 Wimonrat Panpetch1
Wimonrat Panpetch1 Pornpimol Phuengmaung1
Pornpimol Phuengmaung1 Thanya Cheibchalard2
Thanya Cheibchalard2 Naraporn Somboonna2,3
Naraporn Somboonna2,3 Asada Leelahavanichkul1,4*†
Asada Leelahavanichkul1,4*† Somying Tumwasorn1,3*†
Somying Tumwasorn1,3*†- 1Department of Microbiology, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand
- 2Department of Microbiology, Faculty of Science, Chulalongkorn University, Bangkok, Thailand
- 3Microbiome Research Unit for Probiotics in Food and Cosmetics, Chulalongkorn University, Bangkok, Thailand
- 4Department of Microbiology, Faculty of Medicine, Center of Excellence in Immunology and Immune-Mediated Diseases, Bangkok, Thailand
Clostridioides difficile is a major cause of diarrhea in patients with antibiotic administration. Lacticaseibacillus casei T21, isolated from a human gastric biopsy, was tested in a murine C. difficile infection (CDI) model and colonic epithelial cells (Caco-2 and HT-29). Daily administration of L. casei T21 [1 × 108 colony forming units (CFU)/dose] for 4 days starting at 1 day before C. difficile challenge attenuated CDI as demonstrated by a reduction in mortality rate, weight loss, diarrhea, gut leakage, gut dysbiosis, intestinal pathology changes, and levels of pro-inflammatory cytokines [interleukin (IL)-1β, tumor necrosis factor (TNF)-α, macrophage inflammatory protein 2 (MIP-2), and keratinocyte chemoattractant (KC)] in the intestinal tissue and serum. Conditioned media from L. casei T21 exerted biological activities that fight against C. difficile as demonstrated in colonic epithelial cells by the following: (i) suppression of gene expression and production of IL-8, an important chemokine involved in C. difficile pathogenesis, (ii) reduction in the expression of SLC11A1 (solute carrier family 11 member 1) and HuR (human antigen R), important genes for the lethality of C. difficile toxin B, (iii) augmentation of intestinal integrity, and (iv) up-regulation of MUC2, a mucosal protective gene. These results supported the therapeutic potential of L. casei T21 for CDI and the need for further study on the intervention capabilities of CDI.
Introduction
Clostridioides difficile, an anaerobic Gram-positive spore-forming bacillus (Kachrimanidou and Malisiovas, 2011), is one of the important causative organisms of diarrhea in hospitalized patients who receive antibiotics (Kelly et al., 1994b; Bartlett, 2002; Aslam et al., 2005). Clinical symptoms of C. difficile infection (CDI) vary from mild diarrhea (usually self-limited) to pseudomembranous colitis with severe sepsis (Mylonakis et al., 2001) and/or toxic megacolon (Kuehne et al., 2011). The pathogenesis of CDI is associated with antibiotic-induced gut dysbiosis that facilitates C. difficile colonization and toxin production (Mooyottu et al., 2017). Two protein exotoxins referred to as toxin A (TcdA) and toxin B (TcdB) are the major virulence factors contributing to CDI (Lyerly et al., 1988; Voth and Ballard, 2005; Kuehne et al., 2011). Binding of TcdA and TcdB to specific receptors on the surface of intestinal epithelial cells stimulates the secretion of several pro-inflammatory cytokines and chemokines (Hodges and Gill, 2010). Both toxins cause the loss of intestinal epithelial barrier function (gut leakage) by glucosylating Rho GTPases, which causes actin cytoskeleton rearrangement, tight junction disruption, and enterocyte cell death (Pothoulakis, 2000; Aktories and Barbieri, 2005; Jank and Aktories, 2008; Kuehne et al., 2011; Chen et al., 2015). In addition, binary toxin (C. difficile transferase, CDT) is observed in some C. difficile strains that cause severe CDI. This toxin is an ADP-ribosyltransferase that causes depolymerization of F-actin and rearrangement of the actin cytoskeleton (Gerding et al., 2014; Aktories et al., 2018).
The pathogenic effects of TcdA and TcdB have been studied extensively. Epithelial cells demonstrate a decrease in transepithelial electrical resistance (TEER) and an increase in paracellular permeability after toxin activation, indicating that C. difficile toxins disrupt gut tight junctions (Hecht et al., 1988; Nusrat et al., 2001; Zemljic et al., 2010). Toxin-activated intestinal epithelial chemotactic mediators, such as interleukin (IL)-8, cause an accumulation of neutrophils and lymphocytes (inflammatory colitis) and other clinical signs of infectious diarrhea (such as white blood cell in feces) (Viswanathan et al., 2009; Sun et al., 2010). CDI not only causes local intestinal inflammation but also induces systemic inflammation from gut leakage-induced bacteremia. Disruption of gut tight junctions also allows for the transfer of intestinal contents, including TcdA and TcdB from C. difficile, into the circulation, resulting in the activation of various immune cells in the bloodstream (Viswanathan et al., 2009; Sun et al., 2010). Subsequently, the activated immune cells systemically secrete several pro-inflammatory cytokines, which lead to systemic inflammatory responses and sepsis. In addition to bacterial factors, host factors also contribute to the severity of CDI. Notably, a previous report identified solute carrier family 11 member 1 gene (SLC11A1), which enhances TcdB lethality by the increased Rho GTPase glucosylation, and the suppression of SLC11A1 resulted in reduced toxin sensitivity. In addition, the up-regulation of SLC11A1 requires the RNA-binding protein HuR or human antigen R (encoded in HuR) to stabilize the mRNA (Feng and Cohen, 2013).
Probiotics are live microorganisms that, when administered in adequate amounts, confer a health benefit on the host (FAO and WHO, 2001; Hill et al., 2014). Probiotics have been a popular approach for the prevention and improvement of treatment efficacy of human diseases. Several meta-analyses suggest that probiotics, mainly Lactobacillus, are effective for preventing C. difficile-associated diarrhea (Ritchie and Romanuk, 2012; Goldenberg et al., 2017; Shen et al., 2017). It has been suggested that probiotic administration counteracts gut dysbiosis caused by antibiotics or infections (Reid et al., 2011), resulting in the restoration of gut microbiota diversity, which plays a crucial role in the prevention of CDI (Kachrimanidou and Tsintarakis, 2020). Specific strains of Lactobacillus spp. effectively inhibit the pathogenicity of C. difficile both in vitro (Banerjee et al., 2009; Trejo et al., 2010; Spinler et al., 2017) and in vivo (Leelahavanichkul et al., 2016) and secrete several anti-inflammatory substances that attenuate enterocyte injury from several insults (Boonma et al., 2014; Panpetch et al., 2016, 2018).
Despite a variety of probiotics, we speculated that indigenous probiotic strains derived from a specific population might be more suitable for a specific ethnic group. Accordingly, Lacticaseibacillus casei (formerly Lactobacillus casei) strain T21 isolated from a human gastric biopsy might be suitable for use as a probiotic for populations in Southeast Asia. An investigation of the effect of L. casei T21 on C. difficile infection in a mouse model and in colonic epithelial cells was conducted.
Materials and Methods
Bacterial Strains and Culture Conditions
L. casei strain T21 was obtained from the stock culture of the Department of Microbiology, Faculty of Medicine, Chulalongkorn University. Bacterial stock culture was maintained in deMan Rogosa Sharpe (MRS) broth (Oxoid, Hampshire, United Kingdom) containing 20% (vol/vol) glycerol at –80°C. L. casei T21 was cultured on MRS agar under anaerobic conditions using gas generation sachets (Anaero Pack-Anaero, Mitsubishi Gas Chemical, Japan) at 37°C for 48 h. C. difficile ATCC BAA1870 (ATCC, Manassas, VA, United States) was cultured anaerobically on Brucella agar (Becton Dickinson, France) supplemented with 5% (vol/vol) sheep blood at 37°C for 48 h.
C. difficile Infection Mouse Model and L. casei T21 Intervention
The experimental protocol in accordance with the US National Institutes of Health standards (NIH publication no. 85–23, revised 1985) was approved by the Institutional Animal Care and Use Committee of the Faculty of Medicine, Chulalongkorn University (SST006/2560). Male 8-week-old C57BL/6 mice were purchased from the Nomura Siam International Co., Ltd. (Lumphini, Pathumwan, Bangkok, Thailand). CDI mouse model as previously developed (Chen et al., 2008) and recently published (Panpetch et al., 2019) was performed with modifications. Briefly, 500 μL of the antibiotic cocktail (Sigma-Aldrich, St. Louis, MO, United States) containing gentamicin (3.5 mg/kg), colistin (4.2 mg/kg), metronidazole (21.5 mg/kg), and vancomycin (4.5 mg/kg) was administered by oral gavage twice a day from day –6 to day –4 before C. difficile infection (D-6–D-4) (Figure 1A). Mice were free from antibiotic administration for 2 days and received an intraperitoneal injection of a single dose of clindamycin (10 mg/kg) at 1 day before infection (D-1). After the treatment with antibiotics (ATB), mice were gavaged with either 0.5 ml of normal saline solution (NSS) in the ATB-administered uninfected group (ATB uninfected group; n = 12) or 1 × 1010 colony forming units (CFU) of C. difficile vegetative cells in 0.5 ml of NSS once daily for 2 days (D0 and D1) in the C. difficile group (n = 24). Mice were observed and monitored daily for weight, stool consistency, and survival until D7. Blood was collected through tail vein nicking for enumeration of bacteria at D2, D4, and D7. According to our pilot study, C. difficile-infected mice developed severe symptoms for 3 days, some succumbed to infection, and the survivors gradually recovered from CDI. At D3, mice from each group (n = 8) were then tested for gut leakage and sacrificed with cardiac puncture under isoflurane anesthesia for determining CDI severity by using serum pro-inflammatory cytokines as markers. The stool consistency was semi-quantitatively evaluated using the following scoring; 0, normal; 1, soft or loose; and 2, diarrhea, as previously published (Kim et al., 2012).
