 William L. King1
William L. King1 Mirjam Kaestli2
Mirjam Kaestli2 Nachshon Siboni1
Nachshon Siboni1 Anna Padovan2
Anna Padovan2 Keith Christian2
Keith Christian2 David Mills3
David Mills3 Justin Seymour1
Justin Seymour1 Karen Gibb2*
Karen Gibb2*- 1Climate Change Cluster, University of Technology Sydney, Sydney, NSW, Australia
- 2Research Institute for the Environment and Livelihoods, Charles Darwin University, Darwin, NT, Australia
- 3Genecology Research Centre, University of the Sunshine Coast, Sunshine Coast, QLD, Australia
Diseases of bivalves of aquacultural importance, including the valuable Australian silver-lipped pearl oyster (Pinctada maxima), have been increasing in frequency and severity. The bivalve microbiome is linked to health and disease dynamics, particularly in oysters, with putative pathogens within the Vibrio genus commonly implicated in oyster diseases. Previous studies have been biased toward the Pacific oyster because of its global dominance in oyster aquaculture, while much less is known about the microbiome of P. maxima. We sought to address this knowledge gap by characterizing the P. maxima bacterial community, and we hypothesized that bacterial community composition, and specifically the occurrence of Vibrio, will vary according to the sampled microenvironment. We also predicted that the inside shell swab bacterial composition could represent a source of microbial spillover biofilm into the solid pearl oyster tissues, thus providing a useful predictive sampling environment. We found that there was significant heterogeneity in bacterial composition between different pearl oyster tissues, which is consistent with patterns reported in other bivalve species and supports the hypothesis that each tissue type represents a unique microenvironment for bacterial colonization. We suggest that, based on the strong effect of tissue-type on the pearl oyster bacterial community, future studies should apply caution when attempting to compare microbial patterns from different locations, and when searching for disease agents. The lack of association with water at each farm also supported the unique nature of the microbial communities in oyster tissues. In contrast to the whole bacterial community, there was no significant difference in the Vibrio community among tissue types nor location. These results suggest that Vibrio species are shared among different pearl oyster tissues. In particular, the similarity between the haemolymph, inside shell and solid tissues, suggests that the haemolymph and inside shell environment is a source of microbial spillover into the oyster tissues, and a potentially useful tool for non-destructive routine disease testing and early warning surveillance. These data provide important foundational information for future studies identifying the factors that drive microbial assembly in a valuable aquaculture species.
Introduction
There is growing evidence that the microbial communities living in association with a diverse range of animal hosts significantly contribute to host behavior, physiology and health (McFall-Ngai et al., 2013; Raina et al., 2018). Within a host, each tissue represents a unique microenvironment which facilitates distinct host-microbial interactions (Lokmer et al., 2016; King et al., 2020). For example, intestinal-associated microbial communities are commonly involved in nutrient mineralization and uptake for the host (Sonnenburg et al., 2004; Seth and Taga, 2014). For marine organisms, microbial communities contribute to important physiological processes including nutrient uptake and host defenses (Siboni et al., 2008; Glasl et al., 2016; Pita et al., 2018).
In recent years, diseases of marine organisms, particularly bivalves of aquacultural importance, have been increasing in frequency and severity (King et al., 2019a, b). There is a growing body of research that has linked microbiome composition to bivalve health and disease dynamics, particularly within oysters (Trabal et al., 2012; Trabal Fernández et al., 2014). For example, species assigned to the Vibrio genus are commonly implicated in oyster diseases (Wegner et al., 2013; Wendling et al., 2014; King et al., 2019a, b, c, d), whereby during the early stage of disease event, commensal vibrios are often replaced by phylogenetically similar pathogenic vibrios (Lemire et al., 2015). Therefore, it has been proposed that characterizing and understanding shifts in the Vibrio population could be important for predicting disease events (King et al., 2019c).
The silver-lipped pearl oyster (Pinctada maxima) is prized for its ability to produce large, high quality nacreous pearls and forms the basis of the valuable Australian pearling industry. At its peak, this industry was worth $200 million per year, yet is now valued at below $50 million per year, and still falling, due in part to largely unexplained disease events (Joint Select Committee on Northern Australia, 2016). Although there is an imperative to focus on pearl oyster health research to allow the industry to return pearl production as a major Australian Aquaculture industry, studies of bivalve bacterial composition (and the microbiome) are biased toward the Pacific oyster (Crassostrea gigas), because of its global dominance in oyster aquaculture (King et al., 2019b). While less is known about the microbiome of pearl oysters, Dubé et al. (2019) recently characterized the P. margaritifera microbiome using 16S rRNA gene sequencing and reported that the microbial communities were tissue specific.
Vibrios have been implicated in pearl oyster diseases internationally (Wang et al., 2016), including Australia (Negri et al., 2004), and were reported in low abundance in the P. margaritifera (Dubé et al., 2019) and P. fucata martensii bacterial communities (Zheng et al., 2021). It is still not clear what factors govern bacterial assemblage structure within the pearl oyster and to what extent putative pathogens, including Vibrios, influence pearl oyster health and productivity. Pearl oysters are subject to multiple stressors with the increasing adoption of aquaculture techniques (Adzigbli et al., 2020). Thus, to minimize the susceptibility of the oysters to disease, non-destructive, “least-stress” disease monitoring methods, such as inner shell swabs or haemolymph sampling, are needed. However, these approaches would only be of value if the microbiome of these samples were representative of those from other tissues.
Given the importance of microbial communities in physiological processes and disease prediction, our first objective was to provide the first characterization of the bacterial community of P. maxima by defining the bacterial communities in different oyster tissues. A second objective was to determine patterns in Vibrio, given their implication in disease in other oyster species. Based on previous studies, we hypothesized that bacterial and Vibrio composition would vary according to the sampled microenvironment and that the inside shell swab bacterial composition could represent a source of microbial spillover biofilm into the solid pearl oyster tissues, thus providing a useful predictive sampling environment. In addition to providing evidence in support of a non-destructive sampling technique, this study provides foundational information about the microbial assembly in a valuable, understudied aquaculture species.
Meterials and Methods
Sites and Sample Processing
Pinctada maxima tissue and seawater samples were collected in April 2018 from two pearl oyster farm sites in Western Australia, namely Seaflower Bay (SF) and Wargul Wargul Bay (WW), which are located in the Vansittart Bay region (12.438241 S 130.796684 E) of the northern Kimberley. Three unseeded oysters ranging in size from 90 to 95 mm (dorso-ventral) were harvested from SF (numbers limited as they were brood stock) and 10 oysters (seeded 22 months earlier) ranging from 150 to 170 mm (dorso-ventral) were harvested from WW. The outer shell of each oyster was swabbed using sterile Copan Rayon Tip swabs (Interpath Services, catalog:155CIS). A processing knife was used to cleanly sever the adductor muscle of each oyster, before the nacreous inside shell surface was swabbed. The mantle was then detached from one shell valve using a sterile scalpel blade, and using a 22-gauge needle, 200–500 μL of haemolymph was collected from the auricle. Tissue from the mantle, gill, digestive diverticula, large intestine and heart (auricles and ventricle) were dissected from each oyster and all samples were placed into separate, sterile tubes and stored at −80°C until extraction. Seawater (250 mL) collected from each site was filtered through a 0.22 μm mixed cellulose membrane.
Genomic DNA (gDNA) from the tissue and haemolymph samples was extracted using the Qiagen DNeasy Blood and Tissue Kit (catalog: 69504) according to the manufacturer’s instructions. The swabs and filtered water samples were extracted using the MP Biomedicals FastDNATM SPIN Kit for Soil (catalog: 6560200). Purity ratio (260/280) and the DNA concentration in each sample was quantified using the NanoDropTM One (Thermo ScientificTM). A total of 106 samples were processed across the two oyster farms.
