 Joanna Wyrębek1*
Joanna Wyrębek1* Tomasz Molcan2
Tomasz Molcan2 Kamil Myszczyński3
Kamil Myszczyński3 Anne D. van Diepeningen4
Anne D. van Diepeningen4 Alexander A. Stakheev5
Alexander A. Stakheev5 Maciej Żelechowski1
Maciej Żelechowski1 Katarzyna Bilska1
Katarzyna Bilska1 Tomasz Kulik1*
Tomasz Kulik1*- 1Department of Botany and Nature Protection, University of Warmia and Mazury in Olsztyn, Olsztyn, Poland
- 2Department of Bioinformatics, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw, Poland
- 3Molecular Biology Laboratory, Institute of Animal Reproduction and Food Research, Polish Academy of Sciences, Olsztyn, Poland
- 4Biointeractions & Plant Health, Wageningen Plant Research, Wageningen, Netherlands
- 5Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Moscow, Russia
Fungal complexes are often composed of morphologically nearly indistinguishable species with high genetic similarity. However, despite their close relationship, they can exhibit distinct phenotypic differences in pathogenicity and production of mycotoxins. Many plant pathogenic and toxigenic fungi have been shown to consist of such cryptic species. Identification of cryptic species in economically important pathogens has added value in epidemiologic studies and provides opportunities for better control. Analysis of mitochondrial genomes or mitogenomics opens up dimensions for improved diagnostics of fungi, especially when efficient recovery of DNA is problematic. In comparison to nuclear DNA, mitochondrial DNA (mtDNA) can be amplified with improved efficacy due to its multi-copy nature. However, to date, only a few studies have demonstrated the usefulness of mtDNA for identification of cryptic species within fungal complexes. In this study, we explored the value of mtDNA for identification of one of the most important cereal pathogens Fusarium graminearum sensu stricto (F.g.). We found that homing endonucleases (HEGs), which are widely distributed in mitogenomes of fungi, display small indel polymorphism, proven to be potentially species specific. The resulting small differences in their lengths may facilitate further differentiation of F.g. from the other cryptic species belonging to F. graminearum species complex. We also explored the value of SNP analysis of the mitogenome for typing F.g. The success in identifying F.g. strains was estimated at 96%, making this tool an attractive complement to other techniques for identification of F.g.
Introduction
Fusarium graminearum sensu stricto (F.g.) ranks number 4 in the top 10 most important and best-studied species that cause diseases on agriculturally important plants (Dean et al., 2012). The fungus is often involved in two diseases of cereals: fusarium head blight (FHB) of wheat and barley and fusarium ear rot (FER) of maize, which lead to major losses for grain production worldwide. Besides yield reduction, F.g. is able to produce mycotoxins, among which trichothecenes pose a serious hazard to human and animals (Dong et al., 2020; Yao et al., 2020). F.g. belongs to the monophyletic fungal complex referred to as F. graminearum species complex (FGSC). This complex includes 16 genetically characterized cryptic species (Sarver et al., 2011), several of which are involved in cereal diseases in certain agricultural areas. The species are difficult to identify by morphological characters and often share a high DNA sequence similarity (Walkowiak et al., 2016). However, despite their close relationship, FGSC species and even strains within species can exhibit distinct phenotypic differences in pathogenicity and mycotoxin production, while some strains lack pathogenicity on certain hosts (Jianbo, 2014; van der Lee et al., 2015; Walkowiak et al., 2016).
The increasing number of genomic sequences that are deposited in public databases has enabled research in genomic features that contribute to phenotypic variation and niche specialization within FGSC. Genome comparisons revealed significant divergence between them that can be mostly linked to single-nucleotide polymorphisms (SNPs), insertions/deletions (indels), and gene content variation, which is species specific or fixed in certain populations. Comparative genomics provides insights into the evolutionary processes contributing to pathogen divergence at both the macroevolutionary and microevolutionary scales (Walkowiak et al., 2016; Kelly and Ward, 2018).
Besides nuclear genome analyses, analysis of fusarium mitogenomes may provide useful data to study phylogenetic relationships and evolution. Despite high conservation, mitogenomes of fungi within complexes often exhibit a significant degree of polymorphism (Brankovics et al., 2017, 2020). This variation largely results from the irregular distribution of mobile genetic elements (MGEs) such as introns and associated homing endonucleases (HEGs; Basse, 2010; Joardar et al., 2012; Pogoda et al., 2019). MGEs comprise a substantial fraction of fungal mitogenomes and display a different evolutionary history from other mitochondrial loci (Guha et al., 2018; Kolesnikova et al., 2019). Their mosaic distribution throughout evolution can be explained with the “aenaon” model, which combines “intron-early” evolution enriched by “intron-late” events through recombination involving vertical and horizontal gene transfer (Megarioti and Kouvelis, 2020). Notably, variation in MGE content appears to be dependent on taxonomic sampling. This variation can be highly conserved, indicating a low mobility of ancestrally acquired MGEs (Kulik et al., 2020b). In contrast, more recent enrichment events result in increased mosaicism of MGE patterns that can be observed at the population level (Wolters et al., 2015).
Fungi of the genus Fusarium display a high variation in MGE content, from MGE-poor to MGE-rich mitogenomes (Losada et al., 2014; Brankovics et al., 2017). Our previous study performed on a large collection of F.g. strains has shown that the mitogenome of this fungus is MGE rich and mainly includes introns from the group I intron family, which harbor either LAGLIDADG or GIY-YIG HEGs. Comparison of mitogenomes of geographically diverse F.g. strains did not reveal any population-specific profiles, thus supporting the hypothesis on ancestral acquisition of HEGs. Assuming their early acquisition and low mobility, recently diverging cryptic species may thus share a similar content of MGEs (Kulik et al., 2020b). This hypothesis will be tested using mitogenomes obtained in this study.
Variation in mitogenomes of closely related fungi may also be detected by phylogenetic conflict between single-gene trees. This phenomenon was discovered among and within members of closely related fungal complexes: the Fusarium fujikuroi species complex (FFSC) and Fusarium oxysporum species complex (FOSC; Brankovics et al., 2020). Conflicting tree topologies indicate incomplete lineage sorting from ancestral polymorphism or more recent interspecies gene flow since both can result in similar phylogenetic patterns (Fourie et al., 2013, 2018). The phylogenetic discordance complicates interpretation of phylogenetic reconstructions and largely limits clustering-based identification approaches (Wu et al., 2018).
Besides its evolutionary value, mitochondrial DNA (mtDNA) is promising in the diagnostics of fungi, especially with regard to its multi-copy nature, which facilitates its high recovery and amplification success (Santamaria et al., 2009). Both MGE content variation and SNP in mitogenomes could be used for identification purposes of fusaria (Wu et al., 2019; Kulik et al., 2020b). However, discriminating cryptic species based on mitochondrial sequences is challenging due to the high degree of conservation, and detecting species-specific sites requires screening of a large set of genomic data to differentiate between intraspecific and interspecific variation (Kulik et al., 2015, 2020a).
The analysis of SNPs provides an excellent tool for identifying and typing species (Uelze et al., 2020a). In general, SNPs display relatively low mutation rates and are evolutionarily stable (Leekitcharoenphon et al., 2012). Usually, SNPs are identified by mapping sequence data from individual strains against a closely related reference genome. This type of analysis relies on the generation of a certain number of SNPs. It includes all studied genomes, including the reference genome (Uelze et al., 2020b).
The aims of this study are (1) to explore whether MGE polymorphisms can be used for identification of F.g., (2) discover the value of SNP analysis for typing F.g., and (3) confirm the utility of mt-based phylogenetic approach for the determination of F.g. identity.
In total, 122 strains were analyzed: 88 strains of F.g. and 34 strains representing other members of the FGSC. We discovered that HEGs present in mitogenomes of the FGSC display small indel polymorphism, which facilitates recognition of F.g. We also showed that whole-mitogenome SNP analysis enables typing of F.g. with 96% confidence, which can make this tool a valuable complement to other diagnostic tools for F.g. A phylogenetic analyses based on core mitochondrial genes showed different topologies in the reconstructed phylogenetic trees and did not cluster strains of F.g. into single species-specific clades, which neglects the value of this approach for determination of species identity.
