
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 23 December 2021
Sec. Infectious Agents and Disease
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.713724
 Eby M. Sim1*
Eby M. Sim1* Ryan Kim1
Ryan Kim1 Mailie Gall1
Mailie Gall1 Alicia Arnott1Peter Howard1Mary Valcanis2Benjamin P. Howden2
Alicia Arnott1Peter Howard1Mary Valcanis2Benjamin P. Howden2 Vitali Sintchenko1,3,4
Vitali Sintchenko1,3,4
The disease caused by Shiga toxin-producing Escherichia coli (STEC) remains a significant public health challenge globally, but the incidence of human STEC infections in Australia remains relatively low. This study examined the virulence characteristics and diversity of STEC isolates in the state of New South Wales between December 2017 and May 2020. Utilisation of both whole and core genome multi-locus sequence typing (MLST) allowed for the inference of genomic diversity and detection of isolates that were likely to be epidemiologically linked. The most common STEC serotype and stx subtype detected in this study were O157:H7 and stx1a, respectively. A genomic scan of other virulence factors present in STEC suggested interplay between iron uptake system and virulence factors that mediate either iron release or countermeasures against host defence that could result in a reduction of stx1a expression. This reduced expression of the dominant stx genotype could contribute to the reduced incidence of STEC-related illness in Australia. Genomic surveillance of STEC becomes an important part of public health response and ongoing interrogation of virulence factors in STEC offers additional insights for the public health risk assessment.
Escherichia coli is a bacterial commensal in humans, but some strains have evolved to be pathogens by the acquisition of virulence factors. Shiga toxin-producing Escherichia coli (STEC) is a group of predominantly food-borne pathogens that can cause acute gastroenteritis and sometimes haemolytic uraemic syndrome (HUS), neurological complications, and even death (Tarr et al., 2005). This progression to HUS has been linked to the production of Shiga toxin (Stx) encoded by the stx operon consisting of two genes encoding the enzymatically active subunit A and a receptor binding pentameric subunit B of the A1B5 holotoxin, respectively (Melton-Celsa, 2014). STEC can produce two structurally similar but immunologically distinct toxins, namely, Stx1 and Stx2, which can be further subtyped into stx1a, stx1c, stx1d, and stx2a–2g (Melton-Celsa, 2014). This ability to produce Stx is conferred by temperate bacteriophages (stx phages) (Allison, 2007). Not all subtypes are equally pathogenic and infections with STEC harbouring either stx2a, stx2c, or stx2d appeared to be more likely to progress to HUS (Friedrich et al., 2002; Orth et al., 2007). Each stx (pro)phage harbours one stx operon and the detection of multiple stx operons, within an E. coli host, is an indication of multiple integrated stx prophages (Hayashi et al., 2001; Perna et al., 2001; Allison et al., 2003; Fogg et al., 2007; Forde et al., 2019). The stx operon is located in a conserved location within the stx phage, linking Stx production with prophage induction (Neely and Friedman, 1998). This switch to the lytic life cycle via prophage induction releases infectious phage virions, which can disseminate the stx genes to other E. coli cells at the expense of its current host (Allison, 2007).
This horizontal dissemination of stx has allowed for the emergence of hybrid E. coli pathotypes. The most prolific hybrid was the Shiga toxin-producing enteroaggregative E. coli (EAEC) responsible for the large community outbreak in Germany associated with contaminated sprouts (Rohde et al., 2011). Apart from producing Stx, some STEC also harboured the locus of enterocyte effacement (LEE), a pathogenicity island also found in both typical and atypical enteropathogenic E. coli (EPEC) (Schmidt, 2010). This pathogenicity island encodes a type 3 secretion system (T3SS) responsible for both intimate adhesion mediated by intimin (encoded by the eae gene) and its translocated receptor Tir, as well as the effacement of microvilli (Deng et al., 2004; Schmidt, 2010). Not all effector proteins secreted by this T3SS are encoded within the LEE, and these non-LEE effectors can be found either on genomic island or associated with prophages (Deng et al., 2004; Creuzburg et al., 2005; Tobe et al., 2006; Ogura et al., 2009). Some STEC also harboured a plasmid that encodes multiple virulence genes of which the gene ehxA (sometimes also referred to as hlyA), part of the plasmid borne haemolysin operon, has been traditionally used as a diagnostic marker for STEC in culture (Paton and Paton, 1998; Lim et al., 2010). While each individual virulence gene in this plasmid contribute to virulence in their own right, there has yet to be a direct association of between plasmid carriage and severity of disease (Lim et al., 2010).
Horizontal dissemination of stx also enabled virulence among multiple serotypes with STEC serotype O157:H7 being the most commonly recognised as causing human disease (Rangel et al., 2005; Vally et al., 2012; Gould et al., 2013). However, other non-O157 serotypes have also been linked to severe disease, namely, O26, O45, O103, O111, O121, and O145 (Gould et al., 2013; Luna-Gierke et al., 2014). Collectively, these seven serotypes are known as the “Top-7” STEC serotypes, a term used to describe all STEC deemed adulterants by the U.S. Department of Agriculture, Food Safety and Inspection Services (USDA-FSIS, 2011). In addition to serotyping, other molecular techniques, like the lineage specific polymorphism assay (LSPA-6) and the Manning SNP typing scheme, added further resolution but they tended to be biassed to STEC O157:H7 (Yang et al., 2004; Manning et al., 2008). Whole genome sequencing (WGS) has been recently adopted for public health surveillance of STEC and has allowed in silico inference of serotypes and detection of STEC outbreaks as well retrospective epidemiological studies (Joensen et al., 2014; Dallman et al., 2015; Ingle et al., 2019; Jenkins et al., 2019).
The incidence of STEC-associated illness in Australia was historically reported as low (Vally et al., 2012), but with increased surveillance using molecular techniques, incidence was determined to be 2.07 per 100,000 population (Supplementary Table 1). Barring differences in detection methodology, the incidence in Australia was comparable to the United Kingdom and Canada while significantly lower than in New Zealand (Supplementary Table 1). The most common serotype isolated in Australia was STEC O157:H- (Vally et al., 2012; Ingle et al., 2019) with the non-motile phenotype associated with a single base insertion in flgF gene (Pintara et al., 2018). However, these non-motile O157 did encode an intact fliC, the target for in silico H-antigen typing, and as such would be genomically identified as O157:H7 when typed in silico (Pintara et al., 2018). Australian STEC O157 isolates typically harbour stx1a and stx2c either alone or in combination (Fegan and Desmarchelier, 2002; Mellor et al., 2012, 2015) and have been reported to be genetically distinct from international collections of isolates (Leotta et al., 2008; Mellor et al., 2012, 2013, 2015; Ingle et al., 2019; Pintara et al., 2020). It remains unknown if the other non-O157 STEC were also geographically and genetically distinct. In this study, we examined genomic diversity and pathogenic potential of STEC isolates recovered from human cases of gastroenteritis and HUS between December 2017 and May 2020 in New South Wales (NSW), Australia using WGS and downstream analysis. While useful in epidemiology, serotyping is not a reliable indicator for the disease states caused by STEC with the best predictors being stx subtypes and the presence or absence of eae (WHO and FAO, 2018). To that end, we developed a virulence barcode to be used with WGS data, which not only tracks the subtypes of prominent STEC virulence factors eae and stx, but also allows for the inference of multiple, isogenic stx copies derived from short read sequencing data. We also aimed to adapt existing in silico tools currently utilised in pathogen genomics, along with an existing wgMLST schema for routine prospective public health laboratory surveillance of STEC.
Shiga toxin-producing Escherichia coli isolates were cultured from unenriched faecal samples initially subjected to polymerase chain reaction (PCR) testing on the BD-MAX™ platform using the Enteric Bacterial Panel targetting the stx genes (Becton Dickinson). Isolation of STEC colonies was attempted for samples that were PCR positive for stx1 and/or stx2, by picking up to three separate colonies and testing these subcultured colonies for the presence of stx with subsequent somatic O antigen serotyping at the NSW Enteric Reference Laboratory, the Institute of Clinical Pathology and Medical Research (ICPMR)-NSW Health Pathology, NSW, Australia. STEC isolates collected between December 2017 and December 2019 were also forwarded to the Microbiological Diagnostic Unit Public Health Laboratory (MDU PHL), Victoria, Australia for sequencing as previously described (Ingle et al., 2019). Isolates from January 2020 were sequenced at ICPMR-NSW Health Pathology. DNA was extracted from isolates sequenced at ICPMR using QIAGEN DNeasy Blood & Tissue Kit. Sequencing libraries were prepared using Nextera XT and sequenced on the NextSeq500 platform (Illumina) with 150-bp paired-end reads.
Sequencing reads obtained from both ICPMR and MDU PHL were subsequently trimmed using Trimmomatic version 0.36 (Bolger et al., 2014) and assembled using SKESA version 2.3 (Souvorov et al., 2018) on default settings. When required, both Artemis version 18.1.0 (Carver et al., 2012) and Artemis Comparison Tool (ACT) version 18.1.0 (Carver et al., 2005) were used for visualising draft assemblies and pairwise BLASTN comparison, respectively. ABRicate version 1.0.11 was employed to infer presence (minid 90; mincov 90) of antimicrobial resistance genes against the Resfinder database (Zankari et al., 2012), E. coli virulence factors against the ecoli_VF database2, and stx and eae subtypes using a custom database derived from reference sequences in the published literature (Blanco et al., 2005; Scheutz et al., 2012). To aid in data collation post ABRicate screening, E. coli virulence genes were clustered using CD-HIT version 4.8.1 (Fu et al., 2012) to collapse (-c 0.9; -g 1) virulence factors into a single group when multiple homologues exist. Assignation of the virulence factors belonging to the various secretomes was performed as previously reviewed (Monteiro et al., 2016). Type 6 Secretion Systems identified were further subtyped based on the SecReT6 Database (Li et al., 2015). Genomic locations of non-LEE effectors were assigned employing locations reported in reference genomes of STEC O157 (Hayashi et al., 2001; Tobe et al., 2006) and non-O157 (Ogura et al., 2009) isolates.
Mapping of fastq reads in this study was performed using BWA version 0.7.17-r1188 with the MEM algorithm (Li, 2013). Normalisation of stx, to detect potential harbouring of multiple isogenic stx genes (multiple copies of identical stx genes), was achieved by mapping sequencing reads onto a pseudomolecule consisting of the aforementioned stx subtype database and the nucleotide sequence of recA from STEC O157 strain Sakai (NCBI GenBank Accession: BA000007.3) to account for possible interference from the library preparation process. The mean read depth of identified stx subtype was normalised against the mean read depth of the housekeeping gene recA. All normalised values <2 were reported as unlikely to possess additional isogenic stx subtypes. For all values ≥2, the mean read depth of stx was further normalised against the “+1 standard deviation” of the mean read depth over recA gene sequence. Samples with a normalised value of ≥2 for both were reported as inferred plausible for isogenic stx subtypes possession while samples with a value <2 following additional normalisation was reported as inferred possible for multiple isogenic stx subtype possession. Published genomes and/or sequencing reads of STEC O157:H7 strain Sakai (Hayashi et al., 2001), STEC O157:H7 strain TW14359 (Kulasekara et al., 2009), STEC O111:H- strain 95NR1 (McAllister et al., 2016; Forde et al., 2019), STEC O104:H4 strain 2011C-3493 (Ahmed et al., 2012), EPEC O127:H6 strain E2348/69 (Iguchi et al., 2009), and K-12 strain MG1655 (Blattner et al., 1997) were used as either controls or reference genomes for mapping.
