
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 01 November 2021
Sec. Food Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.737979
This article is part of the Research Topic Rapid and Cost-effective Technologies to Detect the Pathogens in Food and Environment View all 14 articles
 Koji Nakae1
Koji Nakae1 Tadasuke Ooka2*
Tadasuke Ooka2* Koichi Murakami3
Koichi Murakami3 Yukiko Hara-Kudo4Naoko Imuta2
Yukiko Hara-Kudo4Naoko Imuta2 Yasuhiro Gotoh5
Yasuhiro Gotoh5 Yoshitoshi Ogura5,6
Yoshitoshi Ogura5,6 Tetsuya Hayashi5Yasuhiro Okamoto1Junichiro Nishi2
Tetsuya Hayashi5Yasuhiro Okamoto1Junichiro Nishi2Escherichia albertii is a recently recognized human enteropathogen that is closely related to Escherichia coli. As E. albertii sometimes causes outbreaks of gastroenteritis, rapid strain typing systems, such as the O- and H-serotyping systems widely used for E. coli, will be useful for outbreak investigation and surveillance. Although an O-genotyping system has recently been developed, the diversity of E. albertii H-antigens (flagellins) encoded by fliC genes remains to be systematically investigated, and no H-serotyping or genotyping system is currently available. Here, we analyzed the fliC genes of 243 genome-sequenced E. albertii strains and identified 73 sequence types, which were grouped into four clearly distinguishable types designated E. albertii H-genotypes 1–4 (EAHg1–EAHg4). Although there was a clear sign of intraspecies transfer of fliC genes in E. albertii, none of the four E. albertii H-genotypes (EAHgs) were closely related to any of the 53 known E. coli H-antigens, indicating the absence or rare occurrence of interspecies transfer of fliC genes between the two species. Although the analysis of more E. albertii strains will be required to confirm the low level of variation in their fliC genes, this finding suggests that E. albertii may exist in limited natural hosts or environments and/or that the flagella of E. albertii may function in a limited stage(s) in their life cycle. Based on the fliC sequences of the four EAHgs, we developed a multiplex PCR-based H-genotyping system for E. albertii (EAH-genotyping PCR), which will be useful for epidemiological studies of E. albertii infections.
Escherichia albertii is a recently recognized human enteropathogen and an avian pathogen (Albert et al., 1992; Huys et al., 2003; Oaks et al., 2010; Ooka et al., 2012). E. albertii is often misidentified as enteropathogenic Escherichia coli (EPEC) or enterohemorrhagic E. coli (EHEC) due to similar phenotypic and genetic characteristics, including similar biochemical properties and possession of a locus of enterocyte effacement (LEE) encoding a type III secretion system (Ooka et al., 2012; Gomes et al., 2020). In addition, as multiple outbreaks of E. albertii have recently been reported (Konno et al., 2012; Ooka et al., 2013; Masuda et al., 2020), rapid strain typing systems, such as the PCR-based O- and H-genotyping systems widely used for E. coli (Iguchi et al., 2015; Banjo et al., 2018), should be useful for E. albertii outbreak investigation and surveillance. In E. albertii, although only a genotyping system based on the variation in O-antigen biosynthesis genes has been developed thus far (Ooka et al., 2019), rapid, and low-cost effective H-genotyping system are useful to increase the discrimination power and to assist epidemiological studies in combination with O-genotyping system.
H-antigens (flagellins) are used for the serotyping of strains in many Gram-negative bacteria (Orskov and Orskov, 1992). In E. coli, flagellin is encoded by the fliC gene in the fliY-T region on the chromosome or its homologs, such as fliK, fliA, and fimA (Ratiner, 1998; Wang et al., 2003; Tominaga, 2004; Feng et al., 2008; Ratiner et al., 2010), and a total of 53 H-antigens have been identified thus far. The flagellar filament is composed of a single protein, flagellin. Flagellin is composed of four major domains: the N- and C-terminal domains (D0 and D1, respectively) form the inner and outer tubules of the flagellum, respectively, and internal D2 and D3 domains are exposed on the surface of the flagellar filament. The D0 and D1 domains are highly conserved among bacterial species, whereas the D2 and D3 domains are highly variable even between strains belonging to the same species (Samatey et al., 2001). In E. albertii, the gene cluster associated with flagellar biosynthesis and its regulation, including the fliY-T region, is conserved in most strains (Ooka et al., 2015). Although the flagellum is not produced under routine culture conditions, it has recently been revealed that its production is induced under conditions of low temperature and nutrient limitation (Ikeda et al., 2020; Murakami et al., 2020). However, the sequence variation of the fliC gene has not yet been examined. To clarify this issue, we systematically analyzed the fliC genes of 243 E. albertii strains sequenced thus far and compared them with the sequences of 53 known E. coli H-antigens. In addition, we attempted to develop a multiplex PCR-based H-genotyping system for E. albertii strains based on the sequence diversity of their fliC genes.
