 Solène Irion1,2
Solène Irion1,2 Oleksandra Rudenko3
Oleksandra Rudenko3 Michael Sweet4
Michael Sweet4 Pascale Chabanet2
Pascale Chabanet2 Andrew C. Barnes3
Andrew C. Barnes3 Pablo Tortosa1
Pablo Tortosa1 Mathieu G. Séré2,4*
Mathieu G. Séré2,4*- 1Université de La Réunion, Unité Mixte de Recherche, Processus Infectieux en Milieu Insulaire Tropical (UMR PIMIT), Inserm1187, CNRS9192, IRD249, Plateforme de Recherche CYROI, Saint Denis, France
- 2Université de La Réunion, Unité Mixte de Recherche, Ecologie marine tropicale des océans Pacifique et Indien (UMR ENTROPIE), CNRS, IRD, Saint Denis, France
- 3School of Biological Sciences, Centre for Marine Science, The University of Queensland, Brisbane, QLD, Australia
- 4Aquatic Research Facility, Environmental Sustainability Research Centre, University of Derby, Derby,United Kingdom
Streptococcus iniae is an emerging zoonotic pathogen of increasing concern for aquaculture and has caused several epizootics in reef fishes from the Caribbean, the Red Sea and the Indian Ocean. To study the population structure, introduction pathways and evolution of S. iniae over recurring epizootics on Reunion Island, we developed and validated a Multi Locus Sequence Typing (MLST) panel using genomic data obtained from 89 isolates sampled during epizootics occurring over the past 40years in Australia, Asia, the United States, Israel and Reunion Island. We selected eight housekeeping loci, which resulted in the greatest variation across the main S. iniae phylogenetic clades highlighted by the whole genomic dataset. We then applied the developed MLST to investigate the origin of S. iniae responsible for four epizootics on Reunion Island, first in inland aquaculture and then on the reefs from 1996 to 2014. Results suggest at least two independent S. iniae emergence events occurred on the island. Molecular data support that the first epizootic resulted from an introduction, with inland freshwater aquaculture facilities acting as a stepping-stone. Such an event may have been facilitated by the ecological flexibility of S. iniae, able to survive in both fresh and marine waters and the ability of the pathogen to infect multiple host species. By contrast, the second epizootic was associated with a distinct ST of cosmopolitan distribution that may have emerged as a result of environment disturbance. This novel tool will be effective at investigating recurrent epizootics occurring within a given environment or country that is despite the fact that S. iniae appears to have low genetic diversity within its lineage.
Introduction
Streptococcal infections underlie disease outbreaks in numerous farmed and wild fish species, causing septicemia, central nervous system damage and meningoencephalitis (Eldar et al., 1994; Toranzo et al., 2005). The pathologic agent, Streptococcus iniae has been directly linked to massive economic losses in both marine and freshwater aquaculture environments, with mortality rates reaching 75% in tilapia farms for example (Perera et al., 1994; Eldar et al., 1997a; Francis et al., 2014). In addition, S. iniae has been known to infect mammals as well, such as dolphins (Pier and Madin, 1976; Agnew and Barnes, 2007; Song et al., 2017) and is occasionally zoonotic, generating soft tissue infections and sepsis in humans (Weinstein et al., 1997; Koh et al., 2004; Lau et al., 2006).
Although first isolated in the 1970s (from abscesses in captive freshwater dolphins), the first recorded S. iniae outbreaks in farmed fish were documented in the 1980s throughout Japan, the United States, Israel, Australia and Asia (Eldar et al., 1994; Perera et al., 1994; Stoffregen et al., 1996; Bromage et al., 1999; Nguyen et al., 2002). Since then, the geographical range of these epidemics has expanded with cases being reported throughout Europe, South America, the Middle East and Africa (El Aamri et al., 2010; Fadaeifard et al., 2011; Figueiredo et al., 2012; Türe and Alp, 2016). Inactivated (or killed) vaccines were developed in 1995 in order to mitigate the economic consequences of these outbreaks (Eldar et al., 1997a; Klesius et al., 2000; Shoemaker et al., 2010). However, the efficacy of vaccination has been challenged as reinfection of vaccinated stock are known to occur, most notably following the emergence of new serotypes bypassing vaccine protection through spontaneous point mutations in genes involved in capsule biosynthesis (Bachrach et al., 2001; Eyngor et al., 2008; Millard et al., 2012; Barnes and Silayeva, 2016).
Although epidemics are usually associated with farmed fish, several outbreaks have been reported in wild populations. Whilst the majority of these were in the vicinity of aquaculture facilities (Zlotkin et al., 1998; Colorni et al., 2002), a result suggesting possible transmission between cultured and wild fish, there are a number of instances where recurrent mass mortalities of reef fish have occurred in the absence of such connection (Ferguson et al., 2000; Keirstead et al., 2014). For example, mass mortalities of reef fish have occurred in 2002 and 2014 on Reunion Island (an overseas department of France and a geographically isolated oceanic island located in the western Indian Ocean; Turquet et al., 2002; Quod et al., 2014). Several fish kill phenomena have been reported during the austral summer from 2001 to 2003 (Pothin et al., 2001; Turquet et al., 2002) and more recently between January and May of 2014 (Quod et al., 2014). The last recorded episode (2014) was the most virulent with thousands of fish killed, including 34 families such as Surgeon fish (Acanthuridae, 12 sp.), Triggerfish (Balistidae, 8 sp.) and Groupers (Serranidae, 6 sp.).
Although the fish kills were linked to S. iniae infection, transmission of the pathogen was unknown, specifically regarding the origin of the causal agent. Molecular epidemiology is often employed to identify the transmission chains of specific pathogens within a given environment. For S. iniae, the diversity and evolutionary history of specific isolates has been assessed using molecular fingerprinting techniques. For example, restriction fragment length polymorphism was used to distinguish isolates responsible for outbreaks in American and Israeli fish farms (Eldar et al., 1997b, 1999). Whilst, other studies focused on using random amplified polymorphic DNA (Dodson et al., 1999; Kvitt and Colorni, 2004; Erfanmanesh et al., 2012), however both methods showed marginal discriminatory power. Pulsed-field gel electrophoresis (PFGE) has also been employed to evaluate strain diversity in S. iniae and shows the greatest discriminative power being able to differentiate between pathogenic and commensal strains (Fuller et al., 2001). PFGE also allows for discrimination between isolates collected within a specific region over a short period of time (Nawawi et al., 2008; Zhou et al., 2008), and strains collected during different epizootics in several host species (Facklam et al., 2005; Nawawi et al., 2008). However, PFGE does not necessarily address the phylogenetic history of the bacteria, as it targets a highly variable region within the microbial genome (Achtman, 2008). Further, PFGE appears to be poorly reproducible from one laboratory to the other and this lack of data transferability impedes analyses at a global scale. Therefore, we sought to explore other options to allow us to understand the potential point of origin of the S. iniae involved in the fish die offs in Reunion.
Multi Locus Sequence Typing (MLST) is a robust and reproducible method that has been widely used to characterize bacterial isolates since its introduction in 1998 (Maiden et al., 1998). It is based on the sequencing of several housekeeping genes in order to characterize a strain through a unique combination of alleles corresponding to an arbitrary sequence type (ST) number (Maiden, 2006). In the era of genomics, MLST is still considered relevant since it provides the overall clonal frame, population structure and diversity of a bacterial taxa (Pérez-Losada et al., 2013). Here, we therefore aimed to develop the first MLST scheme for S. iniae using eight selected housekeeping genes to identify possible origins of four wild and farmed fish epizootics, which have occurred in Reunion from 1996 onwards.
Materials and Methods
Bacterial Strains
Three Reunionese strains of S. iniae were utilized in this study. The first was isolated in 2002 from a grouper (Variola louti). The second was isolated in 2014 from a striped large-eye bream (Gnathodentex aurolineatus) and the third, isolated in 2009 from diseased red drum (Sciaenops ocellatus). This last isolate was attributed as the cause of a fish kill in an offshore marine fish farm (Table 1). In addition, we utilized the type strain CIP 103769T. Figure 1 pinpoints the location of the epizootics investigated on Reunion Island.
