 Xiaonan Zhang
Xiaonan Zhang Xiaomeng Wang
Xiaomeng Wang Min Wu
Min Wu Reena Ghildyal
Reena Ghildyal Zhenghong Yuan
Zhenghong Yuan- 1Centre for Research in Therapeutic Solutions, Faculty of Science and Technology, University of Canberra, Canberra, ACT, Australia
- 2Shanghai Public Health Clinical Center, Fudan University, Shanghai, China
- 3Key Laboratory of Medical Molecular Virology, School of Basic Medical Sciences, Shanghai Medical College, Fudan University, Shanghai, China
Hepatitis B virus (HBV) infection is a global public health problem that plagues approximately 240 million people. Chronic hepatitis B (CHB) often leads to liver inflammation and aberrant repair which results in diseases ranging from liver fibrosis, cirrhosis, to hepatocellular carcinoma. Despite its narrow species tropism, researchers have established various in vivo models for HBV or its related viruses which have provided a wealth of knowledge on viral lifecycle, pathogenesis, and immunity. Here we briefly revisit over five decades of endeavor in animal model development for HBV and summarize their advantages and limitations. We also suggest directions for further improvements that are crucial for elucidation of the viral immune-evasion strategies and for development of novel therapeutics for a functional cure.
Introduction
Over half a century has passed since a precipitin line in an immunodiffusion agar gel was formed between serum of a hemophilia patient receiving transfusion and serum of an Australian aborigine (Blumberg et al., 1965). This serendipitous event turned out to be the first revelation of a new etiology: Hepatitis B Virus (HBV) (Sutnick et al., 1969; Dane et al., 1970; Almeida et al., 1971). The active research surrounding this virus thereafter has yielded in-depth understanding of its natural history, immunobiology, and pathogenesis.
HBV belongs to the hepadnaviridae which is characterized by a small genome (3.2 kilobase) of partially double-stranded DNA. It enters the human hepatocytes via the interaction between viral envelope protein and human sodium taurocholate co-transporting polypeptide (NTCP) in a highly species-specific manner (Yan et al., 2012; He et al., 2015; Burwitz et al., 2017; Takeuchi et al., 2019; Chen et al., 2020). It then reproduces its genome by a transcription—reverse transcription process. The former is done under the genetic instruction of the covalently closed circular DNA (cccDNA) which is formed by repairing the incoming viral genome or recycling of the viral DNA produced in the cytoplasm. The cccDNA serves as the template for transcription of the pregenomic RNA and other mRNAs that encode at least seven proteins. The pregenomic RNA is not only the messenger for viral core and polymerase, but also the template for reverse transcription, which strictly requires polymerase assisted core particle formation. HBV is a hepatotropic virus with strict host and organ tropism.
Individuals chronically infected with HBV can develop a series of liver diseases, from liver fibrosis, cirrhosis to hepatocellular carcinoma. Although great progress has been made in the prevention and treatment of chronic hepatitis B (CHB), there are still over 240 million people infected with HBV and around 650,000 people die of this disease each year globally (World Health Organization [WHO], 2015). To further reduce these figures, besides broader coverage of HBV vaccination, novel approaches of therapy that are able to cure chronic infection are needed. The continuing development of animal models for HBV infection has been instrumental for our understanding of its lifecycle, development of antiviral agents, and testing of preventive measures. A series of models have been developed ever since the “Australian antigen” was discovered. Their utilities and shortcomings are reviewed (Table 1). Possible directions for further development to meet the needs of an HBV cure are also discussed.
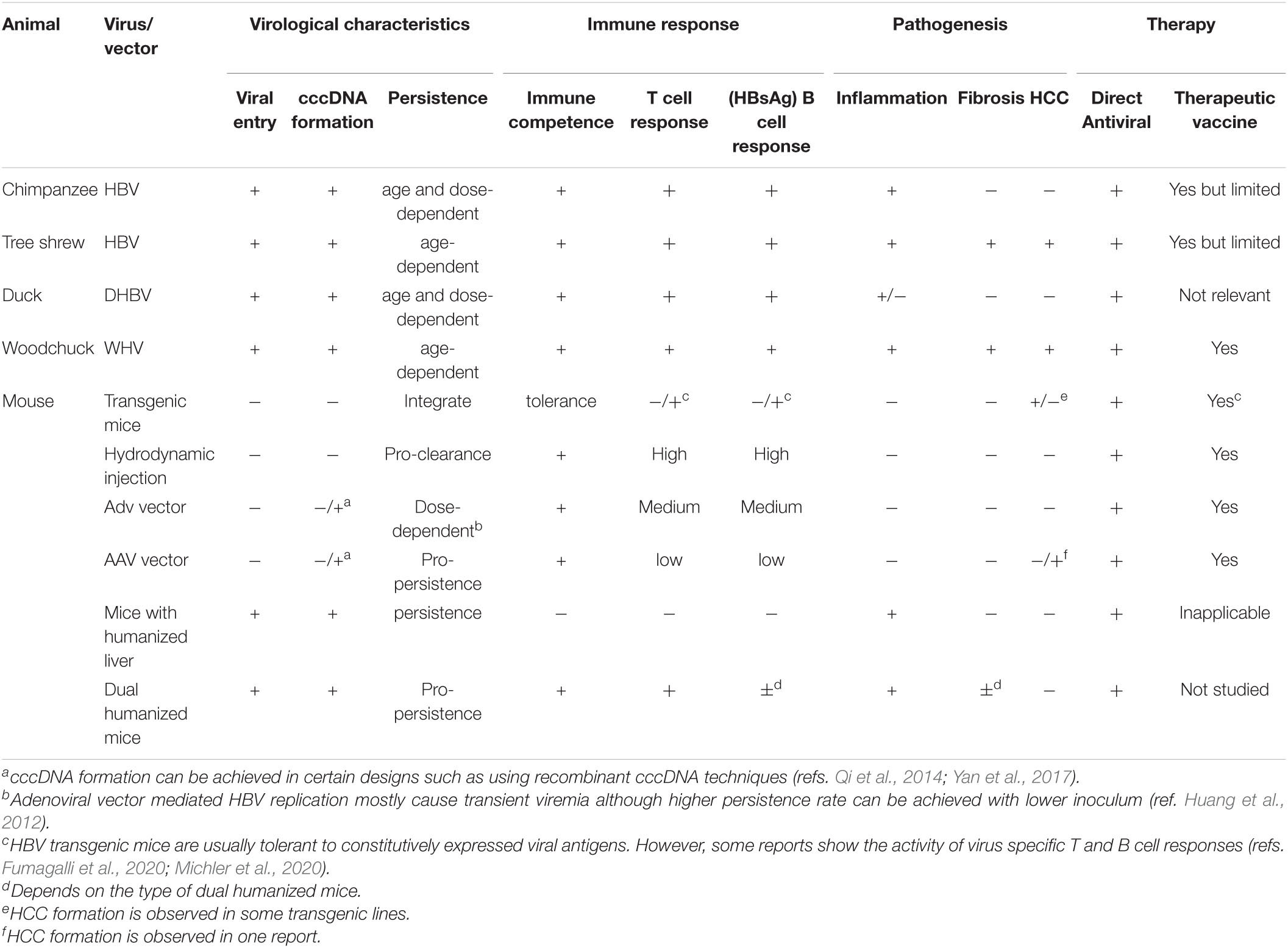
Table 1. Basic features of animal models for major hepadnaviruses.
Chimpanzee and Other Primates
Apart from human subjects, chimpanzees were almost the only model used in the early stages of HBV research (McAuliffe et al., 1980). In as early as 1969, two groups independently reported the appearance of Australian antigen and development of its antibody after inoculation of positive sera in chimpanzees (Hirschman et al., 1969; Lichter, 1969). This was followed by several other studies with more detailed longitudinal observations (Maynard et al., 1971, 1972; Barker et al., 1973). Chimpanzees were also the only feasible subjects for evaluation of the first generation vaccines which were purified from HBV positive sera and inactivated (McAuliffe et al., 1980). A major achievement of molecular virology in 1982 was also made possible by injecting cloned viral genome into chimpanzees thus establishing an acute viral hepatitis (Will et al., 1982). All these studies established the molecular etiology of HBV as the causative agent for hepatitis B.
