 Anna Kopf
Anna Kopf Boyke Bunk
Boyke Bunk Sina M. Coldewey
Sina M. Coldewey Florian Gunzer5
Florian Gunzer5 Thomas Riedel
Thomas Riedel Percy Schröttner
Percy Schröttner- 1Institute of Medical Microbiology and Virology, University Hospital Carl Gustav Carus, Dresden, Germany
- 2Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany
- 3Clinic for Anaesthesiology and Intensive Care Medicine, Jena University Hospital, Jena, Germany
- 4Septomics Research Center, Jena University Hospital, Jena, Germany
- 5Department of Hospital Infection Control, University Hospital Carl Gustav Carus, Dresden, Germany
- 6German Center for Infection Research (DZIF), Partner Site Hannover-Braunschweig, Braunschweig, Germany
In the past 12 years, several case reports have clearly demonstrated that Wohlfahrtiimonas chitiniclastica is capable of causing sepsis and bacteremia in humans. However, since most clinicians are not familiar with this species, little is known about its pathogenicity and treatment options while it is as rare but underestimated human pathogen. Therefore, a larger strain collection is required so that methods can be identified that are most suitable to obtain rapid and reliable identification. Moreover, the antimicrobial resistance profile needs to be elucidated in order to explore possible treatment options. Over a period of 6 years, we therefore have collected a total of 14 W. chitiniclastica isolates in routine diagnostics, which now served as the basis for a comprehensive characterization with respect to identification and antibiotic profiling. We compared the accuracy and convenience of several identification techniques in which MALDI-TOF MS and sequencing of the 16S rRNA gene have proven to be suitable for identification of W. chitiniclastica. In addition, whole genome sequencing (WGS)-based digital DNA-DNA hybridization (dDDH) was used as a reference method for strain identification, and surprised with the detection of a novel W. chitiniclastica subspecies. A combination of in silico and in vitro analyses revealed a first insight into the antimicrobial resistance profile and the molecular basis of antimicrobial resistance. Based on our findings, trimethoprim/sulfamethoxazole, levofloxacin, and cephalosporins (e.g., ceftazidime) may be the best antibiotics to use in order to treat infections caused by W. chitiniclastica, while resistance to fosfomycin, amikacin and tobramycin is observed.
Introduction
The gammaproteobacterium Wohlfahrtiimonas chitiniclastica has first been isolated from larvae of Wohlfahrtia magnifica (Tóth et al., 2008), an obligatory parasitic fly that causes myiasis by depositing eggs and larvae in mammalian wounds both in animals and humans (Robbins and Khachemoune, 2010). Bacteria belonging to this species are described as Gram-negative, strictly aerobic, and non-motile rods. Furthermore, the organism is catalase and oxidase positive, while biochemical tests for urease, indole, and H2S are negative (Tóth et al., 2008; Schröttner et al., 2017). They are also noted to have strong chitinase activity, which may be an indicator for a symbiotic relationship with its host fly while playing an important role in metamorphosis (Schröttner et al., 2017; Snyder et al., 2020).
In April 2021, GenBank (Benson et al., 2013) lists 12 genomes of W. chitiniclastica strains while three draft genome reports were published (Cao et al., 2013; Zhou et al., 2016; Matos et al., 2019). The strains are described to be susceptible to the majority of known antibiotics with the exception of fosfomycin (Schröttner et al., 2017; Matos et al., 2019). First genome annotations revealed genes coding for macrolide-specific efflux pumps (macA and macB) (Matos et al., 2019) and a blaVEB–1 gene cassette which confers resistance to ceftazidime, ampicillin, and tetracycline (Zhou et al., 2016). However, in-depth analysis is still required to generate a comprehensive antimicrobial resistance profile as the available antimicrobial susceptibility data are mostly based on case reports and preliminary genome annotations (Zhou et al., 2016; Schröttner et al., 2017; Matos et al., 2019).
Originally isolated from a homogenate of fly larvae, there is increasing evidence that W. chitiniclastica may be the cause of several diseases in humans. Although the pathogenesis of W. chitiniclastica is not yet fully understood, the bacterium is expected to enter traumatic skin lesions through fly larvae, resulting in severe myiasis and/or wound contamination (Robbins and Khachemoune, 2010; Thaiwong et al., 2014; Schröttner et al., 2017). To date, 23 human case reports from 18 countries across the globe have been published (Table 1 and Figure 1) indicating W. chitiniclastica to be associated with humans sepsis and bacteremia. For example, Almuzara et al. (2011) reported the first case of fulminant sepsis with fatal outcome and Campisi et al. (2015) observed W. chitiniclastica bacteremia associated with myiasis, to name but a few. However, since most clinicians are not familiar with this species, it can be assumed that W. chitiniclastica has hardly been recognized as a possible cause while it has recently been described as a new underestimated human pathogen (Schröttner et al., 2017). Therefore, it is necessary to initiate systematic investigations to gain more knowledge about its virulence and treatment options. For this reason, first methods need to be identified that are most suitable to obtain a fast, reliable and robust species identification. Moreover, we need to shed light on the antimicrobial resistance profile to gain knowledge about primary resistances in order to treat infections successfully. In this study, we therefore compared the accuracy of several routine methods of bacterial identification and performed antimicrobial susceptibility testing of 14 isolates collected from clinical samples. Additionally, we conducted whole genome data to elucidate the molecular basis of antimicrobial resistance and to confirm correct species designation.
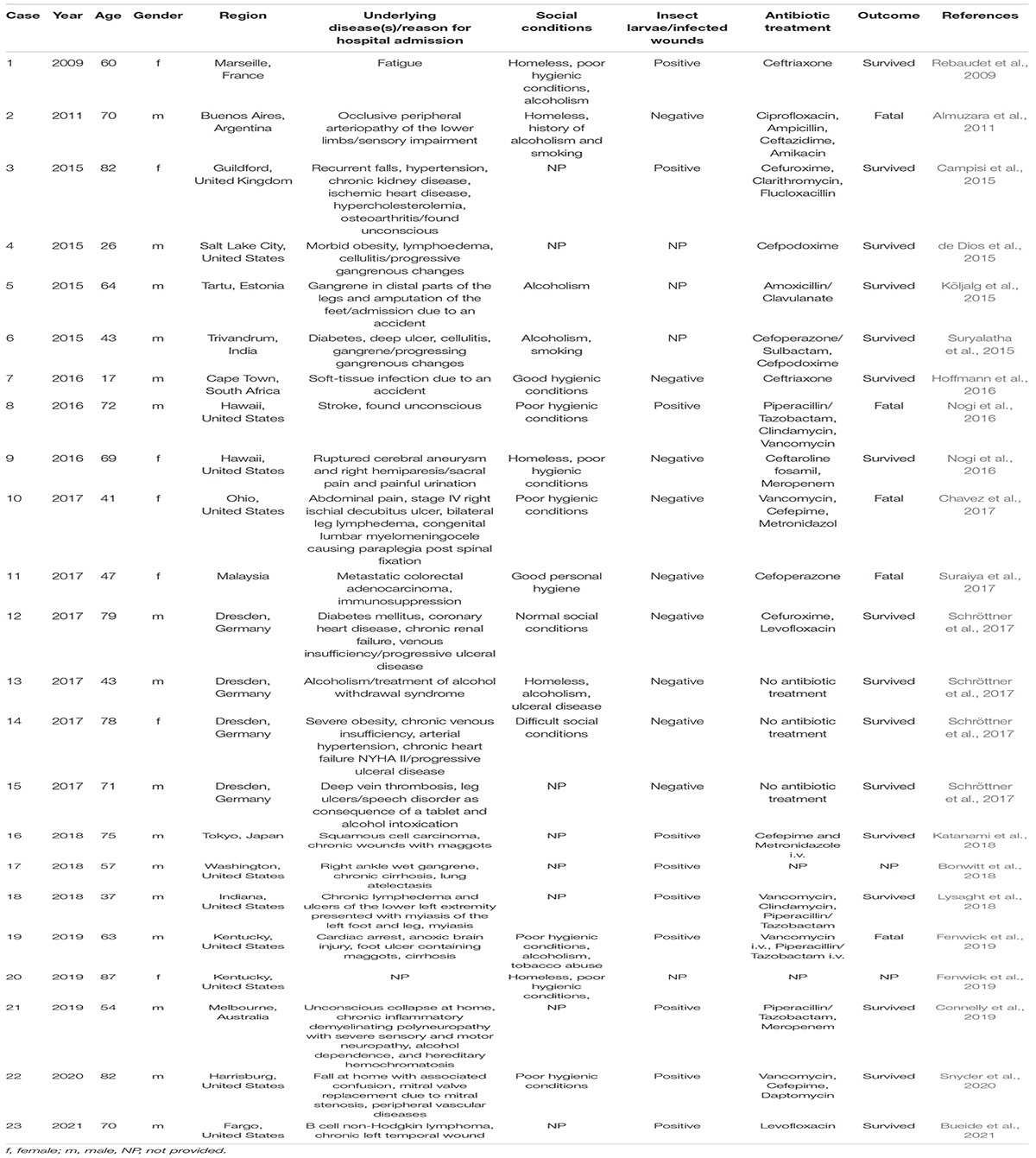
Table 1. Current overview of cases of human infection and colonization with W. chitiniclastica.