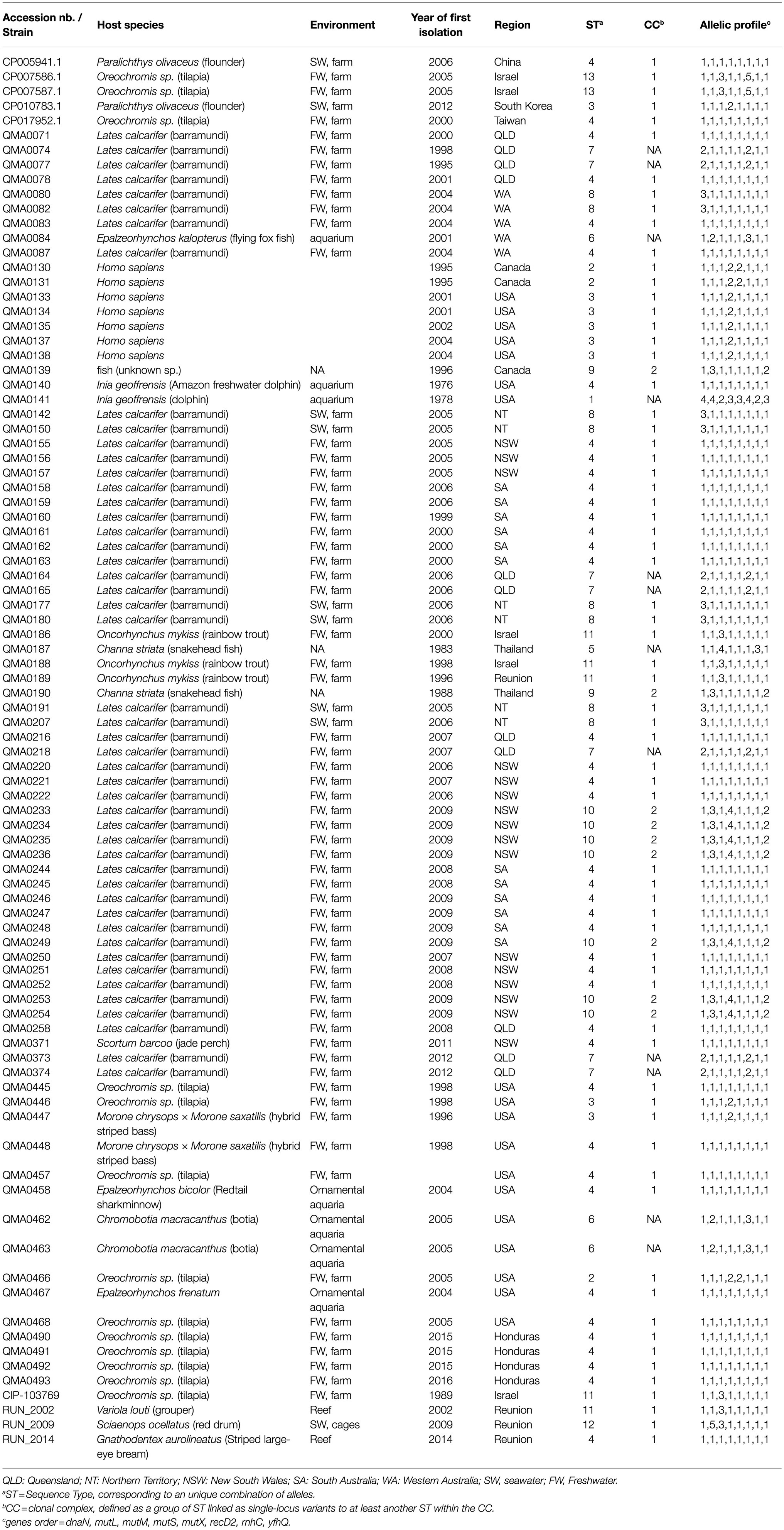
Table 1. Origin and typing of S. iniae strains. For Australian strains, region of isolation is indicated.
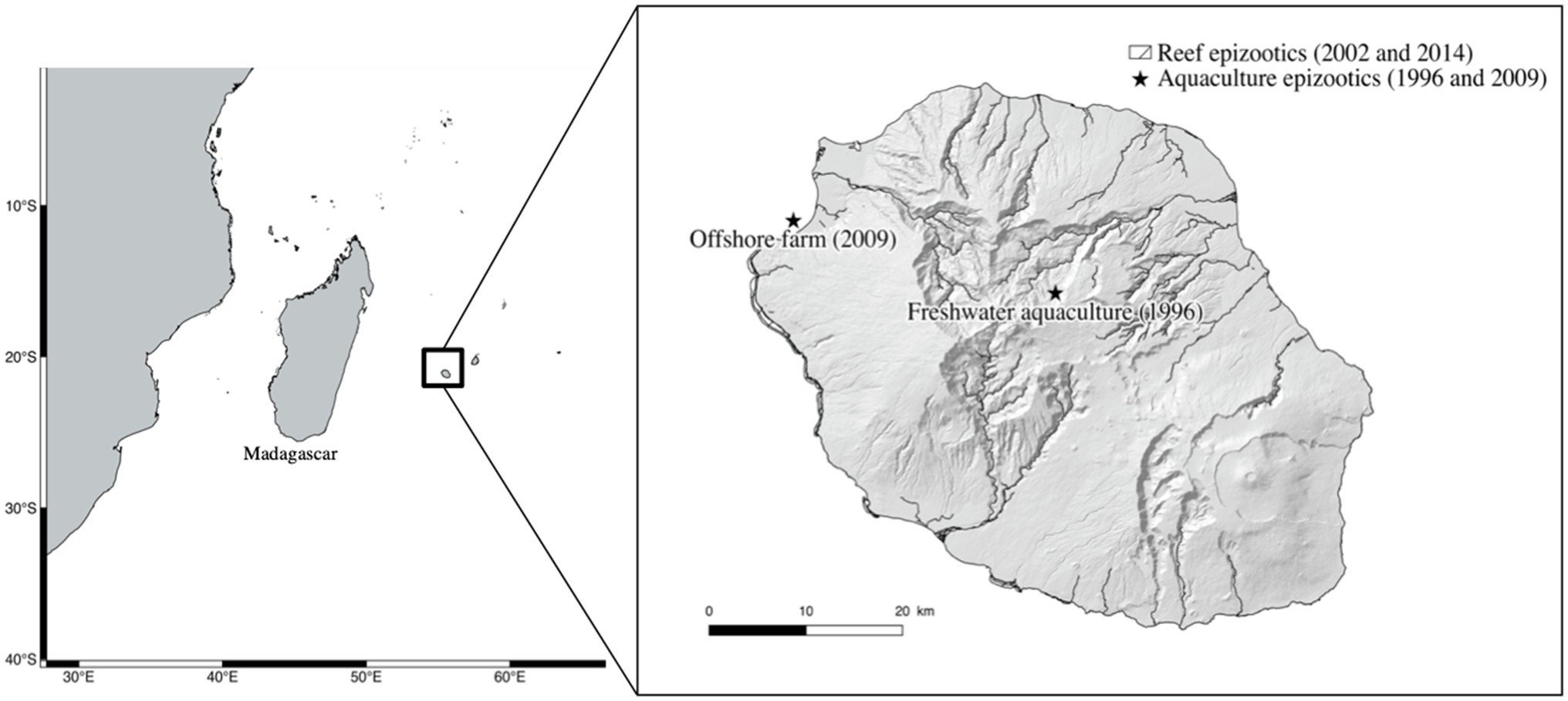
Figure 1. Map of the epizootics investigated on Reunion Island since 1996. Reef epizootics in 2002 and 2014 were spread along the West coast of the island on different coral reefs.
A further 85S. iniae genomes, (76 obtained from fish, two from dolphins, and seven from humans), representing all sequencing data available to date, were also used to assess the performance of the MLST for identifying origins of the three Reunionese strains. The sequences were available on Genbank (genome accession numbers in Table 1) or provided by co-authors (AB and OS). These isolates were temporally (isolated between 1976 and 2016) and geographically diverse, obtained from different countries including North and Central America, Australia, Asia and Israel (Table 1). The collection of isolates has been previously confirmed as belonging to S. iniae using the specific PCR protocol described by (Mata et al., 2004). Details regarding the origin of each isolate are provided in Table 1.