Multi-locus sequence typing (MLST) was determined using mlst version 2.193, a software that utilised resources from PubMLST (Jolley and Maiden, 2010) against the Achtman scheme (Wirth et al., 2006). In silico O:H serotyping was predicted using ABRicate (minid 95; mincov 30) against the EcOH database (Ingle et al., 2016). Whole genome MLST (wgMLST; 2,513 loci) and core genome MLST (cgMLST; 2,248 loci) were determined using the E. coli schema from Ridom4 and chewBBACA version 2.5.5 (Silva et al., 2018). In this study, cgMLST was defined as wgMLST loci present amongst all samples within the dataset. Results of both wgMLST and cgMLST were visualised as a minimum spanning tree generated with GrapeTree using the MSTree V2 algorithm (Zhou et al., 2018). Analysis of single-nucleotide polymorphisms (SNP) in STEC genomes was performed using Snippy version 4.6.05 with different reference genomes, chosen according to their serotypes. SNP distance matrices were generated using snp-dists version 0.66.
In silico LSPA-6 PCR of STEC O157 was performed using previously described oligonucleotides (Yang et al., 2004) using isPcr7. STEC O157:H7 clades according to the Manning scheme (Manning et al., 2008) were visually determined in the draft assemblies using a set of previously described discriminatory positions (Yokoyama et al., 2012). An older version of STEC O157 strain Sakai genome (NCBI GenBank Accession: BA000007.2) was used as a reference for clade typing instead of the newer version (Accession: BA00007.3) as discrepancies in locus tags were previously documented (Ingle et al., 2019) when the genome of STEC O157 strain Sakai was resequenced. Insertion of cytosine within flgF (Pintara et al., 2018), a predicted marker for the non-motile phenotype in Australian STEC O157, was visually interrogated from the draft assembly. Presence of the STEC O157 long polar fimbriae was screened using ABRicate (minid 90; mincov 90) on a custom database consisting of the genes that encoded both the LpfA and the LpfD2 pilus of STEC O157:H7 strain Sakai (Hayashi et al., 2001).
To provide a quick snapshot of pertinent virulence factors, all STEC isolates were assigned a STEC virulence barcode made up of 12 digits in the format of “XX-XX-XX-XX-XX-XX” reflecting their STEC virulence makeup. Each set of two digits represented a particular virulence factor. The first set of two digits represented the presence (to the subtype level) or absence of the eae gene. The next set of two digits represented inference of possible multiple, isogenic stx genes not assembled via short read sequencing. The last four sets of 2-mers each reflected the presence (to the subtype level) or absence of stx. This representation allowed up to four different stx operons to be captured, which is currently the maximum number observed both in vitro (Fogg et al., 2007) and in isolates (McAllister et al., 2016; Forde et al., 2019). The codex for decoding the virulence barcode is listed in Table 1.

Table 1. Codex for decoding the 12-mer STEC virulence barcode and detection methodology.
From December 2017 to May 2020, there were 1,570 cases of STEC notifications nationally and 186 of those notifications were from NSW. Within this time period, NSW Enteric Reference laboratory investigated 67 putative STEC (36% of all notifications); 62 of them were subsequently confirmed as STEC while five isolates did not possess the stx operon. Majority of the STEC isolates, with the exception of two, had accompanying metadata, which included clinical manifestations. Taken together, our genomic dataset covered 33.3% of all STEC cases notified to NSW Health between December 2017 and May 2020.
All 62 draft genomes have sizes ranging between 4.75 Mb and 5.58 Mb, within the bounds of an E. coli genome (Blattner et al., 1997; Hayashi et al., 2001), with the number of contigs (>300 bp) ranging from 70 to 363 (Supplementary Table 2). Nineteen different serotypes were detected in silico, which included three “Top-7 STEC” serotypes, namely, O157:H7, O26:H11, and O111:H8 (Figure 1). Isolates of O157:H7 and O26:H11 were distributed across the study period while some isolates belonging to both O128:H2 and O111:H8 were recovered in close succession to each other. Screening for genes that were involved in antimicrobial resistance (Supplementary Table 3) detected the carriage of mdfA, a gene that encodes an efflux pump that when overexpressed, confer resistance to a broad range of antibiotics (Edgar and Bibi, 1997), in all STEC genomes. Two O157:H7 isolates also screened positive for additional AMR markers. The first isolate contained blaTEM–1B while the second isolate screened positive for blaTEM–1B, strA, strB, sul2, and tetA. Of the 62 assemblies, 10 had stx genes that were not contiguously assembled, despite mapping showing the presence of the associated stx operon. In total, six stx subtypes were documented, namely, stx1a, stx1c, stx2a, stx2b, stx2c, and stx2d (Table 2).
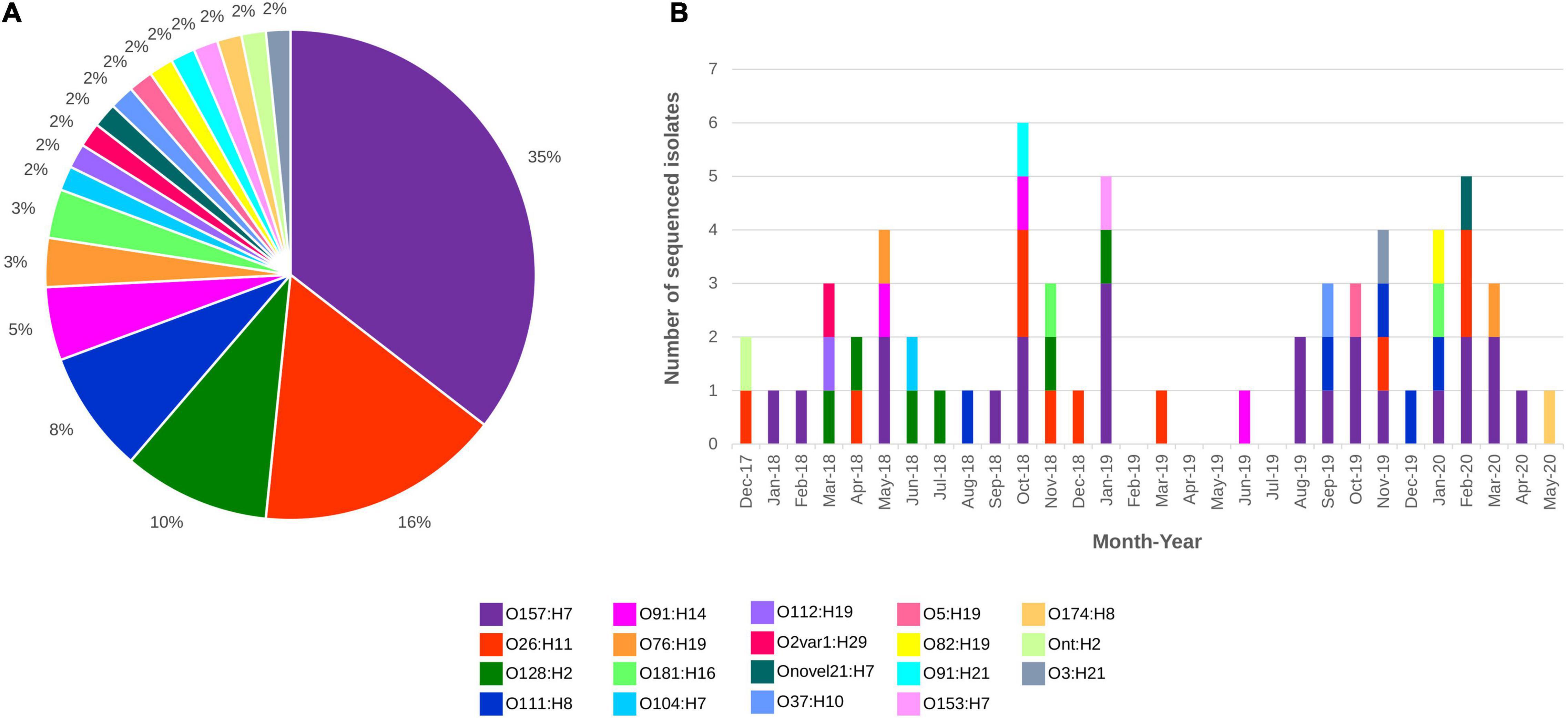
Figure 1. STEC sequenced from December 2017 to May 2020 distributed by (A) Serotype and (B) Serotype and month of isolation. All serotypes are colour coded as listed in the figure key.
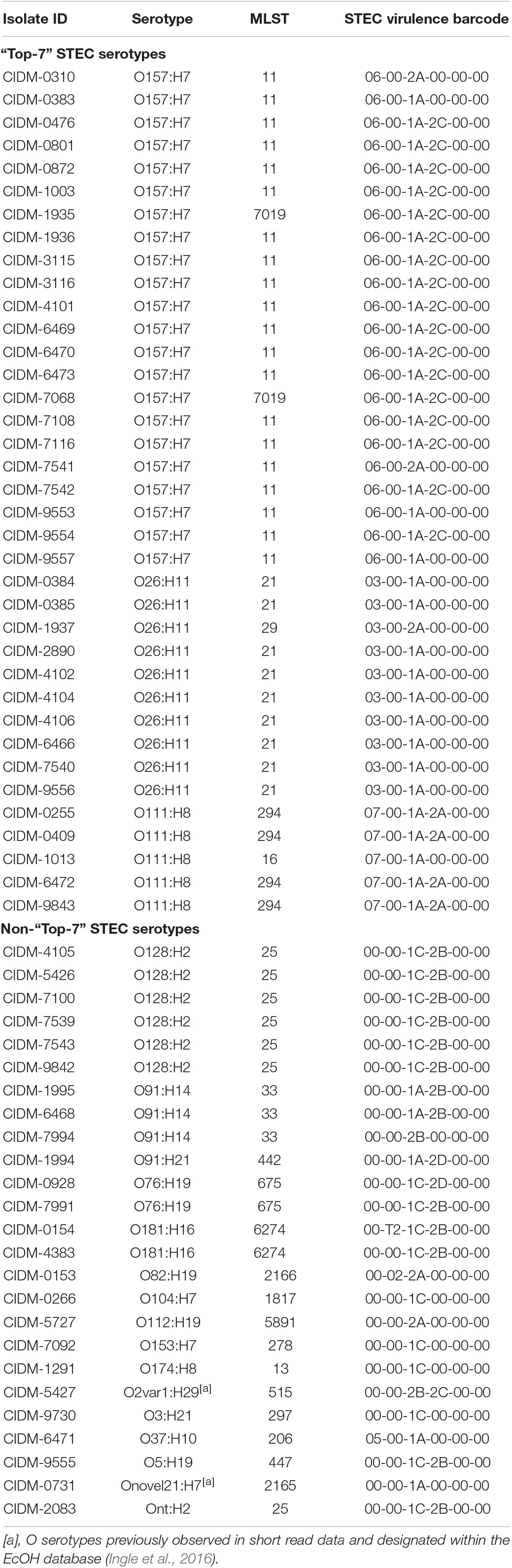
Table 2. In silico predicted serotype and STEC virulence profile of the 62 STEC genomes.