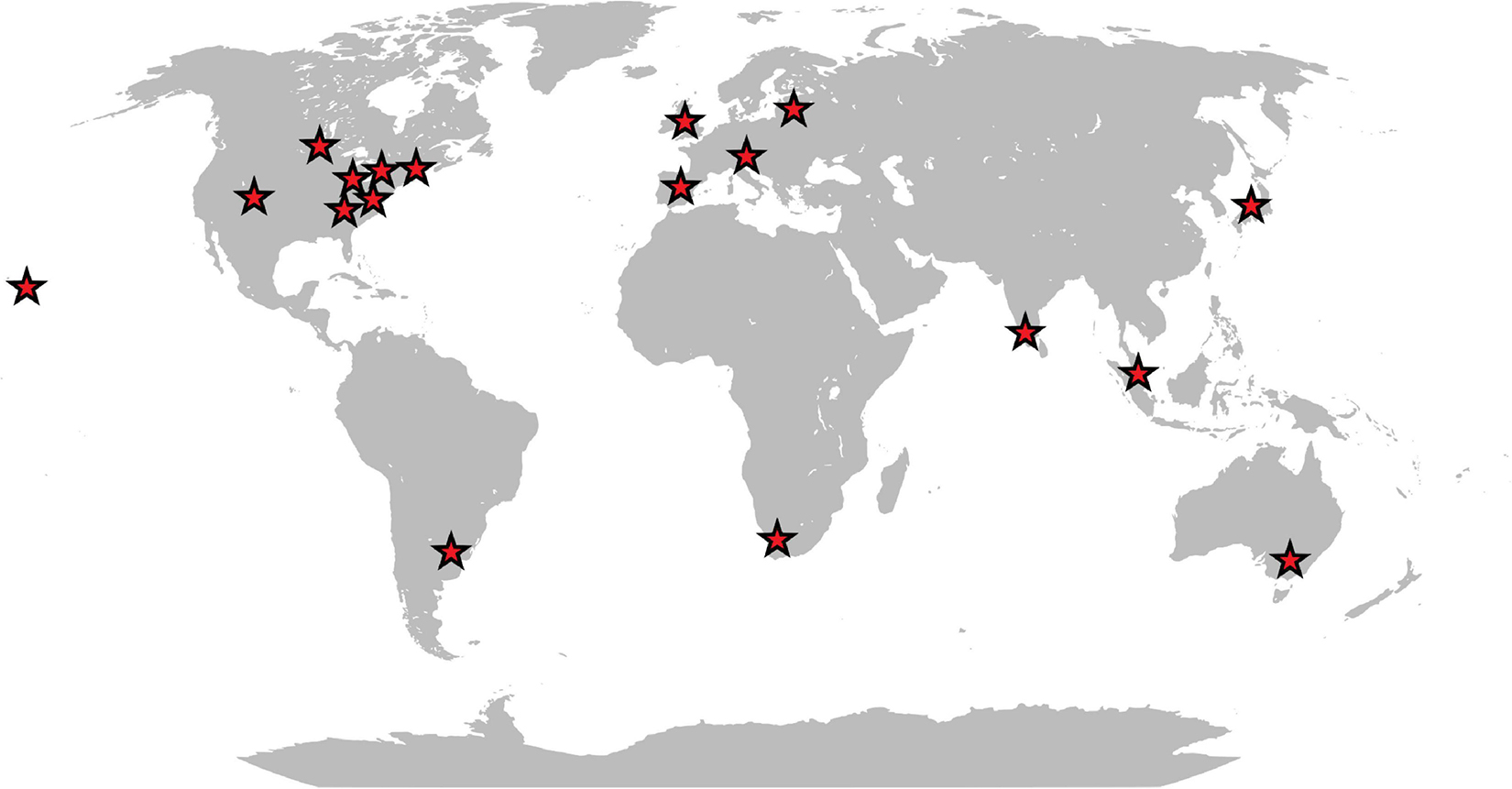
Figure 1. Schematic map showing the geographic distribution of reported human infections caused by W. chitiniclastica.
Materials and Methods
Collection of W. chitiniclastica Strains
Over a period of 6 years, a total of 14 W. chitiniclastica strains have been collected in routine diagnostics (see Table 2). All isolates were recovered exclusively from diagnostic cultures analyzed at the Institute for Medical Microbiology and Virology, University Hospital Carl Gustav Carus (Dresden, Germany). Prior to this publication the isolates DSM 100374, DSM 100374, DSM 100676, and DSM 100917 were briefly described as part of a review article (Schröttner et al., 2017); however a thorough analysis has not been performed. Subsequently, all strains were collected and stored in Pro-Lab DiagnosticsTM MicrobankTM (Fisher Scientific, Schwerte, Germany). The bacteria were additionally deposited at the “Open Collection” of the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). The type strain DSM 18708T was purchased from the DSMZ and included as reference strain in this study.
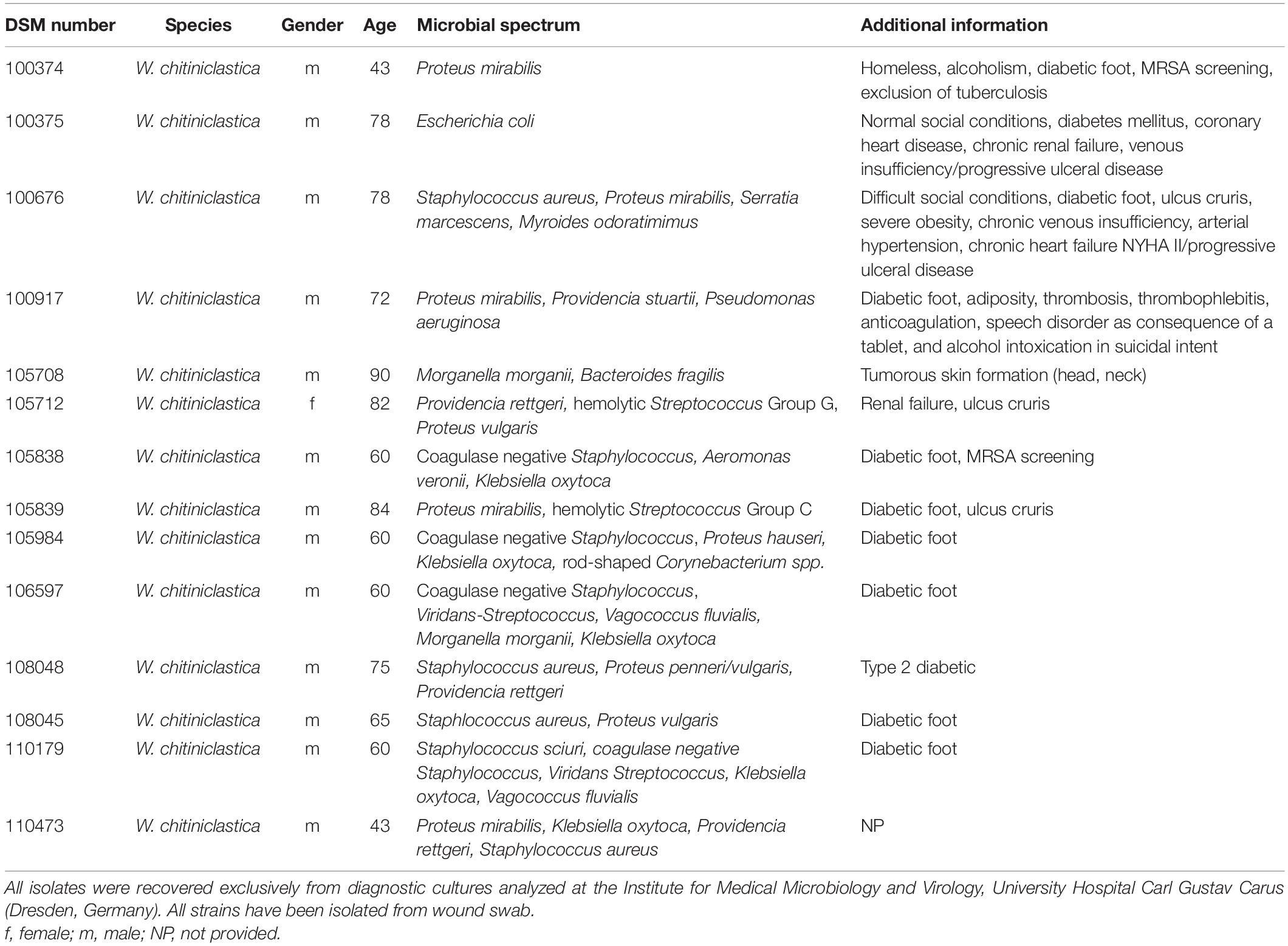
Table 2. Strain sources and patients’ clinical data.
Identification of W. chitiniclastica Using VITEK 2
Frozen colonies were grown on Colombia blood agar plates (bioMérieux, Nürtingen, Germany) for 24 h at 37°C. A single colony from each isolate was picked and transferred to a new Colombia blood agar plate. After another incubation period of 24 h at 37°C, the colonies were suspended in a solution of 3 ml of 0.45% saline. A turbidity of 0.5–0.63 McFarland standard using VITEK DensiCHEK Plus (bioMérieux, Nürtingen, Germany) was established. Bacteria were identified with a VITEK 2 system (bioMérieux, Nürtingen, Germany) using GN ID cards (for analysis of gram-negative bacteria) as described in a previous study (Schröttner et al., 2014). Results are displayed in Table 3 and Supplementary Table 5.
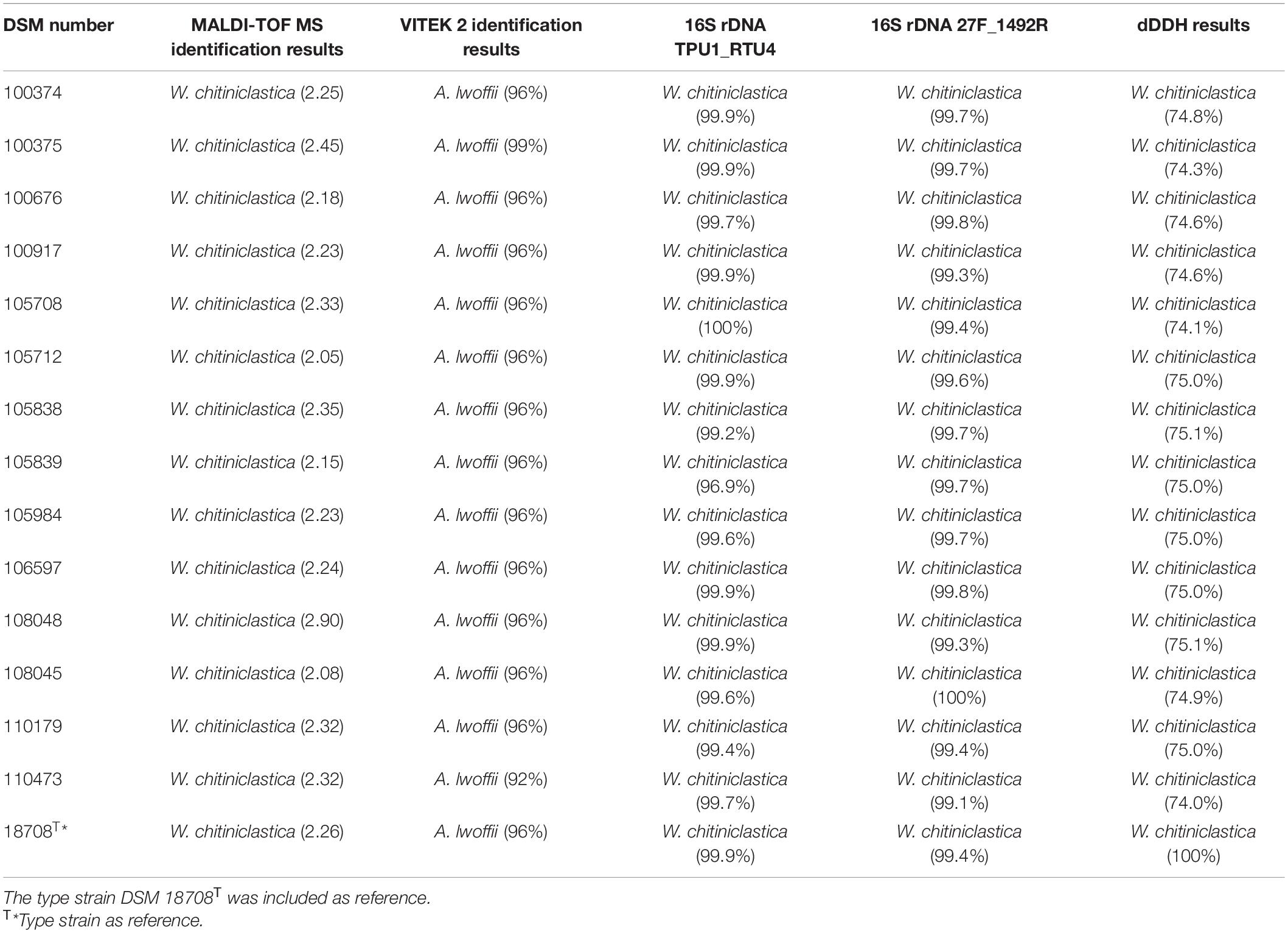
Table 3. Comparison of diagnostic methods applied for identification of W. chitiniclastica.