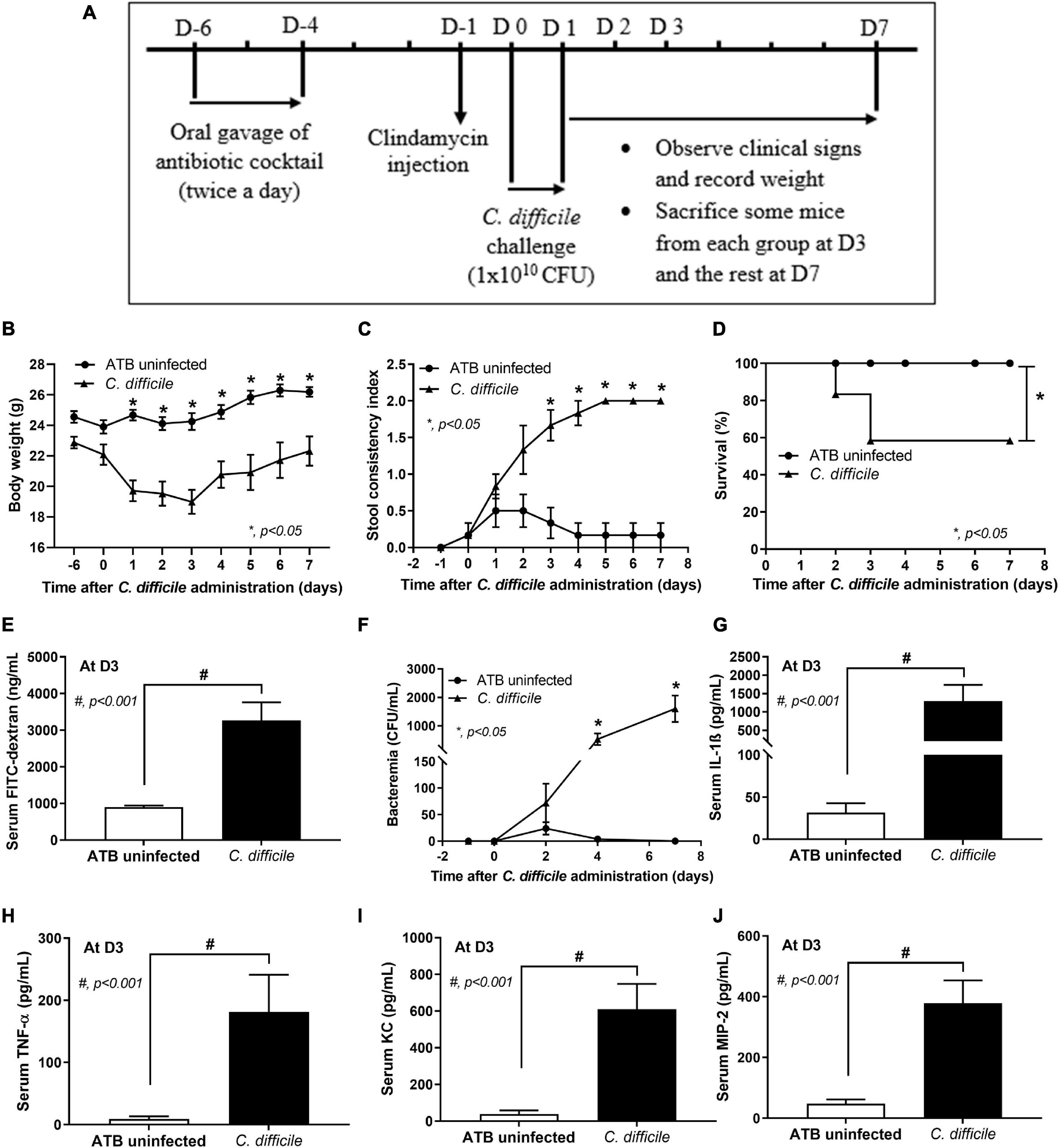
Figure 1. Murine model of C. difficile infection. Mice in the antibiotic-administered uninfected group (ATB uninfected group; n = 12) and C. difficile-infected group (C. difficile group; n = 24) were used in the experiment as in the schematic presentation (A). The severity of C. difficile infection in mice is manifested by weight loss (n = number of mice/group or survivors) (B); stool consistency index (n = number of mice/group or survivors) (C); survival rate (D); bacteremia (n = number of mice/group or survivors) (E); gut leakage by serum FITC-dextran assay monitored at day 3 (n = 8) (F); and levels of serum pro-inflammatory cytokines IL-1β (G), TNF-α (H), KC (I), and MIP-2 (J) at day 3 (n = 8). *p < 0.05; #p < 0.001. FITC, fluorescein isothiocyanate; IL, interleukin; TNF, tumor necrosis factor; KC, keratinocyte chemoattractant; MIP-2, macrophage inflammatory protein 2.
For Lacticaseibacillus treatment in the CDI mouse model, mice were randomly divided into three groups: ATB uninfected group (n = 18), C. difficile-infected mice treated with NSS (NSS group; n = 24), and C. difficile-infected mice treated with L. casei T21 (T21 group; n = 20), which received 1 × 108 CFU of L. casei T21 in 0.5 ml of NSS once daily for 4 days from D-1 (started at 6 h after clindamycin injection), D0 and D1 (together with C. difficile), and D2 (Figure 2A). Mice were observed and monitored daily until D7 as described above. Feces were collected for microbiome analysis at D-6 for baseline and D-1 (before clindamycin injection and gavage with L. casei T21) as time of post-ATB administration. At D3, some mice from each group (n = 8) were tested for gut leakage and sacrificed for determining the parameters of CDI severity. Blood samples and cecal and ascending colonic tissues were collected for the assessment of cytokine levels, which represented systemic and local inflammation, respectively. Cecal and ascending colonic tissues were also used for histopathologic evaluation, while luminal content including feces in the cecum and colon were used for quantitation of C. difficile and microbiome analysis. All mice were sacrificed on D7 at the end of experiment.
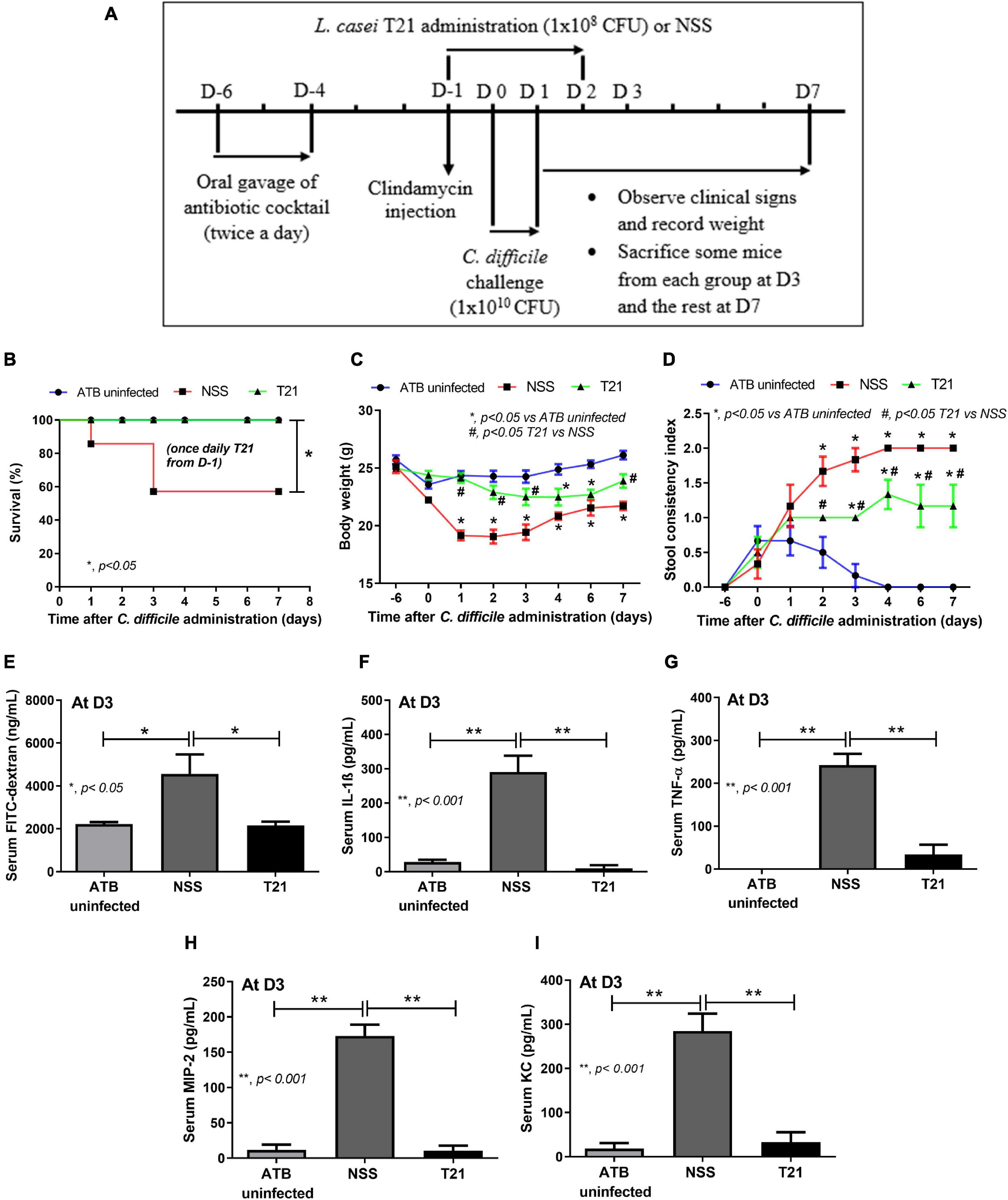
Figure 2. Lacticaseibacillus casei T21 effectively attenuated C. difficile-induced severity in mice. Mice were randomly assigned to three groups: ATB uninfected group (n = 18), NSS group (n = 24), and T21 group (n = 20) as described in the text and used in the experiment (A). The reduction of severity was demonstrated by survival rate (B); body weight (n = number of mice/group or survivors) (C); stool consistency index (n = number of mice/group or survivors) (D); gut leakage by serum FITC-dextran assay monitored at day 3 (n = 8) (E); and levels of serum pro-inflammatory cytokines IL-1β (F), TNF-α (G), MIP-2 (H), and KC (I) at day 3 (n = 8). *p < 0.05; **p < 0.001; #p < 0.05.
Mouse Sample Analysis
The pro-inflammatory cytokines as previously described (Leffler and Lamont, 2015), including IL-1β, tumor necrosis factor (TNF)-α, macrophage inflammatory protein 2 (MIP-2), and keratinocyte chemoattractant (KC), in serum and homogenized tissue of the cecum and colon were measured by an enzyme-linked immunosorbent assay (ELISA) (PeproTech, NJ, United States). For determining tissue pro-inflammatory cytokines, tissue samples (approximately 100 mg) were weighed and homogenized using an Ultra-Turrax homogenizer (IKA, Staufen, Germany) in 500 μL of phosphate-buffered saline (PBS, pH 7.4) containing protease inhibitor and centrifuged at 12,000 × g for 15 min at 4°C to separate the supernatant for analysis.