In this study, we analyzed the 243 E. albertii genome sequences used in our previous study (Ooka et al., 2019). Detailed information of the strains is shown in Supplementary Table 1. The strain information for the 92 E. albertii strains used for the evaluation of EAH-genotyping PCR is provided in Supplementary Table 2.
In previously sequenced E. albertii genomes, the fliC gene has been found to be located between fliA and fliD (Ooka et al., 2015). In the first-step analysis, as it is known that fliC sequences are highly diverse in E. coli (Ratiner et al., 2010), the fliC-containing regions of the 243 E. albertii genomes were identified by blastn search using the fliA and fliD sequences of E. albertii strain CB9786 as queries, with an E-value threshold of 0.01. The fliC gene of each strain was then manually annotated with in silico Molecular Cloning Genomics Edition software version 7.29L (IMC-GE; In Silico Biology, Japan). For the strains not identified the fliC gene in the first-step analysis, we performed the second-step analysis by blastn search using the fliC sequences identified in the first step analysis or the primer sequences designed for EAH-genotyping PCR, which was described below, as queries, with an E-value threshold of 0.01.
Multiple alignment of nucleotide sequences of the fliC gene of E. albertii strains and amino acid sequences of the flagellin of E. albertii and Salmonella Typhimurium strain SJW1103 was prepared using GENETYX (version 15.0.1). After identical sequences showing no SNPs were deduplicated, the nucleotide sequence alignment of E. albertii fliC genes with those of 53 known E. coli H-serotypes (H1–H56, but missing H13, H22, and H50; Supplementary Table 3) was performed using the ClustalW program in MEGA (version 7.0.26) with the default parameters (Kumar et al., 2016).
The phylogenetic analysis of fliC genes was performed with MEGA using the obtained multiple alignment. A phylogenetic tree was reconstructed by the neighbor-joining (NJ) method with the p-distance model. Bootstrap analysis with 1000 replicates was performed to assess the significance of internal branching. The core-gene SNP-based maximum-likelihood (ML) phylogenetic tree of the 243 E. albertii strains was constructed previously (Ooka et al., 2019) using RAxML v8 (Stamatakis, 2014) and displayed and annotated using iTOL v41 (Letunic and Bork, 2016).
Based on the variation in the sequences of the E. albertii fliC genes, we designed four pairs of PCR primers to specifically detect each of the four E. albertii H-genotypes (EAHgs) (Table 1). As a positive control for PCR and a genetic marker of E. albertii, one primer pair targeting an E. albertii-specific region (E_al_1_NF/NR primers) (Ooka et al., 2019) was also included in the primer set. Template DNA for PCR was prepared by the alkaline boiling method. KOD -Multi&Epi- DNA polymerase (TOYOBO, Osaka, Japan) was used for PCR. Each reaction mixture (25 μl) contained 1 μl of template DNA, each primer at 1 μM, and 0.5 U of polymerase. PCR was performed with 25 cycles of 94°C for 2 min for initial denaturation, followed by 10 s at 98°C, 30 s at 60°C, and 60 s at 68°C. The PCR products were analyzed by agarose electrophoresis using 2% agarose S (Nippon Gene, Tokyo, Japan).
