 Nathan A. Krump†
Nathan A. Krump† Jianxin You*
Jianxin You*- Department of Microbiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
Merkel cell polyomavirus (MCPyV) infection causes near-ubiquitous, asymptomatic infection in the skin, but occasionally leads to an aggressive skin cancer called Merkel cell carcinoma (MCC). Epidemiological evidence suggests that poorly controlled MCPyV infection may be a precursor to MCPyV-associated MCC. Clearer understanding of host responses that normally control MCPyV infection could inform prophylactic measures in at-risk groups. Similarly, the presence of MCPyV in most MCCs could imbue them with vulnerabilities that-if better characterized-could yield targeted intervention solutions for metastatic MCC cases. In this review, we discuss recent developments in elucidating the interplay between host cells and MCPyV within the context of viral infection and MCC oncogenesis. We also propose a model in which insufficient restriction of MCPyV infection in aging and chronically UV-damaged skin causes unbridled viral replication that licenses MCC tumorigenesis.
Introduction
Merkel cell polyomavirus (MCPyV) infection can be detected on the skin of most healthy adults (Tolstov et al., 2009; Schowalter et al., 2010), yet details of its virology and infectious cycle remain sparse. Evidence from serological studies suggests that MCPyV infects most people during early childhood with exposure to the virus increasing as populations age (Chen et al., 2011; Viscidi et al., 2011; Martel-Jantin et al., 2013). A vast majority of MCPyV infections are asymptomatic (Tolstov et al., 2011), but some result in an aggressive neuroendocrine skin cancer called Merkel cell carcinoma (MCC) (Feng et al., 2008; Gjoerup and Chang, 2010; Harms, 2017; Schadendorf et al., 2017). The etiology of over 80% of MCC tumors can be traced to MCPyV by the presence of integrated MCPyV genomic sequence in the cellular DNA (Feng et al., 2008; Santos-Juanes et al., 2015).
Though MCC cases are rare, the incidence of MCC has tripled over the last two decades and is projected to increase further in the future (Fitzgerald et al., 2015; Paulson et al., 2018a; Stang et al., 2018; Freeman et al., 2019; Jacobs et al., 2020). MCC has a high rate of mortality with 5-year overall survival around 51% for patients presenting with local disease at the time of diagnosis, and worse prognoses for those with more advanced stages of disease (Harms K. L. et al., 2016). Primary MCC malignancies are combatted by surgical resection, sentinel lymph node biopsy, and/or adjuvant radiation (Cassler et al., 2016). In metastatic MCC, chemotherapies have thus far failed to produce durable responses (Iyer et al., 2016; Cowey et al., 2017). Promisingly, a recent bourgeoning of anti-PD1 and anti-PDL1 treatments for MCC has yielded prolonged responses in patients with advanced disease, though a significant proportion of patients do not respond and the durability of responses varies (Iyer et al., 2016; Becker et al., 2017; D'angelo et al., 2018; Nghiem et al., 2019). The pursuit of more targeted and effective MCC therapies necessitates a better understanding of the oncogenic underpinnings of MCC and the role of MCPyV in tumorigenesis.
Despite the status of MCPyV as a member of the human skin virome and its relevance to human disease, much is unknown about its biology. For instance, while it is clear that MCPyV integration and oncogene expression enable MCC cell growth, the conditions that lead to MCPyV integration are not known. Neither has a consensus been reached regarding the MCC cell of origin. Most pressingly, methods of limiting MCPyV infection and thereby preventing MCC onset have yet to be discovered. In recent years, however, efforts have been aimed at exploiting the presence of MCPyV oncoproteins in MCCs to develop targeted therapies for virus-positive MCC tumors (Chapuis et al., 2014; Gavvovidis et al., 2018; Longino et al., 2019; Sarma et al., 2020). The potential for expanding on these opportunities to provide prophylactic or therapeutic interventions for a highly lethal skin cancer should not be overlooked.
Merkel Cell Carcinoma
MCC was first described in 1972 by Cyril Toker, MD (Toker, 1972). He and his colleagues discovered that the tumors arose in the dermis or lower subcutis layers of the skin and that they metastasized readily via the lymphatic system. Because the malignancies shared neuroendocrine markers with Merkel cells, such as cytokeratin-20 (CK20) and neuron-specific enolase (NSE), the disease was named Merkel cell carcinoma (De Wolff-Peeters et al., 1980; Gu et al., 1983; Moll et al., 1992).
The next breakthrough in characterizing MCC came in 2008 when the Chang and Moore group identified a novel polyomavirus genome monoclonally integrated in the DNA of MCC tumor cells (Feng et al., 2008). Previously, they had correctly predicted the viral etiology of AIDS-associated Kaposi's sarcoma (KS) (Chang et al., 1994), hypothesizing that the altered immune status of these individuals engendered dysbiosis between the host and virus to trigger oncogenesis. Following the same principle, they suspected viral involvement in MCCs given the knowledge that MCCs were far more likely to occur in HIV-positive individuals (Engels et al., 2002). Probing MCC primary tumors and metastases by digital transcriptome subtraction and viral genome walking revealed the presence and sequence of the MCPyV genome, respectively (Feng et al., 2008).
As a result of their discovery, assays to determine the status of MCPyV have provided definitive markers and more assured diagnosis for a subset of MCCs (Buck and Lowy, 2009; Decaprio, 2009; Duncavage et al., 2009; Haugg et al., 2014; Starrett et al., 2020). Moreover, it is now widely acknowledged that immunocompromised individuals are more likely to develop virus-driven cancers (Ellerbrock et al., 2000; Weber et al., 2006; Chadburn et al., 2013; Ponce et al., 2014; Schadendorf et al., 2017). The discovery of MCPyV also opened the opportunity to research the role of MCPyV oncogenes in MCC carcinogenesis and to inform our understanding of human cancer.
Building upon those earlier discoveries, recent research has been aimed at comparing MCPyV-positive and MCPyV-negative MCCs, which despite evidently different etiologies, have similar disease presentation and prognoses (Fischer et al., 2010; Handschel et al., 2010; Schrama et al., 2011). In virus-associated MCC, MCPyV DNA is integrated into the tumor cell genome in a manner that preserves expression of MCPyV genes called tumor (T) antigens (Shuda et al., 2008, 2011; Cheng et al., 2013). Expression of MCPyV T antigens drives oncogenesis in virus-positive MCC tumors and is required for the growth of the tumor cells (Houben et al., 2010, 2012; Shuda et al., 2011, 2014; Spurgeon and Lambert, 2013; Verhaegen et al., 2014; Grundhoff and Fischer, 2015; Wendzicki et al., 2015). MCPyV T antigens can support MCC growth and survival despite otherwise low chromosomal mutation burdens in MCPyV-positive MCCs (Starrett et al., 2017). By contrast, virus-negative MCCs exhibit a high, UV-related, mutation frequency indicating that pro-oncogenic mutations arose as a direct result of chronic exposure to UV-radiation (Harms et al., 2015; Wong et al., 2015; Goh et al., 2016; Starrett et al., 2017). MCPyV-negative MCCs also have higher levels of activation-induced cytidine deaminase (AID) which could contribute to mutagenesis (Matsushita et al., 2017). The gradual selection for transforming mutations may lead to shared traits among MCPyV-negative cancers such as loss-of-function mutations in Rb, NOTCH, PRUNE2, as well as, activating mutations in PI3KCA and HRAS (Sihto et al., 2011; Nardi et al., 2012; Harms et al., 2013, 2015; Cimino et al., 2014; Sahi et al., 2014; Wong et al., 2015; Goh et al., 2016; Harms P. W. et al., 2016).
Disparities between the etiologies of MCPyV-positive and -negative MCCs extend to differences in their morphology. MCPyV-containing malignances are more likely to have regularly-shaped nuclei, low cytoplasm volume, and more homogeneous cell types than those lacking MCPyV (Kuwamoto et al., 2011; Iwasaki et al., 2013). They are also more likely to display classical markers for MCC such as CK20 and neurofilament (Pasternak et al., 2018). Differences in morphological phenotype between the two MCC types could be a reflection of their alternative expression profiles in factors such as cell adhesion molecules or miRNAs (Xie et al., 2014; Iwasaki et al., 2016).
There are also disparities between these two types of cancers that have more immediately discernible implications. Virus-associated MCCs are more likely to occur in non-sun-exposed areas than MCPyV-negative MCC (Dabner et al., 2014; Leroux-Kozal et al., 2015) and on a population scale, virus-negative MCCs occur in greater proportions in Australia, where a significant population of individuals with low-melanin concentrations are exposed to high levels of UV-radiation (Paik et al., 2011). Moreover, MCC tumors in younger patients and females are more likely to be virus-positive (Wang et al., 2017). These trends suggest that while UV-exposure and advanced age increase the incidence of both cancer types, MCPyV oncogenesis is less dependent on these exogenous factors.
Beyond differences in oncogenic mechanism, morphology, and incidence, there are an increasing number of reports that MCPyV-positive MCC patient prognosis is statistically better than those with MCPyV-negative MCC (Sihto et al., 2009; Laude et al., 2010; Higaki-Mori et al., 2012; Leroux-Kozal et al., 2015; Moshiri et al., 2017). One reason for the improved patient survival associated with MCPyV-LT-expressing MCC tumors may be correlated with the presence of foreign T antigens that enhance immunogenicity (Walsh et al., 2016). In MCC patients undergoing PD-1 treatment, there is a greater degree of B and T cell clonality in MCPyV-positive tumor infiltrates than MCPyV-negative, reflecting the greater diversity of neoantigens in the latter case (Miller et al., 2018). MCPyV-positive MCCs also exhibit elevated expression of prokineticin-2, an inflammatory and angiogenic signaling molecule, resulting in enhanced T cell infiltration (Lauttia et al., 2014). Another reason for better prognoses in MCPyV-positive MCC cases could be that lower frequency of somatic mutations equate to expression of wild type tumor suppressors like p53 that support therapeutic interventions and native immune responses in restricting cancer progression.
The juxtaposition of these two MCC subtypes informs our understanding of both through comparative analysis, and can guide the development of novel treatments. In MCPyV-driven MCC, there exists the promise of truly targeted therapies toward an aggressive solid tumor. What we learn about viral MCCs can be applied to the more complicated and divergent cases of MCPyV-negative MCC. These lessons can, in turn, broaden our understanding of this family of rapidly metastasizing skin cancer.
MCPyV Genome and Encoded Genes
MCPyV Genome
MCPyV has a ~5.4 kb circular dsDNA genome (Figure 1). Like other polyomaviruses, existing evidence suggests that the MCPyV genome remains episomal throughout the infectious cycle (Gjoerup and Chang, 2010; Liu et al., 2016a,b). The viral genome is divided into early and late regions by a non-coding control region (NCCR) containing the viral origin of replication and bidirectional promoters that drive early and late gene transcription (Harrison et al., 2011). The early region expresses alternatively spliced tumor antigens, termed large tumor antigen (LT) and small tumor antigen (sT) that support replication, as well as, 57kT and an alternate LT open reading frame (ALTO) with functions that are less defined (Kwun et al., 2009; Carter et al., 2013). Major and minor capsid proteins, VP1 and VP2, respectively, are expressed from the MCPyV late region along with a miRNA that has been proposed to modulate early gene expression (Seo et al., 2009; Schowalter et al., 2011).
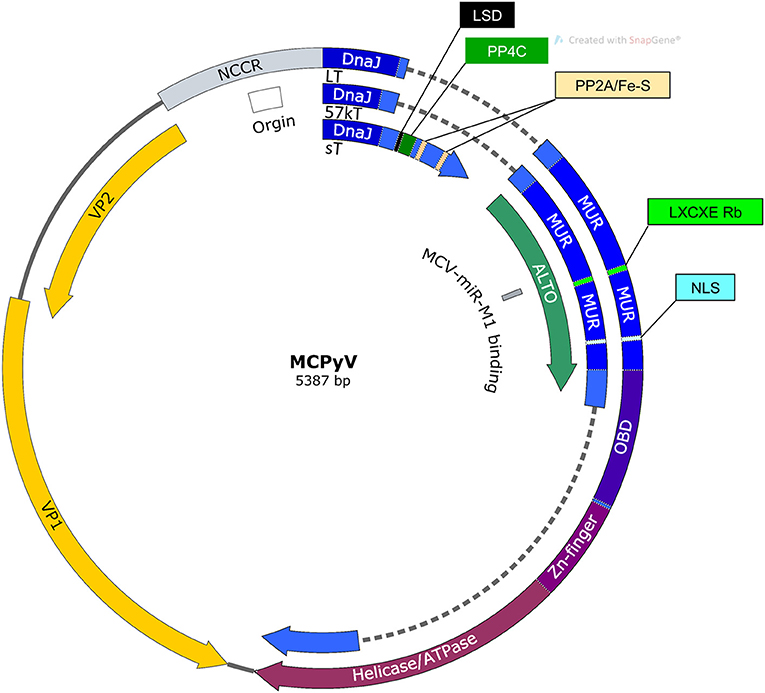
Figure 1. Map of MCPyV genome. NCCR, Non-coding control region; Origin, Origin of replication; LSD, LT stabilization domain; NLS, Nuclear localization signal; MUR, MCPyV unique region; OBD, Origin binding domain.
In MCPyV-positive MCC, the MCPyV genome is integrated into the host DNA such that the functions of its early promoter and partial expression of its T antigens are preserved (Feng et al., 2008). Point mutations in other regions of the genome and truncations of the MCPyV LT C-terminal domain, however, are common in viral MCCs (Liu et al., 2016a). Expression of the viral oncoproteins is largely driven by the preserved MCPyV promoter rather than endogenous promoters, though there are conflicting reports as to whether MCPyV is more likely to integrate into specific regions of chromatin (Doolittle-Hall et al., 2015; Czech-Sioli et al., 2020). Sequencing of integration sites in multiple MCC tumors reveal that initial recombination of a linearized MCPyV genome with the host genome could lead to transient circularization and amplification of the viral genome and neighboring host DNA (Starrett et al., 2017). The amount of amplification and the site of DNA repair accounts for differences in viral genome copy number and duplications of host sequences.
Large Tumor Antigen
In MCPyV infected cells, LT localizes to the nucleus where it performs functions directly and indirectly supporting MCPyV replication (Nakamura et al., 2010). Like T antigens in other polyomaviruses, LT contains an origin binding domain (OBD) and an ATP-dependent helicase domain by which it unwinds MCPyV DNA for replication (Harrison et al., 2011). LT localizes to replication foci containing high concentrations of nascently synthesized MCPyV genomes (Liu et al., 2016b). In replication foci, LT binds to G(A/G)GGC-like pentanucleotide sequences on the MCPyV genome to initiate efficient replication in a manner that requires the LT DnaJ domain and is supported by the presence of sT (Kwun et al., 2009; Harrison et al., 2011).
