 Wei Zhang
Wei Zhang Xuhong Sun
Xuhong Sun Xuyan Wei
Xuyan Wei- Heilongjiang Academy of Agricultural Sciences, Harbin, China
Potato virus A (PVA), a member of the genus Potyvirus, is an important potato pathogen that causes 30%–40% yield reduction to global potato production. Knowledge on the genetic structure and the evolutionary forces shaping the structure of this pathogen is limited but vital in developing effective management strategies. In this study, we investigated the population structure and molecular evolution of PVA by analyzing novel complete genomic sequences from Chinese isolates combined with available sequences from Europe, South America, Oceania, and North America. High nucleotide diversity was discovered among the populations studied. Pairwise FST values between geographical populations of PVA ranged from 0.22 to 0.46, indicating a significant spatial structure for this pathogen. Although purifying selection was detected at the majority of polymorphic sites, significant positive selection was identified in the P1, NIa, and NIb proteins, pointing to adaptive evolution of PVA. Further phylogeny–trait association analysis showed that the clustering of PVA isolates was significantly correlated with geographic regions, suggesting that geography-driven adaptation may be an important determinant of PVA diversification.
Introduction
Potato (Solanum tuberosum L.) is the fourth largest staple crop after rice, wheat, and maize, both worldwide and in China (Qu et al., 2005). Since 1993, China has become the world’s leading potato-producing country (Wang and Zhang, 2004; Jansky et al., 2009), accounting for 26.3% and 22.2% of the global total acreage and yield, respectively (Wang et al., 2011). As a vegetatively propagated crop, potato is prone to infection by more than 50 viruses (Valkonen, 2007; Lesley and Michael, 2020). Among these viruses, six have been recognized as major potato viruses: potato leafroll virus, potato virus Y (PVY), potato virus A (PVA), potato virus M, potato virus X (PVX), and potato virus S (Bai et al., 2007; Zhang et al., 2010; Duan et al., 2018; Mao et al., 2019).
Potato virus A (PVA) has a narrow host range, mainly infecting the members of Solanaceae (Thomas and Nicotiana, 2004). Potato virus A was not officially named until 1932 (Murphy and McKay, 1932), but symptoms suggestive of PVA infection were first reported in 1914 (Orton, 1914). Today, PVA is prevalent in potato production areas worldwide. Normally, the yield loss associated with single PVA infections is moderate, although it can reach 40% in rare cases (Bartels, 1971; German, 2001). However, PVA can infect potato together with many other viruses. In these cases, the yield loss can be much larger (Wang, 1999; Wang et al., 2005; Liu, 2007). For example, double infection of PVA and PVX causes a disease named “potato crinkle” (MacLachlan et al., 1954), which is associated with very severe foliar symptoms and significant yield losses (German, 2001; He et al., 2014; Kreuze et al., 2020). In China, PVA was first discovered on the “Ke shan” variety of potatoes in Heilongjiang Province in 1975. It was subsequently reported in areas including Hunan, Sichuan, Hubei, Zhejiang, Hebei, Fujian, and Guangxi. At present, PVA is prevalent in almost all of the main potato-producing areas in China.
Potato virus A (PVA) causes varying degrees of symptoms, ranging from mild mosaic to severe leaf necrosis, depending on the PVA isolates and potato cultivar (Rajamaki et al., 1998; Nie and Singh, 2001). Relative foliage symptom severity has been used to differentiate PVA isolates into four biological strain groups: very mild, mild, moderately severe, and severe (Bartels, 1971). In addition, Valkonen et al. (1995) and Rajamaki et al. (1998) distinguished four different strain groups (pathotypes) based on whether a PVA isolate caused systemic necrosis (PVA-1), mottle (PVA-2), no infection (PVA-3), or systemic yellowing and stunting (PVA-4) following graft inoculation to the potato cultivar King Edward. A recent study indicated that PVA isolates can be clustered into three monophyletic groups: A, W, and T (Fuentes et al., 2021). Isolates in the A group contain Peruvian potato isolates, whereas the T group comprises three tamarillo isolates from New Zealand. The W group contains isolates with a considerable diversity of sampling locations, and host species (potato and tamarillo). Possibly owing to a fitness advantage over non-recombinants, a substantial increase in the prevalence of A × W recombinant isolates has been observed in South America, Europe and Australia (Fuentes et al., 2021).
As with PVY, the type member of the genus Potyvirus (Lefkowitz et al., 2018), PVA has a single-stranded, positive-sense RNA genome ∼10 kb in size. The genome is translated into a large open reading frame (ORF) consisting of a 3,059 amino-acid polyprotein, which is cleaved to yield 10 mature proteins. In addition, a short protein, known as PIPO (Pretty Interesting Potyviridae ORF), was discovered out of frame in the P3 protein (Chung et al., 2008). Studies on the functions of PVA proteins remain limited, although the main functions of the encoded proteins of the genus Potyvirus have been systematically summarized (Urcuqui-Inchima et al., 2001; Zhang et al., 2013). Among the proteins encoded in the PVA genome, P1 is a transactive accessory factor during genome amplification and is thought to play an essential role in virus adapting to different host species (Verchot and Carrington, 1995; Valli et al., 2007). NIa is the C-terminus of the endosomal protein NIa. It can perform the catalytic cleavage of polyproteins (Wu et al., 2006). The NIb is an RNA-dependent RNA polymerase (Hong and Hunt, 1996) responsible for viral replication (Li et al., 1997; Wu et al., 2006).
Potato virus A (PVA) is transmitted through infected tubers and mechanical friction, besides being transmitted non-persistently by aphids (Li et al., 1997; Zhang et al., 2010). Potato virus A is one of the oldest potato viruses and has been dated to around 1570CE (Hawkes, 1990; Nunn and Qian, 2010). However, our knowledge on the population genetics and evolutionary biology of PVA is relatively limited compared to other potato pathogens such as Phytophthora infestans (Adler et al., 2004; Cardenas et al., 2011) and PVY (Gao et al., 2017; Mao et al., 2019). In this study, we estimated genetic diversity parameters, analyzed population differentiation, identified recombination events, and investigated the role of natural selection during PVA evolution by analyzing the complete genomic sequences of PVA. In addition, we also determined the correlation between the genetic variation and geography of PVA to unveil geography associated adaptation of this virus.
Materials and Methods
Virus Isolates
Potato virus A (PVA) isolates were collected from major potato-growing areas in China. Each isolate was maintained a plant of Nicotiana debneyi in the lab. The presence of PVA was confirmed by DAS-ELISA (Agdia, Elkhart, United States). Total RNA was extracted from each N. debneyi sample using Trizol (Invitrogen, Carlsbad, CA, United States) and reverse-transcribed following the manufacturer’s protocol (Promega, Madison, WI, United States). The complete genome of PVA was obtained by amplifying 10 overlapping fragments (nucleotides 1–409, 200–1428, 1259–2530, 2384–3630, 3476–4730, 4572–5824, 5725–7040, 6889–8160, 7963–9345, and 8986–9567) using 10 pairs of degenerate primers (Supplementary Table 1), which were designed from highly conserved regions of published PVA genomes (accession numbers Nos. KF977085, MT521081, MT435487, MT435489, KM365068, MT502380, and MT502370).
Polymerase chain reaction (PCR) amplifications of cDNA were performed in a total volume of 50.00 μL, containing 2.00 μL of cDNA templates, 25.00 μL of Premix Taq (TaKaRa), 1.00 μL of forward primer (10.00 μmol/L), 1.00 μL of reverse primer (10.00 μmol/L), and 21.00 μL of ddH2O. The PCR program comprised 5 min at 94°C; 35 cycles of 94°C for 30 s, 48°C–57°C for 30 s (Supplementary Table 1), and 72°C for 1 min; followed by a final extension of 10 min at 72°C. Samples were amplified using a DNA Engine Peltier Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, United States). The PCR products were separated on 1.5% agarose gels in Tris-acetate-EDTA (TAE) buffer and visualized by UV illumination.
PCR products were purified and ligated to pESI-T vector, which was provided in the Hieff Clone ® Zero TOPO-TA Cloning Kit (Yeasen, China), and propagated in cells of Escherichia coli strain TOP10. The cloned DNA fragments of recombinant plasmids were sequenced in both directions by Sangon Biological Co. Ltd (Shanghai, China). At least 3–5 independent cDNA clones for each segment were sequenced to assemble consensus sequences.
