
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 02 November 2021
Sec. Microbiotechnology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.744075
This article is part of the Research Topic New Microbial Inoculants for Enhancing Fermentation Quality of Silage View all 17 articles
 Laura Díaz-García1
Laura Díaz-García1 Dayanne Chaparro1
Dayanne Chaparro1 Hugo Jiménez2
Hugo Jiménez2 Luis Fernando Gómez-Ramírez2
Luis Fernando Gómez-Ramírez2 Adriana J. Bernal3Esteban Burbano-Erazo2
Adriana J. Bernal3Esteban Burbano-Erazo2 Diego Javier Jiménez1*
Diego Javier Jiménez1*Traditionally, starting inoculants have been applied to improve ensiling of forage used for livestock feed. Here, we aimed to build up a bioinoculant composed of lactic acid-producing and lignocellulolytic bacteria (LB) derived from the Megathyrsus maximus (guinea grass) phyllosphere. For this, the dilution-to-stimulation approach was used, including a sequential modification of the starting culture medium [Man, Rogosa, and Sharpe (MRS) broth] by addition of plant biomass (PB) and elimination of labile carbon sources. Along 10 growth-dilution steps (T1–T10), slight differences were observed in terms of bacterial diversity and composition. After the sixth subculture, the consortium started to degrade PB, decreasing its growth rate. The co-existence of Enterobacteriales (fast growers and highly abundance), Actinomycetales, Bacillales, and Lactobacillales species was observed at the end of the selection process. However, a significant structural change was noticed when the mixed consortium was cultivated in higher volume (500ml) for 8days, mainly increasing the proportion of Paenibacillaceae populations. Interestingly, Actinomycetales, Bacillales, and Lactobacillales respond positively to a pH decrease (4–5), suggesting a relevant role within a further silage process. Moreover, gene-centric metagenomic analysis showed an increase of (hemi)cellulose-degrading enzymes (HDEs) during the enrichment strategy. Reconstruction of metagenome-assembled genomes (MAGs) revealed that Paenibacillus, Cellulosimicrobium, and Sphingomonas appear as key (hemi)cellulolytic members (harboring endo-glucanases/xylanases, arabinofuranosidases, and esterases), whereas Enterococcus and Cellulosimicrobium have the potential to degrade oligosaccharides, metabolize xylose and might produce lactic acid through the phosphoketolase (PK) pathway. Based on this evidence, we conclude that our innovative top-down strategy enriched a unique bacterial consortium that could be useful in biotechnological applications, including the development/design of a synthetic bioinoculant to improve silage processes.
Ensiling is an ancient technique used to preserve nutrients in roughages (e.g., grass and legumes), generally offered as a ruminant livestock feed (Ávila and Carvalho, 2019). This process is based on spontaneous fermentation, where epiphytic lactic acid bacteria (LAB) metabolize plant-derived sugars, quickly decreasing the pH (between 5 and 4) and preventing silage spoilage (Fabiszewska et al., 2019). Megathyrsus maximus Jacq., a fast-growing, leafy perennial grass, is widely used in the tropics as roughage for livestock farming. Due to its high biomass availability (during both the dry and rainy seasons), protein content (~13%) and livestock digestibility (~60%), this grass has great potential for ensiling (Mojica-Rodríguez and Burbano-Erazo, 2020). However, M. maximus (guinea grass) like many tropical grasses contains a large amount of fiber, including lignin and structural carbohydrates linked to ferulic acid, preventing its efficient utilization directly in feedlots (Grabber et al., 2009). Ensiling of roughages comprises four phases: (1) microbial depletion of oxygen; (2) lactic acid production in anaerobic conditions (here LAB becomes highly abundant); (3) stabilization; and (4) feedout phase where silage is re-exposed to air (Keshri et al., 2019). The type of epiphytic microbiota depends on the forage crop, and is mainly composed of Gammaproteobacteria (e.g., Klebsiella, Citrobacter, Pantoea, and Pseudomonas), Firmicutes (e.g., Clostridium, Bacillus, and Paenibacillus), yeast, and molds, plus a low proportion of LAB (e.g., Lactobacillus, Pediococcus, Enterococcus, Lactococcus, Leuconostoc, and Weissella), actinomycetes and acetic/propionic acid bacteria (Nazar et al., 2021a, b). The phyllosphere of forage could represent an unexplored source of lignocellulolytic microbes that survive in oligotrophic conditions. Regarding ensiling, LAB populations are essential, but they do not exceed 1% of the total epiphytic microbiota (McAllister et al., 2018, Ávila and Carvalho, 2019; Fabiszewska et al., 2019). Therefore, the use of starting homofermentative LAB bioinoculants (e.g., Lactobacillus species) and other additives (e.g., organic acids and enzymes) has been used to improve the quality of the silage, thus benefiting animal productivity (Muck et al., 2018; Su et al., 2019). Additionally, (hemi)cellulases, amylases and proteases have also been added to forage at initiation of ensiling to improve the breakdown of plant polymers, providing labile sugars for lactic acid production and increasing the digestibility of plant cell walls (Windle et al., 2014; Addah et al., 2016; Ogunade et al., 2018). Moreover, it has been suggested that the inoculation of bacterial isolates and/or mixed microbial consortia (e.g., LAB plus lignocellulolytic species) with the potential to produce cellulases, arabinofuranosidases, xylosidases, ferulic acid esterases (FAE; EC 3.1.1.73), and cutinases, could enhance the quality of the silage by increasing the availability of carbohydrates with lower complexity (Muck et al., 2018; Tarraran and Mazzoli, 2018; Bonaldi et al., 2021). FAE has been recently reported in LAB, and its use may show a potential improvement on neutral detergent fiber (Xie et al., 2021).
The design and/or selection of microbial consortia with a desired structure/function can be accomplished by two strategies. In the top-down approach, microbial communities from nature are selected in a specific culture medium that enables the survival of the fittest in sequential transfers (Jiménez et al., 2014a; Díaz-García et al., 2021). On the other hand, in the bottom-up approach, the design of the consortium is carried out by mixing different populations of microbial isolates in specific proportions and compositions (Jiménez et al., 2018, 2020). Interestingly, a synthetic consortium composed of LAB and a cellulolytic fungus has been previously reported (Shahab et al., 2018). In this mutualistic guild, Trichoderma reesei provides the soluble saccharides (by depolymerization of plant polysaccharides), whereas Lactobacillus pentosus utilize them for the production of lactic and acetic acid. Based on these facts, we hypothesize that the selection of consortia composed of LAB and lignocellulolytic bacteria (LB), derived from the epiphytic community of forage biomass, and could be the starting point to design a synthetic bioinoculant that preserves the quality of the plant biomass (PB) during the ensiling process. In addition, the discovery of microbes or a set of mutualistic species that can produce lactic acid directly from PB is interesting from a biotechnological perspective (Tarraran and Mazzoli, 2018). Thus, in this study, we aimed to co-cultivate LAB and LB derived from the M. maximus cv. Agrosavia Sabanera phyllosphere by using a top-down approach (i.e., the dilution-to-stimulation strategy in which easy-to-consume carbon sources are eliminated and growth on the desired substrate is stimulated). During and after the selection process, different modifications of culture conditions were done. Additionally, bacterial 16S rRNA gene and whole metagenome sequencing, including the reconstruction of metagenome assembled genomes (MAGs), was performed to disentangle taxonomic and functional profiles of the obtained consortia. Based on this enrichment and metagenomic analysis, we report here the genomic capacity of a (hemi)cellulolytic Cellulosimicrobium-related species (an actinobacterium) to produce lactic acid from plant-derived xylose.
The Guinea grass M. maximus-associated epiphytic microbial community was the source of inoculum for selection of LAB and LB populations. Firstly, this grass was cut in 1cm2 pieces. Then, in aseptic conditions, 10 fragments were inoculated in 10ml of Man, Rogosa, and Sharpe (MRS) broth (Merck, Darmstadt, Germany) and incubated at 39°C for 24h. After microbial growth, 1ml of the culture was inoculated in 9ml of MRS broth and incubated to the above conditions. This latter procedure was carried out to pre-enrich LAB populations by growth of the community in medium designed to enrich growth of LAB. Subsequently, 25μl of the pre-enriched culture were inoculated in 100ml-flasks with 25ml of MRS-modified broth (i.e., without glucose) containing 1% lignocellulosic substrate (M. maximus cv. Agrosavia Sabanera added as a carbon source; T1; Figure 1; Table 1), vitamin and trace elements solution (Jiménez et al., 2014b). Previously, the lignocellulosic substrate (supplied by Agrosavia) was knife-milled through a 1mm screen and washed twice with distilled water and ethanol 70% (v/v). The grass was collected at regrowth age of 30days in a sub-region of the Colombian dry Caribbean (Motilonia RC, Cesar) with an elevation of 103 masl. The culture flasks were incubated at 28°C under shaking conditions (130rpm). After 4days of incubation, aliquots (25μl) of microbial suspension were transferred to 25ml of fresh liquid medium containing 1% plant biomass (T2), following the dilution-to-stimulation approach (Jiménez et al., 2017). Two negative controls were also set up: (A) microbial inoculum in liquid modified MRS (without plant biomass and glucose) and (B) uninoculated liquid modified MRS (containing plant biomass and without glucose). During the growth-dilution transfers, liquid culture medium was sequentially modified (Table 1) to avoid the assimilation of labile carbon sources (e.g., ammonium citrate, yeast, and/or beef extract), increasing the selection for LB populations. Thus, these modifications were done until no growth in negative control A was observed (Figure 1; Table 1). At the end of each transfer cultures were filtered and the remain substrate was dried for 24h at 45°C to calculate the percentage of weight loss, following the formula proposed by de Lima Brossi et al. (2016). Bacterial growth was determined using optical density (OD) at 600nm and the number of viable cells (CFU/ml) was quantified by plate counting in R2A and MRS agar (Merck, Darmstadt, Germany). Moreover, samples from transfer 10 (T10) were conserved in 20% of glycerol at −20°C. ANOVA and post hoc Tukey-Kramer test were performed using the software R (R Core Team, 2008) to evaluate the significant difference in substrate consumption and bacterial growth along the transfers, with a CI of 99% (α=0.01).