Multi Locus Sequence Typing Development
Loci Selection
To design the Multi Locus Sequence Typing panel, 80 draft genomes assembled by co-authors (OS and AB) as part of a separate study (Silayeva et al., 2020) along with five published assembled genomes (available on Genbank) were aligned. Eight loci or housekeeping genes, including dnaN (encoding DNA polymerase III), rnhC (encoding Ribonuclease HIII), yfhQ (encoding an A/G-specific adenine glycosylase), recD2 (encoding DNA helicase), mutM (encoding the formamidopyrimidine-DNA glycosylase), mutX (encoding the 8-oxo-dGTP diphosphatase), mutL and mutS (both encoding DNA mismatch repair proteins) were selected for MLST analysis. They were chosen because mutations in DNA repair loci are tightly linked to evolutionary history of organisms and, as such, they constitute promising genes for typing bacterial strains (Stich et al., 2010). For S. iniae, a previous study based on draft genomes indicated that these eight loci were correlated with phylogenetic diversification but highly conserved within phylogenetic clades, making them ideal candidates for the development of a MLST (Silayeva et al., 2020). Primer 3 (Untergasser et al., 2012) was then used to design primer pairs for each locus (Table 2).
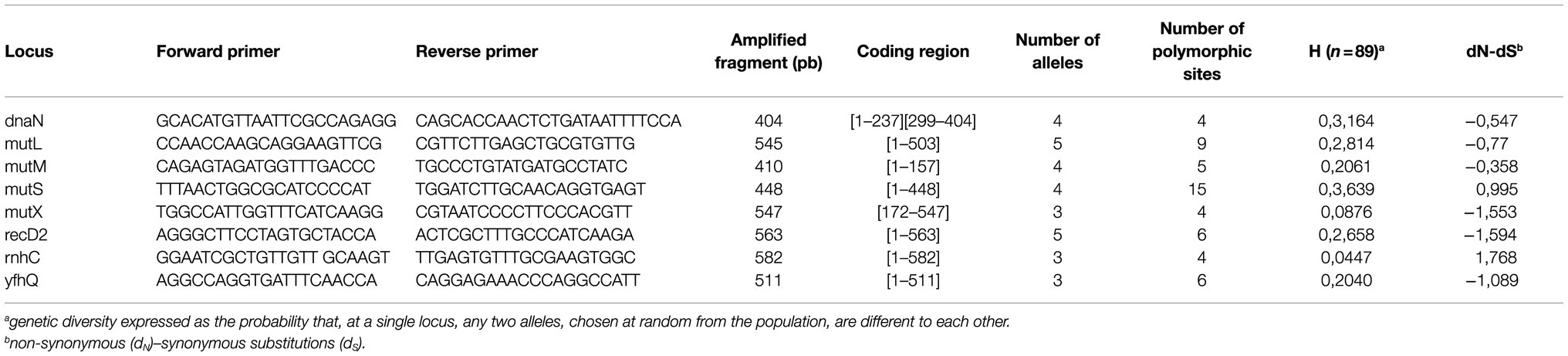
Table 2. Primer sequences and genetic characteristics of the loci used for MLST.
pcr
PCR conditions were tested on four strains for which no sequencing data existed, including the type strain CIP 103769T and three Reunionese strains (collected between 2002 and 2014). The four strains used to test the PCR conditions were grown in Brain Heart Infusion Agar. Single colonies were placed in 100μl of 5mm Tris/HCL at 100°C for 5min. PCR were then run for each of the loci identified above. PCR mixture comprised 1μl of bacterial DNA, 12.5μl of MasterMix (Applied Biosystems, Foster City, United States) and 1μl of each primer (10μm) (Table 2 for the forward and reverse primers for each loci) in a total volume of 25μl. PCR conditions included an initial step of denaturation/lysis at 95°C for 5min, 30cycles of denaturation at 94°C for 30s, annealing at 56°C for 1min, and extension at 72°C for 2min, and a final step of extension at 72°C for 7min. Sanger sequencing (paired end reads) was performed by Genoscreen (Lille, France). Sequences were manually analyzed and trimmed to remove the primers using Geneious 10.1.3 (Kearse et al., 2012).
MLST Typing
The new sequences obtained for each MLST locus for these four strains were then compared with the sequences available from the five assembled genomes (available on Genbank) and 80 available partial genomes (Silayeva et al., 2020). Numbers were assigned arbitrarily to identify each distinct allele type (AT) at each locus. Each unique combination of AT defined a corresponding sequence type (ST), i.e., a group that shares the same combination of alleles at all eight loci. A clonal complex (CC) was defined as a group of ST linked as single-locus variants to at least another ST within the CC. Clonal complexes and evolutionary relationships between isolates were analyzed with a minimum spanning tree using the global optimal eBURST algorithm implemented in PHYLOViZ (Feil et al., 2004; Francisco et al., 2012).
Data Analysis
The discriminatory power of the MLST scheme was measured using the Simpson’s index (Hunter and Gaston, 1988). This index measures the probability for two randomly sampled strains to present different STs. 95% confidence interval for this index was calculated using the Comparing Partitions website.1 The standardized index of association, IAS, was calculated both on the complete data set (89 strains) and on a subset representing each ST only once (13 STs) using LIAN 3.7 (Haubold and Hudson, 2000).2 IAS is used to evaluate the role of recombination in population evolution. A value of zero corresponds to frequent recombination events while clonal populations are identified by an IAs value significantly differing from zero (Smith et al., 1993). LIAN 3.7 was also used to calculate the genetic diversity (H) at each locus, analyzed as a measure of their expected genetic variability. This value ranges from 0 (no diversity) to 1. The number of variable nucleotide sites, non-synonymous (dN) and synonymous substitutions (dS) as well as Z test of selection on coding regions were calculated using Mega7 (Kumar et al., 2016). The Z test evaluates the null hypothesis of strict neutrality (dN =dS) versus the alternative hypotheses of purifying (dN <dS) or positive (dN >dS) selection (Nei and Gojobori, 1986) and was performed on coding regions only, using 1,000 bootstrap replications and Jukes-Cantor adjustments.
Accession Numbers of Sequences
DNA sequences of the alleles determined in this study for each locus were deposited on Genbank under the accession numbers MN327582 to MN327612. Accession numbers for each allele of the MLST are provided in Table 3. The scheme is curated and available to the public via pubMLST (Jolley et al., 2018) at: https://pubmlst.org/organisms/streptococcus-iniae/
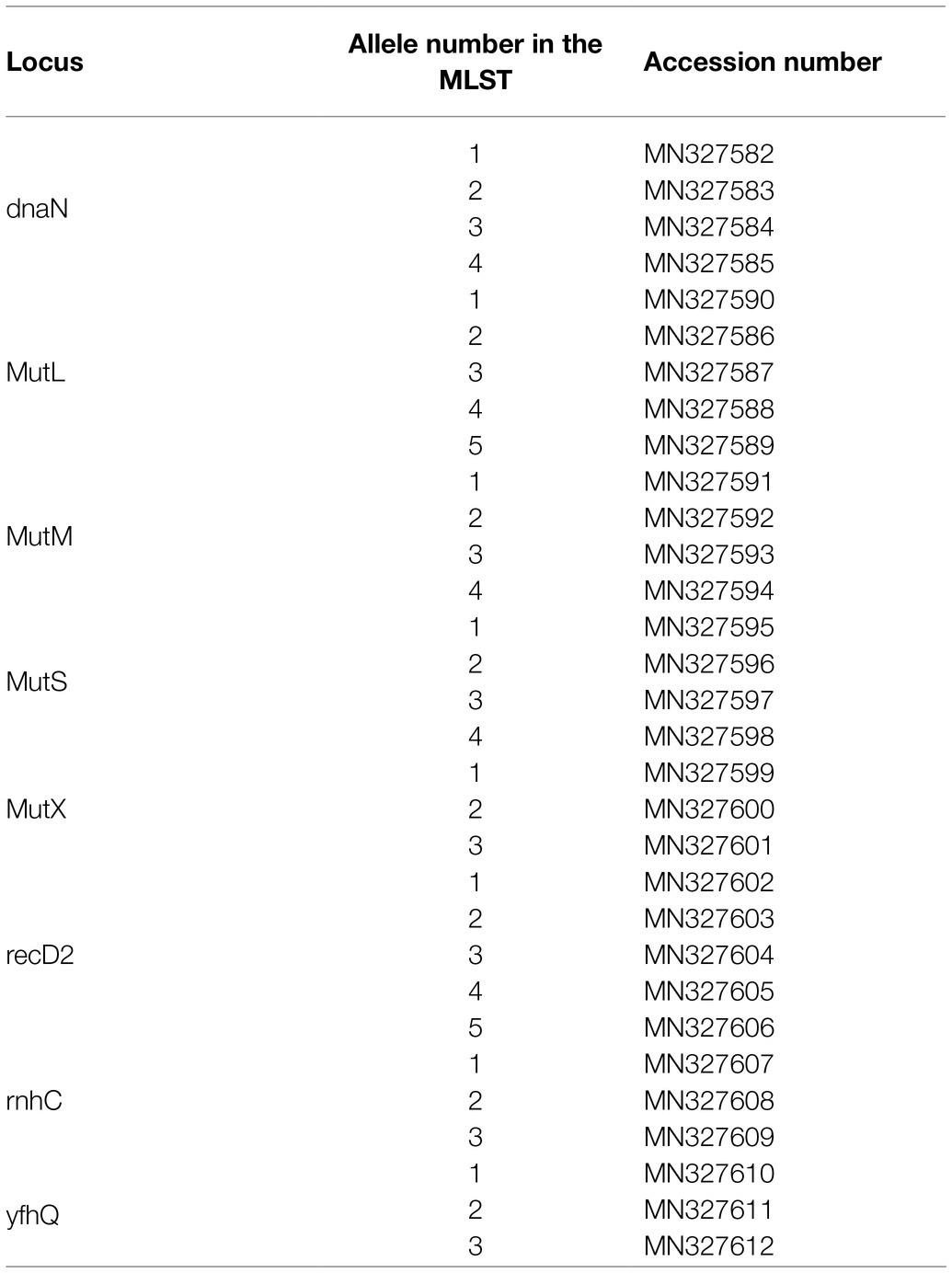
Table 3. Accession number of the MLST scheme.