16S rRNA and Vibrio-Centric hsp60 Amplicon Sequencing
DNA was amplified using: (1) the primers Bakt_341F and Bakt_805R which amplify the V3-V4 region of the bacterial 16S rRNA gene (Herlemann et al., 2011) and (2) the Vibrio-centric hsp60 primers Vib-hspF3-23 and Vib-hspR401-422, as previously described (King et al., 2019c), to characterize the composition and diversity of the entire bacterial assemblage and Vibrio community, respectively. PCR conditions for 16S rRNA amplification were as follows: 95°C for 3 min, 25 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, and a final extension at 72°C for 5 min. For the Vibrio-centric hsp60 assay a 30 μL PCR reaction mixture was prepared using an epMotion 5075l Automated Liquid Handling System (Eppendorf South Pacific) to limit cross sample contamination and the PCR protocol was performed as previously described (King et al., 2019c). Amplicons were sequenced using the Illumina MiSeq platform according to the manufacturer’s guidelines (Ramaciotti Centre for Genomics, Sydney, NSW, Australia). Raw data files in FASTQ format were deposited in NCBI Sequence Read Archive (SRA) under Bioproject number PRJNA594420.
Vibrio-Centric Quantitative PCR (qPCR)
To provide a measure of Vibrio abundance, a quantitative PCR (qPCR) assay was used to quantify Vibrio- specific 16S rRNA gene copies in each sample as previously described (Thompson et al., 2004; Siboni et al., 2016; King et al., 2019c). The resulting data were normalized to milliliters of collected water or milligrams of tissue. Swabs of out- and inside of shells were excluded from this analysis.
Sequence Processing
16S rRNA gene amplicon raw demultiplexed data was processed using the Quantitative Insights into Microbial Ecology (QIIME 2 version 2018.6.0) pipeline (Bolyen et al., 2019). Briefly, paired-ended sequences were imported using the “qiime tools import” command. Sequences were then trimmed and denoised using DADA2 version 1.6, which also removes chimeras (Callahan et al., 2016). Taxonomy was assigned on the rep-set-dada2 output at the single nucleotide level using the sklearn qiime feature classifier against the Silva v132 database (Quast et al., 2013). Sequences identified at the single nucleotide threshold are henceforth denoted as amplicon sequence variants (ASVs). The dataset was further cleaned by removing ASVs which only occurred in one sample and those identified as non-bacterial, chloroplasts or mitochondria. Cleaned data were rarefied at 1,220 reads per sample with 63 samples remaining and 1,263 ASVs. Rarefaction curves indicated that the ASV richness plateaued for the majority of samples at this depth with the exception of outside shell samples.
For the Vibrio-centric hsp60 data, raw pair-ended sequences were joined using FLASH (Magoč and Salzberg, 2011) and trimmed using Mothur (Schloss et al., 2009) (parameters: maxhomop = 5, maxambig = 0, qaverage = 25, minlength = 420, maxlength = 420). These fragments were then clustered at 97% into OTUs and chimeric sequences were removed using vsearch (Rognes et al., 2016). Non-Vibrio sequences were removed by BLASTing cleaned sequences against the Vibrio hsp60 reference dataset and any sequence with similarity lower than 90% was removed. This fasta file was then used to assign taxonomy against the custom Vibrio hsp60 reference dataset with the RDP classifier. OTUs that only occurred in one sample were excluded and samples with less than 10 hsp60 sequences were also excluded. This resulted in 41 samples and 24 hsp60 OTUs. Due to the large spread of sequences per sample (11–29,163), data were not rarefied, rather sequences were normalized to the number of sequences per sample to produce the relative abundance of each taxa for each sample.
Data Analysis
All analyses were conducted in R v4.0.2 and Primer-E (v7, Quest Research Limited). Bacterial richness (observed number of ASVs based on rarefied sequencing data) was compared between locations (# 2) and tissue types (# 8 shell and tissues types) plus farm water using a quasi-Poisson model with sample type (oyster tissue type and water) and location as fixed factors (estimated overdispersion parameter 22). Model residuals were checked for influential outliers and lack of patterns across predictors and fitted values. Oysters (# 12) were added as random intercept in a mixed effect negative binomial model (glmmTMB package – nbinom1 family), but no between-oyster variance in the richness was found and the quasi-Poisson GLM model was used instead.
To visualize differences between bacterial compositions for different oyster tissue types, a Principal Coordinates Analysis (PCoA) was performed in the Phyloseq package on the Bray-Curtis dissimilarity matrix of the square-root transformed and rarefied ASV data (McMurdie and Holmes, 2013). Similarly, in order to examine whether the bacterial compositions differed between tissue types and location, a PERMANOVA test was conducted in Primer-E on the dissimilarity matrix with 999 permutations, type III sums of squares and oysters as random effect nested in location.
To identify taxa that were associated with specific oyster tissue types, a Dufrene-Legendre Indicator Species Analysis (IndVal) was performed (labdsv package) (De Cáceres et al., 2010). P-values were adjusted for multiple testing using the FDR method. Vibrio abundance based on Vibrio 16S rRNA gene qPCR data normalized per mg tissue or mL water (shell swabs excluded) was compared between tissue types and location using a Gamma (log link) mixed effect model with oysters as random intercept (glmmTMB package). No between-oyster variance in the Vibrio abundance was found and the random intercept was dropped. A PERMANOVA analysis on the Vibrio hsp60 composition was conducted as per above, whereby the relative abundances of hsp60 sequences were used in the absence of rarefied data.
Results
Bacterial Richness Was Greatest for the Outside Shell Microbiota
The observed bacterial richness significantly differed between oyster tissue types (Figure 1). Richness was significantly higher in the outer shell than in all other tissue types (P < 0.001 for all) and the inside shell and farm water (P < 0.05 for both). Accounting for oyster tissue type, there was no difference in bacterial community richness between locations.
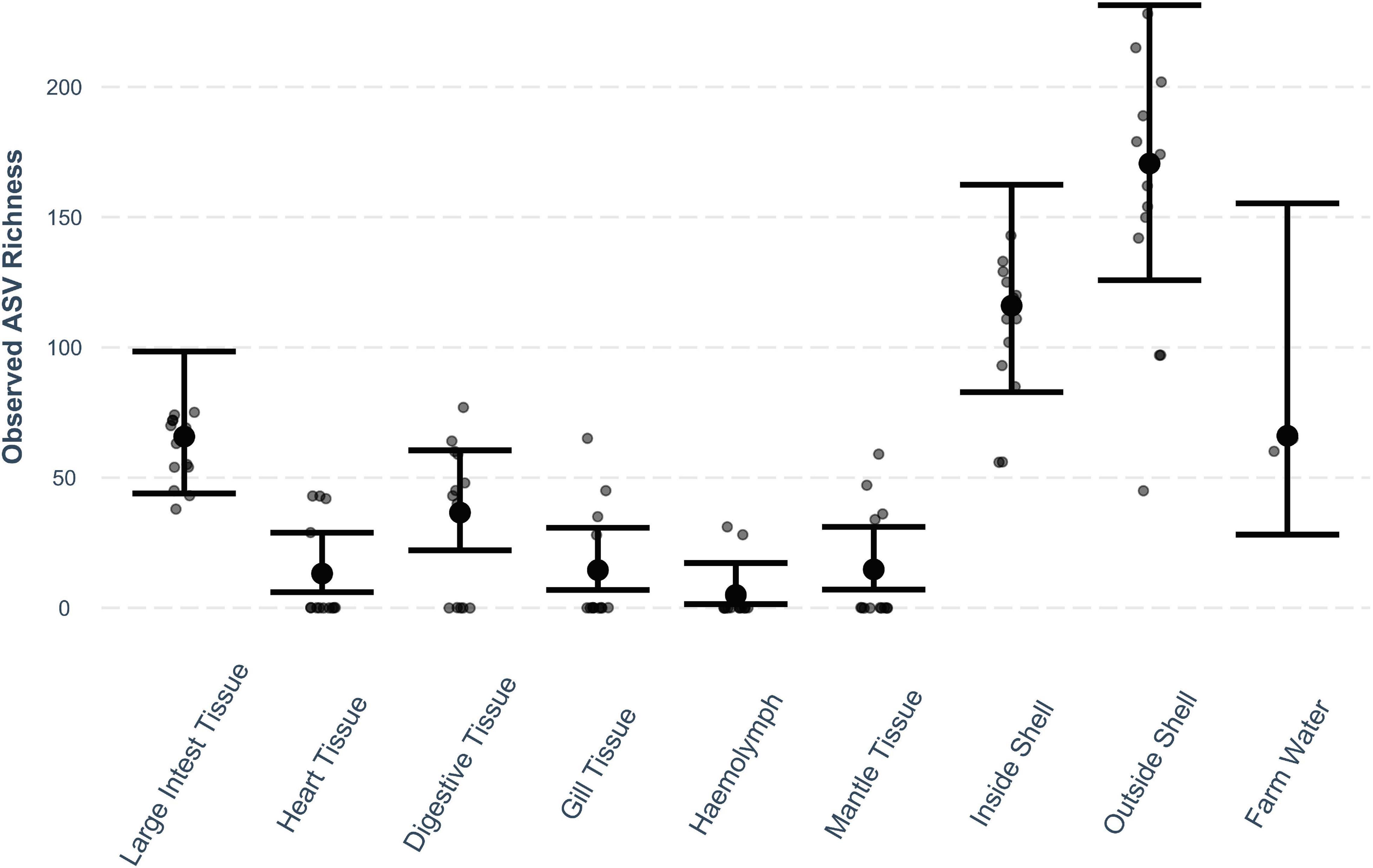
Figure 1. Observed ASV richness (small dots) across oyster tissue types and farm water. Mean estimates (large dots) are based on a quasi-Poisson model with fixed factors oyster tissue type and location. Error bars mark 95% confidence intervals.