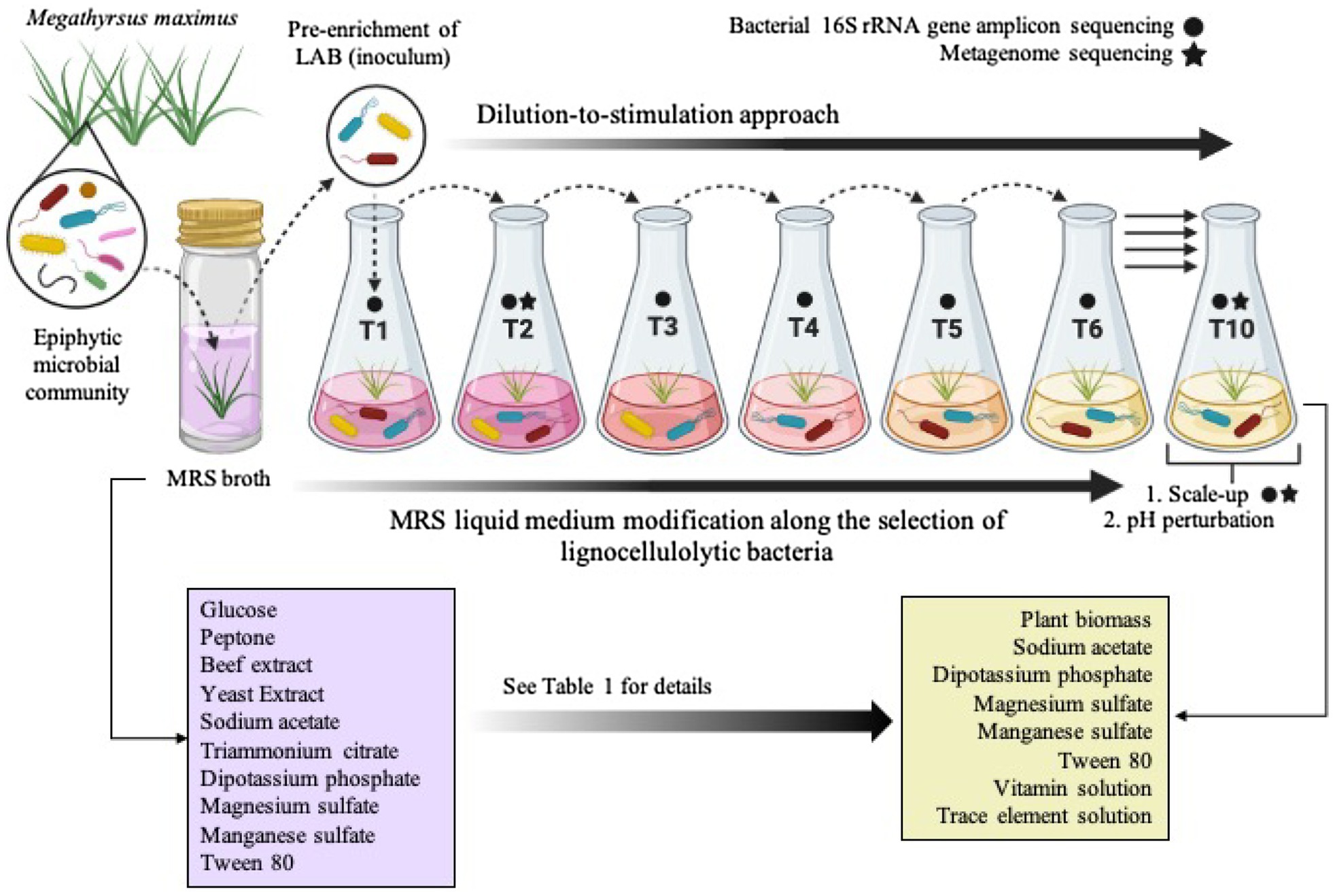
Figure 1. Schematic representation of the top-down enrichment strategy by using the dilution-to-stimulation approach. During the selection process, the liquid Man, Rogosa, and Sharpe (MRS) medium, which is specific for lactic acid bacteria (LAB), was modified as shown in Table 1 in order to select lignocellulolytic bacteria (LB) from the Megathyrsus maximus phyllosphere. The scale-up and pH perturbation were carried out after transfer 10 (T10). Created with BioRender.com.
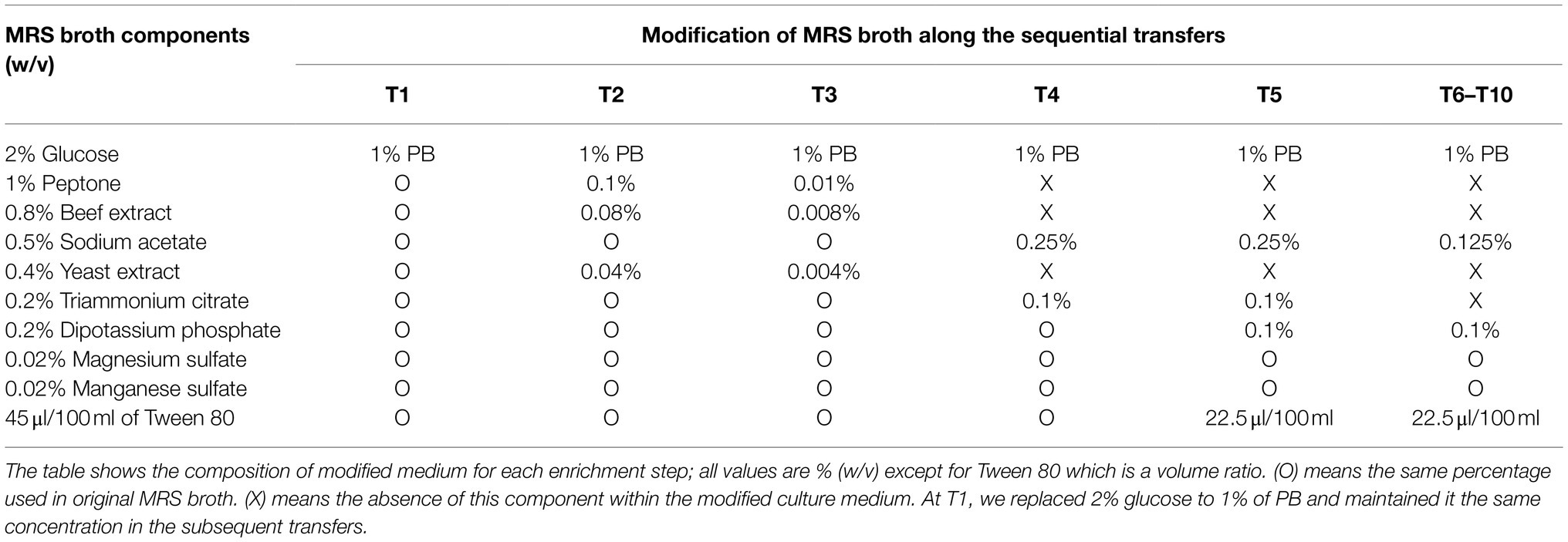
Table 1. Scheme of MRS broth modification during the enrichment strategy (from T1 to T6–T10).
To increase the bacterial biomass, the mixed culture after T10 was transferred to a final volume of 500ml. We carried out this procedure because this consortium was used as a starting bioinoculant in a silage of tropical forages in field experiments (data not shown). The scale-up growth process began with a pre-inoculum from T10 that was cultivated in 25ml of liquid medium (eight replicates), as previously described. Two of these replicates were mixed to produce an inoculum of 50ml. To ensure a high bacterial density, four 2L-flasks with 500ml of fresh medium (same composition as T10) were inoculated with the former 50ml and incubated at 28°C for 8days. Finally, four replicates were mixed and filtered to produce 2L of bacterial culture. Moreover, the consortium obtained at T10 was cultivated (i.e., perturbed) on the modified MRS medium adjusted to different pH values (4, 5, and unmodified pH 6.2) at 28°C for 4days, with shaking at 130rpm (in two sequential transfers). Number of viable cells (CFU/ml) was quantified by plate counting in R2A and MRS agar (Merck, Darmstadt, Germany). OD at 600nm and weight loss of the substrate were calculated with the parameters described above. After microbial growth in different pH values, the secreted fractions of the cultures were tested for the presence of plant polymers-degrading endo-enzymes using a set of six chromogenic polysaccharide hydrogels (CPH; i.e., 2-HE-cellulose, carboxymethylcellulose, arabinoxylan, xylan, xyloglucan, and glucomannan) and two insoluble biomass substrates (ICB; i.e., wheat straw and sugarcane bagasse; Kračun et al., 2015). The semi-quantification of the enzymatic activities was measured as reported Díaz-García et al. (2021) and following the instructions of the manufacturer (GlycoSpot IVS, Farum, Denmark). ANOVA and post hoc Tukey-Kramer test were performed using the software R (R Core Team, 2008) to evaluate significant differences in growth and enzyme activities, with a CI of 99% (α=0.01).
Total microbial DNA was extracted using the DNeasy UltraClean Microbial Kit (Qiagen, Hilden, Germany) according to the instructions of the manufacturer. For the 16S rRNA gene analysis, triplicate samples from the dilution-to-stimulation approach (T1, T2, T3, T4, T5, T6, and T10), after the scale-up process (S) and the pH perturbation (4, 5, and unmodified) were sequenced using the Illumina MiSeq technology (300bp pair-end reads). For each sample, a library of 16S rRNA gene amplicons (hypervariable region V3–V4) was prepared using the primers 341F and 785R (Klindworth et al., 2013). Bioinformatic analysis was done using the software Quantitative Insights Into Microbial Ecology (QIIME2; Bolyen et al., 2019). VSEARCH (Rognes et al., 2016) was used to join raw pair-end reads. Quality control and read correction to obtain amplicon sequence variants (ASVs) were done using Deblur pipeline (Amir et al., 2017). Rarefaction was performed (5,205 and 5,490 reads for the enrichment steps and the pH perturbation samples, respectively) per sample to calculate alpha and beta diversity (including Shannon’s index and weighted UniFrac distances), and for comparisons between samples (e.g., relative abundance of ASVs). MAFFT (Katoh and Standley, 2013) was used to construct multiple alignments of ASVs. A phylogenetic tree was built using FasTree (Price et al., 2009) based on MAFFT alignment. Taxonomic affiliation of ASVs was done using SILVA rRNA gene curated database (Quast et al., 2013).
Based on the 16S rRNA sequencing results, consortia T2, T10, and S (in duplicate) were subjected to Illumina MiSeq technology sequencing (300bp paired-end reads) in order to determine taxonomy profiles and metabolic potential. After whole-metagenome sequencing, the FastQ files were uploaded in the MG-RAST server (Meyer et al., 2008). Overlapping sequence pairs were matched, and non-overlapping reads retained as individual reads, after which, dereplication was performed. Duplicate read-based inferred SE estimation and quality trimming (phred score<20) used default settings. Gene predictions were done using the FragGeneScan software, and subsequently, the proteins were annotated based on BLASTX searches against the RefSeq and KEGG databases using an e-value cutoff of 1e-15, a minimum alignment length of 50 amino acids, and a minimum identity of 50% (Jiménez et al., 2016). Data from MG-RAST annotation were statistically analyzed using the STAMP package (Parks and Beiko, 2010). Moreover, to evaluate the relative abundance of reads per selected enzyme-encoding gene, the counts were normalized to hits, or unique matches, per million reads, thereby accounting for differences in metagenome sizes. Nine genes involved in heterolactic and/or homolactic fermentation of xylose (Tarraran and Mazzoli, 2018) and 10 genes involved in lignocellulose degradation (Jiménez et al., 2014a; Díaz-García et al., 2021) were analyzed. Heat maps were constructed in the web server Heatmapper using row-Z score for each enzyme (Babicki et al., 2016).