Identification of W. chitiniclastica Using MALDI-TOF MS
Identification of the strain collection of W. chitiniclastica using MALDI TOF MS was performed as previously described (Schröttner et al., 2014, 2016). In brief, strains were grown on Colombia blood agar plates for 24 h at 37°C. Single colonies were picked and plated on a 96-well steel target. Bacteria were dried on a laboratory workbench for 10 min and then overlaid with a 1 μl matrix (α-Cyano-4-hydroxycinnamic acid, Bruker Daltonik, Bremen, Germany) dissolved in an organic solvent. Subsequently, MALDI-TOF MS analyses were performed using flexControl software 3.1 (Bruker Daltonik, Bremen, Germany) following the manufacturer’s guidelines. Results are displayed in Table 3.
Identification of W. chitiniclastica Using 16S rRNA Gene Analysis
Prior to PCR amplification, the performance of different primer pairs was evaluated in silico using TestPrime (Klindworth et al., 2012)1 based on the SILVA Reference database (release SSURef 138 NR) (Quast et al., 2013). PCR was carried out using the following primer pair combinations: (i) TPU-1 (5′-AGA GTT TGA TCM TGG CTC AG-3′) and RTU-4 (5′-TAC CAG GGT ATC TAA TCC TGT T-3′) (Funke et al., 2004); and (ii) 27F (5′-AGA GTT TGA TCM TGG CTC AG-3′) (Hongoh et al., 2003) and 1492R (5′-TAC CAG GGT ATC TAA TCC TGT T-3′) (Weisburg et al., 1991). 16S rRNA gene amplification was performed as previously described (Schröttner et al., 2014, 2016). Oligonucleotides were purchased from Biomers.net (Ulm, Germany). PCR products were purified using exonuclease I and shrimp alkaline phosphatase (both enzymes were purchased from New England Biolabs, Frankfurt am Main, Germany). Sanger sequencing was performed by SEQLAB (Sequence Laboratories Göttingen, Göttingen, Germany). Taxonomic identification was done based on BLASTN (Altschul et al., 1990), using the standard database for Nucleotide collection (nt). Results are displayed in Table 3.
Whole Genome Analysis of W. chitiniclastica
Libraries for Whole Genome Sequencing (WGS) on the Illumina platform were prepared from extracted genomic DNA, applying the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, United States) with modifications (Baym et al., 2015). Samples were sequenced on the NextSeqTM 550 with a read length 2 × 150 bp targeting approx. 100× genome coverage followed by short read genome assembly using SpaD ES 3.14 (Bankevich et al., 2012). Whole genome sequences were submitted to NCBI GenBank under Acc. Nos JAGIBR000000000-JAGICE000000000, applying the NCBI Prokaryotic Annotation Pipeline PGAP (Tatusova et al., 2016). Contigs smaller than 300 bp were excluded from the submission. For phylogenomic identification and phylogenomic tree construction, genomic contigs were submitted to the Type Strain Genome Server (TYGS) at tygs.dsmz.de, and the type-based species clustering was done using a 70% dDDH threshold (Meier-Kolthoff and Göker, 2019). Subspecies clustering was based on a 79% dDDH threshold as previously introduced (Meier-Kolthoff et al., 2014). Results are displayed in Table 3 and Figure 2.
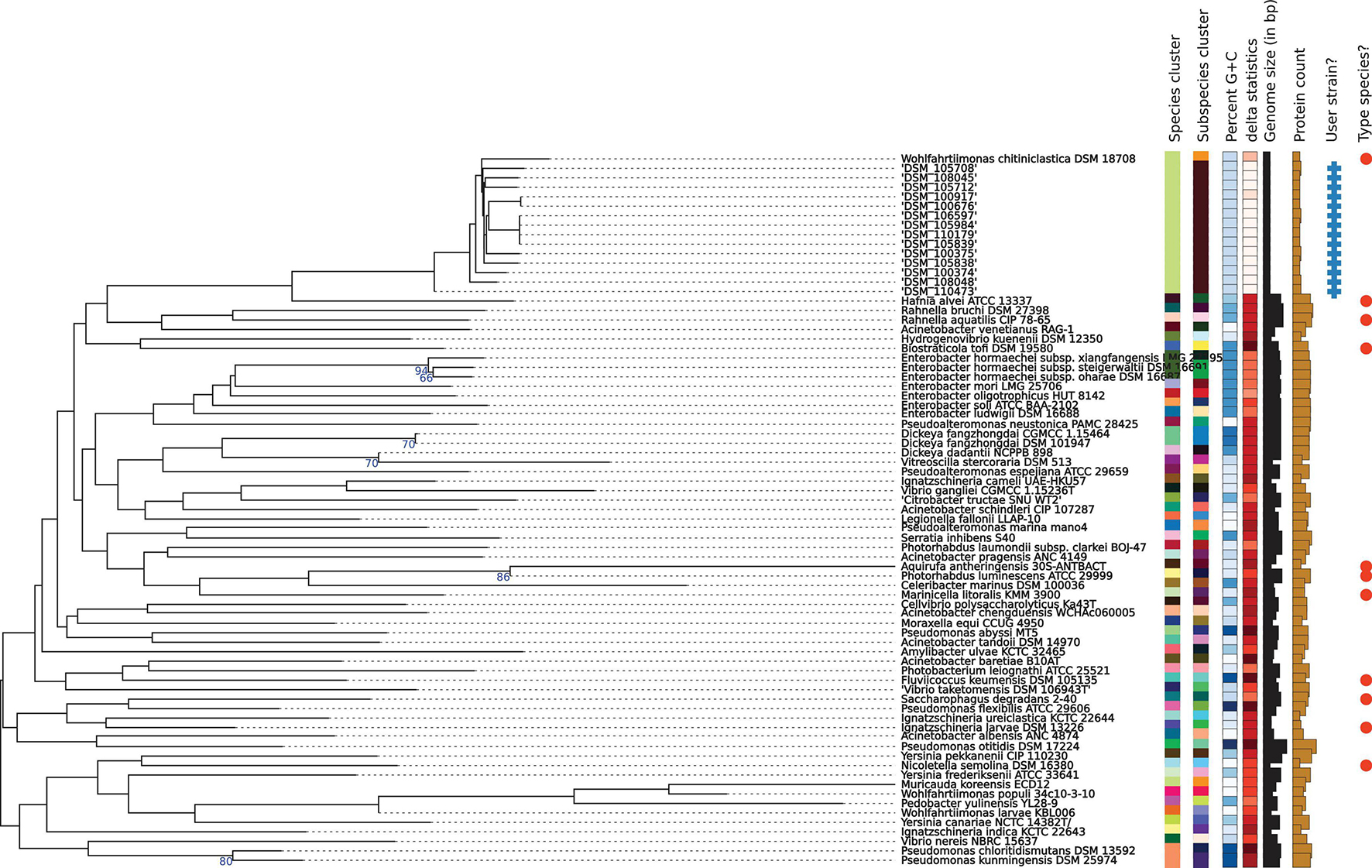
Figure 2. Phylogenomic tree of the W. chitiniclastica species and subspecies delineation based on the GBDP phylogenetic analyses retrieved from the TYGS website. The tree was inferred with FastME 2.1.6.1 (Lefort et al., 2015) from GBDP distances calculated from genome sequences and was subjected to a clustering using established thresholds for delineating species (DDH > 70%) (Meier-Kolthoff et al., 2013a) as well as subspecies (DDH > 79%) (Meier-Kolthoff et al., 2014). The branch lengths are scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values >60% from 100 replications, with an average branch support of 83.0%.
Antibiotic Profiling
The antibiotic susceptibility testing of all W. chitiniclastica isolates was performed using MIC Test Strips (bestbion, Cologne, Germany) according to the manufacturer’s instructions. In brief, a McFarland standard of 0.5 was created for each strain, using NaCl and a DensiCHEK densitometer (bioMérieux, Nürtingen, Germany). The suspended bacteria were plated with a cotton swab on Müller-Hinton Agar (Oxoid Deutschland, Wesel, Germany). Then, MIC Test Strips (bestbion, Cologne, Germany) for each antibiotic were placed on the agar plates. The plates were incubated for 18 ± 2 h at 37°C. The MIC results were evaluated by applying the guidelines for PK/PD (non-species-related) breakpoints according to the criteria published by EUCAST (European Committee on Antimicrobial Susceptibility Testing), using Version 11.0, 01. January 2021.2 Results are displayed in Table 4. For the type strain DSM 18708T, results are represented in Supplementary Table 6. All MIC Test Strips used in this study and their MIC ranges are listed in Supplementary Table 2. The comprehensive antibiotic resistance database CARD3 was used for in silico prediction of antibiotic resistance genes (Alcock et al., 2020). Results are listed in Supplementary Tables 3, 4.