Materials and Methods
Fungal Strains
In total, 122 strains were analyzed in this study, with 88 F.g. strains and 34 strains representing all known cryptic species from the FGSC complex: Fusarium acaciae-mearnsii (three strains), Fusarium aethiopicum (two strains), Fusarium asiaticum (three strains), Fusarium austroamericanum (two strains), Fusarium boothii (three strains), Fusarium brasilicum (two strains), Fusarium cortadariae (three strains), Fusarium gerlachii (two strains), Fusarium louisianense (two strains), Fusarium meridionale (one strain), F. meridionale × F. asiaticum hybrid strain (one strain), Fusarium mesoamericanum (two strains), Fusarium nepalense (two strains), Fusarium ussurianum (two strains), Fusarium vorosii (three strains), and strain CBS 123663, which lacks a Latin binomial. All strains used in this study are included in Supplementary File S1.
DNA Extraction and Sequencing
DNA from fungal strains was extracted from 0.1g of mycelium harvested from PDA medium with the use of the Quick-DNA Plant/Seed Miniprep Kit (Zymo Research, Irvine, CA, United States) and Genomic Mini AX Food (Gdynia, Poland) according to the manufacturer’s protocols. Genome libraries were constructed using either a TruSeq DNA PCR-free library preparation kit (Illumina, San Diego, CA, United States) or a KAPA HyperPlus Kit (Roche Sequencing Solutions, Pleasanton, CA, United States). Whole-genome sequencing was performed by Macrogen (Seoul, South Korea). An Illumina HiSeq X 10 platform was used to sequence the genomes using a paired-end read length of 2×150bp with an insert size of 350bp.
Assembly and Annotation of Mitogenomes
To perform de novo assembly of fungal mitogenomes from whole-genome data, NOVOPlasty 2.7.2 (Dierckxsens et al., 2017) was used. HEGs were identified using NCBI’s ORF Finder, Blast+ (v. 2.9.0; Johnson et al., 2008), and Geneious Prime software (Biomatters Ltd., New Zealand). Annotations were performed using MFannot, InterPro (Mitchell et al., 2015), CD-Search (Marchler-Bauer and Bryant, 2004), and Geneious Prime software (Biomatters Ltd., New Zealand). Annotation of tRNA genes was improved using tRNAscan-SE (Pavesi et al., 1994). Annotations were manually verified and corrected. Complete mitogenomes were deposited in the NCBI database under the GenBank accession numbers given in Supplementary File S1.
Exploration of MGE Diversity
A total of 122 mitogenomes were compared through multiple sequence alignments. The analysis was performed with progressive Mauve (Darling et al., 2010) implemented in Geneious Prime software to examine the distribution of MGEs in the mitogenomes.
SNP Analysis
SNP analysis was performed on the interactive web-based platform Edge bioinformatics.1 SNPs were detected in multiple comparisons of assembled mitogenomes of the strains to the reference whole genome of PH-1 of F.g. (accession number: MH412632). Eight mitogenomes from closely related morphospecies (Fusarium culmorum, Fusarium cerealis, and Fusarium pseudograminearum) were also included in the analysis.
Phylogenic Analysis
Phylogenetic analyses were performed on single-gene alignments of coding sequences of 15 core mitochondrial genes (cox 1–3, cob, atp6, atp8, atp9, nad1-6, nad4L, and ribosomal rnl gene) from 130 strains: 122 mitogenomes of FGSC and eight mitogenomes from closely related morphospecies (F. culmorum, F. cerealis, and F. pseudograminearum). Coding sequence data (CDS) from 15 core mitochondrial genes was first aligned using MAFFT software (v.7.453; Katoh and Toh, 2010) with default settings. Single-gene phylogenetic analyses were performed for atp9, cox1, and cox2. The remaining single-gene alignments were excluded for further phylogenetic analyses due to low sequence polymorphism. The best partition schemes and corresponding substitution models were estimated using PartitionFinder2 (Lanfear et al., 2016). Afterwards, based on the alignment and obtained models, maximum likelihood analysis was conducted using IQ-TREE 2.0.3 with 1,000 ultrafast bootstrap (Minh et al., 2020). F. pseudograminearum was used as an outgroup.
To reveal variation in mitochondrial CDS, core mitochondrial genes were extracted and aligned separately using MAFFT software (v.7.453; Katoh and Standley, 2013). Gene polymorphism analyses were conducted for each CDS based on the alignment of 130 strains. Variation within each CDS was identified as a SNP or indel and counted with the use of an in-house Python script. Additionally, each SNP was characterized as synonymous or nonsynonymous. Nucleotide diversity values (π) for each CDS were calculated with TASSEL software (v.5.2.40; Bradbury et al., 2007). As nucleotide diversity is based only on nucleotide substitutions, the number of indels and percentage of polymorphic sites are given for each CDS. To reveal variation in intronic sequences, conserved introns (found in all studied strains) were extracted and analyzed as described for mitochondrial CDS.
Results
General Characteristics of Fungal Mitogenomes
Mitogenomes of species within the FGSC are highly conserved in terms of a set of 15 protein-coding genes, two rRNA genes (rns and rnl), and 28 tRNA genes, which were localized in the same order and orientation. Their mitogenomes displayed a similar GC content between 31.3 and 32.1%. However, mitogenome comparisons showed considerable differences in their size ranging from the smallest 88.409bp mitogenome of CBS 119183 Fusarium cortaderiae to the largest 106.714bp in CBS 119177 F. vorosii. This variation was mainly associated with a mosaic distribution of MGEs, which is described in one of the following sections.
All FGSC strains contained a large open reading frame with unknown function (LV-uORF; Al-Reedy et al., 2012), which is in every strain located between the rnl and nad2 genes. This LV-uORF differed in size from 4.536 to 6.273bp due to indel polymorphism (Figure 1).

Figure 1. Schematic position of indels in LV-uORF in 122 Fusarium strains.
Mobile Genetic Elements and Their Distribution in FGSC
A total of 45 introns and 56 HEGs were identified in the set of mitochondrial protein-coding genes of the studied strains. Nineteen introns were highly conserved and present in all studied strains. More than half of the introns (n=24) identified were located in the three subunits of the cytochrome c oxidase (cox genes): 14 in cox1, 5 in cox2, and 5 in cox3. Positions of introns were highly conserved among different strains. All introns belonged to the group I intron family except i1 cob found in three strains: F.g. (INRA-156 and 16-462-z) and F. vorosii (CBS 119177), which was classified as belonging to the group II intron. This intron encoded a protein of the reverse transcriptase family. Most of group I introns harbored HEGs, of either the LAGLIDADG or GIY-YIG type, which can be determined based on differences in conserved amino acid motifs. Among 56 distinct HEGs, 33 were assigned as LAGLIDADG endonucleases and 16 as GIY-YIG endonucleases, while seven HEGs could not be precisely predicted as LAGLIDADG or GIY-YIG.
Eight introns showed very high similarity with no size difference variation, while 11 introns displayed some size variation due to small indels mostly below 100bp in size. Nine introns showed large size variation (the i4 nad2 intron, the i1 and i2 cox2 introns, the i2 and i5 cob introns, the i3 cox1 intron, the i1 atp6 intron, and the i1 and i3 cox3 introns), mostly due to the irregular presence of HEGs. HEGs and/or introns that are absent in at least one studied strain are termed “optional” throughout this manuscript.
From the total 46 optional HEGs, 11 were very limited in distribution. A single strain of F. mesoamericanum (CBS 110252) harbored two optional HEGs (144 aa and 170 aa) in the i5 nad2 intron. Due to loss of conserved amino acid motifs reflecting their functional differences, these two HEGs were not precisely determined to either LAGLIDADG or GIY-YIG endonucleases. Similarly, a single strain of F. vorosii (CBS 119177) contained an optional LAGLIDADG (352 aa) found in the i1 atp6 intron. The presence of two optional GIY-YIG (314 aa and 315 aa) and one LAGLIDADG (196 aa) sequences in intron i9 of the cob gene was found in a single strain of F. asiaticum (CBS 110258). More frequent distributions were observed for the remaining MGEs found in the cob gene. An optional LAGLIDADG (474 aa) located in the i2 cob intron was shared by two (CBS 316.73 and CBS 119170) of the three strains of F. boothii. Three strains, CBS 123662 (F. acaciae-mearnsii), CBS 110244 (F. austroamericanum), and CBS 123754 (F. ussurianum), harbored an optional LAGLIDADG (234 aa) in the i8 cob intron. Two other HEGs found in the cob gene were distributed in FGSC at 12.5% frequency and included an optional GIY-YIG (127 aa) in the i2 cob intron of F. brasilicum and F. ussurianum and GIY-YIG (314 aa) as well as LAGLIDADG (192 aa) in the i7 cob intron of all strains of F. nepalense and F. vorosii. The remaining 24 optional HEGs (Supplementary File S1) were more commonly distributed, ranging in frequency from 21.9 to 97.1% (Supplementary File S1; Figures 2–6).