Sequence Dataset
Eleven complete or nearly complete genome sequences of PVA isolates, including nine from China, one from Peru, and one from the Netherlands, were obtained in this study and deposited in GenBank under accession numbers MW592838–MW592842 and MW616801–MW616806 (see Supplementary Table 1 for the list of primers used for sequencing). In addition to the novel sequences, 55 complete genome sequences of PVA isolates were downloaded from GenBank (Supplementary Table 2). The sequences had been collected from 14 countries and had known host species and geographic locations. To increase post-analysis interpretability, the isolates were grouped according to their geographic origins. The combined sequence data included China (n = 10), Europe (n = 15), South America (n = 33), Oceania (n = 6), and North America (n = 2) and were used for the subsequent analyses. Sequences were aligned using the MEGA X (Kumar et al., 2018) and the polyprotein ORF of each sequence was extracted from the alignment. Codon-based sequence alignment was then performed using the MAFFT algorithm (Katoh and Standley, 2013) implemented in PhyloSuite v1.2.2 (Zhang et al., 2020). The program was run using the FFT-NS-I iterative refinement method with the following parameters: mafft –thread 8 –threadtb 5 – threadit 0 –reorder –leavegappyregion. Ambiguously aligned regions were trimmed using the program Gblock 0.91b (Talavera and Castresana, 2007) implemented in PhyloSuite, with the “codon” mode, half gaps allowed, and all other parameters at default settings. The resulting sequence alignment had a length of 9180 nucleotides and used for subsequent population genetics analysis.
Genetic Diversity and Population Differentiation
To assess how the diversity varied across geographical and host populations, haplotype diversity (Hd) and nucleotide diversity (Pi) were calculated using DnaSP v5.0 (Librado and Rozas, 2009). Analysis of molecular variance (AMOVA) was also carried out using Arlequin v3.5 (Excoffier and Lischer, 2010). The significance of φ-statistics was tested by 1023 random permutations of sequences among the population.
Pairwise among-populations fixation indices (FST) were calculated using Arlequin v3.5 (Excoffier and Lischer, 2010), and the significance was obtained with 1000 permutations. A sliding-window analysis was used as an additional approach for evaluating genetic population differentiation. This analysis was performed using the PopGenome package (Ver. 2.7.5; Pfeifer et al., 2014) in R software (ver. 3.5.1), with a window size of 100 nt and a step size of 30 nt. In addition, discriminant analysis of principal components (DAPC) was used to infer clusters of genetically related individuals. This new multivariate method pioneered by Jombart et al. (2010) was designed to investigate the genetic structure of biological populations without assuming panmixia. In this study, we only performed the DAPC analysis based on pre-defined geographic groups using the adegenet package (Ver. 2.0.1; Jombart, 2008) in R software (ver. 3.5.1), and therefore, the populations of North America (two potato isolates) and Oceania (three tamarillo isolates) were excluded from the analysis due to an inadequate sample size (n ≤ 3).
Phylogenetic Network and Recombination Analyses
A recent study found evidence for intragenic recombination within the PVA genome (Fuentes et al., 2021). To investigate the role of recombination, we used two different methods to investigate the occurrence of recombination events in CP sequences. A phylogenetic network was first reconstructed using the neighbor-net method implemented in SplitsTree v4.14.8 (Huson, 1998) with default settings. The pairwise homoplasty index (PHI) test implemented in SplitsTree was also carried out to test signals of recombination (p < 0.05, significant evidence of recombination). In addition, to confirm the occurrence of recombination in our dataset, localization of recombination breakpoints and identification of likely parental sequences were achieved with the RDP v4.101 package, which incorporates the algorithms RDP, Geneconv, Bootscan, Maxchi, Chimaera, Siscan, and 3Seq (Martin et al., 2015). Recombination events supported by at least four different algorithms of analysis and with p values < 1.0 × 10–5, viral isolates were identified as recombinants. Because recombinants may result in misleading results in selection analysis, as reported by Anisimova et al. (2003) and Sironi et al. (2015), the recombinants were excluded from subsequent selection analysis.
Selection Pressure Analysis
To measure the selection pressure in the complete PVA genome, we calculated the ratio (ω) of non-synonymous (dN) to synonymous (dS) substitutions, as it was done in most adaptive evolution studies, using the CodeML program in the PAML package (Yang, 2007) implemented in EasyCodeML v1.4 (Gao et al., 2019). After all recombinants were removed, our selection analysis was based on 58 polyprotein coding-region sequences. The positive selection models (M2a, M8) and their respective null models (M1a, M7) implemented in the site models were used to conduct the adaptive evolution analysis. Likelihood ratio tests (LRTs) were performed twice to compare the difference in the log-likelihoods between the nested codon-based models against an x2 distribution with the degree of freedom equal to the differences in the number of parameters between the models (Yang, 1998). When the LRTs yielded significant results, the Bayes empirical Bayes (BEB) method was used to identify the codons that were the most likely to be under positive selection (Yang et al., 2005).
Phylogeny–Trait Association Analysis
To assess the geographical and host effects on the PVA population, three statistics, the association index (AI), parsimony score (PS), and maximum monophyletic clade size (MC), were calculated from the posterior tree samples using BaTS v2.0 (Parker et al., 2008). For this analysis, phylogenetic uncertainty was used to investigate phylogeny-trait correlations, with 1000 random permutations of tip locations to estimate a null distribution for each statistic. The results that generated a low AI index and PS and high MC scores with p < 0.05 suggested a strong phylogeny–trait association.
Results
High Genetic Diversity in the Potato Virus A Population
A data set consisting of 66 complete sequences was included in the analysis. After trimming the ambiguously regions from the alignment, we found that all mutations in the PVA genome are substations, with two sequences as exceptions, which had one (accession number: AJ131403) and two codon deletions (accession number: MT502380), respectively. The 66 PVA isolates in this study were composed of 66 haplotypes with an overall haplotype diversity of 1.00 and nucleotide diversity of 0.077 (Table 1). When the viral isolates were categorized according to geographic origin, the highest nucleotide diversity (0.103 ± 0.019) was found in the Oceania population and the lowest (0.012 ± 0.002) was discovered in the Chinese population. When the isolates were grouped according to host origin, higher genetic variation was observed in viral isolates collected from tamarillo compared with those isolated from potato (Table 1), although only four isolates isolated from tamarillo were analyzed in this study. Stepwise diversity analysis indicated that the fragment spanning nucleotide 1-189 and 2791-2950 (Supplementary Figure 1), which corresponds to the coding region for P1 and PIPO, respectively, is the most variable and conserved region on the genome of PVA. Sixty haplotypes were identified in the 66 nucleotide sequences of P1 with an overall haplotype diversity of 0.994 and nucleotide diversity of 0.104 (Supplementary Table 3). In PIPO, 28 haplotypes were identified in the 66 sequences, with an overall haplotype diversity of 0.876 and nucleotide diversity of 0.025 (Supplementary Table 3).
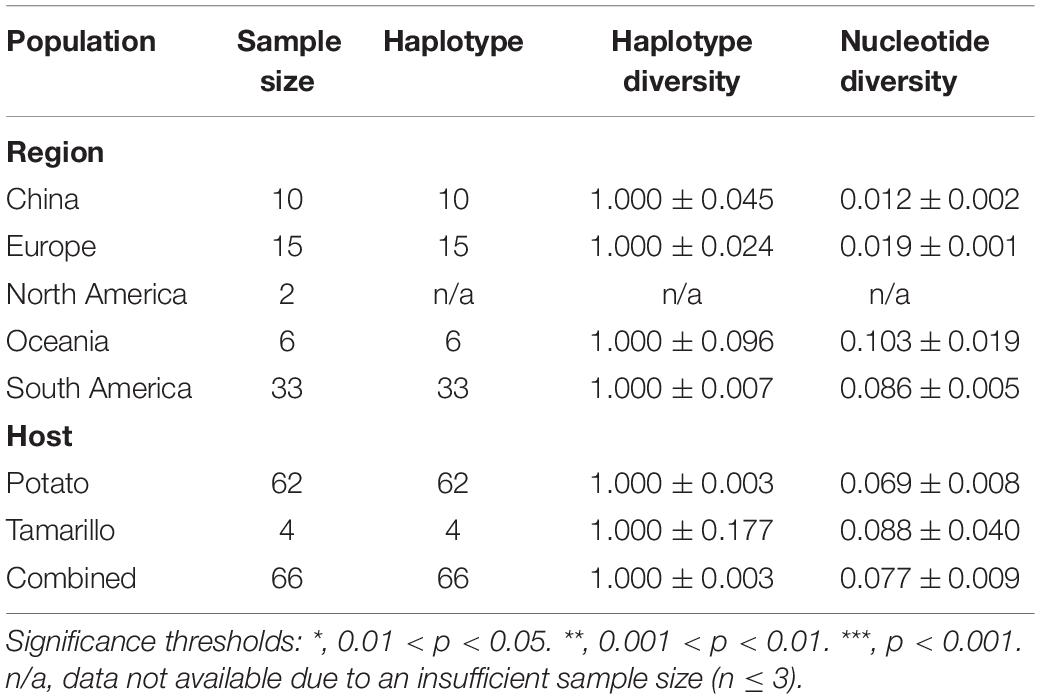
Table 1. Genetic diversity parameter estimates for PVA population.