For the assembly of MAGs, raw sequence data (FastQ files) from T2, T10, and S samples were initially trimmed using the Sickle tool v1.33 with default parameters (available at https://github.com/najoshi/sickle). To avoid misleading results from subsequent binning analysis, carp artifacts were detected. For this, trimmed sequences were filtered against adapter and non-authentic primer sequences originating from the Illumina library preparation (Marter et al., 2021). Metagenome assembly was done using MEGAHIT v1.2.7 (Li et al., 2015) with default parameters. Concoct v1.1.0 (Alneberg et al., 2014), Bowtie v2.3.5 (Langmead and Salzberg, 2012), MetaBAT v2.12.1 (Kang et al., 2015), and MaxBin v2.2.6 (Wu et al., 2016) were used to bin the assembled sequences with default parameters. Binned contigs obtained were subsequently analyzed using DAS tool v1.1.2 (Sieber et al., 2018). To assess the completeness and contamination of the resulting bins, CheckM v1.0.13 was used with the lineage_wf option (Parks et al., 2015). The MAGs were structurally annotated and curated based on rRNAs genes (e.g., 5S, 16S, and 23S) and rpoB gene using DFAST v1.2.6 (Tanizawa et al., 2018). Taxonomic assignment was performed using GTDB v1.4.1 (Parks et al., 2020). Functional annotation was done using the RAST (Aziz et al., 2008) and dbCAN webservers (Yin et al., 2012). Moreover, 24 encoding-genes involved in hetero- or homo-fermentation of pentoses and hexoses (Hatti-Kaul et al., 2018; Tarraran and Mazzoli, 2018) were searched within the MAGs.
The selection of consortia was carried out by the dilution-to-stimulation approach using the epiphytic microbial community of M. maximus as starting inoculum (Figure 1). In the first stage of selection, we aimed to increase the populations of LAB by growing the phyllosphere-derived community in MRS broth. This pre-enrichment was used to inoculate the first transfer (T1), where glucose was removed and 1% (w/v) M. maximus was added as a carbon source. After this stage, increase of LB populations was the main goal. For this purpose, liquid MRS medium was sequentially modified from T2 to T6–T10, removing peptone, yeast extract, beef extract, triammonium citrate, and decreasing the concentration of sodium acetate, dipotassium phosphate, and tween 80 (Table 1). These modifications were carried out until microbial cell growth (measured by OD at 600nm) in the negative control (i.e., without plant biomass) was not observed, indicating that the consortium started to consume the plant polymers (Figure 2A). After T6, the selected consortium was able to consume ~14% of plant biomass and significant differences (value of p<0.01) in this capacity were evidenced compared to T5 (Figure 2B). Moreover, progressive reduction of bacterial growth rate was observed from T1 to T6, suggesting that labile carbon sources were depleted due to the liquid medium modification (Figures 2A,C). LAB populations were abundant at T1–T2. However, they decreased along the enrichment strategy (Figure 2D). At the end of the selective process (T10), around 109 and 105CFU/ml of total and LAB populations were obtained, respectively (Figures 2C,D).
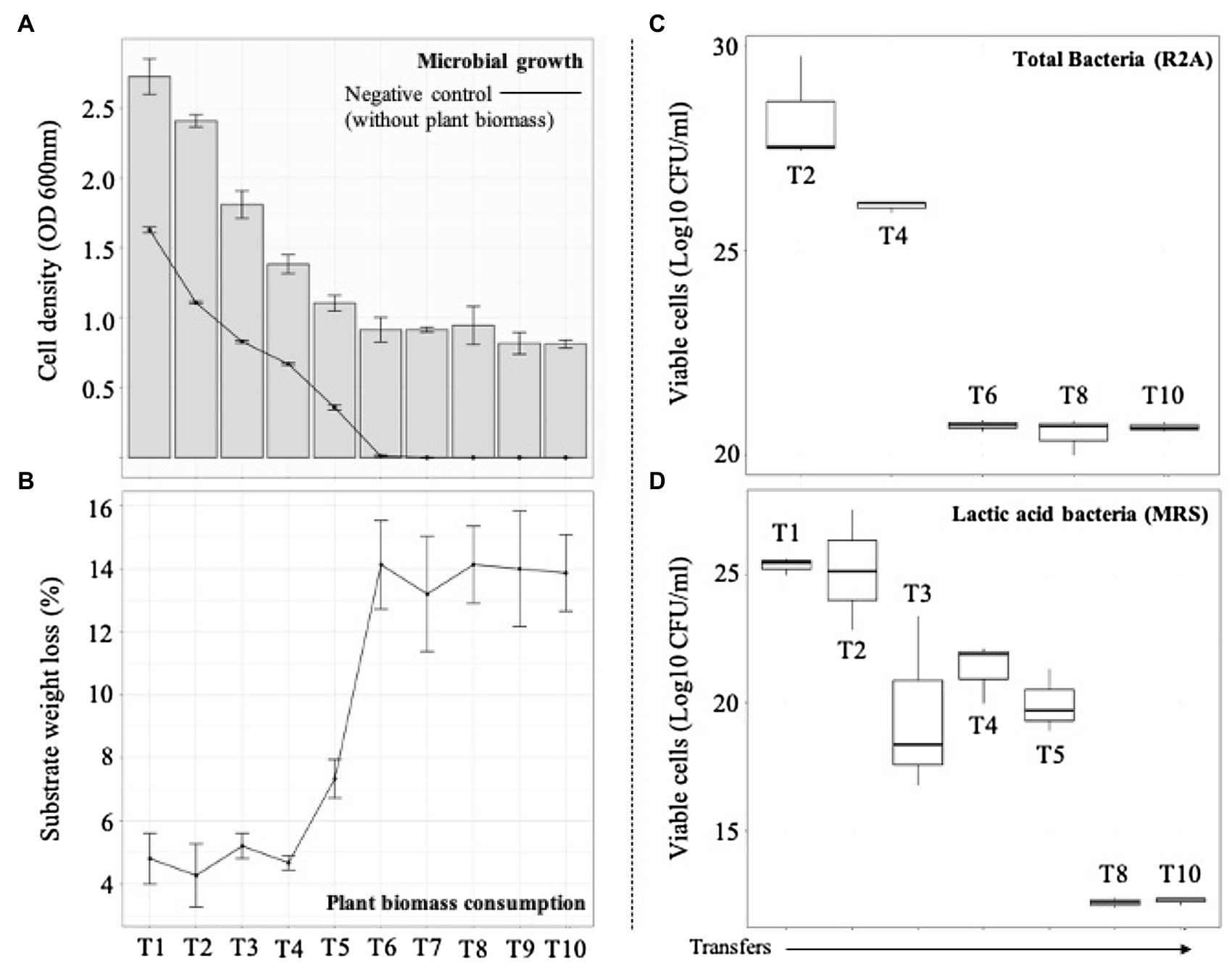
Figure 2. Microbial growth and plant biomass (PB) degradation values after each sequential growth-dilution steps (T1–T10). (A) Bars show cell density [optical density (OD) at 600nm] and the line shows cell density in the control medium containing no PB. (B) Average substrate weight loss (percent). Error bars represent SDs from three biological replicates. Viable cells (Log10 CFU/ml) of (C) total and (D) acid lactic bacteria quantified in agar R2A and MRS, respectively.
The bacterial community changes during the selective process were evaluated by 16S rRNA gene sequencing. In total, this analysis generated 1,623 ASVs and ~1.2Gbp of high-quality reads. The results indicated that bacterial diversity slightly decreased from T1 to T5. However, the number of observed ASVs increased from T5 (44±3.0) to T6 (77±4.1) and T10 (73±4.5). Interestingly, after the scale-up process (to 500ml of culture volume) the number of ASVs increased to 99±4.0 (Figure 3A). The taxonomic affiliation of the 16S rRNA data revealed that Gammaproteobacteria and Bacillales species were mostly selected during the enrichment strategy. Regarding LAB populations, species belonging to the Enterococcaceae family were observed at T1–T2 (~10% of the total community; Figure 3B). They remained in low abundance in the final selected consortium (i.e., T10). Modifications in the MRS liquid medium did not greatly affect the bacterial structural composition between T2 and T10. Remarkably, the final consortium (i.e., T10) showed a significant change after its cultivation in a higher volume (i.e., 500ml; Figure 3C). This scale-up process (denominated S in Figure 3) mostly increased the relative abundance of Paenibacillaceae, Sphingomonadaceae, and Xanthobactereaceae, well known lignocellulolytic families (Figure 3B). At the end, the mixed bioinoculant (S), used in the field silage experiment, contained a higher proportion of LB populations and lower proportion of LAB (mostly species from Enterococcaceae family).