The fact that chimpanzees have the genetic background and immune system that are closest to humans makes them the host of choice for studying the innate and adaptive immune response to HBV infection. One striking feature of HBV acute infection observed in chimpanzees is that the virus does not induce or repress host gene expression in the lag (week 0–2) and logarithmic expansion phase (week 4–6) which is in stark contrast to HCV infection (Wieland S. et al., 2004). The induction of interferon stimulated genes (ISGs) and other inflammatory genes are within the viral clearance phase (week 8–12) which is initiated by infiltration of T cells, B cell, macrophages, and NK cells, etc. Transient antibody-mediated depletion of CD8 + T cells during the logarithmic phase of viral replication caused prolonged viremia and liver damage (Thimme et al., 2003). Meanwhile, the essentiality of the priming of CD4 + T cells in the early phase of the infection was also discovered, as pre-depletion of these cells caused persistent infection with minimal immunopathology (Asabe et al., 2009) reminiscent of the immune tolerance state of CHB. Most interestingly, inoculation of chimpanzees with intermediate viral dose (104 to 107 genome copy) leads to viral clearance whereas high-dose (1010 genome copy) or low-dose (10 copy) leads to persistent infection, indicating that kinetics of viral spread in the early phase determines the fate of the disease (Asabe et al., 2009). In a self-limited infection, an early (week 8–12) non-cytolytic suppression of viremia and a late (week 14–20) cytolytic destruction of HBV positive hepatocytes were both documented by longitudinal observations in chimpanzees, suggestive of a two-phase dynamic process (Guidotti et al., 1999; Wieland S.F. et al., 2004).
The use of chimpanzees has also facilitated the development of novel therapeutics. GS-9620 is a potent, orally active TLR7 agonist in clinical development for treatment of CHB. Its short-term oral administration in HBV infected chimpanzees achieved long-term suppression of viral load by inducing ISGs and cytokines/chemokines (Lanford et al., 2013). Further transcriptomic and histological analyses revealed intrahepatic aggregates comprised of CD8 + T cells and B cells in the portal regions (Li L. et al., 2018). Therapeutic vaccination schemes (Pancholi et al., 2001) and anti-HBV monoclonal antibodies (Eren et al., 2000) were also evaluated in chimpanzees preclinically. Unfortunately, due to increasing concerns over animal ethics, experiments using chimpanzees have been highly restricted (Altevogt et al., 2012).
Apart from chimpanzees, macaques were also suggested to be susceptible to HBV. Inoculation of HBV replicative plasmid into macaques (Macaca Silvanus) caused viremia and pathological changes (Gheit et al., 2002). The same group later found Macaca fasicularis from Mauritius island had a high positive rate of HBV DNA although at low viral load (Dupinay et al., 2013). Genome sequencing revealed that it was a genotype D subtype ayw3 with a substitution at position 67 within preS1. Inoculation of virus-positive pooled serum into Macaca fasicularis caused an acute infection. However, another study failed to establish an infection using the virus harboring this variation and using the same species of macaque (Burwitz et al., 2017). Some other HBV-related viruses were also identified in New World monkeys. The woolly monkey hepatitis B virus (WMHBV) infects its natural host, Lagothrix lagotricha (woolly monkey) (Lanford et al., 1998). Spider monkeys and chimpanzees were also shown to be susceptible to WMHBV in experimental infections (Lanford et al., 1998, 2003). The capuchin monkey hepatitis B virus (CMHBV) was recently identified in Sapajus xanthosternos in Brazil (de Carvalho Dominguez Souza et al., 2018). The surface protein of these two viruses showed high antigenic relatedness as evidenced by cross-reactivity of polyclonal antibody against HBV surface antigen. Furthermore, Hepatitis D Virus pseudotyped with WMHBV and CMHBV surface proteins could infect human hepatocytes suggesting their highly similar cellular entry process. Indeed, molecular substitution assays on key residues on the NTCP polypeptide suggested that amino acid 158 is critical for virus entry (Takeuchi et al., 2019). This residue in New-World monkeys, which include capuchin monkey and woolly monkey, is identical to that of human and chimpanzee. However, these animals not readily available as experimental hosts due to economical and ethical reasons.
Tree Shrew
Tree shrews (Tupaia belangeri) are small mammals closely related to primates. The susceptibility of tree shrews to HBV infection was confirmed both in vivo and ex vivo (Walter et al., 1996; Yan et al., 1996) although a transient and low level of viremia was documented in newborns (Walter et al., 1996). A larger scale, longitudinal research on 46 tree shrews neonatally inoculated with HBV resulted in 6 chronic infections (Wang et al., 2012). Hepatic histopathological changes observed in chronically infected animals were similar to those observed in CHB (Ruan et al., 2013). Continued observation showed that hepatocellular carcinoma occurred in two of the six animals at the late stage of life (Yang et al., 2015). Thus, this model faithfully recapitulates many aspects of the HBV infection in humans. Another contribution made by this model is the study of the viral entry route. Fine mapping of the receptor binding site of PreS1 was made possible using primary hepatocytes isolated from Tupaia (Glebe et al., 2003; Glebe et al., 2005). More importantly, sodium taurocholate co-transporting polypeptide (NTCP) was identified as the functional receptor for HBV using primary culture of Tupaia hepatocytes (Yan et al., 2012).
The Woodchuck and Duck Models
The Woodchuck Hepatitis Virus (WHV) (Summers et al., 1978) was first discovered in the late 1970s. It has significant similarities to HBV in its morphology, genome structure, and replication scheme. Its genome is frequently integrated into the host genome at the N-myc loci (Fourel et al., 1990) and is directly linked with the development of hepatocellular carcinoma (Gouillat et al., 1997). More detailed analysis on HBV showed that integration events are randomly distributed among chromosomes (Mason et al., 2016), the enriched loci found in human liver tumors such as TERT, MLL4, and CCNE1 are the result of clonal expansion (Sung et al., 2012; Zhao et al., 2016). Molecular virology studies revealed that various forms of linear viral DNA can be produced during WHV replication which serves as the substrate for chromosomal integration (Yang et al., 1996). These forms of viral DNA and its integration was also confirmed in CHB (Tu et al., 2021). Woodchuck has also been widely used for the evaluation of therapeutic solutions. The administration of nucleoside analogs on WHV infected animals inhibited viremia but did not inhibit its genetic reservoir (cccDNA) (Moraleda et al., 1997; Dandri et al., 2000). Woodchucks were also used as a testing ground for various combination therapies involving DNA vaccine, antigen-antibody immune complex, immune checkpoint modulators etc. (Roggendorf et al., 2007; Lu et al., 2008; Kosinska et al., 2012, 2013; Liu et al., 2014) with considerable success.
The duck hepatitis B virus (DHBV) (Mason et al., 1980) was first reported to be present in Pekin ducks in 1980 which bears significant resemblance to HBV in its genome, morphology, and replication. Because ducks are much more accessible than other hosts, DHBV was used as a model system for studying the replication scheme of hepadnaviruses. Indeed, Summers and Mason found that DHBV replicates its genome by reverse transcription of an RNA intermediate, i.e., the pregenomic RNA (Summers and Mason, 1982) thus pointing to its evolutionary kinship with retroviruses (Miller and Robinson, 1986). Moreover, ex vivo infection of primary duck hepatocytes revealed the existence of a nuclear cccDNA pool which is not maintained by semiconservative replication but by intracellular recycling of relaxed circular DNA (Tuttleman et al., 1986). The abundance of cccDNA is regulated by the expression level of viral surface protein which determines the relative rate of viral release and intracellular recycling of relaxed circular DNA (Summers et al., 1990). This phenomenon is also corroborated in HBV infection (Lentz and Loeb, 2011; Zhang et al., 2016). By biochemical purification, researchers identified the receptor for DHBV as a 180 kDa glycoprotein (Kuroki et al., 1994; Breiner et al., 1998; Urban et al., 1998), although the entry of HBV does not use human homologs of this protein.
The above-mentioned animal models can be naturally infected with members of hepadnaviruses. There are, however, quite several limitations that restrict further studies on HBV pathogenesis, immune response, and curative strategies. The Pekin duck is a powerful tool for DHBV molecular virology, but its immune system is evolutionarily distant from that of humans. The limited supply of woodchucks and the difficulty in their domestication restrict their larger-scale experimentation. The chimpanzees have been historically crucial for the development of prophylactic vaccine and for studying HBV immunobiology, but their high cost, genetic variations, and ethical regulations have prohibited their further use. Experimental mice have long been the model of choice in immunologic and pathological studies, but they are not susceptible to HBV or other hepadnaviruses. A large number of studies have been performed to establish mouse systems that can address specific aspects of HBV biology.