For quantitation of C. difficile in luminal content including feces, quantitative real-time polymerase chain reaction (qPCR) was performed as previously published (Panpetch et al., 2019). Briefly, genomic DNA of C. difficile was extracted from cecum and colon contents using the High Pure PCR Template Preparation Kit (Roche, NJ, United States), quantified by NanoDrop™ 1000 Spectrophotometer (Thermo Fisher Scientific, Inc., United States), and amplified with tcdB (C. difficile toxin B) primers (forward, 5′-GGAAAAGAGAATGGTTTTATTAA-3′ and reverse, 5′-ATC TTTAGTTATAACTTTGACATCTTT-3′) as previously described (Lemee et al., 2004) in the QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific, Inc., United States) using the QuantiNova® SYBR® Green PCR Kit (QIAGEN, Hilden, Germany). The standard curve was created by using 10-fold serially diluted plasmids containing 1–109 copies of tcdB. The number of C. difficile was calculated using the standard curve and shown as bacterial copy number.
Histopathological analysis was performed by fixing the sample in 10% buffered formalin, embedding in paraffin, sectioning, and staining with hematoxylin and eosin (H&E) before grading (score 0–4) as previously published (Reeves et al., 2011; Erikstrup et al., 2015) based upon epithelial damage, edema, and cellular infiltration by two pathologists in a blinded manner.
Microbiome Analysis
Gut microbiota composition was determined as previously reported (Panpetch et al., 2020). Feces collected at D-6 as baseline, D-1 as time of post-ATB administration, and luminal content including feces at D3 as time of sacrifice were used for microbiome analysis. Briefly, fecal samples or luminal content (0.25 g/mouse; three mice/group) were extracted for total DNA with the DNeasy PowerSoil Kit (Qiagen GmbH, Hilden, Germany). The quality and concentration of the extracted DNA were monitored by agarose gel electrophoresis and NanoDrop spectrophotometry. Libraries of the V4 hypervariable region of 16S rRNA gene were amplified by PCR using primers 515F (forward; 5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (reverse; 5′-GGACTACHVGGGTWTCTAAT-3′), modified with the Illumina adapter and Golay barcode sequences as previously described (Caporaso et al., 2012). PCR was run in triplicate, and the products from the triplicate reactions were pooled and visualized on agarose gel. Amplicons of approximately 381 base pairs were purified by PureDireX PCR Clean-Up & Gel Extraction Kit (BIO-HELIX Co., Ltd., Keelung City, Taiwan) and quantified using PicoGreen fluorescence with the Qubit dsDNA HS assay kit (Invitrogen, Eugene, OR, United States). The amplicon pool was sequenced with the Illumina MiSeq300 platform (Illumina, San Diego, CA, United States) (Caporaso et al., 2012). Sequences were analyzed with Mothur version 1.3 (Schloss et al., 2009). Briefly, quality filtering and trimming were performed to remove low-quality bases and short reads from the raw sequences. Quality-filtered sequences were then aligned to each other and binned into operational taxonomic units (OTUs) with a minimum of 97% similarity. Each representative OTU sequence was compared to the SILVA rDNA sequence database (version 1.32) and assigned a taxonomical annotation. Alpha diversity (total OTUs, Chao1 index, and Shannon diversity) and beta diversity (non-metric multidimensional scaling) were calculated using Mothur (Schloss et al., 2009). The 16S rDNA sequences in this study were deposited in an NCBI open access Sequence Read Archive database with accession number SRP336496.
Gut Leakage Measurement and Enumeration of Bacteria in the Blood
Intestinal epithelial permeability defect (gut leakage) was determined using a single oral administration of 12.5 mg fluorescein isothiocyanate–dextran (FITC-dextran; molecular weight 4.4 kDa) (Sigma-Aldrich, St. Louis, MO, United States), a non-intestinal-absorbable marker, before the determination in serum at 3 h later as previously described (Leelahavanichkul et al., 2016; Panpetch et al., 2018). Serum FITC-dextran was measured by the fluorospectrometry (Thermo Fisher Scientific, Wilmington, DE, United States) with the excitation and emission wavelengths at 485 and 523 nm, respectively, against a standard curve of serially diluted FITC-dextran. For the enumeration of live bacteria, blood (25 μL) was collected through tail vein nicking and spread directly onto blood agar (Oxoid, Hampshire, United Kingdom) and incubated at 37°C for 24 h before counting bacterial colonies.
The Immunomodulatory Effect of L. casei T21 on C. difficile-Stimulated Colonic Epithelial Cells
The conditioned medium of L. casei T21 was tested for immunomodulation of IL-8 production in colonic epithelial cell lines as previously described (Panpetch et al., 2016, 2018). In brief, Lacticaseibacillus-conditioned medium (LCM) was prepared by growing L. casei T21 with an OD600 of 0.1 in MRS broth anaerobically for 48 h. The supernatant was collected and subjected to filtration with a 0.22-μm membrane (Minisart, Sartorius Stedim Biotech GmbH, Goettingen, Germany), concentrated by speed vacuum drying, resuspended in cell culture medium with equal volume, and stored at –20°C until use. In parallel, human colonic epithelial cell lines Caco-2 (ATCC HTB-37) and HT-29 (ATCC HTB-38) were maintained (5 × 104 cells/well) in supplemented Dulbecco’s Modified Eagle Medium (DMEM) and McCoy’s 5A modified medium, respectively. Colonic epithelial cells were then incubated with viable cells of C. difficile ATCC BAA1870 at multiplicity of infection (MOI) 1:300 either alone or with 5% (vol/vol) LCM for 24 h in 5% CO2 at 37°C. Subsequently, the supernatant was collected by centrifugation (125 × g, 4°C for 7 min), and the levels of IL-8 were measured by using a Human CXCL8/IL-8 ELISA kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
In addition, colonic epithelial cells at 2 and 4 h from the incubation time were collected for performing quantitative reverse transcription-polymerase chain reaction (qRT-PCR) as previously described (Panpetch et al., 2018). In short, the total RNA of treated colonic epithelial cells was extracted by TRIzol reagent (Invitrogen, United States), prepared for complementary DNA (cDNA) from total RNA (50 ng) by SuperScript® VILO™ cDNA Synthesis Kit (Invitrogen), and subjected to qPCR measurement in a QuantStudio™ Design & Analysis Software v1.4.3 (Thermo Fisher Scientific) with the following primers: IL-8 (forward 5′-ACACTGCGCCAACACAGAAATTA-3′, reverse 5′-ACACTGCGCCAACACAGAAATTA-3′) and human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward 5′-GCACCGTCAAGGCTGAGAAC-3′, reverse 5′-ATGGTGGTGAAGACGCCAGT-3′) (Imaoka et al., 2008; Panpetch et al., 2016). The expression of IL-8 relative to GAPDH was calculated according to the 2−ΔΔCp method (Pfaffl, 2001).
The Effect of L. casei T21 on the Expression of SLC11A1, HuR, and MUC2 in C. difficile-Stimulated Colonic Epithelial Cells
The conditioned medium of L. casei T21 was tested for its effect on the expression of C. difficile-activated host genes SLC11A1, HuR, and MUC2 by using qRT-PCR as described above with the following primers: SLC11A1 (forward 5′-CTGGACGAATCCCACTCTGG-3′, reverse 5′-CGCGCCACCACATACTCAT-3′), HuR (forward 5′-GCTTGGGCTATGGCTTTGTGAACT-3′, reverse 5′-CGCTG ATGTACAAGTTGGCGTCTT-3′) (Feng and Cohen, 2013), and mucin2 (MUC2) (forward 5′-CCTGCCGACACCTGCTGCAA-3′, reverse 5′-ACACCAGTAGAAGGGACAGCACCT-3′) (Xue et al., 2014). In parallel, the pH of cell culture medium was measured at multiple time points using a pH meter (Orion 4-star, Benchtop pH/Conductivity, Thermo Fisher Scientific).
The Effect of L. casei T21 on Transepithelial Electrical Resistance of Caco-2 Cells
TEER was performed according to a previous report (Gao et al., 2017). In short, Caco-2 cells (ATCC HTB-37) at 5 × 104 cells per well were seeded onto the upper compartment of a 24-well Boyden chamber transwell using high-glucose DMEM supplemented with 20% fetal bovine serum (FBS), 1% HEPES, 1% sodium pyruvate, and 1.3% penicillin/streptomycin under 5% CO2 at 37°C for 15 days with daily medium replacement to establish the confluent monolayer. The cells were then treated with 5% (vol/vol) LCM of L. casei T21 or medium alone together with viable C. difficile cells (5 × 106 CFU/well) with MOI at 1:100 for 24 h. Next, TEER was measured by an EMOM2 Epithelial Volt/Ohm Meter with a chopstick electrode (World Precision Instruments, Inc., Sarasota, United States) that was placed at a 90° angle with one tip in supernatant at the basolateral chamber and another tip at the apical chamber. The TEER value in control media without cells was used as a baseline subtracted from all measurements. The value of TEER was reported as ohm (Ω) × cm2.
Statistical Analysis
Mean ± standard error of mean (SEM) was used for data presentation. The difference between groups was examined for statistical significance by one-way analysis of variance (ANOVA) followed by Tukey’s analysis or unpaired t tests for comparisons of multiple groups or two groups, respectively. Survival analysis was performed by log-rank test. All statistical analyses were performed with GraphPad Prism version 9.0 software (La Jolla, CA, United States). A p-value of < 0.05 was considered statistically significant.
Results
Disease Progression and Severity of Murine C. difficile Infection Model
In the CDI murine model (Figure 1A), C. difficile-infected mice began to develop CDI symptoms such as weight loss and soft stool on day 1 after the first oral gavage with C. difficile on day 0. After the second oral gavage on day 1, CDI symptoms became worse on day 2, and mice were moribund on day 3 with maximum weight loss (Figure 1B) and significant diarrhea (loose stools) as compared with the ATB uninfected group (Figure 1C). By day 3, 41.67% (10/24) of mice succumbed to infection (58.33% survival rate) (Figure 1D). Eight mice were sacrificed on day 3, and the remaining six mice gradually gained weight while still having diarrhea and surviving until the end of the experiment (Figures 1B,C). Additionally, C. difficile damaged intestinal integrity as demonstrated by the increased serum FITC-dextran levels (Figure 1E), causing gut leakage-induced bacteremia (Figure 1F) that enhanced the production of systemic inflammatory cytokines (serum IL-1β, TNF-α, KC, and MIP-2 levels as markers) (Figures 1G–J). In contrast, mice in the ATB uninfected group demonstrated loose stools for a few days (days 1–3) without weight loss, death, gut leakage, bacteremia, or systemic inflammation (Figures 1B–J).