Bacterial Composition Differed According to Location and Pearl Oyster Tissue Type
Pearl oyster-associated bacterial communities differed according to farm location (PERMANOVA: Pseudo F1, 34 = 2.0, P = 0.001) and oyster tissue (Pseudo F7, 34 = 3.0, P = 0.001) (Figure 2). In particular, bacterial communities on the outside shell differed from most other tissues including the inside shell, while bacterial assemblages of the latter also differed from the digestive tissue (P < 0.01 for all). The bacterial communities within the large intestinal tissue showed the lowest variability displaying a similar community structure across all replicates and both locations (Figure 2) at family (Figure 3) and genus (Supplementary Tables 1, 2) levels. A triangle heat map of average inter-group Bray-Curtis similarities between tissue types illustrates that bacterial communities on the outside shell differed from most other tissues including the inside shell, and the bacterial communities within the large intestinal tissue had the most similar community structure across all replicates and both locations (Supplementary Figure 1).
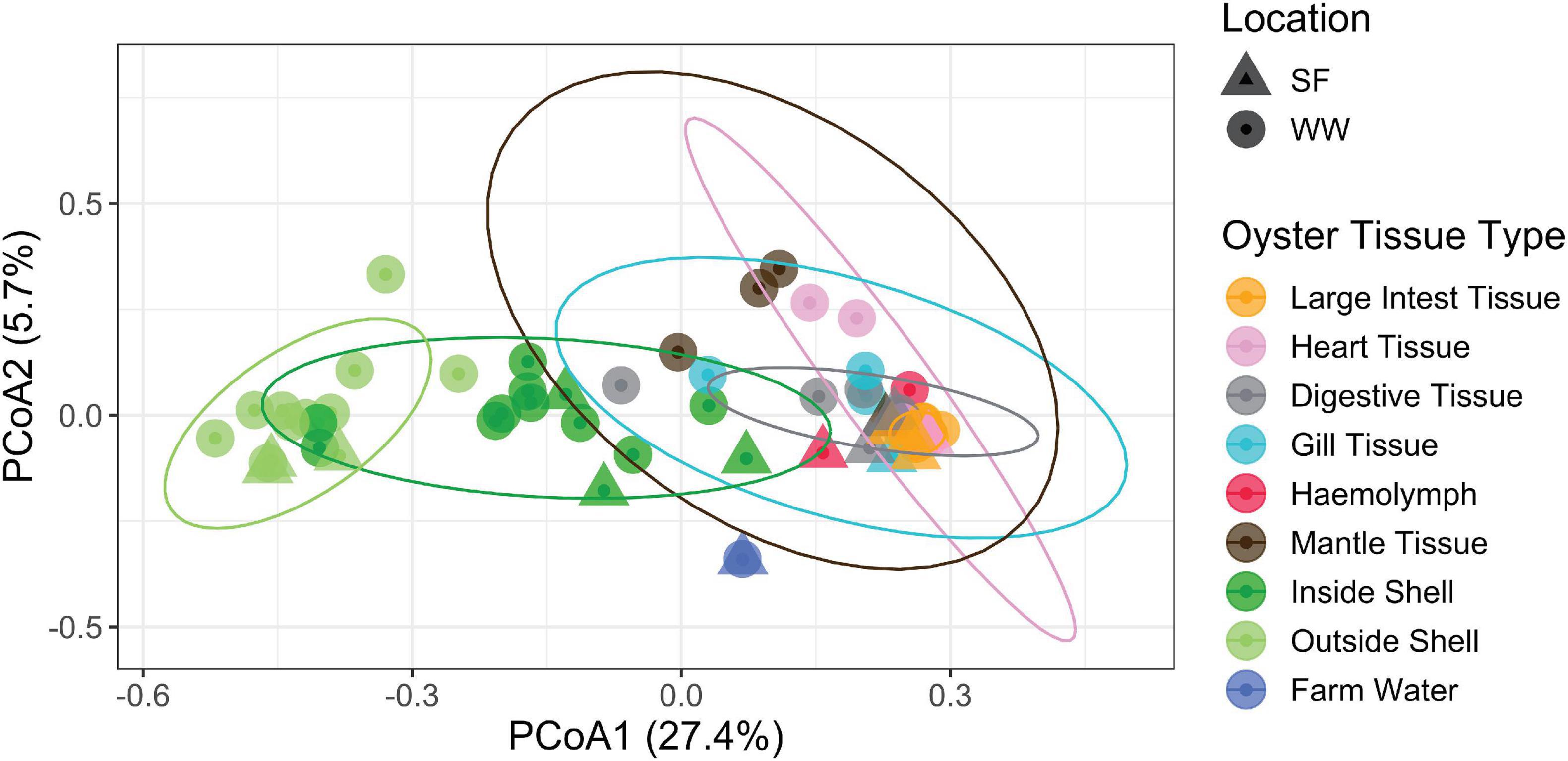
Figure 2. PCoA ordination of bacterial compositions labeled by location and oyster tissue type. The first axis explained 27.4% of the microbiota variance and the second 5.7%. Ellipses show 95% data distribution.