Transgenic Mice
With the development of transgenic mice technology, researchers started to clone HBV sequences into the mouse germline in the 1980s. The resultant mouse strains can be used to study the pathophysiological effects of certain viral gene products. For example, transgenic expression of surface antigen does not cause obvious pathologies (Babinet et al., 1985; Chisari et al., 1985), but the enforced expression of the large surface protein inhibits the release of small surface protein (Chisari et al., 1986), increases proteinaceous cytoplasm containing acidophilic inclusions resembling ground-glass hepatocytes observed in human carriers of HBV, and later develop nodular hyperplasia (Chisari et al., 1987). The oncogenic role of HBx is established by studies on transgenic mice. Kim et al. developed a transgenic mouse line in which HBx was expressed under the control of its native promoter (Kim et al., 1991). Histopathology begins with multifocal preneoplastic lesions followed by benign adenomas and finally malignant carcinomas. They also found that male mice developed disease and died much earlier than females. A similar observation was made in an HBx transgenic mouse lineage with high level of protein expression (Koike et al., 1994). Apart from the reported findings, they suggested that the formation of full-blown carcinoma requires additional genetic events as only small populations of altered foci developed into neoplasia. Indeed, they observed increased DNA synthesis and aneuploid DNA content in a subset of hepatocytes. Although there were a few reports that did not find significant histological alteration in HBx transgenic mice (Lee et al., 1990), the majority of them supported its tumorigenic role (Tsuge et al., 2010; Quetier et al., 2015).
Thanks to the well-developed tools for immunologic studies, HBV transgenic mice were extensively used to elucidate the mechanism of immunopathogenesis and mechanism of virus-mediated tolerization. The knowledge on the functional role of HBeAg has been greatly expanded using e antigen transgenic mice. HBeAg is translated from a precore mRNA a few nucleotides upstream of the initiation codon of the core open reading frame and further processed by proteolytic cleavage. The primary amino acid sequence of HBeAg shows significant identity to that of HBcAg although HBeAg is structurally distinct and is expressed in non-particulate form and secreted into circulation. There are a large number of reports supporting the link between HBeAg and active manipulation of the anti-HBV immune response. For example, HBV G1896A mutation that causes the premature termination of the e antigen is associated with acute fulminant or severe hepatitis (Tong et al., 2013). Using HBeAg-expressing transgenic mice, Milich et al. (1990) found that these mice are immunologically tolerant to not only HBeAg but also HBcAg at the T-cell level. T cells exposed to HBeAg were non-responsive to HBcAg. In addition, after priming with core antigen, in vitro anti-HBc IgG production was greatly reduced in e antigen transgenic mice but IgM production was unaffected suggestive of a reduced Th-cell function. Moreover, non-transgenic mice exposed to HBeAg in utero by their transgenic mother showed reduced T cell response to HBcAg peptides which suggested that HBeAg can pass the placenta to induce fetal tolerance to HBcAg before and around birth. Thus, HBeAg plays a crucial role in restricting T cell response to HBcAg in the early phase of HBV infection and contributes to the establishment of chronic infection.
The transgenic mice expressing individual viral proteins are still limited by the lack of replicative parameters. The establishment of replication-competent HBV transgenic mice has provided a robust system for dissecting the innate and adaptive immune response within the context of persistent HBV infection (Guidotti et al., 1995). In this model, a high level of viral RNA and replicative intermediates are detected in the hepatocytes, and virus particles in serum are morphologically indistinguishable from those in natural infection. Although the host is immune tolerant to the virus, an acute immune response can be initiated by adoptive transfer of specific immune cells. Introduction of HBsAg-specific cytotoxic T lymphocytes (CTLs) in this transgenic mouse results in a transient spike of liver damage (Guidotti et al., 1996) whereby a small fraction of the hepatocytes are killed. Nevertheless, these CTLs clear all traces of HBV gene expression and replication via the antiviral activity of IFN-γ and TNF-α. Indeed, the potent suppression of viral replication is independent of the cytolytic activity as perforin knockout CTLs also abolish viral activity. Nevertheless, the initial cell lysis triggers focal inflammation response in which antigen-non-specific cells such as neutrophils, polymorphonuclear neutrophils, and platelets infiltrate into the hepatic sites (Sitia et al., 2002, 2004; Iannacone et al., 2005). The polymorphonuclear neutrophils are known to express matrix metalloproteinases which facilitate the recruitment of mononuclear cells into the liver and exacerbate liver inflammation (Sitia et al., 2004). In addition to studies on CTL-mediated adaptive immune response, innate immune responses can also be investigated. Kakimi et al. analyzed the antiviral effect of activated natural killer T (NKT) cells by injection of antibody to galactosylceramide (Kakimi et al., 2000). Similarly, the functional role of Interleukin-12 (IL-12) (Cavanaugh et al., 1997), IL-18 (Kimura et al., 2002), was also analyzed. This model is also suitable for evaluation of various antiviral therapies (Julander et al., 2002; Weber et al., 2002; Julander et al., 2003; Uprichard et al., 2005; Ebert et al., 2011; Buchmann et al., 2013).
It is also worth noting that although transgenic mice are thought to be completely tolerant to the transgene-encoded viral protein, spontaneous clearance of HBsAg due to the emergence of antibodies was reported (Fumagalli et al., 2020). Also, a therapeutic vaccination scheme combined with siRNA-mediated knockdown in HBV transgenic mice generated antibodies toward HBsAg (Michler et al., 2020). These results indicate that the transgene-specific B cells are not clonally deleted and may be activated in certain circumstances.
Transfected or Transduced Mice
Although adoptive transfer experiments in HBV transgenic mice can inform the antiviral capability of various cell types, this information does not necessarily translate to a real infection as the cell type under question can be very scarce or functionally tolerized in vivo. Since mouse hepatocytes are not permissive to HBV even after the introduction of the human NTCP gene (He et al., 2015), methods that can introduce viral DNA without germline insertion are needed. The hydrodynamic injection method, which involves rapid, high-pressure tail vein injection of HBV constructs was developed. Yang et al. introduced an overlength replication-competent HBV genome into mice and observed antigen production, viral transcripts, and DNA synthesis within two weeks in wildtype and much longer in NOD/SCID mice (Yang et al., 2002). By optimization of the delivery construct, Huang et al. achieved over 6 months of antigenemia in 40% of the injected immunocompetent mice (Huang et al., 2006). Further studies demonstrated that mouse genetic background has a significant impact on the rate of clearance. BALB/c and NOD/ShiLtJ mice quickly cleared the virus while C3H/HeN mice are more tolerant to viral replication (Chou et al., 2015; Peng et al., 2015). An association with the gut microbiota and age-related viral clearance is proposed (Chou et al., 2015).
Mouse hepatocytes can also be transduced by recombinant viral vectors, e.g., adenovirus (Adv) or adeno-associated virus (AAV), to initiate HBV replication. Transduction by Adv-HBV usually results in a transient antigenemia (Sprinzl et al., 2001; von Freyend et al., 2011) although it can be prolonged by a lower dose inoculation (Huang et al., 2012). By contrast, the introduction of HBV genome via AAV results in an immunotolerant phenotype with long-term antigenemia and minimal liver inflammation and fibrosis (Yang et al., 2014). The strikingly different behavior of these two viral vector systems is thought to derive from the feature of the carriers, the former being an activator of innate immune response and the latter usually stealthy and silent (Greber and Flatt, 2019; Ertl, 2021).
A more recent development in this transient transfer method involves the formation of bona fide cccDNA within the nuclei which may better recapitulate the epigenetic state of the supercoiled episomal cccDNA in natural infection. To achieve this, different approaches were used. Yan et al. utilized a phage φC31 integrase-mediated intramolecular recombination technology to generate a recombinant cccDNA in Escherichia coli (Yan et al., 2017). The resulting DNA molecule is highly similar to the native cccDNA and can initiate viral replication and antigen expression within the mouse liver. Another approach used by Deng’s group utilized the Cre-loxP in vivo recombination system in which the vector backbone was excised by the co-introduced Cre recombinase. The remaining loxP site within the recombinant cccDNA (rcccDNA) is flanked by a pair of splice donor and acceptor sites. Thus, its transcript undergoes a post-transcriptional RNA splicing yielding an RNA pregenome identical to that of a wildtype virus (Qi et al., 2014). Hydrodynamic injection of this vector resulted in the formation of rcccDNA within the nuclei. The same group further improved this system by using a replication-defective adenoviral vector to transfer the rcccDNA into Alb-Cre transgenic mice which led to prominent HBV persistence for over 62 weeks (Li G. et al., 2018). We have also established an rcccDNA model based on the Cre-loxP recombination strategy by using a AAV8 vector that is hepatotropic and achieved long-term antigenemia and cccDNA persistence (Wu et al., 2020).
Humanized Chimeric Mice
Although the HBV transgenic and transfected mice each partially recapitulate some features of CHB, neither of them supports the complete infection cycle of infection due to the lack of cellular receptor. Engraftment of susceptible human hepatocytes into mouse can overcome this barrier. The “trimera” mouse is the result of the earliest attempts in this direction. Using mice that lack mature T, B and NK cells, human liver fragments were transplanted under the kidney capsule. After injection of HBV positive sera, viral DNA and antigens were detected for about two months (Ilan et al., 1999; Eren et al., 2000). Although this model was used for evaluation of antiviral therapy and monoclonal antibodies against HBsAg, the short time window for infection and transient viremia offered limited operational capability. Another easy-to-use mouse model, in which the HBV replication-competent HepAD38 cells were subcutaneously engrafted into nude mice, may serve as a simple platform for antiviral evaluation (Feitelson et al., 2007; Schinazi et al., 2012). It was not until mouse strains that support expansion of human hepatocytes, that humanized chimeric mice became a robust infection model.