Somatic genes also localize to MCPyV replication foci and support LT-mediated replication. Bromodomain protein-4 (BRD4) associates with LT in replication centers where it amplifies MCPyV replication by recruiting replication factor C (RFC) (Wang et al., 2012). Ataxia telangiectasia mutated (ATM) and Rad3-related (ATR) DNA damage response (DDR) factors also co-localize to MCPyV replication centers in a manner dependent on the presence of LT and the MCPyV origin (Tsang et al., 2014). These DDR proteins support efficient viral genomic DNA synthesis, but may also be essential in limiting the transforming potential of MCPyV T antigens. For example, ATM phosphorylates LT in the C-terminal domain at Ser-816, leading to increased apoptosis (Li et al., 2015). Alanine mutagenesis at this site leads to enhanced colony formation in C33A cells. Therefore, this LT-ATM interaction could both promote MCPyV replication and limit rampant cellular growth. The delicate balance of establishing an S-phase-like environment for the production of new virions without causing the terminal fate of cellular transformation can explain the duality of MCPyV traits.
The necessity of MCPyV to avoid terminating its infectious cycle by transforming its host cell is underscored by the fact that unlike other polyomavirus T antigens, LT neither binds nor inhibits p53 (Lilyestrom et al., 2006; Cheng et al., 2013). In fact, the studies by Li and colleagues showed that the helicase activity in full-length LT, in the context of MCPyV DNA synthesis, induces cell cycle arrest in a p53-dependent manner that limits cellular proliferation (Li et al., 2013). This observation provided an evolutionary explanation as to why MCPyV may have lost the ability to inactivate p53, in that allowing p53 to guard cell cycle progression could limit incidental progression to cancer and abortive MCPyV infection. It also suggests why mature MCCs invariably express a truncated variant of LT (LTT) lacking the C-terminal helicase domain. In support of the protective nature of the LT C-terminal domain, expression of LTT, but not full-length LT, sensitizes cells to UV-DNA damage due to impaired cell cycle arrest and DDR (Demetriou et al., 2012). Moreover, expression of LTT promotes cell growth, while expression of the C-terminal domain alone, or full-length LT, negatively regulates cell growth (Cheng et al., 2013).
The ability of MCPyV LTT to promote cellular proliferation has been attributed to its ability to bind and inactivate Rb through an LXCXE domain similar to other polyomaviruses (Houben et al., 2012). This region is present in both wild-type LT and LTT. The LT-Rb interaction results in enhanced E2F-transcriptional activity; promoting growth in MCPyV-positive MCC cells (Sihto et al., 2011; Hesbacher et al., 2016; Schrama et al., 2016). The impact of LT-Rb binding was further illustrated by the fact that silencing LTT expression in a xenograft MCC mouse model resulted in tumor regression in an Rb-binding dependent manner (Houben et al., 2012). The LXCXE-Rb interaction also enhances entry into S-phase, cellular proliferation, and motility in hTERT immortalized BJ human foreskin fibroblasts (BJ-hTERT) (Richards et al., 2015). The functional significance of MCPyV LTT inactivation of Rb in human populations is also supported by the finding that MCPyV-negative MCCs frequently contain mutations in the Rb gene, whereas MCPyV-positive tumors usually express wild type Rb (Sihto et al., 2011).
LT-Rb binding domain could also be responsible for increasing Sox2 and subsequent Atoh expression in MCPyV-positive MCC cells (Harold et al., 2019). This is significant because the activity of these transcription factors can confer cells with markers of the shared phenotype between Merkel cells and MCCs both in vitro and in vivo (Verhaegen et al., 2017). Though sT has garnered more attention regarding cellular transformation, it may be that LT alters the transcriptional landscape of the original cell of MCC to imbue it with its morphological characteristics.
Recently, it has been shown that the Rb inhibitory domain of LT could indirectly activate p53 by upregulating ARF, an inhibitor of the p53-degrading E3 ubiquitin ligase MDM2 (Park et al., 2019). This assertion suggests that even without the helicase domain, LTT would activate p53 in MCC cells, but the investigators propose that the sT-MYC-EP400 transcriptional complex counteracts p53 activity through upregulation of MDM2 and the related enzyme MDM4 (Park et al., 2019).
Small Tumor Antigen
MCPyV sT consists of 186 amino acids, including a C-terminus that is spliced out of the other MCPyV T antigens that confers it with entirely unique functions. MCPyV sT localizes to the nucleus and is able to support LT-mediated replication though the exact mechanism remains unclear (Kwun et al., 2009). The functions carried out by the C-terminal domain unique to MCPyV sT is of critical importance because its expression is necessary for MCC survival and appears to be the primary driver of cellular transformation (Shuda et al., 2011).
MCPyV sT expression is capable of transforming rat fibroblasts and epithelial cells in an in vivo mouse model via a region incorporating amino acids 91–95 termed the LT stabilization domain (LSD) (Shuda et al., 2011; Kwun et al., 2015; Verhaegen et al., 2015). As its name implies, MCPyV sT containing wild-type LSD increases LT protein level, though the underlying mechanism is an area of active investigation (Kwun et al., 2013; Dye et al., 2019). MCPyV sT drives cellular transformation in rat fibroblasts by promoting hyperphosphorylation of eukaryotic translation-initiation factor 4E-binding protein (4E-BP1) (Shuda et al., 2011; Wu et al., 2015; Velasquez et al., 2016). The LSD domain was also linked to activation of the non-canonical NF-κB pathway, induction of a senescence associated secretory phenotype (SASP), and enhanced MCC cell proliferation (Zhao et al., 2020). Besides those functions attributed to the LSD domain, numerous other oncogenic functions have been ascribed to MCPyV sT.
MCPyV sT expression may promote cellular growth by activating c-Jun downstream of MEK/ERK factors (Wu et al., 2016). In addition, MCPyV sT expression in normal fibroblasts elevates aerobic glycolysis via modulation of the host cell transcriptome, including upregulation of monocarboxylate lactate transporter SLC16A1 (MCT1), likely contributing to oncogenic potential (Berrios et al., 2016). Still another way in which sT could enhance metastatic potential is through disruption of inter-cellular junctions via upregulation of A-disintegrase-and-metalloproteinase (ADAM) 10 and 17, which are more highly expressed in MCPyV-positive MCCs (Nwogu et al., 2018). The manner in which MCPyV sT might be affecting transcriptional changes described above is by recruiting a MYCL-MAX heterodimer to the EP400 complex (Cheng et al., 2017). This interaction was elegantly shown to promote cell viability in MCPyV-positive cell lines, MKL-1 and WaGa, as well as, confer a transforming phenotype in keratinocytes (Cheng et al., 2017). Two genes that are upregulated by this transcriptional program are MDM2, which promotes p53 proteasomal degradation, and lysine-specific histone demethylase 1A (LSD1), which is necessary for maintaining plasticity and proliferative capacity of MCC cells (Park et al., 2019, 2020; Leiendecker et al., 2020). Importantly, inhibition of MDM2 and LSD1 induces cell death in MCC cells and reduces the growth of MCC tumor in mice, demonstrating the therapeutic potential of using MDM2 and LSD1 inhibitors in treating this highly aggressive skin cancer (Park et al., 2019, 2020; Leiendecker et al., 2020).
MCPyV sT contains protein phosphatase 2A (PP2A) binding sites similar to other polyomaviruses, but this binding activity is not required for transformation of rat fibroblasts (Kwun et al., 2015). It has been suggested that these PP2A binding sites are involved in the interaction with protein phosphatase 4C (PP4C) (Griffiths et al., 2013; Kwun et al., 2015). In addition to findings mentioned earlier, one group found that sT targets NEMO through interaction with its regulatory subunit (PP4R1) to disrupt NF-κB mediated inflammatory signaling (Griffiths et al., 2013; Abdul-Sada et al., 2017). The sT-PP4C interaction has also been implicated in lowering microtubule stability through altered expression of cellular proteins like stathmin, as well as, Rho GTPase-mediated actin remodeling, leading to an enhanced cell motility phenotype (Knight et al., 2015; Stakaityte et al., 2018). These broad changes resulting from the sT-PP4C interaction have been recently ascribed to upregulated p38 MAPK signaling via MKK4 (Dobson et al., 2020). MCPyV sT related motility might therefore confer transformed cells with invasive and metastatic properties.
Our group found that the proposed PP2A binding sites might also serve as iron-sulfur (Fe/S) cluster domains (Tsang et al., 2015). This discovery had significance because proteins containing Fe/S domains often modulate helicase activity (Pugh et al., 2008; Wu and Brosh, 2012). The function of MCPyV sT Fe/S clusters was linked with its localization to LT-containing replication foci and enhanced MCPyV DNA synthesis without increasing LT protein stability. Moreover, MCPyV sT was able to coordinate Fe/S much more efficiently than sT proteins from SV40, HPyV6, and HPyV7. Another group recently found that expression of MCPyV sT in HEK 293 cells elevated several markers of DNA damage, and at a higher rate than HPyV6 and HPyV7 sT proteins (Wu et al., 2019). Given these findings, the supportive role of sT in LT-mediated MCPyV replication could be direct, through sT activity in replication centers, or indirect, through activation of ATM and subsequent phosphorylation of LT Ser816.
57kT and ALTO
A third alternatively spliced T antigen is that of 57kT (Figure 1). 57kT does not appear to support MCPyV replication, and its specific function in infection or in MCC oncogenesis is unclear (Kwun et al., 2009). Because it shares the MCPyV unique region (MUR) with LT, but lacks the helicase and origin-binding domains, it is reasonable to hypothesize that 57kT indirectly supports MCPyV infection, but does not directly support replication as LT does. Similar to LTT, 57kT could bolster pro-oncogenic functions associated with Rb-binding without helicase-related cell cycle arrest or DNA damage.
Carter and colleagues discovered an overprinting gene product expressed from the early region that they termed alternate Large T open reading frame (ALTO) (Carter et al., 2013) (Figure 1). This transcript utilizes a start codon that is +1 nucleotide frame-shifted relative to the second exon of LT. The authors hypothesize that overprinting genes such as these evolve in viruses as a strategy to maximize the coding potential of relatively small genomes (Carter et al., 2013). Recently, several ALTO-encoding circular RNAs have been identified in MCPyV-positive MCC cell lines and tumor tissues (Yang et al., 2021). MCPyV ALTO translated from these circular RNAs has the ability to transactivate recombinant promoters, as well as, a number of key cellular genes involved in MCPyV pathogenesis. Therefore, MCPyV ALTO protein may be able to modulate MCPyV infectious and tumorigenic potential through transcription regulation (Yang et al., 2021).
VP1, VP2, and miR-M1
The MCPyV late promoter drives expression of the capsid proteins VP1 and VP2, as well as, a miRNA, MCPyV miR-M1 (Figure 1). VP1 and VP2 encapsidate MCPyV DNA during packaging and mediate cell surface interactions that activate entry and trafficking (Schowalter et al., 2011; Neu et al., 2012; Schowalter and Buck, 2013). Attachment to cell surfaces is mediated through interactions between VP1 capsomeres and sulphated polysaccharides while subsequent viral entry requires the interaction between VP1 and sialic acid (Neu et al., 2012; Bayer et al., 2020). In A549 cells, MCPyV entry was mediated through caveolar/lipid raft endocytosis (Becker et al., 2019). Subsequent MCPyV trafficking is carried out by the endosomal-to-ER pathway requiring microtubule transport activity (Becker et al., 2019). By comparison, VP2 mediates post-attachment phases of MCPyV entry and is necessary for native MCPyV infection, if not the formation of pseudo-virus particles (Schowalter and Buck, 2013).
Another late region gene product MCPyV miR-M1 likely regulates MCPyV early gene expression. In MCPyV-transfected neuroectodermal tumor (PFSK-1) cells, highly expressed miR-M1 transcripts down-regulated the expression of MCPyV T antigens and MCPyV replication (Theiss et al., 2015). The function of miRNA-M1 to restrain MCPyV replication and gene expression enabled a low level of detectable MCPyV to persist in cells for several months. Thus, the MCPyV miRNA could drive a persistence mechanism by which the virus maintains a limited infection in human hosts for prolonged periods. In addition, it was found that expression of a synthetic MCPyV-miR-M1 in HEK293 or MCC cells targets the cellular host gene SP100 thereby lowering CXCL8 expression and neutrophil chemotaxis (Akhbari et al., 2018). In principle, this anti-inflammatory effect could be protective in the setting of MCPyV infection. Unlike analogous miRNAs in animal polyomavirus tumors, however, the MCPyV miRNA is not highly expressed in MCC tumors, suggesting that the function for which it is selected in MCPyV infection does not, in turn, promote tumor fitness (Chen et al., 2015).
MCPyV Tropism and a Model of Infection
Epidemiological evidence suggests that MCPyV establishes asymptomatic, persistent infections in most people. As many as 88% of healthy adults are positive for MCPyV-specific antibodies (Kean et al., 2009; Pastrana et al., 2009; Tolstov et al., 2009; Touze et al., 2011). Serological activity against the MCPyV major capsid protein increases as populations age, from about 10% in early childhood to about 80% in adults (Chen et al., 2011; Tolstov et al., 2011; Viscidi et al., 2011). MCPyV-specific antibody titers positively correlate with viral load as measured by MCPyV DNA encapsidated in viral particles shed from healthy skin, suggesting that MCPyV positivity rates could also increase as populations age (Schowalter et al., 2010; Pastrana et al., 2012).
Within a given healthy individual, MCPyV antibody titers remain relatively stable over a period of at least 15 months (Pastrana et al., 2012). By comparison, neutralizing antibody titers to MCPyV, but not other human polyomaviruses, are significantly higher in patients with MCPyV-positive MCC despite the fact that MCC tumors do not express capsid protein (Pastrana et al., 2009). Together, these findings suggest that MCPyV has the capacity to persist, and that MCPyV expansion within a host correlates with disease propensity. Inadequate restriction of MCPyV may be a critical factor in enabling MCC development. This hypothesis is supported by the fact that chronic UV-exposure, advanced age, and HIV-related or iatrogenic immunosuppression pose significant risk for MCC (Heath et al., 2008; Bertrand et al., 2013; Ma and Brewer, 2014).
Direct evidence of the natural host reservoir cells that maintain latent MCPyV infection remains elusive. To take steps in establishing the cellular context of MCPyV infection, we discovered that primary human skin dermal fibroblasts support productive MCPyV infection in vitro and ex vivo (Liu et al., 2016b). While developing the in vitro model, we found that the addition of modulatory factors present in the skin could greatly enhance MCPyV proliferation. Specifically, viral entry is stimulated during the first 2 days of infection in the absence of serum and in the presence of collagenase. This is analogous to the inflammatory skin in which matrix metalloproteinases (MMPs) digest collagen fibers and activate chemokines (Werner and Grose, 2003; Gill and Parks, 2008). Subsequently, MCPyV replication is boosted by priming human dermal fibroblasts with epidermal and fibroblast growth factors and a WNT activator, which induce fibroblast proliferation and MMP expression (Liu et al., 2016b). That these same conditions are found in the skin wounding response suggests that the MCPyV infection cycle could be linked to damage to the skin by way of abrasions or UV-damage.
