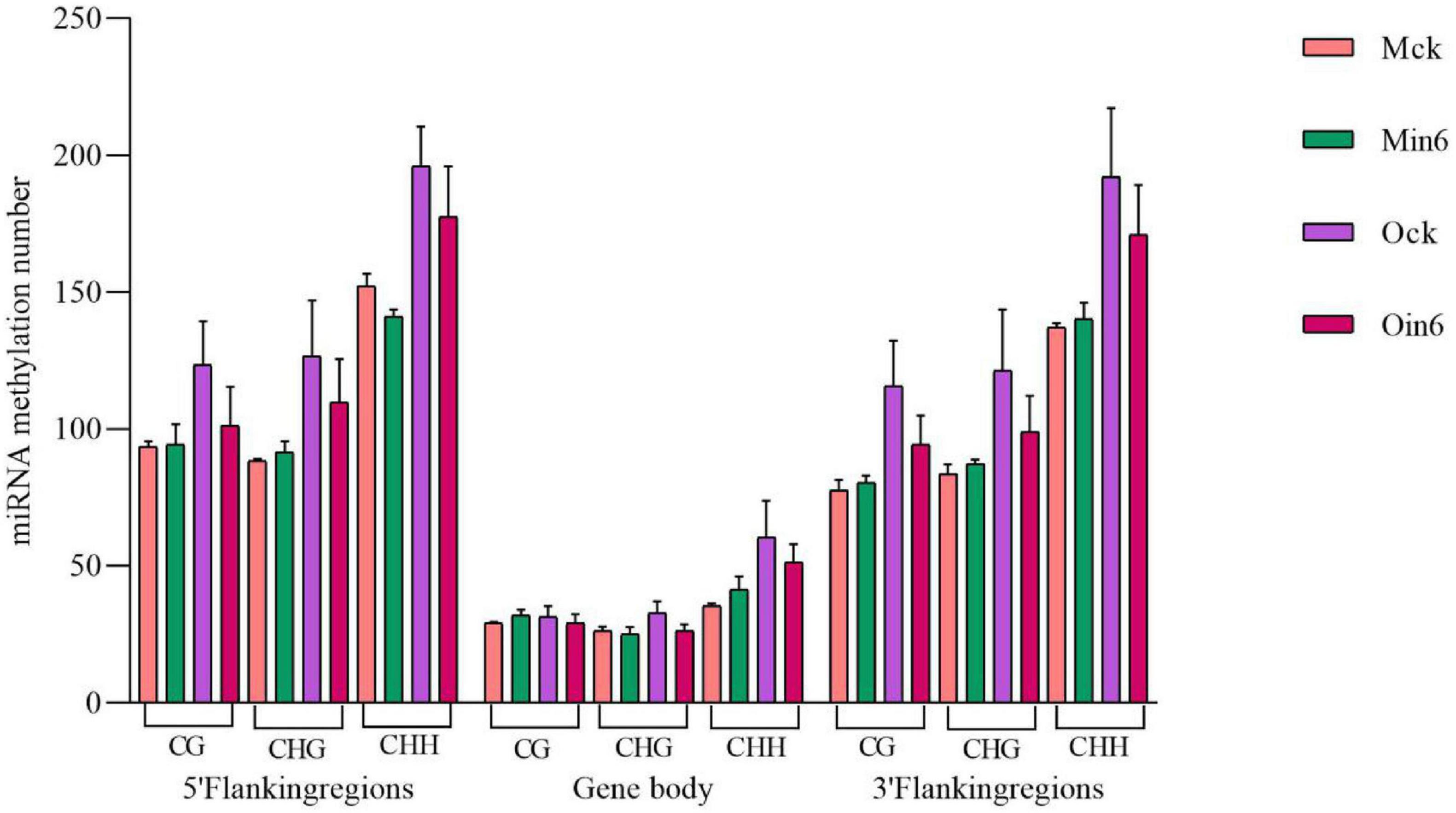
Figure 8. miRNA methylation numbers under different contexts in different regions of Populus tomentosa ‘henan’ and Populus × euramericana ‘74/76’. Error bars indicate SEM.