Screening of short-read assembly (NCBI GenBank accession: GCA_000467695.1) and the complete genome (NCBI GenBank accession: CP021339.1) of 95NR1 revealed that multiple isogenic stx subtypes could be masked from the in silico screen when using draft genomic assemblies as input whereby two additional stx2a genes present in the completed genome were missed (Supplementary Table 2). Normalisation of stx genes against the recA gene demonstrated that majority of the 62 genomes were unlikely to harbour additional isogenic stx genes with the exception of two isolates, CIDM-0153 and CIDM-0154. The normalised value for the mean read depth of the implicated stx to the mean read depth of recA was 2.97 and 2.14 for CIDM-0153 and CIDM-0154, respectively. However, when the + 1 standard deviation value of recA was used for normalisation, the value for CIDM-0154 was <2.0 but CIDM-0153 was ≥2.0. Taken together, our results indicated that CIDM-0153 plausibly harboured an additional isogenic stx2a operon. On the other hand, CIDM-0154 could harbour an additional stx2b but based on described cutoffs, it was equally possible that artefacts resulting from either sequencing or prophage induction during the laboratory cultivation and/or extraction process could have influenced our genome analysis results.
Isolates from the “Top-7” STEC serotypes made up more than half of our dataset with the most dominant serotype being O157:H7 (n = 22; Figure 1). In silico MLST typed 20 STEC O157:H7 isolates as ST11 while two isolates were reported as ST7019. STEC O157:H7 are typically typed as ST11 and a query on EnteroBase (accessed 05/08/2020)8 revealed that O157:H7 of ST7019 has also been sighted (n = 35) with majority of the isolates originating from Australia (n = 32). Three different virulence barcodes (Table 2) were associated with O157:H7 with the majority being 06-00-1A-2C-00-00 (n = 17) followed by 06-00-1A-00-00-00 (n = 3) and 06-00-2A-00-00-00 (n = 2). All O157:H7 isolates harboured a γ1 subtype eae consistent with previous reports (Oswald et al., 2000; Blanco et al., 2005) and majority harboured stx1a either alone or in combination with stx2c, an stx genotype common in Australian STEC O157 (Fegan and Desmarchelier, 2002; Mellor et al., 2012, 2013, 2015; Ingle et al., 2019; Pintara et al., 2020).
All STEC O157:H7 genomes were further interrogated with additional serotype-specific typing schemes. Majority (n = 20) of O157:H7 isolates possessed the cytosine insertion in flgF, suggesting that majority of the isolates were non-motile (Supplementary Table 4). In silico LSPA-6 revealed that one O157:H7 isolate belonged to lineage I/II, characterised by an allelic profile of 211111 (Supplementary Table 4), an LSPA-6 lineage typical of STEC O157 from Australia (Mellor et al., 2012, 2013). The other 21 isolates were assigned an allelic profile of 213111 due to the third amplicon targetting yhcG yielding an amplicon (392 bases) two bases shorter than a Lineage I designation (394 bases). Interrogation of the in silico amplicon sequence revealed that the two base deletion was located at position 91 (A) and 92 (T) of a lineage I yhcG amplicon. SNP typing according to the Manning scheme using a reduced discriminatory panel (Yokoyama et al., 2012) indicated that 21 isolates belonged to clade 7 while one isolate belonged to clade 8 with the latter being one of the O157:H7 isolate with a 06-00-2A-00-00-00 barcode designation (Supplementary Tables 2, 4).
Of the O26:H11 isolates (n = 10) in our dataset (Table 2), nine represented ST21 while one was typed as ST29. Two virulence barcodes were associated with O26:H11, namely, 03-00-1A-00-00-00 (n = 9) and 03-00-2A-00-00-00 (n = 1) for ST21 and ST29, respectively. Similar to O26:H11, the STEC O111:H8 isolates (n = 5) were split between two sequence types, namely, ST294 (n = 4) and ST16 (n = 1). This also resulted in two different STEC barcodes, with the ST294 isolates possessing a STEC virulence barcode of 07-00-2A-00-00-00 while the ST16 isolate possessed a barcode of 07-00-1A-00-00-00. Subtypes of eae for all O26:H11 (subtype: β1) and O111:H8 (subtype: γ2/θ) isolates were in agreement with previous reports (Oswald et al., 2000; Blanco et al., 2005).
The remaining 25 isolates were spread over 16 serotypes, 15 MLST sequence types, and 12 different assigned barcodes with no overlap of previously assigned barcodes to the “Top-7” serotypes (Table 2). Some non-“Top-7” STEC serotypes in our dataset harboured stx subtypes that were detected in their “Top-7” counterparts, with stx1a detected in O37:H10 (n = 1), O91:H14 (n = 2; three isolates in total but one did not harbour stx1a), O91:H21 (n = 1), and Onovel21:H7 (n = 1); stx2a detected in O112:H19 (n = 1) and O82:H19 (n = 1); and stx2c detected in O2var1:H29 (n = 1). Both Onovel21 and O2var1 were novel O serotypes previously observed in short read data and designated within the EcOH database (Ingle et al., 2016). Amongst these eight isolates, four harboured an additional stx subtype not encoded by any of the “Top-7” STEC serotypes with stx2b harboured by isolates of serotype O2var1:H29 and O91:H14 (n = 3) and stx2d harboured by an O91:H21 isolate. The remaining 16 isolates belonging to serotypes O128:H2 (n = 6), O181:H16 (n = 2), O76:H19 (n = 2), Ont:H2 (n = 1), O104:H7 (n = 1), O153:H7 (n = 1), O174:H8 (n = 1), O3:H21 (n = 1), and O5:H19 (n = 1) each harboured stx1c either alone (n = 4) or in combination with stx2b (n = 11) or stx2d (n = 1). Only one isolate from a non-“Top-7” STEC serotype harboured eae (Table 2).
Of the 60 STEC isolates with accompanying data on clinical presentation, 55 patients presented with either bloody diarrhoea (n = 30) or mild clinical manifestations (diarrhoea and/or abdominal pain; n = 25). Majority of the patients with mild disease harboured stx subtypes that were not typically associated with the progression to HUS (Figure 2), with the exception of a STEC O111:H8 isolate (barcode: 07-00-1A-2A-00-00) and a STEC O76:H19 isolate (barcode: 00-00-1C-2D-00-00). Majority of the stx2a-positive STECs were isolated from patients with bloody diarrhoea, and none were isolated from patients with mild disease (Figure 2). Five STECs were isolated from patients that progressed to HUS and majority (n = 4) possessed an stx2 subtype (stx2a or stx2d) while the remaining isolate (serotype O104:H7) possessed stx1c only. A STEC (serotype O78:H-) harbouring stx1c was previously reported to cause HUS in a toddler while other family members remained asymptomatic (Lienemann et al., 2012). Apart from stx subtypes, young age was also a risk factor for HUS (Tarr et al., 2005) and could have been a contributing factor to the development of HUS in the report by Lienemann et al. (2012). Two of the four STECs belonged to serotype O111:H8 while the remaining two belonged to serotype O91:H21 and O82:H19, respectively. Interestingly, the O82:H19 isolate was determined to plausibly harbour two copies of the stx2a operon (barcode: 00-02-2A-00-00), which could have been a contributory factor to severe disease manifestation as previously observed, albeit in a different STEC serotype (Henning et al., 1998; McAllister et al., 2016).
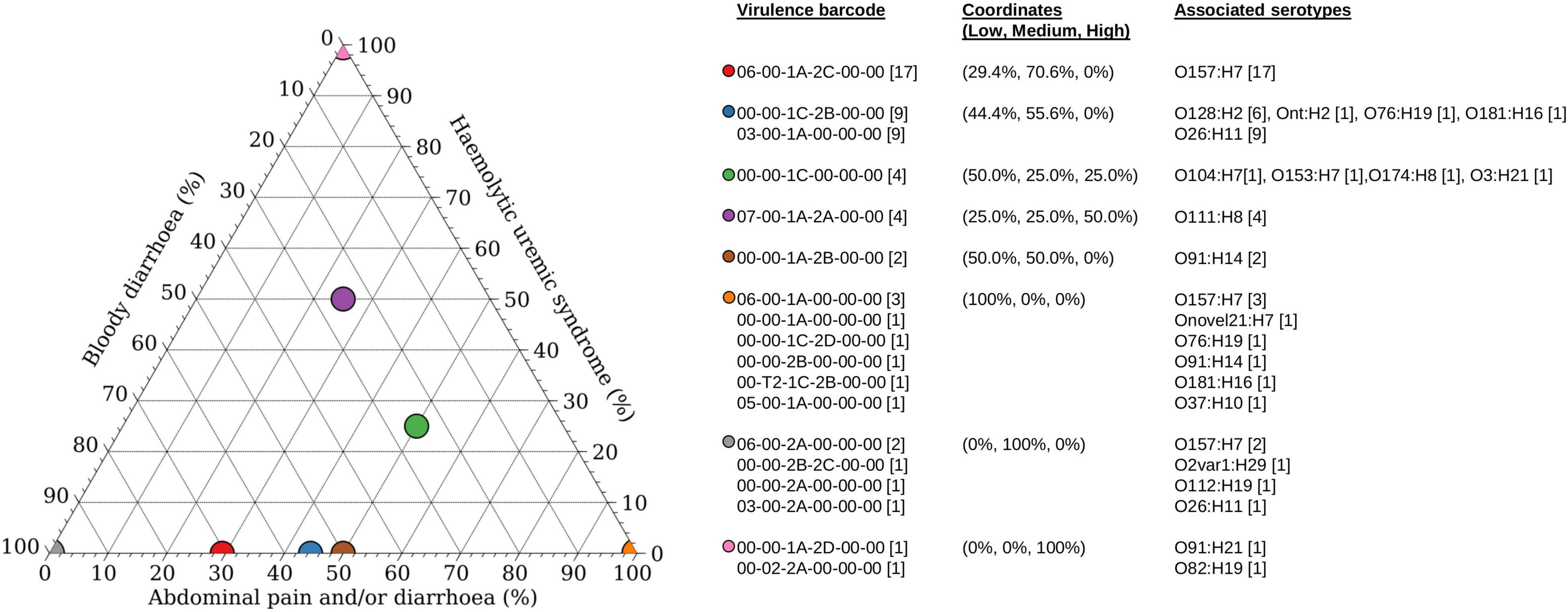
Figure 2. Association of disease outcomes with virulence barcode. Ternary plot of 60 STEC with accompanying clinical information grouped according to virulence barcode and plotted as a percentage against increasing clinical manifestations ranging from low (abdominal pain and/or diarrhoea), medium (bloody diarrhoea), and high (haemolytic uraemic syndrome). Coordinates of each group are listed within parenthesis in the format of (percentage low, percentage medium, and percentage high). Graph was generated using Veusz version 3.2.1 (https://github.com/veusz/veusz).