Results
Locus Genetic Characteristics and Discriminatory Power of MLST
A total combined sequence length of 4,010 nucleotides was obtained when concatenating all eight housekeeping loci. Fragment lengths ranged from 404bp (dnaN) to 582bp (rnhC). Considering all 89 strains, the number of alleles for a locus varied from three (mutX and yfhQ) to five (recD2 and mutL) and the number of polymorphic sites from four (dnaN, mutX and rnhC) to 15 (mutS). The genetic diversity, estimated by the H index, reached a minimum of 0.0447 (rnhC) and a maximum of 0.3639 (mutS). The main characteristics of genetic diversity for each locus are summarized in Table 2.
For most genes, synonymous substitutions (dS) were more frequent than non-synonymous substitutions (dN). Only mutS and rnhC had more non-synonymous substitutions than synonymous substitutions but this result was not statistically significant (Table 2). The higher number of synonymous substitutions over non-synonymous substitutions could indicate that these loci are under neutral or purifying selection. However, the Z test for purifying selection was not significant for any loci. LIAN analysis of linkage disequilibrium based on the eight housekeeping genes suggested that the bacterial population analyzed in this study is in linkage disequilibrium when considering all 89 strains (IAS=0.1348; p<0.01), or a subset consisting of the 13 unique STs (IAS=0.1839; p<0.01). The discriminatory power of this MLST estimated by the Simpson’s index was 0.761 with a confidence interval of 95% between 0.677 and 0.845.
Population Structure
The 89 strains analyzed in this study were grouped into 13 different sequence types (STs) and two clonal complexes (CCs; Figure 2). From the 13 STs, three (ST1, ST5 and ST12) were represented by a single isolate. ST1 corresponds to one of the non-fish hosts, an isolate obtained from a dolphin (Inia geoffrensis) in 1978. This ST was the most genetically distant and shared no alleles with any of the other 12 STs (Table 1 and Figure 2). ST5 was obtained from a giant snakehead (Channa micropeltes) isolated in Thailand in 1983 and ST12 from a red drum (Sciaenops ocellatus) in an offshore aquaculture facility in Reunion Island. The other STs comprised between two and 41 isolates each. CC1 was represented by sixty-eight isolates (76,40% of included isolates) and CC2 by nine isolates (23.60% of included isolates). Within CC1, 41 isolates (46.07% of the collection) belonged to ST4, which is considered by goeBURST analysis as the ancestral type or founder of CC1. As such, ST4 is the most common S. iniae ST in our collection and has been isolated from multiple geographical origins (United States, China, Taiwan, Australia, Reunion Island) over a large period of time (1976 to 2016). This ST has been found associated in our study to different fish species and a dolphin. CC2 comprised two distinct STs: ST10 (seven isolates) and ST9 (two isolates). ST10 is represented by vaccine escape strains sampled in Australian barramundi farms in 2009, considered as hypermutators (Barnes and Silayeva, 2016) whereas ST9 included the only Canadian isolate from fish and one of the two Thai isolates, again obtained from fish.
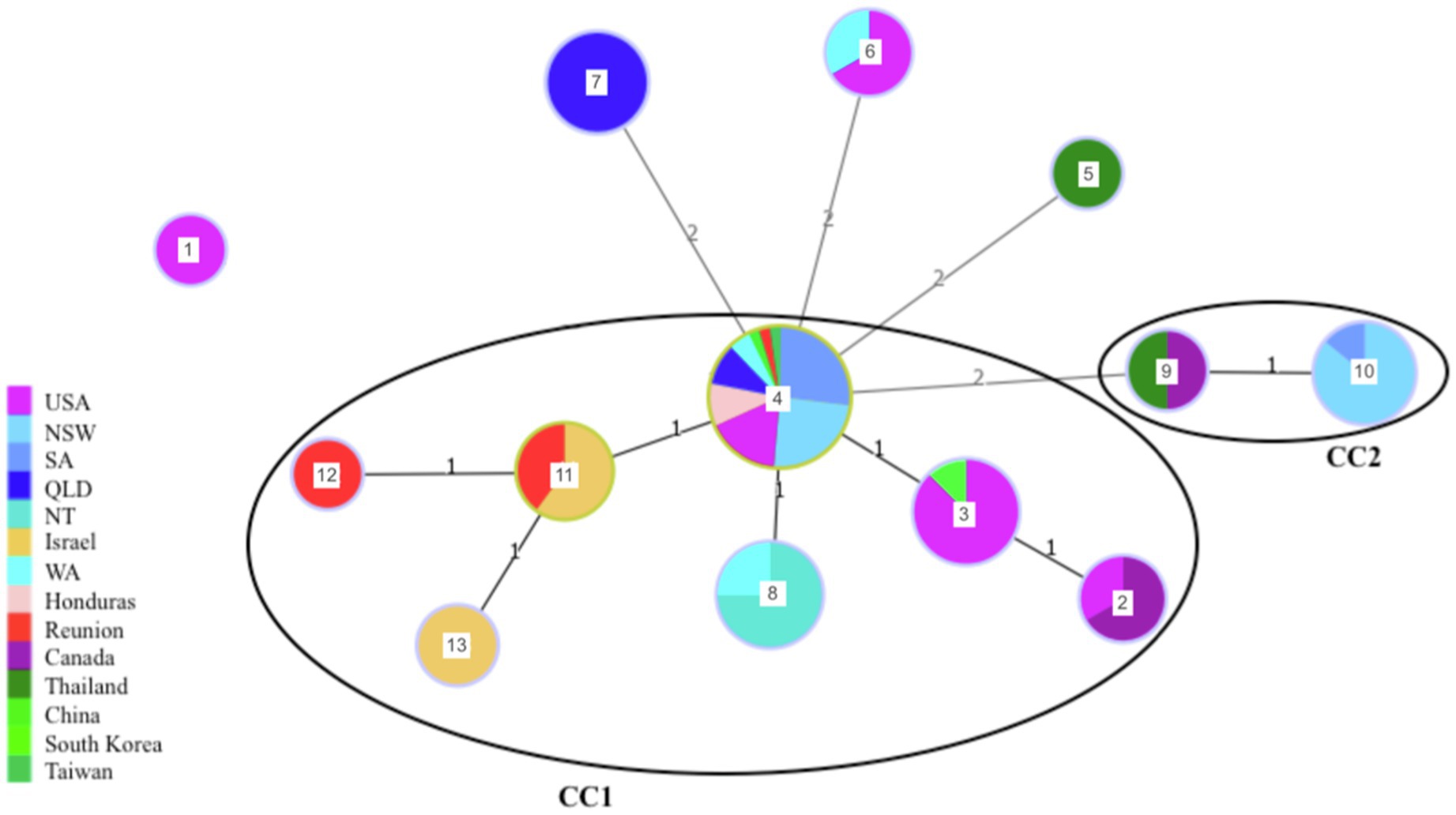
Figure 2. Minimum spanning tree of S. iniae collection illustrating the evolutionary relationships between isolates depending on their geographical origin. Each circle represents a ST and the size of the circle is proportional to the number of isolates within this ST (log scale). ST number is indicated at the center of each circle. Numbers over the connecting lines indicate the number of locus difference between the closest STs (1: SLV; 2: DLV). Clonal complexes are shown (7 of eight alleles shared).