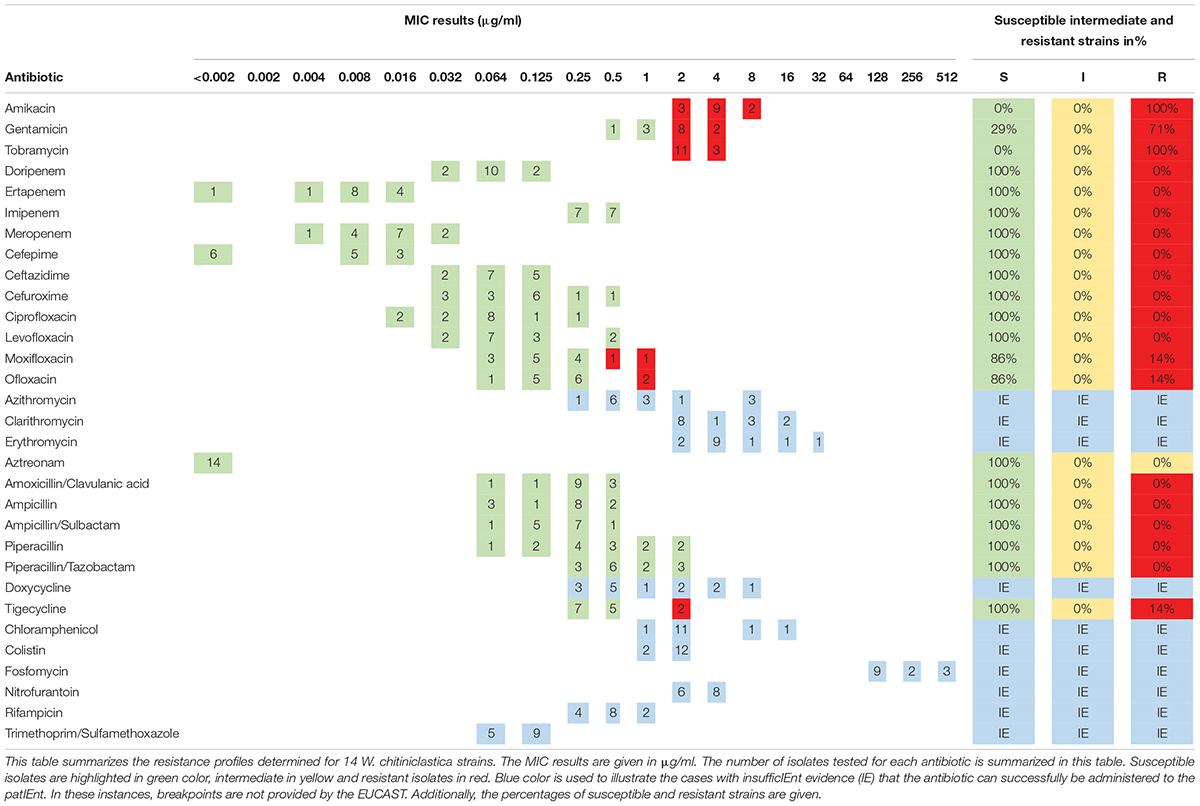
Table 4. Minimum inhibitory concentration (MIC) distribution of 14 W. chitiniclastica strains.
Results
Strain Collection and Its Characteristics
An overview of the W. chitiniclastica strain collection analyzed in this study is given in Table 2. All isolates were collected from wound swabs and patients’ medium age was 67.86 years ranging from 43 to 90 years. The majority were male (n = 13) and suffered from diabetes. There was only one female patient. This patient suffered from renal failure. Information regarding the social situation and living conditions were provided only in three cases (Schröttner et al., 2017). One patient (case 2) was homeless while one (case 3) lived under poor hygienic conditions. In contrast to these two, one patient (case 1) lived under normal conditions, which were not further specified. The associated microbial spectrum consists of Staphylococcus spp., Pseudomonas spp., Proteus spp., and Streptococcus spp. among others. Therefore, it remains unclear if W. chitiniclastica was the causative agent of the diseases or part of the microbiome.
Identification of W. chitiniclastica Based on 16S rRNA Gene Analysis
16S rRNA amplification from pure culture of each isolate was successful and both primer pairs have proven of value (Table 2). In all cases sequence identity for W. chitiniclastica was above ≥98.7% and therefore fulfilling the criteria for bacterial species identification (Meier-Kolthoff et al., 2013b).
Identification of W. chitiniclastica Based on MALDI-TOF MS
Wohlfahrtiimonas chitiniclastica was successfully identified by MALDI-TOF MS (Schröttner et al., 2016). Based on the manufacturer’s guidelines, score values above 2.0 are interpreted as secure identification at both the genus and species levels; scores between 2.0 and 1.7 as reliable identification on the genus, but not at the species level; and scores below 1.7 were regarded as an unreliable identification (Schröttner et al., 2016). In this study, all scores were above 2 (Table 2), allowing us to identify all isolates as W. chitiniclastica with high confidence.
Results Obtained for Identification of W. chitiniclastica From VITEK 2
VITEK 2 results of W. chitiniclastica lead to misidentification as Acinetobacter lwoffii of all strains included in this study (Table 3), which has also been reported in previous case reports (de Dios et al., 2015; Chavez et al., 2017; Snyder et al., 2020). Identification of DSM 110473 resulted in 92% probability for species identification. Based on the manufacturer’s instructions 89–92% probability correlates with a good confidence level. Notably, for the remaining isolates, the results were above 96%, representing an excellent species identification; however, in our study a misidentification as A. lwoffii was evident for all strains. Biochemical characteristics based on the VITEK 2 system showed positive results for tyrosine arylamidase, Ellman’s reagent, L-lactate alkalization, and oxidase (Supplementary Table 5).
Identification of W. chitiniclastica Based on Whole Genome Sequencing
Phylogenomic analysis of all 14 strains revealed correct taxonomic assignment to W. chitiniclastica. Hereby, dDDH values of 74.0–75.1% (Table 3) were computed against the type strain of the species DSM 18708T and therefore fulfilling the criteria for bacterial species identification (Meier-Kolthoff and Göker, 2019). A phylogenomic tree based on whole-genome sequences was constructed using the TYGS web server4 (Figure 2) and all 14 isolates cluster in one subclade with the type strain DSM 18708T. Notably, the isolates from Dresden form a new subspecies using a 79% dDDH threshold (Meier-Kolthoff et al., 2014).
Antibiotic Profiling
The susceptibility profile and the MIC distribution of all strains tested using MIC Test Strips are summarized in Table 4. Additionally, the MIC results of each isolate are provided in Supplementary Table 1. All 14 strains were susceptible to all penicillins, carbapenems, cephalosporines, and monobactams tested in this study. This is in line with the susceptibility profile of the type strain DSM 18708T (Supplementary Table 5).
In addition, all 14 strains were susceptible to the fluoroquinolones tested in this study, apart from two exceptions. The isolates DSM 105984 and DSM 106597 were resistant to ofloxacin and moxifloxacin (Supplementary Table 1). Interestingly, these two strains showed higher MIC values for all tested fluoroquinolones in comparison to the other strains (Supplementary Table 1). A similar picture was obtained for the tetracycline tigecycline. The majority were susceptible while DSM 105984 and DSM 106597 appear to be resistant. In contrast, all strains appear to be resistant to the aminoglycosides amikacin and tobramycin. In addition, 10 strains showed resistance to gentamicin. The type strain DSM 18708T appears to be susceptible to all three aminoglycosides tested in this study (Supplementary Table 5).
No breakpoints were available for trimethoprim/sulfamethoxazole, fosfomycin, doxycycline, colistin, chloramphenicol, nitrofurantoin, rifampicin, and macrolides. However, the MIC results determined for fosfomycin are all at a high range. Therefore, antimicrobial resistance to this antibiotic may be assumed. On the contrary, low MIC results were obtained for trimethoprim/sulfamethoxazole making susceptibility feasible.
The Comprehensive Antibiotic Resistance Database (CARD) (Alcock et al., 2020) was used for in silico identification of potential genes encoding for antibiotic resistance. Using the parameters “Perfect hit and Strict hit only” and “High-quality/coverage,” the strains DSM 100676, DSM 100917, and DSM 105708 each revealed one strict hit for tet(D) with an identity of 52.28% for the matching region (Supplementary Table 3). The “Strict” algorithm of the CARD system represents a flexible sequence variation but lies within the curated BLAST bit score cut-offs (Alcock et al., 2020) and by that making a correct identification still highly feasible. For the remaining isolates, no perfect or strict hits were obtained. However, when the parameter was changed to “Perfect hit, Strict hit, and Loose hit,” about 150 hits per strain were revealed with an identity for the matching region from 21 to 81%. Detailed results are displayed in Supplementary Table 3. However, keeping in mind that the “Loose” algorithm of the CARD system works outside of the detection model cut-offs to provide detection of new and more distant homologs of antimicrobial resistance (AMR) genes (Alcock et al., 2020), in silico results based on loose hits should always be taken with caution and require further research. Nevertheless, it could help us to identify potential resistance genes and/or shed light on new unknown modifications.
With respect to the resistance mechanism, the majority of hits belonged to antibiotic target alteration and efflux systems (Supplementary Table 4). Attention should be paid to genes mentioned as follows: Up to six genes encoding for fosfomycin efflux proteins were identified, and each strain showed a hit for MurA transferase with mutation conferring resistance to fosfomycin, which is involved in antibiotic target alteration of fosfomycin (Fu et al., 2015). In addition, several genes involved in broad range efflux systems of diverse antibiotics such as aminoglycosides, makrolides, fluoroquinolones, and tetracycline were detected including genes coding for macrolide-specific efflux pumps (macA and macB) (Kobayashi et al., 2000; Yum et al., 2009). Each strain also contained genes encoding for aminoglycoside resistant mechanisms such as apmA, baeS, and cpxA. Interestingly, all isolates revealed hits for the trimethoprim resistance gene dfrI (56.63% identity) and the sulfonamide resistance gene sul3 (39.85% identity), as well as several hits for potential genes coding for β-lactamases, such as PNGM-1, NmcR, and GOB-16, although identity of the matching region was less than 36% in all cases.
Discussion
The majority of the W. chitiniclastica isolates collected for the study was associated with chronic open skin wounds and related to comorbidities, such as diabetes. Moreover, poor social and hygienic conditions can be considered a risk factor for an infection with this human pathogen (Schröttner et al., 2017). These findings are in line with previous case studies, which further emphasize the correlation between the emergence of infectious diseases and social and economic inequalities (Campisi et al., 2015; de Dios et al., 2015; Suryalatha et al., 2015; Nogi et al., 2016; Chavez et al., 2017). Other microbial species were detected along with W. chitiniclastica, making it difficult to modulate the pathogenic potential of one or the other and/or appoint the causative agent of the infection in our collection. Although only rudimentary information is available and no thorough characterization of the microbial community took place, it is interesting to note that similarities to the diabetic foot microbiome occur. The genera Staphylococcus, Pseudomonas as well as Streptococcus were recently described as dominant taxa in chronic diabetic foot ulcers (Gardner et al., 2013; Wolcott et al., 2016; Gardiner et al., 2017) while Proteus spp. was specific to individuals (Gardiner et al., 2017). Although W. chitiniclastica occurred as part of polymicrobial infections (Campisi et al., 2015; de Dios et al., 2015; Kõljalg et al., 2015; Nogi et al., 2016), it has not yet been reported to be an abundant member of the diabetic skin and/or wound microbiome. However, further research could uncover its specific role and potential pathogenicity in association with chronic wounds and diseases, such as diabetes.
16S rRNA sequencing has proven to be a good and rapid identification method for bacterial organisms directly from clinical samples. However, the diagnostic power of this technique heavily depends on the choice of primer (Armougom, 2009; Klindworth et al., 2012) as well as the amplicon length and coverage of variable regions (Sune et al., 2020). Suboptimal primers can fail to detect single species or even whole groups (Andersson et al., 2008; Tringe and Hugenholtz, 2008; Wang and Qian, 2009), and the different variable regions within the 16S rRNA gene exhibit varying degrees of sequence diversity while no single region is able to distinguish among all bacteria (Chakravorty et al., 2007). Therefore, careful choice of primer pairing prior to amplification is strongly recommended. In this study, we decided to choose primer pairs, which are well-established in clinical routine diagnostics and satisfy with a good in silico coverage for the genus Wohlfahrtiimonas. Assuming that a standard PCR can tolerate up to two mismatches between the primer and its target (Nossa et al., 2010), the two primer pairs were chosen based on their TestPrime results (Klindworth et al., 2012) with an in silico coverage of 100%, and we are pleased to report that both primer pairs successfully amplified the 16S gene sequence of W. chitiniclastica from pure culture.