The picture of MCPyV infection is far from complete, yet inferences can be made from distinct sources of evidence. In ex vivo skin culture, MCPyV preferentially infects dermal fibroblasts underlying the basal layer of the epidermis and those surrounding hair follicles (Liu et al., 2016b). Furthermore, MCPyV virions are readily detected in eyebrow hair bulbs sampled from healthy human volunteers (Peretti et al., 2014; Bellaud et al., 2015; Hampras et al., 2015). It is possible that MCPyV infects the dermal cells surrounding the hair follicle and subsequently uses the follicular space as a means to disseminate to the skin surface and access new hosts. In addition, migration of MCPyV-infected fibroblasts to wound sites could represent another mode of transmission from reservoir cells into the deeper layers of the skin.
MCPyV may also infect cells at body sites other than the skin to establish a reservoir (Salakova et al., 2016). For instance, MCPyV DNA was detected in buffy coats of healthy blood donors and inflammatory monocytes of MCC patients, indicating that the virus may establish latent infection in peripheral blood leukocytes (Mertz et al., 2010; Pancaldi et al., 2011). In two MCPyV-positive patients with prior history of MCC and active non-melanoma/non-MCC skin cancers respectively, MCPyV DNA was detected in inflammatory, but not resident monocytes (Mertz et al., 2010). Presence of MCPyV in inflammatory monocytes of patients with distinct medical histories suggests that the virus may reside in these cells and use them to spread throughout the body (Mertz et al., 2010).
There is no animal model for MCPyV infection; a goal which has proven to be challenging due to the narrow host range of the virus (Liu et al., 2018). Lack of an animal model for MCPyV infection presents a major obstacle for identifying potential reservoirs and elucidating the MCPyV infectious cycle. Generation of MCPyV chimeras with mammalian polyomaviruses may provide a solution to overcome its narrow host range in the future (Liu et al., 2018).
MCC Cell of Origin
Though named for similarities to Merkel cells, the true original cell of MCC is rigorously debated. The proposition that MCC arises from differentiated Merkel cells is in question because these cells are post-mitotic, and thereby have limited oncogenic potential, and because they arise in the epidermis, while MCCs almost always occur in the dermis or subcutis layers (Toker, 1972). In response, it has been suggested that MCCs could arise from Merkel cell progenitor cells present at the hair follicle (Zur Hausen et al., 2013; Sauer et al., 2017; Narisawa et al., 2019). Similarly, progenitor cells derived from the neural crest have been pointed to since MCPyV-positive MCC cell lines cocultured with keratinocytes undergo neuronal morphological differentiation in a manner dependent on MCPyV LT upregulation of Sox2 and Atoh1 (Harold et al., 2019).
Some also propose that MCC could have an epithelial origin since on rare occasions, epithelial MCCs have been reported (Narisawa et al., 2020; Navarrete-Dechent et al., 2020; Song et al., 2020). One group advocating for an epithelial origin of MCC points to a case of a combined MCC and trichoblastoma tumor that shared somatic mutations (Kervarrec et al., 2019). They propose that this mixed tumor could represent a transition from early to late MCC carcinogenesis in cells with integrated MCPyV genome beginning to predominate. The same group also found that expressing MCPyV sT and GLI1 in keratinocytes results in an MCC-like phenotype, including expression of CK20 (Kervarrec et al., 2020). Another group has developed a mouse model for MCC, by expressing MCPyV sT and Atoh in keratinocytes, that results in the epidermal layer developing several MCC markers and characteristics (Verhaegen et al., 2017).
It has also been argued that pre/pro B cells are the source of MCC because MCC cells consistently express a number of B-lymphoid lineage markers, like Pax5 and TdT (Zur Hausen et al., 2013; Sauer et al., 2017). Cell expression similarities may be coincidental however, since it has also been proposed that epigenetic changes in the cell of origin could lead to a dramatic transcriptional and phenotypic changes culminating in Merkel cell resemblance. It has even been proposed that MCPyV-positive and -negative carcinomas have distinct cells of origin, and that through epigenetic reprogramming they converge on a common phenotype (Sunshine et al., 2018). Under normal developmental conditions, loss of polycomb repressive complex 2 (PRC2) and subsequent reduction H3K27me3 marks enables the differentiation of Merkel cells in mice (Bardot et al., 2013; Perdigoto et al., 2016). Given this information, one group reasoned that the development of MCC may involve a similar change in the epigenome of the unknown cell of origin. They found that pure MCPyV-positive MCCs were more likely to have lower H3K27me3 than MCPyV-negative tumors (Busam et al., 2017). Other researchers, however, found contradicting evidence that virus-negative MCCs, especially those with combined squamous cell carcinomas, had lower H3K27me3 marks than MCPyV-positive MCCs (Matsushita et al., 2019).
Immune Responses to MCC
Immune function is relevant to every aspect of MCC progression. Incidence of MCC is greatly increased in immunocompromised individuals, especially in those who are HIV-positive or recipients of organ transplants (Koljonen et al., 2009; Ma and Brewer, 2014; Cook et al., 2019). Loss of adaptive immune competence increases the risk of both MCPyV-positive and negative tumors, implying that immune surveillance of nascently transformed MCC cells is protective at early stages of the disease. There is also evidence that adequate immune responses can lessen the severity of MCC disease progression. For example, patients with chronic inflammatory disease had higher rates of MCC incidence, and the tumors of those that developed MCC had higher expression of the proliferative marker Ki-67, and a greater tumor size (Sahi et al., 2017). Furthermore, systemic immune suppression has been linked to greater incidence of MCC, as well as, lower rates of survival in MCC patients (Paulson et al., 2013). These epidemiological data suggest that immune restriction of MCC is critical to patient outcomes at each stage of the disease.
Studies of the immunological interface in MCC tumors have revealed further detail regarding the course of disease and the ability of MCC to evade destruction. For example, circulating MCPyV T antigen-specific antibody level positively correlates with MCPyV-positive MCC recurrence and has prognostic value (Samimi et al., 2016; Paulson et al., 2017). In MCC tumors, CD8 T cell and other immune cell infiltration is usually poor, and those cases with better infiltration positively correlate with patient outcomes (Sihto et al., 2012; Lipson et al., 2013; Wheat et al., 2014; Feldmeyer et al., 2016; Miller et al., 2017). Moreover, the MCPyV-specific T cells present in MCC tumors expressed markers of exhaustion like PD-1 and Tim-3 (Afanasiev et al., 2013). The same phenomenon could be found in vitro and in a MCC xenograft mouse-model (Dowlatshahi et al., 2013). Histopathology of MCC tumors indicate that immune cells expressing exhaustion markers like PD-1 and CD33 congregate in the areas surrounding the tumors, suggesting that MCC acquires traits that suppress immune cell migration (Mitteldorf et al., 2017).
Observations that tumor-experienced T cells were unable to infiltrate MCC tumors led to recent efforts to introduce immune checkpoint therapies in metastatic cases. A 2015 case study showed a promising response in a patient with metastatic MCC case treated with anti-PD1 antibodies (Mantripragada and Birnbaum, 2015). In the years that followed, clinical trials exploring safety and efficacy of anti-PD1 and anti-PDL-1 infusions yielded significant response rates and relatively low rates of adverse events given the severity of patient prognosis (Iyer et al., 2016; Kaufman et al., 2016; Nghiem et al., 2019). While the durability of responses varied, they could last well over a year (Kaufman et al., 2018; Nghiem et al., 2019).
Given that there are no effective chemotherapeutic treatments available for metastatic MCC, the introduction of immune checkpoint inhibitors greatly benefitted patients (Colunga et al., 2017; Paulson et al., 2018a). Still, a significant portion of patients do not respond or have responses that are short-lived. Moreover, patients with immunosuppression or autoimmune disease may not be eligible for immune-based therapy. It also became clear that loss of durability or relapse is a problem in long term treatment for MCC, even in MCPyV-positive MCC cases where mutagenesis is low. A longitudinal study revealed that HLA-I components were downregulated in resistant tumor cells at the level of transcription as a result of continued interaction with CD8 T cells (Paulson et al., 2018b). These shortcomings necessitate the development of alternative strategies or combination of existing therapies to modulate immunity to MCC.
One such strategy is to augment innate immunity at the primary tumor site in order to enhance adaptive function systemically. One group was able to achieve improved responses at distal MCC metastases by injecting a TLR-4 agonist intratumorally at the primary MCC site followed by standard-of-care surgery and irradiation (Bhatia et al., 2019). They later delivered IL-12 plasmids to MCC tumors via electroporation that led to enhanced immunogenicity at primary and distal tumors in all patients tested; 25% of which had responses in the progression of their disease (Bhatia et al., 2020). Still other means of activating intralesional innate inflammation have proven effective. Administration of an oncolytic herpesvirus that stimulates granulocyte-macrophage colony-stimulating factor has led to durable complete responses in multiple patients (Nguyen et al., 2019; Westbrook et al., 2019). A recent case study involving an advanced-stage MCC patient resulted in complete remission upon receiving a combination of radiation therapy and anti-PD1 therapy (Bloom et al., 2019). This suggests that targeted DNA damage at accessible lesions combined with activation of immunity against a primary tumor could help train immune responses against distant lesions.
While probing the cause of immune suppression characterizing the MCC tumor microenvironment, our group discovered that STING (stimulator of interferon genes) is dramatically repressed in MCC cell lines and tumor cells (Liu et al., 2020). Among these immortalized and primary MCC cells, the marked reduction in STING expression was unique to those that were MCPyV-positive (Liu et al., 2020). Since SV40 LT has been shown to antagonize the STING signaling pathway (Lau et al., 2015), it would be valuable to determine if MCPyV LTT expressed in virus-positive MCC cells downregulates STING expression. We also showed that rescuing STING expression and activation in MCC cells led to greatly induced cytokine expression, T cell migration, and MCC cell death (Liu et al., 2020). Delivery of a mutant STING vector via AAV transduction and subsequent activation with a selective agonist could achieve the same results. Such a strategy has the potential to be highly specific in humans without the danger of systemic inflammatory pathology because the mutant-specific STING agonist does not interact with native human STING (Liu et al., 2020).
Targeting distal MCC metastases is also the aim of researchers activating or genetically engineering immune cells for autologous infusion. For example, one group showed that activation and administration of genetically engineered T cells expressing TCRs specific to naturally processed MCPyV T epitopes led to tumor regression in mouse MCC xenografts (Gavvovidis et al., 2018). In one human case study, a similar infusion procedure using MCPyV T antigen-specific T cells that were expanded ex vivo resulted in HLA upregulation, T cell recruitment, and responses in most metastases (Chapuis et al., 2014). A recent study developed a means of improving autologous CD8 T cell therapeutic vaccines for MCC by exposing those T cells to cytokine-conditioned dendritic cells presenting LTT peptides on both MHC-I and II surface receptors (Gerer et al., 2017). MCPyV-positive tumor microenvironments can also be engaged by engineered CD4 T cells that recognize the LXCXE epitope of LTT (Longino et al., 2019).
Immune Responses to MCPyV
Given that the risk factors for developing MCC, including advanced age, UV exposure, and compromised adaptive immunity, can each alter the immune environment in the skin, it is possible that unmitigated proliferation of MCPyV encourages oncogenesis (Stang et al., 2018). The possibility that uncontrolled MCPyV activity leads to MCC cases agrees with the ability of Chang and Moore to predict the viral etiology of MCC due to its greater incidence in immune compromised persons (Feng et al., 2008). The transition from low-level MCPyV persistence to rampant proliferation could promote carcinogenesis by increasing the frequency of integration events or entry into the original host cell of MCC. Because nascently transformed cells are also favored by loss of normal immunity, it would be easy to conflate these two factors. Interestingly though, there is increasing evidence that alterations to systemic and skin immunity can exacerbate MCPyV infections.
A study in Japan found that MCPyV DNA prevalence and viral load on sun-exposed skin increased sharply in individuals over the age of 40 and remained high for the oldest groups (Hashida et al., 2016a). Also, HIV-positive men more frequently have detectable MCPyV DNA on their skin and in their sera, and those with poorly-controlled HIV infection have higher MCPyV antibody titers and DNA loads than those with better-controlled infections (Wieland et al., 2011; Fukumoto et al., 2013; Vahabpour et al., 2017). In kidney transplant recipients, MCPyV DNA was more readily detected in the urine of those with BKPyV-DNAemia and with histologically verified polyomavirus-associated nephropathy (Signorini et al., 2014; Wang et al., 2019). MCPyV DNA detection rates and loads were significantly lower in patients with psoriasis compared to the skin of healthy volunteers suggesting a potential inverse relationship between chronic inflammation in the skin and MCPyV proliferation (Hashida et al., 2019). MCPyV-positive MCC patients, however, have higher MCPyV DNA loads shed from their skin and more frequently produce MCPyV VP1-specific circulating antibodies and at higher titers (Pastrana et al., 2009, 2012; Faust et al., 2011; Touze et al., 2011; Hashida et al., 2016b). More recently, it was found that higher MCPyV DNA load correlates with worse survival outcomes in MCC patients (Von Der Grun et al., 2019).
These findings suggest that sudden loss of normal immune functions could swing the balance between host and virus to favor uncontrolled MCPyV proliferation (Figure 2). From the perspective of the virus, however, MCPyV must have evolved mechanisms of its own to evade host immunity since it can persist and remain highly prevalent in the general population asymptomatically (Tolstov et al., 2009; Schowalter et al., 2010; Foulongne et al., 2012). The mechanisms by which MCPyV evades immune detection and/or destruction are not known.