Plants undergo genome-wide DNA methylation changes during infection with pathogens, which has been described in a few annual plants (Zhang et al., 2018). For example, global DNA hypomethylation was suggested to be part of the basal pattern-triggered immunity response in rice and tomato treated with different nematode species or flag22 (Atighi et al., 2020). Similarly, DNA hypomethylation was associated with resistance of P. tomentosa to L. populi, suggesting that pathogen-induced DNA methylation may involve similar mechanisms between annual and perennial plants. In response to Heterodera glycines infection, the susceptible soybean line exhibited reduced global methylation of both protein-coding genes and transposable elements, whereas the resistant line showed the opposite response, with increased global MLs (Rambani et al., 2020). When a resistant near-isogenic line of wheat was compared to a susceptible line, more hypermethylated and fewer hypomethylated genes were found in the former line at both 0 and at 96 h post-inoculation with Puccinia triticina (Saripalli et al., 2020). Those results suggest that regulation of DNA methylation is of great significance for plants facing biotic stress. In this investigation, DNA methylation changes associated with pathogen infection between two poplar types with different resistance levels were investigated through WGBS. Interestingly, both resistant and susceptible poplars showed DNA hypomethylation; the previously reported contrasting DNA methylation patterns were not observed here (Figure 3; Rambani et al., 2020; Saripalli et al., 2020). Intriguingly, the resistant type P. tomentosa showed significantly reduced DNA hypomethylation under pathogen stress, which differed from the DNA hypermethylation patterns observed in resistant plants in previous studies. These results suggested that the DNA methylation patterns of susceptible and resistant types may differ, making it necessary to explore the subtle differences in epigenetic regulation between annual and perennial plants during pathogen infection.
Cyst nematode parasitism induced dynamic changes in the Arabidopsis root epigenome, evidenced by a clear distinction between two infection time points, with increased CG hypermethylation seen at 10 dpi relative to 5 dpi, specifically in protein-coding genes (Hewezi et al., 2017). Similarly, this investigation indicated dynamic changes in DNA methylation of poplars, which were associated with different courses of pathogen infection (Figure 1). Interestingly, our results differed from the DNA methylation changes observed in Arabidopsis, which were largely similar between 3 and 5 dpi after infection with Pseudomonas syringae (Dowen et al., 2012). DNA methylation decreased with prolongation of infection time in poplar (Figure 1), in contrast to the trend reported in the Arabidopsis root epigenome (Hewezi et al., 2017). The change trend of DNA methylation in this investigation could be elucidated more thoroughly using four rather than two infection time points (Dowen et al., 2012; Hewezi et al., 2017). Comprehensively, there might be a specific relationship between the pathogen infection period and global changes in DNA methylation may differ among plant species and stressors.
The methylation maintenance mode and structure of CHH differed from the other two sequence contexts, indicating that the regulation process of CHH methylation in plants might be different with that of CG and CHG. Our results provided compelling evidence that CHH methylation might be the main type of DNA methylation occurring in poplars under biotic stress, and it changed dynamically among infection time points (Figure 1). This was consistent with previous reports that CHH hypomethylation played key roles in plant immune responses (Geng et al., 2019; Sun et al., 2019; Atighi et al., 2020). In this investigation, the number of differential CHH hypermethylation sited in P. × euramericana was roughly double that in P. tomentosa (Figure 5). Moreover, a more significant difference in CHH methylation occurred in P. tomentosa (Figure 3). Together, these results reflected dynamic variation of CHH hypomethylation between different poplar species, demonstrating the key role of CHH hypomethylation in the poplar immune response to pathogen affection. Further annotation of DMR regions revealed that the level of CHH hypomethylation in susceptible and resistant poplars was overwhelmingly higher in repeat regions, followed by promoter regions, which was different with the previous finding that CHH hypomethylation occurred predominantly in gene promoters during the rice immune response to Meloidogyne graminicola infection (Atighi et al., 2020). Similarly, in wheat inoculated with P. triticina, ML was generally abundant in intergenic regions, followed by promoter regions, transcription termination sites, and exons/introns (Saripalli et al., 2020). Overall, these investigations supported the conclusion that the promoter is a crucial region for DNA methylation mediated plant responses to biotic stressors, even though the promoter region may not accumulate the highest level of DNA methylation.
Previous reports have suggested that many CDPKs were associated with plant defense mechanisms against abiotic attacks. CDPK genes and the CDPK-related protein kinases (CRKs) played pivotal roles in the biological processes underlying Arabidopsis immunity to bacteria, fungi, insects, and viruses (Yip Delormel and Boudsocq, 2019). CDPK and CRK genes, which were dramatically induced during Ralstonia solanacearum infection, may act in a coordinated manner to mediate the immune response of pepper plants (Cai et al., 2015). Many rice CDPK genes had been demonstrated to respond to various stresses, including rice blast and chitin stress (Wan et al., 2007). Furthermore, a few CDPK genes were differentially expressed in P. trichocarpa during fungal infection, according to genome-wide analysis of the CDPK gene family (Zuo et al., 2013). The present investigation suggested a crucial role of CDPK24 in the defense response of poplars to pathogen infection, based on analysis of whole-genome DNA methylation and transcriptomic profiles. We found that CHH hypomethylation stimulated the expression of CDPK24, which was involved in the immune response of the resistant poplar to pathogen infection, thereby providing the first insight into the crucial role of DNA methylation in modifying CDPK24 in the poplar response to biotic stress. Considering that CDPKs were related to immune responses in diverse plants, the regulatory roles of CDPKs in plant responses to pathogen infection may be conserved. However, the detailed functions of CDPKs in pathogen resistance remain to be confirmed.