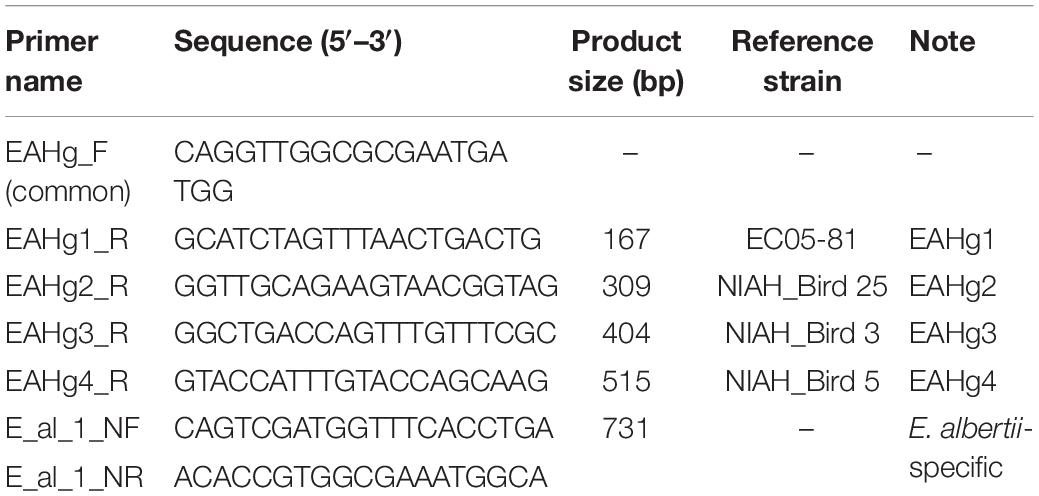
Table 1. Primer information for EAH-genotyping PCR.
Among the 243 E. albertii genomes examined by the first and second step analysis, intact and partial sequence of the fliC genes were identified in 231 and 9 genomes and no sequences were detected in the remaining 3 genomes (Supplementary Table 1). Through the clustering analysis of the 231 of intact fliC gene sequences, we identified a total of 73 sequence types with one or more SNPs (Supplementary Table 1), among which 42 were singletons, and 31 were composed of sequences from multiple genomes (named clusters C1–C31).
The phylogenetic analysis of the 73 fliC sequences identified in E. albertii with 53 fliC sequences of known E. coli H-serotypes as references revealed that the fliC genes of E. albertii formed a monophyletic branch, separate from those of E. coli (Figure 1A). To obtain more detailed information on the sequence variation in E. albertii fliC genes, we performed a phylogenetic analysis of only the E. albertii fliC genes (Figure 1B). This analysis revealed that the E. albertii fliC genes can be divided into four distinct groups, in which the nucleotide sequence identities between the groups were less than 90%, and those within each group were over 97%. We defined these four groups as the genotypes of E. albertii fliC genes and named them E. albertii H-genotypes 1–4 (EAHg1–EAHg4). In addition, by the primer screening analysis for the nine strains with partial sequences of the fliC gene, all nine strains could be genotyped into either of the four EAHgs. Together with the results of in silico analysis of 231 genome-sequenced strains, the most dominant type among the 243 E. albertii genomes was EAHg4 (109 strains; 44.9%), followed by EAHg1 (59 strains; 24.3%), EAHg3 (50 strains; 20.6%), EAHg2 (22 strains; 9.0%), and no fliC gene (3 strains; 1.2%) (Table 2).