In the early 1990s, researchers found that mice with the liver specific expression of the urokinase-type plasminogen activator (Alb-uPA) resulted in elevated uPA concentration, hypofibrinogenemia and neonatal hemorrhaging (Sandgren et al., 1991). This feature was exploited to facilitate the repopulation of xenogenic hepatocytes in various genetic backgrounds of immune deficiency (Rhim et al., 1995; Petersen et al., 1998; Tateno et al., 2004). Dandri et al. established active HBV replication in uPA/RAG-2 mouse repopulated with human hepatocytes although the humanized efficiency is less than 15% (Dandri et al., 2001). Later development of uPA-SCID mice resulted in a more pronounced engraftment (Meuleman et al., 2005). Indeed, a high (1010 copies/ml) and long-lasting viral titre could be achieved (Tsuge et al., 2005). Now the uPA-SCID model has been widely utilized for evaluation of antivirals (Oehler et al., 2014; Mueller et al., 2018), engineered immune therapy (Koh et al., 2018), and mechanism of innate immune response against HBV (Lütgehetmann et al., 2011; Belloni et al., 2012).
A major drawback of the uPA-SCID model is that the uncontrolled expression of the uPA gene in very early life requires human hepatocytes xenograft soon after birth, which is difficult to manipulate and easily causes severe hemorrhage during operation (Meuleman and Leroux-Roels, 2008). The later development of the Fah (Fumaryl acetoacetate hydrolase) knockout mice largely resolved this issue. Fah is an enzyme that catalyze the last step of Tyrosine/Phenylalanine catabolism. The loss of Fah gene causes the accumulation of toxic metabolic intermediates in this pathway. Supplementation of 2-(2-nitro-4-fluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), an inhibitor that blocks the enzymatic conversion of upstream intermediate, to the drinking water prevents the liver toxicity (Grompe et al., 1995). This feature provides much more control over the extent of mouse liver damage and the timing of hepatocyte xenograft. Based on this, several humanized models were established (Azuma et al., 2007; Bissig et al., 2007), among which the Fah−/− mice with additional immune deficiency (Rag2−/− and IL-2rγ−/−), hence named FRG triple KO mice achieved the highest engraftment rate (90%) (Azuma et al., 2007). FRG KO mice have been successfully used to establish HBV and HCV infection (Bissig et al., 2010) and to evaluate the antiviral action of interferon-α subtypes (Chen et al., 2021).
A major limitation of the humanized mice described above is the lack of the adaptive immune system that can recognize the incoming HBV and make responses that contribute to viral clearance or immunopathology. To solve this, an A2/NGS-hu-HSC/Hep mice model was established (Bility et al., 2014). In this model, the human HLA-A2 allel was introduced into NOD/SCID/IL-2Rγ−/− background and transplanted with human CD34+ cells from fetal liver which serve as hematopoietic stem cells and liver progenitor cells for the reconstituted mice (Bility et al., 2014). Injection of anti-Fas agonistic antibody (Jo2) causes mouse hepatocyte damage and makes room for the introduced progenitor cells to settle in the liver. The human HLA-A2 transgene is thought to promote the HLA-restricted human T cell function. This model achieved around 25% repopulation of human hepatocytes and successful infection of HBV although with relatively low viral load (< 5 × 105 copies/ml). Importantly, HBV specific CD8+ T cell response was elicited, and significant liver inflammation and fibrosis was observed. Notably, the authors observed intrahepatic accumulation of M2-like macrophage which is associated with accelerated liver fibrosis in CHB patients. The authors did not report humoral immune response against HBV. In another study, researchers developed the HIS-HUHEP model, in which CD34+ fetal derived human hematopoietic stem cells (HIS), and adult human hepatocytes (HUHEP) were introduced into a BALB/c Rag2/IL2rγ KO NOD, sirpa, uPA transgenic mice (Dusseaux et al., 2017). The introduction of adult hepatocytes caused much higher HBV viral load and antigens. Inflammatory cell infiltration surrounding core antigen positive hepatocytes was observed although no sign of fibrosis was found. The infiltrating lymphocytes included NK cells, CD4+ and CD8+ T cells with activation/exhaustion markers such as PD-1 and CTLA-4. Importantly, antibodies to surface and core antigens were detected. As the access to fetal hematopoietic stem cells and primary hepatocytes became more limited, other sources of stem cells were explored. Yuan et al. used human bone mesenchymal stem cells to repopulate in FRG mice in BALB/c SCID background (Yuan et al., 2019). Depletion of mouse hepatocytes was achieved by anti-Fas agonistic antibody (Jo2) combined with withdrawal of NTBC. Elimination of murine immune cells was secured by the injection of busulfan. This resulted in high liver chimerism and well differentiated hepatocytes. Reconstitution of the human immune cells was also evident with major myeloid and lymphoid cells. Persistent HBV infection with high viral load and antigen titers was observed. A unique feature of this model is that a large number of lymphocytes and macrophages infiltrated the liver after HBV infection, which was accompanied by high level of cytokines and chemokines. This caused progressive liver fibrosis and cirrhosis although viral loads and antigens were not significantly suppressed. Evaluation of the total serum human IgG and IgM revealed significant elevation during infection whereas antibodies to HBV antigens reached a peak at about 12 weeks post infection and then declined. It is possible that viral infection triggered an inflammatory milieu that fostered a polyreactive humoral antibody response in addition to viral antigens. Nevertheless, this dual humanized model constitutes a robust and easy-to-implement system for HBV pathobiology.
Future Directions
With the help of in vivo models, remarkable achievements have been made in the inquiry into how HBV exploits the host molecular and cellular machineries to propagate, and in the development effective preventative and therapeutic measures to contain the spread and progression of this disease. The initial use of chimpanzees as the experimental host established the transmissibility of the “Australian antigen” and unraveled many key features of HBV induced immunologic responses. Studies using woodchuck and pekin duck infected with Hepadnaviridae family members elucidated the framework of viral life cycle and provided surrogate models for antiviral assessment. The endeavor to establish mouse models that can recapitulate different aspects of HBV-mediated disease has also yielded substantial progress.
With the even higher coverage of preventive vaccines and availability of antiviral therapies, the development of a therapeutic scheme that effectively reverses virus-mediated immunotolerance and establishes an immunodominance over HBV without triggering overt liver damage becomes the greatest challenge of our times. Such endeavor will require an immunocompetent small animal model that accommodates most steps of the viral lifecycle and is easy-to-manipulate genetically and immunologically. Obviously, none of the current models fully meet these requirements. The HBV transgenic mouse model is overly tolerant toward the virus while the hydrodynamic injection and Adenoviral transfer model are generally prone to resolution. The AAV model mostly generates a chronic infection reminiscent of a carrier state which seems to be a suitable system for evaluation of therapeutic vaccines. However, it remains to be shown that such an immunotolerant phenotype is characteristic of HBV itself but not of the vector. The dually humanized mice model is regarded as a promising direction. Various immunopathology of CHB, such as inflammation, liver fibrosis and cirrhosis have been recreated by the latest models. However, the engraftment of xenogenic liver and hematopoietic system causes mismatches in the HLA system between the xenograft and the host. Although graft-versus-host disease is not found in the reported systems (Dusseaux et al., 2017; Yuan et al., 2019), there are still concerns over whether the nature of immune recognition and reactions against the virus in such a system is in line with that of natural infection. This will directly affect their suitability in studying the immunologic determinants of viral tolerance and clearance. Indeed, disruptive innovations are required to remove these obstacles before mechanistic details of virus-mediated immunotolerance can be fully unveiled and therapies for a cure can be made.