Genetic Differentiation and Population Structure
The genetic differentiation between all populations of geographic regions was significant, with the FST values ranging from 0.22 to 0.46 (Table 2), indicating significant genetic differentiation between geographic groups of PVA. Similarly, the genetic differentiation between viral isolates with different host species had an FST value of 0.45. The results of sliding-window analysis of the pairwise FST values of population differentiation among geographic regions and host species are illustrated in Figure 1. The pairwise FST values estimated based on the geographic groupings were similar to those based on the host species groupings. This was in agreement with the pairwise FST analysis above (Table 2).
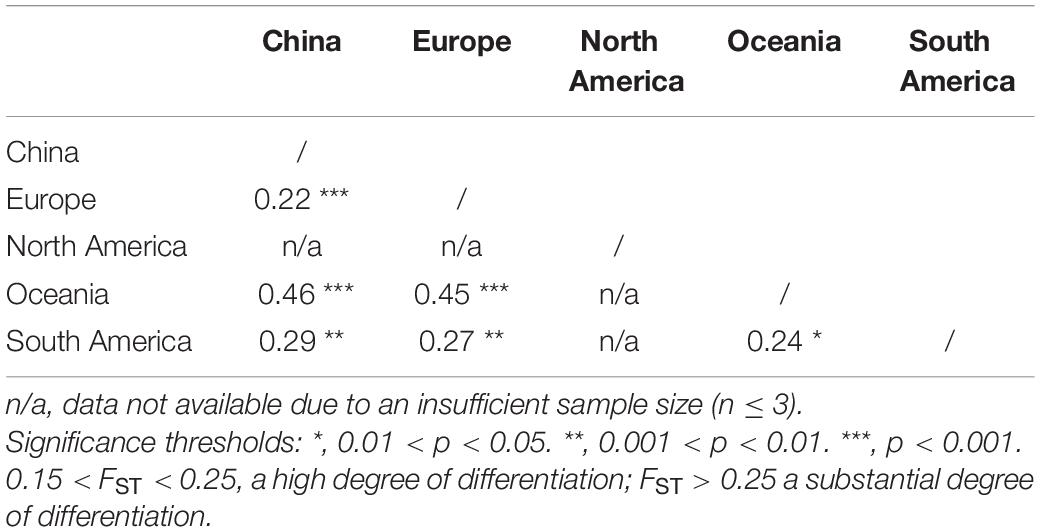
Table 2. Pairwise FST between geographic populations of PVA.
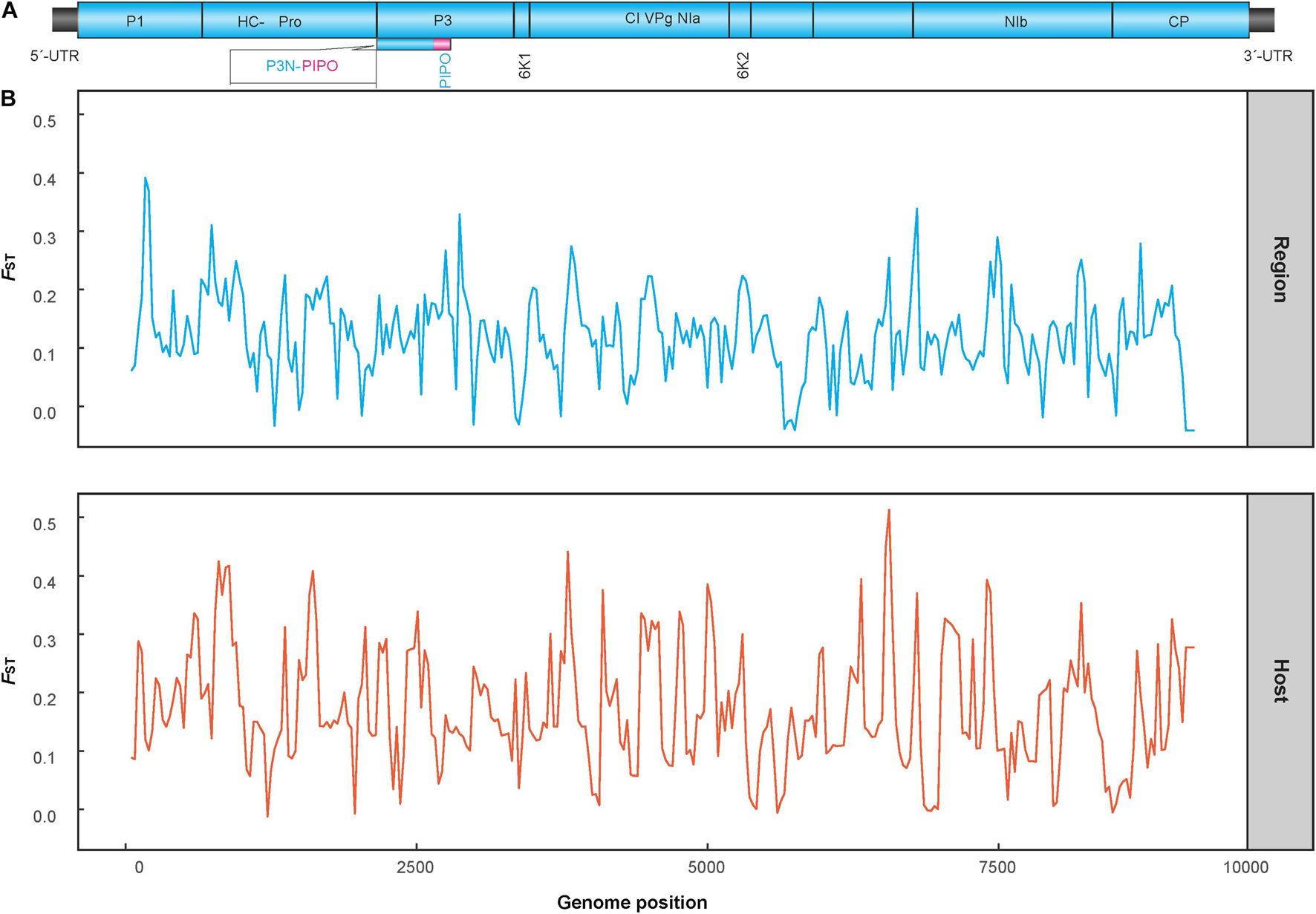
Figure 1. Diagram showing the genomic organization of potato virus A (A) and Sliding-window analysis of population differentiation across geographical and host populations (B). FST values were calculated using the R package of PopGenome. The window size is 100 nucleotides, and the step size is 30 nucleotides.
An AMOVA analysis also revealed a significant level of genetic differentiation between the PVA sequences originating from either different geographic origins or host species. As shown in Table 3, the variation among geographic regions accounted for 28.07% of the total variation (ΦST = 0.281, p < 0.001), while the variation within regions accounted for 71.93%. When performing the AMOVA only on viral isolates from potato and tamarillo, similar results were obtained; significant variation among groups made up 45.81% of the total variation (ΦST = 0.458, p < 0.01, Table 3), which accounted for nearly 50% of the total genetic variance of PVA. Taken together, it seems that the effect of host species on the genetic variance of PVA is greater than that of geography.
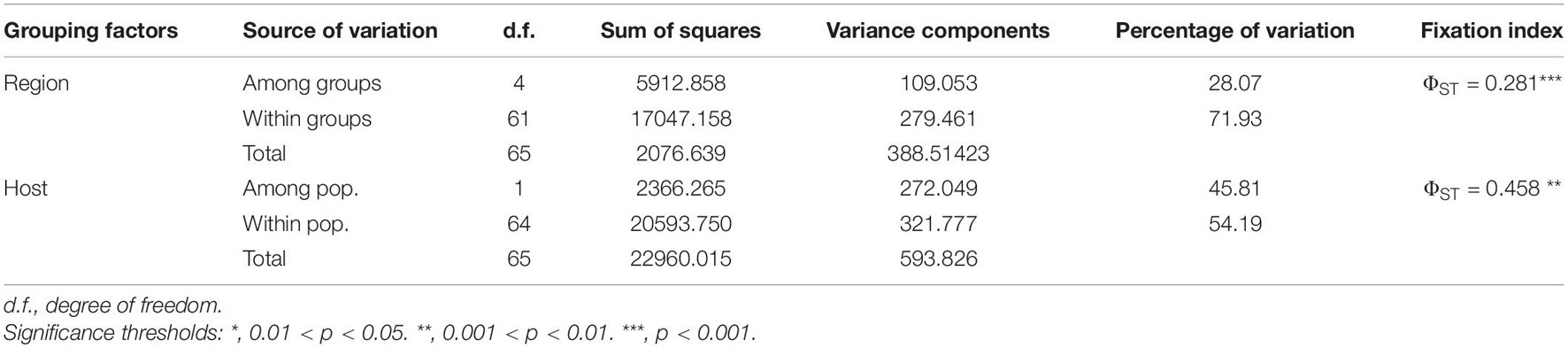
Table 3. Analysis of molecular variance for the effects of geography and host species.