Vitamins are essential nutrients and key enzyme cofactors that regulate cellular metabolism and activate the immune system. Recently, B vitamins have been shown to play roles in the development, stress tolerance, and pathogen resistance of plants (Suzuki et al., 2020). Other studies have obtained evidence through expression profiling of genes involved in VitB6 biosynthesis, showing their involvement in plant disease resistance. Bacillus subtilis CBR05 was reported to induce VitB6 biosynthesis in tomato plants through a de novo pathway, contributing to resistance against Xanthomonas campestris infection (Chandrasekaran et al., 2019). Moreover, Vitamin B6 contributed to disease resistance against Pseudomonas syringae and Botrytis cinerea in Arabidopsis (Zhang et al., 2015; Chandrasekaran et al., 2019). Transcriptome sequencing indicated that the VitB6 biosynthesis pathway was involved in the response of Lilium pumilum to Fusarium oxysporum (He et al., 2019). Two protein families, PDX1s and PDX2, were required for the de novo biosynthesis of VitB6 (Tambasco-Studart et al., 2007). In this investigation, the VitB6 metabolism pathway was found to contribute to the resistance of poplars to pathogen infection. Specifically, this investigation unraveled that DNA methylation modification of PDX was involved in the poplar response to pathogen infection, providing new insights into the connection between the VitB6 metabolism pathway and pathogen resistance in poplars.
Furthermore, two miRNAs targeting CDPK24 were further predicted, namely, ptr-miR477d and ptr-miR169n (Supplementary Table 8). MiR477d-5p was downregulated in P. tomentosa upon pathogen infection, which was consistent with expectation that it showed CHH hypomethylation in the 5’ flanking regions. Previous reports revealed that miR477 was related to plant resistance to pathogen (Hu et al., 2020; Wang et al., 2020). Thus, infection with L. populi in P. × euramericana and P. tomentosa should trigger a battery of plant immune responses. We further postulated that CHH hypomethylation might trigger the suppression of ptc-miR477d-5p, thereby stimulating the induction of CDPK24 and indicating that ptc-miR477d-5p and its target, CDPK24, enhanced the plant immune response to pathogen infection in poplars. Additionally, PDX1.2 was involved in the defense of poplar plants to against biotic stress (Supplementary Figure 10).
Phytohormones had been confirmed to function as regulators of plant immune responses to biotic stress through exogenous hormone treatment (Robert-Seilaniantz et al., 2011). R and PR genes were responsible for plant resistance to multiple diseases (Tyr and Rebecca, 2016). In this investigation, nearly half of the plant hormone genes were associated with DMRs and DEGs, and a negative correlation was found between DNA hypomethylation and differential gene expression (Figure 7 and Supplementary Table 7). In addition, all R and PR genes associated with DEGs and DMRs exhibited DNA hypomethylation and upregulated expression. These R and PR genes and phytohormone related DEGs participated in the immune response through DNA methylation modification, suggesting that DNA methylation modulated poplar defense against pathogen infection through modification of R, PR, and phytohormone genes. In grape berries, melatonin treatment enhanced disease resistance and flavonoid biosynthesis by decreasing the MLs of the promoters of the corresponding genes (Gao et al., 2020). Heterologous expression of the lycopene β-cyclase (lcb) gene in flax was reported to silence its endogenous counterpart due to changes in gene-body methylation and the abscisic acid homeostasis mechanism, thereby increasing plant resistance to fungal pathogens (Boba et al., 2018). Combined together, these results indicated that DNA methylation effectively functioned in plant disease defense through modification of R, PR, and phytohormone genes, thus helping to modulate the molecular epigenetic mechanism.