When the CC definition was relaxed (6/8 shared alleles), all STs except ST1 formed a single network (Figure 2) indicating close evolutionary relationships between the different STs. However, some STs seemed to be specific of a geographical area, such as ST7, which is found only in Queensland. Further, ST12 and ST13 have a limited geographical distribution (Reunion Island and Israel, respectively) and could be considered as descendants of ST11, which included earlier freshwater aquaculture isolates from these regions (trout and tilapia), as well as the first isolate involved in a reef epizootic in 2002 in Reunion Island. Altogether, despite a strong bias toward a few fish species, no clear host specificity could be detected, with isolates retrieved from mammals sharing STs isolated from fish as well (ST2, ST3, ST4, ST6). For instance, ST2 and ST3 were represented by fish and human isolates from North America, ST4 by fish and dolphin isolates, while ST6 included bat and ornamental fish isolates (Table 1).
Discussion
Discriminatory Power of the MLST Scheme
Considering the 89 strains tested within our study, the MLST scheme produced a Simpson’s Index of Diversity (SID) of 0.761, i.e., two strains randomly sampled in our collection have an average probability of 0.761 to display different STs. The genetic diversity estimated by the SID is well below 0.90, considered as the desirable threshold to interpret typing results with confidence (Hunter and Gaston, 1988). The observed lack of diversity in the S. iniae strains assed in this study is in keeping with other studies using MLST and indicates that it may be hard to find sufficient for highly-descriminatory MLST variation in S. iniae core genome genes, despite the remarkable variability in pangenome including virulence and antigenicity regions such as capsule biosynthesis genes (Lowe et al., 2007; Heath et al., 2016; Zeng et al., 2016). As a result of this, one major advantage of the use of MLST over say Multiple-Locus Variable number tandem repeat Analysis (MLVA) for example, is the use of these slowly evolving housekeeping genes (Jenke et al., 2011). This allows researchers to more reliably deduce long-term evolutionary events which may occur. However, that said, MLVA might still have higher discriminatory power and be useful to distinguish closely related serotypes or to monitor vaccine escape strains as and when needed.
Multiple Emergence of This Pathogen on Reunion Island
Although we are unable to ascertain the exact origin of the S. iniae strains responsible for the epizootics on Reunion Island we are able to generate a number of hypotheses from our data. Indeed, the strains isolated during the different epizootics on Reunion Island belonged to different STs. This pattern does not support the emergence of a specific clone adapted to the local environment setup. Early strains isolated in 1996 (from a freshwater aquaculture facility) and 2002 (one of the reef fish epizootics) both belong to ST11, which also comprises three Israeli S. iniae strains isolated in 1989, 1998 and 2000 from Tilapia and rainbow trout aquaculture facilities (Table 1 and Figure 2). The strain isolated in 2009 on Reunion Island was obtained from diseased red drums in an offshore farm and belonged to ST12, itself a direct descendant of ST11. This suggests a common origin of these early Reunionese strains with Israeli aquaculture strains, followed by a local diversification. Even though these two countries are geographically distant, the pathogen could have been introduced to the Island via carrier fish import for aquaculture development on Reunion Island and/or aquarists. Israel was indeed, in the early 1990s, a major exporter of tilapia breeding lines for the growing tilapia industry worldwide (Popma and Lovshin, 1996). Unfortunately, no information regarding the origin of the diseased rainbow trout from the 1996 epizootic in Reunion, or of other fishes coexisting with them at that time in the farm, such as Tilapia, was available to confirm this hypothesis. The likely freshwater origin of the strain isolated during the 2002 epizootic is in keeping with the ecological plasticity of S. iniae, able to survive and adapt from a fresh water aquaculture facility around 900 meters above sea level to coastal environments, and across several host species. The example of ST8 and the probable spread of S. iniae in Australian barramundi, from freshwater aquaculture to sea cages tends to confirm that this hypothesis is plausible. Transmission of S. iniae from caged to wild fish has been suggested in the past (Colorni et al., 2002), nevertheless, to our knowledge, it is the first report of the possible spread of a pathogenic strain from freshwater aquaculture to reef fish. It is important to note that trout from the Reunionese facility affected by S. iniae in 1996 were later released into the rivers surrounding the farm in order to develop recreational fishing (Fédération de la pêche de La Réunion). As the first reef fish epizootic in 2002 originated from the same ST (ST11) as the one affecting trout from the freshwater aquaculture facility in 1996, this underlines the importance of developing robust and sensitive molecular tools to screen animals for pathogens before any release into the wild.
In contrast, the S. iniae strain involved during the last 2014 epizootic was identified as different from those isolated in 1996, 2002 and 2009 and matched the more ancestral and cosmopolitan strain, ST4. A double reversion of MutL and MutM would be needed to obtain the haplotype found in 2014 from the one found in 2009. This seems highly unlikely considering the very low mutation rates in DNA repair genes MutL and MutM (Denamur and Matic, 2006). As a consequence, we propose that this last epidemic results from the local emergence of a S. iniae lineage of cosmopolitan distribution.
Tentative Commentary on Genetic Diversity of S. iniae Worldwide
Only 13 different STs belonging to two CCs were found within our collection of 89 strains. Moreover, a unique ST (ST4) representing 46.07% of our sample was found on all continents (North America, Australia, Asia). Clearly, the choice of critical functional genes as MLST loci will constrain diversification, as they are likely under strong purification selection. Consistently, the genetic diversity of housekeeping loci included into the scheme was low. However, this lack of variability might result from a sampling bias towards pathogenic strains from aquaculture. Indeed, it should be noted that further work on this topic including a greater range of samples may show a greater diversity in housekeeping genes. Of the 89 isolates examined, 62 are from only two species (tilapia and barramundi) and four locations. 48 originate from Australia and found in Barramundi, and 14 are from Tilapia (from Israel, United States and Hondura). That leaves only 13 strains originating from different fish species. We were also only able to map these to seven from human origin and two from dolphins. Therefore, the samples may be biased in both the host species and geographical distribution. We also did not include non-virulent strains or those solely associated with the environment as these were not available. It would be interesting to include more non-pathogenic strains in order to determine whether this lack of diversity is specific to pathogenic strains or common to environmental and/or avirulent strains, less subject to host immune selection. However, at the moment, it is still hard to isolate environmental strains. Indeed, confirmation of isolate identity as S. iniae by commercial bacterial identification kits is still problematic due to the biochemical profile being absent from databases supplied with the kits or variability of some atypical S. iniae strains (Roach et al., 2006). Moreover, most of the epizootics caused by this species occur in developing countries and remain underreported. Nevertheless, atypical strains isolated from mammals were almost always found in an ST also including strains isolated from aquaculture or ornamental fish. As such, increasing the number of host species may not necessarily increase S. iniae’s diversity and the results obtained in this study may constitute an informative baseline to be completed in the future to reassess the diversity of this pathogen on a broader host and geographical range.
Alternatively, we suggest that the low diversity of housekeeping genes may be due to a recent speciation of S. iniae, as hypothesized for other streptococci such as S. thermophiles (Delorme et al., 2010) or S. agalactiae (Brochet et al., 2006), both also known fish pathogens in their own right (Pereira et al., 2010; Bowater et al., 2012). As far as S. agalactiae is concerned, the commonly used MLST of Jones et al. (2003) was not sufficiently resolutive to define the population structure of the pathogen in aquatic hosts, and new typing systems were developed, including combination of MLST (sequencing of up to 15 housekeeping genes), serotyping and presence/absence of virulence associated genes (Sørensen et al., 2010; Delannoy et al., 2013; Godoy et al., 2013). These combined approaches might allow resolving the main phylogenetic events, but are time consuming and costly. The low diversity observed herein may also result from a recent bottleneck history, reducing the size and diversity of S. iniae populations. In many instances, genetically monomorphic pathogens have undergone such bottlenecks, for example following a crucial genetic event, causing a change in ecological niche (Achtman, 2008).
