 Haimei Zhou1,2†
Haimei Zhou1,2† Wei Lu
Wei Lu Yuyong He
Yuyong He
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 26 July 2021
Sec. Microbial Symbioses
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.712212
This article is part of the Research Topic Nutritional Physiology and Gut Microbiome View all 21 articles
Administration of all-trans retinoic acid (ATRA) to pregnant sows improves developmental defects of Hoxa1–/– fetal pigs, and this study aimed to explore the influence of maternal ATRA administration during pregnancy on gut microbiota of neonatal piglets. Samples of jejunal and ileal meconium of neonatal piglets before suckling were collected including 5 Hoxa1–/– and 20 non-Hoxa1–/– (Hoxa1+/+ and Hoxa1+/−) neonatal piglets from the control group and 5 Hoxa1–/– and 7 non-Hoxa1–/– neonatal piglets from the experimental group. Results indicated that Hoxa1 mutation shaped the bacterial composition of the jejunum and ileum of neonatal piglets and Hoxa1–/– neonatal piglets had significantly higher diversity and species richness, higher relative abundance of phylum Bacteroidetes, lower relative abundances of phylum Firmicutes and genus Lactobacillus, and lower ratio of Firmicutes to Bacteroidetes than non-Hoxa1–/– neonatal piglets. After maternal ATRA administration, Hoxa1–/– neonatal piglets had significantly higher diversity and species richness, higher relative abundances of two bacterial phyla (Bacteroidetes and Proteobacteria), and lower relative abundances of phylum Firmicutes and genus Lactobacillus in the jejunum than non-Hoxa1–/– neonatal piglets. Hoxa1–/– neonatal piglets delivered by sows with maternal ATRA administration had lower diversity and species richness and higher relative abundance of phylum Firmicutes in the jejunum than Hoxa1–/– neonatal piglets born by sows with no maternal ATRA administration. Non-Hoxa1–/– neonatal piglets delivered by sows with maternal ATRA administration had higher diversity and species richness and significantly lower relative abundances of phyla Firmicutes and Actinobacteria and genus Lactobacillus in the ileum than non-Hoxa1–/– neonatal piglets born by sows with no maternal ATRA administration. Hoxa1 mutation decreased the expression of bacterial genes involved in ABC transporters, purine metabolism, and aminoacyl-tRNA biosynthesis and increased the expression of bacterial genes involved in two-component system, starch and sucrose metabolism, and arginine and proline metabolism. Maternal ATRA administration decreased the expression of bacterial genes involved in arginine and proline metabolism, peptidoglycan biosynthesis, and fatty acid biosynthesis. Hoxa1 mutation resulted in bacterial dysbiosis of the small intestine of Hoaxa1–/– neonatal piglets, and maternal ATRA administration restored the bacterial dysbiosis of Hoxa1–/– neonatal piglets and altered the bacterial composition of the small intestine of non-Hoxa1–/– neonatal piglets.
Gut microbiota is integral to feed digestion, nutrient absorption and metabolism, immune response, and gastrointestinal development (Morgavi et al., 2015), and the colonization of intestinal microbiota during early life could further influence the subsequent microbiota of adult host (Ben Salem et al., 2005). Many studies demonstrated that the intestine of prenatal animals really has microorganism (Alipour et al., 2018; Stinson et al., 2019; Hummel et al., 2020; Bi et al., 2021; Husso et al., 2021), and at present, no literature on the differences in intestinal microbiota composition between mutant and wild-type fetuses is found, but for postnatal individuals, there are differences in gut microbiota between mutant and wild-type host, for example, nucleotide-binding oligomerization domain-containing protein 2 (NOD2) mutation caused Crohn’s disease (CD) (Hampe et al., 2001; Ogura et al., 2001) and Crohn’s disease individuals had lower bacterial diversity than healthy controls (Joossens et al., 2011). Cystic fibrosis transmembrane conductance regulator (CFTR) mutation resulted in multiorgan defects, and CFTR–/– mice had significantly lower alpha diversity of intestinal bacterial community (p < 0.05) and had reduced relative abundance of protective species such as Acinetobacter lwoffii and Lactobacilliales members compared with wild-type mice (Lynch et al., 2013). Methyl-CpG-binding protein 2 (MeCP2) mutation developed into the Rett syndrome (RTT), and RTT patients had significantly less diversity in gut bacteria community compared with healthy controls (p < 0.01). Meanwhile, RTT individuals had the most abundant phylum of Actinobacteria, but healthy controls had the most abundant phylum of Firmicutes, and a significant decrease of Bacteroidetes was observed in RTT subjects (Strati et al., 2016).
Gene mutation not only can alter intestinal microbiota composition of postnatal animals but also can cause abnormal phenotypes of fetuses: cytochrome C oxidase subunit IV isoform 1 (COX4I1) mutation caused short stature and poor weight gain (Abu-Libdeh et al., 2017); mutations of Huntington (HTT) exhibited fetal ear defects (Murthy et al., 2019); K+ channels Kir4.1 (KCNJ10) gene mutation resulted in seizures and ataxia (Ai Dhaibani et al., 2018); gamma-1 adaptin gene (Ap1g1) mutation developed abnormalities of the inner ear and testes (Johnson et al., 2016); WD repeat domain phosphoinositide-interacting protein 2 (WIPI2) mutation led to skeletal and cardiac abnormalities (Jelani et al., 2019); fibroblast growth factor receptor 2 (FGFR2) mutation induced midfacial hypoplasia and bilateral syndactyly of the hands and feet (Giancotti et al., 2014); solute carrier family 26a member 4 (SLC26A4) mutation developed deafness (Nonose et al., 2018); a Q186K mutation in Hoxa2 resulted in external ear malformation (Alasti et al., 2008); and the Hoxa1 mutation of g.50111251 G > TC developed abnormal auricle and external auditory canal, dyspnea, and even death in newborn piglets (Qiao et al., 2015). Some defects of phenotypes can be rescued by feeding special chemicals to pregnant animals during pregnancy at a specific time: administration of exogenous RA to pregnant mice at a dose of 2.5 mg/kg on day embryonic 7.5 or embryonic 8.5 effectively repaired the Hoxa1 mutant mice from inner ear defects (Pasqualetti et al., 2001), and all-trans retinoic acid (ATRA) administration to pregnant sows at the level of 4 mg/kg body weight on 14 days postcoitum (dpc) was also effective for the repair of ear defects of Hoxa1–/– fetal pigs (Zhou et al., 2021). As mentioned above, the abnormal phenotypes of fetus caused by gene mutation can be rescued via chemical administration during gestation, but there is no information if maternal administration of chemicals can also change the intestinal bacterial composition of mutant and wild-type individuals. Understanding the influence of maternal administration with special chemicals during pregnancy on the community composition and function of the neonatal gut bacteria may help to develop strategies to prevent young animals from suffering from some diseases and to guide the healthy development of the offspring. Our previous studies demonstrated that maternal administration with ATRA at the level of 4 mg/kg body weight on 14 dpc had the best effects in repairing ear defects of Hoxa1–/– fetal pigs, and the aims of this study are (1) to find out if maternal administration with ATRA can alter the intestinal bacterial compositions of Hoxa1–/– and non-Hoxa1–/– fetal piglets and (2) to compare the differences in intestinal bacterial compositions of neonate piglets between Hoxa1–/– and non-Hoxa1–/– genotypes.
Eight Hoxa1+/– sows derived from one Chinese Erhualian founder boar and one Shaziling founder sow were mated to one healthy Hoxa1+/– boar and randomly assigned to a control group (six sows) and an experimental group (two sows). Pregnant sows in the control group were orally administered with ATRA at a level of 0 mg/kg body weight, and pregnant sows in the experimental group were orally administered with ATRA at a level of 4 mg/kg body weight (Zhou et al., 2021).
After birth, all samples were collected before suckling. Samples of ears of all neonatal piglets were collected and stored in EP tubes containing 75% alcohol for Hoxa1 genotyping. Hoxa1–/– piglets delivered by sows from the control group had ear defects, and the ear defects of Hoxa1–/– piglets born by sows from the experimental group were effectively repaired; all non-Hoxa1–/– (Hoxa1+/+, Hoxa1+/–) either from the control group or the experimental group had normal ears (Zhou et al., 2021). Samples of meconium of all neonatal piglets were collected from jejunal and ileal sections by exsanguination after anesthetization with pentobarbital sodium (100 mg/kg body weight) according to the protocol approved by the Animal Ethics Committee of Jiangxi Agricultural University and immediately stored at −80°C for microbiome analysis. A total of 37 neonatal piglets were sampled, namely, 25 piglets (5 Hoxa1–/– and 20 non-Hoxa1–/–) from the control group and 12 piglets (5 Hoxa1–/– and 7 non-Hoxa1–/–) from the experimental group.