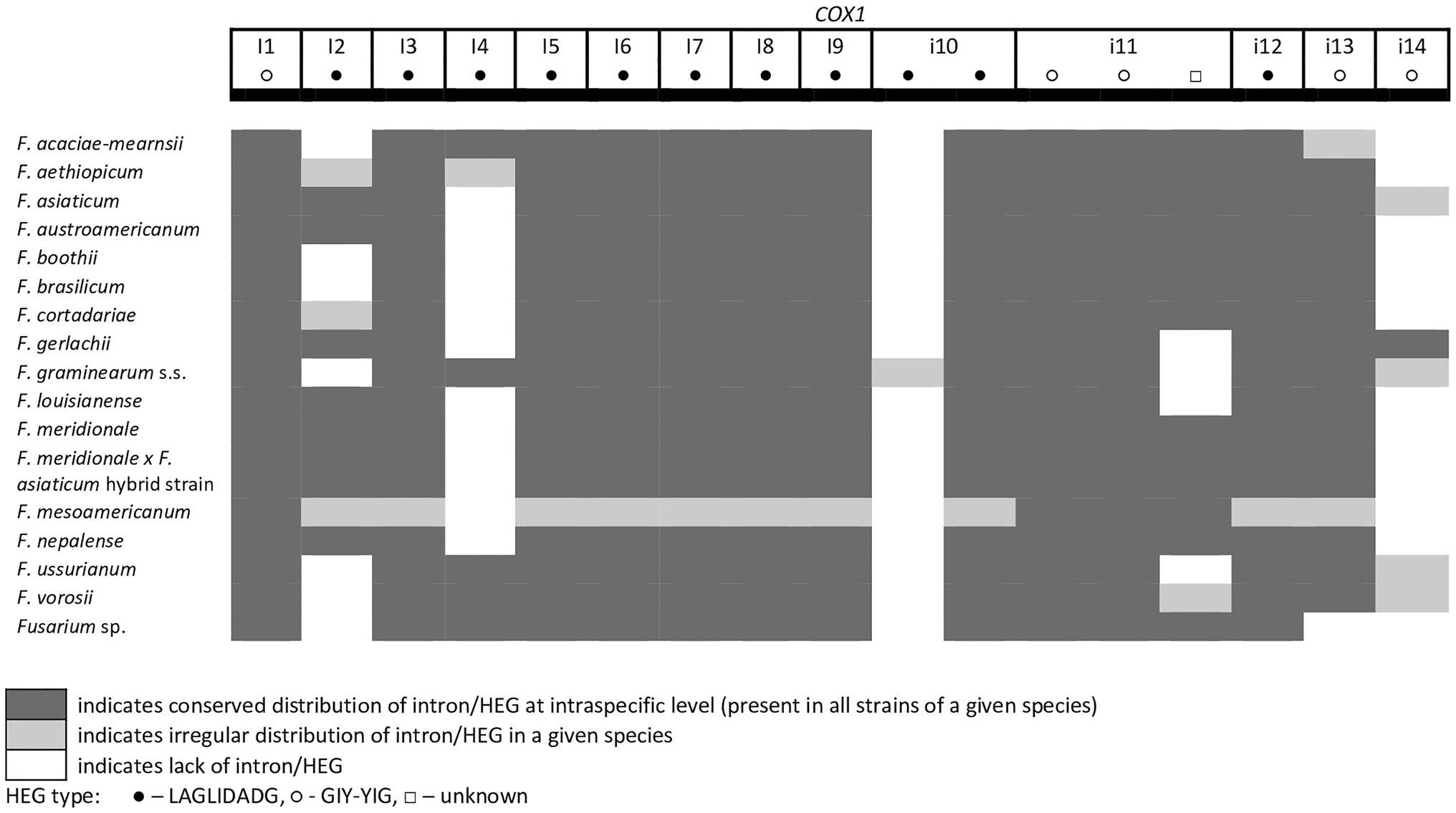
Figure 2. Distribution of introns and associated HEGs found in the COX1 gene.
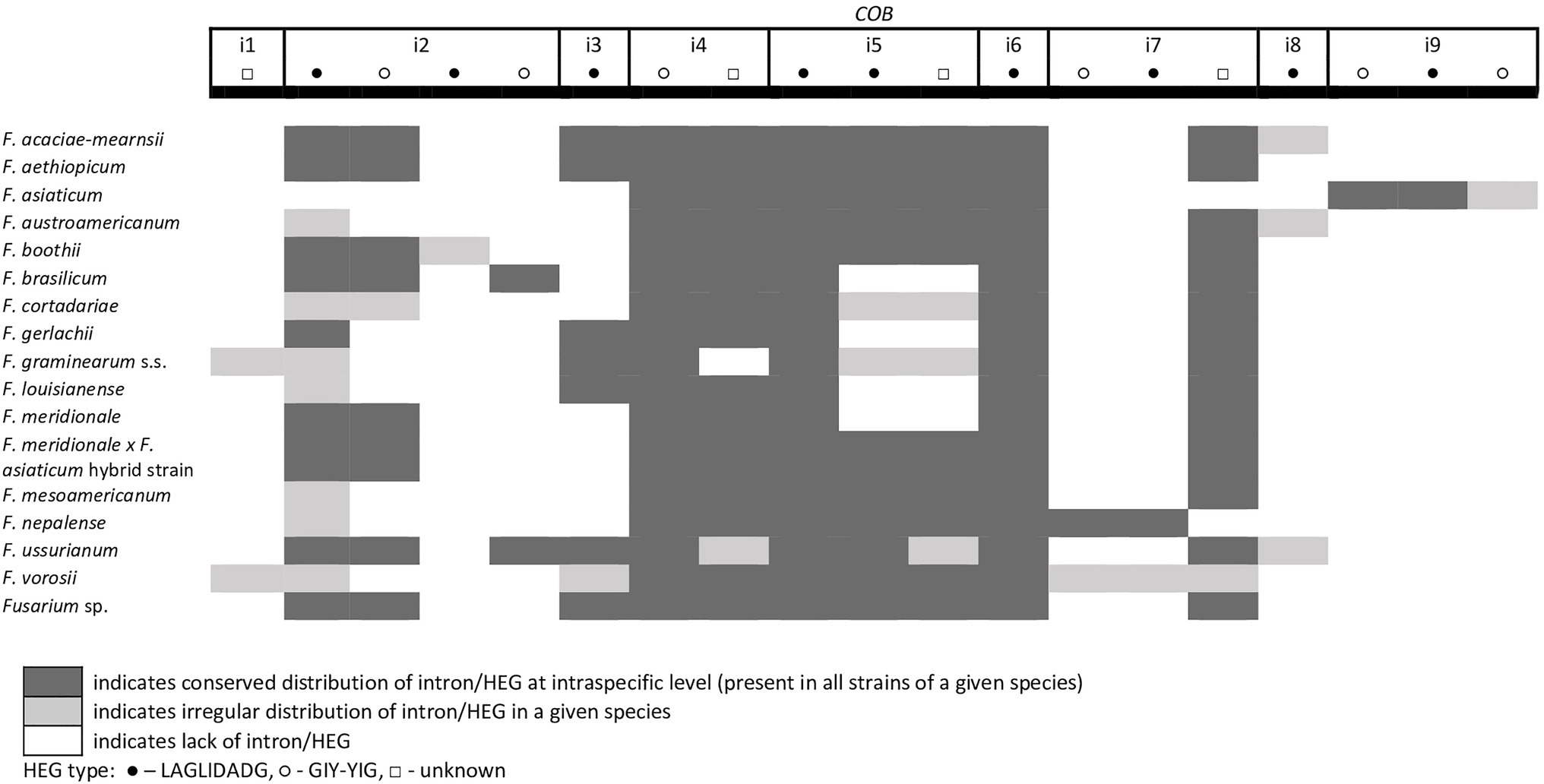
Figure 3. Distribution of introns and associated HEGs found in the COB gene.