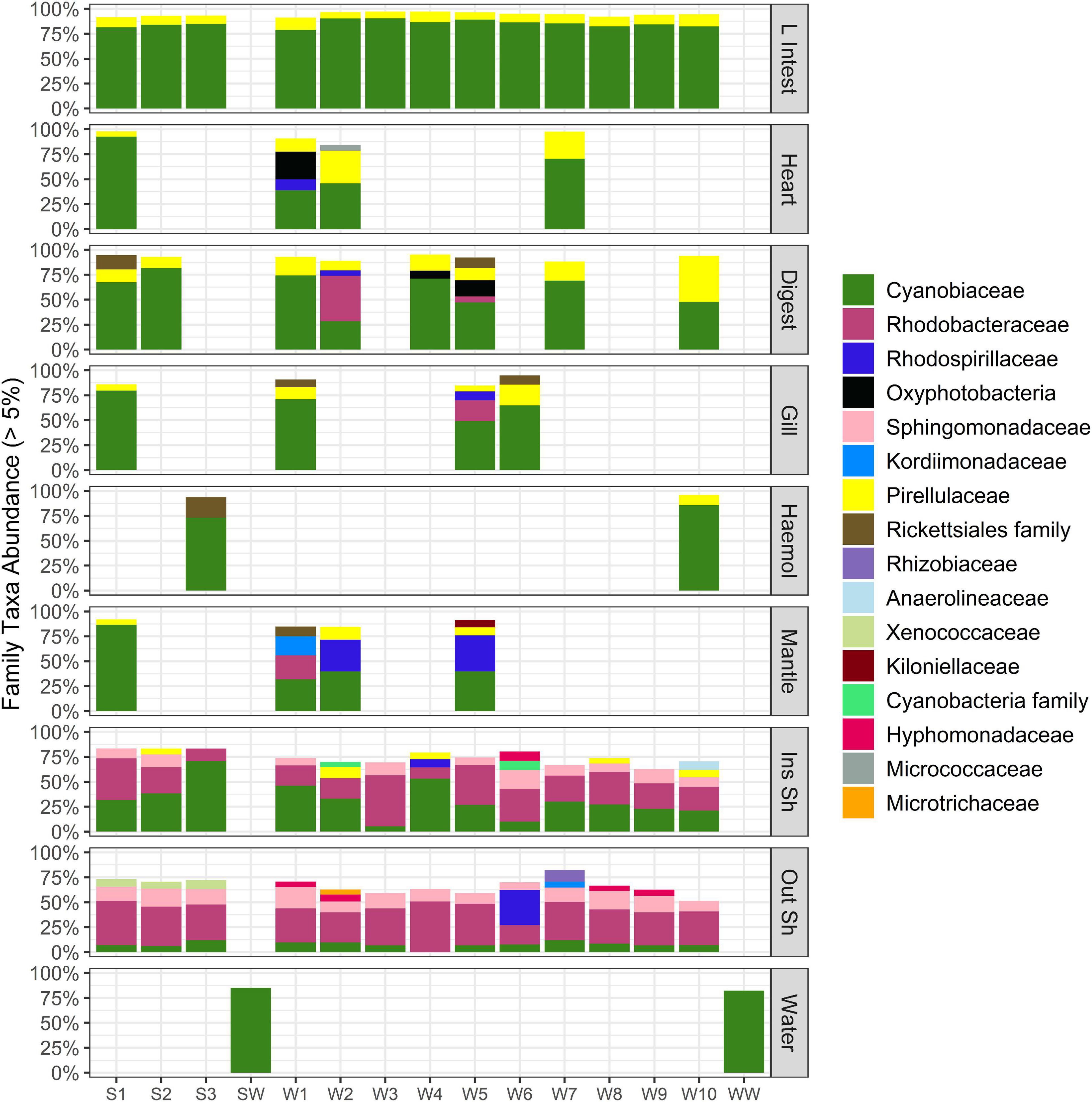
Figure 3. Stacked bar plot of bacterial taxa at family level (relative abundance > 5%) across oyster tissue types and farm water. Each column is a sample. “L Intest” large intestinal tissue, “Digest” digestive tissue, “Haemol” haemolymph, “Ins Sh” inside shell, “Out Sh” outside shell, “Water” farm water. “S,” Seaflower Bay and “W,” Wargul Wargul Bay. Only samples with > 1220 sequence reads were included.
Cyanobiaceae were the most dominant family across all oyster tissue types and farm water, with average relative abundance of 49%. The second and third most dominant families were Rhodobacteraceae and Pirellulaceae representing 15 and 8.5% average relative abundance, respectively (Figure 3). The dominance of the Cyanobiaceae family largely contributed to its over-representation in the oyster tissues (average relative abundance 54%) compared to the inside and outside of the shell (15%) and was largely Synechococcus CC9902 (Supplementary Tables 1, 2). The Pirellulaceae (Planctomycete) were represented by one genus, Blastopirellula (Supplementary Tables 1, 2). Rhodobacteraceae were over-represented in the inside and outside of the shell (24%) relative to the grouped oyster tissues (4%) comprising the genera Ruegeria, Nautella, Silicimonas, and Actibacterium (Supplementary Tables 1, 2).
Indicator Species Analysis was used to identify bacterial families that were associated with a sampled environment (Table 1). The outside shell bacterial composition had the greatest number of taxa which were significantly associated with a single oyster tissue type (9 families). These families occurred in 77–100% of outside shell samples (n = 13) and 41–81% of their occurrence in the dataset was in these samples. In particular, Xenococcaceae and Synechococcales almost exclusively occurred in the outer shell (80% of counts with remaining counts mainly occurring in the inside shell) and more than 90% of outer shell samples contained these taxa. One family each was significantly associated with the inside shell (an uncultured Cyanobacterial family) and the large intestinal tissue (a Planctomycetales family). Seven Alpha- and Deltaproteobacterial taxa were associated with the farm water samples; these were mainly marine SAR11, 116, and 324 clade families.
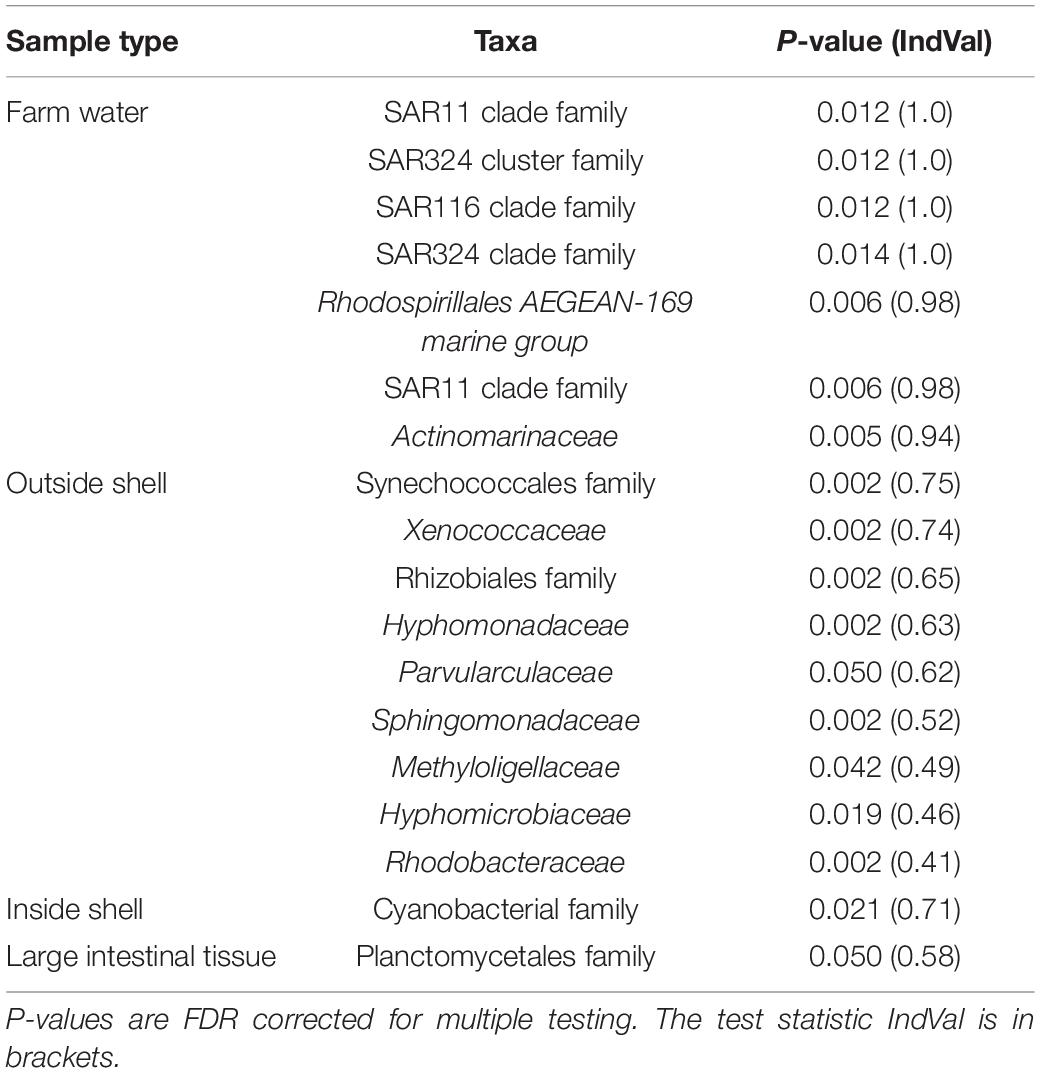
Table 1. Indicator species analysis showing bacterial taxa at family level which were significantly associated with different oyster tissue types or farm water.
Vibrio Abundance Differed According to Pearl Oyster Tissue Type
Vibrio abundance significantly differed according to oyster tissue type (P < 0.001), but did not differ between locations (P = 0.4). Vibrio abundance in the large intestine tissue was greater than in all other tissue types (Tukey adjusted P < 0.01 for all except P = 0.013 for digestive tissue and P = 0.057 for water). More specifically, it was on average 32 times greater than the digestive tissue, 44 and 80 times for gill and haemolymph tissue and 280 and 600 times greater than the heart and mantle tissue.