Genomic DNA of intestinal meconium was extracted with the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the instructions of the manufacturer, and the quantity and quality of DNA were measured with NanoDropTM 2000/2000c Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States). The V3–V4 region of bacterial 16S rRNA genes was amplified using primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 806R (5′-GGACTACHVGGGTATCTAAT-3′). PCR reactions were performed in triplicate 50 μl mixture containing 5 μl of 10 × KOD buffer, 5 μl of 2 mM dNTPs, 3 μl of 25 mM MgSO4, 1.5 μl of each primer (10 μM), 1 μl of KOD polymerase, and 100 ng of template DNA. The PCR conditions consisted of initial denaturation at 94°C for 2 min, followed by 30 cycles of denaturation at 98°C for 10 s, annealing at 65°C for 30 s, elongation at 768°C for 30 s, and finally 68°C for 5 min. The PCR products were subsequently subjected to electrophoresis on 2% agarose gel and stained with ethidium bromide, and the targeted fragment size was purified using the AMPure XP Beads (Beckman Agencourt, Brea, CA, United States) according to the manufacturer’s instructions and quantified using ABI StepOnePlus Real-Time PCR System (Life Technologies, Foster City, CA, United States). Purified amplicons were pooled in equimolar and paired-end sequenced (PE250) on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, United States) according to standard protocols. The raw reads were deposited into the NCBI Sequence Read Archive (SRA) database (accession number: SRP239498).
Raw reads were further filtered using FASTP (version 0.18.0) (Chen et al., 2018), and paired end clean reads were merged as raw tags using FLASH (version 1.2.11) (Magoč and Salzberg, 2011) with a minimum overlap of 10 bp and mismatch error rates of 20%. The clean tags were clustered into operational taxonomic units (OTUs) at 97% sequence similarity using UPARSE (version 9.2.64) pipeline (Edgar, 2013). According to the algorithm principle, the sequences with the highest occurrence frequency were selected as the representative sequence of OTUs. The representative OTU sequences were classified into organisms by a naive Bayesian model using RDP classifier (version 2.2) (Wang et al., 2007) based on the SILVA database (version 132) (Pruesse et al., 2007) with a confidence threshold value of 0.8 to obtain taxonomic information and the community composition of each sample at various classification levels; the abundance statistics of each taxonomy was visualized using Krona (version 2.6) (Ondov et al., 2011).
Chao1 and Shannon index were calculated in QIIME (version 1.9.1) (Caporaso et al., 2010) and alpha index comparison between groups was calculated by Welch’s t-test in R project Vegan package (version 2.5.3) (Oksanen et al., 2010). Bacterial community structure and composition were compared using non-metric multidimensional scaling (NMDS) analysis by means of weighed UniFrac distances in R using the metaMDS function (Caporaso et al., 2012). Permutational multivariate analyses of variance (PERMANOVA; “adonis and anosim” in vegan R package) with 999 random permutations were performed to assess the influence of substrate on the community variances. The functional potentials of intestinal bacteria were predicted using Tax4Fun package in R software based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) terms at level 3 with the observed 16S rRNA gene sequences (Aßhauer et al., 2015; Kanehisa et al., 2015). Analysis of function difference between groups was calculated by Welch’s t-test in R project Vegan package (version 2.5.3) (Oksanen et al., 2010).
Table 1 shows the alteration of richness and diversity of small intestinal bacterial community between Hoxa1–/– and non-Hoxa1–/– neonatal piglets within the same treatment group. Hoxa1–/– neonatal piglets either from the control group or the experimental group had significantly higher OTU (p < 0.01), Chao1 (p < 0.01), and Shannon (p < 0.05) of jejunal bacterial community than non-Hoxa1–/– neonatal piglets, respectively. In the control group, Hoxa1–/– neonatal piglets had significantly higher OTU (p < 0.05), Chao1 (p < 0.05), and Shannon (p < 0.01) of ileal bacterial community than non-Hoxa1–/– neonatal piglets. However, in the experimental group, Hoxa1–/– neonatal piglets had no significantly higher OTU, Chao1, and Shannon of ileal bacterial community than non-Hoxa1–/– neonatal piglets, respectively (p > 0.05).
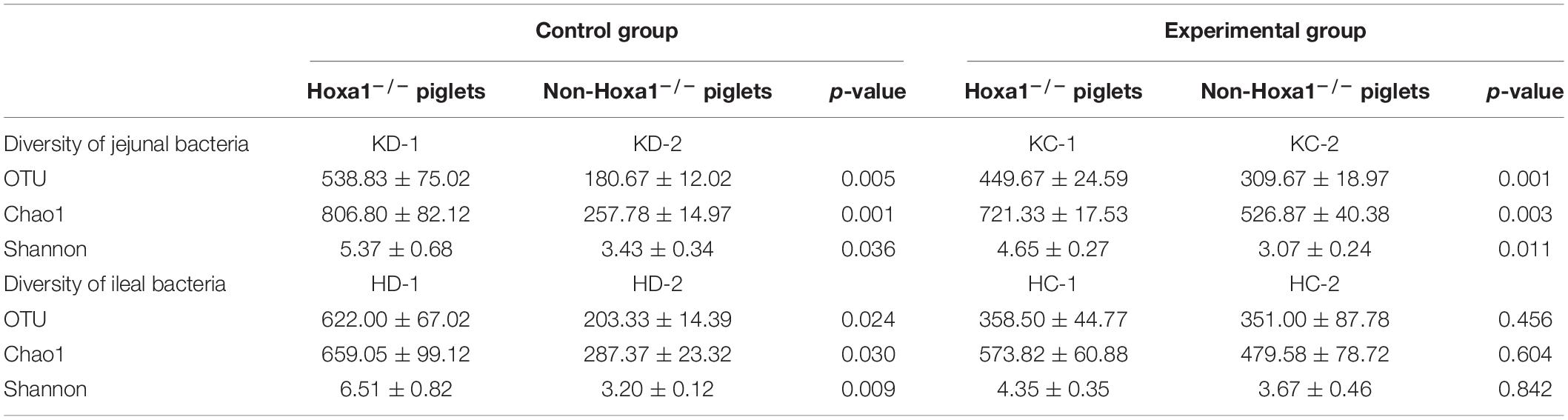
Table 1. Diversity comparison between Hoxa1–/– and Non-Hoxa1–/– newly born piglets in the same treatment group.
Table 2 indicates the change of richness and diversity of intestinal bacterial community of neonatal piglets with the same genotype between the control group and the experimental group. Hoxa1–/– neonatal piglets from the control group had no significantly higher OTU, Chao1, and Shannon of jejunal and ileal bacterial community than Hoxa1–/– neonatal piglets from the experimental group, respectively (p > 0.05). Non-Hoxa1–/– neonatal piglets from the control group had significantly lower OTU (p < 0.01) and Chao1 (p < 0.01) but had no significantly higher Shannon (p > 0.05) of jejunal bacterial community than non-Hoxa1–/– neonatal piglets from the experimental group, respectively. Non-Hoxa1–/– neonatal piglets from the control group had no significant lower OTU, Chao1, and Shannon of ileal bacterial community than that of non-Hoxa1–/– neonatal piglets from the experimental group (p > 0.05).

Table 2. Diversity comparison of newly born piglets with the same genotype between the control group and the experimental group.
These results demonstrate that the g.50111251 G > TC mutation in Hoxa1 significantly increased the OTU, Chao1, and Shannon of jejunal and ileal bacterial community, respectively, when comparing Hoxa1–/– neonatal piglets with non-Hoxa1–/– neonatal piglets. Maternal ATRA administration decreased the OTU, Chao1, and Shannon of jejunal and ileal bacterial community of Hoxa1–/– neonatal piglets but increased the OTU, Chao1, and Shannon of jejunal and ileal bacterial community of non-Hoxa1–/– neonatal piglets with an exception of Shannon in ileal bacteria.