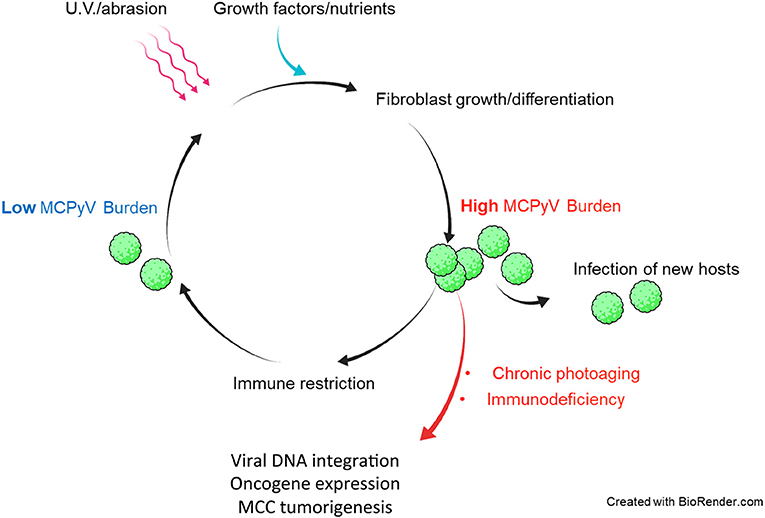
Figure 2. Hypothetical model of in vivo MCPyV persistence and dysbiosis leading to MCPyV-positive MCC. Healthy individuals support both low and high MCPyV loads, depending on external factors. U.V. irradiation or abrasion of the skin could cause infected dermal fibroblasts to upregulate MCPyV gene expression and replication. Damaged and repairing skin could be a path by which MCPyV escapes the dermis to infect new hosts. Healthy immune responses may reduce the MCPyV burden asymptomatically. In immunocompromised patients and those with years of chronic U.V. damage, the microenvironment of the skin may be altered in a way that induces MCPyV entry into the original cell of MCC and integration into the host chromatin to drive MCC tumorigenesis.
Some examples of MCPyV gene products or transfected MCPyV genomes interacting with cellular immunity have been documented. For instance, LT expression reduces TLR-9 expression via downregulation of C/EBP transcription factors in epithelial and MCC cell lines (Shahzad et al., 2013). This observation agrees with in vivo data revealing that TLR-9 expression was significantly lower in MCPyV-positive MCC tumors compared to MCPyV-negative tumors (Jouhi et al., 2015). MCPyV sT expression was shown to inhibit NF-κB inflammatory signaling through its interaction with PP4C and the adapter NEMO (Griffiths et al., 2013; Abdul-Sada et al., 2017). In addition, it has been shown that a MCPyV-miR-M1 mimic specifically targeted SP100 for degradation (Akhbari et al., 2018). This downregulation of SP100 reduced the secretion of CXCL8 in MCC cells treated with TNF-α (Akhbari et al., 2018). Notably, depletion of the nuclear protein SP100 also enhances MCPyV replication in H1299 cells (Neumann et al., 2016).
There are also examples of cells eliciting an immune response to the expression of MCPyV genes and genomes. As mentioned previously, MCPyV LT and sT expression in BJ-hTERT cells led to upregulation of cellular growth and expression of genes that increase motility (Richards et al., 2015). Accompanying this phenotype was the dramatic induction of many inflammatory response genes including interferon-stimulated genes (ISGs) like OAS1 and ISG20; cytokines like IL-1β and IL-6; and chemokines like CXCL1 and CXCL6 (Richards et al., 2015). Whether MCPyV T antigens induce such a response in the context of infection remains undetermined.
Despite the recent progress made in revealing the MCPyV-host interface, the approaches used previously involved transfection or transduction of MCPyV genes into established cancer cell lines. In order to understand how these mechanisms may contribute to MCPyV persistence, our group examined the innate immune response to MCPyV in the context of infection in human dermal fibroblasts, which plausibly model MCPyV infection of human skin (Krump et al., 2021). After establishing the MCPyV gene expression and replication kinetics in infected cells, we found that late events in the infectious cycle activated the cGAS-STING and NF-κB pathways and subsequent expression of anti-viral ISGs as well as innate inflammatory cytokines (Krump et al., 2021). CRISPR knockout of elements of these immune regulatory pathways yielded significantly higher levels of MCPyV replication per cell, suggesting that the innate gene induction has the potential to restrict MCPyV replication, even in the absence of cellular immune factors (Krump et al., 2021).
A Model of MCPyV Dysbiosis Leading to MCC Development
MCPyV infects and sheds from the skin of most people without discernible symptoms (Pastrana et al., 2009; Schowalter et al., 2010). The evolutionary strategies that allow MCPyV to asymptomatically infect a large portion of the population, often for prolonged periods of time, remain a mystery (Chen et al., 2011). The auspicious discovery that human dermal fibroblasts support MCPyV infection enabled our recent characterization of the MCPyV infectious cycle and the consequences it poses for the host cell (Liu et al., 2016b; Krump et al., 2021). By understanding the scenarios in which MCPyV might fail to strike a balance with the host immune system, we may be able to infer the events preceding MCPyV integration and oncogenesis.
While direct observation of the interface between MCPyV and systemic immune responses awaits development of an animal model, it is currently possible to formulate a model of MCPyV infection based on data from several sources: (1) the epidemiology of MCC and MCPyV infections, (2) existing research on fibroblast biology and wounding responses in human skin, and (3) in vitro findings that establish the prerequisites for MCPyV replication and gene expression (Liu et al., 2016b; Krump et al., 2021). In a similar manner, by juxtaposing decades of skin pathology research with observed cellular response to MCPyV infection, we may be able to provide new insights regarding dysregulated conditions that might lead to MCPyV-driven MCC (Walder et al., 1971; Hardie et al., 1980; Ahmad et al., 2014; Krump et al., 2021).
If indeed dermal fibroblasts are capable of supporting MCPyV infection in vivo as they are ex vivo and in vitro (Liu et al., 2016b), then a model of the course of MCPyV infection can begin to take shape (Figure 2). For instance, healthy human hosts may support a low basal rate of MCPyV activity in fibroblasts that avoids immune recognition, as early events like viral entry and trafficking in vitro failed to activate ISGs (Krump et al., 2021). In the event that host skin is abraded or irradiated with UV light, damaged keratinocytes release growth factors and WNT agonists to induce MMP expression and expansion of fibroblasts (Gill and Parks, 2008; Whyte et al., 2012). These tissue changes could stimulate MCPyV early gene expression and DNA synthesis (Liu et al., 2016b). In this scenario, MCPyV PAMPS and/or DAMPs present during later stages of infection upregulate ISGs and inflammatory cytokines in manner that restricts viral replication (Krump et al., 2021). Serological evidence suggests that antibody-related adaptive responses to elevated MCPyV loads could also be a significant restriction factor (Faust et al., 2011). Dermal fibroblasts proliferating and navigating to the wounded tissue or shedding layers of sunburned skin could facilitate MCPyV transmission to new hosts (Figure 2). The antiviral state conferred by ISG induction, innate cytokine signaling, and likely recruited immune cells at the wound site would restrict and clear cells with high levels of MCPyV infection. A return to persistent, low-level MCPyV infection could be achieved by those infected resident skin cells that avoid immune detection (Figure 2).
Fluctuations in MCPyV load in dynamic equilibrium with insults to the skin and immune responses could be evolutionarily advantageous to MCPyV as it would only risk detection when the opportunity to infect new hosts arises (Figure 2). Should MCPyV activity correspond to skin damage and repair, then chronic wounding, inflammation, or altered skin architecture could represent the early events that promote MCPyV DNA amplification and integration into host DNA that lead to MCPyV-associated MCC (Figures 2, 3). This possibility is supported by epidemiological studies showing a strong correlation between immunosuppression, elevated MCPyV genome loads, and increased risk for MCC (Heath et al., 2008; Wieland et al., 2011). These studies suggest that faulty immune control of MCPyV infection could disturb the balance of MCPyV-host interaction to cause unbridled viral replication, which can increase the chance of incidental integration of viral DNA and MCC oncogenesis (Figures 2, 3). These observations would also explain why certain populations have a higher risk of developing MCC.
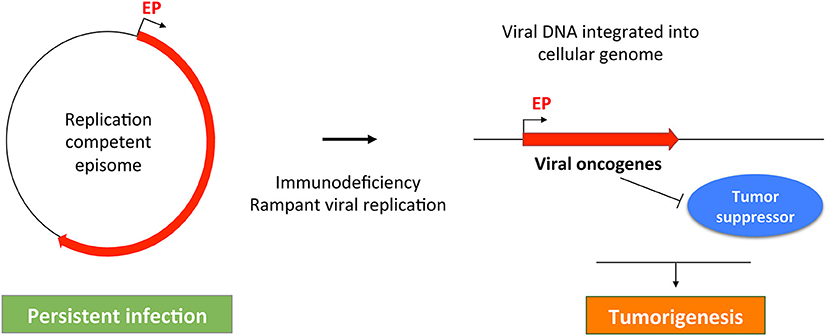
Figure 3. A common theme in MCPyV- and HPV-induced tumorigenesis. MCPyV and HPV both replicate as episomes in persistently infected cell. Failure of host immune system may cause uncontrolled viral replication, which can stimulate viral DNA integration into the host cellular genome. The integrated viral genome expresses viral oncogenes that can inhibit tumor suppressors to induce malignant transformation. Red line indicates the early region of the oncogenic viruses. EP, Early promoter.
The molecular events linking MCPyV infection and MCC development are reminiscent of those underlying the malignancies driven by human papillomaviruses (HPVs). In non-malignant human cells, HPVs, like MCPyV, normally replicate and maintain their genomes as episomes (Longworth and Laimins, 2004; You et al., 2004; You, 2010; Wang et al., 2013) (Figure 3). During the course of persistent infection, a compromised immune system and/or other pathologic conditions could cause rampant viral replication to promote integration of viral genomes into the host DNA. An additional parallel between MCPyV and HPV oncogenic mechanisms is that the integrated viral genomes typically lose the ability to replicate, but the non-replicating viral genomes retain the capacity to express viral oncogenes, such as LTT/sT (encoded by MCPyV) and E6/E7 (encoded by HPV). These viral oncoproteins stimulate cellular proliferation and malignant transformation by inhibiting host tumor suppressor such as RB and p53 (Gaglia and Munger, 2018) (Figure 3). The uncontrolled cellular proliferation could ultimately inflict oncogenesis by allowing virally induced precancerous lesions to persist and expand (Figure 3). In addition, viral oncogenes encoded by the integrated MCPyV and HPV genomes also share the ability to induce genomic instability (Li et al., 2013; Gaglia and Munger, 2018), which can introduce more DNA breaks in the host genome to stimulate viral genome integration. Hyperproliferation and DNA damage induced by viral oncogenes and/or U.V. irradiation may allow the emerging tumor cells to accumulate additional genetic mutations needed to develop into invasive tumors (Figure 3).
Future Perspective
Over 90% of MCC patients are not immune-compromised by clinical definitions, yet almost all are over the age of 50 and have low melanin content in their skin (Heath et al., 2008). Therefore, unraveling the impact of UV-radiation and aging to the skin could reveal key aspects of early events in MCPyV-associated MCC. Further elucidation of tactics employed by MCPyV to manipulate the host immune system for promoting its own propagation and driving cellular transformation will likely offer new clues for understanding the mechanism driving MCPyV tumorigenesis.
The bourgeoning of cancer therapies aimed at activating and directing immune responses to malignancies give much cause for hope for the treatment of MCC and other viral cancers. Still, gaps in our understanding of the biology driving MCPyV-related oncogenesis and MCC immune escape could widen the breadth of patients that respond to treatments and the durability of those responses. Addressing some of our gaps in knowledge will require technological advances like improved detection of low-copy number viral DNA genomes in tissue isolates or accurate animal models of MCPyV infection. Until that time, we can make strides in our investigation using the wealth of knowledge on human skin and cancer biology, and by expanding the model of in vitro MCPyV infection. Applying this knowledge to re-establish asymptomatic equilibrium between host and MCPyV in at-risk individuals has the potential to prevent MCC cases from occurring in the future.
Author Contributions
NK and JY: conceptualization and writing—review and editing. NK: writing—original draft preparation and visualization. JY: supervision, project administration, and funding acquisition. Both authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health (NIH) Grants R01CA187718, R21AR074073, R21AI149761, the NCI Cancer Center Support Grant (NCI P30 CA016520), and the Penn CFAR pilot award (P30 AI 045008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank all members of the JY's laboratory for their feedback and support throughout the course of this project. We apologize to our colleagues whose primary research results could not be cited due to space constraints.