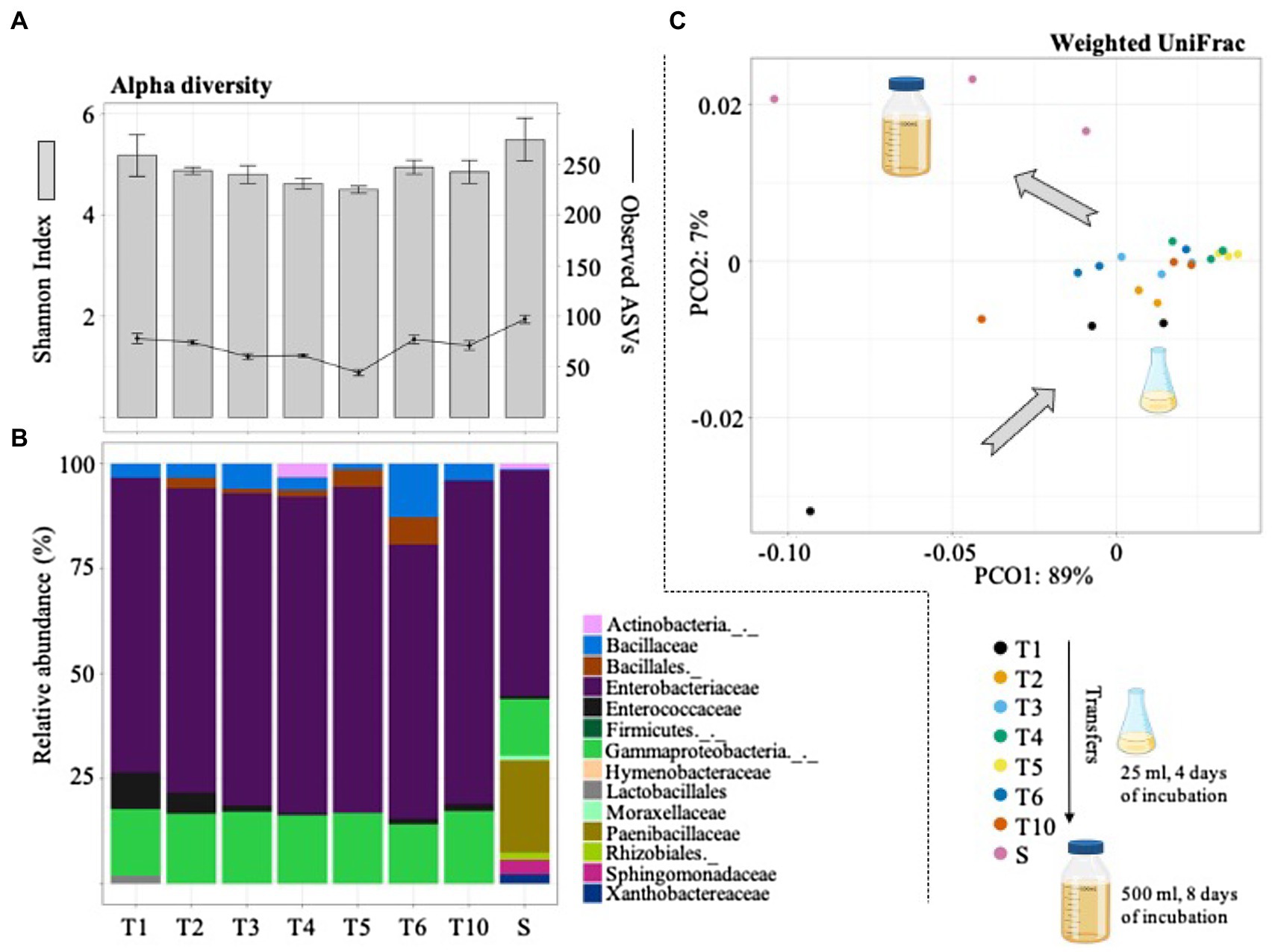
Figure 3. Diversity (alpha and beta) and structural composition of the selected consortia during the sequential growth-dilution steps (T1–T6 and T10) and after its cultivation in a higher volume (i.e., scale-up growth process; S). (A) Observed amplicon sequencing variants (ASVs) and Shannon index values. Error bars represent SDs from three biological replicates. (B) Relative abundance (%) of taxa (family or higher level) detected using the SILVA database. (C) PCoA plot based on weighted UniFrac distances that shows the bacterial community similarity between the sequential transfers and the scale-up growth process (S). Icons were obtained in BioRender.com.
The mixed consortia obtained at T2, T10, and S was subjected to a whole-metagenomic sequencing and analysis. Approximately 1.3, 0.9, and 3.0Gbp were obtained in T2, T10, and S, respectively, with a read average size of 300bp. Based on the taxonomic affiliation of reads, four bacterial classes were highly abundant. In this regard, Enterobacteriales and Lactobacillales decreased from T2 to T10 and S, while Actinomycetales and Bacillales increased from T2 to T10 and S (Figure 4A). Enterobacteriales species were the predominant taxa (between 50 and 90% of total affiliated sequences) within consortia. At T10 and S, the relative abundance of Lactobacillales was lower than at T2, decreasing from about 4% to between 1 and 3% (Figure 4A). Regarding specific members of LAB, Enterococcus sp. was the most abundant genus (between 1 and 5% relative abundance) in T2, T10, and S. Other genera, like Lactobacillus sp., Leuconstoc sp., Pediococcus sp., and Weissella sp. were found in very low abundance (less than 0.15%) in all the stages of selection, although their populations increased in S with respect to T10 (Supplementary Figure S1). Regarding fungal communities, metagenome data revealed that they are found in very low relative abundance (less than 0.001%). Based on the function (KO level), consortia T2 and T10 showed a similar profile, compared with the consortium obtained after scale-up (S) process (Figure 4B). Moreover, the results showed that nine of 10 genes involved in lignocellulose transformation were enriched from T2 to T10 and S (Figure 4C). Regarding genes relevant to lactic acid production, we observed that genes encoding xylose dissimilation activities [e.g., transketolase (TK) and transaldolase (TA)] were highly abundant at T2, whereas others [e.g., 6-phosphofructokinase and L-lactate dehydrogenase (LDH)] were highly abundant at T10 and S (Figure 4C).
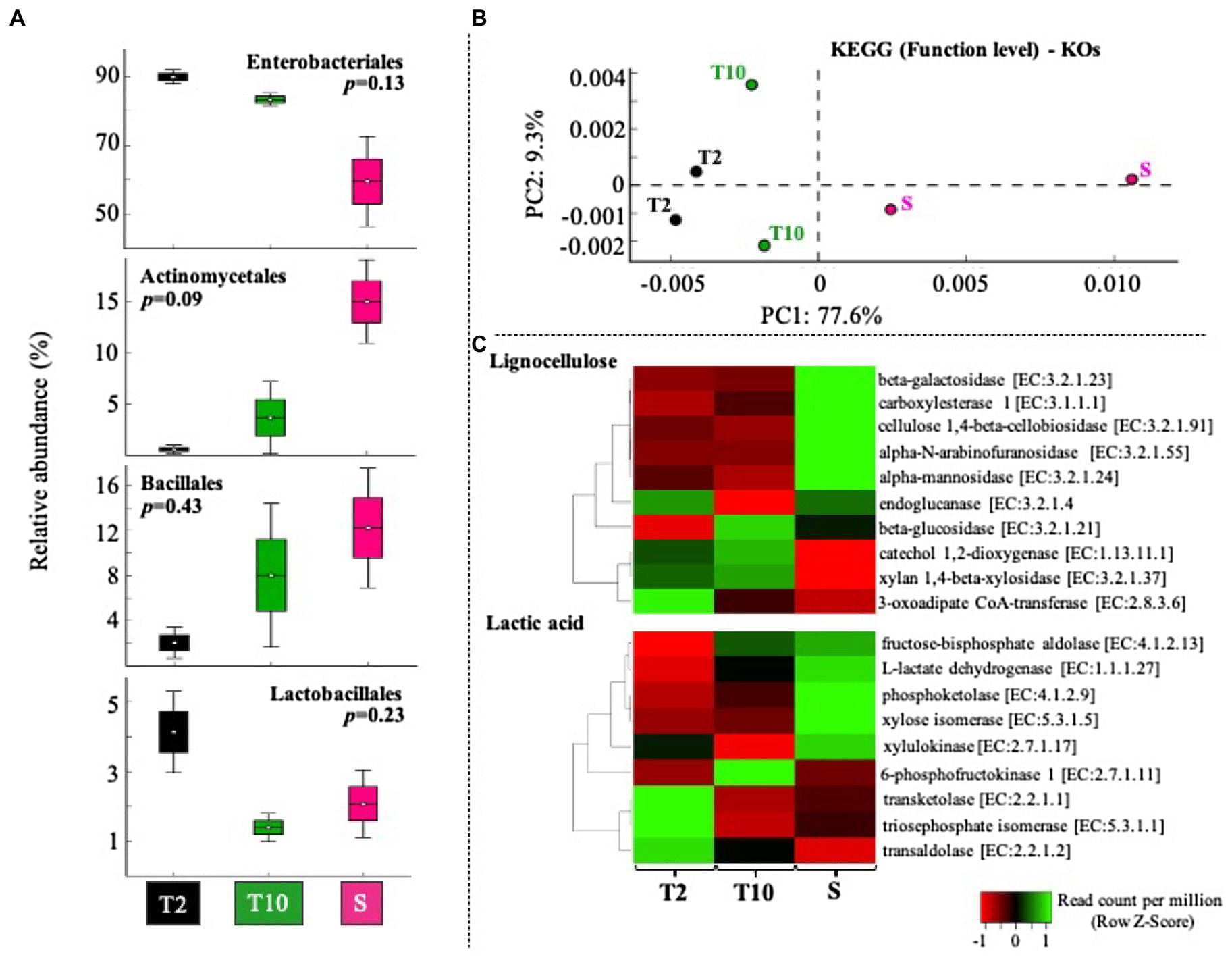
Figure 4. Gene-centric metagenomic analysis of selected consortia after transfer T2, T10, and the scale-up growth process (S). (A) Relative proportion (%) of the most abundant orders (Enterobacteriales>Actinomycetales>Bacillales>Lactobacillales). Sequences taxonomic assignment was carried out by using RefSeq database. (B) Clustering based on functional assignment (KEGG databases) of annotated sequences at KOs level. (C) Heat map of normalized abundance values (Row Z score) obtained using the number of sequences annotated within 10 enzyme-encoding genes involved in lignocellulose degradation and nine enzyme-encoding genes involved in heterolactic and/or homolactic fermentation of xylose.
The ability of the selected consortium (i.e., T10) to grow in acidic silage conditions was evaluated by modifying the initial pH of the liquid medium. At pH 4, a significant (value of p<0.01) increase of LAB was evidenced, showing a viable population density of around 4×108 CFU/ml. In contrast, total bacterial cells decreased at pH 4 and pH 5, compared to the unmodified liquid medium (pH around 6.2; Figure 5A). After the perturbation at different pH values, the metasecretome of the consortium was evaluated for the presence of plant polymer-degrading endo-enzymes. The highest and significant (value of p<0.01) enzymatic activities on a set of CPHs and ICBs were obtained in the metasecretome of the mixed consortium after growth at pH 5 (Figure 5B; Supplementary Figure S2). At this pH, the plant biomass consumption was 17.8±0.6%. Additionally, 16S rRNA gene sequencing analysis were carried out after the perturbation at different pH values. On average, 104 ASVs were generated from 50Mbp of high-quality reads. Alpha diversity results demonstrated a decrease of observed ASVs in the pH gradient, reaching a value of 31±1.0 at pH 4 (Figure 5C). Interestingly, a significant (value of p<0.001) increase in the relative abundance of Actinomycetales and Lactobacillales orders was observed at pH 5 (Figure 5D). However, the highest abundant order was Enterobacteriales (Supplementary Figure S3).