The compositions of bacteria in the jejunal and ileal meconium are presented in Figures 1, 2. The phyla Firmicutes and Proteobacteria were the dominant bacteria in both meconium, and Hoxa1 mutation altered the relative abundances of intestinal bacteria at the phylum level (Figures 1A, 2A). In the control group, Hoxa1–/– neonatal piglets had lower abundance of Firmicutes and higher abundances of Proteobacteria, Bacteroidetes, and Verrucomicrobia than non-Hoxa1–/– neonatal piglets in jejunal (KD-1:KD-2) and ileal meconium (HD-1:HD-2), respectively. In the experimental group, Hoxa1–/– neonatal piglets still had lower abundance of Firmicutes and higher abundances of Proteobacteria and Bacteroidetes than non-Hoxa1–/– neonatal piglets in jejunal (KC-1:KC-2) and ileal (HC-1:HC-2) meconium, respectively, but Hoxa1–/– neonatal piglets from the experimental group had higher abundance of Firmicutes and lower abundances of Bacteroidetes and Verrucomicrobia than Hoxa1–/– neonatal piglets from the control group in jejunal (KC-1:KD-1) and ileal meconium (HC-1:HD-1), respectively. Maternal administration with ATRA also had an influence on the phyla bacteria abundance of non-Hoxa1–/– neonatal piglets, and the data in Figures 1A, 2A indicated that non-Hoxa1–/– neonatal piglets from the experimental group had higher abundance of Firmicutes and lower abundances of Proteobacteria and Bacteroidetes than non-Hoxa1–/– neonatal piglets from the control group in the jejunal meconium (KC-2:KD-2), but had lower abundance of Firmicutes and higher abundances of Proteobacteria, Bacteroidetes, and Verrucomicrobia than non-Hoxa1–/– neonatal piglets from the control group in the ileal meconium (HC-2:HD-2).

Figure 1. Bacterial community composition of jejunal digesta. (A) Phylum level; (B) genus level. KC-1: bacterial composition of jejunal digesta sampled from Hoxa1− /− neonatal piglets delivered by sows administered with ATRA, KC-2: bacterial composition of jejunal digesta sampled from non-Hoxa1− /− neonatal piglets delivered by sows administered with ATRA, KD-1: bacterial composition of jejunal digesta sampled from Hoxa1− /− neonatal piglets delivered by sows administered without ATRA, KD-2: bacterial composition of jejunal digesta sampled from non-Hoxa1− /− neonatal piglets delivered by sows administered without ATRA.
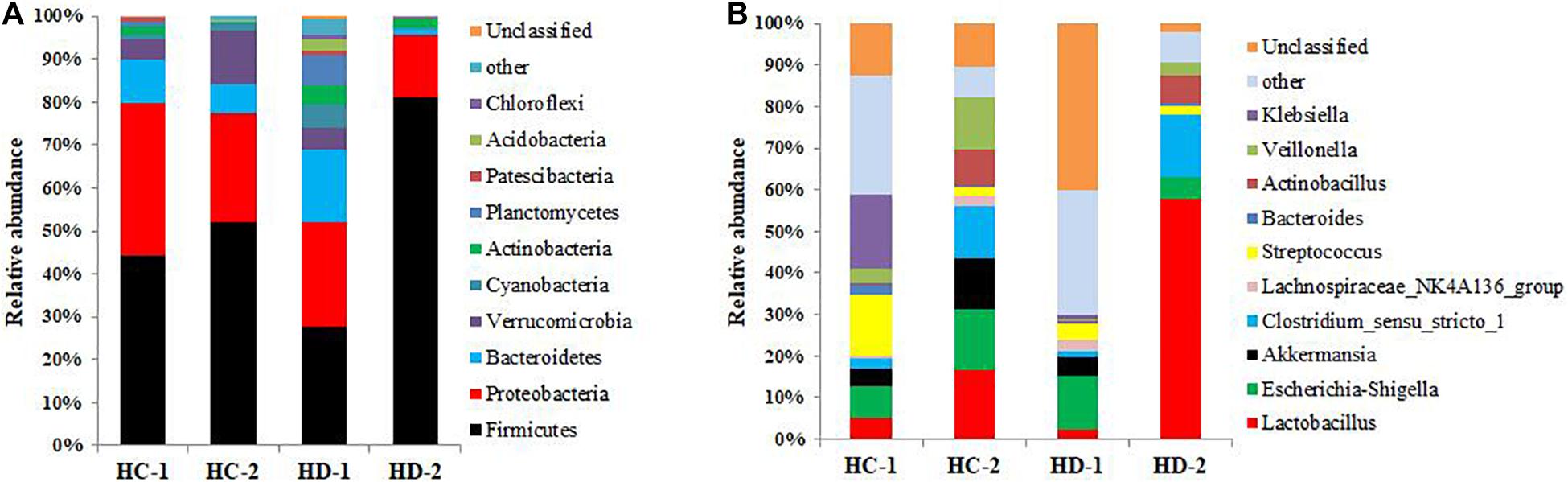
Figure 2. Bacterial community composition of ileal digesta. (A) Phylum level; (B) genus level. HC-1: bacterial composition of ileal digesta sampled from Hoxa1− /− neonatal piglets delivered by sows administered with ATRA, HC-2: bacterial composition of ileal digesta sampled from non-Hoxa1− /− neonatal piglets delivered by sows administered with ATRA, HD-1: bacterial composition of ileal digesta sampled from Hoxa1− /− neonatal piglets delivered by sows administered without ATRA, HD-2: bacterial composition of ileal digesta sampled from non-Hoxa1− /− neonatal piglets delivered by sows administered without ATRA.
At the genus level, data in Figure 1B showed that in the control group, Hoxa1–/– neonatal piglets had lower abundances of Lactobacillus, Actinobacillus, and Streptococcus and higher abundances of Escherichia–Shigella, Akkermansia, and Lachnospiraceae_NK4A136_group than non-Hoxa1–/– neonatal piglets, respectively, in the jenunal meconium (KD-1:KD-2), but in the experimental group, Hoxa1–/– neonatal piglets had lower abundances of Lactobacillus, Clostridium_sensu_stricto_1, and Veillonella and higher abundances of Escherichia–Shigella, Bacteroides, and Lachnospiraceae_NK4A136_group than non-Hoxa1–/– neonatal piglets, respectively, in the jejunal meconium (KC-1:KC-2). Hoxa1–/– neonatal piglets from the experimental group had higher abundances of Lactobacillus, Clostridium_sensu_stricto_1, and Bacteroides and lower abundances of Escherichia–Shigella, Lachnospiraceae_NK4A136_group, and Akkermansia than Hoxa1–/– neonatal piglets from the control group, respectively, in the jejunal meconium (KC-1:KD-1). Results in Figure 2B indicated that in the control group, Hoxa1–/– neonatal piglets had lower abundances of Lactobacillus, Clostridium_sensu_stricto_1, Bacteroides, Actinobacillus, and Veillonella and higher abundances of Escherichia–Shigella, Akkermansia, Streptococcus, and Lachnospiraceae_NK4A136_group than non-Hoxa1–/– neonatal piglets, respectively, in the ileal meconium (HD-1:HD-2), but in the experimental group, Hoxa1–/– neonatal piglets had lower abundances of Lactobacillus, Escherichia–Shigella, Akkermansia, and Clostridium_sensu_stricto_1 and higher abundances of Streptococcus and Klebsiella than non-Hoxa1–/– neonatal piglets, respectively, in the ileal meconium (HC-1:HC-2). Hoxa1–/– neonatal piglets from the experimental group had higher abundances of Lactobacillus, Clostridium_sensu_stricto_1, Streptococcus, Bacteroides, Veillonella, and Klebsiella and lower abundances of Escherichia–Shigella and Akkermansia than Hoxa1–/– neonatal piglets from the control group, respectively, in the ileal digesta (HC-1:HD-1).
The relative abundances of differential bacterial taxa in the jejunal meconium (at least one of the relative abundances is greater than 0.01%) are presented as percentage in Tables 3, 4. The results in Table 3 indicate that four bacterial taxa with differential abundances at the phylum level and 10 bacterial taxa with differential abundances at the genus level were identified in the samples of jejunal meconium between Hoxa1–/– and non-Hoxa1–/– piglets from the control group, and Hoxa1–/– neonatal piglets had significantly lower relative abundances of Firmicutes (p < 0.01) at the phylum level and of Lactobacillus (p < 0.01), Staphylococcus (p < 0.01), and Veillonella (p < 0.01) at the genus level, respectively, in the jejunal digesta than non-Hoxa1–/– neonatal piglets. After maternal ATRA administration, Hoxa1–/– neonatal piglets from the experimental group still had significantly lower relative abundances of Firmicutes (p < 0.01) at the phylum level and of Lactobacillus (p < 0.01) at the genus level, respectively, in the jejunal digesta than non-Hoxa1–/– neonatal piglets.