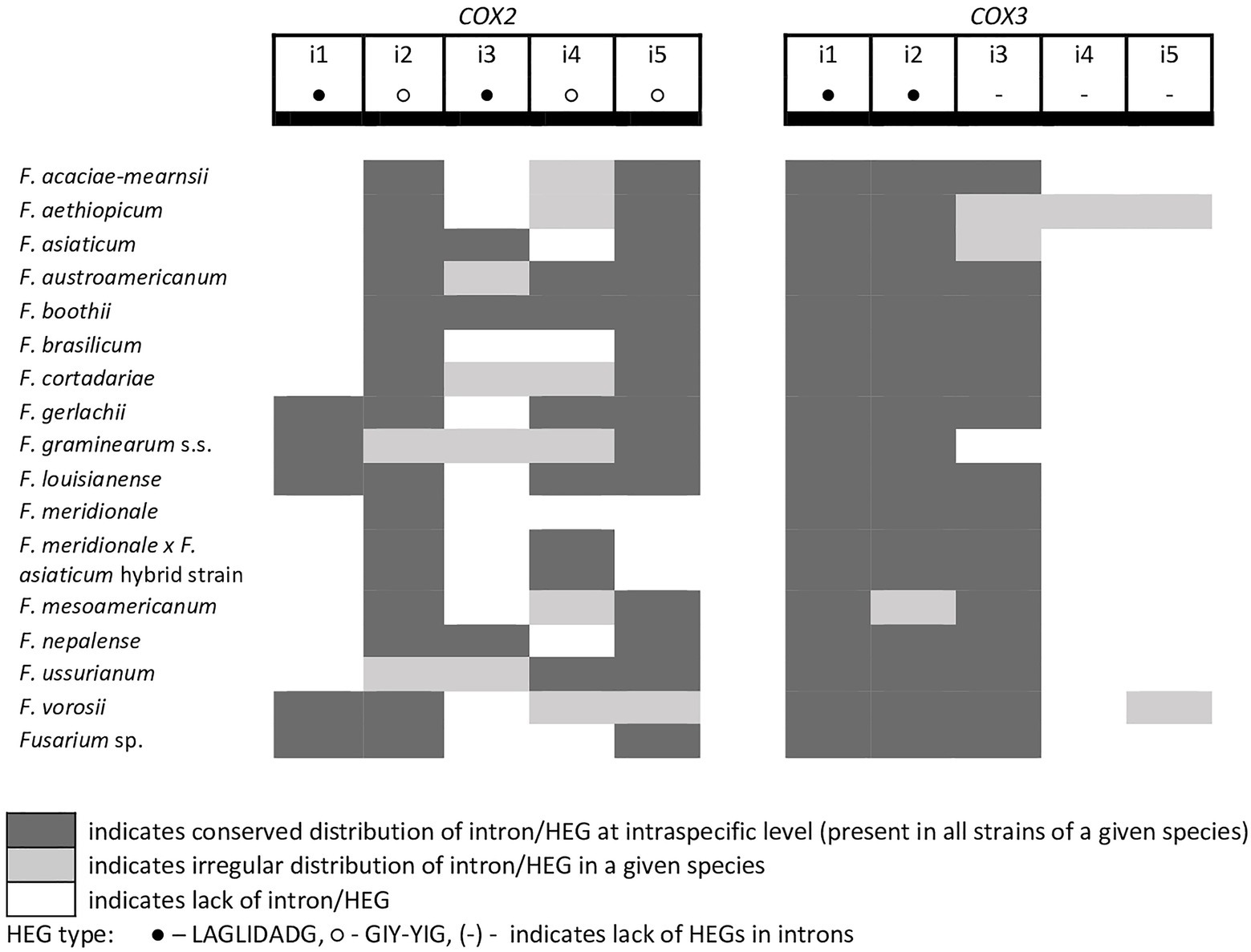
Figure 4. Distribution of introns and associated HEGs found in the COX2 and COX3 genes.
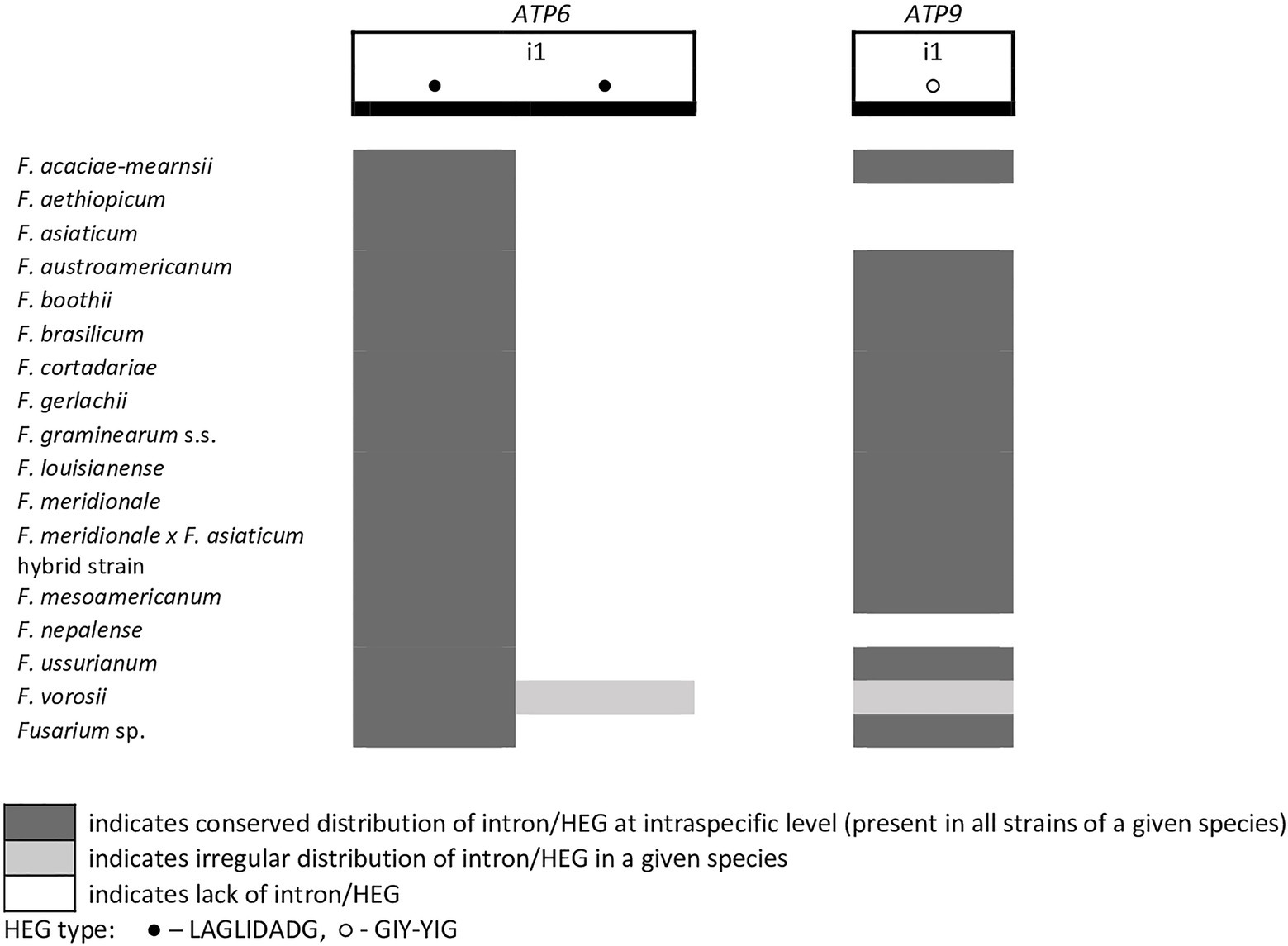
Figure 5. Distribution of introns and associated HEGs found in the ATP6 and ATP9 genes.