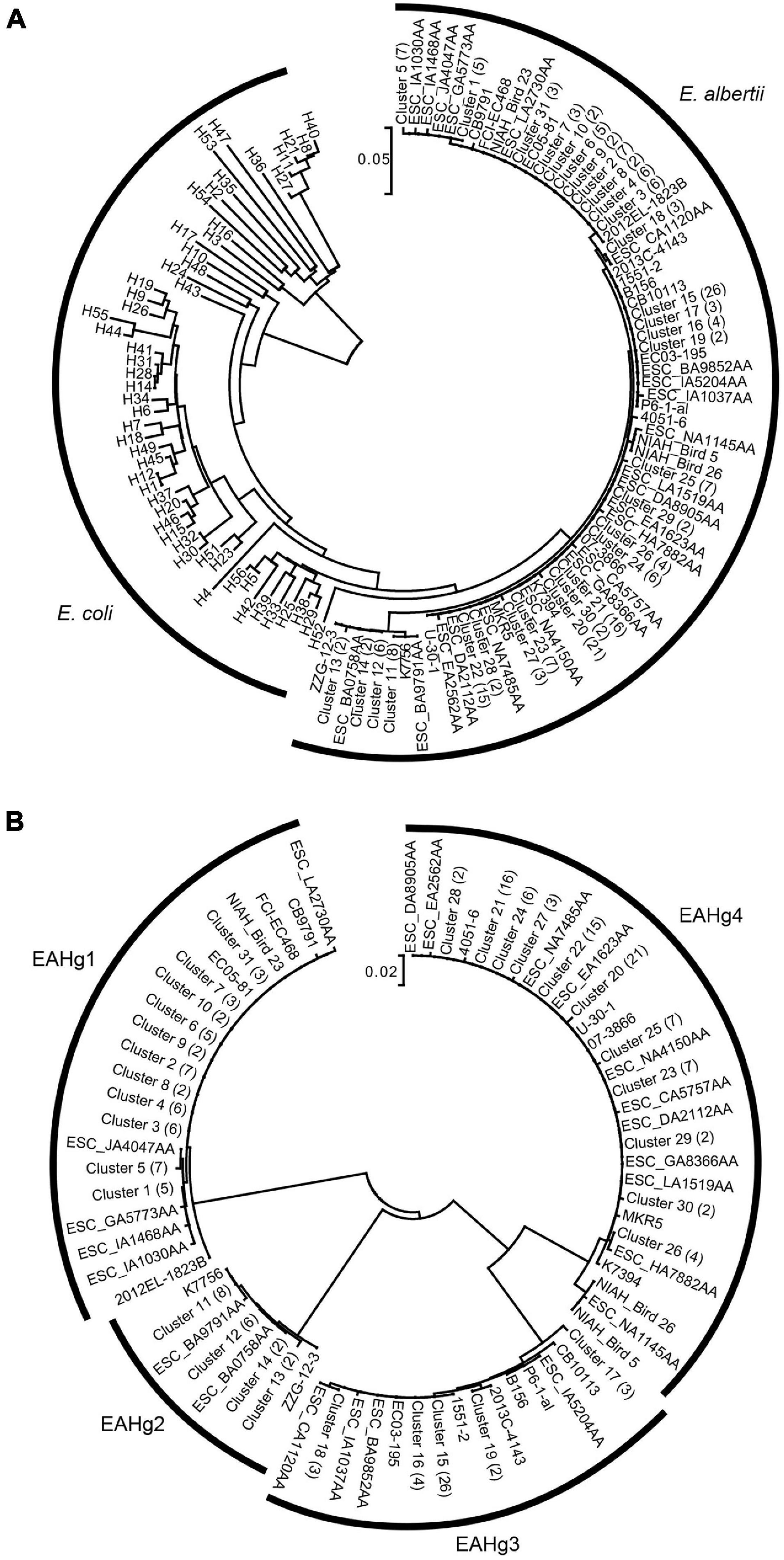
Figure 1. (A) Neighbor-joining tree of 73 fliC sequences identified from 231 E. albertii strains. The sequences of 53 known fliC genotypes of E. coli are included as references. The number of strains with identical fliC sequences is shown in parentheses for each cluster. (B) Neighbor-joining tree of only the 73 fliC sequences of E. albertii. The number of strains with identical fliC sequences is shown in parentheses for each cluster.

Table 2. Summary of in silico- and PCR-based EAH genotyping.
Amino acid sequence comparison of the representative fliC-encoded flagellin of the four genotypes and Salmonella Typhimurium strain SJW1103 revealed that while the sequences of the D0 and D1 domains were highly conserved, those of the D2 and D3 domains were variable (Supplementary Figure 1).