Table 3. The differential jejunal bacterial community of newborn piglets between different genotypes within the same treatment group (%).
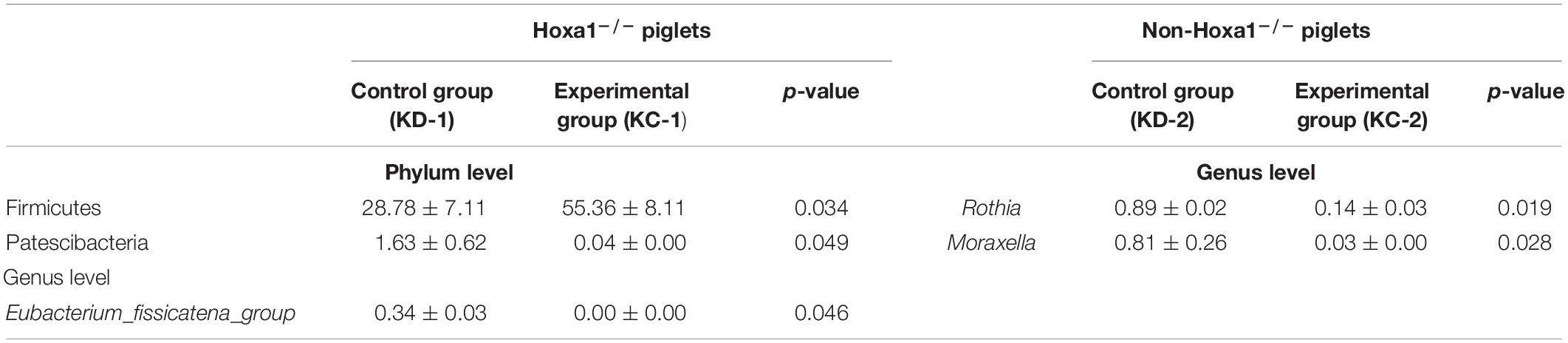
Table 4. The differential jejunal bacterial community of newborn piglets between the control and experimental groups within the same genotype (%).
Data in Table 4 show that Hoxa1–/– neonatal piglets from the experimental group had significantly higher relative abundance of Firmicutes (p < 0.05) and lower relative abundance of Patescibacteria (p < 0.05) at the phylum level and lower relative abundance of Eubacterium_fissicatena_group (p < 0.05) at the genus level, respectively, in the jejunal meconium than Hoxa1–/– neonatal piglets from the control group, and this means that maternal ATRA administration increased the relative abundance of phylum Firmicutes and decreased the relative abundances of phylum Patescibacteria and genus Eubacterium_fissicatena_group of Hoxa1–/– neonatal piglets, respectively. Non-Hoxa1–/– neonatal piglets from the experimental group only had significantly lower relative abundances of Rothia (p < 0.05) and Moraxella (p < 0.05) at the genus level in the jejunal meconium than non-Hoxa1–/– neonatal piglets from the control group.
The differential relative abundances of bacterial taxa of ileal meconium of neonatal piglets between different genotypes within the same treatment group are presented as percentages in Table 5. Three bacterial taxa with differential abundances at the phylum level and 12 bacterial taxa with differential abundances at the genus level were identified in the ileal meconium between Hoxa1–/– and non-Hoxa1–/– neonatal piglets from the control group (Table 5), and Hoxa1–/– neonatal piglets had significantly lower relative abundances of Firmicutes (p < 0.01) at the phylum level and of Lactobacillus (p < 0.01) and Moraxella (p < 0.01) at the genus level in the ileal meconium, respectively, than non-Hoxa1–/– neonatal piglets, but Hoxa1–/– neonatal piglets had significantly higher relative abundances of Bacteroidetes (p < 0.05) and Deferribacteres (p < 0.05) at the phylum level and of Prevotellaceae_UCG-001 (p < 0.05), Ruminococcaceae_UCG-014 (p < 0.05), Eubacterium_xylanophilum_group (p < 0.05), Ruminiclostridium (p < 0.05), Acinetobacter (p < 0.05), Ruminococcus_1 (p < 0.05), Mucispirillum (p < 0.05), Ruminococcaceae_UCG-005 (p < 0.05), and Eubacterium_coprostanoligenes_group (p < 0.05) at the genus level in the ileal meconium, respectively, than non-Hoxa1–/– neonatal piglets. After maternal ATRA administration, Hoxa1–/– neonatal piglets only had significantly lower relative abundance of Spirochaetes at the phylum level in the ileal meconium than non-Hoxa1–/– neonatal piglets (p < 0.05).
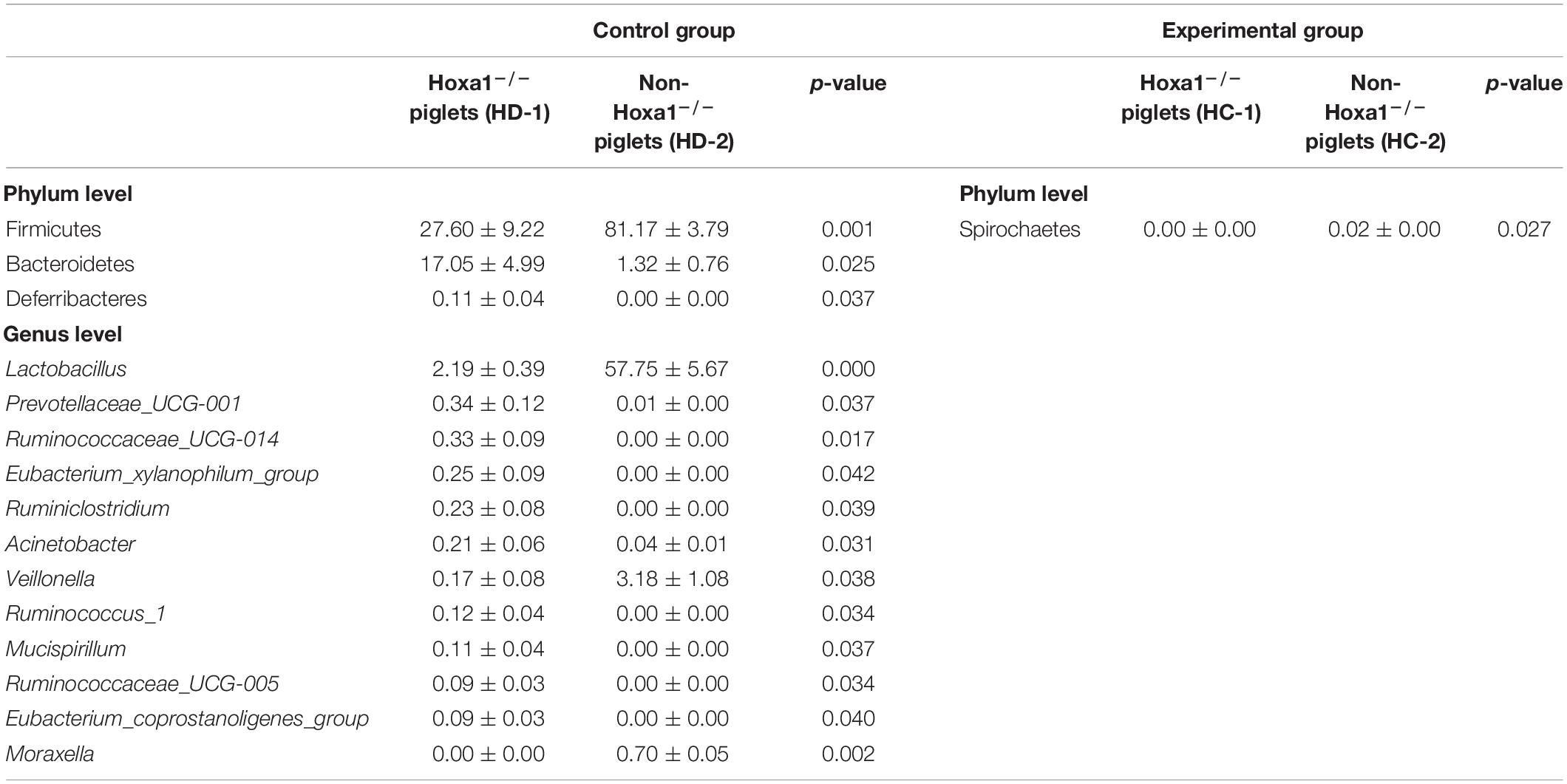
Table 5. The differential ileal bacterial community of newborn piglets between different genotypes within the same treatment group (%).