Ecological Flexibility
The low diversity observed in the MLST seems associated to a great ecological flexibility of this pathogen. Our data confirms that S. iniae does not appear to have any major host specificity (Agnew and Barnes, 2007). Although most of our sampling originates from farmed fish, where it causes the highest mortalities, the few available STs obtained from infections in mammals (including humans) did not cluster together but rather with STs obtained from other fish isolates (Table 1; ST2, ST3), suggesting transmission from farmed fish to humans (Weinstein et al., 1997; Facklam et al., 2005). Genomic data indicate that a host jump might have been facilitated by rapid mutations (Silayeva et al., 2020). This ecological flexibility, underlined by the apparent lack of host specificity of S. iniae, is further emphasized by the ability of a particular clone to survive both in fresh and seawater. As already discussed, ST11 groups with early strains isolated from freshwater aquaculture in Israel and Reunion Island as well as the first strain responsible for a reef fish epizootic on Reunion Island. ST8 is another example of ST genotyped from barramundi freshwater farms in Western Australia (2004) and from sea cages in the Northern Territory (2005–2006; Table 1). This ST has been associated with important fish loss since 2004 in these two regions where S. iniae was not described before (Creeper and Buller, 2006). Although both Western Australia and Northern Territory strains were genotyped as ST8, Nawawi et al. (2009) showed that Northern Territory isolates were further characterized by a mutation on the lactate oxidase gene (lctO) and able to process lactate at a faster rate than the other strains (Nawawi et al., 2009). This change has been attributed to environmental influences of large tidal flows linked with increased swimming activity of the barramundi hosts in seawater, that have led to the evolution of the lctO gene variant, encoding a more efficient enzyme in these specific isolates (Nawawi et al., 2009). Although our MLST does not highlight this kind of rapid discrete mutation, it does enable retracing the probable common origin of isolates from different locations. Further, MLST enables the exploration of ecological flexibility of S. iniae, indicating the ability to switch to various hosts and habitats (freshwater vs. seawater).
Conclusion
Although MLST based on the sequencing of a few housekeeping genes was developed almost 20years ago, it is still a rapid, convenient and relevant method to shed light on the origin and long term evolution of bacteria (Pérez-Losada et al., 2013; Jolley and Maiden, 2014). We selected eight housekeeping genes that were able to distinguish the main S. iniae phylogenetic clades and this enabled us to explore both origin and evolution of S. iniae causing mass fish die offs on the remote Reunion Island. The MLST scheme suggests at least two different origins of the pathogen causing epizootics on the island. The strains involved in 1996, 2002 and 2009 epizootics are genetically closely related and probably share a common ancestor with aquaculture strains from Israel, whereas the strain isolated in 2014 belongs to a more cosmopolitan ST. In addition, results of our MLST seemed to indicate an ecological flexibility of this pathogen, with some strains able to infect mammals as well as fish hosts or colonize both fresh and seawater environments. Despite or maybe because of this ecological flexibility, we observed a low global genetic diversity of this pathogen. That said, we cannot ignore this result may reflect a sampling bias towards aquaculture fish species most affected by this pathogen. However, we still encourage the use of this quick and convenient tool to document the diversity of this pathogen on new host species, regions, and environmental sources when available. Finally, considering the low genetic diversity of S. iniae, the results obtained to investigate the recurrent epizootics on Reunion Island underline the usefulness of this MLST scheme to monitor disease emergence, retrace possible transmission routes and investigate the evolution of this pathogen over recurring epizootics.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics Statement
The animal study was reviewed and approved by Marine Reserve of Reunion Island.
Author Contributions
SI and MS conceived and designed the experiments, performed the experiments, analyzed the data, contributed to reagents, materials, and analysis tools, wrote the paper, prepared figures and/or tables, and reviewed drafts of the paper. MS wrote the paper, prepared figures and/or tables, and reviewed drafts of the paper. OS, PC, and AB conceived and designed the experiments, wrote the paper, prepared figures and/or tables, and reviewed drafts of the paper. PT conceived and designed the experiments, performed the experiments, contributed to reagents, materials, and analysis tools, wrote the paper, prepared figures and/or tables, and reviewed drafts of the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was cofunded in the frame of the REMPOR project by the European Union (EU, FEDER), the Regional Council of Reunion, and the French Department of Ecology, Sustainable Development, Transportation and Housing (DEAL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all staff members of the Natural Marine Reserve of Reunion Island (RNMR) for their assistance in the field. We further acknowledge Dr. Patrick Daniel from Pyrenees and Landes Laboratory for his guidance in the Lab and for providing us with live strains of S. iniae. All authors of this manuscript declare no conflict of interests inherent to this submission.
Footnotes
1. ^http://www.comparingpartitions.info
2. ^http://guanine.evolbio.mpg.de/cgi-bin/lian/lian.cgi.pl/query
References
Achtman, M. (2008). Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu. Rev. Microbiol. 62, 53–70. doi: 10.1146/annurev.micro.62.081307.162832
Agnew, W., and Barnes, A. C. (2007). Streptococcus iniae: an aquatic pathogen of global veterinary significance and a challenging candidate for reliable vaccination. Vet. Microbiol. 122, 1–15. doi: 10.1016/j.vetmic.2007.03.002
Bachrach, G., Zlotkin, A., Hurvitz, A., Evans, D. L., and Eldar, A. (2001). Recovery of streptococcus iniae from diseased fish previously vaccinated with a streptococcus vaccine. Appl. Environ. Microbiol. 67, 3756–3758. doi: 10.1128/AEM.67.8.3756-3758.2001
Barnes, A. C., and Silayeva, O. (2016). Vaccination against streptococcal infections in farmed fish. Microbiol. Aust. 37, 118–121. doi: 10.1071/MA16040
Bowater, R. O., Forbes-Faulkner, J., Anderson, I. G., Condon, K., Robinson, B., Kong, F., et al. (2012). Natural outbreak of Streptococcus agalactiae (GBS) infection in wild giant Queensland grouper, Epinephelus lanceolatus (Bloch), and other wild fish in northern Queensland. Australia. J. Fish Dis. 35, 173–186. doi: 10.1111/j.1365-2761.2011.01332.x
Brochet, M., Couvé, E., Zouine, M., Vallaeys, T., Rusniok, C., Lamy, M.-C., et al. (2006). Genomic diversity and evolution within the species Streptococcus agalactiae. Microbes Infect. 8, 1227–1243. doi: 10.1016/j.micinf.2005.11.010
Bromage, E. S., Thomas, A., and Owens, L. (1999). Streptococcus iniae, a bacterial infection in barramundi Lates calcarifer. Dis. Aquat. Org. 36, 177–181. doi: 10.3354/dao036177
Colorni, A., Diamant, A., Eldar, A., Kvitt, H., and Zlotkin, A. (2002). Streptococcus iniae infections in Red Sea cage-cultured and wild fishes. Dis. Aquat. Org. 49, 165–170. doi: 10.3354/dao049165