Vibrio Community Patterns Did Not Differ According to Pearl Oyster Tissue Type
Due to the significantly elevated Vibrio abundance in the various tissue types, we sought to identify the species present and discern patterns in the Vibrio community using the hsp60 gene as a taxonomic marker. By using this marker gene and after cleaning the dataset, 41 samples were characterized. The majority of these samples were from the inside (n = 13) and outside shell (n = 12), followed by the haemolymph (n = 6), digestive tissue (n = 5), large intestinal tissue (n = 3) and farm water (n = 2). Notably, no hsp60 sequences were recovered from the heart, gill nor mantle tissue. In contrast to the whole bacterial community, there was no specific Vibrio fingerprint or clustering of Vibrio communities by oyster tissue type with the exception of the large intestinal tissue (Figures 4, 5). A triangle heat map of average inter-group Bray-Curtis similarities between tissue types confirmed that with the exception of the large intestinal tissue the levels of similarity were generally evenly distributed within and between tissues types indicating no specific Vibrio fingerprint for a specific tissue type (Supplementary Figure 2).
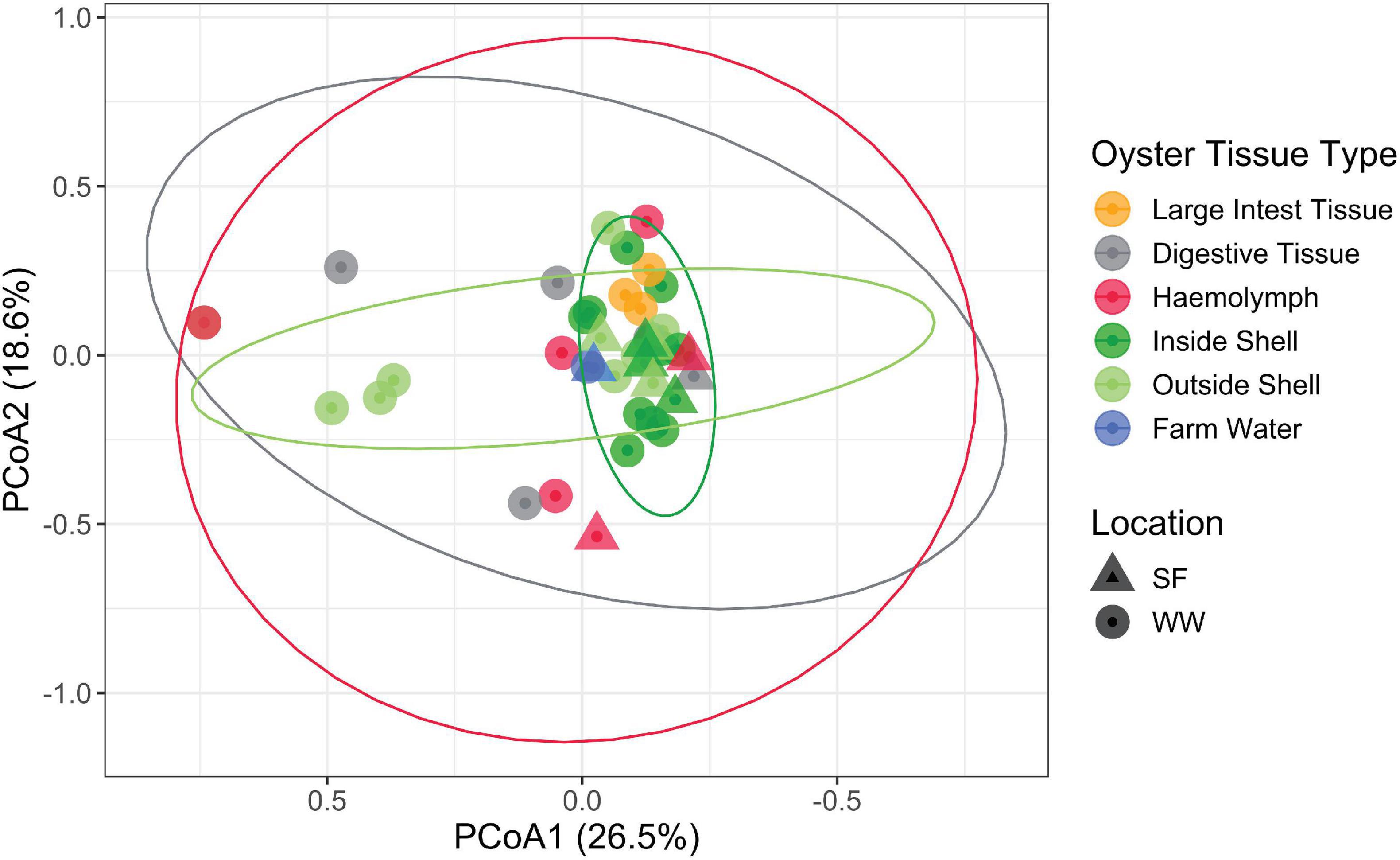
Figure 4. PCoA ordination of the Vibrio compositions based on hsp60 sequences labeled by location and oyster tissue type. The first axis explained 26.5% of the Vibrio community variance and the second 18.6%. Ellipses show 95% data distribution.
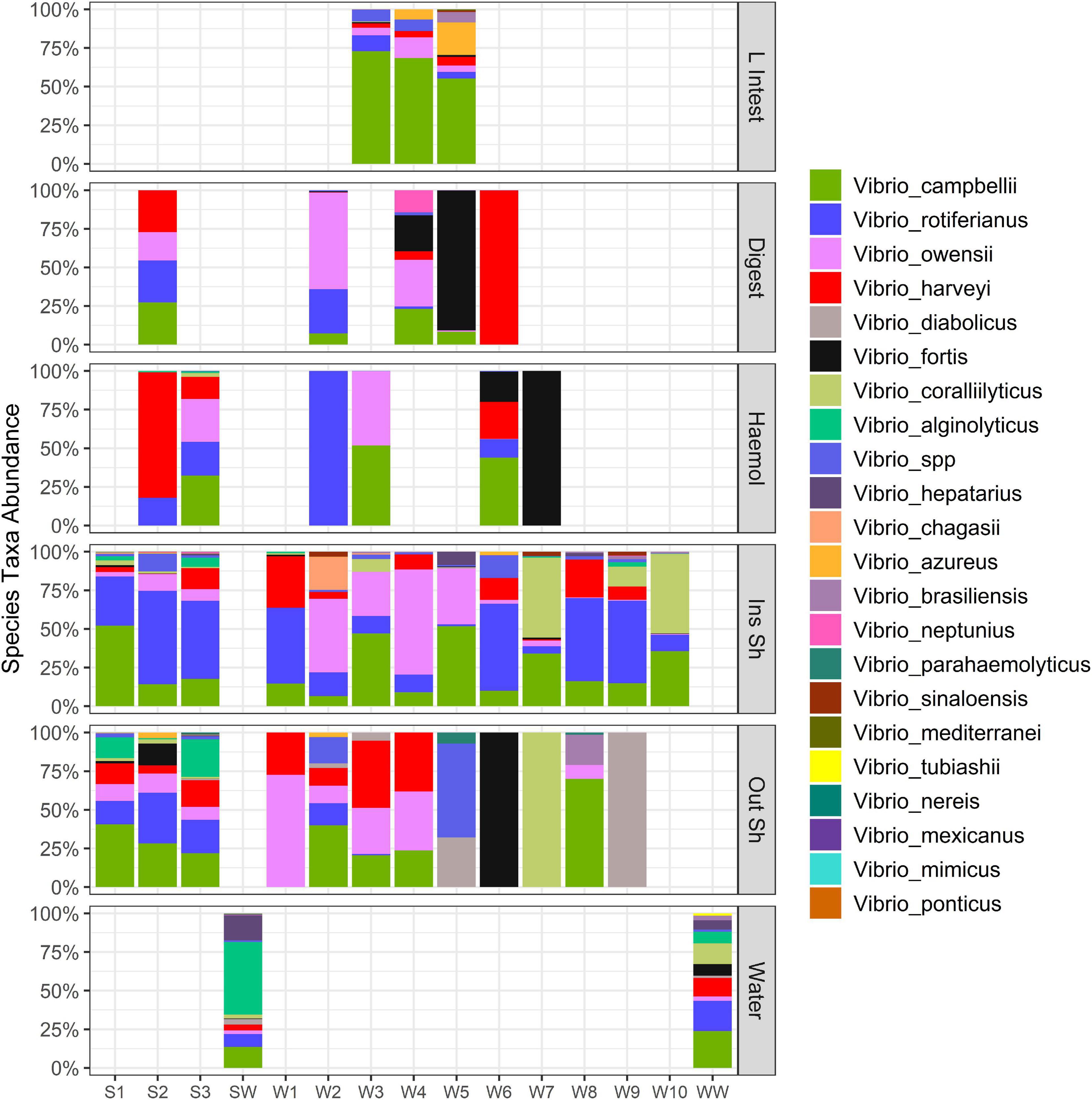
Figure 5. Stacked bar plot of Vibrio species across oyster tissue types and farm water. Each column is a sample. “L Intest” large intestinal tissue, “Digest” digestive tissue, “Haemol” haemolymph, “Ins Sh” inside shell, “Out Sh” outside shell, “Water” farm water. “S,” Seaflower Bay and “W,” Wargul Wargul Bay. Only samples with > 10 sequence reads were included.