The differential relative abundances of bacterial taxa of ileal meconium of neonatal piglets with the same genotype between different treatment groups are presented as percentages in Table 6. Data indicated that Hoxa1–/– piglets from the experimental group had significantly lower relative abundances of Deferribacteres (p < 0.05) at the phylum level and of Mucispirillum (p < 0.05) and Ruminococcaceae_UCG-005 (p < 0.05) at the genus level in the ileal meconium, respectively, than Hoxa1–/– neonatal piglets from the control group, and non-Hoxa1–/– neonatal piglets from the experimental group had significantly lower relative abundances of Firmicutes (p < 0.05) and Actinobacteria (p < 0.05) at the phylum level and of Lactobacillus (p < 0.01), Staphylococcus (p < 0.05), Moraxella (p < 0.01), Rothia (p < 0.01), and Pedobacter (p < 0.05) at the genus level in the ileal meconium, respectively, than non-Hoxa1–/– neonatal piglets from the control group.
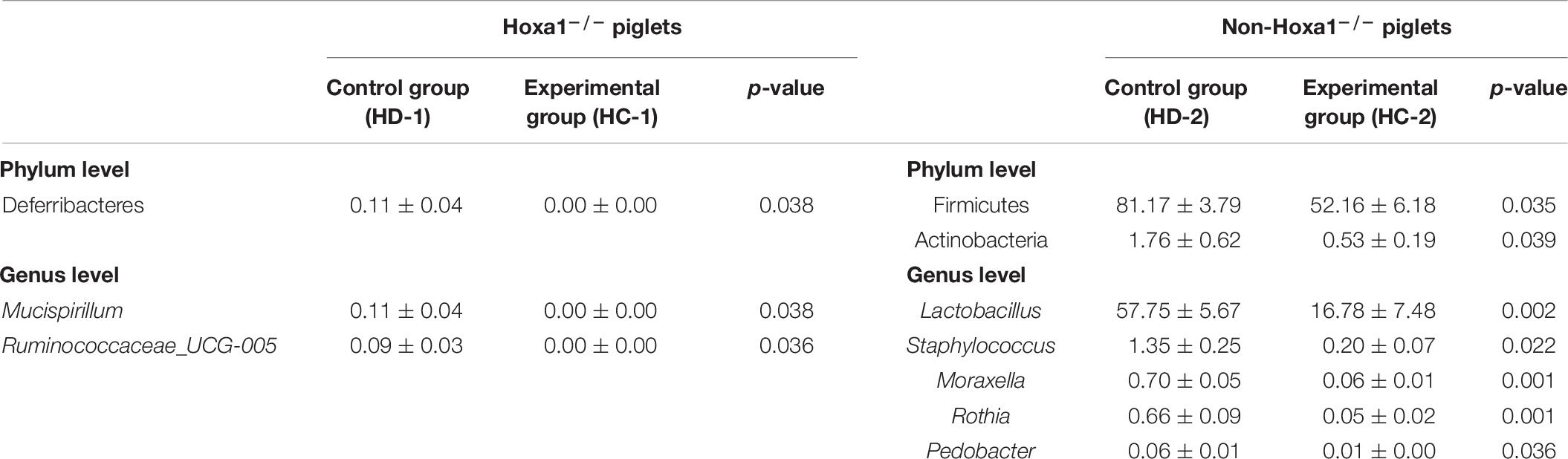
Table 6. The differential ileal bacterial community of newborn piglets with the same genotype between the control and experimental groups (%).
The Tax4Fun package in R software was used to predict the functional potentials of intestinal bacteria based on the KEGG KO terms at level 3 with the observed 16S rRNA gene sequences, and the Welch’s t-test results indicated that there were significant differences in 64 microbial metabolic pathways between jejunal bacteria of non-Hoxa1–/– (KD-2) and Hoxa1–/– (KD-1) neonatal piglets from the control group. The abundances of 22 functions were significantly higher and the abundances of 42 functions were significantly lower in KD-1 compared with KD-2 (Figure 3A). After maternal ATRA administration, a total of 22 microbial metabolic pathways were significantly different between jejunal bacteria of non-Hoxa1–/– (KC-2) and Hoxa1–/– (KC-1) neonatal piglets from the experimental group, and KC-1 had significantly higher expression of genes involved in starch and sucrose metabolism, oxidative phosphorylation, chloroalkane and chloroalkene degradation, meiosis, GABAergic synapse, glutamatergic synapse, retinol metabolism, non-homologous end-joining, and basal transcription factors compared with KC-2 (Figure 3B). KC-1 had significantly higher expression of genes involved in chloroalkane and chloroalkene degradation, retinol metabolism, proximal tubule bicarbonate reclamation, bile secretion, steroid biosynthesis, and hypertrophic cardiomyopathy than KD-1 (Figure 4A), and KC-2 had significantly lower expression of genes involved in nicotinate and nicotinamide metabolism, fatty acid biosynthesis, ribosome biogenesis in eukaryotes, phosphatidylinositol signaling system, mineral absorption, non-homologous end-joining, and betalain biosynthesis than KD-2 (Figure 4B).
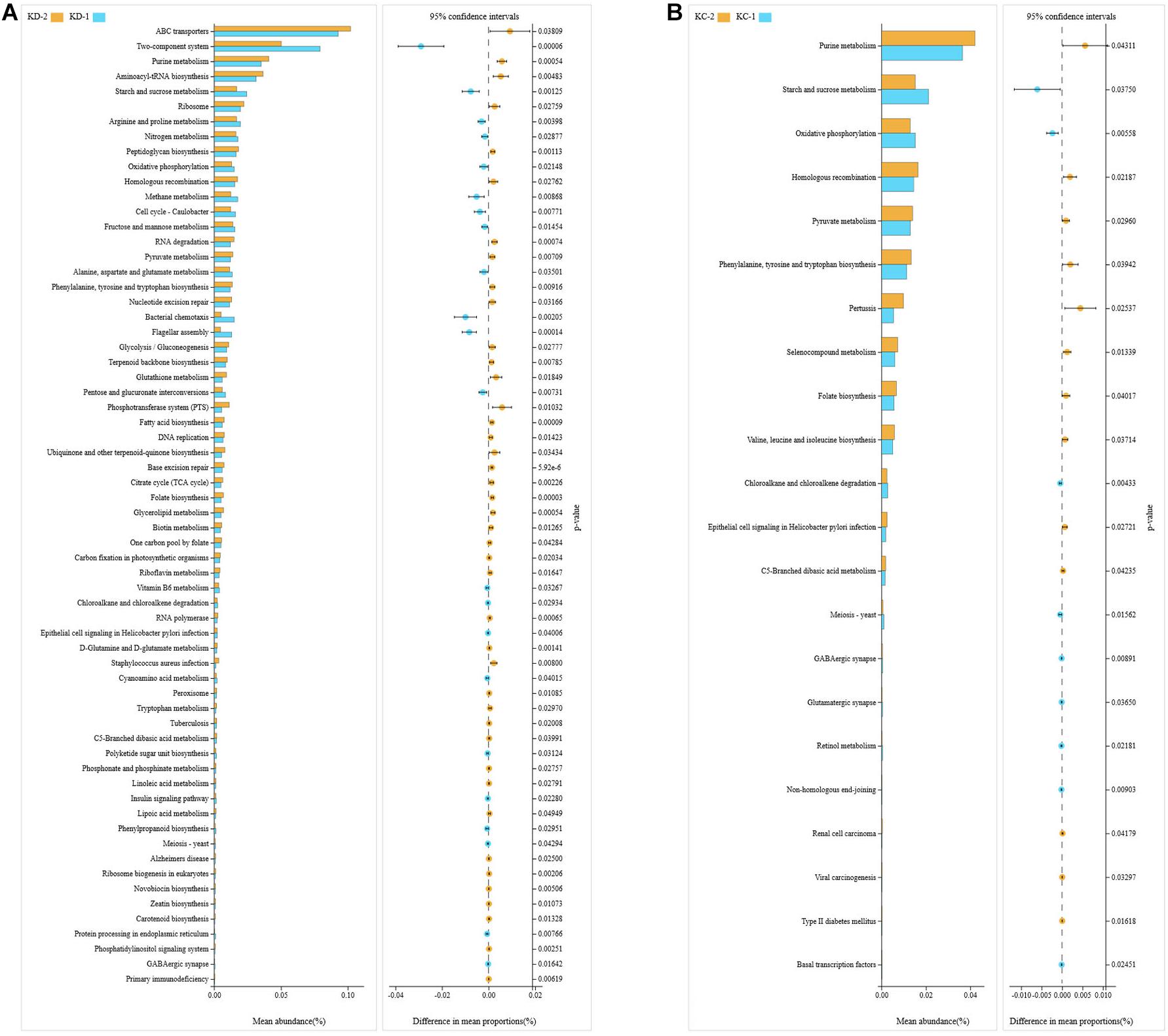
Figure 3. KEGG pathways for bacteria in jejunal meconium of neonatal piglets before suckling. KD-1: bacteria of jejunal meconium of Hoxa1− /− neonatal piglets from the control group. KD-2: bacteria of jejunal meconium of non-Hoxa1− /− neonatal piglets from the control group. KC-1: bacteria of jejunal meconium of Hoxa1− /− neonatal piglets from the experimental group. KC-2: bacteria of jejunal meconium of non-Hoxa1− /− neonatal piglets from the experimental group. (A) Control group. (B) Experimental group.