References
Abdul-Sada, H., Muller, M., Mehta, R., Toth, R., Arthur, J. S. C., Whitehouse, A., et al. (2017). The PP4R1 sub-unit of protein phosphatase PP4 is essential for inhibition of NF-kappaB by merkel polyomavirus small tumour antigen. Oncotarget 8, 25418–25432. doi: 10.18632/oncotarget.15836
Afanasiev, O. K., Yelistratova, L., Miller, N., Nagase, K., Paulson, K., Iyer, J. G., et al. (2013). Merkel polyomavirus-specific T cells fluctuate with merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clin. Cancer Res. 19, 5351–5360. doi: 10.1158/1078-0432.CCR-13-0035
Ahmad, I., Simanyi, E., Guroji, P., Tamimi, I. A., Delarosa, H. J., Nagar, A., et al. (2014). Toll-like receptor-4 deficiency enhances repair of UVR-induced cutaneous DNA damage by nucleotide excision repair mechanism. J. Invest. Dermatol. 134, 1710–1717. doi: 10.1038/jid.2013.530
Akhbari, P., Tobin, D., Poterlowicz, K., Roberts, W., and Boyne, J. R. (2018). MCV-miR-M1 targets the host-cell immune response resulting in the attenuation of neutrophil chemotaxis. J. Invest. Dermatol. 138, 2343–2354. doi: 10.1016/j.jid.2018.03.1527
Bardot, E. S., Valdes, V. J., Zhang, J., Perdigoto, C. N., Nicolis, S., Hearn, S. A., et al. (2013). Polycomb subunits Ezh1 and Ezh2 regulate the Merkel cell differentiation program in skin stem cells. EMBO J. 32, 1990–2000. doi: 10.1038/emboj.2013.110
Bayer, N. J., Januliene, D., Zocher, G., Stehle, T., Moeller, A., and Blaum, B. S. (2020). Structure of merkel cell polyomavirus capsid and interaction with its glycosaminoglycan attachment receptor. J. Virol. 94, e01664–e01619. doi: 10.1128/JVI.01664-19
Becker, J. C., Stang, A., Hausen, A. Z., Fischer, N., Decaprio, J. A., Tothill, R. W., et al. (2017). Epidemiology, biology and therapy of Merkel cell carcinoma: conclusions from the EU project IMMOMEC. Cancer Immunol. Immunother. 67, 341–351. doi: 10.1007/s00262-017-2099-3
Becker, M., Dominguez, M., Greune, L., Soria-Martinez, L., Pfleiderer, M. M., Schowalter, R., et al. (2019). Infectious entry of merkel cell polyomavirus. J. Virol. 93, e02004–e02018. doi: 10.1128/JVI.02004-18
Bellaud, G., Gheit, T., Pugin, A., Pretet, J. L., Tommasino, M., Mougin, C., et al. (2015). Prevalence of human polyomavirus DNA in eyebrow hairs plucked from patients with psoriasis treated with TNF inhibitors. J. Eur. Acad. Dermatol. Venereol. 29, 1019–1021. doi: 10.1111/jdv.12439
Berrios, C., Padi, M., Keibler, M. A., Park, D. E., Molla, V., Cheng, J., et al. (2016). Merkel cell polyomavirus small t antigen promotes pro-glycolytic metabolic perturbations required for transformation. PLoS Pathog. 12:e1006020. doi: 10.1371/journal.ppat.1006020
Bertrand, M., Mirabel, X., Desmedt, E., Vercambre-Darras, S., Martin De Lassalle, E., Bouchindhomme, B., et al. (2013). [Merkel cell carcinoma: a new radiation-induced cancer?]. Ann. Dermatol. Venereol. 140, 41–45. doi: 10.1016/j.annder.2012.10.598
Bhatia, S., Longino, N. V., Miller, N. J., Kulikauskas, R., Iyer, J. G., Ibrani, D., et al. (2020). Intratumoral delivery of plasmid IL12 via electroporation leads to regression of injected and noninjected tumors in merkel cell carcinoma. Clin. Cancer Res. 26, 598–607. doi: 10.1158/1078-0432.CCR-19-0972
Bhatia, S., Miller, N. J., Lu, H., Longino, N. V., Ibrani, D., Shinohara, M. M., et al. (2019). Intratumoral G100, a TLR4 agonist, induces antitumor immune responses and tumor regression in patients with merkel cell carcinoma. Clin. Cancer Res. 25, 1185–1195. doi: 10.1158/1078-0432.CCR-18-0469
Bloom, B. C., Augustyn, A., Pezzi, T. A., Menon, H., Mayo, L. L., Shah, S. J., et al. (2019). Rescue of immunotherapy-refractory metastatic merkel cell carcinoma with conventionally fractionated radiotherapy and concurrent pembrolizumab. Front. Oncol. 9:223. doi: 10.3389/fonc.2019.00223
Buck, C. B., and Lowy, D. R. (2009). Getting stronger: the relationship between a newly identified virus and Merkel cell carcinoma. J. Invest. Dermatol. 129, 9–11. doi: 10.1038/jid.2008.302
Busam, K. J., Pulitzer, M. P., Coit, D. C., Arcila, M., Leng, D., Jungbluth, A. A., et al. (2017). Reduced H3K27me3 expression in Merkel cell polyoma virus-positive tumors. Mod. Pathol. 30, 877–883. doi: 10.1038/modpathol.2017.8
Carter, J. J., Daugherty, M. D., Qi, X., Bheda-Malge, A., Wipf, G. C., Robinson, K., et al. (2013). Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc. Natl. Acad. Sci. U.S.A. 110, 12744–12749. doi: 10.1073/pnas.1303526110
Cassler, N. M., Merrill, D., Bichakjian, C. K., and Brownell, I. (2016). Merkel cell carcinoma therapeutic update. Curr. Treat. Options Oncol. 17, 36. doi: 10.1007/s11864-016-0409-1
Chadburn, A., Abdul-Nabi, A. M., Teruya, B. S., and Lo, A. A. (2013). Lymphoid proliferations associated with human immunodeficiency virus infection. Arch. Pathol. Lab. Med. 137, 360–370. doi: 10.5858/arpa.2012-0095-RA
Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J., Knowles, D. M., et al. (1994). Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266, 1865–1869. doi: 10.1126/science.7997879
Chapuis, A. G., Afanasiev, O. K., Iyer, J. G., Paulson, K. G., Parvathaneni, U., Hwang, J. H., et al. (2014). Regression of metastatic Merkel cell carcinoma following transfer of polyomavirus-specific T cells and therapies capable of re-inducing HLA class-I. Cancer Immunol. Res. 2, 27–36. doi: 10.1158/2326-6066.CIR-13-0087
Chen, C. J., Cox, J. E., Azarm, K. D., Wylie, K. N., Woolard, K. D., Pesavento, P. A., et al. (2015). Identification of a polyomavirus microRNA highly expressed in tumors. Virology 476, 43–53. doi: 10.1016/j.virol.2014.11.021
Chen, T., Hedman, L., Mattila, P. S., Jartti, T., Ruuskanen, O., Soderlund-Venermo, M., et al. (2011). Serological evidence of Merkel cell polyomavirus primary infections in childhood. J. Clin. Virol. 50, 125–129. doi: 10.1016/j.jcv.2010.10.015
Cheng, J., Park, D. E., Berrios, C., White, E. A., Arora, R., Yoon, R., et al. (2017). Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 13:e1006668. doi: 10.1371/journal.ppat.1006668
Cheng, J., Rozenblatt-Rosen, O., Paulson, K. G., Nghiem, P., and Decaprio, J. A. (2013). Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 87, 6118–6126. doi: 10.1128/JVI.00385-13
Cimino, P. J., Robirds, D. H., Tripp, S. R., Pfeifer, J. D., Abel, H. J., and Duncavage, E. J. (2014). Retinoblastoma gene mutations detected by whole exome sequencing of Merkel cell carcinoma. Mod. Pathol. 27, 1073–1087. doi: 10.1038/modpathol.2013.235
Colunga, A., Pulliam, T., and Nghiem, P. (2017). Merkel cell carcinoma in the age of immunotherapy: facts and hopes. Clin Cancer Res. 24, 2035–2043. doi: 10.1158/1078-0432.CCR-17-0439
Cook, M., Baker, K., Redman, M., Lachance, K., Nguyen, M. H., Parvathaneni, U., et al. (2019). Differential outcomes among immunosuppressed patients with merkel cell carcinoma: impact of immunosuppression type on cancer-specific and overall survival. Am. J. Clin. Oncol. 42, 82–88. doi: 10.1097/COC.0000000000000482
Cowey, C. L., Mahnke, L., Espirito, J., Helwig, C., Oksen, D., and Bharmal, M. (2017). Real-world treatment outcomes in patients with metastatic Merkel cell carcinoma treated with chemotherapy in the USA. Future Oncol. 13, 1699–1710. doi: 10.2217/fon-2017-0187
Czech-Sioli, M., Günther, T., Therre, M., Spohn, M., Indenbirken, D., Theiss, J., et al. (2020). High-resolution analysis of Merkel Cell Polyomavirus in Merkel Cell Carcinoma reveals distinct integration patterns and suggests NHEJ and MMBIR as underlying mechanisms. PLoS Pathog. 16:e1008562. doi: 10.1371/journal.ppat.1008562
Dabner, M., Mcclure, R. J., Harvey, N. T., Budgeon, C. A., Beer, T. W., Amanuel, B., et al. (2014). Merkel cell polyomavirus and p63 status in Merkel cell carcinoma by immunohistochemistry: Merkel cell polyomavirus positivity is inversely correlated with sun damage, but neither is correlated with outcome. Pathology 46, 205–210. doi: 10.1097/PAT.0000000000000069
D'angelo, S. P., Russell, J., Lebbe, C., Chmielowski, B., Gambichler, T., Grob, J. J., et al. (2018). Efficacy and safety of first-line avelumab treatment in patients with stage iv metastatic merkel cell carcinoma: a preplanned interim analysis of a clinical trial. JAMA Oncol. 4:e180077. doi: 10.1001/jamaoncol.2018.0077
De Wolff-Peeters, C., Marien, K., Mebis, J., and Desmet, V. (1980). A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer 46, 1810–1816.
Decaprio, J. A. (2009). Does detection of Merkel cell polyomavirus in Merkel cell carcinoma provide prognostic information? J. Natl. Cancer Inst. 101, 905–907. doi: 10.1093/jnci/djp162
Demetriou, S. K., Ona-Vu, K., Sullivan, E. M., Dong, T. K., Hsu, S. W., and Oh, D. H. (2012). Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int. J. Cancer 131, 1818–1827. doi: 10.1002/ijc.27440
Dobson, S. J., Anene, A., Boyne, J. R., Mankouri, J., Macdonald, A., and Whitehouse, A. (2020). Merkel cell polyomavirus small tumour antigen activates the p38 MAPK pathway to enhance cellular motility. Biochem. J. 477, 2721–2733. doi: 10.1042/BCJ20200399
Doolittle-Hall, J. M., Cunningham Glasspoole, D. L., Seaman, W. T., and Webster-Cyriaque, J. (2015). Meta-analysis of DNA tumor-viral integration site selection indicates a role for repeats, gene expression and epigenetics. Cancers (Basel) 7, 2217–2235. doi: 10.3390/cancers7040887
Dowlatshahi, M., Huang, V., Gehad, A. E., Jiang, Y., Calarese, A., Teague, J. E., et al. (2013). Tumor-specific T cells in human Merkel cell carcinomas: a possible role for Tregs and T-cell exhaustion in reducing T-cell responses. J. Invest. Dermatol. 133, 1879–1889. doi: 10.1038/jid.2013.75
Duncavage, E. J., Zehnbauer, B. A., and Pfeifer, J. D. (2009). Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod. Pathol. 22, 516–521. doi: 10.1038/modpathol.2009.3
Dye, K. N., Welcker, M., Clurman, B. E., Roman, A., and Galloway, D. A. (2019). Merkel cell polyomavirus Tumor antigens expressed in Merkel cell carcinoma function independently of the ubiquitin ligases Fbw7 and beta-TrCP. PLoS Pathog. 15:e1007543. doi: 10.1371/journal.ppat.1007543
Ellerbrock, T. V., Chiasson, M. A., Bush, T. J., Sun, X. W., Sawo, D., Brudney, K., et al. (2000). Incidence of cervical squamous intraepithelial lesions in HIV-infected women. JAMA 283, 1031–1037. doi: 10.1001/jama.283.8.1031
Engels, E. A., Frisch, M., Goedert, J. J., Biggar, R. J., and Miller, R. W. (2002). Merkel cell carcinoma and HIV infection. Lancet 359, 497–498. doi: 10.1016/S0140-6736(02)07668-7
Faust, H., Pastrana, D. V., Buck, C. B., Dillner, J., and Ekstrom, J. (2011). Antibodies to Merkel cell polyomavirus correlate to presence of viral DNA in the skin. J. Infect. Dis. 203, 1096–1100. doi: 10.1093/infdis/jiq173
Feldmeyer, L., Hudgens, C. W., Ray-Lyons, G., Nagarajan, P., Aung, P. P., Curry, J. L., et al. (2016). Density, distribution, and composition of immune infiltrates correlate with survival in merkel cell carcinoma. Clin. Cancer Res. 22, 5553–5563. doi: 10.1158/1078-0432.CCR-16-0392
Feng, H., Shuda, M., Chang, Y., and Moore, P. S. (2008). Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319, 1096–1100. doi: 10.1126/science.1152586
Fischer, N., Brandner, J., Fuchs, F., Moll, I., and Grundhoff, A. (2010). Detection of Merkel cell polyomavirus (MCPyV) in Merkel cell carcinoma cell lines: cell morphology and growth phenotype do not reflect presence of the virus. Int. J. Cancer 126, 2133–2142. doi: 10.1002/ijc.24877
Fitzgerald, T. L., Dennis, S., Kachare, S. D., Vohra, N. A., Wong, J. H., and Zervos, E. E. (2015). Dramatic increase in the incidence and mortality from merkel cell carcinoma in the United States. Am. Surg. 81, 802–806. doi: 10.1177/000313481508100819
Foulongne, V., Sauvage, V., Hebert, C., Dereure, O., Cheval, J., Gouilh, M. A., et al. (2012). Human skin microbiota: high diversity of dna viruses identified on the human skin by high throughput sequencing. PLoS ONE 7:e38499. doi: 10.1371/journal.pone.0038499
Freeman, M. B., Holman, D. M., Qin, J., and Lunsford, N. B. (2019). Merkel cell carcinoma incidence, trends, and survival rates among adults aged ≥50 years from United States Cancer Statistics. J. Am. Acad. Dermatol. 80, 1154–1156. doi: 10.1016/j.jaad.2018.10.045
Fukumoto, H., Sato, Y., Hasegawa, H., and Katano, H. (2013). Frequent detection of Merkel cell polyomavirus DNA in sera of HIV-1-positive patients. Virol. J. 10:84. doi: 10.1186/1743-422X-10-84
Gaglia, M. M., and Munger, K. (2018). More than just oncogenes: mechanisms of tumorigenesis by human viruses. Curr. Opin. Virol. 32, 48–59. doi: 10.1016/j.coviro.2018.09.003
Gavvovidis, I., Leisegang, M., Willimsky, G., Miller, N. J., Nghiem, P., and Blankenstein, T. (2018). Targeting Merkel cell carcinoma by engineered T cells specific to T-antigens of Merkel cell polyomavirus. Clin. Cancer Res. 24, 3644–3655. doi: 10.1158/1078-0432.CCR-17-2661
Gerer, K. F., Erdmann, M., Hadrup, S. R., Lyngaa, R., Martin, L. M., Voll, R. E., et al. (2017). Preclinical evaluation of NF-kappaB-triggered dendritic cells expressing the viral oncogenic driver of Merkel cell carcinoma for therapeutic vaccination. Ther. Adv. Med. Oncol. 9, 451–464. doi: 10.1177/1758834017712630
Gill, S. E., and Parks, W. C. (2008). Metalloproteinases and their inhibitors: regulators of wound healing. Int. J. Biochem. Cell Biol. 40, 1334–1347. doi: 10.1016/j.biocel.2007.10.024
Gjoerup, O., and Chang, Y. (2010). “Chapter 1–update on human polyomaviruses and cancer,” in Advances in Cancer Research, eds F.V.W. George and K. George (Cambridge, MA: Academic Press), 1–51.
Goh, G., Walradt, T., Markarov, V., Blom, A., Riaz, N., Doumani, R., et al. (2016). Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 7, 3403–3415. doi: 10.18632/oncotarget.6494
Griffiths, D. A., Abdul-Sada, H., Knight, L. M., Jackson, B. R., Richards, K., Prescott, E. L., et al. (2013). Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J. Virol. 87, 13853–13867. doi: 10.1128/JVI.02159-13
Grundhoff, A., and Fischer, N. (2015). Merkel cell polyomavirus, a highly prevalent virus with tumorigenic potential. Curr. Opin. Virol. 14, 129–137. doi: 10.1016/j.coviro.2015.08.010
Gu, J., Polak, J. M., Van Noorden, S., Pearse, A. G., Marangos, P. J., and Azzopardi, J. G. (1983). Immunostaining of neuron-specific enolase as a diagnostic tool for Merkel cell tumors. Cancer 52, 1039–1043.
Hampras, S. S., Michel, A., Schmitt, M., Waterboer, T., Kranz, L., Gheit, T., et al. (2015). Merkel cell polyomavirus (MCV) T-antigen seroreactivity, MCV DNA in eyebrow hairs, and squamous cell carcinoma. Infect. Agents Cancer 10:35. doi: 10.1186/s13027-015-0030-0
Handschel, J., Muller, D., Depprich, R. A., Ommerborn, M. A., Kubler, N. R., Naujoks, C., et al. (2010). The new polyomavirus (MCPyV) does not affect the clinical course in MCCs. Int. J. Oral Maxillofac. Surg. 39, 1086–1090. doi: 10.1016/j.ijom.2010.06.024
Hardie, I. R., Strong, R. W., Hartley, L. C., Woodruff, P. W., and Clunie, G. J. (1980). Skin cancer in Caucasian renal allograft recipients living in a subtropical climate. Surgery 87, 177–183.