We designed a multiplex PCR primer set (Table 1) based on the sequence variation in the fliC genes of the four EAHgs. A forward universal primer was designed based on the highly conserved sequences of all four EAHgs, and reverse primers were designed based on the variable regions of each EAHg (Supplementary Figure 2), so that the amplicons generated with the combinations of the universal forward primer and the reverse primers exhibited a ladder pattern that ranged from 167 to 515 bp. One E. albertii-specific primer pair (E_al_1_NF/E_al_1_NR) (Ooka et al., 2015) was included in the primer set as a marker to detect E. albertii and as a positive control for PCR. The examination of the primer set in four strains representing each of the four EAHgs confirmed that the primer set yielded PCR products of the expected sizes for each genotype (Figure 2). To validate the PCR-based genotyping system, we performed the comparison of in silico and PCR-based genotyping on the same dataset of genomes. In this analysis, only 42 strains were used because the remaining 201 strains are not available in our laboratories. As shown in Supplementary Figure 3, all of the 42 strains exhibited the same results between in silico and the PCR-based genotyping. In addition, we applied the mixed DNA samples containing four strains with different EAH-genotypes as a template for the system to evaluate the specificity and obtained the primer set yielded PCR products of the expected sizes for four genotypes (Supplementary Figure 4).
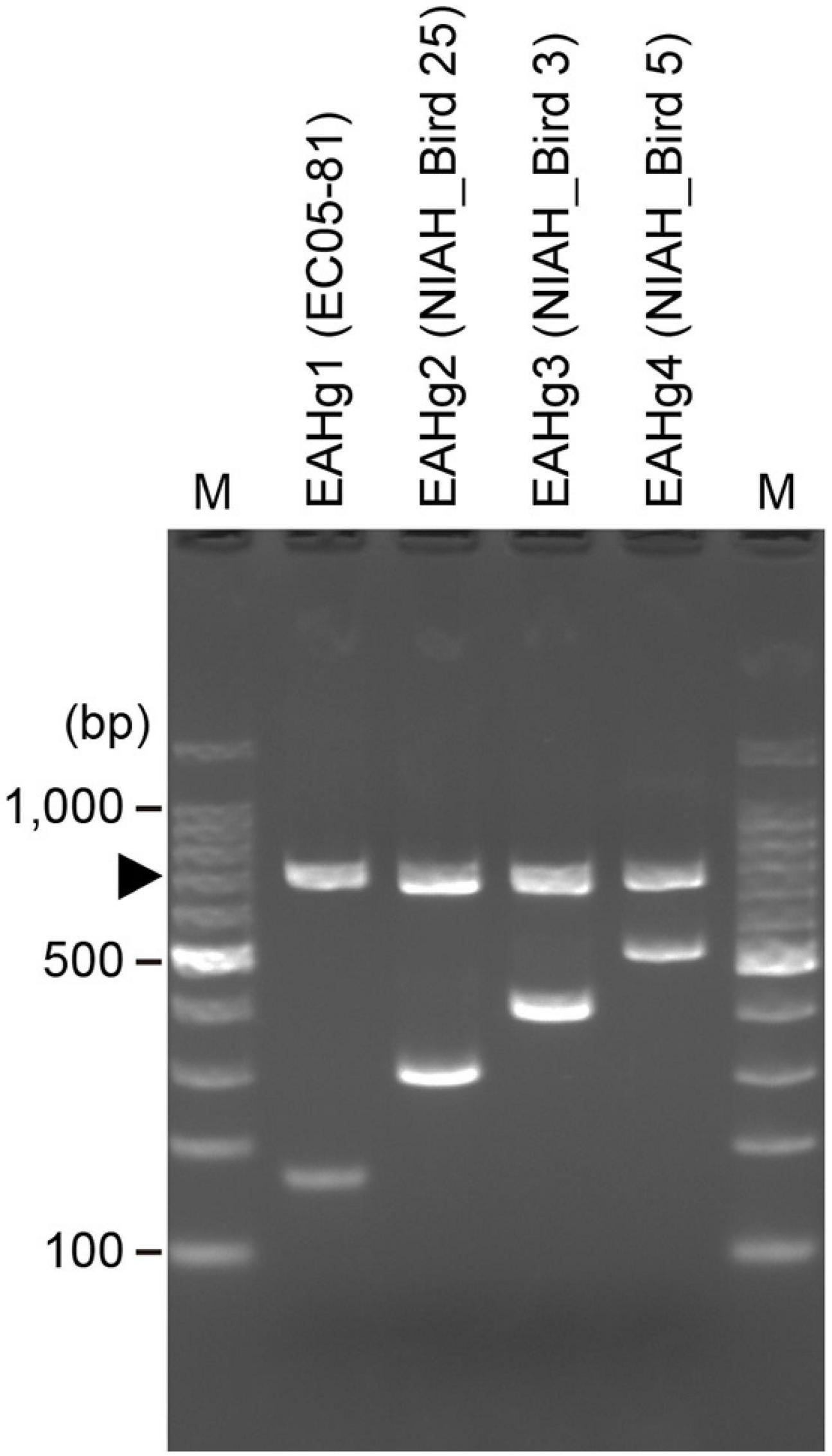
Figure 2. Electrophoresis patterns obtained by EAH-genotyping PCR. A total of four strains representing the four EAHgs were analyzed using a PCR primer mix designed in this study. Strain names are indicated in parentheses. An arrowhead indicates the bands derived from the E. albertii-specific primer pair E_al_1_NF/NR. Lane M, 100 bp DNA ladder.