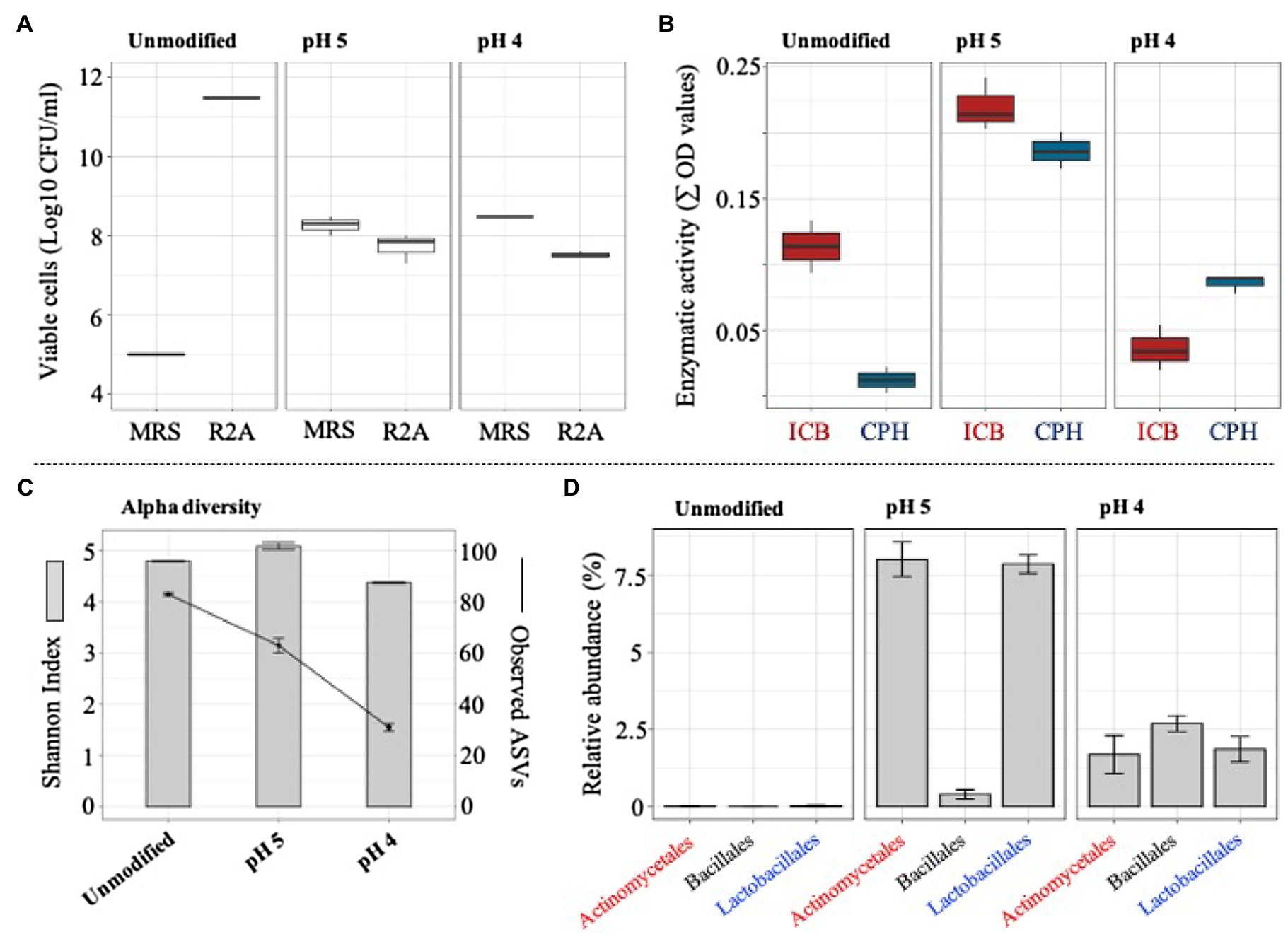
Figure 5. Microbial growth, enzymatic activity, and structural composition of selected consortium (at T10) after its cultivation in lower pH values (unmodified, 5 and 4). (A) Viable cells (Log10 CFU/ml) of total and acid lactic bacteria quantified in agar R2A and MRS, respectively. (B) Secreted endo-enzymatic activities (∑ OD values) on six chromogenic polysaccharide hydrogels (CPH) and two insoluble biomass substrates (ICB) after T10 consortium growth on unmodified pH, pH 5, and pH 4. (C) Observed ASVs and Shannon index values. (D) Relative proportion of taxa (class) that increase their abundance at pH 5 and pH 4 (16S rRNA data). Error bars represent SDs from three biological replicates.
A total of 2, 3, and 9 high quality MAGs (>90% of completeness and <3.5% of contamination) were reconstructed from the T2, T10, and S metagenomes, respectively (Table 2; Supplementary Table S1). The genome lengths ranged from 2.28 to 5.93Mb. At T2 and T10, MAGs belong to Cellulosimicrobium funkei (Bin7_T2 and Bin8_T10) and Klebsiella aerogenes (Bin25_T2 and Bin2_T10) were obtained. Interestingly, the data revealed that three different MAGs from Enterococcus species were obtained. At T2, a single genome associated to Enterococcus faecalis, while in both T10 and S, two genomes associated to Enterococcus casseliflavus. Functional annotation of all reconstructed MAGs showed that Bin7_T2, Bin8_T10, Bin3_T10, Bin11_S, Bin6_S, Bin002_S, and Bin8_S contain more than nine genes-encoding glycosyl hydrolases (GHs) from CAZy families involved in degradation of (hemi)cellulose (Table 2). Moreover, Bin3_T10 and Bin6_S affiliated to E. casseliflavus harbor more than 20 GHs involved in transformation of oligosaccharides (e.g., GH1, GH2, and GH3). In addition, two LDHs encoding-genes were found in those two MAGs (Table 2; Supplementary Table S2).
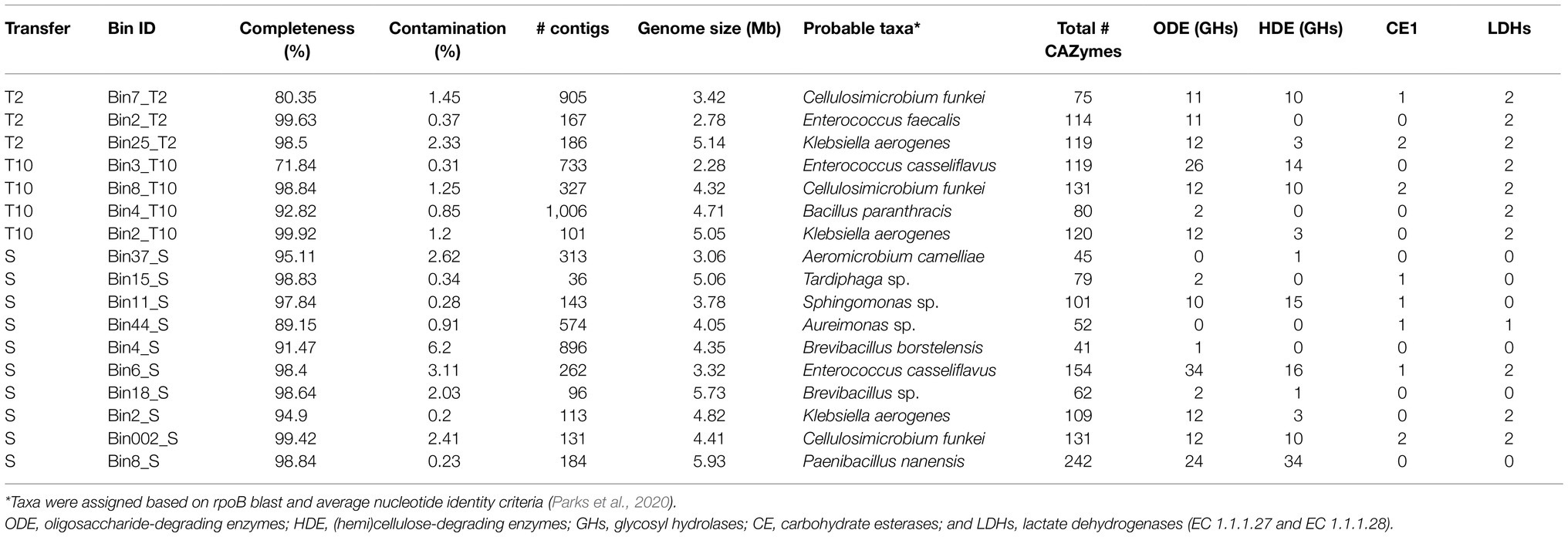
Table 2. Features of metagenome assembled genome (MAGs) reconstructed from T2, T10, and S consortia.
A total of five MAGs (Bin6_S, Bin002_S, Bin2_S, Bin4_T10, and Bin44_S) were selected in order to analyze their potential to produce lactic acid from sugars. These MAGs contained at least one LDH encoding-gene (Table 2). Based on the functional annotation using the RAST server, encoding-genes involved in fermentation of pentoses and hexoses were detected within these five MAGs (Supplementary Table S2). The results showed that Bin6_S (E. casseliflavus), Bin002_S (C. funkei), and Bin2_S (K. aerogenes) encode complete enzymatic machinery to produce lactic acid. Specifically, Bin6_S and Bin002_S contain the genes necessary for heterolactic acid fermentation of pentoses (e.g., xylose and arabinose) through the phosphoketolase (PK) pathway (Figure 6). In addition, Bin002_S has the metabolic potential to carry out a homolactic fermentation of xylose through the pentose phosphate (PP) pathway (Figure 6) and heterolactic fermentation of maltose through the PK pathway. Moreover, Bin2_S contains the gene capability to produce D-lactate from glycerol through an Embden–Meyerhof (EM) glycolytic pathway (Supplementary Table S2).