The results of the DAPC analysis showed similar patterns of population differentiation as those revealed by the pairwise FST analysis. Discriminant analysis of principal components scatter plots indicated that the population of China were relatively distinct from the other populations along the first discriminant function axis, while the population of Europe exhibited more subtle structure along the second discriminant function axis (Figure 2). Discriminant analysis of principal components scatterplots also showed that the analyzed PVA isolates were divided into three genetic clusters (Figure 2), corresponding to the geographic regions. All the three genetic clusters were clearly differentiated. Cluster 1 contained all PVA isolates from China. Cluster 2 contained 15 individuals from Europe, whereas Cluster 3 comprised 33 individuals from South America.
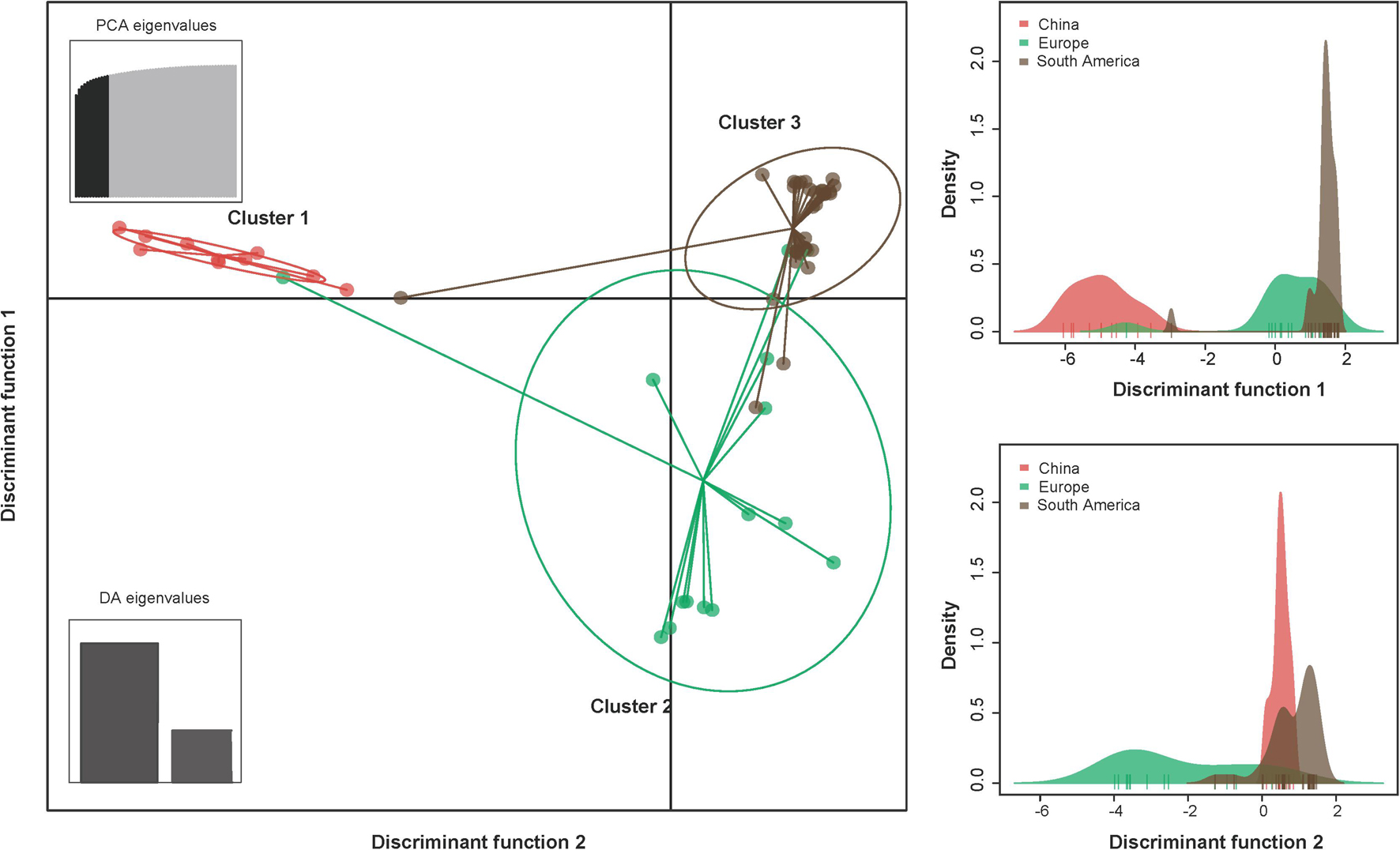
Figure 2. The discriminant analysis of principal components (DAPC) for the geographic structure of PVA isolates from potato. The graph represents the individuals as dots and the groups as inertia ellipses. The bar plots of eigenvalues for the analysis are shown in the inset panel. The density of individuals according to clusters identified along the discriminant function is shown in the right panel. Diagram showing the genomic structure of the PVA genome is shown in the top panel. Viral isolates infecting potato from North America and Oceania were excluded from the analysis due to inadequate sample size (n ≤ 3).
Significant Recombination Signals in the Complete Potato Virus A Genome
Our phylogenetic network analysis showed that PVA isolates were clustered into three lineages (Figure 3), corresponding to the groups W (World), A (Andean), and T (Tamarillo) from the phylogenetic analysis by Fuentes et al. (2021). Three isolates from tamarillo were placed into the T lineage and 14 isolates were placed into the A lineage. The W lineage contained isolates with a considerable diversity of sampling locations. Within the W lineage, all Chinese isolates formed a highly divergent sub-lineage (Figure 3). We also found several conflicting phylogenetic signals that may have been due to recombination (Figure 3), which was supported by the PHI test with statistically significant evidence of recombination (p < 0.001). Using the RDP package, 8 PVA isolates were identified as recombinants by at least 4 algorithms (Table 4). In one recombinant (Apu046, accession number MT502353), the breakpoints were detected in the P3 cistron, whereas in all other 7 recombinants (accession numbers GU144321, MT435486, MT435487, MT435489, MT435495, MT502353, MT502377 and MT521083), the breakpoints were detected in the CP cistron (Table 4). The sequences of these eight recombinants were excluded from the selection analysis presented below.