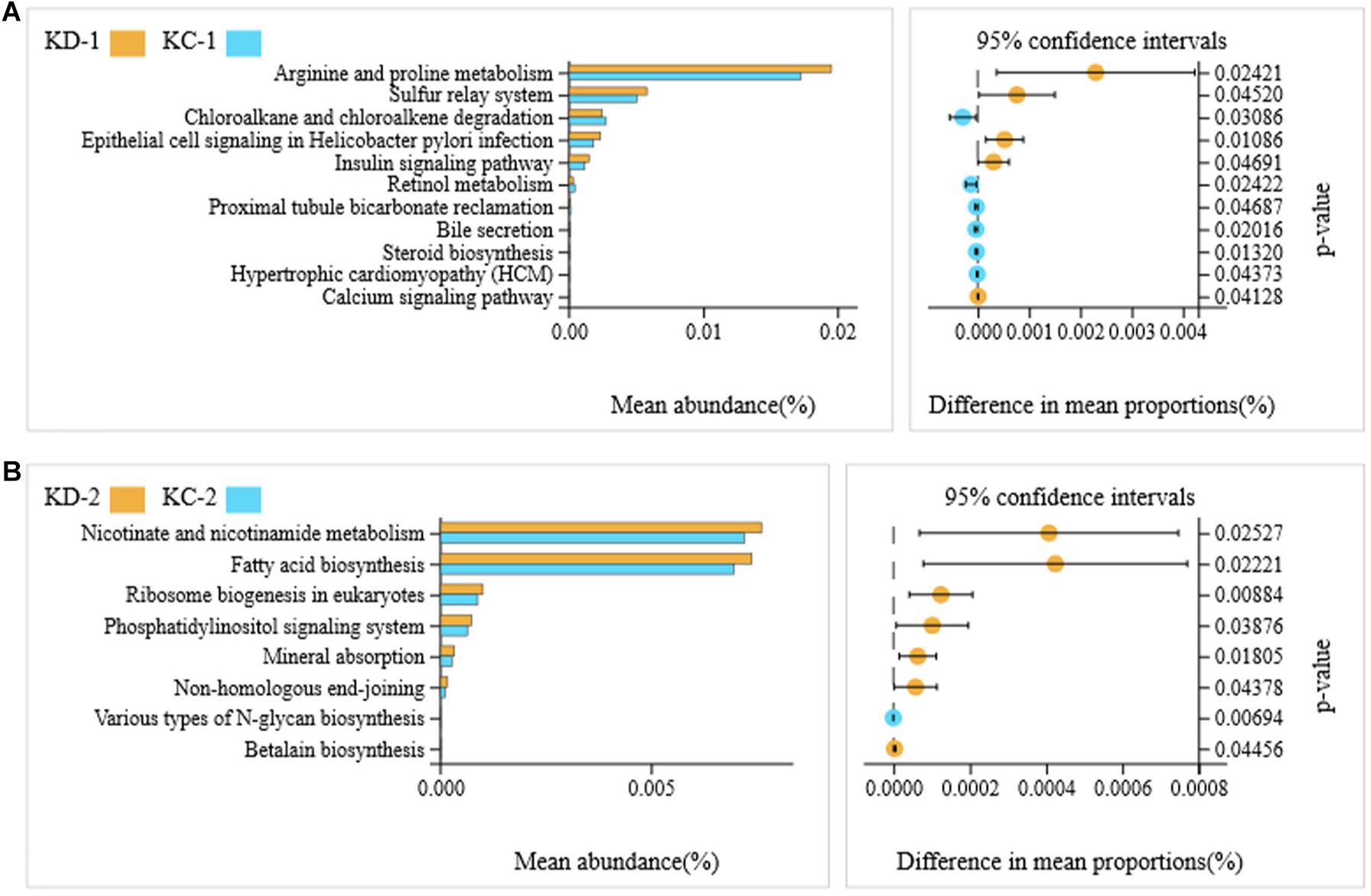
Figure 4. KEGG pathways for bacteria in jejunal meconium of neonatal piglets before suckling. KD-1: bacteria of jejunal meconium of Hoxa1− /− neonatal piglets from the control group. KC-1: bacteria of jejunal meconium of Hoxa1− /− neonatal piglets from the experimental group. KD-2: bacteria of jejunal meconium of non-Hoxa1− /− neonatal piglets from the control group. KC-2: bacteria of jejunal meconium of non-Hoxa1− /− neonatal piglets from the experimental group. (A) Hoxa1–/– piglets. (B) Non-Hoxa1–/–piglets.
A total of 36 microbial metabolic pathways were significantly different between ileal bacteria of non-Hoxa1–/– (HD-2) and Hoxa1–/– (HD-1) neonatal piglets from the control group, and 10 pathways were significantly upregulated and 26 pathways were significantly downregulated in HD-1 compared with HD-2 (Figure 5A). After maternal ATRA administration, 40 microbial metabolic pathways were significantly different between ileal bacteria of non-Hoxa1–/– (HC-2) and Hoxa1–/– (HC-1) neonatal piglets from the experimental group, and 36 pathways were significantly upregulated and 4 pathways were significantly downregulated in HC-1 compared with HC-2 (Figure 5B). HC-1 had significantly lower expression of genes involved in arginine and proline metabolism and significantly higher expression of genes involved in RNA degradation, pyruvate metabolism, renal cell carcinoma, and type II diabetes mellitus than HD-1 (Figure 6A), and HC-2 had significantly higher expression of genes involved in flagellar assembly and Salmonella infection and significantly lower expression of genes involved in peptidoglycan biosynthesis, fatty acid biosynthesis, base excision repair, D-glutamine and D-glutamate metabolism, peroxisome, tuberculosis, peroxisome proliferator-activated receptors (PPAR) signaling pathway, ribosome biogenesis in eukaryotes, adipocytokine signaling pathway, phosphatidylinositol signaling system, steroid degradation, mineral absorption, non-homologous end-joining, renin–angiotensin system, proteasome, sesquiterpenoid and triterpenoid biosynthesis, and biosynthesis of type II polyketide products than HD-2 (Figure 6B).

Figure 5. KEGG pathways for bacteria in ileal meconium of neonatal piglets before suckling. HD-1: bacteria of ileal meconium of Hoxa1− /− neonatal piglets from the control group. HD-2: bacteria of ileal meconium of non-Hoxa1− /− neonatal piglets from the control group. HC−1: bacteria of ileal meconium of Hoxa1− /− neonatal piglets from the experimental group. HC-2: bacteria of ileal meconium of non-Hoxa1− /− neonatal piglets from the experimental group. (A) Control group. (B) Experimental group.