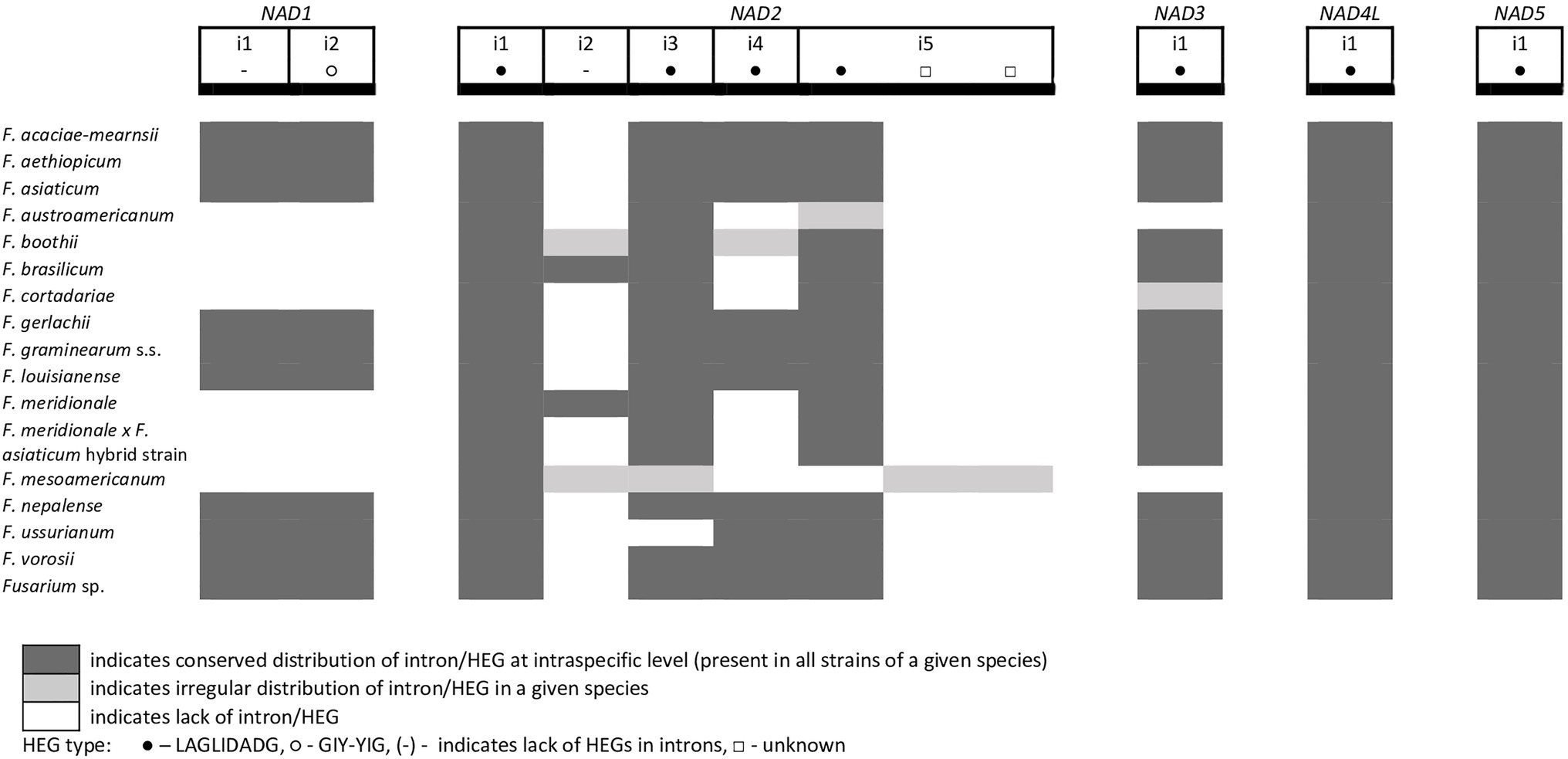
Figure 6. Distribution of introns and associated HEGs found in the NAD genes.
A large collection of F.g. strains enabled the study of MGE distribution in their mitogenomes and possible cryptic divergence. Forty different MGEs were found in F.g.; however, none showed a species-specific conservation. We previously found that a unique MGE pattern in the cox3 gene allows reliable differentiation of F.g. from other closely related morphospecies. This unique pattern mainly concerns the species-specific loss of the i3 intron, which was evident in all strains of F.g., regardless of their geographic origin. In this study, we found that in contrast to F.g., the i3 cox3 intron was present in almost all other members of the FGSC, except for two strains of F. asiaticum (191,317 and 180,363; Yang et al., 2020; Supplementary File S1). However, besides their irregular distribution, HEGs can differ in size due to small indel polymorphism (Table 1). To explore the value of this type of polymorphism for identification of F.g., we compared a total of 1,564 individual HEGs and found that the difference in HEG size observed in eight HEGs (Table 1) can be used as a unique diagnostic feature for F.g.
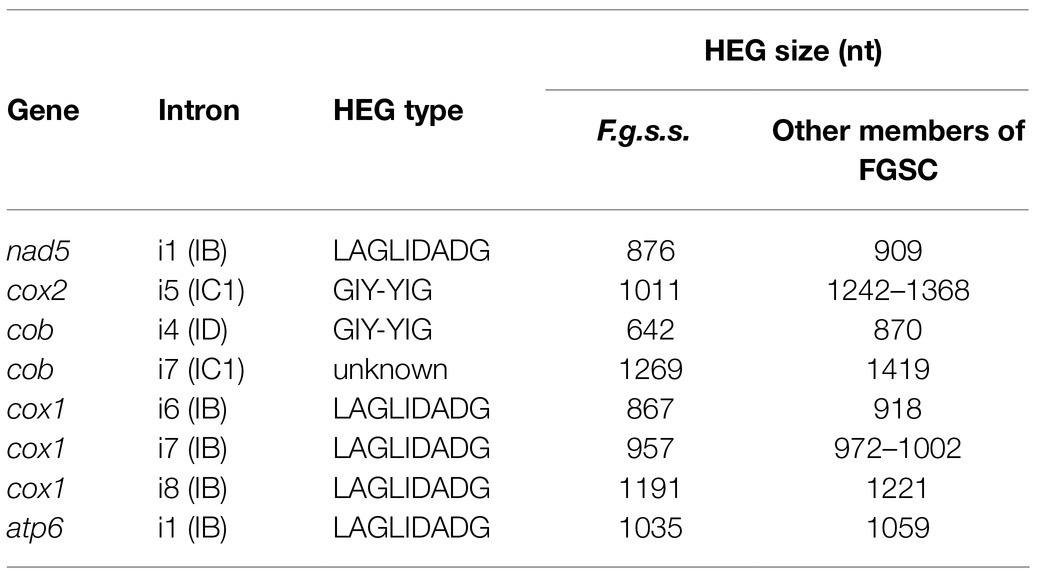
Table 1. List of HEGs allowing differentiation of F.g.s.s. based on size difference.
SNP Analysis
We performed a whole-mitogenome SNP analysis to reveal the utility of this type of approach for differentiation of F.g. from the other species. We calculated the number of SNPs for every mitogenome to reveal if interspecific variability in mtDNA is greater than intraspecific variability, indicating the existence of the so-called “barcoding gap” in the data set. SNPs were detected from the core genome of assembled mitogenomes by multiple comparisons of studied mitogenomes against the reference mitogenome of PH-1 strain (CBS 123657, accession number: MH412632). Analyses of variation in both CDS and conserved introns (Tables 2 and 3) showed that most SNPs could be found in intronic sequences. In general, SNP variability was low, ranging from 9 to 230 SNPs per strain (Supplementary File S2). In most cases, intraspecific variability did not exceed 30 SNPs. However, two exceptional strains of F.g., INRA-156 and CBS 134070, yielded 32 and 36 SNPs, respectively, which exceeded the least interspecies variability (31 SNPs) found for F. vorosii strain CBS 119177. Thus, assuming intraspecific variability below 30 SNPs, two out of 88 strains of F.g. could not be determined based on SNP analysis, which indicates 96% accuracy of this SNP-based approach. Besides exceptional strain (CBS 119177), the least interspecific variability was found for two strains of F. gerlachii, yielding 36 SNPs each. Surprisingly, the highest number of interspecific SNPs was found for two strains of F. ussurianum (227 and 230 SNPs, respectively), which exceeded the number of SNPs found in case of three morphospecies: F. cerealis, F. culmorum, and F. pseudograminearum. This result may indicate that some cryptic species may have an accelerated evolutionary rate with a unique evolutionary pattern in their mtDNA. In addition, the difference in evolutionary rate could be even observed at the strain level. Among the three strains of F. vorosii, CBS 119178 and CBS 123664 exhibited a similar number of interspecies SNPs (147 and 148 SNPs, respectively), which is in contrast to the exceptional strain (CBS 119177), which, as indicated above, yielded only 31 SNPs. An evidence for different evolutionary rates at the strain level was also observed by positioning some strains of the same species into scattered clades on a phylogenetic tree. This has been indicated in the next section.