Harms, K. L., Healy, M. A., Nghiem, P., Sober, A. J., Johnson, T. M., Bichakjian, C. K., et al. (2016). Analysis of Prognostic Factors from 9387 Merkel Cell Carcinoma Cases Forms the Basis for the New 8th Edition AJCC Staging System. Ann. Surg. Oncol. 23, 3564–3571. doi: 10.1245/s10434-016-5266-4
Harms, P. W. (2017). Update on Merkel Cell Carcinoma. Clin. Lab. Med. 37, 485–501. doi: 10.1016/j.cll.2017.05.004
Harms, P. W., Collie, A. M., Hovelson, D. H., Cani, A. K., Verhaegen, M. E., Patel, R. M., et al. (2016). Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod. Pathol. 29, 240–248. doi: 10.1038/modpathol.2015.154
Harms, P. W., Patel, R. M., Verhaegen, M. E., Giordano, T. J., Nash, K. T., Johnson, C. N., et al. (2013). Distinct gene expression profiles of viral- and nonviral-associated merkel cell carcinoma revealed by transcriptome analysis. J. Invest. Dermatol. 133, 936–945. doi: 10.1038/jid.2012.445
Harms, P. W., Vats, P., Verhaegen, M. E., Robinson, D. R., Wu, Y. M., Dhanasekaran, S. M., et al. (2015). The distinctive mutational spectra of polyomavirus-negative merkel cell carcinoma. Cancer Res. 75, 3720–3727. doi: 10.1158/0008-5472.CAN-15-0702
Harold, A., Amako, Y., Hachisuka, J., Bai, Y., Li, M. Y., Kubat, L., et al. (2019). Conversion of Sox2-dependent Merkel cell carcinoma to a differentiated neuron-like phenotype by T antigen inhibition. Proc. Natl. Acad. Sci. U.S.A. 116, 20104–20114. doi: 10.1073/pnas.1907154116
Harrison, C. J., Meinke, G., Kwun, H. J., Rogalin, H., Phelan, P. J., Bullock, P. A., et al. (2011). Asymmetric assembly of Merkel cell polyomavirus large T-antigen origin binding domains at the viral origin. J. Mol. Biol. 409, 529–542. doi: 10.1016/j.jmb.2011.03.051
Hashida, Y., Higuchi, T., Tanaka, M., Shibata, Y., Nakajima, K., Sano, S., et al. (2019). Prevalence and viral loads of cutaneous human polyomaviruses in the skin of patients with chronic inflammatory skin diseases. J. Infect. Dis. 219, 1564–1573. doi: 10.1093/infdis/jiy618
Hashida, Y., Kamioka, M., Tanaka, M., Hosokawa, S., Murakami, M., Nakajima, K., et al. (2016a). Ecology of merkel cell polyomavirus in healthy skin among individuals in an asian cohort. J. Infect. Dis. 213, 1708–1716. doi: 10.1093/infdis/jiw040
Hashida, Y., Nakajima, K., Nakajima, H., Shiga, T., Tanaka, M., Murakami, M., et al. (2016b). High load of Merkel cell polyomavirus DNA detected in the normal skin of Japanese patients with Merkel cell carcinoma. J. Clin. Virol. 82, 101–107. doi: 10.1016/j.jcv.2016.07.011
Haugg, A. M., Rennspiess, D., Zur Hausen, A., Speel, E. J., Cathomas, G., Becker, J. C., et al. (2014). Fluorescence in situ hybridization and qPCR to detect Merkel cell polyomavirus physical status and load in Merkel cell carcinomas. Int. J. Cancer 135, 2804–2815. doi: 10.1002/ijc.28931
Heath, M., Jaimes, N., Lemos, B., Mostaghimi, A., Wang, L. C., Penas, P. F., et al. (2008). Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J. Am. Acad. Dermatol. 58, 375–381. doi: 10.1016/j.jaad.2007.11.020
Hesbacher, S., Pfitzer, L., Wiedorfer, K., Angermeyer, S., Borst, A., Haferkamp, S., et al. (2016). RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 7, 32956–32968. doi: 10.18632/oncotarget.8793
Higaki-Mori, H., Kuwamoto, S., Iwasaki, T., Kato, M., Murakami, I., Nagata, K., et al. (2012). Association of Merkel cell polyomavirus infection with clinicopathological differences in Merkel cell carcinoma. Hum. Pathol. 43, 2282–2291. doi: 10.1016/j.humpath.2012.04.002
Houben, R., Adam, C., Baeurle, A., Hesbacher, S., Grimm, J., Angermeyer, S., et al. (2012). An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int. J. Cancer 130, 847–856. doi: 10.1002/ijc.26076
Houben, R., Shuda, M., Weinkam, R., Schrama, D., Feng, H., Chang, Y., et al. (2010). Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 84, 7064–7072. doi: 10.1128/JVI.02400-09
Iwasaki, T., Matsushita, M., Kuwamoto, S., Kato, M., Murakami, I., Higaki-Mori, H., et al. (2013). Usefulness of significant morphologic characteristics in distinguishing between Merkel cell polyomavirus-positive and Merkel cell polyomavirus-negative Merkel cell carcinomas. Hum. Pathol. 44, 1912–1917. doi: 10.1016/j.humpath.2013.01.026
Iwasaki, T., Matsushita, M., Nonaka, D., Nagata, K., Kato, M., Kuwamoto, S., et al. (2016). Lower expression of CADM1 and higher expression of MAL in Merkel cell carcinomas are associated with Merkel cell polyomavirus infection and better prognosis. Hum. Pathol. 48, 1–8. doi: 10.1016/j.humpath.2015.09.030
Iyer, J. G., Blom, A., Doumani, R., Lewis, C., Tarabadkar, E. S., Anderson, A., et al. (2016). Response rates and durability of chemotherapy among 62 patients with metastatic Merkel cell carcinoma. Cancer Med. 5, 2294–2301. doi: 10.1002/cam4.815
Jacobs, D., Huang, H., Olino, K., Weiss, S., Kluger, H., Judson, B. L., et al. (2020). Assessment of age, period, and birth cohort effects and trends in merkel cell carcinoma incidence in the United States. JAMA Dermatol. 157, 59–65. doi: 10.1001/jamadermatol.2020.4102
Jouhi, L., Koljonen, V., Bohling, T., Haglund, C., and Hagstrom, J. (2015). The expression of Toll-like receptors 2, 4, 5, 7 and 9 in Merkel cell carcinoma. Anticancer Res. 35, 1843–1849.
Kaufman, H. L., Russell, J., Hamid, O., Bhatia, S., Terheyden, P., D'angelo, S. P., et al. (2016). Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 17, 1374–1385. doi: 10.1016/S1470-2045(16)30364-3
Kaufman, H. L., Russell, J. S., Hamid, O., Bhatia, S., Terheyden, P., D'angelo, S. P., et al. (2018). Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after >/=1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J. Immunother. Cancer 6:7. doi: 10.1186/s40425-017-0310-x
Kean, J. M., Rao, S., Wang, M., and Garcea, R. L. (2009). Seroepidemiology of human polyomaviruses. PLoS Pathog. 5:e1000363. doi: 10.1371/journal.ppat.1000363
Kervarrec, T., Aljundi, M., Appenzeller, S., Samimi, M., Maubec, E., Cribier, B., et al. (2019). Polyomavirus-positive merkel cell carcinoma derived from a trichoblastoma suggests an epithelial origin of this merkel cell carcinoma. J. Invest. Dermatol. 140, 976–985. doi: 10.1016/j.jid.2019.09.026
Kervarrec, T., Samimi, M., Hesbacher, S., Berthon, P., Wobser, M., Sallot, A., et al. (2020). Merkel Cell Polyomavirus T Antigens Induce Merkel Cell-Like Differentiation in GLI1-Expressing Epithelial Cells. Cancers (Basel) 12:1989. doi: 10.3390/cancers12071989
Knight, L. M., Stakaityte, G., Wood, J. J., Abdul-Sada, H., Griffiths, D. A., Howell, G. J., et al. (2015). Merkel cell polyomavirus small T antigen mediates microtubule destabilization to promote cell motility and migration. J. Virol. 89, 35–47. doi: 10.1128/JVI.02317-14
Koljonen, V., Kukko, H., Pukkala, E., Sankila, R., Bohling, T., Tukiainen, E., et al. (2009). Chronic lymphocytic leukaemia patients have a high risk of Merkel-cell polyomavirus DNA-positive Merkel-cell carcinoma. Br. J. Cancer 101, 1444–1447. doi: 10.1038/sj.bjc.6605306
Krump, N. A., Wang, R., Liu, W., Yang, J. F., Ma, T., and You, J. (2021). Merkel cell polyomavirus infection induces an antiviral innate immune response in human dermal fibroblasts. J. Virol. 95:e0221120. doi: 10.1128/JVI.02211-20
Kuwamoto, S., Higaki, H., Kanai, K., Iwasaki, T., Sano, H., Nagata, K., et al. (2011). Association of Merkel cell polyomavirus infection with morphologic differences in Merkel cell carcinoma. Hum. Pathol. 42, 632–640. doi: 10.1016/j.humpath.2010.09.011
Kwun, H. J., Guastafierro, A., Shuda, M., Meinke, G., Bohm, A., Moore, P. S., et al. (2009). The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J. Virol. 83, 12118–12128. doi: 10.1128/JVI.01336-09
Kwun, H. J., Shuda, M., Camacho, C. J., Gamper, A. M., Thant, M., Chang, Y., et al. (2015). Restricted protein phosphatase 2A targeting by Merkel cell polyomavirus small T antigen. J. Virol. 89, 4191–4200. doi: 10.1128/JVI.00157-15
Kwun, H. J., Shuda, M., Feng, H., Camacho, C. J., Moore, P. S., and Chang, Y. (2013). Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 14, 125–135. doi: 10.1016/j.chom.2013.06.008
Lau, L., Gray, E. E., Brunette, R. L., and Stetson, D. B. (2015). DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350:568. doi: 10.1126/science.aab3291
Laude, H. C., Jonchere, B., Maubec, E., Carlotti, A., Marinho, E., Couturaud, B., et al. (2010). Distinct merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog. 6:e1001076. doi: 10.1371/journal.ppat.1001076
Lauttia, S., Sihto, H., Kavola, H., Koljonen, V., Bohling, T., and Joensuu, H. (2014). Prokineticins and Merkel cell polyomavirus infection in Merkel cell carcinoma. Br. J. Cancer 110, 1446–1455. doi: 10.1038/bjc.2014.20
Leiendecker, L., Jung, P. S., Krecioch, I., Neumann, T., Schleiffer, A., Mechtler, K., et al. (2020). LSD1 inhibition induces differentiation and cell death in Merkel cell carcinoma. EMBO Mol. Med. 12:e12525. doi: 10.15252/emmm.202012525
Leroux-Kozal, V., Leveque, N., Brodard, V., Lesage, C., Dudez, O., Makeieff, M., et al. (2015). Merkel cell carcinoma: histopathologic and prognostic features according to the immunohistochemical expression of Merkel cell polyomavirus large T antigen correlated with viral load. Hum. Pathol. 46, 443–453. doi: 10.1016/j.humpath.2014.12.001
Li, J., Diaz, J., Wang, X., Tsang, S. H., and You, J. (2015). Phosphorylation of Merkel cell polyomavirus large tumor antigen at serine 816 by ATM kinase induces apoptosis in host cells. J. Biol. Chem. 290, 1874–1884. doi: 10.1074/jbc.M114.594895
Li, J., Wang, X., Diaz, J., Tsang, S. H., Buck, C. B., and You, J. (2013). Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol. 87, 9173–9188. doi: 10.1128/JVI.01216-13
Lilyestrom, W., Klein, M. G., Zhang, R., Joachimiak, A., and Chen, X. S. (2006). Crystal structure of SV40 large T-antigen bound to p53: interplay between a viral oncoprotein and a cellular tumor suppressor. Genes Dev. 20, 2373–2382. doi: 10.1101/gad.1456306
Lipson, E. J., Vincent, J. G., Loyo, M., Kagohara, L. T., Luber, B. S., Wang, H., et al. (2013). PD-L1 expression in the Merkel cell carcinoma microenvironment: association with inflammation, Merkel cell polyomavirus and overall survival. Cancer Immunol Res 1, 54–63. doi: 10.1158/2326-6066.CIR-13-0034
Liu, W., Kim, G. B., Krump, N. A., Zhou, Y., Riley, J. L., and You, J. (2020). Selective reactivation of STING signaling to target Merkel cell carcinoma. Proc. Natl. Acad. Sci. U.S.A. 117, 13730–13739. doi: 10.1073/pnas.1919690117
Liu, W., Krump, N. A., Macdonald, M., and You, J. (2018). Merkel cell polyomavirus infection of animal dermal fibroblasts. J. Virol. 92, e01610–e01617. doi: 10.1128/JVI.01610-17
Liu, W., Macdonald, M., and You, J. (2016a). Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr. Opin. Virol. 20, 20–27. doi: 10.1016/j.coviro.2016.07.011
Liu, W., Yang, R., Payne, A. S., Schowalter, R. M., Spurgeon, M. E., Lambert, P. F., et al. (2016b). Identifying the target cells and mechanisms of merkel cell polyomavirus infection. Cell Host Microbe 19, 775–787. doi: 10.1016/j.chom.2016.04.024
Longino, N. V., Yang, J., Iyer, J. G., Ibrani, D., Chow, I. T., Laing, K. J., et al. (2019). Human CD4(+) T cells specific for merkel cell polyomavirus localize to merkel cell carcinomas and target a required oncogenic domain. Cancer Immunol. Res. 7, 1727–1739. doi: 10.1158/2326-6066.CIR-19-0103
Longworth, M. S., and Laimins, L. A. (2004). Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 68, 362–372. doi: 10.1128/MMBR.68.2.362-372.2004
Ma, J. E., and Brewer, J. D. (2014). Merkel cell carcinoma in immunosuppressed patients. Cancers (Basel) 6, 1328–1350. doi: 10.3390/cancers6031328
Mantripragada, K., and Birnbaum, A. (2015). Response to anti-PD-1 therapy in metastatic merkel cell carcinoma metastatic to the heart and pancreas. Cureus 7:e403. doi: 10.7759/cureus.403
Martel-Jantin, C., Pedergnana, V., Nicol, J. T., Leblond, V., Tregouet, D. A., Tortevoye, P., et al. (2013). Merkel cell polyomavirus infection occurs during early childhood and is transmitted between siblings. J. Clin. Virol. 58, 288–291. doi: 10.1016/j.jcv.2013.06.004
Matsushita, M., Iwasaki, T., Nonaka, D., Kuwamoto, S., Nagata, K., Kato, M., et al. (2017). Higher expression of activation-induced cytidine deaminase is significantly associated with merkel cell polyomavirus-negative merkel cell carcinomas. Yonago Acta Med. 60, 145–153. doi: 10.33160/yam.2017.09.002
Matsushita, M., Iwasaki, T., Wardhani, L. O., Kuwamoto, S., Nonaka, D., Nagata, K., et al. (2019). Decreased H3K27me3 expression is associated with merkel cell polyomavirus-negative merkel cell carcinoma, especially combined with cutaneous squamous cell carcinoma. Anticancer Res. 39, 5573–5579. doi: 10.21873/anticanres.13751
Mertz, K. D., Junt, T., Schmid, M., Pfaltz, M., and Kempf, W. (2010). Inflammatory monocytes are a reservoir for Merkel cell polyomavirus. J. Invest. Dermatol. 130, 1146–1151. doi: 10.1038/jid.2009.392
Miller, N. J., Church, C. D., Dong, L., Crispin, D., Fitzgibbon, M. P., Lachance, K., et al. (2017). Tumor-infiltrating merkel cell polyomavirus-specific T cells are diverse and associated with improved patient survival. Cancer Immunol. Res. 5, 137–147. doi: 10.1158/2326-6066.CIR-16-0210
Miller, N. J., Church, C. D., Fling, S. P., Kulikauskas, R., Ramchurren, N., Shinohara, M. M., et al. (2018). Merkel cell polyomavirus-specific immune responses in patients with Merkel cell carcinoma receiving anti-PD-1 therapy. J. Immunother. Cancer 6:131. doi: 10.1186/s40425-018-0450-7
Mitteldorf, C., Berisha, A., Tronnier, M., Pfaltz, M. C., and Kempf, W. (2017). PD-1 and PD-L1 in neoplastic cells and the tumor microenvironment of Merkel cell carcinoma. J. Cutan. Pathol. 44, 740–746. doi: 10.1111/cup.12973
Moll, R., Lowe, A., Laufer, J., and Franke, W. W. (1992). Cytokeratin 20 in human carcinomas. a new histodiagnostic marker detected by monoclonal antibodies. Am. J. Pathol. 140, 427–447.