Author Contributions
XZ and XW conceived and drafted the manuscript. MW, RG, and ZY provided additional content to the manuscript and critically reviewed the text. All the authors read through the manuscript and agreed on the final test.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Almeida, J. D., Rubenstein, D., and Stott, E. J. (1971). New antigen-antibody system in Australia-antigen-positive hepatitis. Lancet 2, 1225–1227. doi: 10.1016/s0140-6736(71)90543-5
Altevogt, B. M., Pankevich, D. E., Pope, A. M., and Kahn, J. P. (2012). Research agenda. Guiding limited use of chimpanzees in research. Science 335, 41–42. doi: 10.1126/science.1217521
Asabe, S., Wieland, S. F., Chattopadhyay, P. K., Roederer, M., Engle, R. E., Purcell, R. H., et al. (2009). The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J. Virol. 83, 9652–9662. doi: 10.1128/jvi.00867-09
Azuma, H., Paulk, N., Ranade, A., Dorrell, C., Al-Dhalimy, M., Ellis, E., et al. (2007). Robust expansion of human hepatocytes in Fah-/-/Rag2-/-/Il2rg-/- mice. Nat. Biotechnol. 25, 903–910. doi: 10.1038/nbt1326
Babinet, C., Farza, H., Morello, D., Hadchouel, M., and Pourcel, C. (1985). Specific expression of hepatitis B surface antigen (HBsAg) in transgenic mice. Science 230, 1160–1163. doi: 10.1126/science.3865370
Barker, L. F., Chisari, F. V., McGrath, P. P., Dalgard, D. W., Kirschstein, R. L., Almeida, J. D., et al. (1973). Transmission of type B viral hepatitis to chimpanzees. J. Infect. Dis. 127, 648–662.
Belloni, L., Allweiss, L., Guerrieri, F., Pediconi, N., Volz, T., Pollicino, T., et al. (2012). IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Invest. 122, 529–537. doi: 10.1172/jci58847
Bility, M. T., Cheng, L., Zhang, Z., Luan, Y., Li, F., Chi, L., et al. (2014). Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog. 10:e1004032. doi: 10.1371/journal.ppat.1004032
Bissig, K. D., Le, T. T., Woods, N. B., and Verma, I. M. (2007). Repopulation of adult and neonatal mice with human hepatocytes: a chimeric animal model. Proc. Natl. Acad. Sci. U.S.A. 104, 20507–20511. doi: 10.1073/pnas.0710528105
Bissig, K. D., Wieland, S. F., Tran, P., Isogawa, M., Le, T. T., Chisari, F. V., et al. (2010). Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J. Clin. Invest. 120, 924–930. doi: 10.1172/jci40094
Blumberg, B. S., Alter, H. J., and Visnich, S. (1965). A “New” antigen in leukemia sera. JAMA 191, 541–546. doi: 10.1001/jama.1965.03080070025007
Breiner, K. M., Urban, S., and Schaller, H. (1998). Carboxypeptidase D (gp180), a Golgi-resident protein, functions in the attachment and entry of avian hepatitis B viruses. J. Virol. 72, 8098–8104. doi: 10.1128/jvi.72.10.8098-8104.1998
Buchmann, P., Dembek, C., Kuklick, L., Jager, C., Tedjokusumo, R., von Freyend, M. J., et al. (2013). A novel therapeutic hepatitis B vaccine induces cellular and humoral immune responses and breaks tolerance in hepatitis B virus (HBV) transgenic mice. Vaccine 31, 1197–1203. doi: 10.1016/j.vaccine.2012.12.074
Burwitz, B. J., Wettengel, J. M., Muck-Hausl, M. A., Ringelhan, M., Ko, C., Festag, M. M., et al. (2017). Hepatocytic expression of human sodium-taurocholate cotransporting polypeptide enables hepatitis B virus infection of macaques. Nat. Commun. 8:2146.
Cavanaugh, V. J., Guidotti, L. G., and Chisari, F. V. (1997). Interleukin-12 inhibits hepatitis B virus replication in transgenic mice. J. Virol. 71, 3236–3243. doi: 10.1128/jvi.71.4.3236-3243.1997
Chen, C. Y., Winer, B. Y., Chavez, D., Guerra, B., Brasky, K. M., Eng, S., et al. (2020). Woolly monkey-HBV infection in squirrel monkeys as a surrogate nonhuman primate model of HBV infection. Hepatol. Commun. 4, 371–386. doi: 10.1002/hep4.1471
Chen, J., Li, Y., Lai, F., Wang, Y., Sutter, K., Dittmer, U., et al. (2021). Functional comparison of interferon-alpha subtypes reveals potent hepatitis B virus suppression by a concerted action of interferon-alpha and interferon-gamma signaling. Hepatology 73, 486–502. doi: 10.1002/hep.31282
Chisari, F. V., Filippi, P., Buras, J., McLachlan, A., Popper, H., Pinkert, C. A., et al. (1987). Structural and pathological effects of synthesis of hepatitis B virus large envelope polypeptide in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 84, 6909–6913. doi: 10.1073/pnas.84.19.6909
Chisari, F. V., Filippi, P., McLachlan, A., Milich, D. R., Riggs, M., Lee, S., et al. (1986). Expression of hepatitis B virus large envelope polypeptide inhibits hepatitis B surface antigen secretion in transgenic mice. J. Virol. 60, 880–887. doi: 10.1128/jvi.60.3.880-887.1986
Chisari, F. V., Pinkert, C. A., Milich, D. R., Filippi, P., McLachlan, A., Palmiter, R. D., et al. (1985). A transgenic mouse model of the chronic hepatitis B surface antigen carrier state. Science 230, 1157–1160. doi: 10.1126/science.3865369
Chou, H. H., Chien, W. H., Wu, L. L., Cheng, C. H., Chung, C. H., Horng, J. H., et al. (2015). Age-related immune clearance of hepatitis B virus infection requires the establishment of gut microbiota. Proc. Natl. Acad. Sci. U.S.A. 112, 2175–2180. doi: 10.1073/pnas.1424775112
Dandri, M., Burda, M. R., Torok, E., Pollok, J. M., Iwanska, A., Sommer, G., et al. (2001). Repopulation of mouse liver with human hepatocytes and in vivo infection with hepatitis B virus. Hepatology 33, 981–988. doi: 10.1053/jhep.2001.23314
Dandri, M., Burda, M. R., Will, H., and Petersen, J. (2000). Increased hepatocyte turnover and inhibition of woodchuck hepatitis B virus replication by adefovir in vitro do not lead to reduction of the closed circular DNA. Hepatology 32, 139–146. doi: 10.1053/jhep.2000.8701
Dane, D. S., Cameron, C. H., and Briggs, M. (1970). Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet 1, 695–698. doi: 10.1016/s0140-6736(70)90926-8
de Carvalho Dominguez Souza, B. F., König, A., Rasche, A., de Oliveira Carneiro, I., Stephan, N., Corman, V. M., et al. (2018). A novel hepatitis B virus species discovered in capuchin monkeys sheds new light on the evolution of primate hepadnaviruses. J. Hepatol. 68, 1114–1122. doi: 10.1016/j.jhep.2018.01.029
Dupinay, T., Gheit, T., Roques, P., Cova, L., Chevallier-Queyron, P., Tasahsu, S. I., et al. (2013). Discovery of naturally occurring transmissible chronic hepatitis B virus infection among Macaca fascicularis from Mauritius Island. Hepatology 58, 1610–1620. doi: 10.1002/hep.26428
Dusseaux, M., Masse-Ranson, G., Darche, S., Ahodantin, J., Li, Y., Fiquet, O., et al. (2017). Viral load affects the immune response to HBV in mice with humanized immune system and liver. Gastroenterology 153, 1647.e9–1661.e9.
Ebert, G., Poeck, H., Lucifora, J., Baschuk, N., Esser, K., Esposito, I., et al. (2011). 5′ triphosphorylated small interfering RNAs control replication of hepatitis B virus and induce an interferon response in human liver cells and mice. Gastroenterology 141, 696.e4–706.e4.
Eren, R., Ilan, E., Nussbaum, O., Lubin, I., Terkieltaub, D., Arazi, Y., et al. (2000). Preclinical evaluation of two human anti-hepatitis B virus (HBV) monoclonal antibodies in the HBV-trimera mouse model and in HBV chronic carrier chimpanzees. Hepatology 32, 588–596. doi: 10.1053/jhep.2000.9632
Ertl, H. C. J. (2021). T cell-mediated immune responses to AAV and AAV vectors. Front. Immunol. 12:666666. doi: 10.3389/fimmu.2021.666666
Feitelson, M. A., Clayton, M. M., Sun, B., and Schinazi, R. F. (2007). Development of a novel mouse model to evaluate drug candidates against hepatitis B virus. Antivir. Chem. Chemother. 18, 213–223. doi: 10.1177/095632020701800405
Fourel, G., Trepo, C., Bougueleret, L., Henglein, B., Ponzetto, A., Tiollais, P., et al. (1990). Frequent activation of N-myc genes by hepadnavirus insertion in woodchuck liver tumours. Nature 347, 294–298. doi: 10.1038/347294a0
Fumagalli, V., Di Lucia, P., Venzin, V., Bono, E. B., Jordan, R., Frey, C. R., et al. (2020). Serum HBsAg clearance has minimal impact on CD8+ T cell responses in mouse models of HBV infection. J. Exp. Med. 217:e20200298.