To systematically understand the crosstalk of DNA methylation and poplar response to biotic stress, we further postulated a putative regulation model (Figure 9). Biotic stress triggered DNA methylation changes, which was followed by a wide range of response activities. The plant-pathogen interaction pathway was activated by CDPK. Similarly, the VitB6 metabolism pathway was activated by PDX. CDPK and PDX may regulate a burst of reactive oxygen species (ROS), leading to the hypersensitive response (HR) (Tambasco-Studart et al., 2007). Appropriate activation of HR by pathogens may cause, or have an association with, plant disease resistance (Balint-Kurti, 2019). Enrichment of the monoterpenoid biosynthesis pathway in the KEGG results indicated SAR through ROS and AZELAIC ACID INDUCED1 (AZI1), which likely functioned as infochemicals in plant-to-plant signaling, thereby allowing defense signals to propagate between neighboring plants (Riedlmeier et al., 2017). Effector-triggered immunity (ETI) is triggered by the activation of R genes, resulting in halting further colonization and attenuating disease resistance (Jones and Dangl, 2006). Phytohormone-regulating signal cascades were involved in poplar responses to biotic stresses (Bari and Jones, 2009; Verma et al., 2016). Taken together, as perennial and sessile organisms, poplars are equipped with a sophisticated multilayered immune system based on DNA methylation to win the arm race with pathogen infection.
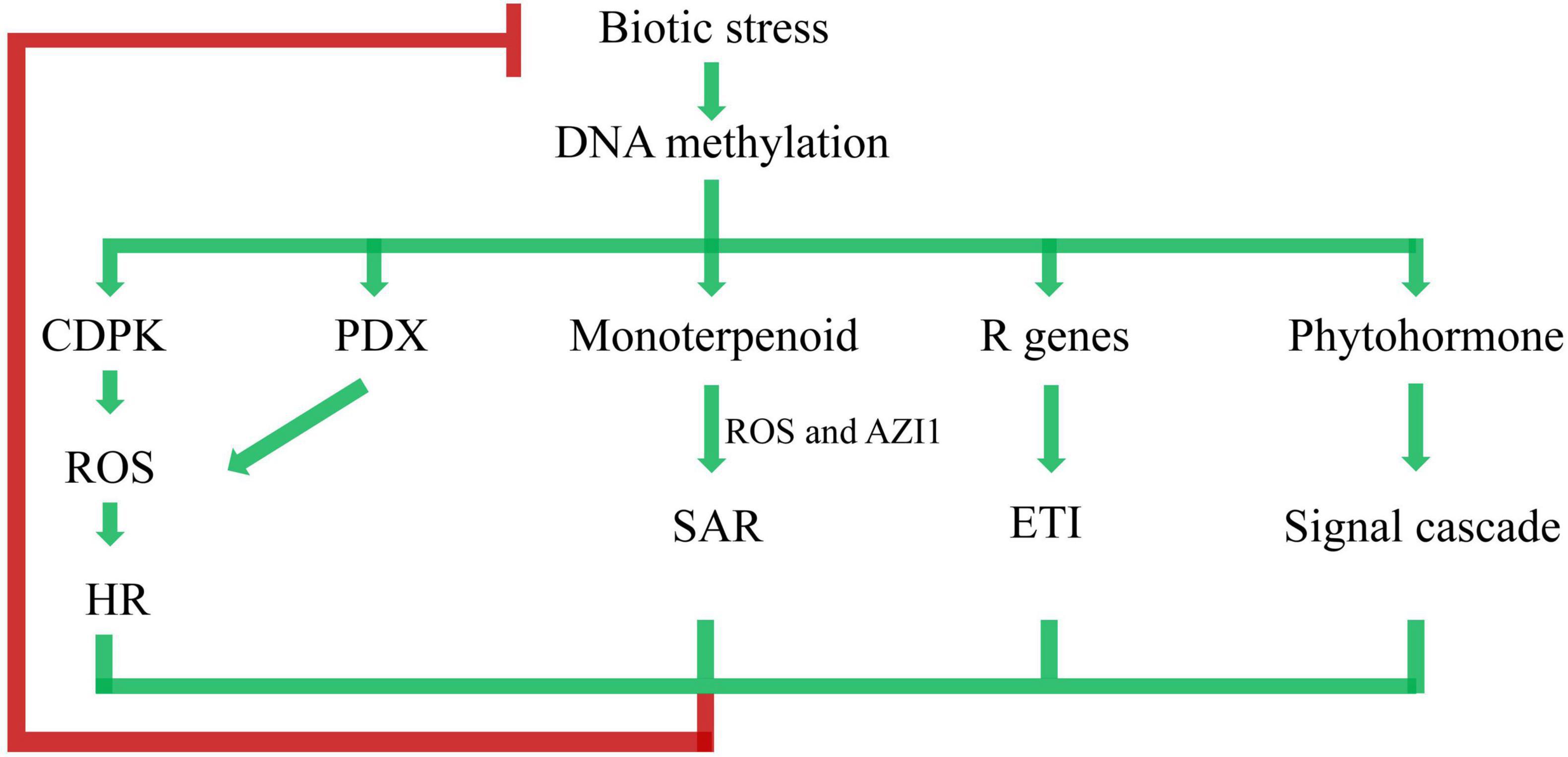
Figure 9. Network of immune pathways associated with plant–pathogen interaction. ROS represents reactive oxygen species, HR represents hypersensitive response, AZI1 represents AZELAIC ACID INDUCED1, SAR represents systemic acquired resistance, ETI represents effector-triggered immunity. Arrows represent positive regulation (accumulation of transcripts, proteins, or hormones), and blocked arrow represents negative regulation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI SRA BioProject, accession numbers: PRJNA778554, PRJNA778562, PRJNA778625, PRJNA778867, PRJNA779116, and PRJNA779165.