L. casei Strain T21 Reduced Mortality, Clinical Symptoms, and Disease Severity of C. difficile-Infected Mice
For the treatment of L. casei T21 in the murine model of CDI, C. difficile-infected mice in the T21 group received 1 × 108 CFU of L. casei T21 once daily for 4 days (D-1–D-2), whereas infected mice in the NSS group received NSS (Figure 2A). All mice in the T21 and the ATB uninfected group survived, while only 54.17% (13/24) of the NSS group survived by day 3 (Figure 2B). Compared to the NSS group, the T21 group had significantly lesser weight loss (except at days 4 and 5) (Figure 2C) and diarrhea (Figure 2D), which were monitored for 7 days. The average weight at each day (except at days 4 and 5) between the T21 group and the ATB uninfected group showed no significant difference (Figure 2C). However, mice in the T21 group still had soft stool with a stool consistency index significantly different from the ATB uninfected group (Figure 2D). Treatment with T21 also reduced gut leakage and systemic inflammation. The T21 group had significantly decreased levels of FITC-dextran (Figure 2E) and pro-inflammatory cytokines IL-1β, TNF-α, MIP-2, and KC in sera (Figures 2F–I). Likewise, L. casei T21 also attenuated intestinal injury in the cecum and colon as evaluated by histopathology (Figures 3A,B) and reduced the levels of pro-inflammatory cytokines in the intestinal tissue (Figures 3C–F). Notably, C. difficile-infected mice without T21 treatment (NSS group) demonstrated several characteristics of severe intestinal injury, including loss of villi, villous edema, numerous neutrophil infiltration (Figure 3A), and neutrophils in feces (data not shown). In addition, T21 treatment reduced the abundance of C. difficile in the intestinal content. The analysis of luminal content including feces by q-PCR of the tcdB gene demonstrated that the T21 group had significantly decreased copies compared to the NSS group (Figure 3G). Moreover, the absence of the tcdB gene in the ATB uninfected mice (Figure 3G) revealed the reliability of the experiment.
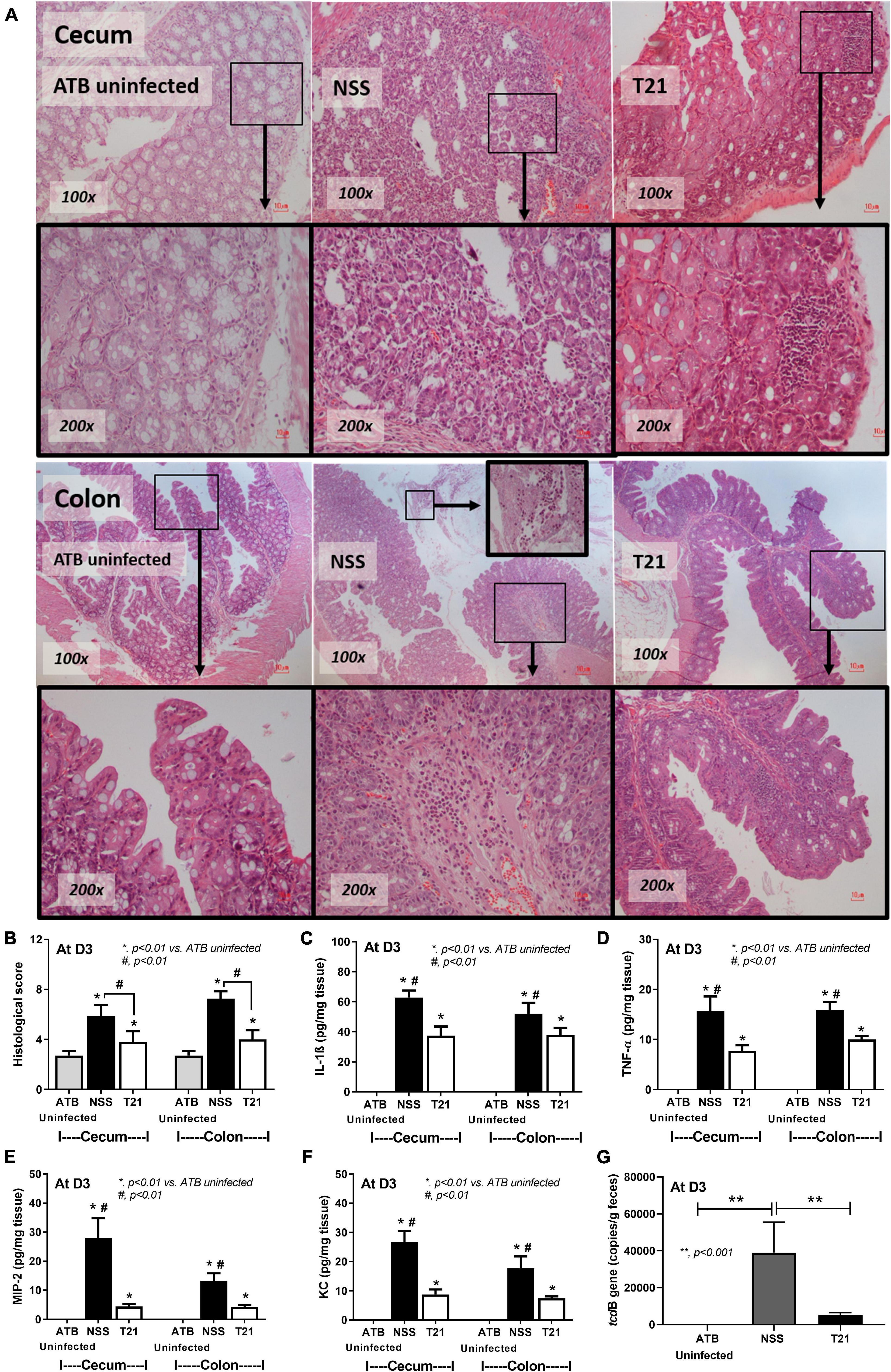
Figure 3. Lacticaseibacillus casei T21 attenuated C. difficile-induced cecal and colonic tissue damage. Three groups of mice (n = 8 for each group) as described in Figure 2 were sacrificed at day 3 and examined for histopathology and local inflammation. Representative image of hematoxylin and eosin (H&E) staining sections (A), histological scores (B), and levels of intestinal pro-inflammatory cytokines (C–F) are shown. The copy number of tcdB, which represents C. difficile abundance in each group of mice, is demonstrated (G). *p < 0.01; **p < 0.001; #p < 0.01.
L. casei T21 Slightly Attenuated Gut Dysbiosis in C. difficile-Infected Mice
Gut dysbiosis of the model was evaluated by a fecal microbiome analysis of mice in the ATB uninfected, NSS, and T21 groups at multiple time points. Alpha diversity measures used in this study included total OTUs (the simplest measure of richness), Chao1 (a measure of richness that gives more weight to rare taxon), and Shannon (a measure of richness and evenness). Antibiotic cocktail treatment significantly reduced the diversity of fecal bacteria as the values of the total OTUs (Figure 4A) and Chao1 index (Figure 4B) were significantly lower in all groups of mice at post-ATB compared to baseline. After clindamycin injection, antibiotic-induced dysbiosis worsened at day 3 of the experiment as determined by total OTUs (Figure 4A), but not by Chao1 index (Figure 4B). C. difficile infection did not lead to a significant decrease of bacterial diversity as demonstrated by total OTUs (Figure 4A) and Chao1 index (Figure 4B). Surprisingly, T21 treatment did not increase the diversity of fecal bacteria as the values of total OTUs (Figure 4A) and Chao1 index (Figure 4B) of the NSS and T21 groups were not significantly different. The T21 group did show an increased Chao1 index, although the value was not significantly different from that of the NSS group. The values of Chao1 index were also not significantly different in the T21 and ATB uninfected groups (Figure 4B). Notably, the bacterial diversity in all groups of mice at day 3, post-ATB, and baseline was not significantly different as determined by the Shannon index (Figure 4C). To examine the beta diversity, non-metric multidimensional scaling (NMDS) based on Thetayc dissimilarity was performed. The NMDS (Figure 4D) demonstrated similar results in all groups at baseline (D-6) (blue-colored symbols at the upper left quadrant) and at post-ATB (D-1) (red-colored symbols at the lower right quadrant). In contrast, there were some differences at D3 from the experiments between groups of mice with or without L. casei T21 (green-colored symbols of the NSS and T21 groups), suggesting a possible impact of L. casei T21 on gut microbiota. The ATB uninfected and T21 groups showed similar results.
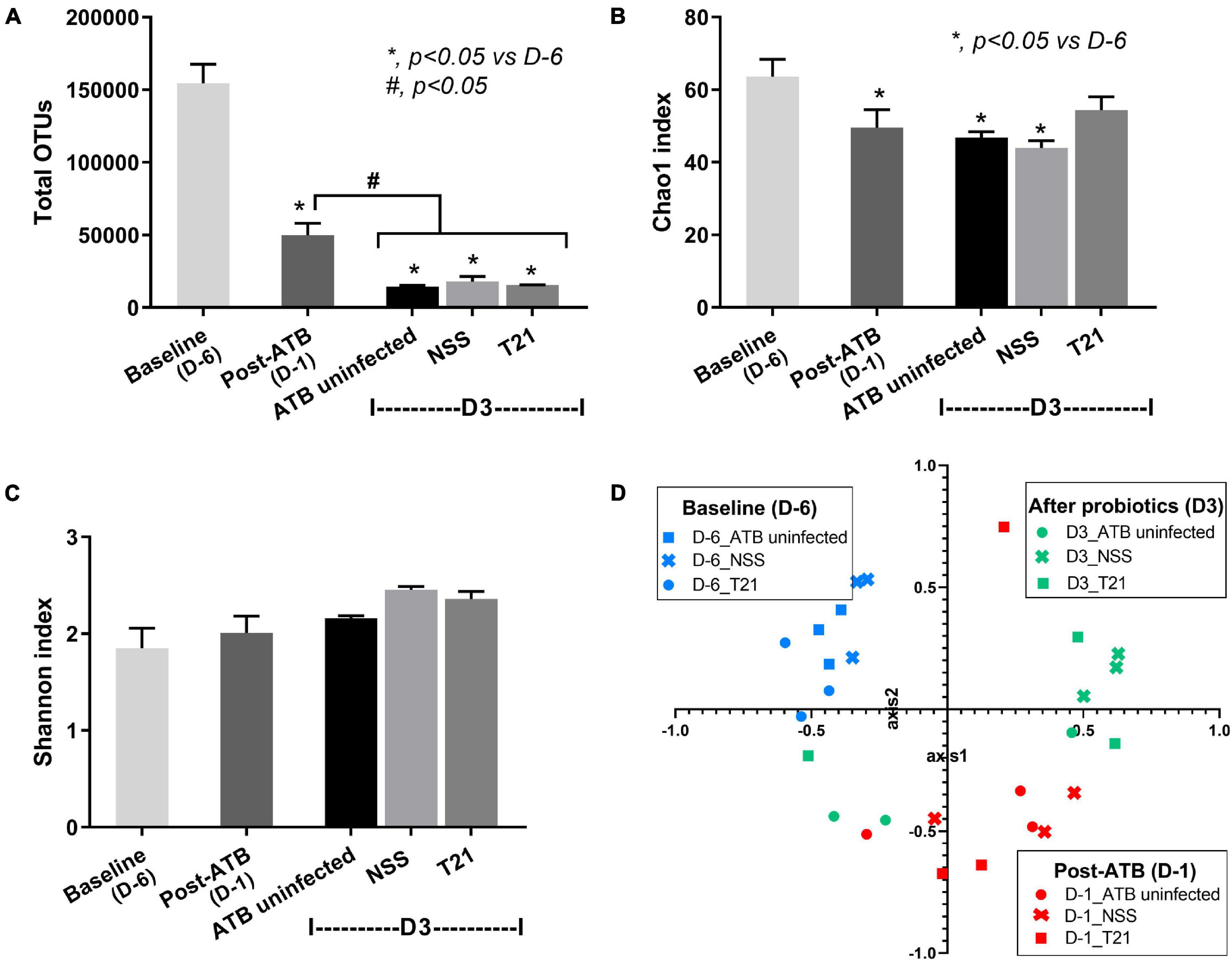
Figure 4. Diversity of gut microbiota in each group of mice at multiple time points. Fecal microbiome data of the ATB uninfected, NSS, and T21 groups (n = 3/group) at baseline, post-ATB, and at sacrifice were calculated for microbial diversity and presented as total operational taxonomic units (OTUs) (A), Chao1 index (B), Shannon index (C), and the non-metric multidimensional scaling (NMDS) based on Thetayc dissimilarity (D). Independent triplicate experiments were performed. *p < 0.05 vs. D-6, #p < 0.05.