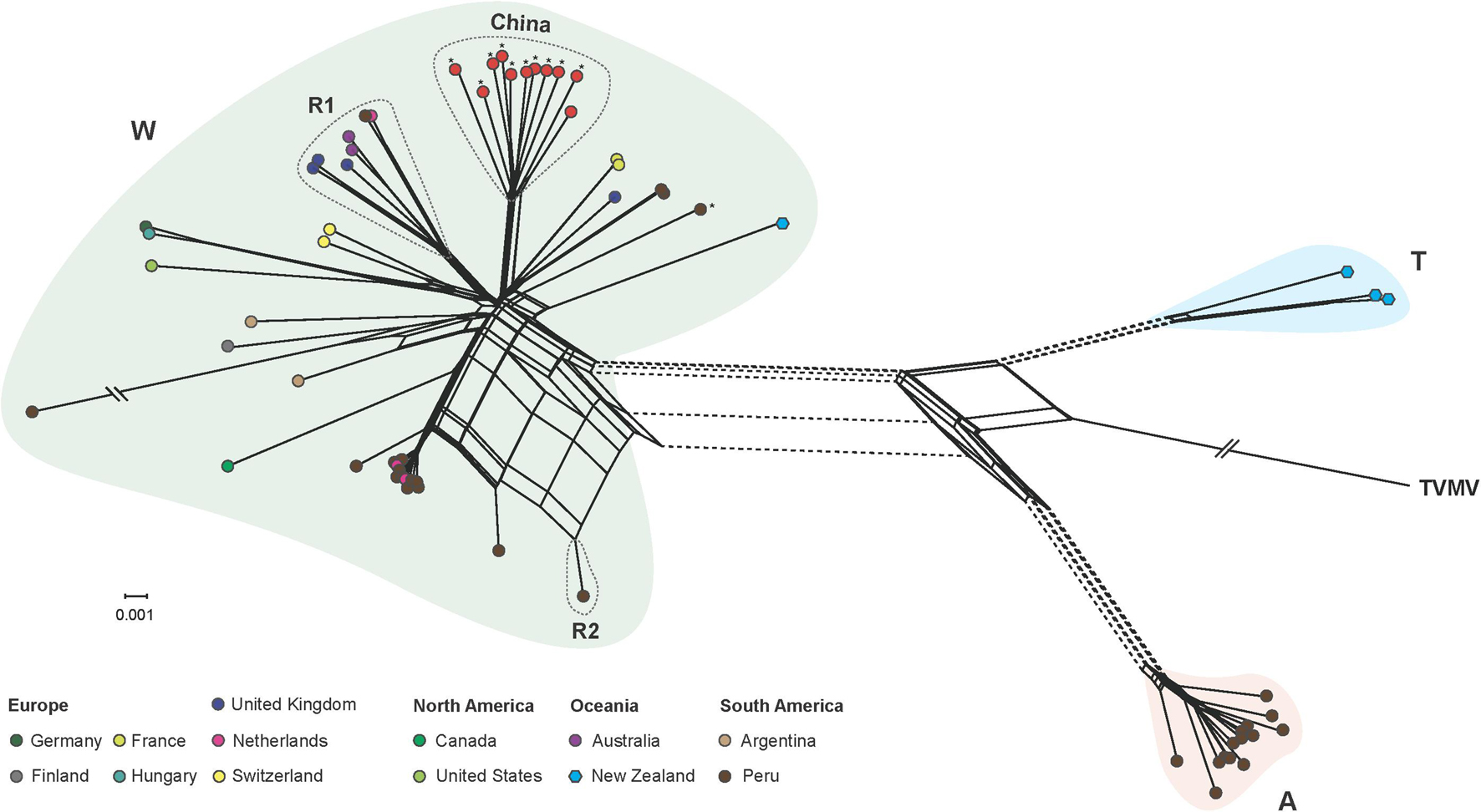
Figure 3. Phylogenetic network inferred from the complete genome of 66 potato virus A isolates. Unique color indicates the geographic origin, as shown in the color key. Phylogenetic groups W (world), A (Andean), and T (tamarillo) were proposed by Fuentes et al. (2021). PVA isolates infecting potato in different regions are indicated by circles, as shown in the color key, and those infecting taramillo are shown in blue hexagons. Isolates sequenced in the study are marked by asterisks. Parallel slashes indicate the branch lengths that were pruned to fit the image size. R1 and R2 indicate recombination group 1 and 2, respectively, which contained recombinants identified by the RDP package. See Table 4 for details of the recombinants. A tobacco vein mottling virus (accession number NC_001768) isolate was used as an outgroup.
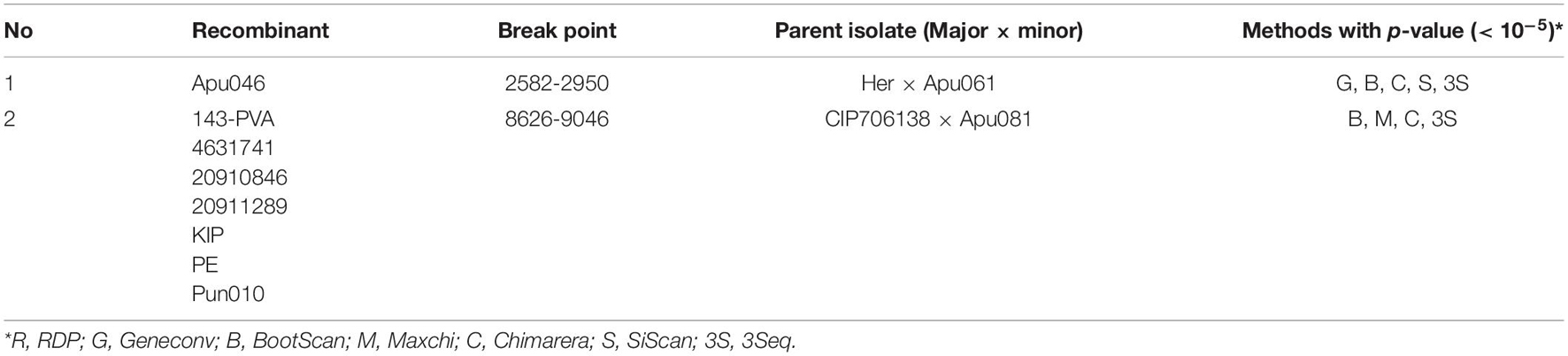
Table 4. Recombination events detected in the genome of potato virus A by RDP4 Suites.
Selection Pressure
Fifty-eight non-recombinant sequences were included in the selection and phylogeny-trait association analyses. The ratio of mean dN/dS (less than 1) of the polyprotein coding region showed that the majority of polymorphic sites (98.85%) were under purifying selection (Figure 4), suggesting that most of mutations in the genome were deleterious and consequently being weeded out by natural selection. However, the LRT indicated that the positive selection models (M2a and M8) were significantly better than the control models (M1a and M7), providing evidence for the presence of codons under positive selection. Further analysis from BEB scores indicated a strong positive selection pressure on nine codons, including the P1 (codon sites 34, 46, and 146), NIa (codon site 2268), and NIb cistrons (codon sites 2557, 2560, 2561, 2563, and 2591), with high posterior probability ≥0.95 (Table 5).
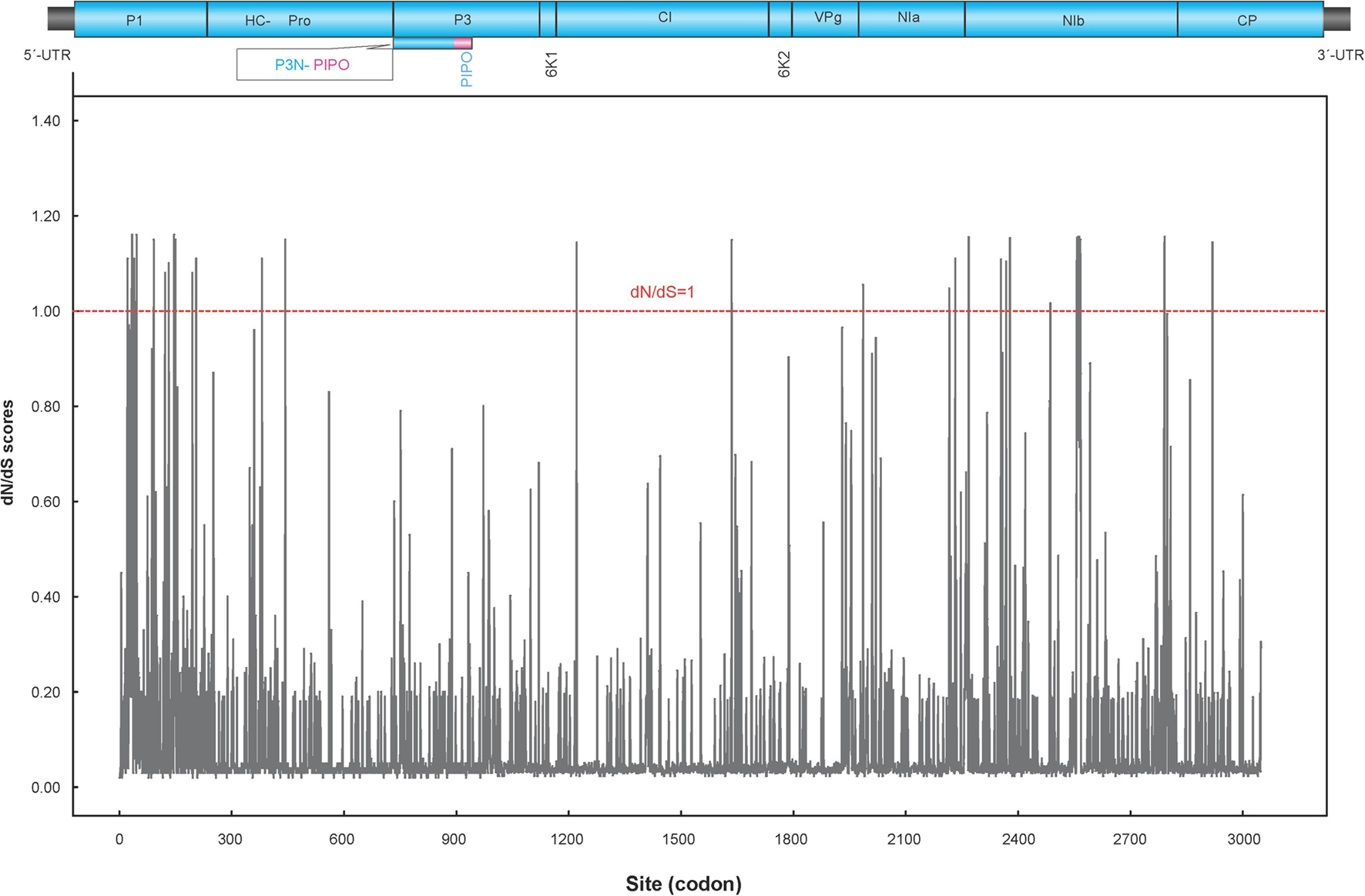
Figure 4. Sliding window plot of dN/dS ratios across the complete PVA genome. The red dotted line indicates sites under neutral selection (dN/dS = 1). A diagram showing the genomic structure of the PVA genome is shown in the top panel.