Creeper, J. H., and Buller, N. B. (2006). An outbreak of streptococcus iniae in barramundi (Lates calcarifera) in freshwater cage culture. Aust. Vet. J. 84, 408–411. doi: 10.1111/j.1751-0813.2006.00058.x
Delannoy, C. M., Crumlish, M., Fontaine, M. C., Pollock, J., Foster, G., Dagleish, M. P., et al. (2013). Human Streptococcus agalactiae strains in aquatic mammals and fish. BMC Microbiol. 13:41. doi: 10.1186/1471-2180-13-41
Delorme, C., Bartholini, C., Bolotine, A., Ehrlich, S. D., and Renault, P. (2010). Emergence of a cell wall protease in the Streptococcus thermophilus population. Appl. Environ. Microbiol. 76, 451–460. doi: 10.1128/AEM.01018-09
Denamur, E., and Matic, I. (2006). Evolution of mutation rates in bacteria. Mol. Microbiol. 60, 820–827. doi: 10.1111/j.1365-2958.2006.05150.x
Dodson, S. V., Maurer, J. J., and Shotts, E. B. (1999). Biochemical and molecular typing of streptococcus iniae isolated from fish and human cases. J. Fish Dis. 22, 331–336. doi: 10.1046/j.1365-2761.1999.00170.x
El Aamri, F., Padilla, D., Acosta, F., Caballero, M. J., Roo, J., Bravo, J., et al. (2010). First report of streptococcus iniae in red porgy (Pagrus pagrus, L.). J. Fish Dis. 33, 901–905. doi: 10.1111/j.1365-2761.2010.01191.x
Eldar, A., Bejerano, Y., and Bercovier, H. (1994). Streptococcus shiloi and Streptococcus difficile: two new streptococcal species causing a meningoencephalitis in fish. Curr. Microbiol. 28, 139–143. doi: 10.1007/BF01571054
Eldar, A., Horovitcz, A., and Bercovier, H. (1997a). Development and efficacy of a vaccine against streptococcus iniae infection in farmed rainbow trout. Vet. Immunol. Immunopathol. 56, 175–183. doi: 10.1016/S0165-2427(96)05738-8
Eldar, A., Lawhon, S., Frelier, F. P., Assenta, L., Simpson, B. R., Varner, P. W., et al. (1997b). Restriction fragment length polymorphisms of 16S rDNA and of whole rRNA genes (ribotyping) of streptococcus iniae strains from the United States and Israel. FEMS Microbiol. Lett. 151, 155–162. doi: 10.1111/j.1574-6968.1997.tb12564.x
Eldar, A., Perl, S., Frelier, P. F., and Bercovier, H. (1999). Red drum Sciaenops ocellatus mortalities associated with streptococcus iniae infection. Dis. Aquat. Org. 36, 121–127. doi: 10.3354/dao036121
Erfanmanesh, A., Soltani, M., Pirali, E., Mohammadian, S., and Taherimirghaed, A. (2012). Genetic characterization of streptococcus iniae in diseased farmed rainbow trout (Onchorhynchus mykiss) in Iran. Sci. World J. 2012:594073. doi: 10.1100/2012/594073
Eyngor, M., Tekoah, Y., Shapira, R., Hurvitz, A., Zlotkin, A., Lublin, A., et al. (2008). Emergence of novel streptococcus iniae exopolysaccharide-producing strains following vaccination with nonproducing strains. Appl. Environ. Microbiol. 74, 6892–6897. doi: 10.1128/AEM.00853-08
Facklam, R., Elliott, J., Shewmaker, L., and Reingold, A. (2005). Identification and characterization of sporadic isolates of streptococcus iniae isolated from humans. J. Clin. Microbiol. 43, 933–937. doi: 10.1128/JCM.43.2.933-937.2005
Fadaeifard, F., Raissy, M., Momtaz, H., and Zahedi, R. (2011). Detection of streptococcus iniae by polymerase chain reaction in rainbow trout (Oncorhynchus mykiss) in West Iran. Afr. J. Microbiol. Res. 5, 4722–4724. doi: 10.5897/AJMR11.674
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004
Ferguson, H. W., John, V. S. S., Roach, C. J., Willoughby, S., Parker, C., and Ryan, R. (2000). Caribbean reef fish mortality associated with streptococcus iniae. Vet. Rec. 147, 662–664.
Figueiredo, H. C. P., Nobrega Netto, L., Leal, C. A. G., Pereira, U. P., and Mian, G. F. (2012). Streptococcus iniae outbreaks in Brazilian Nile tilapia (Oreochromis niloticus L) farms. Braz. J. Microbiol. 43, 576–580. doi: 10.1590/S1517-83822012000200019
Francis, P. M., Suming, Z., Jiting, Z., Xiaoye, Z., Shuwei, A., and Guoliang, W. (2014). Massive mortality associated with streptococcus iniae infection in cage-cultured red drum (Sciaenops ocellatus) in eastern China. Afr. J. Microbiol. Res. 8, 1722–1729. doi: 10.5897/AJMR2014.6659
Francisco, A. P., Vaz, C., Monteiro, P. T., Melo-Cristino, J., Ramirez, M., and Carriço, J. A. (2012). PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinform. 13:87. doi: 10.1186/1471-2105-13-87
Fuller, J. D., Bast, D. J., Nizet, V., Low, D. E., and de Azavedo, J. C. S. (2001). Streptococcus iniae virulence is associated with a distinct genetic profile. Infect. Immun. 69, 1994–2000. doi: 10.1128/IAI.69.4.1994-2000.2001
Godoy, D. T., Carvalho-Castro, G. A., Leal, C. A. G., Pereira, U. P., Leite, R. C., and Figueiredo, H. C. P. (2013). Genetic diversity and new genotyping scheme for fish pathogenic Streptococcus agalactiae. Lett. Appl. Microbiol. 57, 476–483. doi: 10.1111/lam.12138
Haubold, B., and Hudson, R. R. (2000). LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics 16, 847–849. doi: 10.1093/bioinformatics/16.9.847
Heath, C., Gillen, C. M., Chrysanthopoulos, P., Walker, M. J., and Barnes, A. C. (2016). Streptococcus iniae cpsG alters capsular carbohydrate composition and is a cause of serotype switching in vaccinated fish. Vet. Microbiol. 193, 116–124. doi: 10.1016/j.vetmic.2016.08.011
Hunter, P. R., and Gaston, M. A. (1988). Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J. Clin. Microbiol. 26, 2465–2466. doi: 10.1128/jcm.26.11.2465-2466.1988
Jenke, C., Lindstedt, B. A., Harmsen, D., Karch, H., Brandal, L. T., and Mellmann, A. (2011). Comparison of multilocus variable-number tandem-repeat analysis and multilocus sequence typing for differentiation of hemolytic-uremic syndrome-associated Escherichia coli (HUSEC) collection strains. J. Clin. Microbiol. 49, 3644–3646. doi: 10.1128/JCM.05035-11
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Welcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Jolley, K., and Maiden, M. (2014). Using MLST to study bacterial variation: prospects in the genomic era. Future Microbiol. 9, 623–630. doi: 10.2217/fmb.14.24
Jones, N., Bohnsack, J. F., Takahashi, S., Oliver, K. A., Chan, M.-S., Kunst, F., et al. (2003). Multilocus sequence typing system for group B streptococcus. J. Clin. Microbiol. 41, 2530–2536. doi: 10.1128/JCM.41.6.2530-2536.2003
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Keirstead, N. D., Brake, J. W., Griffin, M. J., Halliday-Simmonds, I., Thrall, M. A., and Soto, E. (2014). Fatal septicemia caused by the zoonotic bacterium streptococcus iniae during an outbreak in Caribbean reef fish. Vet. Pathol. 51, 1035–1041. doi: 10.1177/0300985813505876
Klesius, P. H., Shoemaker, C. A., and Evans, J. J. (2000). Efficacy of single and combined streptococcus iniae isolate vaccine administered by intraperitoneal and intramuscular routes in tilapia (Oreochromis niloticus). Aquaculture 188, 237–246. doi: 10.1016/S0044-8486(00)00345-8
Koh, T. H., Kurup, A., and Chen, J. (2004). Streptococcus iniae discitis in Singapore. Emerg. Infect. Dis. 10, 1694–1696. doi: 10.3201/eid1009.040029
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kvitt, H., and Colorni, A. (2004). Strain variation and geographic endemism in streptococcus iniae. Dis. Aquat. Org. 61, 67–73. doi: 10.3354/dao061067
Lau, S. K. P., Woo, P. C. Y., Luk, W., Fung, A. M. Y., Hui, W., Fong, A. H. C., et al. (2006). Clinical isolates of streptococcus iniae from Asia are more mucoid and β-hemolytic than those from North America. Diagn. Microbiol. Infect. Dis. 54, 177–181. doi: 10.1016/j.diagmicrobio.2005.09.012