DX was responsible for conceptualization, bioinformatic analysis, data interpretation, and drafting of the manuscript. KZ performed the pathogen culture and inoculation and was responsible for sequencing. XY and YM assisted with the data analysis and critical evaluation of the manuscript. YY contributed to the plant growth, sampling, and DNA extraction. YW was responsible for supervision, project administration, and funding acquisition to support this research. All authors have read and agreed to submit the manuscript.
This work was supported by the National Natural Science Foundation of China (32071504 and 31670671) and Beijing Forestry University Undergraduate Innovation and Entrepreneurship Training Program (X201910022077).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the Novogene Company for assisting with the sequencing analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.750089/full#supplementary-material
Atighi, M. R., Verstraeten, B., De Meyer, T., and Kyndt, T. (2020). Genome-wide DNA hypomethylation shapes nematode pattern-triggered immunity in plants. New Phytol. 227, 545–558. doi: 10.1111/nph.16532
Balint-Kurti, P. (2019). The plant hypersensitive response: concepts, control and consequences. Mol. Plant Pathol. 20, 1163–1178. doi: 10.1111/mpp.12821
Bari, R., and Jones, J. D. G. (2009). Role of plant hormones in plant defence responses. Plant Mol. Biol. 69, 473–488. doi: 10.1007/s11103-008-9435-0
Boba, A., Kostyn, K., Preisner, M., Wojtasik, W., Szopa, J., and Kulma, A. (2018). Expression of heterologous lycopene β-cyclase gene in flax can cause silencing of its endogenous counterpart by changes in gene-body methylation and in ABA homeostasis mechanism. Plant Physiol. Biochem. 127, 143–151. doi: 10.1016/j.plaphy.2018.03.023
Cai, H., Cheng, J., Yan, Y., Xiao, Z., Li, J., Mou, S., et al. (2015). Genome-wide identification and expression analysis of calcium-dependent protein kinase and its closely related kinase genes in Capsicum annuum. Front. Plant Sci. 6:737. doi: 10.3389/fpls.2015.00737
Chandrasekaran, M., Paramasivan, M., and Chun, S. (2019). Bacillus subtilis CBR05 induces vitamin B6 biosynthesis in tomato through the de novo pathway in contributing disease resistance against Xanthomonas campestris pv. vesicatoria. Sci. Rep. 9:6495. doi: 10.1038/s41598-019-41888-6
Derksen, H., Rampitsch, C., and Daayf, F. (2013). Signaling cross-talk in plant disease resistance. Plant Sci. 207, 79–87. doi: 10.1016/j.plantsci.2013.03.004
Ding, G., Guo, D., Guan, Y., Chi, C., and Liu, B. (2019). Changes of DNA methylation of Isoetes sinensis under Pb and Cd stress. Environ. Sci. Pollut. R. 26, 3428–3435. doi: 10.1007/s11356-018-3864-3
Dowen, R. H., Pelizzola, M., Schmitz, R. J., Lister, R., Dowen, J. M., Nery, J. R., et al. (2012). Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. U.S.A. 109, E2183–E2191. doi: 10.1073/pnas.1209329109
Elhamamsy, A. R. (2016). DNA methylation dynamics in plants and mammals: overview of regulation and dysregulation. Cell Biochem. Funct. 34, 289–298. doi: 10.1002/cbf.3183
Feng, H., Conneely, K. N., and Wu, H. (2014). A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 42:e69. doi: 10.1093/nar/gku154
Gao, S., Ma, W., Lyu, X., Cao, X., and Yao, Y. (2020). Melatonin may increase disease resistance and flavonoid biosynthesis through effects on DNA methylation and gene expression in grape berries. BMC Plant Biol. 20:231. doi: 10.1186/s12870-020-02445-w
Geng, S., Kong, X., Song, G., Jia, M., Guan, J., Wang, F., et al. (2019). DNA methylation dynamics during the interaction of wheat progenitor Aegilops tauschii with the obligate biotrophic fungus Blumeria graminis f. sp. tritici. New Phytol. 221, 1023–1035. doi: 10.1111/nph.15432
Goll, M. G., and Bestor, T. H. (2005). Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74:5.