To further evaluate the performance of the system, we determined the H-genotypes of 92 E. albertii strains using this system. These strains were isolated from diarrheal patients and birds in various regions of Japan. In this analysis, we were able to genotype all strains (Supplementary Table 2). Similar to the results of in silico analysis of the 240 genome-sequenced strains, EAHg4 (48 strains; 43.6%) was found to be predominant, followed by EAHg1 (29 strains; 26.4%), EAHg2 (18 strains; 16.4%), and EAHg3 (15 strains; 13.6%) (Table 2). These results suggest that our system is useful for the H-genotyping of E. albertii and that the four genotypes cover the most of the diversity of H-genotypes in the E. albertii lineage.
Finally, we investigated the relationship between the phylogeny of the genome-sequenced strains used in this study and the distribution of the four EAHgs and the 40 Escherichia albertii O-genotypes (EAOgs), identified in our previous study (Ooka et al., 2019), in these strains by mapping H- and O-genotype information in a core-gene sequence-based ML phylogenetic tree of the strains (Figure 3). This analysis revealed that although very closely related strains shared the same H-genotype, each EAHg appeared in multiple sublineages in both clades 1 and 2, suggesting relatively frequent within-species transfer of fliC genes in E. albertii. In addition, there is no correlation between the combination of the H- and O-genotypes and their phylogenetic relationship.

Figure 3. Phylogenetic view of the 225 E. albertii strains that have been genome sequenced thus far (9) and the distribution of the four EAHgs and the 40 EAOgs in these strains. Strain names are indicated at each tip, and the 14 completely sequenced strains are indicated. Information on the distribution of the four EAHgs and the 40 EAOgs are also shown.
In this study, we analyzed the sequence variation of fliC genes among 231 genome-sequenced E. albertii strains and identified 73 sequence types, which were grouped into four clearly distinguishable genotypes (EAHg1–EAHg4). The four genotypes showed >97% sequence identity within each group and <90% identity between groups. As seen in E. coli fliC genes (Samatey et al., 2001), the sequences encoding domains D0 and D1 (corresponding to the N- and C-terminal regions of flagellin, respectively) were highly conserved, but those of domains D2 and D3 (forming the surface exposed region) were variable between H-genotypes (Supplementary Figure 1). In our previous analysis (Ooka et al., 2015), we revealed that flagellar biosynthesis-related genes other than fliC show high conservation of nucleotide sequences (>90% identity). Therefore, it appears that the fliC gene is under a certain amount of immunological selection in its hosts or environmental selection. However, it was notable that only four H-genotypes were identified in E. albertii, in sharp contrast to the situation in E. coli, in which as many as 53 H-genotypes have been identified. Although the analysis of more E. albertii strains will be required to confirm the low level of variation in their fliC genes, this finding may suggest the possibility that this species is living in limited natural hosts or environments and/or that their flagella are required in a limited stage in their life cycle. This possibility may receive some support from the recent finding that flagella are produced only at lower temperatures and under nutrient-limited conditions (Ikeda et al., 2020; Murakami et al., 2020). It is also noteworthy that although a low level of sequence variation was observed for E. albertii fliC genes, we detected a sign of relatively frequent within-species transfer of this gene in E. albertii (Figure 3). However, none of the four H-genotypes identified in this study were closely related to any of the 53 H-genotypes of E. coli, suggesting the absence or very rare occurrence of interspecies transfer of fliC between E. albertii and E. coli. This is in marked contrast to the situation for O-antigen biosynthesis loci, which show clear signs of interspecies transfer between the two species (Ooka et al., 2019). Although the mechanism(s) generating such a difference is currently unknown, there may be some restriction of the flagellin sequence of E. albertii.