Moshiri, A. S., Doumani, R., Yelistratova, L., Blom, A., Lachance, K., Shinohara, M. M., et al. (2017). Polyomavirus-negative merkel cell carcinoma: a more aggressive subtype based on analysis of 282 cases using multimodal tumor virus detection. J. Invest. Dermatol. 137, 819–827. doi: 10.1016/j.jid.2016.10.028
Nakamura, T., Sato, Y., Watanabe, D., Ito, H., Shimonohara, N., Tsuji, T., et al. (2010). Nuclear localization of Merkel cell polyomavirus large T antigen in Merkel cell carcinoma. Virology 398, 273–279. doi: 10.1016/j.virol.2009.12.024
Nardi, V., Song, Y., Santamaria-Barria, J. A., Cosper, A. K., Lam, Q., Faber, A. C., et al. (2012). Activation of PI3K signaling in Merkel cell carcinoma. Clin. Cancer Res. 18, 1227–1236. doi: 10.1158/1078-0432.CCR-11-2308
Narisawa, Y., Inoue, T., and Nagase, K. (2019). Evidence of proliferative activity in human Merkel cells: implications in the histogenesis of Merkel cell carcinoma. Arch. Dermatol. Res. 311, 37–43. doi: 10.1007/s00403-018-1877-x
Narisawa, Y., Inoue, T., and Nagase, K. (2020). Dermal and intraepidermal merkel cell carcinoma with squamous cell carcinoma: a report of a rare case with special reference to the touch dome. Am. J. Dermatopathol. 43, 15–20. doi: 10.1097/DAD.0000000000001618
Navarrete-Dechent, C., Cordova, M., Aleissa, S., Battle, L. R., Ganly, I., Pulitzer, M., et al. (2020). Dermoscopy and reflectance confocal microscopy of intraepidermal Merkel cell carcinoma. Australas J. Dermatol. 62, 238–241. doi: 10.1111/ajd.13513
Neu, U., Hengel, H., Blaum, B. S., Schowalter, R. M., Macejak, D., Gilbert, M., et al. (2012). Structures of Merkel cell polyomavirus VP1 complexes define a sialic acid binding site required for infection. PLoS Pathog. 8:e1002738. doi: 10.1371/journal.ppat.1002738
Neumann, F., Czech-Sioli, M., Dobner, T., Grundhoff, A., Schreiner, S., and Fischer, N. (2016). Replication of Merkel cell polyomavirus induces reorganization of promyelocytic leukemia nuclear bodies. J. Gen. Virol. 97, 2926–2938. doi: 10.1099/jgv.0.000593
Nghiem, P., Bhatia, S., Lipson, E. J., Sharfman, W. H., Kudchadkar, R. R., Brohl, A. S., et al. (2019). Durable tumor regression and overall survival in patients with advanced merkel cell carcinoma receiving pembrolizumab as first-line therapy. J. Clin. Oncol. 37, 693–702. doi: 10.1200/JCO.18.01896
Nguyen, M. H. K., Leong, S. P., Abendroth, R., Kashani-Sabet, M., and Kim, K. B. (2019). Complete clinical response to intralesional talimogene laherparepvec injection in a patient with recurrent, regionally advanced Merkel cell carcinoma. JAAD Case Rep. 5, 849–851. doi: 10.1016/j.jdcr.2019.07.006
Nwogu, N., Boyne, J. R., Dobson, S. J., Poterlowicz, K., Blair, G. E., Macdonald, A., et al. (2018). Cellular sheddases are induced by Merkel cell polyomavirus small tumour antigen to mediate cell dissociation and invasiveness. PLoS Pathog. 14:e1007276. doi: 10.1371/journal.ppat.1007276
Paik, J. Y., Hall, G., Clarkson, A., Lee, L., Toon, C., Colebatch, A., et al. (2011). Immunohistochemistry for Merkel cell polyomavirus is highly specific but not sensitive for the diagnosis of Merkel cell carcinoma in the Australian population. Hum. Pathol. 42, 1385–1390. doi: 10.1016/j.humpath.2010.12.013
Pancaldi, C., Corazzari, V., Maniero, S., Mazzoni, E., Comar, M., Martini, F., et al. (2011). Merkel cell polyomavirus DNA sequences in the buffy coats of healthy blood donors. Blood 117, 7099–7101. doi: 10.1182/blood-2010-09-310557
Park, D. E., Cheng, J., Berrios, C., Montero, J., Cortes-Cros, M., Ferretti, S., et al. (2019). Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. U.S.A. 116, 1027–1032. doi: 10.1073/pnas.1818798116
Park, D. E., Cheng, J., Mcgrath, J. P., Lim, M. Y., Cushman, C., Swanson, S. K., et al. (2020). Merkel cell polyomavirus activates LSD1-mediated blockade of non-canonical BAF to regulate transformation and tumorigenesis. Nat. Cell Biol. 22, 603–615. doi: 10.1038/s41556-020-0503-2
Pasternak, S., Carter, M. D., Ly, T. Y., Doucette, S., and Walsh, N. M. (2018). Immunohistochemical profiles of different subsets of Merkel cell carcinoma. Hum. Pathol. 82, 232–238. doi: 10.1016/j.humpath.2018.07.022
Pastrana, D. V., Tolstov, Y. L., Becker, J. C., Moore, P. S., Chang, Y., and Buck, C. B. (2009). Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog. 5:e1000578. doi: 10.1371/journal.ppat.1000578
Pastrana, D. V., Wieland, U., Silling, S., Buck, C. B., and Pfister, H. (2012). Positive correlation between Merkel cell polyomavirus viral load and capsid-specific antibody titer. Med. Microbiol. Immunol. 201, 17–23. doi: 10.1007/s00430-011-0200-7
Paulson, K. G., Iyer, J. G., Blom, A., Warton, E. M., Sokil, M., Yelistratova, L., et al. (2013). Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J. Invest. Dermatol. 133, 642–646. doi: 10.1038/jid.2012.388
Paulson, K. G., Lewis, C. W., Redman, M. W., Simonson, W. T., Lisberg, A., Ritter, D., et al. (2017). Viral oncoprotein antibodies as a marker for recurrence of Merkel cell carcinoma: a prospective validation study. Cancer 123, 1464–1474. doi: 10.1002/cncr.30475
Paulson, K. G., Park, S. Y., Vandeven, N. A., Lachance, K., Thomas, H., Chapuis, A. G., et al. (2018a). Merkel cell carcinoma: current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 78, 457–463.e2. doi: 10.1016/j.jaad.2017.10.028
Paulson, K. G., Voillet, V., Mcafee, M. S., Hunter, D. S., Wagener, F. D., Perdicchio, M., et al. (2018b). Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun. 9:3868. doi: 10.1038/s41467-018-06300-3
Perdigoto, C. N., Dauber, K. L., Bar, C., Tsai, P. C., Valdes, V. J., Cohen, I., et al. (2016). Polycomb-mediated repression and sonic hedgehog signaling interact to regulate merkel cell specification during skin development. PLoS Genet. 12:e1006151. doi: 10.1371/journal.pgen.1006151
Peretti, A., Borgogna, C., Rossi, D., De Paoli, L., Bawadekar, M., Zavattaro, E., et al. (2014). Analysis of human beta-papillomavirus and Merkel cell polyomavirus infection in skin lesions and eyebrow hair bulbs from a cohort of patients with chronic lymphocytic leukaemia. Br. J. Dermatol. 171, 1525–1528. doi: 10.1111/bjd.13215
Ponce, R. A., Gelzleichter, T., Haggerty, H. G., Heidel, S., Holdren, M. S., Lebrec, H., et al. (2014). Immunomodulation and lymphoma in humans. J. Immunotoxicol. 11, 1–12. doi: 10.3109/1547691X.2013.798388
Pugh, R. A., Honda, M., Leesley, H., Thomas, A., Lin, Y., Nilges, M. J., et al. (2008). The iron-containing domain is essential in Rad3 helicases for coupling of ATP hydrolysis to DNA translocation and for targeting the helicase to the single-stranded DNA-double-stranded DNA junction. J. Biol. Chem. 283, 1732–1743. doi: 10.1074/jbc.M707064200
Richards, K. F., Guastafierro, A., Shuda, M., Toptan, T., Moore, P. S., and Chang, Y. (2015). Merkel cell polyomavirus T antigens promote cell proliferation and inflammatory cytokine gene expression. J. Gen. Virol. 96, 3532–3544. doi: 10.1099/jgv.0.000287
Sahi, H., Savola, S., Sihto, H., Koljonen, V., Bohling, T., and Knuutila, S. (2014). RB1 gene in Merkel cell carcinoma: hypermethylation in all tumors and concurrent heterozygous deletions in the polyomavirus-negative subgroup. APMIS 122, 1157–1166. doi: 10.1111/apm.12274
Sahi, H., Sihto, H., Artama, M., Koljonen, V., Bohling, T., and Pukkala, E. (2017). History of chronic inflammatory disorders increases the risk of Merkel cell carcinoma, but does not correlate with Merkel cell polyomavirus infection. Br. J. Cancer 116, 260–264. doi: 10.1038/bjc.2016.391
Salakova, M., Koslabova, E., Vojtechova, Z., Tachezy, R., and Sroller, V. (2016). Detection of human polyomaviruses MCPyV, HPyV6, and HPyV7 in malignant and non-malignant tonsillar tissues. J. Med. Virol. 88, 695–702. doi: 10.1002/jmv.24385
Samimi, M., Molet, L., Fleury, M., Laude, H., Carlotti, A., Gardair, C., et al. (2016). Prognostic value of antibodies to Merkel cell polyomavirus T antigens and VP1 protein in patients with Merkel cell carcinoma. Br. J. Dermatol. 174, 813–822. doi: 10.1111/bjd.14313
Santos-Juanes, J., Fernandez-Vega, I., Fuentes, N., Galache, C., Coto-Segura, P., Vivanco, B., et al. (2015). Merkel cell carcinoma and Merkel cell polyomavirus: a systematic review and meta-analysis. Br. J. Dermatol. 173, 42–49. doi: 10.1111/bjd.13870
Sarma, B., Willmes, C., Angerer, L., Adam, C., Becker, J. C., Kervarrec, T., et al. (2020). Artesunate affects T antigen expression and survival of virus-positive merkel cell carcinoma. Cancers (Basel) 12:919. doi: 10.3390/cancers12040919
Sauer, C. M., Haugg, A. M., Chteinberg, E., Rennspiess, D., Winnepenninckx, V., Speel, E. J., et al. (2017). Reviewing the current evidence supporting early B-cells as the cellular origin of Merkel cell carcinoma. Crit. Rev. Oncol. Hematol. 116, 99–105. doi: 10.1016/j.critrevonc.2017.05.009
Schadendorf, D., Lebbe, C., Zur Hausen, A., Avril, M. F., Hariharan, S., Bharmal, M., et al. (2017). Merkel cell carcinoma: epidemiology, prognosis, therapy and unmet medical needs. Eur. J. Cancer 71, 53–69. doi: 10.1016/j.ejca.2016.10.022
Schowalter, R. M., and Buck, C. B. (2013). The Merkel cell polyomavirus minor capsid protein. PLoS Pathog. 9:e1003558. doi: 10.1371/journal.ppat.1003558
Schowalter, R. M., Pastrana, D. V., and Buck, C. B. (2011). Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS Pathog. 7:e1002161. doi: 10.1371/journal.ppat.1002161
Schowalter, R. M., Pastrana, D. V., Pumphrey, K. A., Moyer, A. L., and Buck, C. B. (2010). Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe 7, 509–515. doi: 10.1016/j.chom.2010.05.006
Schrama, D., Hesbacher, S., Angermeyer, S., Schlosser, A., Haferkamp, S., Aue, A., et al. (2016). Serine 220 phosphorylation of the Merkel cell polyomavirus large T antigen crucially supports growth of Merkel cell carcinoma cells. Int. J. Cancer 138, 1153–1162. doi: 10.1002/ijc.29862
Schrama, D., Peitsch, W. K., Zapatka, M., Kneitz, H., Houben, R., Eib, S., et al. (2011). Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J. Invest. Dermatol. 131, 1631–1638. doi: 10.1038/jid.2011.115
Seo, G. J., Chen, C. J., and Sullivan, C. S. (2009). Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology 383, 183–187. doi: 10.1016/j.virol.2008.11.001
Shahzad, N., Shuda, M., Gheit, T., Kwun, H. J., Cornet, I., Saidj, D., et al. (2013). The T antigen locus of Merkel cell polyomavirus downregulates human Toll-like receptor 9 expression. J. Virol. 87, 13009–13019. doi: 10.1128/JVI.01786-13
Shuda, M., Chang, Y., and Moore, P. S. (2014). Merkel cell polyomavirus-positive Merkel cell carcinoma requires viral small T-antigen for cell proliferation. J. Invest. Dermatol. 134, 1479–1481. doi: 10.1038/jid.2013.483
Shuda, M., Feng, H., Kwun, H. J., Rosen, S. T., Gjoerup, O., Moore, P. S., et al. (2008). T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. U.S.A. 105, 16272–16277. doi: 10.1073/pnas.0806526105
Shuda, M., Kwun, H. J., Feng, H., Chang, Y., and Moore, P. S. (2011). Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Invest. 121, 3623–3634. doi: 10.1172/JCI46323
Signorini, L., Belingheri, M., Ambrogi, F., Pagani, E., Binda, S., Ticozzi, R., et al. (2014). High frequency of Merkel cell polyomavirus DNA in the urine of kidney transplant recipients and healthy controls. J. Clin. Virol. 61, 565–570. doi: 10.1016/j.jcv.2014.10.012