He, X., Li, W., Zhang, W., Jin, X., Shenkute, A. G., Aynalem, T., et al. (2019). Transcriptome sequencing analysis provides insights into the response to Fusarium oxysporum in Lilium pumilum. Evol. Bioinform. Online 15:1611585122. doi: 10.1177/1176934319838818
Hewezi, T., Lane, T., Piya, S., Rambani, A., Rice, J. H., and Staton, M. (2017). Cyst nematode parasitism induces dynamic changes in the root epigenome. Plant Physiol. 174, 405–420. doi: 10.1104/pp.16.01948
Hou, J., Wu, Q., Zuo, T., Guo, L., Chang, J., Chen, J., et al. (2016). Genome-wide transcriptomic profiles reveal multiple regulatory responses of poplar to Lonsdalea quercina infection. Trees 30, 1389–1402. doi: 10.1007/s00468-016-1376-7
Hu, G., Hao, M., Wang, L., Liu, J., Zhang, Z., Tang, Y., et al. (2020). The cotton miR477-CBP60A module participates in plant defense against Verticillium dahlia. Mol. Plant Microbe Interact. 33, 624–636. doi: 10.1094/MPMI-10-19-0302-R
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484.
Krueger, F., and Andrews, S. R. (2011). The new phytologist. Bioinformatics 27, 1571–1572. doi: 10.1093/bioinformatics/btr167
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Law, J. A., and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. doi: 10.1038/nrg2719
Li, A., and He, W. (2019). Molecular aspects of an emerging poplar canker caused by Lonsdalea populi. Front. Microbiol. 10:2496. doi: 10.3389/fmicb.2019.02496
Li, Y., He, W., Ren, F., Guo, L., Chang, J., Cleenwerck, I., et al. (2014). A canker disease of Populus × euramericana in China caused by Lonsdalea quercina subsp. populi. Plant Dis. 98, 368–378. doi: 10.1094/PDIS-01-13-0115-RE
Liang, D., Zhang, Z., Wu, H., Huang, C., Shuai, P., Ye, C. Y., et al. (2014). Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 15 (Suppl. 1):S9. doi: 10.1186/1471-2156-15-S1-S9
Lister, R., Mukamel, E. A., Nery, J. R., Urich, M., Puddifoot, C. A., Johnson, N. D., et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341:1237905. doi: 10.1126/science.1237905
Liu, M., Gao, J., Yin, F., Gong, G., Qin, C., Ye, K., et al. (2015). Transcriptome analysis of maize leaf systemic symptom infected by Bipolaris zeicola. PLoS One 10:e119858. doi: 10.1371/journal.pone.0119858
Liu, T., Li, Y., Duan, W., Huang, F., and Hou, X. (2017). Cold acclimation alters DNA methylation patterns and confers tolerance to heat and increases growth rate in Brassica rapa. J. Exp. Bot. 68, 1213–1224. doi: 10.1093/jxb/erw496
Liu, Z., Su, Y., Zuo, T., Chang, J., Guo, L., He, W., et al. (2015). The differential expression analysis of the SA and JA signal transduction related genes in poplar varieties susceptible and resistant to canker. Chin. Agric. Sci. Bull. 31, 156–163.
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L. (2005). Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Meng, L., Bregitzer, P., Zhang, S., and Lemaux, P. G. (2003). Methylation of the exon/intron region in the Ubi1 promoter complex correlates with transgene silencing in barley. Plant Mol. Biol. 53, 327–340. doi: 10.1023/B:PLAN.0000006942.00464.e3
Park, Y., and Wu, H. (2016). Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 32, 1446–1453. doi: 10.1093/bioinformatics/btw026
Pavet, V., Quintero, C., Cecchini, N. M., Rosa, A. L., and Alvarez, M. E. (2006). Arabidopsis displays centromeric DNA hypomethylation and cytological alterations of heterochromatin upon attack by Pseudomonas syringae. Mol. Plant Microbe Interact. 19, 577–587.