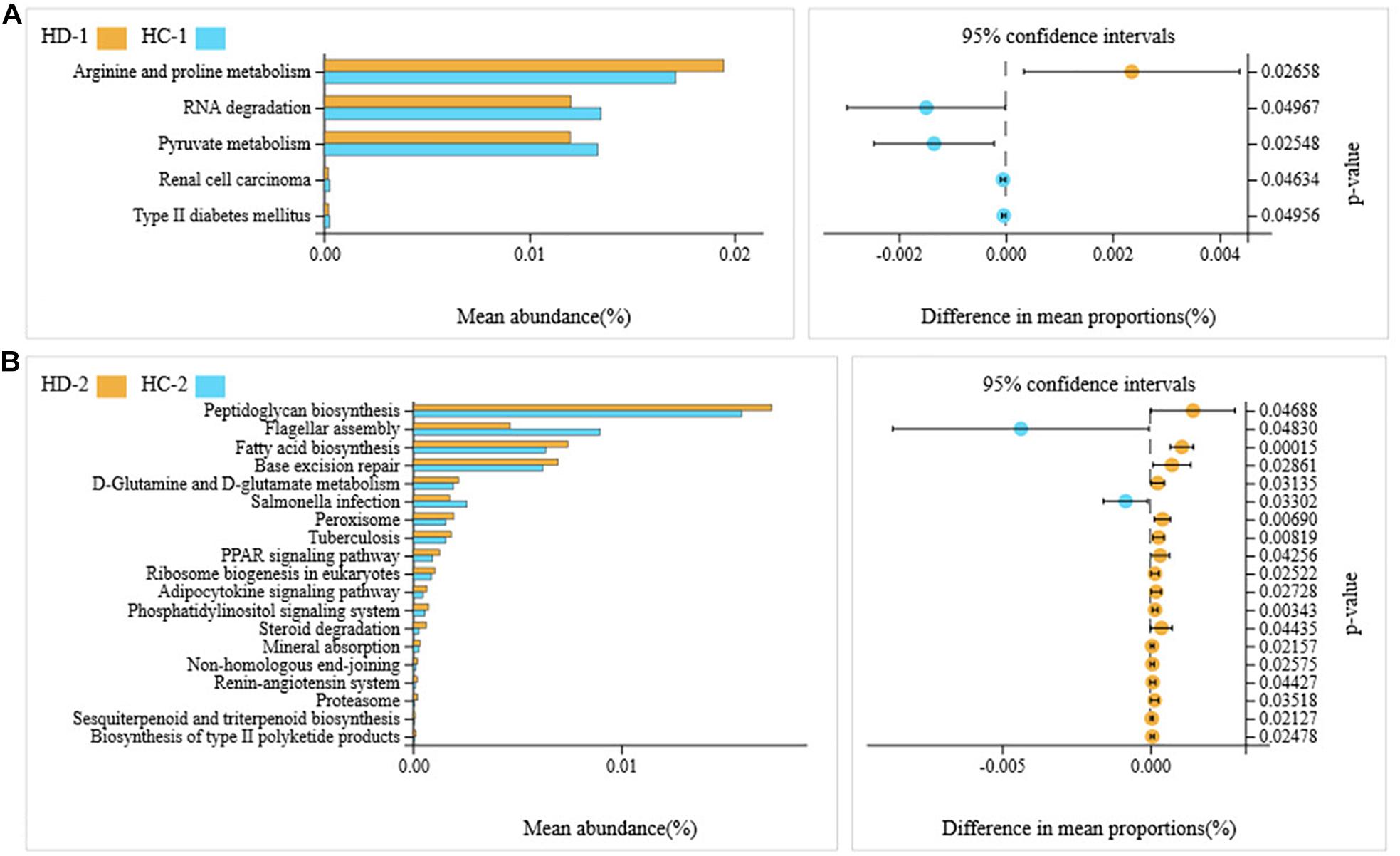
Figure 6. KEGG pathways for bacteria in ileal meconium of neonatal piglets before suckling. HD-1: bacteria of ileal meconium of Hoxa1− /− neonatal piglets from the control group. HC-1: bacteria of ileal meconium of Hoxa1− /− neonatal piglets from the experimental group. HD-2: bacteria of ileal meconium of non-Hoxa1− /− neonatal piglets from the control group. HC-2: bacteria of ileal meconium of non-Hoxa1− /− neonatal piglets from the experimental group. (A) Hoxa1–/– piglets. (B) Non-Hoxa1–/–piglets.
The normal colonization and development of intestinal microbiota is crucial for the normal function of the physiology and immunity of the host (Chung et al., 2012; Furusawa et al., 2013). The highly diverse intestine microbiota is generally considered beneficial for host health and is also regarded as a sign of mature intestine microbiota (Turnbaugh et al., 2007; Le Chatelier et al., 2013), but some studies found that premature development and diversification of the gut microbiota may negatively impact immune function and the highly diverse and rich bacterial community is probably not beneficial for the immature intestinal tract of young animals (Nylund et al., 2013; Wood et al., 2015). The data in our study also indicated that decreasing the number and alpha diversity of small intestinal bacterial community is beneficial to the neonatal piglets, because Hoxa1–/– neonatal piglets delivered by sows with ATRA administration during pregnancy had lower number and alpha diversity of small intestinal bacterial community, heavier birth live weight, and less symptom with dyspnea than Hoxa1–/– neonatal piglets born by sows with no ATRA administration.
Maternal genetics (Goodrich et al., 2014), gene mutation (Strati et al., 2016), delivery methods (Bi et al., 2021; Husso et al., 2021), age (Yatsunenko et al., 2012), disease (Alkanani et al., 2015), diet (David et al., 2014; Angoa-Pérez et al., 2020), and medication (Forslund et al., 2013) are the dominant factors in shaping the composition and abundance of intestinal microbiota. It is reported that the composition of intestinal bacterial communities in human and other mammals after suckling is mainly dominated by the phyla Firmicutes and Bacteroidetes, followed by the phyla Proteobacteria, Actinobacteria, and Verrucomicrobia (Gill et al., 2006; Duncan et al., 2008; Ley et al., 2008); however, the compositions of bacteria in the meconium of fetal bovine and lamb delivered by cesarean section were primarily composed of the phyla Proteobacteria and Firmicutes instead of Firmicutes and Bacteroidetes (Bi et al., 2021; Husso et al., 2021), and data in our study also indicated that Firmicutes and Proteobacteria were the most abundant phyla in the meconium of vaginal-delivered neonatal piglets before suckling.
Gene mutation can cause dysbiosis of gut microbiota by altering the relative abundance of gut microbiota and often develop a wide variety of diseases (Johansson et al., 2011; Alkanani et al., 2015; Strati et al., 2016). Microbial dysbiosis of the gut can destroy body immunity and intestinal mucosal barrier and increase gut permeability via the outgrowth of pathogens or the imbalance production of chemical substances in the gut (Earley et al., 2015). Mutations in the MECP2 gene decreased the relative abundances of Firmicutes and Bacteroidetes and increased the relative abundance of Actinobacteria at the phyla level of patients compared with healthy controls (Strati et al., 2016). Our study showed that Hoxa1 mutation changed the relative abundances of small intestinal microbiota, Hoxa1–/– neonatal piglets had lower relative abundances of phylum Firmicutes and genera Lactobacillus, Clostridium_sensu_stricto_1, Rothia, and Veillonella and higher relative abundances of phyla Proteobacteria, Bacteroidetes, and Verrucomicrobia and genera Escherichia–Shigella and Akkermansia than non-Hoxa1–/– neonate piglets.
The normal ratio of Firmicutes to Bacteroidetes and the relative abundance of microbiota are essential for host health, and gene mutation often changes the Firmicutes to Bacteroidetes ratio and the microbial relative abundance. Tougaard et al. (2015) reported that tumor necrosis factor-like ligand 1A gene knockout mice had lower Firmicutes/Bacteroidetes ratio in the cecum content compared with wild-type mice (Tougaard et al., 2015), and the results of this study also indicated that Hoxa1 mutation resulted in a significantly lower ratio of Firmicutes to Bacteroidetes in the small intestine of Hoxa1–/– neonatal piglets than that of non-Hoxa1–/– neonatal piglets. Alteration in the ratio of Firmicutes to Bacteroidetes and the relative abundances of microbiota often develop into different kinds of diseases in the host. The increased ratio of Firmicutes to Bacteroidetes was reported in obese animals (Bäckhed et al., 2004), and the decreased ratio between Firmicutes and Bacteroidetes was associated with a higher risk of developing type 1 diabetes or celiac disease (Murri et al., 2013; Calderón de la Barca et al., 2020) or autism (Finegold et al., 2010) in children. The combination of significantly decreased ratio of Firmicutes to Bacteroidetes; the lower relative abundances of Firmicutes, Lactobacillus, Clostridium_sensu_stricto_1, Staphylococcus, Rothia, and Veillonella; and the higher relative abundances of Bacteroidetes and Patescibacteria might be some of the important factors to develop in Hoxa1–/– neonatal piglets undesirable symptoms of bad birth live weight, dyspnea, and death. Previous studies reported that the significantly decreased relative abundances of phylum Firmicutes and of genus Lactobacillus were correlated with prenatal stress (Zijlmans et al., 2015), multiple sclerosis (Chen et al., 2016), type 1 diabetes (Murri et al., 2013; Alkanani et al., 2015), and diarrheal disease (Zhuang et al., 2017). The decreased levels of Veillonella, Lachnospira, Rothia, Roseburia, and Faecalibacterium were also associated with asthma in children (Hilty et al., 2010; Hufnagl et al., 2020). Decreasing the numbers of Firmicutes, Lactobacillus, Veillonella, Rothia, and Clostridium_sensu_stricto_1 will increase the growth and colonization of bacterial pathogens (Kim et al., 2017; Vitetta et al., 2017) and weaken gastrointestinal digestion, development, and immune functions (Edwards et al., 2017; Granja-Salcedo et al., 2017). The increased relative abundance of Bacteroidetes can generate more propionate, and the surplus of propionate can lower food intake, increase energy expenditures (Chambers et al., 2015, 2018), and facilitate the absorption of iron (Sfera et al., 2020); chronic iron overload increases the risk of reactive oxygen species and DNA damage (Sfera et al., 2020) and decreases weight gain (Calarge et al., 2016).