Gheit, T., Sekkat, S., Cova, L., Chevallier, M., Petit, M. A., Hantz, O., et al. (2002). Experimental transfection of Macaca sylvanus with cloned human hepatitis B virus. J. Gen. Virol. 83, 1645–1649. doi: 10.1099/0022-1317-83-7-1645
Glebe, D., Aliakbari, M., Krass, P., Knoop, E. V., Valerius, K. P., and Gerlich, W. H. (2003). Pre-s1 antigen-dependent infection of Tupaia hepatocyte cultures with human hepatitis B virus. J. Virol. 77, 9511–9521. doi: 10.1128/jvi.77.17.9511-9521.2003
Glebe, D., Urban, S., Knoop, E. V., Cag, N., Krass, P., Grun, S., et al. (2005). Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology 129, 234–245. doi: 10.1053/j.gastro.2005.03.090
Gouillat, C., Manganas, D., Zoulim, F., Vitrey, D., Saguier, G., Guillaud, M., et al. (1997). Woodchuck hepatitis virus-induced carcinoma as a relevant natural model for therapy of human hepatoma. J. Hepatol. 26, 1324–1330. doi: 10.1016/s0168-8278(97)80468-0
Greber, U. F., and Flatt, J. W. (2019). Adenovirus entry: from infection to immunity. Annu. Rev. Virol. 6, 177–197. doi: 10.1146/annurev-virology-092818-015550
Grompe, M., Lindstedt, S., Al-Dhalimy, M., Kennaway, N. G., Papaconstantinou, J., Torres-Ramos, C. A., et al. (1995). Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat. Genet. 10, 453–460. doi: 10.1038/ng0895-453
Guidotti, L. G., Ishikawa, T., Hobbs, M. V., Matzke, B., Schreiber, R., and Chisari, F. V. (1996). Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 4, 25–36. doi: 10.1016/s1074-7613(00)80295-2
Guidotti, L. G., Matzke, B., Schaller, H., and Chisari, F. V. (1995). High-level hepatitis B virus replication in transgenic mice. J. Virol. 69, 6158–6169. doi: 10.1128/jvi.69.10.6158-6169.1995
Guidotti, L. G., Rochford, R., Chung, J., Shapiro, M., Purcell, R., and Chisari, F. V. (1999). Viral clearance without destruction of infected cells during acute HBV infection. Science 284, 825–829. doi: 10.1126/science.284.5415.825
He, W., Ren, B., Mao, F., Jing, Z., Li, Y., Liu, Y., et al. (2015). Hepatitis D virus infection of mice expressing human sodium taurocholate co-transporting polypeptide. PLoS Pathog. 11:e1004840. doi: 10.1371/journal.ppat.1004840
Hirschman, R. J., Shulman, N. R., Barker, L. F., and Smith, K. O. (1969). Virus-like particles in sera of patients with infectious and serum hepatitis. JAMA 208, 1667–1670. doi: 10.1001/jama.1969.03160090027006
Huang, L. R., Gabel, Y. A., Graf, S., Arzberger, S., Kurts, C., Heikenwalder, M., et al. (2012). Transfer of HBV genomes using low doses of adenovirus vectors leads to persistent infection in immune competent mice. Gastroenterology 142, 1447.e3–1450.e3.
Huang, L. R., Wu, H. L., Chen, P. J., and Chen, D. S. (2006). An immunocompetent mouse model for the tolerance of human chronic hepatitis B virus infection. Proc. Natl. Acad. Sci. U.S.A. 103, 17862–17867. doi: 10.1073/pnas.0608578103
Iannacone, M., Sitia, G., Isogawa, M., Marchese, P., Castro, M. G., Lowenstein, P. R., et al. (2005). Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat. Med. 11, 1167–1169. doi: 10.1038/nm1317
Ilan, E., Burakova, T., Dagan, S., Nussbaum, O., Lubin, I., Eren, R., et al. (1999). The hepatitis B virus-trimera mouse: a model for human HBV infection and evaluation of anti-HBV therapeutic agents. Hepatology 29, 553–562. doi: 10.1002/hep.510290228
Julander, J. G., Colonno, R. J., Sidwell, R. W., and Morrey, J. D. (2003). Characterization of antiviral activity of entecavir in transgenic mice expressing hepatitis B virus. Antiviral Res. 59, 155–161. doi: 10.1016/s0166-3542(03)00109-8
Julander, J. G., Sidwell, R. W., and Morrey, J. D. (2002). Characterizing antiviral activity of adefovir dipivoxil in transgenic mice expressing hepatitis B virus. Antiviral Res. 55, 27–40. doi: 10.1016/s0166-3542(01)00223-6
Kakimi, K., Guidotti, L. G., Koezuka, Y., and Chisari, F. V. (2000). Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J. Exp. Med. 192, 921–930. doi: 10.1084/jem.192.7.921
Kim, C. M., Koike, K., Saito, I., Miyamura, T., and Jay, G. (1991). HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 351, 317–320. doi: 10.1038/351317a0
Kimura, K., Kakimi, K., Wieland, S., Guidotti, L. G., and Chisari, F. V. (2002). Interleukin-18 inhibits hepatitis B virus replication in the livers of transgenic mice. J. Virol. 76, 10702–10707. doi: 10.1128/jvi.76.21.10702-10707.2002
Koh, S., Kah, J., Tham, C. Y. L., Yang, N., Ceccarello, E., Chia, A., et al. (2018). Nonlytic lymphocytes engineered to express virus-specific T-cell receptors limit HBV infection by activating APOBEC3. Gastroenterology 155, 180.e6–193.e6.
Koike, K., Moriya, K., Iino, S., Yotsuyanagi, H., Endo, Y., Miyamura, T., et al. (1994). High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 19, 810–819. doi: 10.1002/hep.1840190403
Kosinska, A. D., Johrden, L., Zhang, E., Fiedler, M., Mayer, A., Wildner, O., et al. (2012). DNA prime-adenovirus boost immunization induces a vigorous and multifunctional T-cell response against hepadnaviral proteins in the mouse and woodchuck model. J. Virol. 86, 9297–9310. doi: 10.1128/jvi.00506-12
Kosinska, A. D., Zhang, E., Johrden, L., Liu, J., Seiz, P. L., Zhang, X., et al. (2013). Combination of DNA prime–adenovirus boost immunization with entecavir elicits sustained control of chronic hepatitis B in the woodchuck model. PLoS Pathog. 9:e1003391. doi: 10.1371/journal.ppat.1003391
Kuroki, K., Cheung, R., Marion, P. L., and Ganem, D. (1994). A cell surface protein that binds avian hepatitis B virus particles. J. Virol. 68, 2091–2096. doi: 10.1128/jvi.68.4.2091-2096.1994
Lanford, R. E., Chavez, D., Barrera, A., and Brasky, K. M. (2003). An infectious clone of woolly monkey hepatitis B virus. J. Virol. 77, 7814–7819. doi: 10.1128/jvi.77.14.7814-7819.2003
Lanford, R. E., Chavez, D., Brasky, K. M., Burns, R. B. III, and Rico-Hesse, R. (1998). Isolation of a hepadnavirus from the woolly monkey, a New World primate. Proc. Natl. Acad. Sci. U.S.A. 95, 5757–5761. doi: 10.1073/pnas.95.10.5757
Lanford, R. E., Guerra, B., Chavez, D., Giavedoni, L., Hodara, V. L., Brasky, K. M., et al. (2013). GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 144, 1508–1517. doi: 10.1053/j.gastro.2013.02.003
Lee, T. H., Finegold, M. J., Shen, R. F., DeMayo, J. L., Woo, S. L., and Butel, J. S. (1990). Hepatitis B virus transactivator X protein is not tumorigenic in transgenic mice. J. Virol. 64, 5939–5947. doi: 10.1128/jvi.64.12.5939-5947.1990
Lentz, T. B., and Loeb, D. D. (2011). Roles of the envelope proteins in the amplification of covalently closed circular DNA and completion of synthesis of the plus-strand DNA in hepatitis B virus. J. Virol. 85, 11916–11927. doi: 10.1128/jvi.05373-11
Li, G., Zhu, Y., Shao, D., Chang, H., Zhang, X., Zhou, D., et al. (2018). Recombinant covalently closed circular DNA of hepatitis B virus induces long-term viral persistence with chronic hepatitis in a mouse model. Hepatology 67, 56–70. doi: 10.1002/hep.29406
Li, L., Barry, V., Daffis, S., Niu, C., Huntzicker, E., French, D. M., et al. (2018). Anti-HBV response to toll-like receptor 7 agonist GS-9620 is associated with intrahepatic aggregates of T cells and B cells. J. Hepatol. 68, 912–921. doi: 10.1016/j.jhep.2017.12.008
Lichter, E. A. (1969). Chimpanzee antibodies to Australia antigen. Nature 224, 810–811. doi: 10.1038/224810a0
Liu, J., Zhang, E., Ma, Z., Wu, W., Kosinska, A., Zhang, X., et al. (2014). Enhancing virus-specific immunity in vivo by combining therapeutic vaccination and PD-L1 blockade in chronic hepadnaviral infection. PLoS Pathog. 10:e1003856. doi: 10.1371/journal.ppat.1003856
Lu, M., Yao, X., Xu, Y., Lorenz, H., Dahmen, U., Chi, H., et al. (2008). Combination of an antiviral drug and immunomodulation against hepadnaviral infection in the woodchuck model. J. Virol. 82, 2598–2603. doi: 10.1128/jvi.01613-07
Lütgehetmann, M., Bornscheuer, T., Volz, T., Allweiss, L., Bockmann, J. H., Pollok, J. M., et al. (2011). Hepatitis B virus limits response of human hepatocytes to interferon-α in chimeric mice. Gastroenterology 140, 2074–2083. doi: 10.1053/j.gastro.2011.02.057
Mason, W. S., Gill, U. S., Litwin, S., Zhou, Y., Peri, S., Pop, O., et al. (2016). HBV DNA integration and clonal hepatocyte expansion in chronic hepatitis B patients considered immune tolerant. Gastroenterology 151, 986.e4–998.e4.