To characterize the microbiome composition in each group of mice, the relative taxa abundances at each time point were compared. The gut microbiome of mice at baseline was predominated by Bacteriodetes, followed by Firmicutes and Proteobacteria, as shown by the average relative abundances of microbiota at the phylum level in each group of mice (Figure 5A) and the relative abundances of Bacteroidetes (Figure 5D), Firmicutes (Figure 5E), and Proteobacteria (Figure 5F) at each time point. In contrast, antibiotic cocktail treatment induced a significant decrease in the relative abundance of Bacteriodetes and Firmicutes and a significant increase in the abundance of Proteobacteria and Verrucomicrobia at D-1 (Figures 5A,D–G). Antibiotic pre-conditioning of the model thus caused fecal dysbiosis as indicated by a decrease in Firmicutes and Bacteroidetes together with an increase in Proteobacteria and Verrucomicrobia. By day 3, microbiota composition at the phylum level almost turned to the baseline without T21 treatment. The relative abundance of Bacteroidetes (Figure 5D), Firmicutes (Figure 5E), and Verrucomicrobia (Figure 5G) in the ATB uninfected group and the NSS group was not significantly different from the baseline, whereas the abundance of Proteobacteria was still significantly different from the baseline (Figure 5F). L. casei T21 treatment led to an increase in the relative abundance of Firmicutes in the T21 group with a significant difference compared to post-ATB, but not from the NSS group (Figure 5E). In addition, T21 treatment resulted in a significant decrease in the relative abundance of Verrucomicrobia in the T21 group compared to the NSS group and post-ATB, and the abundance of Verrucomicrobia in the T21 group was not significantly different from the baseline (Figure 5G). Although the relative abundance of Proteobacteria in the T21 group significantly decreased compared to post-ATB, this change also occurred in the NSS group (Figure 5F).
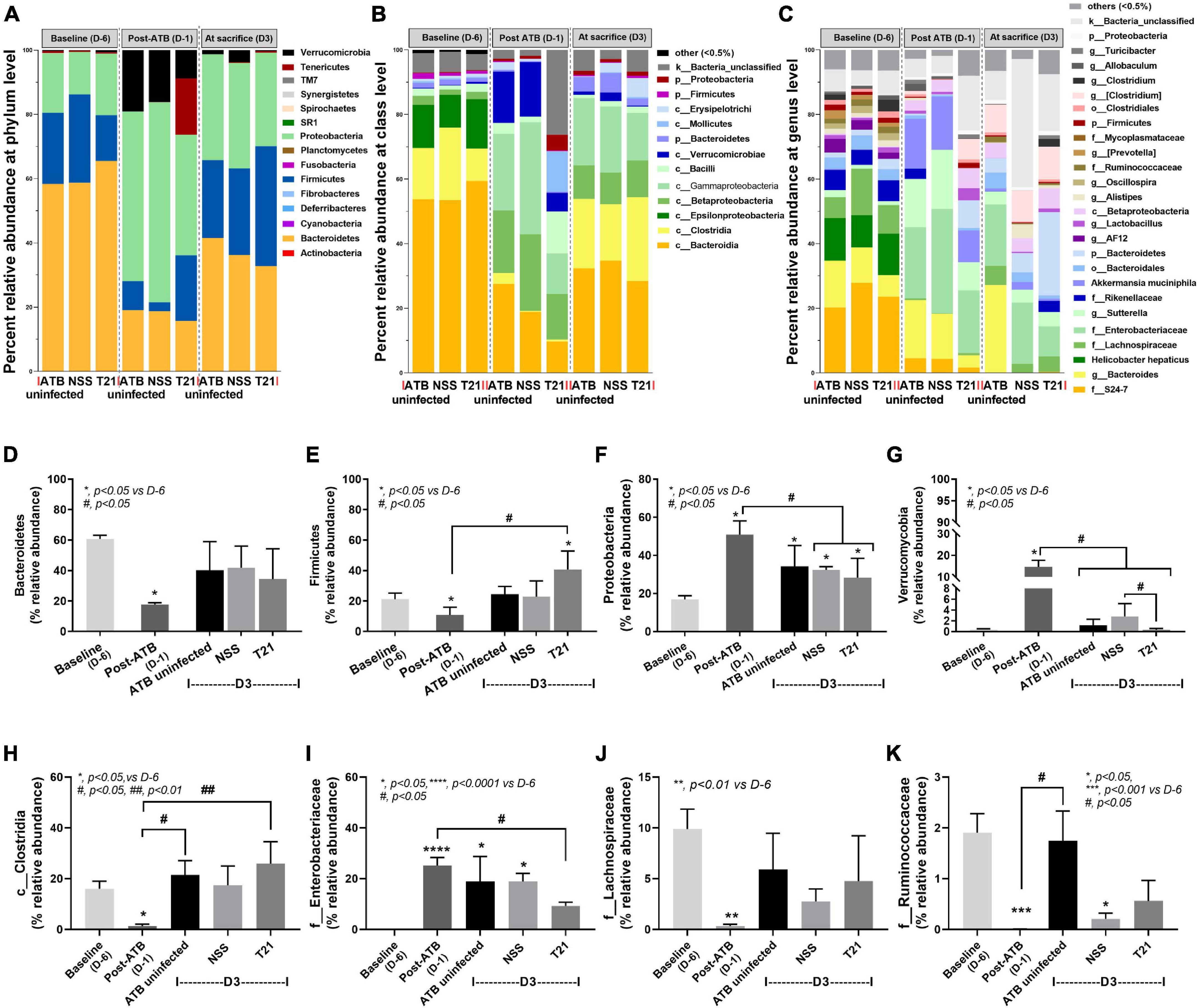
Figure 5. Lacticaseibacillus casei T21 slightly attenuated gut dysbiosis in C. difficile-infected mice. Gut dysbiosis of the C. difficile infection (CDI) model was evaluated by fecal microbiome analysis of mice in the ATB uninfected, NSS, and T21 groups at multiple time points. The average relative abundances of microbiota at the phylum level (A), class level (B), and genus level (C) are demonstrated. The average relative abundances of individual taxon are shown: Bacteroidetes (D), Firmicutes (E), Proteobacteria (F), Verrucomicrobia (G), Clostridia (H), Enterobacteriaceae (I), Lachnospiraceae (J), and Ruminococaceae (K). Notably, data of all mice at day –6 (D-6) and day –1 (D-1) of the experiments are combined into baseline and post-antibiotic administration (post-ATB), respectively, due to the non-different procedures in these mice. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; #p < 0.01; ##p < 0.01.
The analysis of microbiome at the lower taxon levels (class, order, family, genus, and species) was performed. The average relative abundances of microbiota at baseline, post-ATB, and among groups of mice at sacrifice (day 3) are shown at the class level (Figure 5B) and the genus level (Figure 5C). There were significant changes in class Clostridia (Figure 5H) and family Enterobacteriaceae (Figure 5I) in the T21 group at sacrifice (D3) as compared with mice at post-ATB. Although the NSS group had changes in the relative abundance of these taxa as compared with mice at post-ATB, the differences were not statistically significant. Microorganisms of interest in class Clostridia (phylum Firmicutes) were the families Lachnospiraceae and Ruminococcaceae (Figure 5C) that have been reported to protect C. difficile colonization (Reeves et al., 2012; Lee et al., 2017). The relative abundances of the family Lachnospiraceae (Figure 5J) were not significantly different among the ATB uninfected, the NSS, and the T21 groups, while there was a slight increase in the abundance of the family Ruminococcaceae in the T21 group compared to the NSS group, although the difference was not statistically significant (Figure 5K). The relative abundances of other taxa (at the class and genus levels) were also not significantly different among these groups of mice at sacrifice (data not shown). Data on the relative abundances of microbiota from individual mouse are shown at the levels of phylum (Supplementary Figure 1), class (Supplementary Figure 2), and genus (Supplementary Figure 3) in Supplementary Material.
L. casei T21 Suppressed IL-8 Production, Modulated Host Gene Expression, and Increased Transepithelial Electrical Resistance of C. difficile-Activated Colonic Epithelial Cells
Probiotic bacteria, such as Lactobacillus spp., have been reported to produce biologically active compounds that can suppress inflammation (Thomas et al., 2012, 2016; Panpetch et al., 2016, 2018). To strengthen the beneficial effects of L. casei T21 in the murine model of CDI, the LCM of T21 was tested for its ability to attenuate inflammation and modulate the expression of important host genes involved in the pathogenesis of CDI in colonic epithelial cell lines. The LCM of T21 suppressed the production of IL-8 (Figures 6A,B) and down-regulated the expression of IL-8 (Figures 6C,D) in C. difficile-stimulated Caco-2 and HT-29 colonic epithelial cells, respectively. In C. difficile-stimulated Caco-2 cells, the LCM of T21 down-regulated the expression of associated genes of toxin lethality SLC11A1 (Figure 6E) and HuR (Figure 6F) while up-regulated a mucosal protective gene MUC2 (Figure 6G). Similar benefits exerted by the LCM of T21 were also demonstrated in HT-29 cells (Figures 6H–J). Additionally, the LCM of T21 strengthened mucosal integrity as shown by TEER values in C. difficile-stimulated differentiated Caco-2 cells (Figure 6K). However, TEER could not be determined in the HT-29 cell line due to limitation in the generation of polarized monolayers (Le Bivic et al., 1988). Since the enhanced mucin production by enterocytes is also influenced by the acidity in gut content (Velcich and Augenlicht, 1993; Shekels et al., 1996), lactic acid produced from L. casei T21 might stimulate the up-regulation of MUC2 expression. To determine whether the up-regulated MUC2 could be attributable to the acidity, the pH of the cell culture medium (McCoy’s 5A modified medium) was measured at multiple time points. The LCM of T21 did not significantly reduce the pH of the cell culture medium at 2 and 4 h after incubation with HT-29 cells as compared to controls (Figure 6L). A simple acidification of the cell culture medium with lactic acid also did not upregulate MUC2 expression (data not shown). This suggested that mucin production was induced by other substances produced by L. casei T21.