Although only four H-genotypes were identified in E. albertii, the multiplex PCR system that we constructed to identify H-genotypes in E. albertii will be a useful tool for epidemiological studies of E. albertii infections, particularly if used in combination with the O-genotyping system that we previously constructed (Ooka et al., 2019). In addition, the H-genotyping system will be useful for further analyzing the diversity of fliC genes in E. albertii or searching for additional genotypes.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
KN and TO designed the study and wrote the manuscript. KM and YH-K provided the samples. KN, TO, YG, NI, and YoO analyzed the data. TO, YaO, TH, and JN were responsible for supervision and management of the study. All authors contributed to the article and approved the submitted version.
This work was supported by the Japan Society for the Promotion of Science KAKENHI (Grant Numbers 25460539, 16K08781, and 20K07498 to TO, and 20310116 and 221S0002 to TH), and a Health Labour Sciences Research Grant (H30-Shokuhin-Ippan-001 to YH-K).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank F. Funakura, S. Yamada, and T. Oku for providing technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.737979/full#supplementary-material
Albert, M. J., Faruque, S. M., Ansaruzzaman, M., Islam, M. M., Haider, K., Alam, K., et al. (1992). Sharing of virulence-associated properties at the phenotypic and genetic levels between enteropathogenic Escherichia coli and Hafnia alvei. J. Med. Microbiol. 37, 310–314. doi: 10.1099/00222615-37-5-310
Banjo, M., Iguchi, A., Seto, K., Kikuchi, T., Harada, T., Scheutz, F., et al. (2018). Escherichia coli H-Genotyping PCR: a Complete and Practical Platform for Molecular H Typing. J. Clin. Microbiol. 56, e00190–18. doi: 10.1128/JCM.00190-18
Feng, L., Liu, B., Liu, Y., Ratiner, Y. A., Hu, B., Li, D., et al. (2008). A genomic islet mediates flagellar phase variation in Escherichia coli strains carrying the flagellin-specifying locus flk. J. Bacteriol. 190, 4470–4477. doi: 10.1128/JB.01937-07
Gomes, T. A. T., Ooka, T., Hernandes, R. T., Yamamoto, D., and Hayashi, T. (2020). Escherichia albertii Pathogenesis. EcoSal Plus 9, 1–18. doi: 10.1128/ecosalplus.ESP-0015-2019
Huys, G., Cnockaert, M., Janda, J. M., and Swings, J. (2003). Escherichia albertii sp. nov., a diarrhoeagenic species isolated from stool specimens of Bangladeshi children. Int. J. Syst. Evol. Microbiol. 53, 807–810. doi: 10.1099/ijs.0.02475-0
Iguchi, A., Iyoda, S., Seto, K., Morita-Ishihara, T., Scheutz, F., Ohnishi, M., et al. (2015). Escherichia coli O-Genotyping PCR: a Comprehensive and Practical Platform for Molecular O Serogrouping. J. Clin. Microbiol. 53, 2427–2432. doi: 10.1128/JCM.00321-15
Ikeda, T., Shinagawa, T., Ito, T., Ohno, Y., Kubo, A., Nishi, J., et al. (2020). Hypoosmotic stress induces flagellar biosynthesis and swimming motility in Escherichia albertii. Commun. Biol. 3:87. doi: 10.1038/s42003-020-0816-5
Konno, T., Yatsuyanagi, J., Takahashi, S., Kumagai, Y., Wada, E., Chiba, M., et al. (2012). Isolation and identification of Escherichia albertii from a patient in an outbreak of gastroenteritis. Jpn. J. Infect. Dis. 65, 203–207. doi: 10.7883/yoken.65.203
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Masuda, K., Ooka, T., Akita, H., Hiratsuka, T., Takao, S., Fukada, M., et al. (2020). Epidemiological Aspects of Escherichia albertii Outbreaks in Japan and Genetic Characteristics of the Causative Pathogen. Foodborne Pathog. Dis. 17, 144–150. doi: 10.1089/fpd.2019.2654