Lowe, B. A., Miller, J. D., and Neely, M. N. (2007). Analysis of the polysaccharide capsule of the systemic pathogen streptococcus iniae and its implications in virulence. Infect. Immun. 75, 1255–1264. doi: 10.1128/IAI.01484-06
Maiden, M. C. J. (2006). Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 60, 561–588. doi: 10.1146/annurev.micro.59.030804.121325
Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U. S. A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Mata, A. I., Blanco, M. M., Dominguez, L., Fernández-Garayzábal, J. F., and Gibello, A. (2004). Development of a PCR assay for streptococcus iniae based on the lactate oxidase (lctO) gene with potential diagnostic value. Vet. Microbiol. 101, 109–116. doi: 10.1016/j.vetmic.2004.03.012
Millard, C. M., Baiano, J. C. F., Chan, C., Yuen, B., Aviles, F., Landos, M., et al. (2012). Evolution of the capsular operon of streptococcus iniae in response to vaccination. Appl. Environ. Microbiol. 78, 8219–8226. doi: 10.1128/AEM.02216-12
Nawawi, R. A., Baiano, J., and Barnes, A. C. (2008). Genetic variability amongst streptococcus iniae isolates from Australia. J. Fish Dis. 31, 305–309. doi: 10.1111/j.1365-2761.2007.00880.x
Nawawi, R. A., Baiano, J. C. F., Kvennefors, E. C. E., and Barnes, A. C. (2009). Host-directed evolution of a novel lactate oxidase in streptococcus iniae isolates from barramundi (Lates calcarifer). Appl. Environ. Microbiol. 75, 2908–2919. doi: 10.1128/AEM.02147-08
Nei, M., and Gojobori, T. (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426. doi: 10.1093/oxfordjournals.molbev.a040410
Nguyen, H. T., Kanai, K., and Yoshikoshi, K. (2002). Ecological investigation of streptococcus iniae in cultured Japanese flounder (Paralichthys olivaceus) using selective isolation procedures. Aquaculture 205, 7–17. doi: 10.1016/S0044-8486(01)00667-6
Pereira, U. P., Mian, G. F., Oliveira, I. C. M., Benchetrit, L. C., Costa, G. M., and Figueiredo, H. C. P. (2010). Genotyping of Streptococcus agalactiae strains isolated from fish, human and cattle and their virulence potential in Nile tilapia. Vet. Microbiol. 140, 186–192. doi: 10.1016/j.vetmic.2009.07.025
Perera, R. P., Johnson, S. K., Collins, M. D., and Lewis, D. H. (1994). Streptococcus iniae associated with mortality of Tilapia nilotica × T. aurea hybrids. J. Aquat. Anim. Health 6, 335–340. doi: 10.1577/1548-8667(1994)006<0335:SIAWMO>2.3.CO;2
Pérez-Losada, M., Cabezas, P., Castro-Nallar, E., and Crandall, K. A. (2013). Pathogen typing in the genomics era: MLST and the future of molecular epidemiology. Infect. Genet. Evol. 16, 38–53. doi: 10.1016/j.meegid.2013.01.009
Pier, G. B., and Madin, S. H. (1976). Streptococcus iniae sp. nov., a beta-hemolytic streptococcus isolated from an Amazon freshwater dolphin, Inia geoffrensis. Int. J. Syst. Evol. Microbiol. 26, 545–553. doi: 10.1099/00207713-26-4-545
Popma, T. J., and Lovshin, L. L. (1996). Worldwide Prospects for Commercial Production of Tilapia. United States: International Center for Aquaculture and Aquatic Environments Alabama.
Pothin, K., Quod, J.-P., Cauvin, B., Melot, P., Turquet, J., and Conéjero, S. (2001). Recherche des causes des mortalités de poissons de fond dans le secteur Ouest de La Réunion (du 8/11 au 31/12/2000); December 31, 2000.
Quod, J.-P., Turquet, J., Cambert, H., Mulochau, T., and Durville, P. (2014). Episode 2014 de mortalité massive des poissons de la côte Ouest de La Réunion. Direction régionale de l’Environnement de la Réunion, Saint-Denis, Réunion.
Roach, J. C. M., Levett, P. N., and Lavoie, M. C. (2006). Identification of streptococcus iniae by commercial bacterial identification systems. J. Microbiol. Methods 67, 20–26. doi: 10.1016/j.mimet.2006.02.012
Shoemaker, C. A., LaFrentz, B. R., Klesius, P. H., and Evans, J. J. (2010). Protection against heterologous streptococcus iniae isolates using a modified bacterin vaccine in Nile tilapia, Oreochromis niloticus (L.). J. Fish Dis. 33, 537–544. doi: 10.1111/j.1365-2761.2010.01148.x
Silayeva, O., Engelstädter, J., and Barnes, A. C. (2020). Evolutionary epidemiology of streptococcus iniae: linking mutation rate dynamics with adaptation to novel immunological landscapes. Infect. Genet. Evol. 85:104435. doi: 10.1016/j.meegid.2020.104435
Smith, J. M., Smith, N. H., O’Rourke, M., and Spratt, B. G. (1993). How clonal are bacteria? Proc. Natl. Acad. Sci. U. S. A. 90, 4384–4388.
Song, Z., Yue, R., Sun, Y., Liu, C., Khan, S. H., Li, C., et al. (2017). Fatal bacterial septicemia in a bottlenose dolphin Tursiops truncatus caused by streptococcus iniae. Dis. Aquat. Org. 122, 195–203. doi: 10.3354/dao03069
Sørensen, U. B. S., Poulsen, K., Ghezzo, C., Margarit, I., and Kilian, M. (2010). Emergence and global dissemination of host-specific Streptococcus agalactiae clones. MBio 24:e00178-10. doi: 10.1128/mBio.00178-10
Stich, M., Manrubia, S. C., and Lázaro, E. (2010). Variable mutation rates as an adaptive strategy in replicator populations. PLoS One 5:e11186. doi: 10.1371/journal.pone.0011186
Stoffregen, D. A., Backman, S. C., Perham, R. E., Bowser, P. R., and Babish, J. G. (1996). Initial disease report of streptococcus iniae infection in hybrid striped (sunshine) bass and successful therapeutic intervention with the fluoroquinolone antibacterial enrofloxacin. J. World Aquac. Soc. 27, 420–434. doi: 10.1111/j.1749-7345.1996.tb00626.x
Toranzo, A. E., Magariños, B., and Romalde, J. L. (2005). A review of the main bacterial fish diseases in mariculture systems. Aquaculture 246, 37–61. doi: 10.1016/j.aquaculture.2005.01.002
Türe, M., and Alp, H. (2016). Identification of bacterial pathogens and determination of their antibacterial resistance profiles in some cultured fish in Turkey. J. Vet. Res. 60, 141–146. doi: 10.1515/jvetres-2016-0020
Turquet, J., Quod, J.-P., and Wickel, J. (2002). Mortalité massive de poissons coralliens de fond de mars à mai 2002 sur le littoral ouest de La Réunion. Direction régionale de l’Environnement de la Réunion, Saint-Denis, Réunion.
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res. 40:e115. doi: 10.1093/nar/gks596
Weinstein, M. R., Litt, M., Kertesz, D. A., Wyper, P., Rose, D., Coulter, M., et al. (1997). Invasive infections due to a fish pathogen, streptococcus iniae. N. Engl. J. Med. 337, 589–594. doi: 10.1056/NEJM199708283370902
Zeng, Y., He, Y., Wang, K.-Y., Wang, J., Zeng, Y.-K., Chen, Y.-X., et al. (2016). cpsJ gene of streptococcus iniae is involved in capsular polysaccharide synthesis and virulence. Antonie Van Leeuwenhoek 109, 1483–1492. doi: 10.1007/s10482-016-0750-1
Zhou, S. M., Xie, M. Q., Zhu, X. Q., Ma, Y., Tan, Z. L., and Li, A. X. (2008). Identification and genetic characterization of streptococcus iniae strains isolated from diseased fish in China. J. Fish Dis. 31, 869–875. doi: 10.1111/j.1365-2761.2008.00954.x
Keywords: Streptococcus iniae, MLST, epizootics, reef fish, aquaculture
Citation: Irion S, Rudenko O, Sweet M, Chabanet P, Barnes AC, Tortosa P and Séré MG (2021) Molecular Investigation of Recurrent Streptococcus iniae Epizootics Affecting Coral Reef Fish on an Oceanic Island Suggests at Least Two Distinct Emergence Events. Front. Microbiol. 12:749734. doi: 10.3389/fmicb.2021.749734
Edited by:
Carlos Angulo, Centro de Investigación Biológica del Noroeste (CIBNOR), MexicoReviewed by:
Henrique César Pereira Figueiredo, School of Veterinary Medicine, Federal University of Minas Gerais, BrazilPaul Carl Sikkel, Arkansas State University, United States
Copyright © 2021 Irion, Rudenko, Sweet, Chabanet, Barnes, Tortosa and Séré. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mathieu G. Séré, bWF0aGlldS5zZXJlQGdtYWlsLmNvbQ==