Only minor genomic diversity within and amongst serotypes based on both MLST and the presence/absence of key STEC virulence factors was documented. Further genomic diversity was interrogated using wg/cgMLST. The difference in the number of loci between the wgMLST and cgMLST schema was 265 fewer loci in the latter scheme. In both wgMLST and cgMLST, each of the 62 isolates possessed a distinct genotype and all isolates belonging to the same serotype clustered together (Figure 3A). There were minor differences in the topology of the minimum spanning tree (MST) between wgMLST (Figure 3) and cgMLST (Supplementary Figure 1) but the general trend of serotype clustering remained the same. When the virulence barcode was overlaid over the wgMLST MST (Figure 3B), it provided a visual snapshot that echoed the aforementioned observation of STECs belonging to the “Top-7” serotypes being more likely to harbour eae than non-“Top-7” STEC serotypes and that both stx1c and stx2b were more likely to be harboured by non-“Top-7” STEC than the “Top-7” serotypes (Table 2 and Figure 3B).
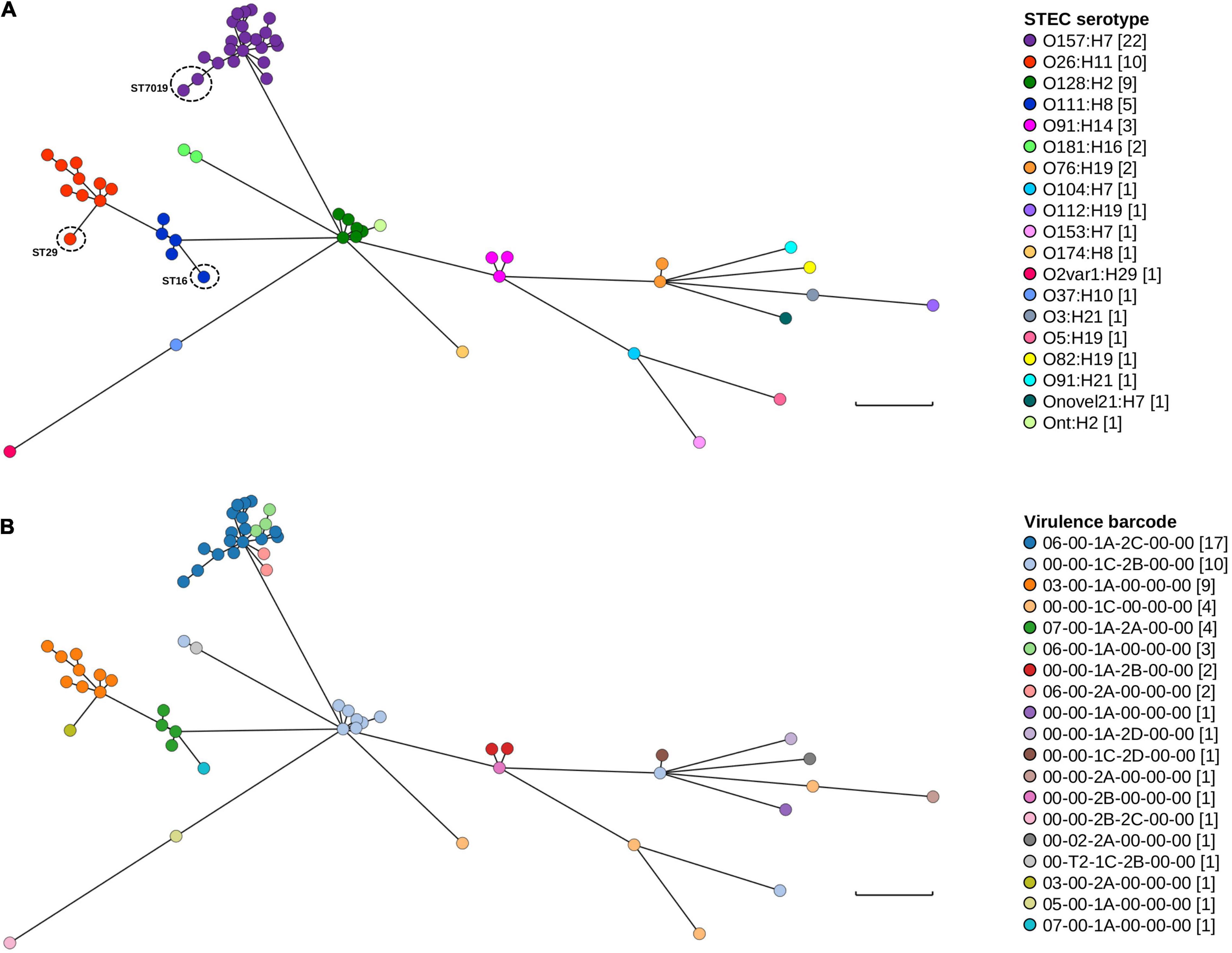
Figure 3. Genomic and STEC virulence diversity of STEC isolates in this study. (A) Minimum spanning tree of the 62 STEC genomes generated from wgMLST. (B) Minimum spanning tree (wgMLST) with STEC virulence barcode overlaid. Each isolate is represented on the minimal spanning tree as nodes, colour coded according either to serotype or STEC virulence barcode as per the figure key. Branch length between nodes denote number of allelic differences and scale bar represents 900 differences. Tree was generated and visualised on GrapeTree (Zhou et al., 2018).
Clustering according to serotype was expected as wzx and wzy genes were loci for both wgMLST and cgMLST. One STEC genome could not be typed in silico (Table 2) but wg/cgMLST grouped it amongst O128:H2, a serogroup that possessed the same MLST sequence type as this particular untypeable STEC. Pairwise comparison (BLASTN) between the draft assembly of this isolate and both wzxO128 and wzyO128 revealed that both genes were not assembled. Furthermore, read mapping revealed a mean read depth of 5.61× and 7.56× over wzxO128 (94.4% coverage) and wzyO128 (97.8% coverage), respectively. For continuity, this particular isolate was still referred to as Ont:H2. For serotypes that contained multiple MLST sequence types, these isolates could also be easily observed from both wgMLST and cgMLST MST, evident from their longer branch length within their respective serotype cluster (Figure 3 and Supplementary Figure 1). The number of allelic differences within serotypes ranged from 10 to 429 and 1 to 364 while allelic difference between serotypes ranged from 854 to 2,341 and 743 to 2,129 for wgMLST and cgMLST, respectively. The shortest branch length for wgMLST and cgMLST was between two O157:H7 isolates and two O128:H2 isolates, respectively. When typed by wgMLST, the two O128:H2 isolates possessed 12 allelic differences between them while the two O157:H7 isolates possessed six allelic differences when typed by cgMLST. Further interrogation of metadata revealed that both O128:H2 isolates were cultured from patients in the same household in close succession (<1 month) while the two O157:H7 isolates were recovered from unrelated cases 13 months apart in different geographical locations (>200 km).
Core SNP analysis was also performed on isolates belonging to both the “Top-7” serotypes and STEC O128:H2 to supplement wgMLST and cgMLST data. The wide disparity of allelic differences between different serotypes suggested that the utilisation of a single reference genome for all STEC, at least in our dataset, could be problematic and a different, appropriate reference genome was used for each serotype. SNP distance matrices (Supplementary Tables 5–8) showed that the aforementioned two STEC O128:H2 genomes were over 10 SNPs apart while the two STEC O157:H7 isolates were 17 SNPs apart. Taken together, our results suggested that closely related strains could either be inferred from wgMLST followed by core SNP analysis confirmation or inferred directly from cgMLST at the cost of shrinking the number of loci for future analysis.
As eae was typically used as a proxy marker for the LEE (Paton and Paton, 1998), presence of the entire core LEE sequence was subsequently confirmed by mapping. Trimmed reads from 38 eae-positive STEC were mapped onto the genome of STEC O157 strain Sakai (BA000007.3; positions: 4,589,382–4,623,687). All eae-positive genomes had reads covering majority of the LEE albeit at variable read depths (Figure 4A). Coverage over the entire core LEE of Sakai ranged from 99.8 to 100%, 86.41 to 87.11%, 84.26 to 85.19%, and 81.60% for serotypes O157:H7, O26:H11, O111:H8, and O37:H10, respectively. Genes that were required to build the T3SS apparatus all had read coverage confirming that the T3SS apparatus of these 38 isolates were intact. The main regions of non-coverage were over the 3′ end of eae, which was discriminatory for subtypes (Blanco et al., 2005) and over genes encoding effector proteins (Figure 4A).