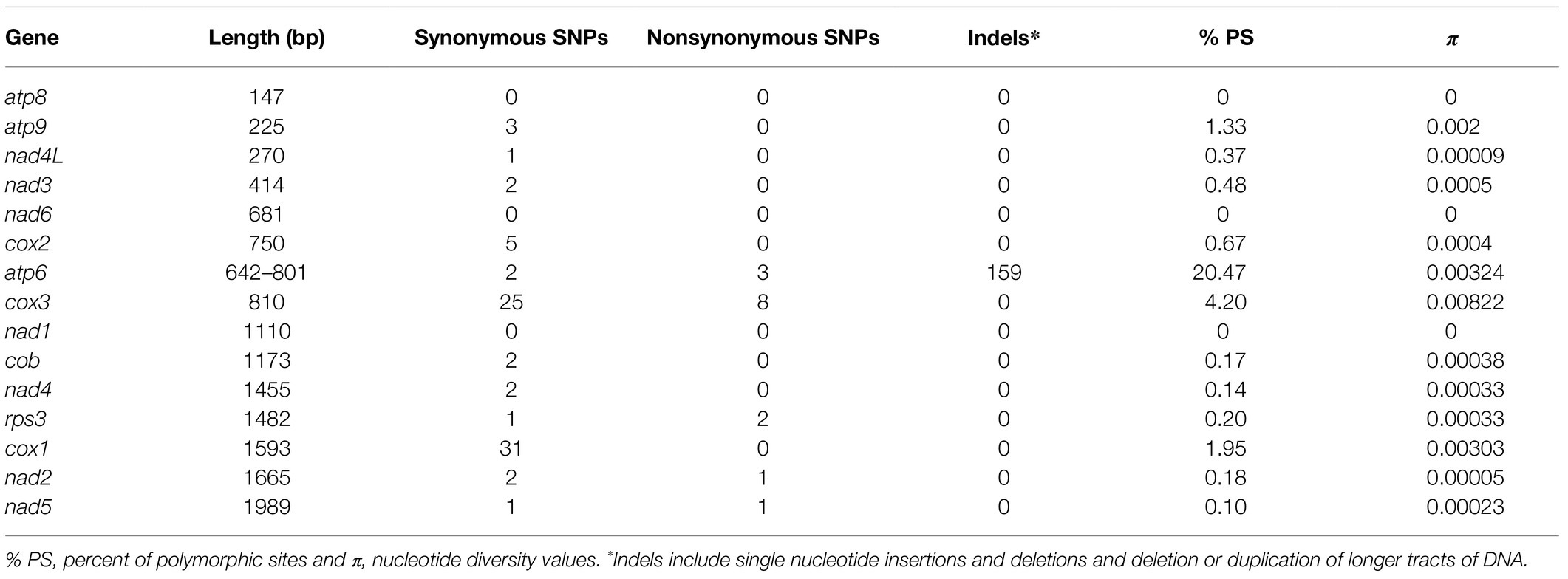
Table 2. Variation in mitochondrial CDS among fungal strains.
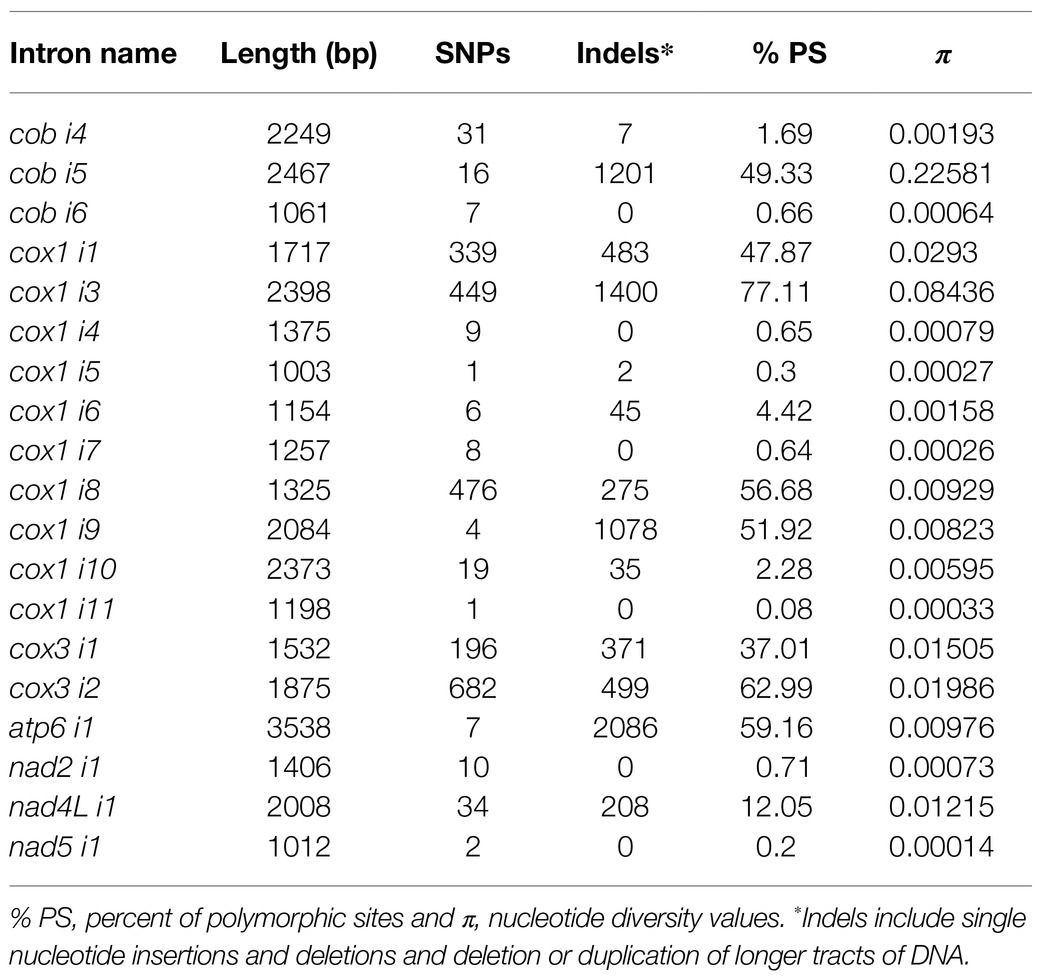
Table 3. Variation in conserved mitochondrial introns among fungal strains.
Phylogenetic Analyses
Phylogenetic trees of atp9, cox1, and cox2 genes possess different topologies, which can be explained by their different evolutionary histories. In addition, majority of the well-supported clades were multispecies. These results arise from low CDS variation in the studied strains, which were further confirmed by calculating nucleotide diversity values, number of polymorphic sites, and SNPs (Table 2). We also found that some strains of the same species were scattered in different clades of the tree. This was mostly evident in the cox1 tree for strains from F.g. and other cryptic species such as F. boothii, F. mesoamericanum, F. cortadariae, and F. vorosii (Supplementary File S4), suggesting incomplete lineage sorting and introgression in the course of the evolution of the studied Fusaria.
Discussion
The introduction of molecular tools has revolutionized the diagnostics of microorganisms, including fungal plant pathogens. The use of DNA-based markers opens possibilities for improved control of specific pathogens and pathotypes (Luchi et al., 2020). Identification of fungi based on mtDNA provides a valuable alternative to existing molecular approaches based on nuclear data, especially when efficient recovery of DNA is problematic. mtDNA can be amplified with higher success than nuclear DNA due to its high copy number in fungal cells (Krimitzas et al., 2013; Franco et al., 2017). However, exploration of mtDNA for diagnostic purposes may be challenging due to difficulties in detecting species-specific polymorphism. These difficulties are mostly reflected by (1) the highly conserved nature of mtDNA (Zhang et al., 2020), (2) intrastrain mosaicism in MGE patterns (Losada et al., 2014), and (3) phylogenetic discordance between different mitochondrial genes (Brankovics et al., 2017; Fourie et al., 2018). A previous comparative analysis of a large set of F.g. strains underlined the high conservation and similarity of mitogenomes in closely related morphospecies: F. cerealis, F. culmorum, and F. pseudograminearum. In general, mitogenomes of these closely related species were MGE rich, and their irregular distribution among strains determines, to a large extent, mitogenome variation. MGEs that do not display mobility among other lineages can be fixed in species. Such targets are good candidates for developing diagnostic tools for identification of species; however, their identification requires comparison of a large set of target and nontarget strains. On the other hand, frequent mobility of MGEs could be incorporated into population genetic studies illustrating the local genetic variation and gene flow (Wolters et al., 2015; Wu et al., 2019).