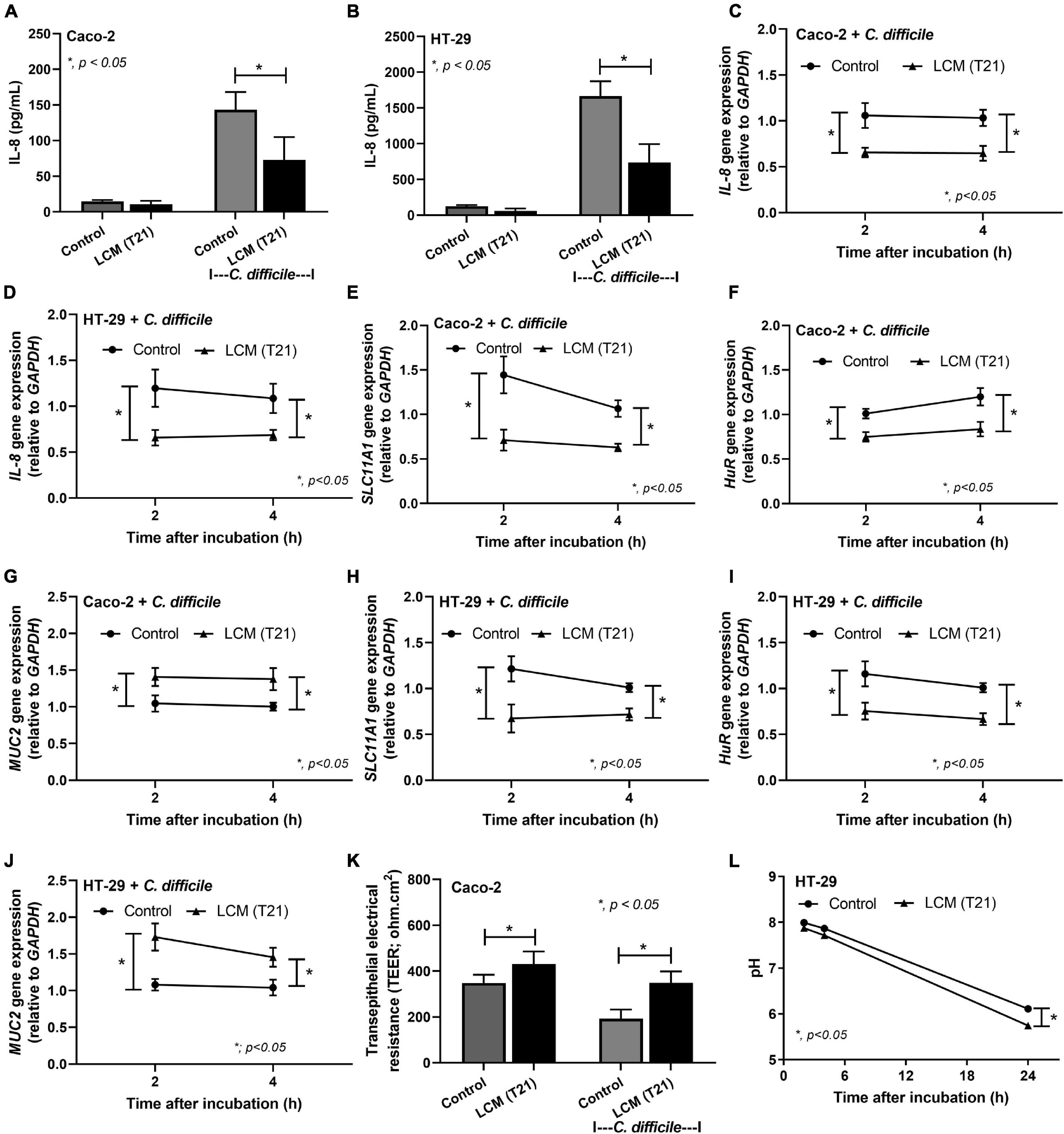
Figure 6. The Lacticaseibacillus casei-conditioned medium (LCM) of L. casei T21 attenuated C. difficile-induced IL-8 production, host gene expression, and increased transepithelial electrical resistance of C. difficile-stimulated colonic epithelial cells. The results are shown as IL-8 production in Caco-2 and HT-29 cells, respectively (A,B); IL-8 gene expression (relative to GAPDH) in Caco-2 and HT-29 cells, respectively (C,D); the expression of SLC11A1, HuR, and MUC-2 in Caco-2 cells, respectively (E–G); the expression of SLC11A1, HuR, and MUC-2 in HT-29 cells, respectively (H–J); the transepithelial electrical resistance (TEER) values of Caco-2 cells (K); and the pH of cell culture medium (McCoy’s 5A modified medium for HT-29 cells) (L). The results were from three independent experiments each in triplicate and expressed as the mean ± SEM. *p < 0.05.
Discussion
In this study, the effect of L. casei T21 treatment on CDI was investigated in a murine model of C. difficile infection with antibiotic pre-conditioning before C. difficile oral gavage. The clinical features of CDI are primarily mediated by TcdA and TcdB, which are the major virulence factors of C. difficile (Voth and Ballard, 2005; Sun et al., 2010). Clinical symptoms including less weight loss and diarrhea in L. casei T21-treated mice are correlated with mild intestinal pathology characterized by decreased epithelial damage, edema, and neutrophil infiltration. Due to the importance of intestinal neutrophil accumulation in C. difficile pathogenesis, the anti-inflammatory properties of probiotics is of utmost importance for the alleviation of CDI symptoms. Neutrophil infiltration, which leads to the congestion and edema of colonic mucosa and epithelial cell damage (Kelly et al., 1994a; Castagliuolo et al., 1998), results from the stimulation by pro-inflammatory cytokines secreted from C. difficile toxin-induced intestinal epithelial cells and immune cells (Kim et al., 2002; Savidge et al., 2003; Sun et al., 2010). Interestingly, L. casei T21 administration markedly reduced the levels of IL-1β, TNF-α, MIP-2, and KC (mouse homolog of IL-8) in the colon and cecum of mice as well as in sera. The anti-inflammatory properties of L. casei T21was also supported by in vitro results that found the conditioned medium of L. casei T21 suppressed gene expression and production of IL-8 in C. difficile-stimulated colonic epithelial cells Caco-2 and HT-29. Our findings were consistent with other reports showing that the administration of specific strains of probiotics ameliorates intestinal pathology with the reduction in the levels of pro-inflammatory cytokines in tissue and/or sera in animal models of CDI (Koon et al., 2016; Roychowdhury et al., 2018; Wei et al., 2018; Xu et al., 2018). The production of anti-inflammatory substances from lactobacilli is well known (Thomas et al., 2012, 2016; Boonma et al., 2014; Panpetch et al., 2016, 2018), although the nature of the substances varies depending on the strain of probiotic bacteria.
Our microbiome results regarding antibiotic-induced dysbiosis is in agreement with findings from previous reports (Mooyottu et al., 2017; Xu et al., 2018; Li et al., 2019) that showed antibiotic treatment induced a decrease in the dominant bacterial phyla Firmicutes and Bacteroidetes and an increase in phylum Proteobacteria. Although the influence of L. casei T21 on gut dysbiosis attenuation was subtle, with an increase in members of phylum Firmicutes (class Clostridia) and a decrease in family Enterobacteriaceae (phylum Proteobacteria), the abundance of C. difficile in cecum and colon luminal content was decreased as determined by quantitative analysis of C. difficile toxin B gene (tcdB). Members of class Clostridia, which have been reported to protect C. difficile colonization, include the family Lachnospiraceae (Reeves et al., 2012; Lee et al., 2017) and the family Ruminococcaceae (Lee et al., 2017; Li et al., 2019). However, our results showed only a slight increase in Ruminococcaceae in mice treated with L. casei T21. We speculate that members of class Clostridia either alone or in combination might mediate colonization resistance against C. difficile.
Since C. difficile toxins are mainly responsible for the pathogenesis of CDI, an interference in toxin effect might be another mechanism to be considered for C. difficile attenuation (Kolling et al., 2012; Panpetch et al., 2019; Yong et al., 2019). C. difficile toxins inactivate Rho GTPases resulting in gut leakage, intestinal inflammation, and cell death (Chen et al., 2015). The interference with the expression of toxin lethality-associated genes SLC11A1 and HuR may lead to the reduction of gut leakage, inflammation, and mortality in C. difficile-infected mice treated with L. casei T21. While L. casei T21 reduced the toxin effect by down-regulation of the expression of SLC11A1 and HuR, which enhances TcdB action, other probiotics interfere with the activity of C. difficile toxins by other mechanisms. For example, Saccharomyces boulardii interferes the binding between the toxins and intestinal brush borders (Castagliuolo et al., 1999), L. delbrueckii directly inhibits the cytotoxicity (Banerjee et al., 2009), and Streptococcus thermophilus reduced toxin production through potent lactic acid generation (Kolling et al., 2012).
L. casei T21 was found to up-regulate the expression of MUC2, which codes for mucin, an intestinal mucosal protective factor, referred to as “mucin barrier” (Dharmani et al., 2009; Paone and Cani, 2020), which can promote gut integrity. Similarly, Lactobacillus plantarum induces the expression of MUC2 and MUC3 that inhibit Escherichia coli adherence to intestinal epithelium cells (Mack et al., 1999). The mucin-binding protein in several strains of lactobacilli also implies an association between lactobacilli and intestinal mucin (Cornick et al., 2015). Although the enterocyte stimulation by lactic acid might theoretically enhance mucin production (Shekels et al., 1996), the direct incubation of lactic acid-containing cell culture medium with HT-29 cell line did not result in the up-regulation of MUC2 expression. Other substances produced by L. casei T21 are possibly associated with MUC2 gene up-regulation. More studies on this topic are required. Our findings revealed that L. casei T21 had a protective effect against C. difficile infection and suggested a great potential of L. casei T21 as a probiotic for humans, especially in Southeast Asian populations.
Conclusion
Our study demonstrated that L. casei strain T21 attenuated C. difficile infection in mice through anti-inflammation, attenuation of gut leakage and dysbiosis, interference with toxin lethality by down-regulation of the toxin enhancer gene, and augmentation of mucin production by up-regulation of mucin-producing gene.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: SRP336496).