Of the 24 unique Vibrio species detected, Vibrio campbellii, Vibrio rotiferianus, Vibrio owensii, and Vibrio harveyi were the most dominant members recovered from oysters, representing an average relative abundance of 25, 18, 16, and 14%, respectively (Figure 5). Indicator species analysis identified V. hepatarius, V. diabolicus, and V. tubiashii were associated with farm water (IndVal > 0.75, FDR adjusted P-value < 0.05), however in keeping with the observed high variability of the Vibrio community within oyster tissue types (Figures 4, 5), no Vibrio species was significantly associated with a specific oyster tissue.
Discussion
Here we provide the first characterization of the bacterial community associated with the silver-lipped pearl oyster (Pinctada maxima). This study revealed that, as has been observed in other oyster species (King et al., 2020), discrete bacterial communities are associated with different pearl oyster tissue types. This finding supports the hypothesis that each tissue type represents a unique microenvironment for bacterial colonization and offers differences in niche space available for bacterial colonization (Dubé et al., 2019; Pathirana et al., 2019; King et al., 2020). The lack of association with water at each farm also supports the unique nature of the microbial communities in oyster tissues. The dominance of the Cyanobiaceae genus Synechococcus in both oyster tissue type and farm water has been reported elsewhere including in the pearl oyster intestine and surrounding water environment (Zheng et al., 2021). The higher relative abundance of the Rhodobacteraceae family in the shell swab samples relative to the pearl oyster tissues samples suggests the preferential colonization of these species on the shell. This family included the genera Nautella and Ruegeria reported in other pearl oyster species (Zheng et al., 2021) and members of the genus Ruegeria can produce the broad-spectrum antibiotic tropodithietic acid (Beyersmann et al., 2017). The dominance in the shell microbiome supports their reported ability to rapidly colonize surfaces and produce antibacterial components, preventing other bacteria from growing (Arfken et al., 2017). Ruegeria spp. have been used in aquaculture to suppress growth of marine pathogens including Vibrio sp., and have potential as probiotic or antifouling agents (Berger et al., 2011). Notably, some genera within the Rhodobacteraceae family are also known for their ability to metabolize calcium compounds and could be using the shell as a source of nutrients (Pujalte et al., 2014).
The pearl oyster large intestinal tissue was remarkably different from all other tested tissue types and was dominated by an uncultured family in the Planctomycetales. In agreement with our data, Planctomycete bacteria have been identified as dominant members within the intestinal and gut environment in many bivalve species including pearl oysters (Dubé et al., 2019; Zheng et al., 2021). In our study, the dominant genus Blastopirellula is in the family Pirellulaceae, members of which are dominant in pearl oyster alimentary tissue (Dubé et al., 2019). Members of this family can reportedly exploit sulfated algal polysaccharides that might be commonly ingested by oysters as a consequence of phytoplankton consumption. All Blastopirellula characterized to date use a wide range of simple non-sulfated sugars, at least some of which are likely to occur in the oyster digestive tract as algal biomass is hydrolyzed (King et al., 2012). Because of the low between-replicate variability and species evenness in the intestinal tissue, it suggests that this tissue microenvironment is relatively stable and could be a suitable environment for bacteria with long generation times.
When we measured tissue specificity for the Vibrio community using the hsp60 Vibrio taxonomic marker we found that for those tissues with hsp60 genes detected, unlike the whole bacterial community analysis, there was no tissue specificity, with the exception of the large intestine. V. campbellii was the dominant member across all samples, primarily driven by its over-representation in the intestinal tissue. V. campbellii has been implicated in shrimp diseases (Haldar et al., 2011; Vicente et al., 2020), but little is known about its associations with oysters. In addition, V. rotiferianus, V. owensii and V. harveyi were the most dominant members recovered from oysters and these were present in outer and inner shell swabs, haemolymph, digestive tissue, large intestine and seawater, but not heart, gill nor mantle tissue. These three species have been isolated from moribund Pacific oysters (Wang et al., 2021) and while V. harveyi is a known oyster pathogen, implicated in Pacific oyster mortality events (King et al., 2019c), less is known about V. rotiferianus and V. owensii. Wang et al. (2021) demonstrated that V. owensii had low pathogenicity in Pacific oysters, however, this species is implicated in an emergent shrimp disease (Liu et al., 2021). Vibrio fortis was dominant in some samples and interestingly this species and V. harveyi increased dramatically in simulated heat wave experiments associated with Pacific oyster mortality, which implicates them as pathogens, cooperatively or independently (Green et al., 2019).
There is a paucity of studies characterizing whole Vibrio community using culture-independent techniques in bivalves and we are not aware of studies that have described multiple Vibrio species distribution in pearl oyster tissue because the 16S rRNA approach generally does not resolve vibrios (Dubé et al., 2019). Our finding that vibrios colonize a wide variety of pearl oyster tissues, including the shell, may reflect the fact that oysters represent a potentially important ecological niche for these bacteria. Furthermore, vibrios are known for their expansive metabolic capabilities and are able to colonize a multitude of environments (Thompson et al., 2004; Grimes et al., 2009). Consequently, these vibrio communities are potentially not host specific and rather reflect a random assemblage of Vibrio spp. influenced by the surrounding environment (Wendling et al., 2014; Wendling and Wegner, 2015). It may also be the case that the Vibrio community, including those in the haemolymph can persist in the oyster tissues due to a lack of sensitivity to the bactericidal activity of the haemolymph (Pruzzo et al., 2005).
It has been suggested that the oyster haemolymph provides a good indication of overall oyster health and is where disease is likely to manifest (Lokmer and Wegner, 2015; King et al., 2019b). Shifts in the oyster haemolymph microbiome have been linked to disease (Lemire et al., 2015; Lokmer and Wegner, 2015). Routine haemolymph sampling could be an early detection, disease prevention tool. For example, Lokmer et al. (2016) reported a spillover of cultivable Vibrionaceae from the haemolymph into solid tissues during a disease event. Furthermore, we have demonstrated that several known and potential pathogenic Vibrio spp. were detected in P. maxima haemolymph which supports a capacity for routine and early detection in the event of disease symptoms.
The similarity between the inside shell and pearl oyster tissues vibrio compositions could suggest that the inside shell environment is also a source of microbial spillover into the oyster tissues. In support of this observation, a bacterial pathogen of the Pacific oyster preferentially colonizes the inner shell and causes tissue pathology from this microenvironment (Boardman et al., 2008). This may also be relevant for non-vibrio pathogens. For example, studies of a bacterial pathogen (Roseovarius crassostreae) of the cultured eastern oysters (Crassostrea virginica) indicate colonization on the inside shell microenvironment and movement and pathology on solid oyster tissues (Boardman et al., 2008), suggesting that microbial spillover from the inside-shell microbial biofilm into the oyster is plausible. Understanding the links between the shell and oyster tissue bacterial community is important, because analysis of the shell bacterial community may provide opportunities for non-invasive sampling of the oyster bacterial community in situ. Our results show that the inner shell swab also has potential as a less stressful routine surveillance tool and is easily sampled when the shell is being subjected to routine farming techniques.