TPU-1/RTU-4 has been widely used in medical research as well as routine diagnostics (Funke et al., 2004; Haanperä et al., 2007; Broecker et al., 2016; Rudolph et al., 2019) and generates a shorter ∼800 bp fragment spanning hypervariable region V1 through V5, respectively. Hereby, V1-V3 is expected to provide reasonable taxonomic resolution to discriminate between taxa (Johnson et al., 2019) and enabled us to identify each isolate as W. chitiniclastica. However, it has been shown that taxonomic analysis based on short amplicons cannot achieve the taxonomic resolution afforded by sequencing the entire (∼1,500 bp) gene (Johnson et al., 2019), which still poses a problem for in-depth phylogenetic analysis (Tringe and Hugenholtz, 2008; Nossa et al., 2010; Johnson et al., 2019). The combination 27F/1492R overcomes this limitation by generating amplicons, spanning all nine hypervariable regions, which we would recommend when accurate classification of individual organisms at very high taxonomic resolution is required. In summary, TPU-1/RTU-4 as well as 27F/1492R is suitable for amplification of the 16S rRNA gene of W. chitiniclastica from pure culture and leads to correct identification. Although this has not been tested yet, we believe that both primer pairs are also suitable to detect W. chitiniclastica within the scope of a thorough microbial community profiling.
MALDI-TOF MS-based microbial identification is a well-established method in routine diagnostics (Mellmann et al., 2008; Schröttner et al., 2016). Although limitation on species level identification due to missing spectra in the database of unknown species might occur (Timperio et al., 2017; Strejcek et al., 2018), it has the advantage of speed and low cost, which most likely have priority in daily clinical practice (Seng et al., 2009). In case of an infection with W. chitiniclastica it excels as a fast and inexpensive identification tool.
The VITEK 2 system proved to be ineffective for identification of W. chitiniclastica isolates as previously reported (de Dios et al., 2015; Chavez et al., 2017; Snyder et al., 2020). The information about this bacterium appears to be missing the VITEK 2 database, leading to misidentification as A. lwoffii. Both organisms are Gram-negative, rod-shaped and show almost identical biochemical characteristics. Based on the VITEK 2 system, differences occur only with respect to the oxidase reaction. A. lwoffii is oxidase negative unlike W. chitiniclastica (Tóth et al., 2008; de Dios et al., 2015). Little is known about a potential natural habitat of W. chitiniclastica in humans apart from its association to parasitic flies, unlike A. lwoffii, which is described to be part of the physiological skin flora (Seifert et al., 1997; Berlau et al., 1999) but could also cause severe infections in humans (Ku et al., 2000). The latter could be problematic since Acinetobacter species are known for inherent resistance against many of the available antimicrobial agents (Tripathi et al., 2014), leading for example to multidrug-resistant clinical isolates of A. lwoffii (Hu et al., 2011). Although most Acinetobacter strains are still susceptible to carbapenems (Peleg et al., 2008), first resistances have emerged (Eliopoulos et al., 2008), making it a significant challenge to treat possible infections. W. chitiniclastica, on the other hand, appears to be susceptible to the majority of known antibiotics (Schröttner et al., 2017; Matos et al., 2019). Due to this contrary picture in terms of susceptibility for these two organisms, appropriate identification of the causing microorganisms and their resistance profiles is crucial to limit formation of multidrug-resistant species. With the currently available database, the VITEK 2 system appears not to be reliable and we therefore do not advocate for this method. For the identification of W. chitiniclastica isolates, we would rather recommend using MALDI-TOF MS and 16S rRNA gene sequencing. Although both serve as reliable identification tools, we would endorse a combination of both methods as it leads to a higher reliability and more robust identification accuracy (Schröttner et al., 2016), which is especially beneficial in case of doubtful results (Schröttner et al., 2014). Keeping in mind that neither MALDI-TOF MS nor 16S rRNA gene sequencing might be in use for routine diagnostics in every diagnostic laboratory, W. chitiniclastica might be even more common but misidentified due to a suboptimal diagnostic method. Moreover, with 23 human case reports from 18 different locations a clear geographical clustering appears to be missing (Table 1 and Figure 1) suggesting a potential spread and transmission. This further emphasize the hypothesis that W. chitiniclastica might be not as rare as originally anticipated.
Our study indicates that dDDH has proven a worthy identification method for W. chitiniclastica; however, since dDDH is a very costly and time-consuming technique and requires access to next generation sequencing technology (NGS), it is most likely irrelevant in daily clinical routine diagnostics. Notably, our 14 isolates from Dresden cluster in a subspecies, a fact that has not been described for any W. chitiniclastica strain yet. Subspecies are known to show adaptation to different environments (Biller et al., 2015; Lamas et al., 2018) and geographical locations (Truong et al., 2015; Costea et al., 2017). For example, recent studies of the human gut showed that discrete subspecies of the species Agathobacter rectalis and Prevotella copri are associated with geographically distinct human populations (Truong et al., 2015; Costea et al., 2017), whereas few strains occurred in multiple unrelated cohorts (Truong et al., 2015). Keeping in mind that the type strain DSM 18708T has been isolated from animal source in Hungary (Tóth et al., 2008), the formation of a novel subspecies within the 14 isolates from Dresden most likely represents the adaptation to a human environment or a different geographic location. With hopefully increasing numbers of available genomes associated to human cases preferentially from various locations, large-scale genome analysis is recommended to unravel phenotypic and/or genotypic differences within the W. chitiniclastica clade.
Wohlfahrtiimonas chitiniclastica are described to be susceptible to the majority of known antibiotics with the exception of fosfomycin (Schröttner et al., 2017; Matos et al., 2019); however, no comprehensive and comparative antimicrobial resistance profiling of a larger strain collection has been performed so far. Based on our in vitro susceptibility testing, all W. chitiniclastica isolates appear to be susceptible to β-lactam antibiotics such as penicillins, cephalosporines, monobactams, and carbapenems. This is in contrast to the complete genome sequence analysis of the W. chitiniclastica strain BM-Y, which carried a blaVEB–1 gene cassette, thus conferring resistance to ceftazidime and ampicillin, among others (Zhou et al., 2016). Interesting to note is that our in silico analysis also revealed potential genes coding for β-lactamases such as PNGM-1 (Park et al., 2018), NmcR (Naas and Nordmann, 1994) and GOB-16 (Morán-Barrio et al., 2007); however, the identity of the matching region was less than 36% in all cases. Therefore, we believe that W. chitiniclastica contains at most an incomplete beta-lactamase or a homologous protein with a yet unknown function. This is in line with previous case studies, where cephalosporins, such as cefuroxime, have proven to be successful to treat infections caused by W. chitiniclastica (Rebaudet et al., 2009; Campisi et al., 2015; Suryalatha et al., 2015; Snyder et al., 2020; Bueide et al., 2021), and by that supporting our results of a comprehensive susceptibility against a wide majority of β-lactam antibiotics.
All 14 strains were susceptible to the fluoroquinolones ciprofloxacin and levofloxacin. This is congruent with two case studies, where an infection caused by W. chitiniclastica was successfully treated with levofloxacin (Schröttner et al., 2017; Bueide et al., 2021) as well as brief antibiotic susceptibility tests within individual clinical reports (de Dios et al., 2015; Kõljalg et al., 2015; Chavez et al., 2017; Katanami et al., 2018; Snyder et al., 2020). In this study, the majority of the isolates was also susceptible to moxifloxacin and ofloxacin apart from two exceptions. The isolates, DSM 105984 and DSM 106597, appear to be resistant. Interestingly, based on our in silico analysis, all isolates contained a point mutation in gyrB (58.73% identity), known to confer resistance to moxifloxacin in Clostridioides difficile (Walkty et al., 2010). However, a previous study showed that not all mutations leading to amino acid substitution in GyrB seem to be relevant, as at least one was also detected in susceptible strains (Spigaglia et al., 2008). Further research will be necessary to gain a better understanding whether this point mutation plays any role in resistance or not. Our in silico results could serve as basis for identifying potential target genes for a thorough sequence analysis and/or genetic mutations. Until then, ciprofloxacin and levofloxacin may be the best fluoroquinolones to use.
In vitro analysis with respect to tetracycline resistances showed a rather diverse picture. We obtained diversified MIC results for doxycycline, ranging from 0.35 up to 8 μg/ml. Unfortunately, no EUCAST guideline is available, and to the best of our knowledge, no case reports have been published so far. In the case of tigecycline, 12 isolates were susceptible while DSM 105984 and DSM 106597 were resistant. This rather diverse resistance profile for tetracycline is also reflected in the literature. In some case studies, the isolate was susceptible to tetracyclines (Almuzara et al., 2011; Nogi et al., 2016), and in another report, it was resistant (Snyder et al., 2020). This rather varying picture continues within the present in silico study. Resistance to tetracyclines can be governed by tet genes, such as tet(D), which encode for a tetracycline antibiotic efflux pump (Levy et al., 1999; Hedayatianfard et al., 2014). Interestingly, the strains DSM 100676, DSM 100917, and DSM 105708 revealed one strict hit each for tet(D) with an identity of 52.28% for the matching region. Unfortunately, this is not congruent with the in vitro results for tigecycline, in which the strains DSM 100676, DSM 100917, and DSM 105708 were susceptible. Interestingly, for doxycycline, these three strains showed comparatively high MIC values with 4 and 8 μg/ml, respectively (Supplementary Table 1). Keeping in mind that the “Strict” algorithm of the CARD system represents a flexible sequence variation but lies within the curated BLAST bit score cut-offs (Alcock et al., 2020), resistance to doxycycline governed by tet(D) among others therefore appears to be feasible. The two tigecycline resistant isolates, DSM 106584 and DSM 106597, on the other hand, had no hits to tet(D) but loose hits to several efflux pumps and to a gene encoding for the mobile Tet(X) ortholog, which is described to confer high-level tigecycline resistance (Fang et al., 2020). However, since we only found loose hits, these in silico results should be taken with caution and require further research. This leaves us with a rather questionable picture regarding the tetracycline resistance profile, which still needs to be resolved.