Mason, W. S., Seal, G., and Summers, J. (1980). Virus of Pekin ducks with structural and biological relatedness to human hepatitis B virus. J. Virol. 36, 829–836. doi: 10.1128/jvi.36.3.829-836.1980
Maynard, J. E., Berquist, K. R., Krushak, D. H., and Purcell, R. H. (1972). Experimental infection of chimpanzees with the virus of hepatitis B. Nature 237, 514–515. doi: 10.1038/237514a0
Maynard, J. E., Hartwell, W. V., and Berquist, K. R. (1971). Hepatitis-associated antigen in chimpanzees. J. Infect. Dis. 123, 660–664. doi: 10.1093/infdis/123.6.660
McAuliffe, V. J., Purcell, R. H., and Gerin, J. L. (1980). Type B hepatitis: a review of current prospects for a safe and effective vaccine. Rev. Infect. Dis. 2, 470–492. doi: 10.1093/clinids/2.3.470
Meuleman, P., and Leroux-Roels, G. (2008). The human liver-uPA-SCID mouse: a model for the evaluation of antiviral compounds against HBV and HCV. Antivir. Res. 80, 231–238. doi: 10.1016/j.antiviral.2008.07.006
Meuleman, P., Libbrecht, L., De Vos, R., de Hemptinne, B., Gevaert, K., Vandekerckhove, J., et al. (2005). Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology 41, 847–856. doi: 10.1002/hep.20657
Michler, T., Kosinska, A. D., Festag, J., Bunse, T., Su, J., Ringelhan, M., et al. (2020). Knockdown of virus antigen expression increases therapeutic vaccine efficacy in high-titer hepatitis b virus carrier mice. Gastroenterology 158, 1762.e9–1775.e9.
Milich, D. R., Jones, J. E., Hughes, J. L., Price, J., Raney, A. K., and McLachlan, A. (1990). Is a function of the secreted hepatitis B e antigen to induce immunologic tolerance in utero? Proc. Natl. Acad. Sci. U.S.A. 87, 6599–6603. doi: 10.1073/pnas.87.17.6599
Miller, R. H., and Robinson, W. S. (1986). Common evolutionary origin of hepatitis B virus and retroviruses. Proc. Natl. Acad. Sci. U.S.A. 83, 2531–2535. doi: 10.1073/pnas.83.8.2531
Moraleda, G., Saputelli, J., Aldrich, C. E., Averett, D., Condreay, L., and Mason, W. S. (1997). Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J. Virol. 71, 9392–9399. doi: 10.1128/jvi.71.12.9392-9399.1997
Mueller, H., Wildum, S., Luangsay, S., Walther, J., Lopez, A., Tropberger, P., et al. (2018). A novel orally available small molecule that inhibits hepatitis B virus expression. J. Hepatol. 68, 412–420. doi: 10.1016/j.jhep.2017.10.014
Oehler, N., Volz, T., Bhadra, O. D., Kah, J., Allweiss, L., Giersch, K., et al. (2014). Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology 60, 1483–1493. doi: 10.1002/hep.27159
Pancholi, P., Lee, D. H., Liu, Q., Tackney, C., Taylor, P., Perkus, M., et al. (2001). DNA prime/canarypox boost-based immunotherapy of chronic hepatitis B virus infection in a chimpanzee. Hepatology 33, 448–454. doi: 10.1053/jhep.2001.21594
Peng, X. H., Ren, X. N., Chen, L. X., Shi, B. S., Xu, C. H., Fang, Z., et al. (2015). High persistence rate of hepatitis B virus in a hydrodynamic injection-based transfection model in C3H/HeN mice. World J. Gastroenterol. 21, 3527–3536. doi: 10.3748/wjg.v21.i12.3527
Petersen, J., Dandri, M., Gupta, S., and Rogler, C. E. (1998). Liver repopulation with xenogenic hepatocytes in B and T cell-deficient mice leads to chronic hepadnavirus infection and clonal growth of hepatocellular carcinoma. Proc. Natl. Acad. Sci. U.S.A. 95, 310–315. doi: 10.1073/pnas.95.1.310
Qi, Z., Li, G., Hu, H., Yang, C., Zhang, X., Leng, Q., et al. (2014). Recombinant covalently closed circular hepatitis B virus DNA induces prolonged viral persistence in immunocompetent mice. J. Virol. 88, 8045–8056. doi: 10.1128/jvi.01024-14
Quetier, I., Brezillon, N., Revaud, J., Ahodantin, J., DaSilva, L., Soussan, P., et al. (2015). C-terminal-truncated hepatitis B virus X protein enhances the development of diethylnitrosamine-induced hepatocellular carcinogenesis. J. Gen. Virol. 96, 614–625. doi: 10.1099/vir.0.070680-0
Rhim, J. A., Sandgren, E. P., Palmiter, R. D., and Brinster, R. L. (1995). Complete reconstitution of mouse liver with xenogeneic hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 92, 4942–4946. doi: 10.1073/pnas.92.11.4942
Roggendorf, M., Schulte, I., Xu, Y., and Lu, M. (2007). Therapeutic vaccination in chronic hepatitis B: preclinical studies in the woodchuck model. J. Viral Hepat. 14(Suppl. 1), 51–57. doi: 10.1111/j.1365-2893.2007.00914.x
Ruan, P., Yang, C., Su, J., Cao, J., Ou, C., Luo, C., et al. (2013). Histopathological changes in the liver of tree shrew (Tupaia belangeri chinensis) persistently infected with hepatitis B virus. Virol. J. 10:333. doi: 10.1186/1743-422x-10-333
Sandgren, E. P., Palmiter, R. D., Heckel, J. L., Daugherty, C. C., Brinster, R. L., and Degen, J. L. (1991). Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell 66, 245–256. doi: 10.1016/0092-8674(91)90615-6
Schinazi, R. F., Bassit, L., Clayton, M. M., Sun, B., Kohler, J. J., Obikhod, A., et al. (2012). Evaluation of single and combination therapies with tenofovir disoproxil fumarate and emtricitabine in vitro and in a robust mouse model supporting high levels of hepatitis B virus replication. Antimicrob. Agents Chemother. 56, 6186–6191. doi: 10.1128/aac.01483-12
Sitia, G., Isogawa, M., Iannacone, M., Campbell, I. L., Chisari, F. V., and Guidotti, L. G. (2004). MMPs are required for recruitment of antigen-nonspecific mononuclear cells into the liver by CTLs. J. Clin. Investig. 113, 1158–1167. doi: 10.1172/jci200421087
Sitia, G., Isogawa, M., Kakimi, K., Wieland, S. F., Chisari, F. V., and Guidotti, L. G. (2002). Depletion of neutrophils blocks the recruitment of antigen-nonspecific cells into the liver without affecting the antiviral activity of hepatitis B virus-specific cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 99, 13717–13722. doi: 10.1073/pnas.172521999
Sprinzl, M. F., Oberwinkler, H., Schaller, H., and Protzer, U. (2001). Transfer of hepatitis B virus genome by adenovirus vectors into cultured cells and mice: crossing the species barrier. J. Virol. 75, 5108–5118. doi: 10.1128/jvi.75.11.5108-5118.2001
Summers, J., and Mason, W. S. (1982). Replication of the genome of a hepatitis B–like virus by reverse transcription of an RNA intermediate. Cell 29, 403–415. doi: 10.1016/0092-8674(82)90157-x
Summers, J., Smith, P. M., and Horwich, A. L. (1990). Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J. Virol. 64, 2819–2824. doi: 10.1128/jvi.64.6.2819-2824.1990
Summers, J., Smolec, J. M., and Snyder, R. (1978). A virus similar to human hepatitis B virus associated with hepatitis and hepatoma in woodchucks. Proc. Natl. Acad. Sci. U.S.A. 75, 4533–4537. doi: 10.1073/pnas.75.9.4533
Sung, W. K., Zheng, H., Li, S., Chen, R., Liu, X., Li, Y., et al. (2012). Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 44, 765–769.