Conclusion
In recent years, interest in using the bivalve microbiome to detangle disease dynamics has exponentially increased. However, it is necessary to build a foundation of the factors that govern bivalve microbiome assembly to understand how the bivalve microbiome could contribute to disease processes. Our bacterial community diversity study has addressed a knowledge gap for the commercially important pearl oyster, P. maxima, whereby we have shown that pearl oyster bacterial composition is governed by both location and tissue-type, which is consistent with observations in other bivalve species. Interestingly, there was no significant difference in the Vibrio community between tissue types nor location. These results imply that Vibrio species are shared among different pearl oyster tissues. In particular, the similarity between the haemolymph, inside shell and solid tissues suggests that they are a source of microbial spillover into the oyster tissues, and a potentially useful tool for non-destructive routine disease testing and early warning surveillance. Based on the strong effect of tissue-type on the pearl oyster bacterial community, future studies should apply caution when attempting to compare microbial patterns with current literature, particularly from different locations and microenvironments, and when searching for disease agents. The bacterial community analyses and conclusions are based on 63 samples and 1,263 ASVs (following rarefaction), and while larger sample sets could be analyzed in the future, this work provides important foundational information for future studies identifying the factors that drive microbial assembly in a valuable aquaculture species.
Data Availability Statement
The raw data files in FASTQ format were deposited in NCBI Sequence Read Archive (SRA) under BioProject ID: PRJNA594420. Data analysis workflow, reference data set and taxonomy file are available at https://doi.org/10.17605/OSF.IO/4798P.
Author Contributions
WK and NS created the degenerate primers and optimized the PCR. MK and WK analyzed the data. DM provided samples. AP was responsible for laboratory work. KG, AP, DM, and KC conceived and designed the study. KG, WK, MK, AP, KC, and JS wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by a Charles Darwin University internal grant, a Paspaley Pearls Group small grant and an Australian Research Council Linkage grant (LP 160101795) to JS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Angela Williams (Paspaley Pearls Group) for sample collection, Kitman Dyrting (Northern Territory Government) for oyster tissue dissection, Zarah Tinning and Dion Lambrinidis (CDU) for laboratory technical support, and Alea Rose for data analysis support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.723649/full#supplementary-material
Supplementary Figure 1 | Heat map triangle showing average inter-group Bray-Curtis similarities of the bacterial communities between tissue types. The circles show the average within-tissue type similarities and larger circles and numbers represent greater similarities within and between tissue types.
Supplementary Figure 2 | Heat map triangle showing average inter-group Bray-Curtis similarities of the Vibrio communities between tissue types. The circles show the within-tissue type similarities and larger circles and numbers represent greater similarities within and between tissue types.
Supplementary Table 1 | Relative abundance (>1%) of bacterial taxa to genus level associated with different oyster tissue types.
Supplementary Table 2 | Relative abundance (>1%) of bacterial taxa to genus level associated with different oyster tissue types grouped by location.
References
Adzigbli, L., Hao, R., Jiao, Y., Deng, Y., Du, X., Wang, Q., et al. (2020). Immune response of pearl oysters to stress and diseases. Rev. Aquac. 12, 513–523. doi: 10.1111/raq.12329
Arfken, A., Song, B., Bowman, J. S., and Piehler, M. (2017). Denitrification potential of the eastern oyster microbiome using a 16S rRNA gene based metabolic inference approach. PLoS One 12:e0185071. doi: 10.1371/journal.pone.0185071
Berger, M., Neumann, A., Schulz, S., Simon, M., and Brinkhoff, T. (2011). Tropodithietic acid production in Phaeobacter gallaeciensis is regulated by N-acyl homoserine lactone-mediated quorum sensing. J. Bacteriol. 193, 6576–6585. doi: 10.1128/jb.05818-11
Beyersmann, P. G., Tomasch, J., Son, K., Stocker, R., Göker, M., Wagner-Döbler, I., et al. (2017). Dual function of tropodithietic acid as antibiotic and signaling molecule in global gene regulation of the probiotic bacterium Phaeobacter inhibens. Sci. Rep. 7, 1–9. doi: 10.1038/s41598-017-00784-7
Boardman, C. L., Maloy, A. P., and Boettcher, K. J. (2008). Localization of the bacterial agent of juvenile oyster disease (Roseovarius crassostreae) within affected eastern oysters (Crassostrea virginica). J. Invertebr. Pathol. 97, 150–158. doi: 10.1016/j.jip.2007.08.007
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857.
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
De Cáceres, M., Legendre, P., and Moretti, M. (2010). Improving indicator species analysis by combining groups of sites. Oikos 119, 1674–1684. doi: 10.1111/j.1600-0706.2010.18334.x
Dubé, C. E., Ky, C.-L., and Planes, S. (2019). Microbiome of the Black-Lipped Pearl Oyster Pinctada margaritifera, a Multi-Tissue Description With Functional Profiling. Front. Microbiol. 10:1548. doi: 10.3389/fmicb.2019.01548
Glasl, B., Herndl, G. J., and Frade, P. R. (2016). The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 10, 2280–2292.
Green, T. J., Siboni, N., King, W. L., Labbate, M., Seymour, J. R., and Raftos, D. (2019). Simulated marine heat wave alters abundance and structure of Vibrio populations associated with the Pacific Oyster resulting in a mass mortality event. Microb. Ecol. 77, 736–747. doi: 10.1007/s00248-018-1242-9
Grimes, D. J., Johnson, C. N., Dillon, K. S., Flowers, A. R., Noriea, N. F., and Berutti, T. (2009). What Genomic Sequence Information Has Revealed About Vibrio Ecology in the Ocean—A Review. Microb. Ecol. 58, 447–460. doi: 10.1007/s00248-009-9578-9
Haldar, S., Chatterjee, S., Sugimoto, N., Das, S., Chowdhury, N., Hinenoya, A., et al. (2011). Identification of Vibrio campbellii isolated from diseased farm-shrimps from south India and establishment of its pathogenic potential in an Artemia model. Microbiology 157, 179–188. doi: 10.1099/mic.0.041475-0
Herlemann, D. P. R., Labrenz, M., Jürgens, K., Bertilsson, S., Waniek, J. J., and Andersson, A. F. (2011). Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5, 1571–1579. doi: 10.1038/ismej.2011.41
Joint Select Committee on Northern Australia (2016). Joint Select Committee on Northern Australia. Available Online at: https://www.aph.gov.au/Parliamentary_Business/Committees/Joint/Former_Committees/Northern_Australia/Aquaculture/Report [Accessed Mar 15, 2021]
King, G. M., Judd, C., Kuske, C. R., and Smith, C. (2012). Analysis of Stomach and Gut Microbiomes of the Eastern Oyster (Crassostrea virginica) from Coastal Louisiana, USA. PLoS One 7:e51475. doi: 10.1371/journal.pone.0051475
King, W. L., Jenkins, C., Go, J., Siboni, N., Seymour, J. R., and Labbate, M. (2019a). Characterisation of the Pacific oyster microbiome during a summer mortality event. Microb. Ecol. 77, 502–512. doi: 10.1007/s00248-018-1226-9
King, W. L., Jenkins, C., Seymour, J. R., and Labbate, M. (2019b). Oyster disease in a changing environment: decrypting the link between pathogen, microbiome and environment. Mar. Environ. Res. 143, 124–140. doi: 10.1016/j.marenvres.2018.11.007