Ethics Statement
The animal study was reviewed and approved by the experimental protocol in accordance with the US National Institutes of Health standards (NIH publication No. 85-23, revised 1985) was approved by the Institutional Animal Care and Use Committee of the Faculty of Medicine, Chulalongkorn University (SST006/2560).
Author Contributions
ST, AL, and WP designed the study. WP performed in vitro and in vivo experiments. PP performed in vitro experiments. WP, TC, and NS designed and performed the microbiome analysis. WP, AL, and ST analyzed the data, discussed the results, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Thai Government Research Budget for Fiscal years 2016 and 2017 from the National Research Council of Thailand (GB-B_60_080_30_24) to ST, the Fundamental Fund 2565, Program Management Unit for Human Resources and Institutional Development Research and Innovation-CU (Global Partnership B16F630071 and Flagship B05F630073), TSRI Fund (CU_FRB640001_01_23_1) to AL, and the National Research Council of Thailand (NRCT5-RGJ63001). WP was supported by Rachadapisek Sompote Fund for Postdoctoral Fellowship, Chulalongkorn University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to express their gratitude to the Translational Research in Inflammation and Immunology Research Unit (TRIRU), Department of Microbiology, Chulalongkorn University, Bangkok, Thailand.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.745299/full#supplementary-material
References
Aktories, K., and Barbieri, J. T. (2005). Bacterial cytotoxins: targeting eukaryotic switches. Nat. Rev. Microbiol. 3, 397–410.
Aktories, K., Papatheodorou, P., and Schwan, C. (2018). Binary Clostridium difficile toxin (CDT) - A virulence factor disturbing the cytoskeleton. Anaerobe 53, 21–29. doi: 10.1016/j.anaerobe.2018.03.001
Aslam, S., Hamill, R. J., and Musher, D. M. (2005). Treatment of Clostridium difficile-associated disease: old therapies and new strategies. Lancet Infect. Dis. 5, 549–557. doi: 10.1016/S1473-3099(05)70215-2
Banerjee, P., Merkel, G. J., and Bhunia, A. K. (2009). Lactobacillus delbrueckii ssp. bulgaricus B-30892 can inhibit cytotoxic effects and adhesion of pathogenic Clostridium difficile to Caco-2 cells. Gut Pathog. 1:8. doi: 10.1186/1757-4749-1-8
Bartlett, J. G. (2002). Clinical practice. Antibiotic-associated diarrhea. N. Engl. J. Med. 346, 334–339.
Boonma, P., Spinler, J. K., Venable, S. F., Versalovic, J., and Tumwasorn, S. (2014). Lactobacillus rhamnosus L34 and Lactobacillus casei L39 suppress Clostridium difficile-induced IL-8 production by colonic epithelial cells. BMC Microbiol. 14:177. doi: 10.1186/1471-2180-14-177
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624.
Castagliuolo, I., Keates, A. C., Wang, C. C., Pasha, A., Valenick, L., Kelly, C. P., et al. (1998). Clostridium difficile toxin A stimulates macrophage-inflammatory protein-2 production in rat intestinal epithelial cells. J. Immunol. 160, 6039–6045.
Castagliuolo, I., Riegler, M. F., Valenick, L., Lamont, J. T., and Pothoulakis, C. (1999). Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect. Immun. 67, 302–307. doi: 10.1128/IAI.67.1.302-307.1999
Chen, S., Sun, C., Wang, H., and Wang, J. (2015). The role of Rho GTPases in toxicity of Clostridium difficile toxins. Toxins (Basel) 7, 5254–5267. doi: 10.3390/toxins7124874
Chen, X., Katchar, K., Goldsmith, J. D., Nanthakumar, N., Cheknis, A., Gerding, D. N., et al. (2008). A mouse model of Clostridium difficile-associated disease. Gastroenterology 135, 1984–1992. doi: 10.1053/j.gastro.2008.09.002
Cornick, S., Tawiah, A., and Chadee, K. (2015). Roles and regulation of the mucus barrier in the gut. Tissue Barriers 3:e982426.
Dharmani, P., Srivastava, V., Kissoon-Singh, V., and Chadee, K. (2009). Role of intestinal mucins in innate host defense mechanisms against pathogens. J. Innate Immun. 1, 123–135.
Erikstrup, L. T., Aarup, M., Hagemann-Madsen, R., Dagnaes-Hansen, F., Kristensen, B., Olsen, K. E., et al. (2015). Treatment of Clostridium difficile infection in mice with vancomycin alone is as effective as treatment with vancomycin and metronidazole in combination. BMJ Open Gastroenterol. 2:e000038. doi: 10.1136/bmjgast-2015-000038
FAO and WHO (2001). Report of a Joint FAO/WHO Expert Consultation on Evaluation of Health and Nutritional Properties of Probiotics in Food Including Powder Milk With Live Lactic Acid Bacteria. Rome: Food and Agriculture Organization of the United Nations, World Health Organization.
Feng, Y., and Cohen, S. N. (2013). Upregulation of the host SLC11A1 gene by Clostridium difficile toxin B facilitates glucosylation of Rho GTPases and enhances toxin lethality. Infect. Immun. 81, 2724–2732. doi: 10.1128/IAI.01177-12
Gao, Y., Li, S., Wang, J., Luo, C., Zhao, S., and Zheng, N. (2017). Modulation of intestinal epithelial permeability in differentiated Caco-2 cells exposed to Aflatoxin M1 and Ochratoxin A individually or collectively. Toxins (Basel) 10:13.
Gerding, D. N., Johnson, S., Rupnik, M., and Aktories, K. (2014). Clostridium difficile binary toxin CDT: mechanism, epidemiology, and potential clinical importance. Gut Microbes 5, 15–27. doi: 10.4161/gmic.26854
Goldenberg, J. Z., Yap, C., Lytvyn, L., Lo, C. K., Beardsley, J., Mertz, D., et al. (2017). Probiotics for the prevention of Clostridium difficile-associated diarrhea in adults and children. Cochrane Datab. Syst. Rev. 12:CD006095.
Hecht, G., Pothoulakis, C., Lamont, J. T., and Madara, J. L. (1988). Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J. Clin. Invest. 82, 1516–1524. doi: 10.1172/JCI113760
Hill, C., Guarner, F., Reid, G., Gibson, G. R., Merenstein, D. J., Pot, B., et al. (2014). Expert consensus document. The international scientific association for probiotics and prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 11, 506–514. doi: 10.1038/nrgastro.2014.66
Hodges, K., and Gill, R. (2010). Infectious diarrhea: cellular and molecular mechanisms. Gut Microbes 1, 4–21. doi: 10.4161/gmic.1.1.11036
Imaoka, A., Shima, T., Kato, K., Mizuno, S., Uehara, T., Matsumoto, S., et al. (2008). Anti-inflammatory activity of probiotic Bifidobacterium: enhancement of IL-10 production in peripheral blood mononuclear cells from ulcerative colitis patients and inhibition of IL-8 secretion in HT-29 cells. World J. Gastroenterol. 14, 2511–2516. doi: 10.3748/wjg.14.2511
Jank, T., and Aktories, K. (2008). Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 16, 222–229. doi: 10.1016/j.tim.2008.01.011
Kachrimanidou, M., and Malisiovas, N. (2011). Clostridium difficile infection: a comprehensive review. Crit. Rev. Microbiol. 37, 178–187.
Kachrimanidou, M., and Tsintarakis, E. (2020). Insights into the role of human gut microbiota in Clostridioides difficile infection. Microorganisms 8:200.
Kelly, C. P., Pothoulakis, C., and Lamont, J. T. (1994b). Clostridium difficile colitis. N. Engl. J. Med. 330, 257–262.
Kelly, C. P., Keates, S., Siegenberg, D., Linevsky, J. K., Pothoulakis, C., and Brady, H. R. (1994a). IL-8 secretion and neutrophil activation by HT-29 colonic epithelial cells. Am. J. Physiol. 267, G991–G997.
Kim, J. J., Shajib, M. S., Manocha, M. M., and Khan, W. I. (2012). Investigating intestinal inflammation in DSS-induced model of IBD. J. Vis. Exp. 3678.
Kim, J. M., Kim, J. S., Jun, H. C., Oh, Y. K., Song, I. S., and Kim, C. Y. (2002). Differential expression and polarized secretion of CXC and CC chemokines by human intestinal epithelial cancer cell lines in response to Clostridium difficile toxin A. Microbiol. Immunol. 46, 333–342. doi: 10.1111/j.1348-0421.2002.tb02704.x
Kolling, G. L., Wu, M., Warren, C. A., Durmaz, E., Klaenhammer, T. R., Timko, M. P., et al. (2012). Lactic acid production by Streptococcus thermophilus alters Clostridium difficile infection and in vitro toxin A production. Gut Microbes 3, 523–529. doi: 10.4161/gmic.21757
Koon, H. W., Su, B., Xu, C., Mussatto, C. C., Tran, D. H., Lee, E. C., et al. (2016). Probiotic Saccharomyces boulardii CNCM I-745 prevents outbreak-associated Clostridium difficile-associated cecal inflammation in hamsters. Am. J. Physiol. Gastrointest. Liver Physiol. 311, G610–G623. doi: 10.1152/ajpgi.00150.2016
Kuehne, S. A., Cartman, S. T., and Minton, N. P. (2011). Both, toxin A and toxin B, are important in Clostridium difficile infection. Gut Microbes 2, 252–255. doi: 10.4161/gmic.2.4.16109
Le Bivic, A., Hirn, M., and Reggio, H. (1988). HT-29 cells are an in vitro model for the generation of cell polarity in epithelia during embryonic differentiation. Proc. Natl. Acad. Sci. U.S.A. 85, 136–140. doi: 10.1073/pnas.85.1.136
Lee, Y. J., Arguello, E. S., Jenq, R. R., Littmann, E., Kim, G. J., Miller, L. C., et al. (2017). Protective factors in the intestinal microbiome against Clostridium difficile infection in recipients of allogeneic hematopoietic stem cell transplantation. J. Infect. Dis. 215, 1117–1123. doi: 10.1093/infdis/jix011
Leelahavanichkul, A., Panpetch, W., Worasilchai, N., Somparn, P., Chancharoenthana, W., Nilgate, S., et al. (2016). Evaluation of gastrointestinal leakage using serum (1–>3)-beta-D-glucan in a Clostridium difficile murine model. FEMS Microbiol. Lett. 363:fnw204. doi: 10.1093/femsle/fnw204
Leffler, D. A., and Lamont, J. T. (2015). Clostridium difficile infection. N. Engl. J. Med. 372, 1539–1548.