Sihto, H., Bohling, T., Kavola, H., Koljonen, V., Salmi, M., Jalkanen, S., et al. (2012). Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin. Cancer Res. 18, 2872–2881. doi: 10.1158/1078-0432.CCR-11-3020
Sihto, H., Kukko, H., Koljonen, V., Sankila, R., Bohling, T., and Joensuu, H. (2009). Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J. Natl. Cancer Inst. 101, 938–945. doi: 10.1093/jnci/djp139
Sihto, H., Kukko, H., Koljonen, V., Sankila, R., Bohling, T., and Joensuu, H. (2011). Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin. Cancer Res. 17, 4806–4813. doi: 10.1158/1078-0432.CCR-10-3363
Song, Y., Zheng, S., and Guo, Y. (2020). Intraepidermal merkel cell carcinoma mimicking melanoma in situ: a case report. Am. J. Dermatopathol. 42, 977–980. doi: 10.1097/DAD.0000000000001793
Spurgeon, M. E., and Lambert, P. F. (2013). Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology 435, 118–130. doi: 10.1016/j.virol.2012.09.029
Stakaityte, G., Nwogu, N., Dobson, S. J., Knight, L. M., Wasson, C. W., Salguero, F. J., et al. (2018). Merkel cell polyomavirus small T antigen drives cell motility via Rho-GTPase-induced filopodium formation. J. Virol. 92:e00940-17. doi: 10.1128/JVI.00940-17
Stang, A., Becker, J. C., Nghiem, P., and Ferlay, J. (2018). The association between geographic location and incidence of Merkel cell carcinoma in comparison to melanoma: an international assessment. Eur. J. Cancer 94, 47–60. doi: 10.1016/j.ejca.2018.02.003
Starrett, G. J., Marcelus, C., Cantalupo, P. G., Katz, J. P., Cheng, J., Akagi, K., et al. (2017). Merkel cell polyomavirus exhibits dominant control of the tumor genome and transcriptome in virus-associated merkel cell carcinoma. MBio 8:e02079-16. doi: 10.1128/mBio.02079-16
Starrett, G. J., Thakuria, M., Chen, T., Marcelus, C., Cheng, J., Nomburg, J., et al. (2020). Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 12:30. doi: 10.1186/s13073-020-00727-4
Sunshine, J. C., Jahchan, N. S., Sage, J., and Choi, J. (2018). Are there multiple cells of origin of Merkel cell carcinoma? Oncogene 37, 1409–1416. doi: 10.1038/s41388-017-0073-3
Theiss, J. M., Gunther, T., Alawi, M., Neumann, F., Tessmer, U., Fischer, N., et al. (2015). A Comprehensive analysis of replicating merkel cell polyomavirus genomes delineates the viral transcription program and suggests a role for mcv-miR-M1 in episomal persistence. PLoS Pathog. 11:e1004974. doi: 10.1371/journal.ppat.1004974
Toker, C. (1972). Trabecular carcinoma of the skin. Arch. Dermatol. 105, 107–110. doi: 10.1001/archderm.1972.01620040075020
Tolstov, Y. L., Pastrana, D. V., Feng, H., Becker, J. C., Jenkins, F. J., Moschos, S., et al. (2009). Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int. J. Cancer 125, 1250–1256. doi: 10.1002/ijc.24509
Tolstov, Y. L., Knauer, A., Chen, J. G., Kensler, T. W., Kingsley, L. A., Moore, P. S., et al. (2011). Asymptomatic primary Merkel cell polyomavirus infection among adults. Emerging Infect. Dis. 17, 1371–1380. doi: 10.3201/eid1708.110079
Touze, A., Le Bidre, E., Laude, H., Fleury, M. J., Cazal, R., Arnold, F., et al. (2011). High levels of antibodies against merkel cell polyomavirus identify a subset of patients with merkel cell carcinoma with better clinical outcome. J. Clin. Oncol. 29, 1612–1619. doi: 10.1200/JCO.2010.31.1704
Tsang, S. H., Wang, R., Nakamaru-Ogiso, E., Knight, S. A., Buck, C. B., and You, J. (2015). The oncogenic small tumor antigen of merkel cell polyomavirus is an iron-sulfur cluster protein that enhances viral DNA replication. J. Virol. 90, 1544–1556. doi: 10.1128/JVI.02121-15
Tsang, S. H., Wang, X., Li, J., Buck, C. B., and You, J. (2014). Host DNA damage response factors localize to merkel cell polyomavirus DNA replication sites to support efficient viral DNA replication. J. Virol. 88, 3285–3297. doi: 10.1128/JVI.03656-13
Vahabpour, R., Nasimi, M., Naderi, N., Salehi-Vaziri, M., Mohajel, N., Sadeghi, F., et al. (2017). Merkel cell polyomavirus IgG antibody levels are associated with progression to AIDS among HIV-infected individuals. Arch. Virol. 162, 963–969. doi: 10.1007/s00705-016-3186-z
Velasquez, C., Cheng, E., Shuda, M., Lee-Oesterreich, P. J., Pogge Von Strandmann, L., Gritsenko, M. A., et al. (2016). Mitotic protein kinase CDK1 phosphorylation of mRNA translation regulator 4E-BP1 Ser83 may contribute to cell transformation. Proc. Natl. Acad. Sci. U.S.A. 113, 8466–8471. doi: 10.1073/pnas.1607768113
Verhaegen, M. E., Mangelberger, D., Harms, P. W., Eberl, M., Wilbert, D. M., Meireles, J., et al. (2017). Merkel cell polyomavirus small T antigen initiates merkel cell carcinoma-like tumor development in mice. Cancer Res. 77, 3151–3157. doi: 10.1158/0008-5472.CAN-17-0035
Verhaegen, M. E., Mangelberger, D., Harms, P. W., Vozheiko, T. D., Weick, J. W., Wilbert, D. M., et al. (2014). Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J. Invest. Dermatol. 135, 1415–1424. doi: 10.1038/jid.2014.446
Verhaegen, M. E., Mangelberger, D., Harms, P. W., Vozheiko, T. D., Weick, J. W., Wilbert, D. M., et al. (2015). Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J. Invest. Dermatol. 135, 1415–1424.
Viscidi, R. P., Rollison, D. E., Sondak, V. K., Silver, B., Messina, J. L., Giuliano, A. R., et al. (2011). Age-specific seroprevalence of Merkel cell polyomavirus, BK virus, and JC virus. Clin. Vaccine Immunol. 18, 1737–1743. doi: 10.1128/CVI.05175-11
Von Der Grun, J., Winkelmann, R., Meissner, M., Wieland, U., Silling, S., Martin, D., et al. (2019). Merkel cell polyoma viral load and intratumoral cd8+ lymphocyte infiltration predict overall survival in patients with merkel cell carcinoma. Front. Oncol. 9:20. doi: 10.3389/fonc.2019.00020
Walder, B. K., Robertson, M. R., and Jeremy, D. (1971). Skin cancer and immunosuppression. Lancet 2, 1282–1283. doi: 10.1016/S0140-6736(71)90602-7
Walsh, N. M., Fleming, K. E., Hanly, J. G., Dakin Hache, K., Doucette, S., Ferrara, G., et al. (2016). A morphological and immunophenotypic map of the immune response in Merkel cell carcinoma. Hum. Pathol. 52, 190–196. doi: 10.1016/j.humpath.2016.02.002
Wang, L., Harms, P. W., Palanisamy, N., Carskadon, S., Cao, X., Siddiqui, J., et al. (2017). Age and Gender associations of virus positivity in merkel cell carcinoma characterized using a novel RNA in situ hybridization assay. Clin. Cancer Res. 23, 5622–5630. doi: 10.1158/1078-0432.CCR-17-0299
Wang, X., Helfer, C. M., Pancholi, N., Bradner, J. E., and You, J. (2013). Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J. Virol. 87, 3871–3884. doi: 10.1128/JVI.03068-12
Wang, X., Li, J., Schowalter, R. M., Jiao, J., Buck, C. B., and You, J. (2012). Bromodomain protein Brd4 plays a key role in Merkel cell polyomavirus DNA replication. PLoS Pathog. 8:e1003021. doi: 10.1371/journal.ppat.1003021
Wang, Y., Strassl, R., Helantera, I., Aberle, S. W., Bond, G., Hedman, K., et al. (2019). Multiplex analysis of Human Polyomavirus diversity in kidney transplant recipients with BK virus replication. J. Clin. Virol. 120, 6–11. doi: 10.1016/j.jcv.2019.08.012
Weber, R., Sabin, C. A., Friis-Moller, N., Reiss, P., El-Sadr, W. M., Kirk, O., et al. (2006). Liver-related deaths in persons infected with the human immunodeficiency virus: the D:A:D study. Arch. Intern. Med. 166, 1632–1641. doi: 10.1001/archinte.166.15.1632
Wendzicki, J. A., Moore, P. S., and Chang, Y. (2015). Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol. 11, 38–43. doi: 10.1016/j.coviro.2015.01.009
Werner, S., and Grose, R. (2003). Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 83, 835–870. doi: 10.1152/physrev.2003.83.3.835
Westbrook, B. C., Norwood, T. G., Terry, N. L. J., Mckee, S. B., and Conry, R. M. (2019). Talimogene laherparepvec induces durable response of regionally advanced Merkel cell carcinoma in 4 consecutive patients. JAAD Case Rep. 5, 782–786. doi: 10.1016/j.jdcr.2019.06.034
Wheat, R., Roberts, C., Waterboer, T., Steele, J., Marsden, J., Steven, N. M., et al. (2014). Inflammatory cell distribution in primary merkel cell carcinoma. Cancers (Basel) 6, 1047–1064. doi: 10.3390/cancers6021047
Whyte, J. L., Smith, A. A., and Helms, J. A. (2012). Wnt signaling and injury repair. Cold Spring Harb. Perspect. Biol. 4:a008078. doi: 10.1101/cshperspect.a008078
Wieland, U., Silling, S., Scola, N., Potthoff, A., Gambichler, T., Brockmeyer, N. H., et al. (2011). Merkel cell polyomavirus infection in HIV-positive men. Arch. Dermatol. 147, 401–406. doi: 10.1001/archdermatol.2011.42
Wong, S. Q., Waldeck, K., Vergara, I. A., Schroder, J., Madore, J., Wilmott, J. S., et al. (2015). UV-associated mutations underlie the etiology of MCV-negative merkel cell carcinomas. Cancer Res. 75, 5228–5234. doi: 10.1158/0008-5472.CAN-15-1877
Wu, J. H., Narayanan, D., Limmer, A. L., Simonette, R. A., Rady, P. L., and Tyring, S. K. (2019). Merkel cell polyomavirus small t antigen induces DNA damage response. Intervirology 62, 96–100. doi: 10.1159/000501419
Wu, J. H., Simonette, R. A., Hsiao, T., Doan, H. Q., Rady, P. L., and Tyring, S. K. (2015). Cutaneous human polyomavirus small T antigens and 4E-BP1 targeting. Intervirology 58, 382–385. doi: 10.1159/000444921
Wu, J. H., Simonette, R. A., Nguyen, H. P., Rady, P. L., and Tyring, S. K. (2016). Merkel cell polyomavirus in Merkel cell carcinogenesis: small T antigen-mediates c-Jun phosphorylation. Virus Genes 52, 397–399. doi: 10.1007/s11262-016-1304-3
Wu, Y., and Brosh, R. M. Jr. (2012). DNA helicase and helicase-nuclease enzymes with a conserved iron-sulfur cluster. Nucleic Acids Res. 40, 4247–4260. doi: 10.1093/nar/gks039
Xie, H., Lee, L., Caramuta, S., Hoog, A., Browaldh, N., Bjornhagen, V., et al. (2014). MicroRNA expression patterns related to merkel cell polyomavirus infection in human merkel cell carcinoma. J. Invest. Dermatol. 134, 507–517. doi: 10.1038/jid.2013.355
Yang, R., Lee, E. E., Kim, J., Choi, J. H., Kolitz, E., Chen, Y., et al. (2021). Characterization of ALTO-encoding circular RNAs expressed by Merkel cell polyomavirus and trichodysplasia spinulosa polyomavirus. PLoS Pathog. 17:e1009582. doi: 10.1371/journal.ppat.1009582
You, J. (2010). Papillomavirus interaction with cellular chromatin. Biochim. Biophys. Acta 1799, 192–199. doi: 10.1016/j.bbagrm.2009.09.009
You, J., Croyle, J. L., Nishimura, A., Ozato, K., and Howley, P. M. (2004). Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 117, 349–360. doi: 10.1016/S0092-8674(04)00402-7
Zhao, J., Jia, Y., Shen, S., Kim, J., Wang, X., Lee, E., et al. (2020). Merkel cell polyomavirus small T antigen activates noncanonical NF-κB signaling to promote tumorigenesis. Mol. Cancer Res. 18, 1623–1637. doi: 10.1158/1541-7786.MCR-20-0587
Keywords: Merkel cell polyomavirus, Merkel cell carcinoma, persistence, innate immune response, integration, dysbiosis, oncogenesis
Citation: Krump NA and You J (2021) From Merkel Cell Polyomavirus Infection to Merkel Cell Carcinoma Oncogenesis. Front. Microbiol. 12:739695. doi: 10.3389/fmicb.2021.739695
Received: 11 July 2021; Accepted: 09 August 2021;
Published: 08 September 2021.
Edited by:
Herbert Johannes Pfister, University of Cologne, GermanyReviewed by:
James A. DeCaprio, Dana–Farber Cancer Institute, United StatesNicole Fischer, University Medical Center Hamburg-Eppendorf, Germany
Copyright © 2021 Krump and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianxin You, amlhbnlvdUBwZW5ubWVkaWNpbmUudXBlbm4uZWR1
†Present address: Nathan A. Krump, Integral Molecular, Inc., Philadelphia, PA, United States