In this study, we confirmed that the irregular distribution of MGEs does not follow divergence of cryptic species within the FGSC. This finding supports the hypothesis of low mobility of ancestrally acquired MGEs that are consequently shared by geographically diverse cryptic species (Kulik et al., 2020b). These ancestral elements may undergo changes throughout evolution, and assuming the lack of horizontal gene flow, these alternations should, at least in part, exhibit species-specific polymorphism. Indeed, in this study, we found that HEGs shared by different species can display small indel polymorphism that is species specific. The small differences in lengths of certain HEGs (Table 1) may facilitate further differentiation of F.g. from the other members of the FGSC.
In this study, we also explored the value of SNP analysis for typing F.g. This approach is increasingly used in the epidemiologic analyses of bacterial (Schürch et al., 2018) and fungal pathogens (Uelze et al., 2020b); however, to our knowledge, it has not yet been tested using fungal mtDNA. Successful identification of strains to the species level is achieved based on difference in intraspecific and interspecific SNP divergences (Bae et al., 2021). We found that intraspecific variation among F.g. strains was very low. Based on our assumption that intraspecific variation may not exceed 30 SNPs, the success rate in identifying strains of F.g. was estimated at 96%. The high confidence of this approach, its simplicity to perform, and the limited time needed for the analysis make this tool an attractive complement to other more refined techniques for identifying of F.g. (Wu et al., 2019; Kulik et al., 2020b).
Mitochondrial sequences can contribute to species identification due to their efficacy in revealing phylogenetic relationships among the studied taxa. However, we found that for F.g. and the other cryptic species within the FGSC, this approach is not discriminative enough mostly due to the highly conserved nature of mtDNA and lack of concordance between the different gene genealogies, which presumably results from incomplete ancestral lineage sorting. Our findings on the lack of concordance between different phylogenies of mitochondrial genes are in line with previous studies by Fourie et al. (2013, 2018) and Brankovics et al. (2020) who detected interspecies gene flow among mitogenomes of closely related members of the FFSC and FOSC. The success in detecting ancestral gene flow events through mitochondrial comparisons is due to the greater levels of conservation and synteny of fungal mitogenomes than observed in nuclear compartments (Brankovics et al., 2020). However, from a diagnostic point of view, this phylogenetic discordance largely limits clustering-based identification approaches (Wu et al., 2018).
Overall, the results presented in this study showed that the use of mtDNA provides valuable information for identification of cryptic species within fungal complexes. Although fungal mitogenomes lack a “universal barcode” for tagging cryptic species, they might display other patterns of species-specific conservation. Uncovering of these sites requires testing a large collection of geographically diverse strains to differentiate between strain- and species-specific variation.
Conclusion
Mitogenomes show promise for identification purposes of important cryptic species like F.g. in the FGSC. Improved identification could be achieved by the combination of intronic variation analysis and whole-mitogenome SNP analysis. However, mitogenomes also show evidence of ancestral gene flow among other members of the FGSC, which largely limits clustering-based identification.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
JW and TK wrote the manuscript. JW, AD, MŻ, AS, and KB contributed to isolation and strain growth. JW, MŻ, and KB extracted the fungal DNA and prepared libraries for whole-genome sequencing. TK assembled the mitogenomes and was responsible for the study design. KM and TM performed the mitogenome annotation. TM performed the phylogenetic analysis. AD and AS edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Science Center, Poland, grant numbers 2015/19/B/NZ9/01329 and 2018/31/B/NZ9/00546, and “Development Program of the University of Warmia and Mazury in Olsztyn,” co-financed by the European Union under the European Social Fund from the Operational Program Knowledge Education Development. JW is a recipient of a scholarship from the program “Interdisciplinary Doctoral Studies in Biology and Biotechnology” (project number POWR.03.05.00-00-Z310/17), which is funded by the European Social Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.714651/full#supplementary-material
Footnotes
References
Al-Reedy, R. M., Malireddy, R., Dillman, C. B., and Kennell, J. C. (2012). Comparative analysis of Fusarium mitochondrial genomes reveals a highly variable region that encodes an exceptionally large open reading frame. Fungal Genet. Biol. 49, 2–14. doi: 10.1016/j.fgb.2011.11.008
Bae, S. H., Oh, J. H., and Lee, J. (2021). Identification of interspecific and intraspecific single nucleotide polymorphisms in Papaver spp. Plant Breed Biotechnol. 9, 55–64. doi: 10.9787/PBB.2021.9.1.55
Basse, C. W. (2010). Mitochondrial inheritance in fungi. Curr. Opin. Microbiol. 13, 712–719. doi: 10.1016/j.mib.2010.09.003
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Brankovics, B., van Dam, P., Rep, M., de Hoog, G. S., van der Lee, T. A. J., Waalwijk, C., et al. (2017). Mitochondrial genomes reveal recombination in the presumed asexual Fusarium oxysporum species complex. BMC Genomics 18:735. doi: 10.1186/s12864-017-4116-5
Brankovics, B., van Diepeningen, A. D., de Hoog, G. S., van der Lee, T. A. J., and Waalwijk, C. (2020). Detecting introgression between members of the Fusarium fujikuroi and F. oxysporum species complexes by comparative mitogenomics. Front. Microbiol. 11:1092. doi: 10.3389/fmicb.2020.01092
Darling, A. E., Mau, B., and Perna, N. T. (2010). Progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Dean, R., Van Kan, J. A. L., Pretorius, Z. A., Hammond-Kosack, K. E., Di Pietro, A., Spanu, P. D., et al. (2012). The top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13, 414–430. doi: 10.1111/j.1364-3703.2011.00783.x
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:18. doi: 10.1093/nar/gkw955
Dong, F., Xu, J., Zhang, X., Wang, S., Xing, Y., Mokoena, M. P., et al. (2020). Gramineous weeds near paddy fields are alternative hosts for the Fusarium graminearum species complex that causes fusarium head blight in rice. Plant Pathol. 69, 433–441. doi: 10.1111/ppa.13143
Fourie, G., van der Merwe, N. A., Wingfield, B. D., Bogale, M., Tudzynski, B., Wingfield, M. J., et al. (2013). Evidence for inter-specific recombination among the mitochondrial genomes of Fusarium species in the Gibberella fujikuroi complex. BMC Genomics 14:605. doi: 10.1186/1471-2164-14-605
Fourie, G., van der Merwe, N. A., Wingfield, B. D., Bogale, M., Wingfield, M. J., and Steenkamp, E. T. (2018). Mitochondrial introgression and interspecies recombination in the Fusarium fujikuroi species complex. IMA Fungus 9, 37–63. doi: 10.5598/imafungus.2018.09.01.04
Franco, M. E. E., López, S. M. Y., Medina, R., Lucentini, C. G., Troncozo, M. I., Pastorino, G. N., et al. (2017). The mitochondrial genome of the plant-pathogenic fungus Stemphylium lycopersici uncovers a dynamic structure due to repetitive and mobile elements. PLoS One 12:e0185545. doi: 10.1371/journal.pone.0185545
Guha, T. K., Wai, A., Mullineux, S. T., and Hausner, G. (2018). The intron landscape of the mtDNA cytb gene among the Ascomycota: introns and intron-encoded open reading frames. Mitochondrial DNA part A DNA mapping. Seq. Anal. 29, 1015–1024. doi: 10.1080/24701394.2017.1404042
Jianbo, Q. (2014). Molecular characterization of the Fusarium graminearum species complex in eastern China. Artic. Eur. J. Plant Pathol. 139, 811–823. doi: 10.1007/s10658-014-0435-4