Lemee, L., Dhalluin, A., Testelin, S., Mattrat, M. A., Maillard, K., Lemeland, J. F., et al. (2004). Multiplex PCR targeting tpi (triose phosphate isomerase), tcdA (Toxin A), and tcdB (Toxin B) genes for toxigenic culture of Clostridium difficile. J. Clin. Microbiol. 42, 5710–5714. doi: 10.1128/JCM.42.12.5710-5714.2004
Li, X., Chu, Q., Huang, Y., Xiao, Y., Song, L., Zhu, S., et al. (2019). Consortium of probiotics attenuates colonization of Clostridioides difficile. Front. Microbiol. 10:2871. doi: 10.3389/fmicb.2019.02871
Lyerly, D. M., Krivan, H. C., and Wilkins, T. D. (1988). Clostridium difficile: its disease and toxins. Clin. Microbiol. Rev. 1, 1–18. doi: 10.1128/CMR.1.1.1
Mack, D. R., Michail, S., Wei, S., Mcdougall, L., and Hollingsworth, M. A. (1999). Probiotics inhibit enteropathogenic E. coli adherence in vitro by inducing intestinal mucin gene expression. Am. J. Physiol. 276, G941–G950.
Mooyottu, S., Flock, G., Upadhyay, A., Upadhyaya, I., Maas, K., and Venkitanarayanan, K. (2017). Protective effect of Carvacrol against gut dysbiosis and Clostridium difficile associated disease in a mouse model. Front. Microbiol. 8:625. doi: 10.3389/fmicb.2017.00625
Mylonakis, E., Ryan, E. T., and Calderwood, S. B. (2001). Clostridium difficile–associated diarrhea: a review. Arch. Intern. Med. 161, 525–533.
Nusrat, A., Von Eichel-Streiber, C., Turner, J. R., Verkade, P., Madara, J. L., and Parkos, C. A. (2001). Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect. Immun. 69, 1329–1336. doi: 10.1128/IAI.69.3.1329-1336.2001
Panpetch, W., Chancharoenthana, W., Bootdee, K., Nilgate, S., Finkelman, M., Tumwasorn, S., et al. (2018). Lactobacillus rhamnosus L34 attenuates gut translocation-induced bacterial sepsis in murine models of leaky gut. Infect. Immun. 86, e700–e717. doi: 10.1128/IAI.00700-17
Panpetch, W., Hiengrach, P., Nilgate, S., Tumwasorn, S., Somboonna, N., Wilantho, A., et al. (2020). Additional Candida albicans administration enhances the severity of dextran sulfate solution induced colitis mouse model through leaky gut-enhanced systemic inflammation and gut-dysbiosis but attenuated by Lactobacillus rhamnosus L34. Gut Microbes 11, 465–480. doi: 10.1080/19490976.2019.1662712
Panpetch, W., Somboonna, N., Palasuk, M., Hiengrach, P., Finkelman, M., Tumwasorn, S., et al. (2019). Oral Candida administration in a Clostridium difficile mouse model worsens disease severity but is attenuated by Bifidobacterium. PLoS One 14:e0210798. doi: 10.1371/journal.pone.0210798
Panpetch, W., Spinler, J. K., Versalovic, J., and Tumwasorn, S. (2016). Characterization of Lactobacillus salivarius strains B37 and B60 capable of inhibiting IL-8 production in Helicobacter pylori-stimulated gastric epithelial cells. BMC Microbiol. 16:242. doi: 10.1186/s12866-016-0861-x
Paone, P., and Cani, P. D. (2020). Mucus barrier, mucins and gut microbiota: the expected slimy partners? Gut 69, 2232–2243. doi: 10.1136/gutjnl-2020-322260
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45.
Pothoulakis, C. (2000). Effects of Clostridium difficile toxins on epithelial cell barrier. Ann. N. Y. Acad. Sci. 915, 347–356.
Reeves, A. E., Koenigsknecht, M. J., Bergin, I. L., and Young, V. B. (2012). Suppression of Clostridium difficile in the gastrointestinal tracts of germfree mice inoculated with a murine isolate from the family Lachnospiraceae. Infect. Immun. 80, 3786–3794. doi: 10.1128/IAI.00647-12
Reeves, A. E., Theriot, C. M., Bergin, I. L., Huffnagle, G. B., Schloss, P. D., and Young, V. B. (2011). The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes 2, 145–158. doi: 10.4161/gmic.2.3.16333
Reid, G., Younes, J. A., Van Der Mei, H. C., Gloor, G. B., Knight, R., and Busscher, H. J. (2011). Microbiota restoration: natural and supplemented recovery of human microbial communities. Nat. Rev. Microbiol. 9, 27–38. doi: 10.1038/nrmicro2473
Ritchie, M. L., and Romanuk, T. N. (2012). A meta-analysis of probiotic efficacy for gastrointestinal diseases. PLoS One 7:e34938.
Roychowdhury, S., Cadnum, J., Glueck, B., Obrenovich, M., Donskey, C., and Cresci, G. A. M. (2018). Faecalibacterium prausnitzii and a prebiotic protect intestinal health in a mouse model of antibiotic and Clostridium difficile exposure. JPEN J. Parenter. Enteral Nutr. 42, 1156–1167. doi: 10.1002/jpen.1053
Savidge, T. C., Pan, W. H., Newman, P., O’brien, M., Anton, P. M., and Pothoulakis, C. (2003). Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology 125, 413–420. doi: 10.1016/s0016-5085(03)00902-8
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Shekels, L. L., Lyftogt, C. T., and Ho, S. B. (1996). Bile acid-induced alterations of mucin production in differentiated human colon cancer cell lines. Int. J. Biochem. Cell Biol. 28, 193–201. doi: 10.1016/1357-2725(95)00125-5
Shen, N. T., Maw, A., Tmanova, L. L., Pino, A., Ancy, K., Crawford, C. V., et al. (2017). Timely use of probiotics in hospitalized adults prevents Clostridium difficile infection: a systematic review with meta-regression analysis. Gastroenterology 152, 1889–1900.e1889. doi: 10.1053/j.gastro.2017.02.003
Spinler, J. K., Auchtung, J., Brown, A., Boonma, P., Oezguen, N., Ross, C. L., et al. (2017). Next-generation probiotics targeting Clostridium difficile through precursor-directed antimicrobial biosynthesis. Infect. Immun. 85, e303–e317. doi: 10.1128/IAI.00303-17
Sun, X., Savidge, T., and Feng, H. (2010). The enterotoxicity of Clostridium difficile toxins. Toxins (Basel) 2, 1848–1880. doi: 10.3390/toxins2071848
Thomas, C. M., Hong, T., Van Pijkeren, J. P., Hemarajata, P., Trinh, D. V., Hu, W., et al. (2012). Histamine derived from probiotic Lactobacillus reuteri suppresses TNF via modulation of PKA and ERK signaling. PLoS One 7:e31951. doi: 10.1371/journal.pone.0031951
Thomas, C. M., Saulnier, D. M., Spinler, J. K., Hemarajata, P., Gao, C., Jones, S. E., et al. (2016). FolC2-mediated folate metabolism contributes to suppression of inflammation by probiotic Lactobacillus reuteri. Microbiologyopen 5, 802–818. doi: 10.1002/mbo3.371
Trejo, F. M., Perez, P. F., and De Antoni, G. L. (2010). Co-culture with potentially probiotic microorganisms antagonises virulence factors of Clostridium difficile in vitro. Antonie Van Leeuwenhoek 98, 19–29. doi: 10.1007/s10482-010-9424-6
Velcich, A., and Augenlicht, L. H. (1993). Regulated expression of an intestinal mucin gene in HT29 colonic carcinoma cells. J. Biol. Chem. 268, 13956–13961.
Viswanathan, V. K., Hodges, K., and Hecht, G. (2009). Enteric infection meets intestinal function: how bacterial pathogens cause diarrhoea. Nat. Rev. Microbiol. 7, 110–119.
Voth, D. E., and Ballard, J. D. (2005). Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18, 247–263. doi: 10.1128/CMR.18.2.247-263.2005
Wei, Y., Yang, F., Wu, Q., Gao, J., Liu, W., Liu, C., et al. (2018). Protective effects of bifidobacterial strains against toxigenic Clostridium difficile. Front. Microbiol. 9:888. doi: 10.3389/fmicb.2018.00888
Xu, Q., Gu, S., Chen, Y., Quan, J., Lv, L., Chen, D., et al. (2018). Protective effect of Pediococcus pentosaceus LI05 against Clostridium difficile infection in a mouse model. Front. Microbiol. 9:2396.
Xue, Y., Zhang, H., Wang, H., Hu, J., Du, M., and Zhu, M. J. (2014). Host inflammatory response inhibits Escherichia coli O157:H7 adhesion to gut epithelium through augmentation of mucin expression. Infect. Immun. 82, 1921–1930. doi: 10.1128/IAI.01589-13
Yong, C. C., Lim, J., Kim, B. K., Park, D. J., and Oh, S. (2019). Suppressive effect of Lactobacillus fermentum Lim2 on Clostridioides difficile 027 toxin production. Lett. Appl. Microbiol. 68, 386–393. doi: 10.1111/lam.13124
Zemljic, M., Rupnik, M., Scarpa, M., Anderluh, G., Palu, G., and Castagliuolo, I. (2010). Repetitive domain of Clostridium difficile toxin B exhibits cytotoxic effects on human intestinal epithelial cells and decreases epithelial barrier function. Anaerobe 16, 527–532. doi: 10.1016/j.anaerobe.2010.06.010
Keywords: Lacticaseibacillus casei T21, probiotics, proinflammatory cytokines, Clostridioides difficile, gut dysbiosis, inflammation, toxin lethality
Citation: Panpetch W, Phuengmaung P, Cheibchalard T, Somboonna N, Leelahavanichkul A and Tumwasorn S (2021) Lacticaseibacillus casei Strain T21 Attenuates Clostridioides difficile Infection in a Murine Model Through Reduction of Inflammation and Gut Dysbiosis With Decreased Toxin Lethality and Enhanced Mucin Production. Front. Microbiol. 12:745299. doi: 10.3389/fmicb.2021.745299
Received: 21 July 2021; Accepted: 02 November 2021;
Published: 01 December 2021.
Edited by:
Axel Cloeckaert, Institut National de Recherche pour l’Agriculture, l’Alimentation et l’Environnement (INRAE), FranceReviewed by:
Marjorie Pizarro-Guajardo, Texas A&M University, United StatesMaria de los Angeles Serradell, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina
Copyright © 2021 Panpetch, Phuengmaung, Cheibchalard, Somboonna, Leelahavanichkul and Tumwasorn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Asada Leelahavanichkul, YWxlZWxhaGF2YW5pdEBnbWFpbC5jb20=; Somying Tumwasorn, c29teWluZy5UQGNodWxhLmFjLnRo
†These authors have contributed equally to this work