Rambani, A., Pantalone, V., Yang, S., Rice, J. H., Song, Q., Mazarei, M., et al. (2020). Identification of introduced and stably inherited DNA methylation variants in soybean associated with soybean cyst nematode parasitism. New Phytol. 227, 168–184. doi: 10.1111/nph.16511
Riedlmeier, M., Ghirardo, A., Wenig, M., Knappe, C., Koch, K., Georgii, E., et al. (2017). Monoterpenes support systemic acquired resistance within and between plants. Plant Cell 29, 1440–1459. doi: 10.1105/tpc.16.00898
Robert-Seilaniantz, A., Grant, M., and Jones, J. D. (2011). Hormone crosstalk in plant disease and defense: more than just jasmonate-salicylate antagonism. Annu. Rev. Phytopathol. 49, 317–343. doi: 10.1146/annurev-phyto-073009-114447
Saripalli, G., Sharma, C., Gautam, T., Singh, K., Jain, N., Prasad, P., et al. (2020). Complex relationship between DNA methylation and gene expression due to Lr28 in wheat-leaf rust pathosystem. Mol. Biol. Rep. 47, 1339–1360. doi: 10.1007/s11033-019-05236-1
Shang, J., Li, Y., Li, A., He, W., Guo, L., and Chang, J. (2014). Dynamic changes of Lonsdalea quercina subsp.populi in infection process of Populus × euramericana. J. Northeast For. Univ. 42, 120–123. doi: 10.3969/j.issn.1000-5382.2014.11.027
Sherman, J. D., and Talbert, L. E. (2002). Vernalization-induced changes of the DNA methylation pattern in winter wheat. Genome 45, 253–260. doi: 10.1139/g01-147
Shipony, Z., Mukamel, Z., Cohen, N. M., Landan, G., Chomsky, E., Zeliger, S. R., et al. (2014). Dynamic and static maintenance of epigenetic memory in pluripotent and somatic cells. Nature 513, 115–119. doi: 10.1038/nature13458
Song, Y., Ci, D., Tian, M., and Zhang, D. (2016). Stable methylation of a non-coding RNA gene regulates gene expression in response to abiotic stress in Populus simonii. J. Exp. Bot. 67, 1477–1492. doi: 10.1093/jxb/erv543
Sun, Y., Fan, M., and He, Y. (2019). DNA methylation analysis of the Citrullus lanatus response to Cucumber green mottle mosaic virus infection by Whole-Genome bisulfite sequencing. Genes 10:344. doi: 10.3390/genes10050344
Suzuki, M., Wu, S., Mimura, M., Alseekh, S., Fernie, A. R., Hanson, A. D., et al. (2020). Construction and applications of a B vitamin genetic resource for investigation of vitamin-dependent metabolism in maize. Plant J. 101, 442–454. doi: 10.1111/tpj.14535
Tambasco-Studart, M., Tews, I., Amrhein, N., and Fitzpatrick, T. B. (2007). Functional analysis of PDX2 from Arabidopsis, a glutaminase involved in vitamin B6 biosynthesis. Plant Physiol. 144, 915–925. doi: 10.1104/pp.107.096784
Tirnaz, S., and Batley, J. (2019). DNA methylation: toward crop disease resistance improvement. Trends Plant Sci. 24, 1137–1150. doi: 10.1016/j.tplants.2019.08.007
Tyr, W., and Rebecca, N. (2016). Multiple disease resistance in plants. Annu. Rev. Phytopathol. 54, 229–252. doi: 10.1146/annurev-phyto-080615-100037
Van Dooren, T. J. M., Silveira, A. B., Gilbault, E., Jiménez-Gómez, J. M., Martin, A., Bach, L., et al. (2020). Mild drought in the vegetative stage induces phenotypic, gene expression, and DNA methylation plasticity in Arabidopsis but no transgenerational effects. J. Exp. Bot. 71, 3588–3602. doi: 10.1093/jxb/eraa132
Verma, V., Ravindran, P., and Kumar, P. P. (2016). Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 16:86. doi: 10.1186/s12870-016-0771-y
Wada, Y., Miyamoto, K., Kusano, T., and Sano, H. (2004). Association between up-regulation of stress-responsive genes and hypomethylation of genomic DNA in tobacco plants. Mol. Genet. Genomics 271, 658–666. doi: 10.1007/s00438-004-1018-4
Wan, B., Lin, Y., and Mou, T. (2007). Expression of rice Ca2+-dependent protein kinases (CDPKs) genes under different environmental stresses. FEBS Lett. 581, 1179–1189. doi: 10.1016/j.febslet.2007.02.030
Wang, D., Sha, Y., Hu, J., Yang, T., Piao, X., and Zhang, X. (2018). Genetic signatures of plant resistance genes with known function within and between species. Genetica 146, 517–528. doi: 10.1007/s10709-018-0044-9