Joardar, V., Abrams, N. F., Hostetler, J., Paukstelis, P. J., Pakala, S., Pakala, S. B., et al. (2012). Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genomics 13:698. doi: 10.1186/1471-2164-13-698
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., and Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36, 5–9. doi: 10.1093/nar/gkn201
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/MOLBEV/MST010
Katoh, K., and Toh, H. (2010). Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26, 1899–1900. doi: 10.1093/bioinformatics/btq224
Kelly, A. C., and Ward, T. J. (2018). Population genomics of Fusarium graminearum reveals signatures of divergent evolution within a major cereal pathogen. PLoS One 13:e0194616. doi: 10.1371/journal.pone.0194616
Kolesnikova, A. I., Putintseva, Y. A., Simonov, E. P., Biriukov, V. V., Oreshkova, N. V., Pavlov, I. N., et al. (2019). Mobile genetic elements explain size variation in the mitochondrial genomes of four closely-related Armillaria species. BMC Genomics 20:351. doi: 10.1186/s12864-019-5732-z
Krimitzas, A., Pyrri, I., Kouvelis, V. N., Kapsanaki-Gotsi, E., and Typas, M. A. (2013). A phylogenetic analysis of greek isolates of Aspergillus species based on morphology and nuclear and mitochondrial gene sequences. Biomed. Res. Int. 2013, 1–18. doi: 10.1155/2013/260395
Kulik, T., Bilska, K., and Żelechowski, M. (2020a). Promising perspectives for detection, identification, and quantification of plant pathogenic fungi and oomycetes through targeting mitochondrial DNA. Int. J. Mol. Sci. 21:2645. doi: 10.3390/ijms21072645
Kulik, T., Brankovics, B., van Diepeningen, A. D., Bilska, K., Żelechowski, M., Myszczyński, K., et al. (2020b). Diversity of mobile genetic elements in the mitogenomes of closely related Fusarium culmorum and F. graminearum sensu stricto strains and its implication for diagnostic purposes. Front. Microbiol. 11:1002. doi: 10.3389/fmicb.2020.01002
Kulik, T., Ostrowska, A., Buśko, M., Pasquali, M., Beyer, M., Stenglein, S., et al. (2015). Development of an FgMito assay: a highly sensitive mitochondrial based qPCR assay for quantification of Fusarium graminearum sensu stricto. Int. J. Food Microbiol. 210, 16–23. doi: 10.1016/j.ijfoodmicro.2015.06.012
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2016). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. doi: 10.1093/molbev/msw260
Leekitcharoenphon, P., Kaas, R. S., Thomsen, M. C. F., Friis, C., Rasmussen, S., and Aarestrup, F. M. (2012). snpTree--a web-server to identify and construct SNP trees from whole genome sequence data. BMC Genomics 13(Suppl. 7):S6. doi: 10.1186/1471-2164-13-S7-S6
Losada, L., Pakala, S. B., Fedorova, N. D., Joardar, V., Shabalina, S. A., Hostetler, J., et al. (2014). Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiol. Lett. 352, 165–173. doi: 10.1111/1574-6968.12387
Luchi, N., Ioos, R., and Santini, A. (2020). Fast and reliable molecular methods to detect fungal pathogens in woody plants. Appl. Microbiol. Biotechnol. 104, 2453–2468. doi: 10.1007/s00253-020-10395-4
Marchler-Bauer, A., and Bryant, S. H. (2004). CD-search: protein domain annotations on the fly. Nucleic Acids Res. 32, W327–W331. doi: 10.1093/nar/gkh454
Megarioti, A. H., and Kouvelis, V. N. (2020). The coevolution of fungal mitochondrial introns and their homing endonucleases (GIY-YIG and LAGLIDADG). Genome Biol. Evol. 12, 1337–1354. doi: 10.1093/gbe/evaa126
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Mitchell, A., Chang, H. Y., Daugherty, L., Fraser, M., Hunter, S., Lopez, R., et al. (2015). The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221. doi: 10.1093/nar/gku1243
Pavesi, A., Conterio, F., Bolchi, A., Dieci, G., and Ottonello, S. (1994). Identification of new eukaryotic tRNA genes in genomic DNA databases by a multistep weight matrix analysis of transcriptional control regions. Nucleic Acids Res. 22, 1247–1256. doi: 10.1093/nar/22.7.1247
Pogoda, C. S., Keepers, K. G., Nadiadi, A. Y., Bailey, D. W., Lendemer, J. C., Tripp, E. A., et al. (2019). Genome streamlining via complete loss of introns has occurred multiple times in lichenized fungal mitochondria. Ecol. Evol. 9, 4245–4263. doi: 10.1002/ece3.5056
Santamaria, M., Vicario, S., Pappadà, G., Scioscia, G., Scazzocchio, C., and Saccone, C. (2009). Towards barcode markers in fungi: an intron map of Ascomycota mitochondria. BMC Bioinform. 10(Suppl. 6):S15. doi: 10.1186/1471-2105-10-S6-S15
Sarver, B. A. J., Ward, T. J., Gale, L. R., Broz, K., Corby Kistler, H., Aoki, T., et al. (2011). Novel Fusarium head blight pathogens from Nepal and Louisiana revealed by multilocus genealogical concordance. Fungal Genet. Biol. 48, 1096–1107. doi: 10.1016/j.fgb.2011.09.002
Schürch, A. C., Arredondo-Alonso, S., Willems, R. J. L., and Goering, R. V. (2018). Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene–based approaches. Clin. Microbiol. Infect. 24, 350–354. doi: 10.1016/j.cmi.2017.12.016
Uelze, L., Borowiak, M., Bönn, M., Brinks, E., Deneke, C., Hankeln, T., et al. (2020a). German-wide interlaboratory study compares consistency, accuracy and reproducibility of whole-genome short read sequencing. Front. Microbiol. 11:573972. doi: 10.3389/fmicb.2020.573972
Uelze, L., Grützke, J., Borowiak, M., Hammerl, J. A., Juraschek, K., Deneke, C., et al. (2020b). Typing methods based on whole genome sequencing data. One Heal. Outlook 2, 1–19. doi: 10.1186/s42522-020-0010-1
van der Lee, T., Zhang, H., van Diepeningen, A., and Waalwijk, C. (2015). Biogeography of Fusarium graminearum species complex and chemotypes: a review. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess 32, 453–460. doi: 10.1080/19440049.2014.984244
Walkowiak, S., Rowland, O., Rodrigue, N., and Subramaniam, R. (2016). Whole genome sequencing and comparative genomics of closely related Fusarium head blight fungi: Fusarium graminearum, F. meridionale and F. asiaticum. BMC Genomics 17:1014. doi: 10.1186/s12864-016-3371-1
Wolters, J. F., Chiu, K., and Fiumera, H. L. (2015). Population structure of mitochondrial genomes in Saccharomyces cerevisiae. BMC Genomics 16:451. doi: 10.1186/s12864-015-1664-4
Wu, K.-L., Chen, W.-Z., Yang, S., Wen, Y., Zheng, Y.-R., Anjago, W. M., et al. (2019). Isolation and identification of Fusarium oxysporum f. sp. cubense in Fujian Province, China. J. Integr. Agric. 18, 1905–1913. doi: 10.1016/S2095-3119(18)62149-5
Wu, M., Kostyun, J. L., Hahn, M. W., and Moyle, L. C. (2018). Dissecting the basis of novel trait evolution in a radiation with widespread phylogenetic discordance. Mol. Ecol. 27, 3301–3316. doi: 10.1111/mec.14780
Yang, M., Zhang, H., van der Lee, T. A. J., Waalwijk, C., van Diepeningen, A. D., Feng, J., et al. (2020). Population genomic analysis reveals a highly conserved mitochondrial genome in Fusarium asiaticum. Front. Microbiol. 11:839. doi: 10.3389/fmicb.2020.00839
Yao, L., Li, Y., Ma, C., Tong, L., Du, F., and Xu, M. (2020). Combined genome-wide association study and transcriptome analysis reveal candidate genes for resistance to Fusarium ear rot in maize. J. Integr. Plant Biol. 62, 1535–1551. doi: 10.1111/jipb.12911
Keywords: Fusarium graminearum sensu stricto, Fusarium graminearum species complex, homing endonucleases, introns, mobile genetic elements, identification
Citation: Wyrębek J, Molcan T, Myszczyński K, van Diepeningen AD, Stakheev AA, Żelechowski M, Bilska K and Kulik T (2021) Uncovering Diagnostic Value of Mitogenome for Identification of Cryptic Species Fusarium graminearum Sensu Stricto. Front. Microbiol. 12:714651. doi: 10.3389/fmicb.2021.714651
Edited by:
Vaibhav Srivastava, Royal Institute of Technology, SwedenReviewed by:
Kerry O’Donnell, National Center for Agricultural Utilization Research (NCAUR), United StatesJohn Carlyle Kennell, Saint Louis University, United States
Copyright © 2021 Wyrębek, Molcan, Myszczyński, van Diepeningen, Stakheev, Żelechowski, Bilska and Kulik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanna Wyrębek, am9hbm5hLnd5cmViZWtAdXdtLmVkdS5wbA==; Tomasz Kulik, dG9tYXN6a3VsaWs3NkBnbWFpbC5jb20=