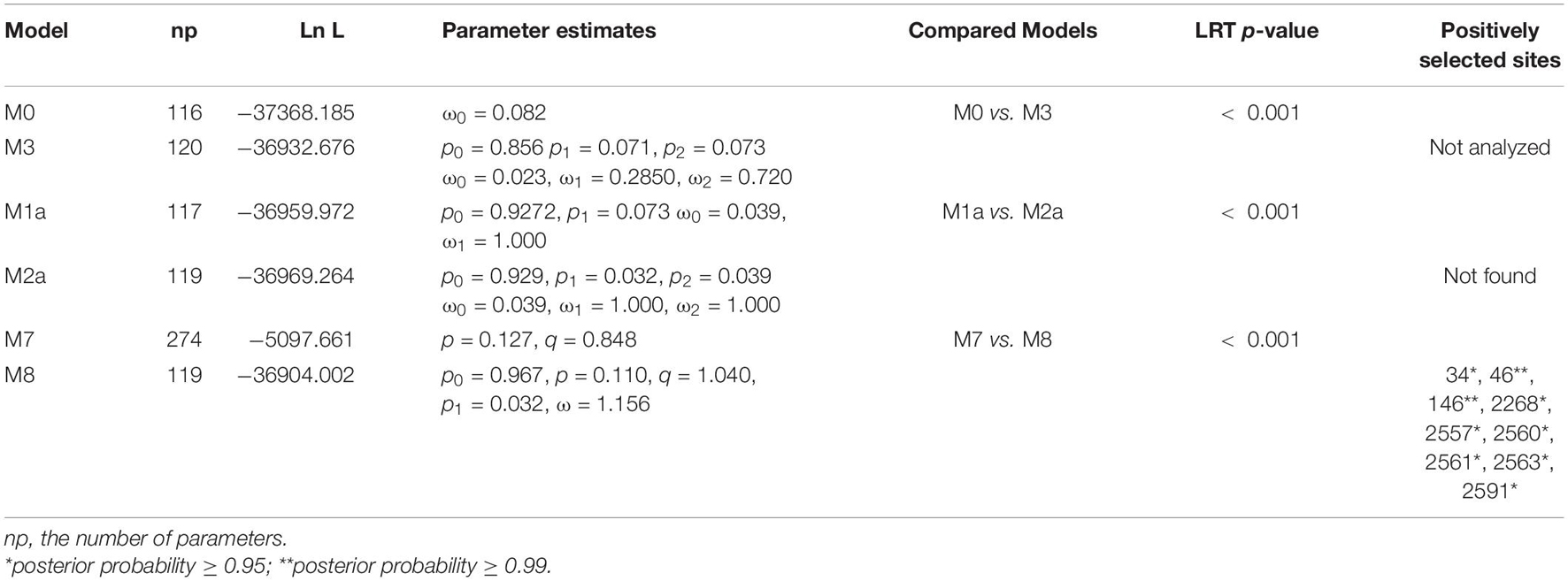
Table 5. Site model tests on the complete genome of PVA.
Geography-Driven Adaptation of Potato Virus A
With the exception of viral isolates from Europe (MC = 2.00, p > 0.05), significant signal for geographic clustering was found when PVA isolates were grouped by their sampling regions based on tests of phylogeny-trait association analysis (MC: p < 0.05, Table 6), indicting a great spatial structure of the pathogen. However, we accepted the null hypothesis of no association between host species and phylogenetic relationships when the PVA isolates were grouped by their host origins (MC: p > 0.05, Table 6). Taken together, the BaTS results suggested that geographic effects contributed to the diversification of the virus, which could be explained by geography-driven adaptation.
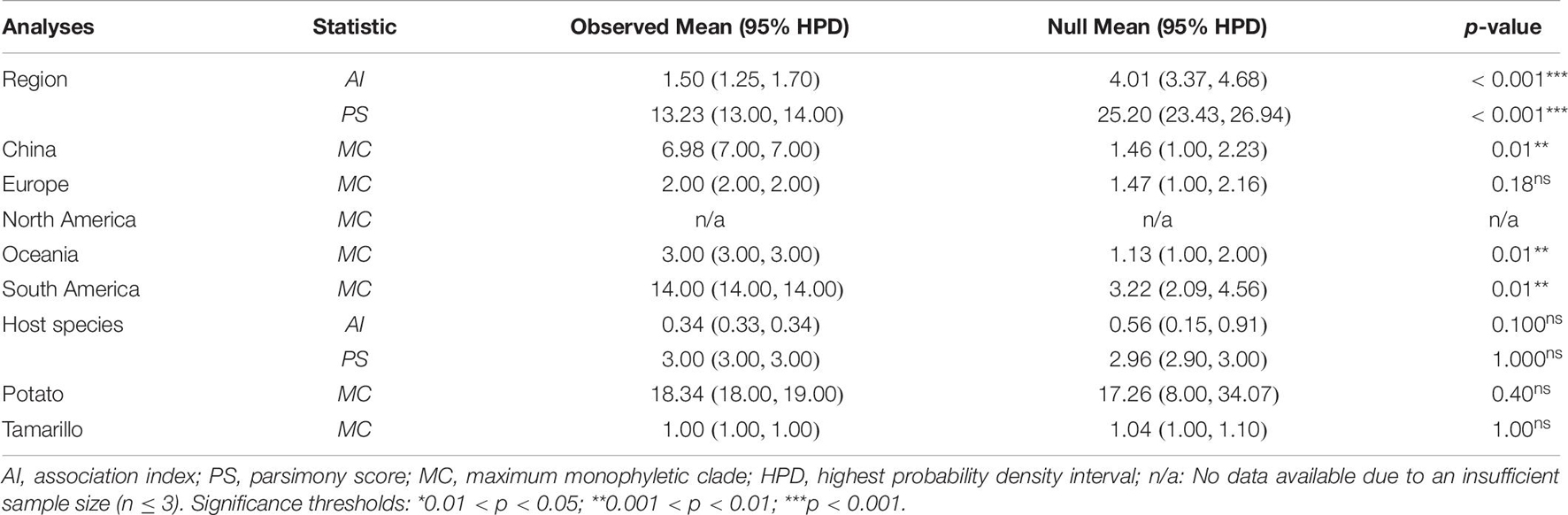
Table 6. Analysis of the geographic and host effects on the population structure of PVA.
Discussion
In this study, we obtained new sequence data for 11 PVA isolates from China, Peru, and the Netherlands. Combing these data with available sequences retrieved from GenBank, we investigated the genetic diversity and population structure of PVA.
Due to high mutation rates, short generation times, and large population sizes, RNA viruses exhibit extreme evolutionary dynamics (Garcia-Arenal et al., 2001). Consistent with previous studies by Rajamaki et al. (1998) and Kekarainen et al. (1999), a high level of genetic diversity was found for PVA (Table 1) in the current study. This high genetic diversity allows plant RNA viruses, including PVA, to rapidly evolve and adapt to the changing environment (Holmes, 2009).
Recombination plays a major role in shaping genome variation (Gibbs and Ohshima, 2010; Pérez-Losada et al., 2015). Recombinants have also been reported in members of the genus Potyvirus, including PVY (Quenouille et al., 2013) and PVA (Fuentes et al., 2021). In this study, similar recombinants were found in the W group proposed by Fuentes et al. (2021). However, no recombinants were identified in PVA isolates from China, which were clustered into a subgroup, showing distinct geographic features (Figure 3). There is one possible explanation for this observation. One is that there is strong selective pressure against the survival of new PVA recombinants of Chinese isolates. Indeed, we found that the large majority of codons in the PVA genome were under purifying selection, suggesting that there are very strong evolutionary constraints acting on PVA and most mutations in the genome were harmful and were subsequently removed by natural selection through reduced survival. However, nine codons in the P1, NIa and NIb proteins were detected to be under positive selection with high confidence levels (posterior probability > 0.95, Table 5). A previous study indicated that most positively selected amino acid sites in the genome of a potyvirus were located to cistrons with hypervariable nucleotides (Wokorach et al., 2020). Consistent with this, the positively selected sites of PVA detected in this study were located to P1, NIa and NIb cistrons. P1, the first protein of the polyprotein, is the most variable protein among potyviruses or within a specific potyviral species (Adams et al., 2005). It is suggested that P1 is involved in adaptation of a potyvirus to a new host species (Valli et al., 2007). Similarly, NIa and NIb also show higher than average genomic variation in PVA (Supplementary Table 3). However, our inferences were drawn solely from the genomic analysis of PVA sequences. Further investigations combining the pathology and biology of this virus will lead to a more comprehensive view of its evolutionary history.