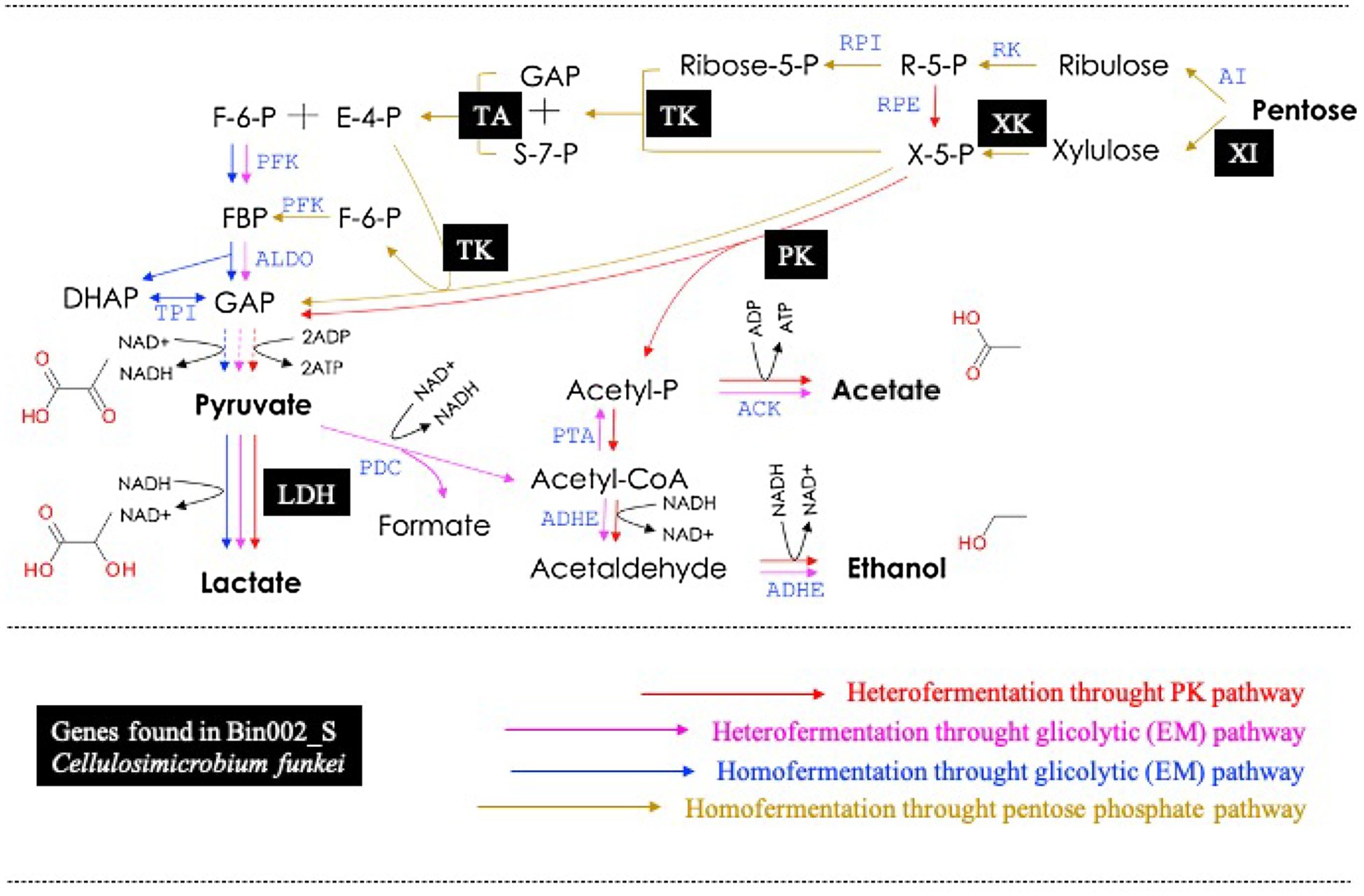
Figure 6. Metabolic pathway for pentoses dissimilation in lactic acid bacteria (modified from Hatti-Kaul et al., 2018). Black squares represent the genes found in the Bin002_S (Cellulosimicrobium funkei). XI, xylose isomerase; XK, xylulose kinase; TK, transketolase; TA, transaldolase; PK, phosphoketolase; and LDH, lactate dehydrogenase. For details in all abbreviations see Hatti-Kaul et al. (2018).
The ensiling of forage crops is a relevant process that enhances feed quality and productivity in livestock systems. The design, application, and testing of starting inoculants is a world-wide priority of research within this field (Muck et al., 2018; Fabiszewska et al., 2019; Nazar et al., 2021a). Thus, this study aimed to build up a unique bioinoculant composed of LAB and LB derived from the phyllosphere of M. maximum cv. Agrosavia Sabanera. This Guinea grass is recognized as one of the best species that improves beef and milk production, especially in tropical countries (Villegas et al., 2020; Carvajal-Tapia et al., 2021). In addition, this pasture crop has an excellent nutritional quality and the ability to tolerate adverse abiotic conditions (e.g., drought), being a species of interest for arid regions (Benabderrahim and Elfalleh, 2021). However, the information about its epiphytic microbial community as a source of LAB and LB is null. Ruminal-derived microbial communities could contain higher proportion of LAB and LB. However, we have selected roughage phyllosphere because its microbial communities are the starter inoculant in a silage process.
Previously, the design of consortia composed of lactic acid-producing and lignocellulolytic microorganisms has been achieved by the co-culture of axenic strains (Li et al., 2018; Shahab et al., 2018). In the current work, we developed an innovative top-down strategy to co-cultivate LAB and LB populations using a rational and sequential modification of MRS broth throughout the dilution-to-stimulation approach (Figure 1; Table 1). During the enrichment process, a high abundance of species from Enterobacteriaceae family was noticed. Within this enriched system, they could take advantage due to their evolutionary adaptation to the M. maximum phyllosphere and also the capacity to grow quickly in liquid environments with labile and plant-derived carbon sources (e.g., glucose; Goldford et al., 2018; Díaz-García et al., 2021). We hypothesized that Enterobacteriaceae species could compete with LAB and LB for the uptake of oligo or monosaccharides, having a secondary role in the deconstruction of lignocellulose, where they could be considered as sugar cheaters (Jiménez et al., 2017). In silage, the proliferation of Enterobacteriaceae species is undesirable because they compete with LAB, can degrade proteins to produce biogenic amines and branched fatty acids (Ávila and Carvalho, 2019).
Although, the modification of MRS broth did not significantly affect the bacterial composition on a taxonomic basis, the substrate weight loss values (after T6) and the enrichment of some enzyme encoding-genes (i.e., from T2 to T10 and S) suggested that these changes were effective to increase the proportion of LB. However, at the end of the selective process (i.e., T10), a co-existence of LAB (mostly belonged to Lactobacillales) and LB (e.g., Actinomycetales and Bacillales) was observed. In silage, we hypothesized that the action of lignocellulolytic enzymes could improves plant polysaccharides degradation, proving fermentable sugars for the entire microbial community, including LAB. In this regard, the supplementation of cellulases and LAB has shown positive effects in the silage of tropical grasses (Khota et al., 2016). In our enrichment strategy, LAB can grow more slowly because they are metabolically less efficient, but they are competitive and can thrive as a minor fraction of the population with an important ecological role by employing several mechanisms to inhibit the growth of other species (e.g., production of organic acids and hydrogen peroxide; Ávila and Carvalho, 2019). Other mechanism that could back up the coexistence of LB and LAB is the cross-feeding. In this scenario, LB can growth using plant-derived monosaccharides, secreting molecules that LAB might consume (Seth and Taga, 2014).
Many factors, such as nutrient availability, biological interactions, temperature and pH modulate population dynamics in microbial systems (Ratzke and Gore, 2018; Xu et al., 2020; Estrela et al., 2021). In this study, drastic changes in the structure/function of the final selected consortium (i.e., T10) were observed after its growth in a higher volume (500ml) and longer time of incubation (8days; i.e., the scale up growth process). We hypothesized that incubation time, oxygen availability, nutrient content/exchange, and spatial co-localization of the bacterial consortium members (in above mentioned culture conditions) could be factors that drive the populations dynamics in this system (Takahashi and Aoyagi, 2018; Jawed et al., 2019; van Tatenhove-Pel et al., 2021). In addition, these results suggested that our mixed consortium, composed of around 70 bacterial species, is still unstable. A microbial inoculant with less diversity could be more stable (Jiménez et al., 2017). After the scale up growth process, an increase of species from Paenibacillaceae family was observed. These types of bacteria could have lower growth rates in a competitive environment and dominate later stages of carbon decomposition (Zhang et al., 2020; Díaz-García et al., 2021). Within silage, Paenibacillus species (spore-forming bacteria) could enhance growth of LAB, tolerate acidic conditions and produce bacteriocins that inhibit yeast and molds (Xu et al., 2018; Singh et al., 2019). However, it is important to note that in some cases Paenibacillus can compete with LAB for fermentable sugars, limiting its growth (Li et al., 2020). In addition, they have a vast lignocellulolytic potential that may improve the digestion of complex carbohydrates in the bovine rumen (Ticona et al., 2021). Paenibacillus species can survive harsh environmental conditions and can be detected even in the last stages of ensiling (Ning et al., 2017). On the other hand, the pH has also modulated the structure, diversity, and metabolic activity of the selected consortium (i.e., T10). In this regard, an increase of lignocellulose consumption and enzymatic activities was observed at pH 5. These results were correlated with a significant increase of Actinomycetales and Lactobacillales species (Figure 5), suggesting that these taxa could be involved in plant biomass transformation and might be metabolically active in a further ensiling process. It has been reported that species from the phylum Actinobacteria are highly abundant in fresh forages (e.g., alfalfa, barley, triticale, and oat), but decrease in abundance during the ensiling processes (Duniere et al., 2017; Hu et al., 2020). Low abundance of Actinobacteria and high abundance of Protobacteria phylum has been observed in Napier grass silage (Nazar et al., 2021a). Species from Actinobacteria generally do not grow below pH 4.5 under microaerophilic conditions, and therefore its growth is unlikely to occur when the pH of the silo stabilizes (Hill et al., 2001). Thus, we presumed that Actinomycetales with the capacity to thrive at lower pH values were selected in our mixed consortium.
Based on MAGs annotation, Paenibacillus, Cellulosimicrobium, and Sphingomonas appear as key (hemi)cellulolytic members, while Cellulosimicrobium, Enterococcus, and Klebsiella are the lactic acid producers within the mixed consortium. Their coexistence can be related to the preferential metabolism of different plant-derived hexoses (i.e., glucose or galactose) and pentoses (i.e., xylose and arabinose; Hatti-Kaul et al., 2018). Within the mixed consortium, Enterococcus species have the potential to transform xylooligosaccharides and/or arabinoxylans. Although rare, the presence of GH1, GH2, GH3, GH5, GH30, GH43, and GH67 families were found in Bin6_S and Bin3_T10. This information could add new perspectives on the carbohydrate metabolism of LAB species involved in the fermentation of hemicellulose-containing substrates (Michlmayr and Kneifel, 2014). To highlight, we found that C. funkei (Bin002_S) has the genomic potential to produce lactic acid from xylose and maltose (through the PK and PP metabolic pathways). Two different pathways are proposed for pentose metabolism, the PK pathway yields 1mol lactic acid/mol sugar, whereas the pentose phosphate pathway provides a theoretical lactic acid yield of 1.67mol/mol sugar (Tanaka et al., 2002). Based on the CAZyme annotation, C. funkei could produce enzymes involved in (hemi)cellulose degradation (e.g., GH16, GH30, GH43, and GH51; Ventorino et al., 2015; Dou et al., 2020). To the best of our knowledge, no native (hemi)cellulolytic LAB from Actinobacteria phylum has been previously reported. Interestingly, we also found that Bin002_S contained two carbohydrate esterase (CE1) enzymes that may improve the digestibility of forage within the rumen (Li et al., 2021). However, the presence of FAE was not detected within the reconstructed MAGs. Interestingly, Cellulosimicrobium and Paenibacillus species have been cataloged as desirable microorganisms in cassava foliage silage (Li et al., 2020). Finally, this study reported the design and characterization of a mixed consortium, obtained from the M. maximus phyllosphere, composed of LAB and LB. Based on the results, we conclude that a synthetic bioinoculant composed of strains of Cellulosimicrobium, Enterococcus, and Paenibacillus species could be used to improve the quality of silage processes. Further studies with our mixed consortium will: (i) apply it as starting inoculant in an ensiling process and evaluate the consequences in terms of quality and the dynamics of microbial populations; (ii) isolate the key bacterial members; (iii) design a synthetic consortium using bacterial axenic cultures (bottom-up approach) and test it in an ensiling process; and (iv) evaluate lactic acid production by Cellulosimicrobium via genome-based metabolic reconstruction using Bin002_S.