Aminoglycosides (AG), such as amikacin, gentamicin, and tobramycin, are broad-spectrum antibiotics and interfere with the bacterial protein translation by binding to the bacterial ribosome. Common AG resistance mechanism include modification of the AG binding site by 16S rRNA methyltransferases (RMTases) and antibiotic target alteration by aminoglycoside phosphotransferases (APHs), aminoglycoside nucleotidyltransferases (ANTs), and aminoglycoside acetyltransferases (AACs) (Yokoyama et al., 2003; Fessler et al., 2011). Based on our in silico analysis, all strains contain a homolog to the apmA gene encoding for an AAC with 47.92% identity for the matching region. In addition, several hits for efflux pumps specific for aminoglycoside and aminocoumarin, such as baeS (Baranova and Nikaido, 2002; Nishino et al., 2005) and cpxA (Srinivasan et al., 2012), were identified, but the result should be viewed with caution since only loose hits are present and the percentage of the matching region was comparatively low, with 27.93 and 33.45%, respectively. Nevertheless, the in vitro experiments support the in silico analysis, which showed that all strains were resistant to amikacin and tobramycin. Similar results were obtained for the type strain DSM 18708T (Supplementary Table 5), suggesting a natural aminoglycoside resistance profile. Moreover, other case studies detected analogous MIC results for amikacin and tobramycin (Kõljalg et al., 2015; Chavez et al., 2017; Katanami et al., 2018), but the interpretation of antibiotic susceptibility was based on the MIC breakpoints for other non-Enterobacteriaceae described in the M100 Performance Standards for Antimicrobial Susceptibility Testing, determined by the Clinical and Laboratory Standards Institute (CLSI) (Wayne and Clinical and Laboratory Standards Institute (CLSI), 2016). Therefore, those isolates were described as susceptible. Although previous studies showed an acceptable level of comparability between EUCAST and CLSI (Kassim et al., 2016; Akdoğan et al., 2021), one should always keep in mind that even small differences might influence the therapeutic option. We rather consider W. chitiniclastica as resistant to amikacin and tobramycin and would therefore not recommend aminoglycosides as first line treatment. However, reevaluation is strongly recommended when updated EUCAST breakpoints become available.
No EUCAST guidelines exist for trimethoprim/sulfamethoxazole, rifampicin, nitrofurantoin, fosfomycin, colistin, doxycyclin, erythromycin, azithromycin, and clarithromycin. However, for some antimicrobial substances, the in vitro analysis revealed comparatively low or high MIC values, respectively. This allows us to generate a hypothesis regarding the resistance profile for some isolates. For instance, we observed comparatively low MIC values for trimethoprim/sulfamethoxazole. This is in line with recent case reports, in which W. chitiniclastica were susceptible to trimethoprim/sulfamethoxazole (Chavez et al., 2017; Katanami et al., 2018; Connelly et al., 2019; Snyder et al., 2020; Bueide et al., 2021). Interestingly, all isolates revealed in silico hits for the trimethoprim resistant gene dfrI (Welch et al., 2007) (56.63% identity) and the sulfonamide resistance gene sul3 (Perreten and Boerlin, 2003) (39.85% identity), assuming that resistance may be feasible. However, previous studies of Escherichia coli reported that trimethoprim resistant genes were not expressed due to defective promotors (Mazurek et al., 2015). Therefore, we believe that susceptibility toward trimethoprim/sulfamethoxazole of all isolates tested in this study appears to be feasible, despite the presence of potential resistance genes. Nevertheless, antimicrobial resistome analysis prior to treatment is always recommended as newly emerged resistance might arise quickly. For example, the first reported case of W. chitiniclastica infection in South Africa surprised with trimethoprim/sulfamethoxazole resistance (Hoffmann et al., 2016). To the best of our knowledge, this is the first report in which an isolate appears to be resistant.
Moderate MIC values for clarithromycin and erythromycin in combination with in silico detection of homologs genes coding for macrolide-specific efflux pumps (macA and macB) (Kobayashi et al., 2000; Yum et al., 2009) make resistance feasible. This is congruent with a preliminary genome report of a W. chitiniclastica strain, which also contained macA and macB genes (Matos et al., 2019). Nevertheless, further research is still required to uncover the macrolide resistance profile fully.
Finally, yet importantly, our in vitro analysis showed very high MIC values for fosfomycin for all isolates. This is in line with previous reports (Schröttner et al., 2017; Matos et al., 2019), making natural resistance very likely. Based on our in silico analysis, all isolates contained a gene homolog encoding for a potential MurA transferase with mutation conferring resistance to fosfomycin (Fu et al., 2015) (50.48% identity) and for the fosfomycin modifying glutathione transferase FosC2 (Wachino et al., 2010) (26.62% identity), among several other hits for efflux proteins. In addition, DSM 110473 carried with 62.67% identity a gene homologous to mdtG, which has been reported, when overexpressed, to increase fosfomycin resistances (Nishino and Yamaguchi, 2001). We strongly believe that W. chitiniclastica features a natural fosfomycin resistance, and maybe a yet unknown resistance mechanism might be present since no strong hits with high percentage identity were detected.
In conclusion, W. chitiniclastica has recently been described as a rare but potential new emerging human pathogen. However, with intensive usage of MALDI-TOF MS and 16S rRNA gene for identification, it might turn out that this species is even more common than currently anticipated. In case of infection, trimethoprim/sulfamethoxazole, levofloxacin, and cephalosporins, such as cefuroxime, may be the best antibiotics to use. Keeping in mind that exposure of many antibiotics lead to enormous selective pressures including resistome expansion (Wright, 2007), constant reevaluation is strongly recommended, in particular when updated EUCAST guidelines become available and/or new case studies are published. Further research is also required in order to identify genes or mutations that are responsible for antimicrobial resistance. The results of our in silico analysis could offer advantages in order to identify potential candidates for target specific manipulations, which will be a crucial component to unravel the genetic resistance profile of W. chitiniclastica.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, JAGIBR000000000; JAGIBS000000000; JAGIBT000000000; JAGIBU000000000; JAGIBV000000000; JAGIBW000000000; JAGIBX000000000; JAGIBY000000000; JAGIBZ000000000; JAGICA000000000; JAGICB000000000; JAGICC000000000; JAGICD000000000; JAGICE000000000.
Ethics Statement
The study was approved by the Ethics Committee at the Technical University of Dresden (EK 61022019). Written informed consent was not obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
PS had the idea and the concept for the study. AK performed the experiments, analyzed the data, and wrote the first version of the manuscript. BB provided the bioinformatic data from the whole genome sequences. TR performed the curation of the bacteria and the inclusion into the DSMZ “Open Collection”. BB, TR, SC, FG, and PS contributed text passages for the manuscript. All authors contributed to the revision of the manuscript and approved the present version.
Funding
This work was supported by the Federal Ministry of Education and Research, Germany (BMBF; ZIK Septomics Research Centre, Translational Septomics, award no. 03Z22JN12 to SC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Franziska Burkhart and Stefan Tiede for excellent technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.712775/full#supplementary-material
Footnotes
- ^ https://www.arb-silva.de/search/testprime/
- ^ http://www.eucast.org/clinical_breakpoints/
- ^ https://card.mcmaster.ca/
- ^ https://tygs.dsmz.de/
References
Akdoğan, D., Güzel, M., Bahçe, Y. G., Aksoy, A., and Akpınar, O. (2021). Comparative antimicrobial susceptibility profiles of uropathogenic extended-spectrum ßlactamase producing strains of Klebsiella pneumonia and Escherichia coli by the CLSI and EUCAST methodologies. Gazi Med. J. 32, 88–93. doi: 10.12996/GMJ.2021.16
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Almuzara, M. N., Palombarani, S., Tuduri, A., Figueroa, S., Gianecini, A., Sabater, L., et al. (2011). First case of fulminant sepsis due to Wohlfahrtiimonas chitiniclastica. J. Clin. Microbiol. 49, 2333–2335. doi: 10.1128/JCM.00001-11
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andersson, A. F., Lindberg, M., Jakobsson, H., Bäckhed, F., Nyrén, P., and Engstrand, L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One 3:e2836. doi: 10.1371/journal.pone.0002836
Armougom, F. (2009). Exploring microbial diversity using 16S rRNA high-throughput methods. J. Comput. Sci. Syst. Biol. 2009, 74–92. doi: 10.4172/jcsb.1000019
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Baranova, N., and Nikaido, H. (2002). The baeSR two-component regulatory system activates transcription of the yegMNOB (mdtABCD) transporter gene cluster in Escherichia coli and increases its resistance to novobiocin and deoxycholate. J. Bacteriol. 184, 4168–4176. doi: 10.1128/jb.184.15.4168-4176.2002
Baym, M., Kryazhimskiy, S., Lieberman, T. D., Chung, H., Desai, M. M., and Kishony, R. (2015). Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One 10:e0128036. doi: 10.1371/journal.pone.0128036
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., et al. (2013). GenBank. Nucleic Acids Res. 41, D36–D42. doi: 10.1093/nar/gks1195