Geographic and host factors were major contributors to the evolutionary dynamics of viruses. The phenomenon is also prevalent in the potyviruses, including PVY (Cuevas et al., 2012), chilli veinal mottle virus (Gao et al., 2016), and Ornithogalum mosaic virus (Gao et al., 2018). Although the phylogenetic network (Figure 3) did not seem to show a clear geography-specific or host species-specific clustering, significant geographical differentiation of PVA was found by more robust AMOVA and sequence-geography association analyses (Tables 3, 6). One explanation for the geographical differentiation is that PVA is a quarantine pest for many countries and agencies. In China, for example, it has been considered a potentially dangerous pest species since 1992. This may have imposed a significant limitation to the international dispersal of PVA.
It should be noted that there are many limitations to this study. For example, the dataset is small and the sequences are very unevenly distributed with respect to their geographical and host origin. Nevertheless, this study represents the first attempt to understand the genetic diversity of PVA at a global level.
Conclusion
In summary, the present study examined the genetic diversity and population structure of PVA and investigated the role of natural selection during the evolution of PVA. We found that genetic variations were correlated with geographic regions and may have been caused by geographically driven adaptation. In addition, we found evidence of diversifying selection in the genome of this pathogen. These results will be helpful in further studies on the molecular biology of PVA and are essential to understanding the adaptive evolution of this virus.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
YB conceived the study. WZ, XS, and XW performed the experiments. WZ, XS, and YB analyzed the data and interpreted the results. WZ and YB led the writing of the manuscript. All authors contributed to the manuscript and agreed on the manuscript before review.
Funding
This work was supported by grants from the China Agriculture Research System of MOF and MARA.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Fangluan Gao at the Fujian Agriculture and Forestry University (FAFU) for his generous help in analyzing the data and Zhenguo Du at FAFU for his comments and suggestions that improved the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.738646/full#supplementary-material
Supplementary Table 1 | Sequences of degenerate primers used to amplify overlapping segments of potato virus A genome.
Supplementary Table 2 | Isolates of potato virus A used in this study.
Supplementary Table 3 | Genetic diversity parameter estimates for difference protein coding regions gene in the genome of potato virus A.
References
Adams, M. J., Antoniw, J. F., and Fauquet, C. M. (2005). Molecular criteria for genus and species discrimination within the family Potyviridae. Ach. Virol. 150, 459–479. doi: 10.1007/s00705-004-0440-6
Adler, N. E., Erselius, L. J., Chacón, M. G., Flier, W. G., Ordoñez, M. E., Kroon, L. P., et al. (2004). Genetic diversity of Phytophthora infestans sensu lato in Ecuador provides new insight into the origin of this important plant pathogen. Phytopathology 94, 154–162. doi: 10.1094/PHYTO.2004.94.2.154
Anisimova, M., Nielsen, R., and Yang, Z. (2003). Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics 164, 1229–1236. doi: 10.1093/genetics/164.3.1229
Bai, Y., Wen, J. Z., Yang, M. X., Yu, D. C., Gao, Y. L., Fan, G. Q., et al. (2007). Comparison of incidence of major potato viruses in southwest and northeast potato-producing regions in China. J. Northeast Agric. Univ. 38, 733–736.
Bartels, R. (1971). “Potato virus A. Description No. 54,” in Descriptions of Plant Viruses, eds K. Crabtree and M. Dallwitz (Wellesbourne, U.K: Commonwealth Mycological Institute/Association of Applied Biologists).
Cardenas, M., Grajales, A., Sierra, R., Rojas, A., Gonzalez-Almario, A., Vargas, A., et al. (2011). Genetic diversity of Phytophthora infestans in the Northern Andean region. BMC Genet. 12:23. doi: 10.1186/%2F1471-2156-12-23
Chung, B. Y., Miller, W. A., Atkins, J. F., and Firth, A. E. (2008). An overlapping essential gene in the Potyviridae. Proc. Natl. Acad. Sci. U.S.A. 105, 5897–5902. doi: 10.1073/pnas.0800468105
Cuevas, J. M., Delaunay, A., Visser, J. C., Bellstedt, D. U., Jacquot, E., and Elena, S. F. (2012). Phylogeography and molecular evolution of potato virus Y. PLoS One 7:e37853. doi: 10.1371/journal.pone.0037853
Duan, G., Zhan, F., Du, Z., Ho, S. Y. W., and Gao, F. (2018). Europe was a hub for the global spread of potato virus S in the 19th century. Virology 525, 200–204. doi: 10.1016/j.virol.2018.09.022
Excoffier, L., and Lischer, H. E. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Fuentes, S., Gibbs, A. J., Adams, I. P., Wilson, C., Botermans, M., Fox, A., et al. (2021). Potato virus A isolates from three continents: their biological properties, phylogenetics, and prehistory. Phytopathology 111, 217–226. doi: 10.1094/PHYTO-08-20-0354-FI
Gao, F., Chen, C., Arab, D. A., Du, Z., He, Y., and Ho, S. Y. W. (2019). EasyCodeML: a visual tool for analysis of selection using CodeML. Ecol. Evol. 9, 3891–3898. doi: 10.1002/ece3.5015
Gao, F., Du, Z., Shen, J., Yang, H., and Liao, F. (2018). Genetic diversity and molecular evolution of Ornithogalum mosaic virus based on the coat protein gene sequence. PeerJ 6:e4550. doi: 10.7717/peerj.4550
Gao, F., Jin, J., Zou, W., Liao, F., and Shen, J. (2016). Geographically driven adaptation of chilli veinal mottle virus revealed by genetic diversity analysis of the coat protein gene. Arch. Virol. 161, 1329–1333. doi: 10.1007/s00705-016-2761-7
Gao, F., Zou, W., Xie, L., and Zhan, J. (2017). Adaptive evolution and demographic history contribute to the divergent population genetic structure of potato virus Y between China and Japan. Evol. Applic. 10, 379–390. doi: 10.1111/eva.12459
Garcia-Arenal, F., Fraile, A., and Malpica, J. M. (2001). Variability and genetic structure of plant virus populations. Annu. Rev. Phytopathol. 39, 157–186. doi: 10.1146/annurev.phyto.39.1.157
German, T. L. (2001). “Potato virus A,” in Compendium of Potato Diseases, 2nd Edn, eds W. R. Stevenson, R. Loria, G. D. Franc, and D. P. Weingartner (St. Paul: APS Press), 66–67.
Gibbs, A., and Ohshima, K. (2010). Potyviruses and the digital revolution. Annu. Rev. Phytopathol. 48, 205–223. doi: 10.1146/annurev-phyto-073009-114404
Hawkes, J. G. (1990). The Potato: Evolution, Biodiversity and Genetic Resources. London: Belhaven Press.