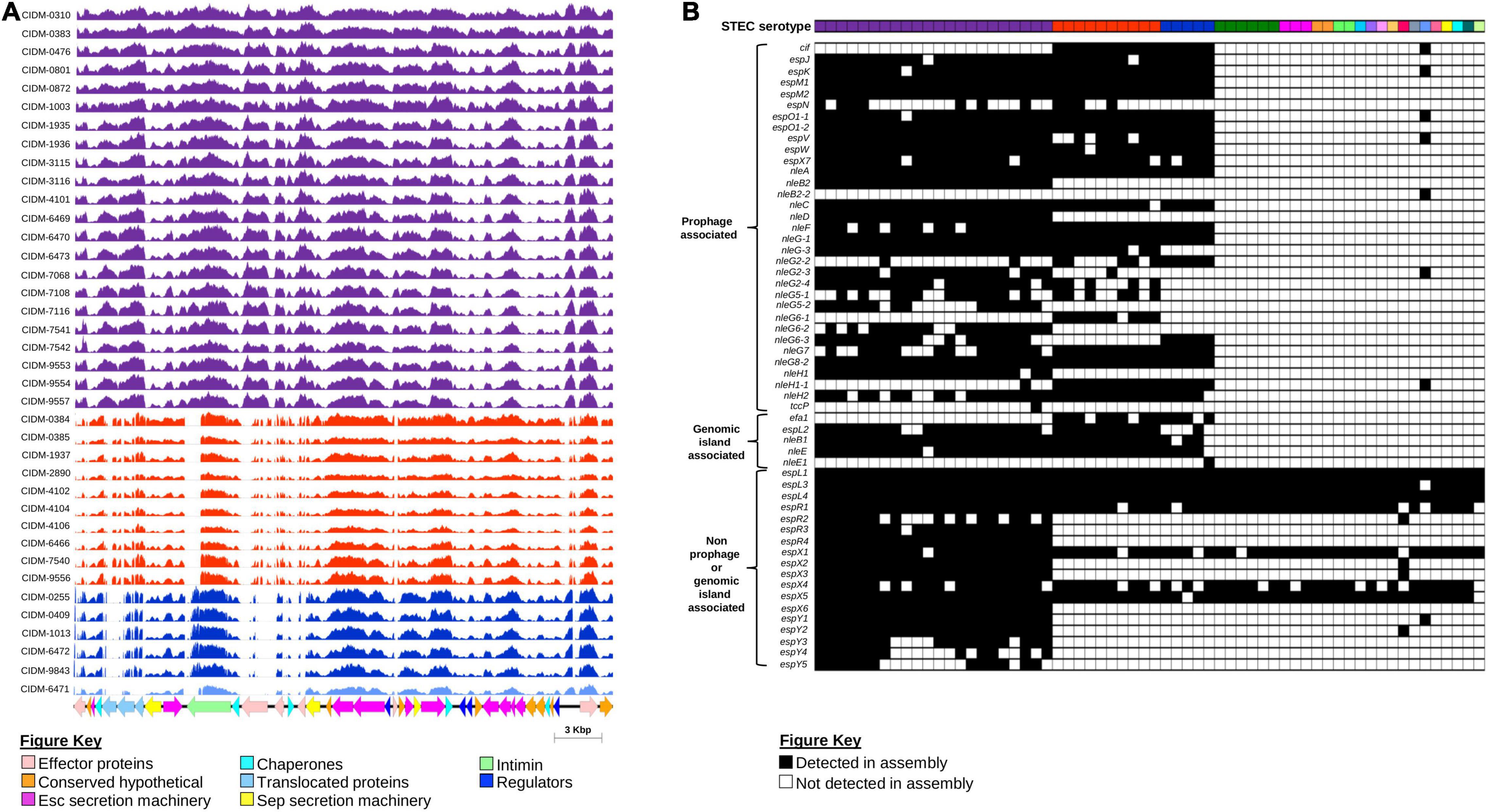
Figure 4. Presence of the LEE Pathogenicity Island and associated effectors amongst the 62 STEC isolates. (A) Coverage histogram of LEE-positive isolates across the core LEE sequence of STEC O157 strain Sakai (BA0000007.3) with nucleotide positions on the x-axis and coverage per base on the y-axis. Histogram bars are colour coded based on serotype listed as per Figures 1, 3. Corresponding coding sequences of the core LEE are colour coded according to function as listed on the Figure key. Figure was generated using Easyfig (Sullivan et al., 2011). (B) Presence and absence of non-LEE encoded effectors. All 62 isolates are colour coded according to serotype as per Figures 1, 3. Presence or absence of genes that encode non-LEE effectors are colour coded black and white, respectively.
While the LEE pathogenicity island harboured genes that encoded effector proteins, there were other effector proteins not encoded within the LEE but were secreted by this T3SS (Tobe et al., 2006). All LEE-positive “Top-7” STECs harboured significantly more non-LEE effector genes than both the sole LEE-positive non-“Top-7” STEC and LEE-negative isolates (Figure 4B and Supplementary Table 9). Some non-LEE effector genes were also detected in LEE-negative STEC, but these were effector genes not previously associated with either prophages or known genomic islands (Figure 4B). On the other hand, only LEE-positive STEC possessed a suite of non-LEE effector genes that were prophage associated (Figure 4B).
Presence of additional virulence factors was inferred from the assemblies of the 62 STEC against the Ecoli_VF Database9 (Figure 5 and Supplementary Table 9). Apart from genes encoded by the LEE pathogenicity island harboured by EPEC, none of our STEC harboured genes were associated with signature virulence factors of other E. coli pathotypes. Interestingly, the additional virulence genes detected were stratified according to serotype (Figure 5). Similar to eae, ehxA was used as a proxy marker for the presence of a pO157-like virulence plasmid in STEC (Paton and Paton, 1998). Amongst the 62 STEC, ehxABCD was detected in the genomes of 52 isolates, indicative that majority of our STEC likely harboured a STEC virulence plasmid (Figure 5). Virulence genes associated with the STEC virulence plasmid were also detected, but majority of these virulence genes were present in isolates belonging to serotype O157:H7.

Figure 5. Presence/absence of other virulence factors amongst the 62 STEC isolates. All 62 isolates, represented on the first row, are colour coded according to serotype as per Figures 1, 3. Presence or absence of virulence factor-associated genes are colour coded black and white, respectively. Virulence factors that consisted of multiple genes were considered present when ≥80% of the associated genes listed in the ecoli_vf database are present. If only 50–79% of the associated genes were present, the virulence factor was colour coded with black dots. If less than 50% of associated genes were present, the virulence factor was inferred as absent and colour coded in white. Associated clinical manifestations of abdominal pain and/or diarrhoea (L), bloody diarrhoea (M), and haemolytic uraemic syndrome (H) for each isolate are listed above serotype. Isolates with no appended data of clinical manifestation data are listed as (U).
Apart from the production of Stx, other toxin genes (hlyE, astA, cnf3, and senB) that could either contribute to the manifestation of diarrhoea (Yatsuyanagi et al., 2003) or invasion (Knust and Schmidt, 2010) were also detected in the genomes of the STEC in our dataset (Figure 5). Majority of the non-“Top-7” serotypes (n = 17) also contained the genes (subAB) that encode the subtilase cytotoxin (Paton et al., 2004). This holotoxin is typically associated with STEC that do not harbour the LEE pathogenicity island and binds to a sialic acid that is present in most mammals but not synthesised by humans (Paton et al., 2004; Byres et al., 2008). However, this sialic acid could be incorporated by humans via diet, and it has been suggested that dietary choices could contribute to disease in STEC producing this toxin (Byres et al., 2008).
A suite of virulence genes that contributed to colonisation were also detected in the genomes in the 62 STEC. These included genes associated with the secretome, as well as outer membrane proteins that mediated adhesion to host cells, invasion, and binding to host produced extracellular matrix (Figure 5 and Supplementary Table 8). While the genes associated with the Type I fimbriae was detected in all STEC O157:H7 isolates, interrogation of the switch upstream of fimA revealed that all STEC O157:H7 isolates possessed the 16-bp deletion consistent with published observations that the Type I fimbriae was not expressed in this serotype (Shaikh et al., 2007). While genes associated with the ETT2 T3SS was detected in majority of our STEC, this secretion system has been reported to be defective due to mutational attrition (Ren et al., 2004), and it was likely to be defective in our STECs as well. Apart from colonisation, a pathogen would also need to be able to scavenge for iron as restriction of iron availability is a mechanism of the innate immunity. In addition, iron is also a negative regulator for Stx1a production (Calderwood and Mekalanos, 1987) and the ability to effectively scavenge iron could have implications on toxin production amongst Stx1a-positive STEC. All STEC isolates possessed the genes involved in the biosynthesis of the Enterobactin siderophore (Figure 5 and Supplementary Table 8) while some also harboured the genes for stealth siderophore synthesis (Fischbach et al., 2006), namely, aerobactin (n = 13, 20.97%), salmochelin (n = 1, 1.61%), and yersiniabactin (n = 18, 29.03%). Apart from siderophores, some of our STECs also harboured genes for other iron uptake mechanisms ranging from a receptor for haem acquisition to systems for direct haem uptake (Figure 5).
This study examined the added value of WGS in improving the resolution of public health laboratory surveillance of STEC in a low-incidence country. It has been previously shown that WGS can be used to great effect in tracking outbreaks of STEC O157 (Dallman et al., 2015; Jenkins et al., 2019). As there were no large outbreaks of STEC during our study period, we were unable to provide additional commentary on the usefulness of WGS in outbreak tracking. Routine public health surveillance has been focussed on utilising SNP-based analysis for the transmission tracking of pathogens of interest. As STEC are a group of pathogens spanning multiple serotypes, MLST types, and possibly being geographically and genetically distinct, the utility of a potentially distant reference genome via SNP analysis could perturb the resolution afforded by SNP analysis. To address this, we employed and adapted a publicly available wgMLST/cgMLST scheme to infer the genomic diversity of STEC in our dataset. The utility of cgMLST has been previously shown to be a scalable and a viable alternative approach for high-resolution analysis (Pearce et al., 2018) and has been utilised to infer genetic relatedness of STEC of interest to public health (Ferdous et al., 2016; Lang et al., 2019; Smith et al., 2019). Despite our small dataset, this approach was able to corroborate with metadata on isolates that have an epidemiological link, albeit clearer at the cgMLST level rather than the wgMLST level as the latter could also capture isolates that were unlikely to be linked but still in circulation in the environment. Regardless, wgMLST/cgMLST may inform further downstream core SNP analysis, which is typically used in public health surveillance and outbreak tracking.
Shiga toxin-producing Escherichia coli O157:H7 dominated our dataset, a result concordant with previous epidemiological studies performed in Australia (Vally et al., 2012; Ingle et al., 2019). Our STEC O157:H7 predominantly carried stx1a only, stx2c only, or both stx1a and stx2c in combination, similar to previous reports (Fegan and Desmarchelier, 2002; Mellor et al., 2012, 2013, 2015). Interestingly, majority of the STEC O157:H7 isolates showed an allelic pattern of 213111 instead of the 211111 typical of Australian O157:H7 when typed by LSPA-6 (Mellor et al., 2013), a difference attributed to a 2-bp deletion in yhcG. We inferred hypervirulence for STEC O157:H7 using the Manning SNP typing scheme whereby hypervirulent isolates belonged to clade 8 (Manning et al., 2008). Similar to previous reports (Mellor et al., 2012), majority of the STEC O157:H7 in our study were typed as clade 7 of the Manning scheme while a single isolate was typed as a hypervirulent clade 8 strain. Hypervirulent STEC O26:H11 strains belonging to ST29 have been shown to harbour the stx2a operon (Bielaszewska et al., 2013). The ST29 O26:H11 in our dataset also harboured the stx2a gene. While MLST is generally embedded in routine genomic analysis in prokaryotic pipelines, the Manning SNP clade is not, and valuable information on potential hypervirulent STEC O157:H7 could be lost.