Berlau, J., Aucken, H., Malnick, H., and Pitt, T. (1999). Distribution of Acinetobacter species on skin of healthy humans. Eur. J. Clin. Microbiol. Infect. Dis. 18, 179–183. doi: 10.1007/s100960050254
Biller, S. J., Berube, P. M., Lindell, D., and Chisholm, S. W. (2015). Prochlorococcus: the structure and function of collective diversity. Nat. Rev. Microbiol. 13, 13–27. doi: 10.1038/nrmicro3378
Bonwitt, J. H., Tran, M., Dykstra, E. A., Eckmann, K., Bell, M. E., Leadon, M., et al. (2018). Fly reservoir associated with wohlfahrtiimonas bacteremia in a human. Emerg. Infect. Dis. 24, 370–373. doi: 10.3201/eid2402.170913
Broecker, F., Klumpp, J., Schuppler, M., Russo, G., Biedermann, L., Hombach, M., et al. (2016). Long-term changes of bacterial and viral compositions in the intestine of a recovered Clostridium difficile patient after fecal microbiota transplantation. Cold Spring Harb. Mol. Case Stud. 2:a000448. doi: 10.1101/mcs.a000448
Bueide, P., Hunt, J., Bande, D., and Guerrero, D. M. (2021). Maggot wound therapy associated with wohlfahrtiimonas chitiniclastica blood infection. Cureus 13, 10–13. doi: 10.7759/cureus.12471
Campisi, L., Mahobia, N., and Clayton, J. J. (2015). Wohlfahrtiimonas chitiniclastica Bacteremia associated with Myiasis, United Kingdom. Emerg. Infect. Dis. 21, 1068–1069. doi: 10.3201/eid2106.140007
Cao, X.-M., Yan, Q.-L., Xu, B.-L., Chen, T., Zhang, X.-L., Wang, J., et al. (2013). Complete genome sequence of wohlfahrtiimonas chitiniclastica strain SH04, isolated from chrysomya megacephala collected from Pudong international airport in China. Genome Announc 1, 4–5. doi: 10.1128/genomea.00119-13
Chakravorty, S., Helb, D., Burday, M., Connell, N., and Alland, D. (2007). A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 69, 330–339. doi: 10.1016/j.mimet.2007.02.005
Chavez, J. A., Alexander, A. J., Balada-Llasat, J. M., and Pancholi, P. (2017). A case of Wohlfahrtiimonas chitiniclastica bacteremia in continental United States. JMM Case Rep. 4, 10–12. doi: 10.1099/jmmcr.0.005134
Connelly, K., Freeman, E., Smibert, O., and Lin, B. (2019). Wohlfahrtiimonas chitiniclastica bloodstream infection due to a maggot-infested wound in a 54-year-old male. J. Glob. Infect. Dis. 11, 125–126. doi: 10.4103/jgid.jgid_58_18
Costea, P. I., Coelho, L. P., Sunagawa, S., Munch, R., Huerta-Cepas, J., Forslund, K., et al. (2017). Subspecies in the global human gut microbiome. Mol. Syst. Biol. 13:960. doi: 10.15252/msb.20177589
de Dios, A., Fisher, M. A., Dingle, T. C., Hamula, C. L., Tayal, A., and Jacob, S. (2015). First report of wohlfahrtiimonas chitiniclastica isolation from a patient with cellulitis in the United States. J. Clin. Microbiol. 53, 3942–3944. doi: 10.1128/jcm.01534-15
Eliopoulos, G. M., Maragakis, L. L., and Perl, T. M. (2008). Acinetobacter baumannii: epidemiology, antimicrobial resistance, and treatment options. Clin. Infect. Dis. 46, 1254–1263. doi: 10.1086/529198
Fang, L.-X., Chen, C., Cui, C.-Y., Li, X.-P., Zhang, Y., Liao, X.-P., et al. (2020). Emerging high-level tigecycline resistance: novel tetracycline destructases spread via the mobile tet(X). Bioessays 42:e2000014. doi: 10.1002/bies.202000014
Fenwick, A. J., Arora, V., and Ribes, J. A. (2019). Wohlfahrtiimonas chitiniclastica: two clinical cases and a review of the literature. Clin. Microbiol. Newsl. 41, 33–38. doi: 10.1016/j.clinmicnews.2019.01.006
Fessler, A. T., Kadlec, K., and Schwarz, S. (2011). Novel apramycin resistance gene apmA in bovine and porcine methicillin-resistant Staphylococcus aureus ST398 isolates. Antimicrob. Agents Chemother. 55, 373–375. doi: 10.1128/AAC.01124-10
Fu, Z., Ma, Y., Chen, C., Guo, Y., Hu, F., Liu, Y., et al. (2015). Prevalence of fosfomycin resistance and mutations in murA, glpT, and uhpT in methicillin-resistant staphylococcus aureus strains isolated from blood and cerebrospinal fluid samples. Front. Microbiol. 6:1544. doi: 10.3389/fmicb.2015.01544
Funke, G., Frod, R., and Sommer, H. (2004). First comprehensively documented case of Paracoccus yeei infection in a human. J. Clin. Microbiol. 42, 3366–3368. doi: 10.1128/JCM.42.7.3366-3368.2004
Gardiner, M., Vicaretti, M., Sparks, J., Bansal, S., Bush, S., Liu, M., et al. (2017). A longitudinal study of the diabetic skin and wound microbiome. PeerJ 2017:3543. doi: 10.7717/peerj.3543
Gardner, S. E., Hillis, S. L., Heilmann, K., Segre, J. A., and Grice, E. A. (2013). The neuropathic diabetic foot ulcer microbiome is associated with clinical factors. Diabetes 62, 923L–930L. doi: 10.2337/db12-0771
Haanperä, M., Jalava, J., Huovinen, P., Meurman, O., and Rantakokko-Jalava, K. (2007). Identification of alpha-hemolytic streptococci by pyrosequencing the 16S rRNA gene and by use of VITEK 2. J. Clin. Microbiol. 45, 762–770. doi: 10.1128/JCM.01342-06
Hedayatianfard, K., Akhlaghi, M., and Sharifiyazdi, H. (2014). Detection of tetracycline resistance genes in bacteria isolated from fish farms using polymerase chain reaction. Vet. Res. Forum Int. Q. J. 5, 269–275.
Hoffmann, R., Fortuin, F., Newton-Foot, M., and Singh, S. (2016). First report of Wohlfahrtiimonas chitiniclastica bacteraemia in South Africa. SAMJ South Afr. Med. J. 106:1062.
Hongoh, Y., Ohkuma, M., and Kudo, T. (2003). Molecular analysis of bacterial microbiota in the gut of the termite Reticulitermes speratus (Isoptera; Rhinotermitidae). FEMS Microbiol. Ecol. 44, 231–242. doi: 10.1016/S0168-6496(03)00026-6
Hu, Y., Zhang, W., Liang, H., Liu, L., Peng, G., Pan, Y., et al. (2011). Whole-genome sequence of a multidrug-resistant clinical isolate of Acinetobacter lwoffii. J. Bacteriol. 193, 5549–5550. doi: 10.1128/JB.05617-11
Johnson, J. S., Spakowicz, D. J., Hong, B. Y., Petersen, L. M., Demkowicz, P., Chen, L., et al. (2019). Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 10, 1–11. doi: 10.1038/s41467-019-13036-1
Kassim, A., Omuse, G., Premji, Z., and Revathi, G. (2016). Comparison of Clinical Laboratory Standards Institute and European Committee on Antimicrobial Susceptibility Testing guidelines for the interpretation of antibiotic susceptibility at a University teaching hospital in Nairobi, Kenya: a cross-sectional stud. Ann. Clin. Microbiol. Antimicrob. 15, 1–7. doi: 10.1186/s12941-016-0135-3
Katanami, Y., Kutsuna, S., Nagashima, M., Takaya, S., Yamamoto, K., Takeshita, N., et al. (2018). Wohlfahrtiimonas chitiniclastica bacteremia hospitalized homeless man with squamous cell carcinoma. Emerg. Infect. Dis. 24, 1746–1748. doi: 10.3201/eid2409.170080
Klindworth, A., Peplies, J., Pruesse, E., Schweer, T., Glöckner, F. O., Quast, C., et al. (2012). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Kobayashi, S., Kuzuyama, T., and Seto, H. (2000). Characterization of the fomA and fomB gene products from Streptomyces wedmorensis, which confer fosfomycin resistance on Escherichia coli. Antimicrob. Agents Chemother. 44, 647–650. doi: 10.1128/aac.44.3.647-650.2000
Kõljalg, S., Telling, K., Huik, K., Murruste, M., Saarevet, V., Pauskar, M., et al. (2015). First report of Wohlfahrtiimonas chitiniclastica from soft tissue and bone infection at an unusually high northern latitude. Folia Microbiol. (Praha) 60, 155–158. doi: 10.1007/s12223-014-0355-x
Ku, S. C., Hsueh, P. R., Yang, P. C., and Luh, K. T. (2000). Clinical and microbiological characteristics of bacteremia caused by acinetobacter lwoffii. Eur. J. Clin. Microbiol. Infect. Dis. 19, 501–505. doi: 10.1007/s100960000315
Lamas, A., Miranda, J. M., Regal, P., Vázquez, B., Franco, C. M., and Cepeda, A. (2018). A comprehensive review of non-enterica subspecies of Salmonella enterica. Microbiol. Res. 206, 60–73. doi: 10.1016/j.micres.2017.09.010
Lefort, V., Desper, R., and Gascuel, O. (2015). FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32, 2798–2800. doi: 10.1093/molbev/msv150
Levy, S. B., McMurry, L. M., Barbosa, T. M., Burdett, V., Courvalin, P., Hillen, W., et al. (1999). Nomenclature for new tetracycline resistance determinants. Antimicrob. Agents Chemother. 43, 1523–1524. doi: 10.1128/aac.43.6.1523
Lysaght, T. B., Wooster, M. E., Jenkins, P. C., and Koniaris, L. G. (2018). Myiasis-induced sepsis: a rare case report of Wohlfahrtiimonas chitiniclastica and Ignatzschineria indica bacteremia in the continental United States. Medicine (Baltimore) 97:e13627. doi: 10.1097/MD.0000000000013627
Matos, J., Faria, A. R., Carvalho Assef, A. P. D., de Freitas-Almeida, A. C., Albano, R. M., and Queiroz, M. L. P. (2019). Draft genome sequence of a wohlfahrtiimonas chitiniclastica strain isolated from frozen chicken in Rio De Janeiro, Brazil. Microbiol. Resour. Announc. 8, 1–2. doi: 10.1128/mra.00352-19
Mazurek, J., Bok, E., Stosik, M., and Baldy-Chudzik, K. (2015). Antimicrobial resistance in commensal Escherichia coli from pigs during metaphylactic trimethoprim and sulfamethoxazole treatment and in the post-exposure period. Int. J. Environ. Res. Public Health 12, 2150–2163. doi: 10.3390/ijerph120202150
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013a). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., and Göker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10:2182. doi: 10.1038/s41467-019-10210-3
Meier-Kolthoff, J. P., Göker, M., Spröer, C., and Klenk, H. P. (2013b). When should a DDH experiment be mandatory in microbial taxonomy? Arch. Microbiol. 195, 413–418. doi: 10.1007/s00203-013-0888-4
Meier-Kolthoff, J. P., Hahnke, R. L., Petersen, J., Scheuner, C., Michael, V., Fiebig, A., et al. (2014). Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand Genomic Sci. 9:2. doi: 10.1186/1944-3277-9-2
Mellmann, A., Cloud, J., Maier, T., Keckevoet, U., Ramminger, I., Iwen, P., et al. (2008). Evaluation of matrix-assisted laser desorption ionization-time-of-flight mass spectrometry in comparison to 16S rRNA gene sequencing for species identification of nonfermenting bacteria. J. Clin. Microbiol. 46, 1946–1954. doi: 10.1128/JCM.00157-08
Morán-Barrio, J., González, J. M., Lisa, M. N., Costello, A. L., Peraro, M. D., Carloni, P., et al. (2007). The metallo-beta-lactamase GOB is a mono-Zn(II) enzyme with a novel active site. J. Biol. Chem. 282, 18286–18293. doi: 10.1074/jbc.M700467200