The bacterial 16S rRNA gene amplicon sequencing data obtained in this study have been deposited under NCBI BioProject accession number PRJNA734654. All metagenome sequences are publicly accessible on the MG-RAST server (Metagenome IDs mgm4912842.3 to mgm4912847.3).
LD-G carried out all wet-lab experiments and bioinformatic analysis and helped to build up the text draft. DC assisted in all wet-lab experiments. HJ provided the pre-enrichment material. AB, HJ, LG-R, and EB-E contributed to the project design and helped in drafting the manuscript. EB-E coordinated the project and gave financial support. DJ coordinated and conceived the project and drafted the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the FAPA project (number PR.3.2018.5287) obtained by DJ at the Universidad de los Andes (Bogotá, Colombia) and a granted project (Number EXT-2019-64-1677) obtained within the cooperation Agrosavia-UniAndes. All authors acknowledge financial support provided by the Vice Presidency for Research and Creation at the Universidad de los Andes.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Juan Camilo Ovalle for technical support and Nancy N. Nichols for all relevant comments about the manuscript. In addition, we thank the Faculty of Sciences at the Universidad de los Andes and Agrosavia for providing administrative support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.744075/full#supplementary-material
Supplementary Figure S1 | Relative proportion (%) of the most common lactic acid bacteria (LAB) found in silage processes. Metagenomic sequence taxonomic assignment was carried out by using RefSeq database.
Supplementary Figure S2 | Secreted endo-enzymatic activities (OD values) on (A) six chromogenic polysaccharide hydrogels (CPH) and (B) two insoluble biomass substrates (ICB) after T10 consortium growth on unmodified pH 6.2, pH 5, and pH 4.
Supplementary Figure S3 | Relative proportion (16S rRNA data) of taxa (class) after incubation of the consortium at T10 in a range of pH values. Error bars represent SDs from three biological replicates.
Supplementary Table S1 | Detailed features of metagenome assembled genomes (MAGs) reconstructed from T2, T10, and S consortia.
Supplementary Table S2 | Number of genes involved in lactic acid production found within Bin6_S, Bin002_S, Bin2_S, Bin4_T10, and Bin44_S.
Addah, W., Baah, J., and McAllister, T. A. (2016). Effects of an exogenous enzyme-containing inoculant on fermentation characteristics of barley silage and on growth performance of feedlot steers. Can. J. Anim. Sci. 96, 1–10. doi: 10.1139/cjas-2015-0079
Alneberg, J., Bjarnason, B. S., de Bruijn, I., Schirmer, M., Quick, J., Ijaz, U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Amir, A., McDonald, D., Navas-Molina, J. A., Kopylova, E., Morton, J. T., Zech Xu, Z., et al. (2017). Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2, e00191–e00216. doi: 10.1128/mSystems.00191-16
Ávila, C. L. S., and Carvalho, B. F. (2019). Silage fermentation-updates focusing on the performance of microorganisms. J. Appl. Microbiol. 128, 966–984. doi: 10.1111/jam.14450
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Babicki, S., Arndt, D., Marcu, A., Liang, Y., Grant, J. R., Maciejewski, A., et al. (2016). Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 44, W147–W153. doi: 10.1093/nar/gkw419
Benabderrahim, M. A., and Elfalleh, W. (2021). Forage potential of non-native Guinea grass in north African agroecosystems: genetic, agronomic, and adaptive traits. Agronomy 11:1071. doi: 10.3390/agronomy11061071
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bonaldi, D. S., Carvalho, B. F., Ávila, C. L. D. S., and Silva, C. F. (2021). Effects of Bacillus subtilis and its metabolites on corn silage quality. Lett. Appl. Microbiol. 73, 46–53. doi: 10.1111/lam.13474
Carvajal-Tapia, J., Velasco, S. M., Villegas, D. M., Arango, J., and Quila, N. J. V. (2021). Biological nitrification inhibition and forage productivity of Megathyrsus maximus in Colombian dry tropics. Plant Soil Environ. 67, 270–277. doi: 10.17221/445/2020-PSE
de Lima Brossi, M. J., Jiménez, D. J., Cortes-Tolalpa, L., and van Elsas, J. D. (2016). Soil-derived microbial consortia enriched with different plant biomass reveal distinct players acting in lignocellulose degradation. Microb. Ecol. 71, 616–627. doi: 10.1007/s00248-015-0683-7
Díaz-García, L., Huang, S., Spröer, C., Sierra-Ramírez, R., Bunk, B., Overmann, J., et al. (2021). Dilution-to-stimulation/extinction method: a combination enrichment strategy to develop a minimal and versatile lignocellulolytic bacterial consortium. Appl. Environ. Microbiol. 87, e02427–e02420. doi: 10.1128/AEM.02427-20
Dou, T. Y., Chen, J., and Liu, C. (2020). Isolation and subunit compositions of the xylanosome complexes produced by Cellulosimicrobium species. Enzym. Microb. Technol. 133:109445. doi: 10.1016/j.enzmictec.2019.109445
Duniere, L., Xu, S., Long, J., Elekwachi, C., Wang, Y., Turkington, K., et al. (2017). Bacterial and fungal core microbiomes associated with small grain silages during ensiling and aerobic spoilage. BMC Microbiol. 17:50. doi: 10.1186/s12866-017-0947-0
Estrela, S., Sanchez-Gorostiaga, A., Vila, J. C., and Sanchez, A. (2021). Nutrient dominance governs the assembly of microbial communities in mixed nutrient environments. eLife 10:e65948. doi: 10.7554/eLife.65948
Fabiszewska, A. U., Zielińska, K. J., and Wróbel, B. (2019). Trends in designing microbial silage quality by biotechnological methods using lactic acid bacteria inoculants: a minireview. World J. Microbiol. Biotechnol. 35:76. doi: 10.1007/s11274-019-2649-2
Goldford, J. E., Lu, N., Bajić, D., Estrela, S., Tikhonov, M., Sanchez-Gorostiaga, A., et al. (2018). Emergent simplicity in microbial community assembly. Science 361, 469–474. doi: 10.1126/science.aat1168
Grabber, J. H., Mertens, D. R., Kim, H., Funk, C., Lu, F., and Ralph, J. (2009). Cell wall fermentation kinetics are impacted more by lignin content and ferulate cross-linking than by lignin composition. J. Sci. Food Agric. 89, 122–129. doi: 10.1002/jsfa.3418
Hatti-Kaul, R., Chen, L., Dishisha, T., and Enshasy, H. E. (2018). Lactic acid bacteria: from starter cultures to producers of chemicals. FEMS Microbiol. Lett. 365:fny213. doi: 10.1093/femsle/fny213
Hill, J., Xiao, G. Q., and Ball, A. S. (2001). Effect of inoculation of herbage prior to ensiling with Streptomyces achromogenes ISP 5028 on chemical composition of silage. Anim. Feed Sci. Technol. 89, 83–96. doi: 10.1016/S0377-8401(00)00239-X
Hu, Z., Niu, H., Tong, Q., Chang, J., Yu, J., Li, S., et al. (2020). The microbiota dynamics of alfalfa silage during ensiling and after air exposure, and the metabolomics after air exposure are affected by Lactobacillus casei and cellulase addition. Front. Microbiol. 11:519121. doi: 10.3389/fmicb.2020.519121
Jawed, K., Yazdani, S. S., and Koffas, M. A. (2019). Advances in the development and application of microbial consortia for metabolic engineering. Metab. Eng. Commun. 352, 1–10. doi: 10.1016/j.mec.2019.e00095
Jiménez, D. J., de Lima Brossi, M. J., Schückel, J., Kračun, S. K., Willats, W. G., and van Elsas, J. D. (2016). Characterization of three plant biomass-degrading microbial consortia by metagenomics- and metasecretomics-based approaches. Appl. Microbiol. Biotechnol. 100, 10463–10477. doi: 10.1007/s00253-016-7713-3
Jiménez, D. J., Dini-Andreote, F., DeAngelis, K. M., Singer, S. W., Salles, J. F., and van Elsas, J. D. (2017). Ecological insights into the dynamics of plant biomass-degrading microbial consortia. Trends Microbiol. 25, 788–796. doi: 10.1016/j.tim.2017.05.012
Jiménez, D. J., Dini-Andreote, F., and van Elsas, J. D. (2014a). Metataxonomic profiling and prediction of functional behaviour of wheat straw degrading microbial consortia. Biotechnol. Biofuels 7:92. doi: 10.1186/1754-6834-7-92
Jiménez, D. J., Korenblum, E., and van Elsas, J. D. (2014b). Novel multispecies microbial consortia involved in lignocellulose and 5-hydroxymethylfurfural bioconversion. Appl. Microbiol. Biotechnol. 98, 2789–2803. doi: 10.1007/s00253-013-5253-7
Jiménez, D. J., Mares, M. C. D., and Salles, J. F. (2018). Temporal expression dynamics of plant biomass-degrading enzymes by a synthetic bacterial consortium growing on sugarcane bagasse. Front. Microbiol. 9:299. doi: 10.3389/fmicb.2018.00299
Jiménez, D. J., Wang, Y., Chaib de Mares, M., Cortes-Tolalpa, L., Mertens, J. A., Hector, R. E., et al. (2020). Defining the eco-enzymological role of the fungal strain Coniochaeta sp. 2T2.1 in a tripartite lignocellulolytic microbial consortium. FEMS Microbiol. Ecol. 96:fiz186. doi: 10.1093/femsec/fiz186
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keshri, J., Chen, Y., Pinto, R., Kroupitski, Y., Weinberg, Z. G., and Saldinger, S. S. (2019). Bacterial dynamics of wheat silage. Front. Microbiol. 10:1532. doi: 10.3389/fmicb.2019.01532
Khota, W., Pholsen, S., Higgs, D., and Cai, Y. (2016). Natural lactic acid bacteria population of tropical grasses and their fermentation factor analysis of silage prepared with cellulase and inoculant. J. Dairy Sci. 99, 9768–9781. doi: 10.3168/jds.2016-11180
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Kračun, S. K., Schückel, J., Westereng, B., Thygesen, L. G., Monrad, R. N., Eijsink, V. G. H., et al. (2015). A new generation of versatile chromogenic substrates for high-throughput analysis of biomass-degrading enzymes. Biotechnol. Biofuels 8:70. doi: 10.1186/s13068-015-0250-y
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, F. H., Ding, Z. T., Chen, X. Z., Zhang, Y. X., Ke, W. C., Zhang, X., et al. (2021). The effects of Lactobacillus plantarum with feruloyl esterase-producing ability or high antioxidant activity on the fermentation, chemical composition, and antioxidant status of alfalfa silage. Anim. Feed Sci. Technol. 273, 1–14. doi: 10.1016/j.anifeedsci.2021.114835
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, D. X., Ni, K. K., Zhang, Y. C., Lin, Y. L., and Yang, F. Y. (2018). Influence of lactic acid bacteria, cellulase, cellulase-producing Bacillus pumilus and their combinations on alfalfa silage quality. J. Integr. Agric. 7, 2768–2782. doi: 10.1016/s2095-3119(18)62060-x
Li, M., Zhang, L., Zhang, Q., Zi, X., Lv, R., Tang, J., et al. (2020). Impacts of citric acid and malic acid on fermentation quality and bacterial community of cassava foliage silage. Front. Microbiol. 11:595622. doi: 10.3389/fmicb.2020.595622
Marter, P., Huang, S., Brinkmann, H., Pradella, S., Jarek, M., Rohde, M., et al. (2021). Filling the gaps in the cyanobacterial tree of life—metagenome analysis of Stigonema ocellatum DSM 106950, Chlorogloea purpurea SAG 13.99 and Gomphosphaeria aponina DSM 107014. Genes 12:389. doi: 10.3390/genes12030389
McAllister, T. A., Dunière, L., Drouin, P., Xu, S., Wang, Y., Munns, K., et al. (2018). Silage review: using molecular approaches to define the microbial ecology of silage. J. Dairy Sci. 101, 4060–4074. doi: 10.3168/jds.2017-13704
Meyer, F., Paarmann, D., D'Souza, M., Olson, R., Glass, E. M., Kubal, M., et al. (2008). The metagenomics RAST server-a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386
Michlmayr, H., and Kneifel, W. (2014). β-Glucosidase activities of lactic acid bacteria: mechanisms, impact on fermented food and human health. FEMS Microbiol. Lett. 352, 1–10. doi: 10.1111/1574-6968.12348
Mojica-Rodríguez, J. E., and Burbano-Erazo, E. (2020). Efecto de dos cultivares de Megathyrsus maximus (Jacq.) en la producción y composición de la leche bovina. Pastos y Forrajes 43, 177–183.