Whole genome sequencing has improved the resolution of subtyping of both stx1 and stx2, which supported additional risk assessment as certain stx subtypes, particularly stx2, are more likely to lead to HUS (Friedrich et al., 2002; Orth et al., 2007). Indeed, our STEC isolated from patients that progressed to HUS predominantly harboured either stx2a or stx2d subtypes, which had been previously associated with a higher likelihood for progression to HUS (Friedrich et al., 2002; Orth et al., 2007). The most common stx subtype detected amongst our STEC dataset was stx1a, which was detected in both “Top-7” and non-“Top-7” serotypes. In addition, subtypes stx1c and stx2b were more associated with the non-“Top-7” STEC in our dataset. This led to the development and evaluation of a STEC virulence barcode, which, when used in tandem with wgMLST/cgMLST visualisation, provided a visual snapshot of pertinent virulence factors relevant for routine surveillance and tracking the stx genotypes. In addition, this barcode also helps to infer multiple isogenic stx in a genome from short read sequencing data. The encoding of multiple isogenic stx has been implicated with disease severity such as in the latter phase of the 1996 STEC O111:H- outbreak in South Australia, when the isolates appeared to be more virulent (Henning et al., 1998). This increase in virulence was subsequently attributed to the carriage of additional stx2a operons, which was confirmed via sequencing (McAllister et al., 2016; Forde et al., 2019). Similarly, one STEC isolated from a HUS patient in our dataset also plausibly harboured an additional stx2a operon as determined by short read sequencing. However, the limits of utilising short read sequencing meant that we were unable to definitively confirm the multiple carriage of stx2a prophages, with confirmation only possible with the utilisation of long read sequencing (Forde et al., 2019). In addition, we are also unable to definitively comment if this was the sole reason that led to HUS. Nonetheless, this aforementioned case, along with 1996 outbreak, provided the impetus that while tracking stx subtypes is important, tracking or inference of multiple copies of the same stx subtypes, especially of the more virulent subtype, could be equally valuable.
While the STEC virulence barcode could be useful for surveillance and risk assessment of virulence, it is unlikely to function as a molecular predictor of disease outcome. Additional factors that could also contribute to the overall pathogenicity for a STEC infection should be acknowledged. Host risk factors, like age and immune system status, have been previously associated with adverse outcomes (Tarr et al., 2005). In addition, an individual’s gut microbiota could also serve as a genetic pool for new STEC as it has been previously shown in germ-free mice models that co-colonisation with STEC and commensal E. coli led to a synergistic increase in disease state (Goswami et al., 2015), a testament to horizontal transfer of its namesake virulence factor.
With particular stx subtypes (stx2a and stx2d) more likely to progress to HUS present in the minority of our dataset, our study corroborated with previous reports on STEC, albeit with a STEC O157 bias, that reduced virulence could be due to both stx genotype and E. coli genetic background (Mellor et al., 2015). The predominant subtype detected in our dataset was Stx1a, which is known to be less cytotoxic than the subtypes Stx2a, Stx2d, and Stx2c (Friedrich et al., 2002; Orth et al., 2007). Previous reports, albeit with a STEC O157:H7 bias, have also shown that Stx1a was the predominant Stx subtype circulating in Australia (Mellor et al., 2012, 2015; Ingle et al., 2019; Pintara et al., 2020). While the lower potency of Stx1a could explain the lower virulence of STEC in Australia, we would like to posit a possible genomic contributing factor to reduced virulence, at least in Stx1a-producing STEC. Expression of Stx1a has been previously shown to be also under the influence of iron, where iron limitation leads to increased expression of the toxin (Calderwood and Mekalanos, 1987). Our STEC all possessed genes encoding the synthesis of the enterobactin siderophore, an iron scavenging molecule to obtain iron from its environment. As a countermeasure, lipocalin-2 is produced by the human host, which binds iron-bound enterobactin, preventing iron uptake by bacteria (Goetz et al., 2002). Majority of our STEC isolates, however, also encoded genes that could synthesise other stealth siderophores, which would facilitate the evasion of lipocalin-2-mediated iron starvation (Fischbach et al., 2006). However, an antimicrobial peptide produced by humans, LL-37, has been shown to be able target stealth siderophores (Zsila and Beke-Somfai, 2020). A counter to LL-37 activity lies in the production of the outer membrane protease OmpT, which can degrade LL-37 (Thomassin et al., 2012), of which its associated gene was detected in majority of our STEC. The detection of genes encoding toxins capable of releasing haem, along with systems for the direct uptake of haem in our STECs, also provided an additional layer for iron sequestering required for biological activity. Indeed, the interplay of various virulence factors indicated that iron starvation could be evaded, perturbing the production of Stx1a via the iron regulation pathway in the process. In a geographical location whereby the most clinically dominant serotype typically harbours the genes encoding Stx1a, this possible negation of toxin production, along with the lower potency of Stx1a, could explain why the incidence of STEC related illness in Australia is lower than our international counterparts.
To our knowledge, this is the first report of prophage-associated non-LEE effectors in STEC serotype O37:H10. Isolates belonging to serotype O37:H10 has been previously observed to harbour the LEE encoded eae gene but were not further subtyped (Brett et al., 2003). Our results showed that similar to the eae-positive “Top-7” serotypes, this particular isolate also harboured non-LEE effector genes that were associated with prophages. These prophage-associated effector genes are typically located in a conserved location, downstream of genes encoding the phage tail fibres (Tobe et al., 2006), and could be transferred to other susceptible E. coli (Creuzburg et al., 2005). The presence of these genes in this non-prolific STEC serotype furthered the support that the (pro)phage metagenome is indeed linked to the pathogenesis of bacteria harbouring an injectisome Type 3 secretion system (Tobe et al., 2006).
It is important to note that our dataset only represented 33.33% of STEC notifications across NSW. This limitation reflects challenges of the recovery of STEC isolates from stool samples further complicated by the horizontal acquisition and potential loss of its namesake virulence factor in an E. coli genomic background (Senthakumaran et al., 2018). In addition, our current laboratory standard operating procedure limits the number of colonies selected for further stx detection to three. This approach could also contribute to relatively low recovery of STEC and an increase in the number of isolates picked could have a positive effect on STEC recovery (van Duynhoven et al., 2008). Nonetheless, a systemic update focussing on improvements to STEC culturing could provide additional circumstantial evidence to strengthen our postulation of genomic factors working in concert, leading to reduced virulence.
The genomic investigation of STEC over a period of 30 months in New South Wales, Australia highlighted the added value of high-resolution laboratory surveillance in a low-incidence setting. It offered detailed characterisation of the predominant harbouring of stx genes and hypervirulent clades/sequence types, which formed the minority of STEC isolates. The genetic background of the iron uptake systems, along with interactions with other virulence factors, could be a contributing factor in limiting the virulence in Stx1a-producing STEC, which made up the majority of the STEC in our study. Our findings emphasise the importance of sustained culture-based genomic surveillance for this group of genomically malleable pathogens of public health interest.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
VS, AA, PH, RK, and ES: study concept and design. ES, RK, MG, and AA: data analysis. ES, MG, RK, AA, MV, BH, and VS: manuscript writing, editing, and/or reviewing. All authors read and approved the manuscript.
This study was supported by funding from NSW Health and distributed through the Centre for Infectious Diseases and Microbiology-Public Health. The funding body had no role in study design, analysis, and decision to publish.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to acknowledge the Microbiological Diagnostic Unit Public Health Laboratory for sequencing STEC genomes for this study. The authors would also like to acknowledge the staff of the Centre for Infectious Diseases and Microbiology Laboratory Services, NSW Health Pathology for sample preparation, STEC CIDT, culture, and isolation of STEC. In addition, the authors thank Nathan Bachmann, Carl Banting, Rajat Dhakal, Jenny Draper, Andrew Ginn, Aneliese Goodwin, Karen-Ann Gray, Connie Lam, Dean Jalocon, Elena Martinez, Marc Ramsperger, Rebecca Rockett, and Verlaine Timms at the Microbial Genomics Reference Laboratory, NSW Health Pathology for their assistance with genome sequencing and bioinformatics analysis. The authors also thank Rosemarie Sadsad and The Sydney Informatics Hub along with The University of Sydney High Performance Computing Cluster for bioinformatics assistance and computing capacity.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.713724/full#supplementary-material
Supplementary Figure 1 | Minimum spanning tree of the of the 62 STEC genomes generated from cgMLST. Each isolate is represented on the minimal spanning tree as nodes, colour coded according either to serotype or STEC virulence barcode as per the figure key. Branch length between nodes denote number of allelic differences with scale bar represent 1,000 differences. Tree was generated and visualised on GrapeTree (Zhou et al., 2018).