He, C., Zhang, W., Hu, X., Singh, M., Xiong, X., and Nie, X. (2014). Molecular characterization of a Chinese isolate of potato virus A (PVA) and evidence of a genome recombination event between PVA variants at the 39-proximal end of the genome. Arch. Virol. 159, 2457–2462. doi: 10.1007/s00705-014-2053-z
Holmes, E. C. (2009). The evolutionary genetics of emerging viruses. Annu. Rev. Ecol. Evol. Syst. 40, 353–372. doi: 10.1146/annurev.ecolsys.110308.120248
Hong, Y., and Hunt, A. G. (1996). RNA polymerase activity catalyzed by a potyvirus-encoded RNA-dependent RNA polymerase. Virology 226, 146–151. doi: 10.1006/viro.1996.0639
Huson, D. H. (1998). SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14, 68–73. doi: 10.1093/bioinformatics/14.1.68
Jansky, S. H., Jin, L. P., Xie, K. Y., Xie, C. H., and Spooner, D. M. (2009). Potato production and breeding in China. Potato Res. 52, 57–65. doi: 10.1007/s11540-008-9121-2
Jombart, T. (2008). adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Jombart, T., Devillard, S., and Balloux, F. (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11:94. doi: 10.1186/1471-2156-11-94
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kekarainen, T., Merits, A., Oruetxebarria, I., Rajamaki, M.-L., and Valkonen, J. P. T. (1999). Comparison of the complete sequences of five different isolates of potato virus A (PVA), genus Potyvirus. Arch. Virol. 144, 2355–2366. doi: 10.1007/s007050050649
Kreuze, J. F., Souza-Dias, J. A. C., Jeevalatha, A., Figueira, A. R., Valkonen, J. P. T., and Jones, R. A. C. (2020). “Viral diseases in potato,” in The Potato Crop, eds H. Campos and O. Ortiz (Cham: Springer), 389–430. doi: 10.1007/978-3-030-28683-5_11
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lefkowitz, E. J., Dempsey, D. M., Hendrickson, R. C., Orton, R. J., Siddell, S. G., and Smith, D. B. (2018). Virus taxonomy: the database of the international committee on taxonomy of viruses (ICTV). Nucleic Acids Res. 46, D708–D717. doi: 10.1093/nar/gkx932
Lesley, T., and Michael, E. T. (2020). Potato virus Y emergence and evolution from the andes of south america to become a major destructive pathogen of potato and other solanaceous crops worldwide. Viruses 12, 1–14. doi: 10.3390/v12121430
Li, X. H., Valdez, P., Olvera, R. E., and Carrington, J. C. (1997). Functions of the tobacco etch virus RNA polymerase (NIb): subcellular transport and protein-protein interaction with VPg/proteinase (NIa). J. Virol. 71, 1598–1607. doi: 10.1128/jvi.71.2.1598-1607.1997
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, W. P. (2007). Synergistic effect of potato virus Y (PVY) and potato spindle tuber viroid (PSTVd) on tuber yield of potato. J. Heilongjiang August First Land Reclamation Univ. 19, 40–43.
MacLachlan, D. S., Larson, R. H., and Walker, J. C. (1954). Potato virus A. Am. Potato J. 31, 67–72. doi: 10.1007/BF02859999
Mao, Y., Sun, X., Shen, J., Gao, F., Qiu, G., Wang, T., et al. (2019). Molecular evolutionary analysis of potato virus Y infecting potato based on the VPg gene. Front. Microbiol. 10:1708. 10.3389/fmicb.2019.01708
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., and Muhire, B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1:vev003. doi: 10.1093/ve/vev003
Murphy, P. A., and McKay, R. (1932). A comparison of some European and American virus diseases of the potato[J]. R. Dublin Soc. Sci. Proc. 20, 347–358.
Nie, X., and Singh, R. P. (2001). Differential accumulation of potato virus A and expression of pathogenesis-related genes in resistant potato cv. Shepody upon graft inoculation. Phytopathology 91, 197–203. doi: 10.1094/PHYTO.2001.91.2.197
Nunn, N., and Qian, N. (2010). The columbian exchange: a history of disease, food and ideas. J. Econ. Perspect. 24, 163–188. doi: 10.1257/jep.24.2.163
Orton, W. A. (1914). Potato wilt, leaf-roll and related diseases. U.S. Dep. Agric. Bull. 64, 1–47. doi: 10.5962/bhl.title.108864
Parker, J., Rambaut, A., and Pybus, O. G. (2008). Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect. Genet. Evol. 8, 239–246. doi: 10.1016/j.meegid.2007.08.001
Pérez-Losada, M., Arenas, M., Galán, J. C., Palero, F., and González-Candelas, F. (2015). Recombination in viruses: mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 30, 296–307. doi: 10.1016/j.meegid.2014.12.022
Pfeifer, B., Wittelsbürger, U., Ramos-Onsins, S. E., and Lercher, M. J. (2014). PopGenome: an efficient swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936. doi: 10.1093/molbev/msu136
Qu, D. Y., Xie, K. J., Jin, L. P., Pang, W. F., Bian, C. S., and Duan, S. G. (2005). Development of potato industry and food security in China. Sci. Agric. Sinica 38, 358–362.
Quenouille, J., Vassilakos, N., and Moury, B. (2013). Potato virus Y: a major crop pathogen that has provided major insights into the evolution of viral pathogenicity. Mol. Plant Pathol. 14, 439–452. doi: 10.1111/mpp.12024
Rajamaki, M., Merits, A., Rabenstein, F., Andrejeva, J., Paulin, L., Kekarainen, T., et al. (1998). Biological, serological, and molecular differences among isolates of potato A potyvirus. Phytopathology 88, 311–321. doi: 10.1094/PHYTO.1998.88.4.311
Sironi, M., Cagliani, R., Forni, D., and Clerici, M. (2015). Evolutionary insights into host-pathogen interactions from mammalian sequence data. Nat. Rev. Genet. 16, 224–236. doi: 10.1038/nrg3905
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Thomas, P. E., and Nicotiana, M. (2004). A highly susceptible new and useful host for potato virus A. Plant Dis. 88, 1160–1160. doi: 10.1094/PDIS.2004.88.10.1160B
Urcuqui-Inchima, S., Haenni, A. L., and Bernardi, F. (2001). Potyvirus proteins: a wealth of functions. Virus Res. 74, 157–175. doi: 10.1016/S0168-1702(01)00220-9
Valkonen, J. P. T. (2007). “Viruses: economical losses and biotechnological potential,” in Potato Biology and Biotechnology Advances and Perspectives, ed. D. Vreugdenhil (Amsterdam: Elsevier), 619–641. doi: 10.1094/PD-79-0748
Valkonen, J. P. T., Puurand, Ü, Slack, S. A., Mäkinen, K., and Saarma, M. (1995). Three strain groups of potato A potyvirus based on hypersensitive responses in potato, serological properties, and coat protein sequences. Plant Dis. 79, 748–753. doi: 10.1094/PD-79-0748
Valli, A., López-Moya, J. J., and García, J. A. (2007). Recombination and gene duplication in the evolutionary diversification of P1 proteins in the family Potyviridae. J. Gen. Virol. 88, 1016–1028. doi: 10.1099/vir.0.82402-0
Verchot, J., and Carrington, J. C. (1995). Evidence that potyvirus P1 protein functions as an accessory factor for genome amplification. J. Virol. 69, 3668–3674. doi: 10.1128/jvi.69.6.3668-3674.1995
Wang, B., Ma, Y., Zhang, Z., Wu, Z., Wu, Y., Wang, Q., et al. (2011). Potato viruses in China. Crop Protect. 30, 1117–1123. doi: 10.1016/j.cropro.2011.04.001
Wang, Q., and Zhang, W. (2004). China’s potato industry and potential impacts on the global market. Am. J. Potato Res. 81, 101–109. doi: 10.1007/BF02853607
Wang, X. M., Jing, L. P., and Yi, H. (2005). Advances in breeding of potato virus-resistant cultivars. Chininse Potato 19, 285–289.
Wang, X. W. (1999). Effect of Infection Status of Potato Virus Y and Potato Virus X on Tuber Yield of Potato. Wuhan: China’s Potato Association, 285–289.
Wokorach, G., Otim, G., Njuguna, J., Edema, H., Njung’e, V., Machuka, E. M., et al. (2020). Genomic analysis of sweet potato feathery mottle virus from East Africa. Physiol. Mol. Plant Pathol. 110:101473. doi: 10.1016/j.pmpp.2020.101473
Wu, X., Tan, X., and Chen, S. (2006). Research progress of replication-related proteins of Potato virus A. Chinese Potato 20, 231–234.
Yang, Z. (1998). Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15, 568–573. doi: 10.1093/oxfordjournals.molbev.a025957
Yang, Z. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., Wong, W. S., and Nielsen, R. (2005). Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. doi: 10.1093/molbev/msi097
Zhang, D., Gao, F., Li, W. X., Jakovliæ, I., Zou, H., Zhang, J., et al. (2020). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, W., Bai, Y. Q., Gao, Y. L., Sheng, Y., Fan, G. Q., Gen, H. W., et al. (2010). A survey on occurrence frequencies of potato viruses in major potato-producing provinces in China. Heilongjiang Agric. Sci. 4, 71–73.
Keywords: potato virus A, genetic diversity, positive selection, phylogeny-trait association analysis, population structure
Citation: Zhang W, Sun X, Wei X, Gao Y, Song J and Bai Y (2021) Geography-Driven Evolution of Potato Virus A Revealed by Genetic Diversity Analysis of the Complete Genome. Front. Microbiol. 12:738646. doi: 10.3389/fmicb.2021.738646
Received: 09 July 2021; Accepted: 09 September 2021;
Published: 01 October 2021.
Edited by:
Richard Allen White III, University of North Carolina at Charlotte, United StatesReviewed by:
Rajarshi Kumar Gaur, Deen Dayal Upadhyay Gorakhpur University, IndiaDenis Jacob Machado, University of North Carolina at Charlotte, United States
Copyright © 2021 Zhang, Sun, Wei, Gao, Song and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanju Bai, eWFuanViYWlAMTYzLmNvbQ==
†These authors have contributed equally to this work