Naas, T., and Nordmann, P. (1994). Analysis of a carbapenem-hydrolyzing class A beta-lactamase from Enterobacter cloacae and of its LysR-type regulatory protein. Proc. Natl. Acad. Sci. U.S.A. 91, 7693–7697. doi: 10.1073/pnas.91.16.7693
Nishino, K., Honda, T., and Yamaguchi, A. (2005). Genome-wide analyses of Escherichia coli gene expression responsive to the BaeSR two-component regulatory system. J. Bacteriol. 187, 1763–1772. doi: 10.1128/JB.187.5.1763-1772.2005
Nishino, K., and Yamaguchi, A. (2001). Analysis of a complete library of putative drug transporter genes in Escherichia coli. J. Bacteriol. 183, 5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001
Nogi, M., Bankowski, M. J., and Pien, F. D. (2016). Wohlfahrtiimonas chitiniclastica infections in 2 elderly patients, Hawaii, USA. Emerg. Infect. Dis. 22, 567–568. doi: 10.3201/eid2203.151701
Nossa, C. W., Oberdorf, W. E., Yang, L., Aas, J. A., Paster, B. J., de Santis, T. Z., et al. (2010). Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World J. Gastroenterol. 16, 4135–4144. doi: 10.3748/wjg.v16.i33.4135
Park, K. S., Kim, T. Y., Kim, J. H., Lee, J. H., Jeon, J. H., Karim, A. M., et al. (2018). PNGM-1, a novel subclass B3 metallo-β-lactamase from a deep-sea sediment metagenome. J. Glob. Antimicrob. Resist. 14, 302–305. doi: 10.1016/j.jgar.2018.05.021
Peleg, A. Y., Seifert, H., and Paterson, D. L. (2008). Acinetobacter baumannii: emergence of a successful pathogen. Clin. Microbiol. Rev. 21, 538–582. doi: 10.1128/CMR.00058-07
Perreten, V., and Boerlin, P. (2003). A new sulfonamide resistance gene (sul3) in Escherichia coli is widespread in the pig population of Switzerland. Antimicrob. Agents Chemother. 47, 1169–1172. doi: 10.1128/aac.47.3.1169-1172.2003
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Rebaudet, S., Genot, S., Renvoise, A., Fournier, P. E., and Stein, A. (2009). Wohlfahrtiimonas chitiniclastica bacteremia in homeless woman. Emerg. Infect. Dis. 15, 985–987. doi: 10.3201/eid1506.080232
Robbins, K., and Khachemoune, A. (2010). Cutaneous myiasis: a review of the common types of myiasis. Int. J. Dermatol. 49, 1092–1098. doi: 10.1111/j.1365-4632.2010.04577.x
Rudolph, W. W., Gunzer, F., Trauth, M., Bunk, B., Bigge, R., and Schröttner, P. (2019). Comparison of VITEK 2, MALDI-TOF MS, 16S rRNA gene sequencing, and whole-genome sequencing for identification of Roseomonas mucosa. Microb. Pathog. 134:103576. doi: 10.1016/j.micpath.2019.103576
Schröttner, P., Gunzer, F., Schüppel, J., and Rudolph, W. W. (2016). Identification of rare bacterial pathogens by 16S rRNA gene sequencing and MALDI-TOF MS. J. Vis. Exp. 2016:53176. doi: 10.3791/53176
Schröttner, P., Rudolph, W. W., Damme, U., Lotz, C., Jacobs, E., and Gunzer, S. (2017). Wohlfahrtiimonas chitiniclastica: current insights into an emerging human pathogen. Epidemiol. Infect. 145, 1292–1303. doi: 10.1017/S0950268816003411
Schröttner, P., Rudolph, W. W., Eing, B. R., Bertram, S., and Gunzer, F. (2014). Comparison of VITEK2, MALDI-TOF MS, and 16S rDNA sequencing for identification of Myroides odoratus and Myroides odoratimimus. Diagn. Microbiol. Infect. Dis. 79, 155–159. doi: 10.1016/j.diagmicrobio.2014.02.002
Seifert, H., Dijkshoorn, L., Gerner-Smidt, P., Pelzer, N., Tjernberg, I., and Vaneechoutte, M. (1997). Distribution of Acinetobacter species on human skin: comparison of phenotypic and genotypic identification methods. J. Clin. Microbiol. 35, 2819–2825. doi: 10.1128/jcm.35.11.2819-2825.1997
Seng, P., Drancourt, M., Gouriet, F., Scola, B., Fournier, P. E., Rolain, J. M., et al. (2009). Ongoing revolution in bacteriology: routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin. Infect. Dis. 49, 543–551. doi: 10.1086/600885
Snyder, S., Singh, P., and Goldman, J. (2020). Emerging pathogens: a case of Wohlfahrtiimonas chitiniclastica and Ignatzschineria indica bacteremia. IDCases 19:e00723. doi: 10.1016/j.idcr.2020.e00723
Spigaglia, P., Barbanti, F., Mastrantonio, P., Brazier, J. S., Barbut, F., Delmée, M., et al. (2008). Fluoroquinolone resistance in Clostridium difficile isolates from a prospective study of C. difficile infections in Europe. J. Med. Microbiol. 57, 784–789. doi: 10.1099/jmm.0.47738-0
Srinivasan, V. B., Vaidyanathan, V., Mondal, A., and Rajamohan, G. (2012). Role of the two component signal transduction system CpxAR in conferring cefepime and chloramphenicol resistance in Klebsiella pneumoniae NTUH-K2044. PLoS One 7:e33777. doi: 10.1371/journal.pone.0033777
Strejcek, M., Smrhova, T., Junkova, P., and Uhlik, O. (2018). Whole-cell MALDI-TOF MS versus 16S rRNA gene analysis for identification and dereplication of recurrent bacterial isolates. Front. Microbiol. 9:1294. doi: 10.3389/fmicb.2018.01294
Sune, D., Rydberg, H., Augustinsson, A. N., Serrander, L., and Jungeström, M. B. (2020). Optimization of 16S rRNA gene analysis for use in the diagnostic clinical microbiology service. J. Microbiol. Methods 170:105854. doi: 10.1016/j.mimet.2020.105854
Suraiya, S., Zuraina, N., Ahmad, F., and Rahman, Z. A. (2017). Fatal wohlfahrtiimonas chitiniclastica bacteremia in an immunocompromised patient. Clin. Microbiol. Newsl. 39, 172–173. doi: 10.1016/j.clinmicnews.2017.07.003
Suryalatha, K., John, J., and Thomas, S. (2015). Wohlfahrtiimonas chitiniclastica-associated osteomyelitis: a rare case report. Future Microbiol. 10, 1107–1109. doi: 10.2217/fmb.15.44
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Thaiwong, T., Kettler, N. M., Lim, A., Dirkse, H., and Kiupel, M. (2014). First report of emerging zoonotic pathogen Wohlfahrtiimonas chitiniclastica in the United States. J. Clin. Microbiol. 52, 2245–2247. doi: 10.1128/JCM.00382-14
Timperio, A. M., Gorrasi, S., Zolla, L., and Fenice, M. (2017). Evaluation of MALDI-TOF mass spectrometry and MALDI BioTyper in comparison to 16S rDNA sequencing for the identification of bacteria isolated from Arctic sea water. PLoS One 12:e181860. doi: 10.1371/journal.pone.0181860
Tóth, E. M., Schumann, P., Borsodi, A. K., Kéki, Z., Kovács, A. L., and Márialigeti, K. (2008). Wohlfahrtiimonas chitiniclastica gen. nov., sp. nov., a new gammaproteobacterium isolated from Wohlfahrtia magnifica (Diptera: sarcophagidae). Int. J. Syst. Evol. Microbiol. 58, 976–981. doi: 10.1099/ijs.0.65324-0
Tringe, S. G., and Hugenholtz, P. (2008). A renaissance for the pioneering 16S rRNA gene. Curr. Opin. Microbiol. 11, 442–446. doi: 10.1016/j.mib.2008.09.011
Tripathi, P. C., Gajbhiye, S. R., and Agrawal, G. N. (2014). Clinical and antimicrobial profile of Acinetobacter spp.: an emerging nosocomial superbug. Adv. Biomed. Res. 3:13. doi: 10.4103/2277-9175.124642
Truong, D. T., Tett, A., Pasolli, E., and Huttenhower, C. (2015). IMicrobial strain-level population structure and genetic diversity from metagenomes. ISME J. 9, 68–80. doi: 10.1101/gr.216242.116.Freely
Wachino, J., Yamane, K., Suzuki, S., Kimura, K., and Arakawa, Y. (2010). Prevalence of fosfomycin resistance among CTX-M-producing Escherichia coli clinical isolates in Japan and identification of novel plasmid-mediated fosfomycin-modifying enzymes. Antimicrob. Agents Chemother. 54, 3061–3064. doi: 10.1128/AAC.01834-09
Walkty, A., Boyd, D. A., Gravel, D., Hutchinson, J., McGeer, A., Moore, D., et al. (2010). Molecular characterization of moxifloxacin resistance from Canadian Clostridium difficile clinical isolates. Diagn. Microbiol. Infect. Dis. 66, 419–424. doi: 10.1016/j.diagmicrobio.2009.12.002
Wang, Y., and Qian, P.-Y. (2009). Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One 4:e7401. doi: 10.1371/journal.pone.0007401
Wayne, P. C., and Clinical and Laboratory Standards Institute (CLSI) (2016). CLSI. Methods for Antimicrobial Dilution and Disk Susceptibility Testing of Infrequently Isolated or Fastidious Bacteria. CLSI Guideline M45, 3rd Edn. Pittsburgh, PA: CLSI.
Weisburg, W. G., Barns, S. M., Pelletier, D. A., and Lane, D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703.
Welch, T. J., Fricke, W. F., McDermott, P. F., White, D. G., Rosso, M.-L., Rasko, D. A., et al. (2007). Multiple antimicrobial resistance in plague: an emerging public health risk. PLoS One 2:e309. doi: 10.1371/journal.pone.0000309
Wolcott, R. D., Hanson, J. D., Rees, E. J., Koenig, L. D., Phillips, C. D., Wolcott, R. A., et al. (2016). Analysis of the chronic wound microbiota of 2,963 patients by 16S rDNA pyrosequencing. Wound Repair Regen. 24, 163–174. doi: 10.1111/wrr.12370
Wright, G. D. (2007). The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 5, 175–186. doi: 10.1038/nrmicro1614
Yokoyama, K., Doi, Y., Yamane, K., Kurokawa, H., Shibata, N., Shibayama, K., et al. (2003). Acquisition of 16S rRNA methylase gene in Pseudomonas aeruginosa. Lancet (London, England) 362, 1888–1893. doi: 10.1016/S0140-6736(03)14959-8
Yum, S., Xu, Y., Piao, S., Sim, S.-H., Kim, H.-M., Jo, W.-S., et al. (2009). Crystal structure of the periplasmic component of a tripartite macrolide-specific efflux pump. J. Mol. Biol. 387, 1286–1297. doi: 10.1016/j.jmb.2009.02.048
Keywords: Wohlfahrtiimonas chitiniclastica, antibiotic profiling, MALDI-TOF MS, 16S rRNA, primer, VITEK 2, digital DNA-DNA hybridization
Citation: Kopf A, Bunk B, Coldewey SM, Gunzer F, Riedel T and Schröttner P (2021) Identification and Antibiotic Profiling of Wohlfahrtiimonas chitiniclastica, an Underestimated Human Pathogen. Front. Microbiol. 12:712775. doi: 10.3389/fmicb.2021.712775
Received: 21 May 2021; Accepted: 27 August 2021;
Published: 22 September 2021.
Edited by:
Fabian Cieplik, University Medical Center Regensburg, GermanyReviewed by:
Aaron Lynne, Sam Houston State University, United StatesTim Maisch, University of Regensburg, Germany
Copyright © 2021 Kopf, Bunk, Coldewey, Gunzer, Riedel and Schröttner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Percy Schröttner, cGVyY3kuc2Nocm9ldHRuZXJAdHUtZHJlc2Rlbi5kZQ==