Ahmed, S. A., Awosika, J., Baldwin, C., Bishop-Lilly, K. A., Biswas, B., Broomall, S., et al. (2012). Genomic comparison of Escherichia coli O104:H4 isolates from 2009 and 2011 reveals plasmid, and prophage heterogeneity, including Shiga toxin encoding phage stx2. PLoS One 7:e48228. doi: 10.1371/journal.pone.0048228
Allison, H. E. (2007). Stx-phages: drivers and mediators of the evolution of STEC and STEC-like pathogens. Future Microbiol. 2, 165–174. doi: 10.2217/17460913.2.2.165
Allison, H. E., Sergeant, M. J., James, C. E., Saunders, J. R., Smith, D. L., Sharp, R. J., et al. (2003). Immunity profiles of wild-type and recombinant Shiga-like toxin-encoding bacteriophages and characterization of novel double lysogens. Infect. Immun. 71, 3409–3418. doi: 10.1128/iai.71.6.3409-3418.2003
Bielaszewska, M., Mellmann, A., Bletz, S., Zhang, W., Köck, R., Kossow, A., et al. (2013). Enterohemorrhagic Escherichia coli O26:H11/H-: a new virulent clone emerges in Europe. Clin. Infect. Dis. 56, 1373–1381. doi: 10.1093/cid/cit055
Blanco, M., Schumacher, S., Tasara, T., Zweifel, C., Blanco, J. E., Dahbi, G., et al. (2005). Serotypes, intimin variants and other virulence factors of eae positive Escherichia coli strains isolated from healthy cattle in Switzerland. Identification of a new intimin variant gene (eae-eta2). BMC Microbiol. 5:23. doi: 10.1186/1471-2180-5-23
Blattner, F. R., Plunkett, G. III, Bloch, C. A., Perna, N. T., Burland, V., Riley, M., et al. (1997). The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462. doi: 10.1126/science.277.5331.1453
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brett, K. N., Ramachandran, V., Hornitzky, M. A., Bettelheim, K. A., Walker, M. J., and Djordjevic, S. P. (2003). stx1c Is the most common Shiga toxin 1 subtype among Shiga toxin-producing Escherichia coli isolates from sheep but not among isolates from cattle. J. Clin. Microbiol. 41, 926–936. doi: 10.1128/jcm.41.3.926-936.2003
Byres, E., Paton, A. W., Paton, J. C., Löfling, J. C., Smith, D. F., Wilce, M. C., et al. (2008). Incorporation of a non-human glycan mediates human susceptibility to a bacterial toxin. Nature 456, 648–652. doi: 10.1038/nature07428
Calderwood, S. B., and Mekalanos, J. J. (1987). Iron regulation of Shiga-like toxin expression in Escherichia coli is mediated by the fur locus. J. Bacteriol. 169, 4759–4764. doi: 10.1128/jb.169.10.4759-4764.1987
Carver, T. J., Rutherford, K. M., Berriman, M., Rajandream, M. A., Barrell, B. G., and Parkhill, J. (2005). ACT: the Artemis Comparison Tool. Bioinformatics 21, 3422–3423. doi: 10.1093/bioinformatics/bti553
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and McQuillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
Creuzburg, K., Recktenwald, J., Kuhle, V., Herold, S., Hensel, M., and Schmidt, H. (2005). The Shiga toxin 1-converting bacteriophage BP-4795 encodes an NleA-like type III effector protein. J. Bacteriol. 187, 8494–8498. doi: 10.1128/jb.187.24.8494-8498.2005
Dallman, T. J., Byrne, L., Ashton, P. M., Cowley, L. A., Perry, N. T., Adak, G., et al. (2015). Whole-genome sequencing for national surveillance of Shiga toxin-producing Escherichia coli O157. Clin. Infect. Dis. 61, 305–312. doi: 10.1093/cid/civ318
Deng, W., Puente, J. L., Gruenheid, S., Li, Y., Vallance, B. A., Vázquez, A., et al. (2004). Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. U.S.A. 101, 3597–3602. doi: 10.1073/pnas.0400326101
Edgar, R., and Bibi, E. (1997). MdfA, an Escherichia coli multidrug resistance protein with an extraordinarily broad spectrum of drug recognition. J. Bacteriol. 179, 2274–2280. doi: 10.1128/jb.179.7.2274-2280.1997
Fegan, N., and Desmarchelier, P. (2002). Comparison between human and animal isolates of Shiga toxin-producing Escherichia coli O157 from Australia. Epidemiol. Infect. 128, 357–362. doi: 10.1017/s0950268801006689
Ferdous, M., Friedrich, A. W., Grundmann, H., de Boer, R. F., Croughs, P. D., Islam, M. A., et al. (2016). Molecular characterization and phylogeny of Shiga toxin-producing Escherichia coli isolates obtained from two Dutch regions using whole genome sequencing. Clin. Microbiol. Infect. 22, .e641–.e649. doi: 10.1016/j.cmi.2016.03.028
Fischbach, M. A., Lin, H., Liu, D. R., and Walsh, C. T. (2006). How pathogenic bacteria evade mammalian sabotage in the battle for iron. Nat. Chem. Biol. 2, 132–138. doi: 10.1038/nchembio771
Fogg, P. C. M., Gossage, S. M., Smith, D. L., Saunders, J. R., McCarthy, A. J., and Allison, H. E. (2007). Identification of multiple integration sites for Stx-phage Phi24B in the Escherichia coli genome, description of a novel integrase and evidence for a functional anti-repressor. Microbiology 153, 4098–4110. doi: 10.1099/mic.0.2007/011205-0
Forde, B. M., McAllister, L. J., Paton, J. C., Paton, A. W., and Beatson, S. A. (2019). SMRT sequencing reveals differential patterns of methylation in two O111:H- STEC isolates from a hemolytic uremic syndrome outbreak in Australia. Sci. Rep. 9:9436. doi: 10.1038/s41598-019-45760-5
Friedrich, A. W., Bielaszewska, M., Zhang, W. L., Pulz, M., Kuczius, T., Ammon, A., et al. (2002). Escherichia coli harboring Shiga toxin 2 gene variants: frequency and association with clinical symptoms. J. Infect. Dis. 185, 74–84. doi: 10.1086/338115
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Goetz, D. H., Holmes, M. A., Borregaard, N., Bluhm, M. E., Raymond, K. N., and Strong, R. K. (2002). The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 10, 1033–1043. doi: 10.1016/s1097-2765(02)00708-6
Goswami, K., Chen, C., Xiaoli, L., Eaton, K. A., and Dudley, E. G. (2015). Coculture of Escherichia coli O157:H7 with a nonpathogenic E. coli strain increases toxin production and virulence in a germfree mouse model. Infect. Immun. 83, 4185–4193. doi: 10.1128/iai.00663-15
Gould, L. H., Mody, R. K., Ong, K. L., Clogher, P., Cronquist, A. B., Garman, K. N., et al. (2013). Increased recognition of non-O157 Shiga toxin-producing Escherichia coli infections in the United States during 2000-2010: epidemiologic features and comparison with E. coli O157 infections. Foodborne Pathog. Dis. 10, 453–460. doi: 10.1089/fpd.2012.1401
Hayashi, T., Makino, K., Ohnishi, M., Kurokawa, K., Ishii, K., Yokoyama, K., et al. (2001). Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8, 11–22. doi: 10.1093/dnares/8.1.11
Henning, P. H., Tham, E. B., Martin, A. A., Beare, T. H., and Jureidini, K. F. (1998). Haemolytic-uraemic syndrome outbreak caused by Escherichia coli O111:H-: clinical outcomes. Med. J. Aust. 168, 552–555. doi: 10.5694/j.1326-5377.1998.tb139083.x
Iguchi, A., Thomson, N. R., Ogura, Y., Saunders, D., Ooka, T., Henderson, I. R., et al. (2009). Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127:H6 strain E2348/69. J. Bacteriol. 191, 347–354. doi: 10.1128/jb.01238-08
Ingle, D. J., Gonçalves, da Silva, A., Valcanis, M., Ballard, S. A., Seemann, T., et al. (2019). Emergence and divergence of major lineages of Shiga-toxin-producing Escherichia coli in Australia. Microb. Genom. 5:e000268. doi: 10.1099/mgen.0.000268
Ingle, D. J., Valcanis, M., Kuzevski, A., Tauschek, M., Inouye, M., Stinear, T., et al. (2016). In silico serotyping of E. coli from short read data identifies limited novel O-loci but extensive diversity of O:H serotype combinations within and between pathogenic lineages. Microb. Genom. 2:e000064. doi: 10.1099/mgen.0.000064
Jenkins, C., Dallman, T. J., and Grant, K. A. (2019). Impact of whole genome sequencing on the investigation of food-borne outbreaks of Shiga toxin-producing Escherichia coli serogroup O157:H7, England, 2013 to 2017. Euro Surveill. 24:1800346. doi: 10.2807/1560-7917.Es.2019.24.4.1800346
Joensen, K. G., Scheutz, F., Lund, O., Hasman, H., Kaas, R. S., Nielsen, E. M., et al. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. doi: 10.1128/jcm.03617-13
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Knust, Z., and Schmidt, G. (2010). Cytotoxic necrotizing factors (CNFs)-a growing toxin family. Toxins 2, 116–127. doi: 10.3390/toxins2010116
Kulasekara, B. R., Jacobs, M., Zhou, Y., Wu, Z., Sims, E., Saenphimmachak, C., et al. (2009). Analysis of the genome of the Escherichia coli O157:H7 2006 spinach-associated outbreak isolate indicates candidate genes that may enhance virulence. Infect. Immun. 77, 3713–3721. doi: 10.1128/iai.00198-09
Lang, C., Hiller, M., Konrad, R., Fruth, A., and Flieger, A. (2019). Whole-genome-based public health surveillance of less common Shiga toxin-producing Escherichia coli serovars and untypeable strains identifies four novel O genotypes. J. Clin. Microbiol. 57:e768-19. doi: 10.1128/jcm.00768-19
Leotta, G. A., Miliwebsky, E. S., Chinen, I., Espinosa, E. M., Azzopardi, K., Tennant, S. M., et al. (2008). Characterisation of Shiga toxin-producing Escherichia coli O157 strains isolated from humans in Argentina, Australia and New Zealand. BMC Microbiol. 8:46. doi: 10.1186/1471-2180-8-46
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [Preprint]. arXiv:1303.3997.
Li, J., Yao, Y., Xu, H. H., Hao, L., Deng, Z., Rajakumar, K., et al. (2015). SecReT6: a web-based resource for type VI secretion systems found in bacteria. Environ. Microbiol. 17, 2196–2202. doi: 10.1111/1462-2920.12794
Lienemann, T., Salo, E., Rimhanen-Finne, R., Rönnholm, K., Taimisto, M., Hirvonen, J. J., et al. (2012). Shiga toxin-producing Escherichia coli serotype O78:H(-) in family, Finland, 2009. Emerg. Infect. Dis. 18, 577–581. doi: 10.3201/eid1804.111310
Lim, J. Y., Yoon, J., and Hovde, C. J. (2010). A brief overview of Escherichia coli O157:H7 and its plasmid O157. J. Microbiol. Biotechnol. 20, 5–14.