Sutnick, A. I., London, W. T., and Blumberg, B. S. (1969). Australia antigen and the quest for a hepatitis virus. Am. J. Dig. Dis. 14, 189–194.
Takeuchi, J. S., Fukano, K., Iwamoto, M., Tsukuda, S., Suzuki, R., Aizaki, H., et al. (2019). A single adaptive mutation in sodium taurocholate cotransporting polypeptide induced by hepadnaviruses determines virus species specificity. J. Virol. 93:e01432-18.
Tateno, C., Yoshizane, Y., Saito, N., Kataoka, M., Utoh, R., Yamasaki, C., et al. (2004). Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am. J. Pathol. 165, 901–912. doi: 10.1016/s0002-9440(10)63352-4
Thimme, R., Wieland, S., Steiger, C., Ghrayeb, J., Reimann, K. A., Purcell, R. H., et al. (2003). CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 77, 68–76. doi: 10.1128/jvi.77.1.68-76.2003
Tong, S., Li, J., Wands, J. R., and Wen, Y. M. (2013). Hepatitis B virus genetic variants: biological properties and clinical implications. Emerg. Microbes Infect. 2:e10.
Tsuge, M., Hiraga, N., Akiyama, R., Tanaka, S., Matsushita, M., Mitsui, F., et al. (2010). HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J. Gen. Virol. 91, 1854–1864. doi: 10.1099/vir.0.019224-0
Tsuge, M., Hiraga, N., Takaishi, H., Noguchi, C., Oga, H., Imamura, M., et al. (2005). Infection of human hepatocyte chimeric mouse with genetically engineered hepatitis B virus. Hepatology 42, 1046–1054. doi: 10.1002/hep.20892
Tu, T., Zhang, H., and Urban, S. (2021). Hepatitis B virus DNA integration: in vitro models for investigating viral pathogenesis and persistence. Viruses 13:180. doi: 10.3390/v13020180
Tuttleman, J. S., Pourcel, C., and Summers, J. (1986). Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 47, 451–460. doi: 10.1016/0092-8674(86)90602-1
Uprichard, S. L., Boyd, B., Althage, A., and Chisari, F. V. (2005). Clearance of hepatitis B virus from the liver of transgenic mice by short hairpin RNA. Proc. Natl. Acad. Sci. U.S.A. 102, 773–778. doi: 10.1073/pnas.0409028102
Urban, S., Breiner, K. M., Fehler, F., Klingmuller, U., and Schaller, H. (1998). Avian hepatitis B virus infection is initiated by the interaction of a distinct pre-S subdomain with the cellular receptor gp180. J. Virol. 72, 8089–8097. doi: 10.1128/jvi.72.10.8089-8097.1998
von Freyend, M. J., Untergasser, A., Arzberger, S., Oberwinkler, H., Drebber, U., Schirmacher, P., et al. (2011). Sequential control of hepatitis B virus in a mouse model of acute, self-resolving hepatitis B. J. Viral Hepat. 18, 216–226. doi: 10.1111/j.1365-2893.2010.01302.x
Walter, E., Keist, R., Niederost, B., Pult, I., and Blum, H. E. (1996). Hepatitis B virus infection of tupaia hepatocytes in vitro and in vivo. Hepatology 24, 1–5. doi: 10.1053/jhep.1996.v24.pm0008707245
Wang, Q., Schwarzenberger, P., Yang, F., Zhang, J., Su, J., Yang, C., et al. (2012). Experimental chronic hepatitis B infection of neonatal tree shrews (Tupaia belangeri chinensis): a model to study molecular causes for susceptibility and disease progression to chronic hepatitis in humans. Virol. J. 9:170. doi: 10.1186/1743-422x-9-170
Weber, O., Schlemmer, K. H., Hartmann, E., Hagelschuer, I., Paessens, A., Graef, E., et al. (2002). Inhibition of human hepatitis B virus (HBV) by a novel non-nucleosidic compound in a transgenic mouse model. Antiviral Res. 54, 69–78. doi: 10.1016/s0166-3542(01)00216-9
Wieland, S., Thimme, R., Purcell, R. H., and Chisari, F. V. (2004). Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. U.S.A. 101, 6669–6674. doi: 10.1073/pnas.0401771101
Wieland, S. F., Spangenberg, H. C., Thimme, R., Purcell, R. H., and Chisari, F. V. (2004). Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc. Natl. Acad. Sci. U.S.A. 101, 2129–2134. doi: 10.1073/pnas.0308478100
Will, H., Cattaneo, R., Koch, H. G., Darai, G., Schaller, H., Schellekens, H., et al. (1982). Cloned HBV DNA causes hepatitis in chimpanzees. Nature 299, 740–742. doi: 10.1038/299740a0
World Health Organization [WHO] (2015). Guidelines for the prevention, care and treatment of persons with chronic hepatitis B infection. Geneva: WHO.
Wu, M., Wang, C., Shi, B., Fang, Z., Qin, B., Zhou, X., et al. (2020). A novel recombinant cccDNA-based mouse model with long term maintenance of rcccDNA and antigenemia. Antiviral Res. 180:104826. doi: 10.1016/j.antiviral.2020.104826
Yan, H., Zhong, G., Xu, G., He, W., Jing, Z., Gao, Z., et al. (2012). Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1:e00049.
Yan, R. Q., Su, J. J., Huang, D. R., Gan, Y. C., Yang, C., and Huang, G. H. (1996). Human hepatitis B virus and hepatocellular carcinoma. 1. Experimental infection of tree shrews with hepatitis B virus. J. Cancer Res. Clin. Oncol. 122, 283–288. doi: 10.1007/bf01261404
Yan, Z., Zeng, J., Yu, Y., Xiang, K., Hu, H., Zhou, X., et al. (2017). HBVcircle: a novel tool to investigate hepatitis B virus covalently closed circular DNA. J. Hepatol. 66, 1149–1157. doi: 10.1016/j.jhep.2017.02.004
Yang, C., Ruan, P., Ou, C., Su, J., Cao, J., Luo, C., et al. (2015). Chronic hepatitis B virus infection and occurrence of hepatocellular carcinoma in tree shrews (Tupaia belangeri chinensis). Virol. J. 12:26. doi: 10.1186/s12985-015-0256-x
Yang, D., Liu, L., Zhu, D., Peng, H., Su, L., Fu, Y. X., et al. (2014). A mouse model for HBV immunotolerance and immunotherapy. Cell. Mol. Immunol. 11, 71–78. doi: 10.1038/cmi.2013.43
Yang, P. L., Althage, A., Chung, J., and Chisari, F. V. (2002). Hydrodynamic injection of viral DNA: a mouse model of acute hepatitis B virus infection. Proc. Natl. Acad. Sci. U.S.A. 99, 13825–13830. doi: 10.1073/pnas.202398599
Yang, W., Mason, W. S., and Summers, J. (1996). Covalently closed circular viral DNA formed from two types of linear DNA in woodchuck hepatitis virus-infected liver. J. Virol. 70, 4567–4575. doi: 10.1128/jvi.70.7.4567-4575.1996
Yuan, L., Jiang, J., Liu, X., Zhang, Y., Zhang, L., Xin, J., et al. (2019). HBV infection-induced liver cirrhosis development in dual-humanised mice with human bone mesenchymal stem cell transplantation. Gut 68, 2044–2056. doi: 10.1136/gutjnl-2018-316091
Zhang, X., Lu, W., Zheng, Y., Wang, W., Bai, L., Chen, L., et al. (2016). In situ analysis of intrahepatic virological events in chronic hepatitis B virus infection. J. Clin. Invest. 126, 1079–1092. doi: 10.1172/jci83339
Keywords: HBV, animal model, CccDNA, chronic hepatitis B, humanized mice
Citation: Zhang X, Wang X, Wu M, Ghildyal R and Yuan Z (2021) Animal Models for the Study of Hepatitis B Virus Pathobiology and Immunity: Past, Present, and Future. Front. Microbiol. 12:715450. doi: 10.3389/fmicb.2021.715450
Received: 27 May 2021; Accepted: 18 June 2021;
Published: 16 July 2021.
Edited by:
Haitao Guo, University of Pittsburgh, United StatesReviewed by:
Moses Turkle Bility, University of Pittsburgh, United StatesBen Burwitz, Oregon Health and Science University, United States
Copyright © 2021 Zhang, Wang, Wu, Ghildyal and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaonan Zhang, WGlhb25hbi5aaGFuZ0BjYW5iZXJyYS5lZHUuYXU=