King, W. L., Siboni, N., Kahlke, T., Green, T. J., Labbate, M., and Seymour, J. R. (2019c). A New High Throughput Sequencing Assay for Characterizing the Diversity of Natural Vibrio Communities and Its Application to a Pacific Oyster Mortality Event. Front. Microbiol. 10:2907. doi: 10.3389/fmicb.2019.02907
King, W. L., Siboni, N., Williams, N. L., Kahlke, T., Nguyen, K. V., Jenkins, C., et al. (2019d). Variability in the composition of Pacific oyster microbiomes across oyster families exhibiting different levels of susceptibility to OsHV-1 μvar disease. Front. Microbiol. 10:473. doi: 10.3389/fmicb.2019.00473
King, W. L., Siboni, N., Kahlke, T., Dove, M., O’Connor, W., Mahbub, K. R., et al. (2020). Regional and oyster microenvironmental scale heterogeneity in the Pacific oyster bacterial community. FEMS Microbiol. Ecol. 96:fiaa054. doi: 10.1093/femsec/fiaa054
Lemire, A., Goudenège, D., Versigny, T., Petton, B., Calteau, A., Labreuche, Y., et al. (2015). Populations, not clones, are the unit of vibrio pathogenesis in naturally infected oysters. ISME J. 9, 1523–1531. doi: 10.1038/ismej.2014.233
Liu, F., Li, S., Yu, Y., Yuan, J., Yu, K., and Li, F. (2021). Pathogenicity of a Vibrio owensii strain isolated from Fenneropenaeus chinensis carrying pirAB genes and causing AHPND. Aquaculture 530:735747. doi: 10.1016/j.aquaculture.2020.735747
Lokmer, A., Kuenzel, S., Baines, J. F., and Wegner, K. M. (2016). The role of tissue-specific microbiota in initial establishment success of Pacific oysters. Environ. Microbiol. 18, 970–987. doi: 10.1111/1462-2920.13163
Lokmer, A., and Wegner, K. M. (2015). Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J. 9, 670–682. doi: 10.1038/ismej.2014.160
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
McFall-Ngai, M., Hadfield, M. G., Bosch, T. C., Carey, H. V., Domazet-Lošo, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U. S. A. 110, 3229–3236. doi: 10.1073/pnas.1218525110
McMurdie, P. J., and Holmes, S. (2013). phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Negri, A. P., Bunter, O., Jones, B., and Llewellyn, L. (2004). Effects of the bloom-forming alga Trichodesmium erythraeum on the pearl oyster Pinctada maxima. Aquaculture 232, 91–102. doi: 10.1016/s0044-8486(03)00487-3
Pathirana, E., McPherson, A., Whittington, R., and Hick, P. (2019). The role of tissue type, sampling and nucleic acid purification methodology on the inferred composition of Pacific oyster (Crassostrea gigas) microbiome. J. Appl. Microbiol. 127, 429–444. doi: 10.1111/jam.14326
Pita, L., Rix, L., Slaby, B. M., Franke, A., and Hentschel, U. (2018). The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6:46. doi: 10.1186/s40168-018-0428-1
Pruzzo, C., Gallo, G., and Canesi, L. (2005). Persistence of vibrios in marine bivalves: the role of interactions with haemolymph components. Environ. Microbiol. 7, 761–772. doi: 10.1111/j.1462-2920.2005.00792.x
Pujalte, M. J., Lucena, T., Ruvira, M. A., Arahal, D. R., and Macián, M. C. (2014). “The Family Rhodobacteraceae,” in The Prokaryotes: Alphaproteobacteria and Betaproteobacteria, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin, Heidelberg: Springer Berlin Heidelberg), 439–512. doi: 10.1007/978-3-642-30197-1_377
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596.
Raina, J. B., Eme, L., Pollock, F. J., Spang, A., Archibald, J. M., and Williams, T. A. (2018). Symbiosis in the microbial world: from ecology to genome evolution. Biol. Open 7:bio032524. doi: 10.1242/bio.032524
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., and Hollister, E. B. (2009). Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microb. 75, 7537–41. doi: 10.1128/aem.01541-09
Seth, E. C., and Taga, M. E. (2014). Nutrient cross-feeding in the microbial world. Front. Microbiol. 5:350. doi: 10.3389/fmicb.2014.00350
Siboni, N., Balaraju, V., Carney, R., Labbate, M., and Seymour, J. R. (2016). Spatiotemporal Dynamics of Vibrio spp. within the Sydney Harbour Estuary. Front. Microbiol. 7:460. doi: 10.3389/fmicb.2016.00460
Siboni, N., Ben-Dov, E., Sivan, A., and Kushmaro, A. (2008). Global distribution and diversity of coral-associated Archaea and their possible role in the coral holobiont nitrogen cycle. Environ. Microbiol. 10, 2979–2990. doi: 10.1111/j.1462-2920.2008.01718.x
Sonnenburg, J. L., Angenent, L. T., and Gordon, J. I. (2004). Getting a grip on things: how do communities of bacterial symbionts become established in our intestine? Nat. Immunol. 5, 569–573. doi: 10.1038/ni1079
Thompson, J. R., Randa, M. A., Marcelino, L. A., Tomita-Mitchell, A., Lim, E., and Polz, M. F. (2004). Diversity and Dynamics of a North Atlantic Coastal Vibrio Community. Appl. Environ. Microb. 70, 4103–10. doi: 10.1128/aem.70.7.4103-4110.2004
Trabal, N., Mazón-Suástegui, J. M., Vázquez-Juárez, R., Asencio-Valle, F., Morales-Bojórquez, E., and Romero, J. (2012). Molecular analysis of bacterial microbiota associated with oysters (Crassostrea gigas and Crassostrea corteziensis) in different growth phases at two cultivation sites. Microb. Ecol. 64, 555–569. doi: 10.1007/s00248-012-0039-5
Trabal Fernández, N., Mazón-Suástegui, J. M., Vázquez-Juárez, R., Ascencio-Valle, F., and Romero, J. (2014). Changes in the composition and diversity of the bacterial microbiota associated with oysters (Crassostrea corteziensis, Crassostrea gigas and Crassostrea sikamea) during commercial production. FEMS Microbiol. Ecol. 88, 69–83. doi: 10.1111/1574-6941.12270
Vicente, A., Taengphu, S., Hung, A. L., Mora, C. M., Dong, H. T., and Senapin, S. (2020). Detection of Vibrio campbellii and V. parahaemolyticus carrying full-length pirABVp but only V. campbellii produces PirVp toxins. Aquaculture 519:734708. doi: 10.1016/j.aquaculture.2019.734708
Wang, H., Yang, B., Li, X., Li, Q., and Shikai, L. (2021). Screening of bacterial pathogens associated with mass summer mortality of the Pacific oyster, Crassostrea gigas, in China. Aquacult. Rep. 20:100672. doi: 10.1016/j.aqrep.2021.100672
Wang, Z. L., Wang, B., Chen, G., Jian, J. C., Lu, Y. S., Xu, Y. H., et al. (2016). Transcriptome analysis of the pearl oyster (Pinctada fucata) hemocytes in response to Vibrio alginolyticus infection. Gene 575, 421–428. doi: 10.1016/j.gene.2015.09.014
Wegner, K. M., Volkenborn, N., Peter, H., and Eiler, A. (2013). Disturbance induced decoupling between host genetics and composition of the associated microbiome. BMC Microbiol. 13:252. doi: 10.1186/1471-2180-13-252
Wendling, C. C., Batista, F. M., and Wegner, K. M. (2014). Persistence, Seasonal Dynamics and Pathogenic Potential of Vibrio Communities from Pacific Oyster Hemolymph. PLoS One 9:e94256. doi: 10.1371/journal.pone.0094256
Wendling, C. C., and Wegner, K. M. (2015). Adaptation to enemy shifts: rapid resistance evolution to local Vibrio spp. in invasive Pacific oysters. Proc. Biol. Sci. 282:20142244. doi: 10.1098/rspb.2014.2244
Keywords: pearl oyster (Pinctada maxima), Vibrio, bacterial communities, tissue-type, haemolymph, hsp60
Citation: King WL, Kaestli M, Siboni N, Padovan A, Christian K, Mills D, Seymour J and Gibb K (2021) Pearl Oyster Bacterial Community Structure Is Governed by Location and Tissue-Type, but Vibrio Species Are Shared Among Oyster Tissues. Front. Microbiol. 12:723649. doi: 10.3389/fmicb.2021.723649
Received: 11 June 2021; Accepted: 21 July 2021;
Published: 09 August 2021.
Edited by:
Alfonso Benítez-Páez, Principe Felipe Research Center (CIPF), SpainReviewed by:
Aide Lasa, University of Santiago de Compostela, SpainKeri Ann Lydon, Gulf Coast Seafood Lab, US Food and Drug Administration, United States
Nolwenn Callac, Institut Français de Recherche pour l’Exploitation de la Mer, France
Copyright © 2021 King, Kaestli, Siboni, Padovan, Christian, Mills, Seymour and Gibb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karen Gibb, a2FyZW4uZ2liYkBjZHUuZWR1LmF1