Luna-Gierke, R. E., Griffin, P. M., Gould, L. H., Herman, K., Bopp, C. A., Strockbine, N., et al. (2014). Outbreaks of non-O157 Shiga toxin-producing Escherichia coli infection: USA. Epidemiol. Infect. 142, 2270–2280. doi: 10.1017/s0950268813003233
Manning, S. D., Motiwala, A. S., Springman, A. C., Qi, W., Lacher, D. W., Ouellette, L. M., et al. (2008). Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc. Natl. Acad. Sci. U.S.A. 105, 4868–4873. doi: 10.1073/pnas.0710834105
McAllister, L. J., Bent, S. J., Petty, N. K., Skippington, E., Beatson, S. A., Paton, J. C., et al. (2016). Genomic comparison of two O111:H- enterohemorrhagic Escherichia coli isolates from a historic hemolytic-uremic syndrome outbreak in Australia. Infect. Immun. 84, 775–781. doi: 10.1128/iai.01229-15
Mellor, G. E., Besser, T. E., Davis, M. A., Beavis, B., Jung, W., Smith, H. V., et al. (2013). Multilocus genotype analysis of Escherichia coli O157 isolates from Australia and the United States provides evidence of geographic divergence. Appl. Environ. Microbiol. 79, 5050–5058. doi: 10.1128/aem.01525-13
Mellor, G. E., Fegan, N., Gobius, K. S., Smith, H. V., Jennison, A. V., D’Astek, B. A., et al. (2015). Geographically distinct Escherichia coli O157 isolates differ by lineage, Shiga toxin genotype, and total Shiga toxin production. J. Clin. Microbiol. 53, 579–586. doi: 10.1128/jcm.01532-14
Mellor, G. E., Sim, E. M., Barlow, R. S., D’Astek, B. A., Galli, L., Chinen, I., et al. (2012). Phylogenetically related argentinean and Australian Escherichia coli O157 isolates are distinguished by virulence clades and alternative Shiga toxin 1 and 2 prophages. Appl. Environ. Microbiol. 78, 4724–4731. doi: 10.1128/aem.00365-12
Melton-Celsa, A. R. (2014). Shiga toxin (Stx) classification, structure, and function. Microbiol. Spectr. 2:EHEC-0024-2013. doi: 10.1128/microbiolspec.EHEC-0024-2013
Monteiro, R., Ageorges, V., Rojas-Lopez, M., Schmidt, H., Weiss, A., Bertin, Y., et al. (2016). A secretome view of colonisation factors in Shiga toxin-encoding Escherichia coli (STEC): from enterohaemorrhagic E. coli (EHEC) to related enteropathotypes. FEMS Microbiol. Lett. 363:fnw179. doi: 10.1093/femsle/fnw179
Neely, M. N., and Friedman, D. I. (1998). Functional and genetic analysis of regulatory regions of coliphage H-19B: location of Shiga-like toxin and lysis genes suggest a role for phage functions in toxin release. Mol. Microbiol. 28, 1255–1267. doi: 10.1046/j.1365-2958.1998.00890.x
Ogura, Y., Ooka, T., Iguchi, A., Toh, H., Asadulghani, M., Oshima, K., et al. (2009). Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 106, 17939–17944. doi: 10.1073/pnas.0903585106
Orth, D., Grif, K., Khan, A. B., Naim, A., Dierich, M. P., and Würzner, R. (2007). The Shiga toxin genotype rather than the amount of Shiga toxin or the cytotoxicity of Shiga toxin in vitro correlates with the appearance of the hemolytic uremic syndrome. Diagn. Microbiol. Infect. Dis. 59, 235–242. doi: 10.1016/j.diagmicrobio.2007.04.013
Oswald, E., Schmidt, H., Morabito, S., Karch, H., Marchès, O., and Caprioli, A. (2000). Typing of intimin genes in human and animal enterohemorrhagic and enteropathogenic Escherichia coli: characterization of a new intimin variant. Infect. Immun. 68, 64–71. doi: 10.1128/iai.68.1.64-71.2000
Paton, A. W., and Paton, J. C. (1998). Detection and characterization of Shiga toxigenic Escherichia coli by using multiplex PCR assays for stx1, stx2, eaeA, enterohemorrhagic E. coli hlyA, rfbO111, and rfbO157. J. Clin. Microbiol. 36, 598–602. doi: 10.1128/jcm.36.2.598-602.1998
Paton, A. W., Srimanote, P., Talbot, U. M., Wang, H., and Paton, J. C. (2004). A new family of potent AB5 cytotoxins produced by Shiga toxigenic Escherichia coli. J. Exp. Med. 200, 35–46. doi: 10.1084/jem.20040392
Pearce, M. E., Alikhan, N. F., Dallman, T. J., Zhou, Z., Grant, K., and Maiden, M. C. J. (2018). Comparative analysis of core genome MLST and SNP typing within a European Salmonella serovar Enteritidis outbreak. Int. J. Food Microbiol. 274, 1–11. doi: 10.1016/j.ijfoodmicro.2018.02.023
Perna, N. T., Plunkett, G. III, Burland, V., Mau, B., Glasner, J. D., Rose, D. J., et al. (2001). Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409, 529–533. doi: 10.1038/35054089
Pintara, A. P., Guglielmino, C. J. D., Rathnayake, I. U., Huygens, F., and Jennison, A. V. (2018). Molecular prediction of the O157:H-negative phenotype prevalent in Australian Shiga toxin-producing Escherichia coli cases improves concordance of in silico serotyping with phenotypic motility. J. Clin. Microbiol. 56:e1906-17. doi: 10.1128/jcm.01906-17
Pintara, A., Jennison, A., Rathnayake, I. U., Mellor, G., and Huygens, F. (2020). Core and accessory genome comparison of Australian and international strains of O157 Shiga toxin-producing Escherichia coli. Front. Microbiol. 11:566415. doi: 10.3389/fmicb.2020.566415
Rangel, J. M., Sparling, P. H., Crowe, C., Griffin, P. M., and Swerdlow, D. L. (2005). Epidemiology of Escherichia coli O157:H7 outbreaks, United States, 1982-2002. Emerg. Infect. Dis. 11, 603–609. doi: 10.3201/eid1104.040739
Ren, C. P., Chaudhuri, R. R., Fivian, A., Bailey, C. M., Antonio, M., Barnes, W. M., et al. (2004). The ETT2 gene cluster, encoding a second type III secretion system from Escherichia coli, is present in the majority of strains but has undergone widespread mutational attrition. J. Bacteriol. 186, 3547–3560. doi: 10.1128/jb.186.11.3547-3560.2004
Rohde, H., Qin, J., Cui, Y., Li, D., Loman, N. J., Hentschke, M., et al. (2011). Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. N. Engl. J. Med. 365, 718–724. doi: 10.1056/NEJMoa1107643
Scheutz, F., Teel, L. D., Beutin, L., Piérard, D., Buvens, G., Karch, H., et al. (2012). Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 50, 2951–2963. doi: 10.1128/jcm.00860-12
Schmidt, M. A. (2010). LEEways: tales of EPEC, ATEC and EHEC. Cell Microbiol. 12, 1544–1552. doi: 10.1111/j.1462-5822.2010.01518.x
Senthakumaran, T., Brandal, L. T., Lindstedt, B. A., Jørgensen, S. B., Charnock, C., and Tunsjø, H. S. (2018). Implications of stx loss for clinical diagnostics of Shiga toxin-producing Escherichia coli. Eur. J. Clin. Microbiol. Infect. Dis. 37, 2361–2370. doi: 10.1007/s10096-018-3384-6
Shaikh, N., Holt, N. J., Johnson, J. R., and Tarr, P. I. (2007). Fim operon variation in the emergence of Enterohemorrhagic Escherichia coli: an evolutionary and functional analysis. FEMS Microbiol. Lett. 273, 58–63. doi: 10.1111/j.1574-6968.2007.00781.x
Silva, M., Machado, M. P., Silva, D. N., Rossi, M., Moran-Gilad, J., Santos, S., et al. (2018). chewBBACA: a complete suite for gene-by-gene schema creation and strain identification. Microb. Genom. 4:e000166. doi: 10.1099/mgen.0.000166
Smith, A. M., Tau, N. P., Kalule, B. J., Nicol, M. P., McCulloch, M., Jacobs, C. A., et al. (2019). Shiga toxin-producing Escherichia coli O26:H11 associated with a cluster of haemolytic uraemic syndrome cases in South Africa, 2017. Access Microbiol. 1:e000061. doi: 10.1099/acmi.0.000061
Souvorov, A., Agarwala, R., and Lipman, D. J. (2018). SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 19:153. doi: 10.1186/s13059-018-1540-z
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Tarr, P. I., Gordon, C. A., and Chandler, W. L. (2005). Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365, 1073–1086. doi: 10.1016/s0140-6736(05)71144-2
Thomassin, J. L., Brannon, J. R., Gibbs, B. F., Gruenheid, S., and Le Moual, H. (2012). OmpT outer membrane proteases of enterohemorrhagic and enteropathogenic Escherichia coli contribute differently to the degradation of human LL-37. Infect. Immun. 80, 483–492. doi: 10.1128/iai.05674-11
Tobe, T., Beatson, S. A., Taniguchi, H., Abe, H., Bailey, C. M., Fivian, A., et al. (2006). An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl. Acad. Sci. U.S.A. 103, 14941–14946. doi: 10.1073/pnas.0604891103
USDA-FSIS (2011). 76 FR 72331- Shiga Toxin-Producing Escherichia coli in certain raw beef products. Federal Register Document number: 2011-30271. Maryland: Food Safety and Inspection Service.
Vally, H., Hall, G., Dyda, A., Raupach, J., Knope, K., Combs, B., et al. (2012). Epidemiology of Shiga toxin producing Escherichia coli in Australia, 2000-2010. BMC Public Health 12:63. doi: 10.1186/1471-2458-12-63
van Duynhoven, Y. T., Friesema, I. H., Schuurman, T., Roovers, A., van Zwet, A. A., Sabbe, L. J., et al. (2008). Prevalence, characterisation and clinical profiles of Shiga toxin-producing Escherichia coli in The Netherlands. Clin. Microbiol. Infect. 14, 437–445. doi: 10.1111/j.1469-0691.2008.01963.x
WHO and FAO (2018). Shiga Toxin-Producing Escherichia coli (STEC) and Food: Attribution, Characterization, and Monitoring: Report in Microbiological Risk Assessment Series. 31.. WHO: Rome.
Wirth, T., Falush, D., Lan, R., Colles, F., Mensa, P., Wieler, L. H., et al. (2006). Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 60, 1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x
Yang, Z., Kovar, J., Kim, J., Nietfeldt, J., Smith, D. R., Moxley, R. A., et al. (2004). Identification of common subpopulations of non-sorbitol-fermenting, beta-glucuronidase-negative Escherichia coli O157:H7 from bovine production environments and human clinical samples. Appl. Environ. Microbiol. 70, 6846–6854. doi: 10.1128/aem.70.11.6846-6854.2004
Yatsuyanagi, J., Saito, S., Miyajima, Y., Amano, K., and Enomoto, K. (2003). Characterization of atypical enteropathogenic Escherichia coli strains harboring the astA gene that were associated with a waterborne outbreak of diarrhea in Japan. J. Clin. Microbiol. 41, 2033–2039. doi: 10.1128/jcm.41.5.2033-2039.2003
Yokoyama, E., Hirai, S., Hashimoto, R., and Uchimura, M. (2012). Clade analysis of enterohemorrhagic Escherichia coli serotype O157:H7/H- strains and hierarchy of their phylogenetic relationships. Infect. Genet. Evol. 12, 1724–1728. doi: 10.1016/j.meegid.2012.07.003
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zhou, Z., Alikhan, N. F., Sergeant, M. J., Luhmann, N., Vaz, C., Francisco, A. P., et al. (2018). GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 28, 1395–1404. doi: 10.1101/gr.232397.117
Keywords: STEC, Top-7 serotypes, stx subtyping, virulence analysis, cgMLST
Citation: Sim EM, Kim R, Gall M, Arnott A, Howard P, Valcanis M, Howden BP and Sintchenko V (2021) Added Value of Genomic Surveillance of Virulence Factors in Shiga Toxin-Producing Escherichia coli in New South Wales, Australia. Front. Microbiol. 12:713724. doi: 10.3389/fmicb.2021.713724
Received: 24 May 2021; Accepted: 22 November 2021;
Published: 23 December 2021.
Edited by:
Thomas Dandekar, Julius Maximilian University of Würzburg, GermanyReviewed by:
Peter Feng, Retired, Rockville, MD, United StatesCopyright © 2021 Sim, Kim, Gall, Arnott, Howard, Valcanis, Howden and Sintchenko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eby M. Sim, ZWJ5LnNpbUBoZWFsdGgubnN3Lmdvdi5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.