Muck, R. E., Nadeau, E. M. G., McAllister, T. A., Contreras-Govea, F. E., Santos, M. C., and Kung, L. (2018). Silage review: recent advances and future uses of silage additives. J. Dairy Sci. 101, 3980–4000. doi: 10.3168/jds.2017-13839
Nazar, M., Wang, S., Zhao, J., Dong, Z., Li, J., Kaka, N. A., et al. (2021a). Effects of various epiphytic microbiota inoculation on the fermentation quality and microbial community dynamics during the ensiling of sterile Napier grass. J. Appl. Microbiol. 130, 1466–1480. doi: 10.1111/jam.14896
Nazar, M., Wang, S., Zhao, J., Dong, Z., Li, J., Kaka, N. A., et al. (2021b). Abundance and diversity of epiphytic microbiota on forage crops and their fermentation characteristic during the ensiling of sterile Sudan grass. World J. Microbiol. Biotechnol. 37:27. doi: 10.1007/s11274-020-02991-3
Ning, T., Wang, H., Zheng, M., Niu, D., Zuo, S., and Xu, C. (2017). Effects of microbial enzymes on starch and hemicellulose degradation in total mixed ration silages. Asian-Australas. J. Anim. Sci. 30, 171–180. doi: 10.5713/ajas.16.0046
Ogunade, I. M., Jiang, Y., Cervantes, A. A. P., Kim, D. H., Oliveira, A. S., Vyas, D., et al. (2018). Bacterial diversity and composition of alfalfa silage as analyzed by Illumina MiSeq sequencing: effects of Escherichia coli O157:H7 and silage additives. J. Dairy Sci. 101, 2048–2059. doi: 10.3168/jds.2017-12876
Parks, D. H., and Beiko, R. G. (2010). Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26, 715–721. doi: 10.1093/bioinformatics/btq041
Parks, D. H., Chuvochina, M., Chaumeil, P. A., Rinke, C., Mussig, A. J., and Hugenholtz, P. (2020). A complete domain-to-species taxonomy for bacteria and archaea. Nat. Biotechnol. 38, 1079–1086. doi: 10.1038/s41587-020-0501-8
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 5, 1043–1055. doi: 10.1101/gr.186072.114
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ratzke, C., and Gore, J. (2018). Modifying and reacting to the environmental pH can drive bacterial interactions. PLoS Biol. 16:e2004248. doi: 10.1371/journal.pbio.2004248
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Seth, E. C., and Taga, M. E. (2014). Nutrient cross-feeding in the microbial world. Front. Microbiol. 5:350. doi: 10.3389/fmicb.2014.00350
Shahab, R. L., Luterbacher, J. S., Brethauer, S., and Studer, M. H. (2018). Consolidated bioprocessing of lignocellulosic biomass to lactic acid by a synthetic fungal-bacterial consortium. Biotechnol. Bioeng. 115, 1207–1215. doi: 10.1002/bit.26541
Sieber, C. M. K., Probst, A. J., Sharrar, A., Thomas, B. C., Hess, M., Tringe, S. G., et al. (2018). Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843. doi: 10.1038/s41564-018-0171-1
Singh, H., Kaur, M., Jangra, M., Mishra, S., Nandanwar, H., and Pinnaka, A. K. (2019). Antimicrobial properties of the novel bacterial isolate Paenibacillus sp. SMB1 from a halo-alkaline lake in India. Sci. Rep. 9:11561. doi: 10.1038/s41598-019-47879-x
Su, R., Ni, K., Wang, T., Yang, X., Zhang, J., Liu, Y., et al. (2019). Effects of ferulic acid esterase-producing Lactobacillus fermentum and cellulase additives on the fermentation quality and microbial community of alfalfa silage. PeerJ 7:e7712. doi: 10.7717/peerj.7712
Takahashi, M., and Aoyagi, H. (2018). Effect of intermittent opening of breathable culture plugs and aeration of headspace on the structure of microbial communities in shake-flask culture. J. Biosci. Bioeng. 126, 96–101. doi: 10.1016/j.jbiosc.2018.01.009
Tanaka, K., Komiyama, A., Sonomoto, K., Ishizaki, A., Hall, S. J., and Stanbury, P. F. (2002). Two different pathways for D-xylose metabolism and the effect of xylose concentration on the yield coefficient of L-lactate in mixed-acid fermentation by the lactic acid bacterium Lactococcus lactis IO-1. Appl. Microbiol. Biotechnol. 60, 160–167. doi: 10.1007/s00253-002-1078-5
Tanizawa, Y., Fujisawa, T., and Nakamura, Y. (2018). DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 34, 1037–1039. doi: 10.1093/bioinformatics/btx713
Tarraran, L., and Mazzoli, R. (2018). Alternative strategies for lignocellulose fermentation through lactic acid bacteria: the state of the art and perspectives. FEMS Microbiol. Lett. 365:fny126. doi: 10.1093/femsle/fny126
Ticona, A. R. P., Ullah, S. F., Hamann, P. R. V., Lopes, F. A. C., and Noronha, E. F. (2021). Paenibacillus barengoltzii A1_50L2 as a source of plant cell wall degrading enzymes and its use on lignocellulosic biomass hydrolysis. Waste Biomass Valor. 12, 393–405. doi: 10.1007/s12649-020-00975-w
van Tatenhove-Pel, R. J., de Groot, D. H., Bisseswar, A. S., Teusink, B., and Bachmann, H. (2021). Population dynamics of microbial cross-feeding are determined by co-localization probabilities and cooperation-independent cheater growth. ISME J. 15, 3050–3061. doi: 10.1038/s41396-021-00986-y
Ventorino, V., Aliberti, A., Faraco, V., Robertiello, A., Giacobbe, S., Ercolini, D., et al. (2015). Exploring the microbiota dynamics related to vegetable biomasses degradation and study of lignocellulose-degrading bacteria for industrial biotechnological application. Sci. Rep. 5:8161. doi: 10.1038/srep08161
Villegas, D., Arevalo, A., Nuñez, J., Mazabel, J., Subbarao, G., Rao, I., et al. (2020). Biological nitrification inhibition (BNI): phenotyping of a core germplasm collection of the tropical forage grass Megathyrsus maximus under greenhouse conditions. Front. Plant Sci. 11:820. doi: 10.3389/fpls.2020.00820
Windle, M. C., Walker, N., and Kung, L. Jr. (2014). Effects of exogenous protease on the fermentation and nutritive value of corn silage harvested at different dry matter contents and ensiled for various lengths of times. J. Dairy Sci. 97, 3053–3060. doi: 10.3168/jds.2013-7586
Wu, Y. W., Simmons, B. A., and Singer, S. W. (2016). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Xie, Y., Guo, J., Li, W., Wu, Z., and Yu, Z. (2021). Effects of ferulic acid esterase-producing lactic acid bacteria and storage temperature on the fermentation quality, in vitro digestibility and phenolic acid extraction yields of sorghum (Sorghum bicolor L.) silage. Microorganisms 9:114. doi: 10.3390/microorganisms9010114
Xu, J., Bu, F., Zhu, W., Luo, G., and Xie, L. (2020). Microbial consortiums of hydrogenotrophic methanogenic mixed cultures in lab-scale ex-situ biogas upgrading systems under different conditions of temperature, pH and CO. Microorganisms 8:772. doi: 10.3390/microorganisms8050772
Xu, Z., Zhang, S., Mu, Y., and Kong, J. (2018). Paenibacillus panacisoli enhances growth of Lactobacillus spp. by producing xylooligosaccharides in corn stover ensilages. Carbohydr. Polym. 184, 435–444. doi: 10.1016/j.carbpol.2017.12.044
Yin, Y., Mao, X., Yang, J., Chen, X., Mao, F., and Xu, Y. (2012). dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, W445–W451. doi: 10.1093/nar/gks479
Keywords: bacterial consortia, lignocellulose, metagenome-assembled genomes, silage, bioinoculant, Cellulosimicrobium
Citation: Díaz-García L, Chaparro D, Jiménez H, Gómez-Ramírez LF, Bernal AJ, Burbano-Erazo E and Jiménez DJ (2021) Top-Down Enrichment Strategy to Co-cultivate Lactic Acid and Lignocellulolytic Bacteria From the Megathyrsus maximus Phyllosphere. Front. Microbiol. 12:744075. doi: 10.3389/fmicb.2021.744075
Edited by:
X. S. Guo, Lanzhou University, ChinaReviewed by:
Marcia Franco, Natural Resources Institute Finland (Luke), FinlandCopyright © 2021 Díaz-García, Chaparro, Jiménez, Gómez-Ramírez, Bernal, Burbano-Erazo and Jiménez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diego Javier Jiménez, ZGouamltZW5lekB1bmlhbmRlcy5lZHUuY28=;ZGppbWVuZXoxOTA5QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.