Murakami, K., Kimura, S., Nagafuchi, O., Sekizuka, T., Onozuka, D., Mizukoshi, F., et al. (2020). Flagellum expression and swimming activity by the zoonotic pathogen Escherichia albertii. Environ. Microbiol. Rep. 12, 92–96. doi: 10.1111/1758-2229.12818
Oaks, J. L., Besser, T. E., Walk, S. T., Gordon, D. M., Beckmen, K. B., Burek, K. A., et al. (2010). Escherichia albertii in wild and domestic birds. Emerg. Infect. Dis. 16, 638–646. doi: 10.3201/eid1604.090695
Ooka, T., Ogura, Y., Katsura, K., Seto, K., Kobayashi, H., Kawano, K., et al. (2015). Defining the Genome Features of Escherichia albertii, an Emerging Enteropathogen Closely Related to Escherichia coli. Genome Biol. Evol. 7, 3170–3179. doi: 10.1093/gbe/evv211
Ooka, T., Seto, K., Kawano, K., Kobayashi, H., Etoh, Y., Ichihara, S., et al. (2012). Clinical significance of Escherichia albertii. Emerg. Infect. Dis. 18, 488–492. doi: 10.3201/eid1803.111401
Ooka, T., Seto, K., Ogura, Y., Nakamura, K., Iguchi, A., Gotoh, Y., et al. (2019). O-antigen biosynthesis gene clusters of Escherichia albertii: their diversity and similarity to Escherichia coli gene clusters and the development of an O-genotyping method. Microb. Genom. 5:e000314. doi: 10.1099/mgen.0.000314
Ooka, T., Tokuoka, E., Furukawa, M., Nagamura, T., Ogura, Y., Arisawa, K., et al. (2013). Human gastroenteritis outbreak associated with Escherichia albertii, Japan. Emerg. Infect. Dis. 19, 144–146. doi: 10.3201/eid1901.120646
Orskov, F., and Orskov, I. (1992). Escherichia coli serotyping and disease in man and animals. Can. J. Microbiol. 38, 699–704.
Ratiner, Y. A. (1998). New flagellin-specifying genes in some Escherichia coli strains. J. Bacteriol. 180, 979–984. doi: 10.1128/JB.180.4.979-984.1998
Ratiner, Y. A., Sihvonen, L. M., Liu, Y., Wang, L., and Siitonen, A. (2010). Alteration of flagellar phenotype of Escherichia coli strain P12b, the standard type strain for flagellar antigen H17, possessing a new non-fliC flagellin gene flnA, and possible loss of original flagellar phenotype and genotype in the course of subculturing through semisolid media. Arch. Microbiol. 192, 267–278. doi: 10.1007/s00203-010-0556-x
Samatey, F. A., Imada, K., Nagashima, S., Vonderviszt, F., Kumasaka, T., Yamamoto, M., et al. (2001). Structure of the bacterial flagellar protofilament and implications for a switch for supercoiling. Nature 410, 331–337. doi: 10.1038/35066504
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tominaga, A. (2004). Characterization of six flagellin genes in the H3, H53 and H54 standard strains of Escherichia coli. Genes Genet. Syst. 79, 1–8. doi: 10.1266/ggs.79.1
Keywords: Escherichia albertii, flagellin, fliC, H-antigen, genotyping
Citation: Nakae K, Ooka T, Murakami K, Hara-Kudo Y, Imuta N, Gotoh Y, Ogura Y, Hayashi T, Okamoto Y and Nishi J (2021) Diversification of Escherichia albertii H-Antigens and Development of H-Genotyping PCR. Front. Microbiol. 12:737979. doi: 10.3389/fmicb.2021.737979
Received: 08 July 2021; Accepted: 07 October 2021;
Published: 01 November 2021.
Edited by:
Xiyang Wu, Jinan University, ChinaReviewed by:
Marianne De Paepe, Institut National de Recherche pour l’Agriculture, l’Alimentation et l’Environnement (INRAE), FranceCopyright © 2021 Nakae, Ooka, Murakami, Hara-Kudo, Imuta, Gotoh, Ogura, Hayashi, Okamoto and Nishi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tadasuke Ooka, dGFvaG9rYTFAbS5rdWZtLmthZ29zaGltYS11LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.