Wang, H., Beyene, G., Zhai, J., Feng, S., Fahlgren, N., Taylor, N. J., et al. (2015). CG gene body DNA methylation changes and evolution of duplicated genes in cassava. Proc. Natl. Acad. Sci. U.S.A. 112, 13729–13734. doi: 10.1073/pnas.1519067112
Wang, S., Liu, S., Liu, L., Li, R., Guo, R., Xia, X., et al. (2020). MiR477 targets the phenylalanine ammonia-lyase gene and enhances the susceptibility of the tea plant (Camellia sinensis) to disease during Pseudopestalotiopsis species infection. Planta 251:59. doi: 10.1007/s00425-020-03353-x
Wu, H., Xu, T., Feng, H., Chen, L., Li, B., Yao, B., et al. (2015). Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 43:e141. doi: 10.1093/nar/gkv715
Xin, Y., Ma, B., Zeng, Q., He, W., Qin, M., and He, N. (2021). Dynamic changes in transposable element and gene methylation in mulberry (Morus notabilis) in response to Botrytis cinerea. Hortic. Res. 8:154. doi: 10.1038/s41438-021-00588-x
Yang, R., Deng, C., Wei, J., He, W., Li, A., and Qian, W. (2018). A large-scale mutational analysis of two-component signaling systems of Lonsdalea quercina revealed that KdpD-KdpE regulates bacterial virulence against host poplar trees. Mol. Plant Microbe Interact. 31, 724–736. doi: 10.1094/MPMI-10-17-0248-R
Yang, X., Zhang, L., Yang, Y., Schmid, M., and Wang, Y. (2021). MiRNA mediated regulation and interaction between plants and pathogens. Int. J. Mol. Sci. 22:2913. doi: 10.3390/ijms22062913
Yip Delormel, T., and Boudsocq, M. (2019). Properties and functions of calcium-dependent protein kinases and their relatives in Arabidopsis thaliana. New Phytol. 224, 585–604. doi: 10.1111/nph.16088
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11:R14. doi: 10.1186/gb-2010-11-2-r14
Zemach, A., McDaniel, IE., Silva, P., and Zilberman, D. (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 14, 916–919. doi: 10.1038/emboj.2010.65
Zhang, H., Lang, Z., and Zhu, J. (2018). Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19, 489–506. doi: 10.1038/s41580-018-0016-z
Zhang, M., Zhang, X., Guo, L., Qi, T., Liu, G., Feng, J., et al. (2019). Single-base resolution methylomes of cotton CMS system reveal epigenomic changes in response to high-temperature stress during anther development. J. Exp. Bot. 71, 951–969. doi: 10.1093/jxb/erz470
Zhang, Y., Jin, X., Ouyang, Z., Li, X., Liu, B., Huang, L., et al. (2015). Vitamin B6 contributes to disease resistance against Pseudomonas syringae pv. tomato DC3000 and Botrytis cinerea in Arabidopsis thaliana. J. Plant Physiol. 175, 21–25. doi: 10.1016/j.jplph.2014.06.023
Zhao, L., Fan, D., Li, Y., Yan, X., and Huang, L. (2016). The extraction of metagenom DNA in branch bark. Biotechnol. Bull. 32, 74–79.
Zheng, X., Chen, L., Xia, H., Wei, H., Lou, Q., Li, M., et al. (2017). Transgenerational epimutations induced by multi-generation drought imposition mediate rice plant’s adaptation to drought condition. Sci. Rep. 7:39843. doi: 10.1038/srep39843
Zhu, C., Zhang, S., Zhou, C., Chen, L., Fu, H., Li, X., et al. (2020). Genome-wide investigation and transcriptional analysis of cytosine-5 DNA methyltransferase and DNA demethylase gene families in tea plant (Camellia sinensis) under abiotic stress and withering processing. PeerJ 8:e8432. doi: 10.7717/peerj.8432
Zou, S., Wang, H., Li, Y., Kong, Z., and Tang, D. (2018). The NB-LRR gene Pm60 confers powdery mildew resistance in wheat. New Phytol. 218, 298–309. doi: 10.1111/nph.14964
Keywords: DNA methylation, poplar, Lonsdalea populi, infection, resistance
Citation: Xiao D, Zhou K, Yang X, Yang Y, Ma Y and Wang Y (2021) Crosstalk of DNA Methylation Triggered by Pathogen in Poplars With Different Resistances. Front. Microbiol. 12:750089. doi: 10.3389/fmicb.2021.750089
Received: 30 July 2021; Accepted: 07 December 2021;
Published: 28 December 2021.
Edited by:
Quan Lu, Chinese Academy of Forestry, ChinaReviewed by:
Marco Catoni, University of Birmingham, United KingdomCopyright © 2021 Xiao, Zhou, Yang, Yang, Ma and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanwei Wang, eXd3YW5nQGJqZnUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.