Preventing microbial dysbiosis in early life can reduce the risk of diseases such as intrauterine growth retardation (IUGR), allergic asthma, type 1 diabetes, and diarrhea. Studies showed that increased Lactobacillus can modulate gut microbiota dysbiosis (Wu et al., 2019), and early colonization with Lactobacillus during the infant period can reduce the risk of allergic asthma (Johansson et al., 2011). The data of our studies demonstrated that maternal ATRA administration increased the birth live weight of Hoxa1–/– neonatal piglets (Zhou et al., 2021). The increased ratio of Firmicutes to Bacteroidetes; the higher relative abundances of Firmicutes, Proteobacteria, and Lactobacillus; and the lower relative abundance of Bacteroidetes were observed in the meconium of the small intestine of Hoxa1–/– neonatal piglets, and the symptom of respiratory distress was not observed in Hoxa1–/– neonatal piglets delivered by sows with ATRA administration.
Gut microbiota may play important roles in the health and nutrient metabolism of the host, and prenatal colonization of a metabolically active microbiome is clinically vital for the health development of the fetus (Bi et al., 2021). A previous study found that microbiota in the gut of fetal lambs had high enrichment of KEGG pathways related to carbohydrate metabolism, energy metabolism, signal transduction, and amino acid metabolism (Bi et al., 2021). Our study indicated that meconium bacteria of the small intestine of neonatal piglets have functional enrichments mainly in membrane transport (ABC transporters), signal transduction (two-component system), nucleotide metabolism (purine metabolism), translation (aminoacyl-tRNA biosynthesis, ribosome), carbohydrate metabolism (starch and sucrose metabolism, fructose and mannose metabolism, pyruvate metabolism), amino acid metabolism (arginine and proline metabolism; alanine, aspartate, and glutamate metabolism; phenylalanine, tyrosine, and tryptophan biosynthesis), energy metabolism (nitrogen metabolism, oxidative phosphorylation, methane metabolism), and cell motility (bacterial chemotaxis). Hoxa1–/– neonatal piglets had lower functional enrichments of small intestinal bacteria in ABC transporters, purine metabolism, aminoacyl-tRNA biosynthesis, and ribosome and pyruvate metabolism and higher functional enrichments of small intestinal bacteria in two-component system; starch and sucrose metabolism; fructose and mannose metabolism; arginine and proline metabolism; alanine, aspartate, and glutamate metabolism; nitrogen metabolism; and bacterial chemotaxis than non-Hoxa1–/– neonatal piglets. In addition, maternal ATRA administration during pregnancy not only can shape the functional enrichments of small intestinal bacteria of Hoxa1–/– neonatal piglets but also can alter the functional enrichments of small intestinal bacteria of non-Hoxa1–/– neonatal piglets. Data showed that non-Hoxa1–/– neonatal piglets delivered by sows with ATRA administration had significantly lower functional enrichments of small intestinal bacteria in nicotinate and nicotinamide metabolism, fatty acid biosynthesis, ribosome biogenesis in eukaryotes, phosphatidylinositol signaling system, mineral absorption, peptidoglycan biosynthesis, base excision repair, D-glutamine and D-glutamate metabolism, tuberculosis, and PPAR signaling pathway and significantly higher functional enrichments of small intestinal bacteria in flagellar assembly and Salmonella infection than non-Hoxa1–/– neonatal piglets born by sows with no ATRA administration.
Hoxa1 mutation altered the diversity of the bacterial community and the relative abundances of several dominant taxa; Hoxa1–/– neonatal piglets had significantly higher alpha diversity of bacterial community, lower phylum Firmicutes and genus Lactobacillus, and higher phylum Bacteroidetes than non-Hoxa1–/– neonatal piglets in the meconium of the jejunum and ileum. Maternal ATRA administration altered the bacterial diversity and the relative abundances of dominant taxa of Hoxa1–/– and non-Hoxa1–/– neonatal piglets; Hoxa1–/– neonatal piglets delivered by sows with ATRA administration had lower alpha diversity of bacterial community and significantly higher relative abundance of phylum Firmicutes than Hoxa1–/– neonatal piglets born by sows with no ATRA administration, but had higher alpha diversity of bacterial community, higher relative abundances of phyla Proteobacteria and Bacteroidetes, and lower relative abundances of phylum Firmicutes and genus Lactobacillus than non-Hoxa1–/– neonatal piglets delivered by sows with ATRA administration. Compared with non-Honx1–/– neonatal piglets delivered by sows with no ATRA administration, non-Hoxa1–/– neonatal piglets born by sows with ATRA administration had higher alpha diversity of bacterial community and lower relative abundances of the genera Rothia and Moraxella.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
The animal study was reviewed and approved by the Ethics Committee for Animal Experimentation of Jiangxi Agricultural University. Written informed consent was obtained from the owners for the participation of their animals in this study.
YH conceived the study. YH and WL designed the study. HZ, HW, YC, and WZ performed the experiments. HZ performed the data analysis. YH and HZ writing the manuscript. All authors read and approved the final manuscript.
This study was supported by the National Natural Science Foundation of China (31560303).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abu-Libdeh, B., Douiev, L., Amro, S., Shahrour, M., Ta-Shma, A., Miller, C., et al. (2017). Mutation in the COX4I1 gene is associated with short stature, poor weight gain and increased chromosomal breaks, simulating Fanconi anemia. Eur. J. Hum. Genet. 25, 1142–1146. doi: 10.1038/ejhg.2017.112
Ai Dhaibani, M. A., El-Hattab, A. W., Holroyd, K. B., Orthmann-Murphy, J., Larson, V. A., Siddiqui, K. A., et al. (2018). Novel mutation in the KCNJ10 gene in three siblings with seizures, ataxia and no electrolyte abnormalities. J. Neurogenet. 32, 1–5. doi: 10.1080/01677063.2017.1404057
Alasti, F., Sadeghi, A., Sanati, M. H., Farhadi, M., Stollar, E., Somers, T., et al. (2008). A mutation in Hoxa2 is responsible for autosomal-recessive microtia in an Iranian family. Am. J. Hum. Genet. 82, 982–991. doi: 10.1016/j.ajhg.2008.02.015
Alipour, M. J., Jalanka, J., Pessa-Morikawa, T., Kokkonen, T., Satokari, R., Hynönen, U., et al. (2018). The composition of the perinatal intestinal microbiota in cattle. Sci. Rep. 8:10437. doi: 10.1038/s41598-018-31494-3
Alkanani, A. K., Hara, N., Gottlieb, P. A., Ir, D., Robertson, C. E., Wagner, B. D., et al. (2015). Alterations in intestinal microbiota correlate with susceptibility to type 1 diabetes. Diabetes 64, 3510–3520. doi: 10.2337/db14-1847
Angoa-Pérez, M., Zagorac, B., Francescutti, D. M., Winters, A. D., Greenberg, J., Ahmad, M. M., et al. (2020). Effects of a high fat diet on gut microbiome dysbiosis in a mouse model of gulf war illness. Sci. Rep. 10:9529. doi: 10.1038/s41598-020-66833-w
Aßhauer, K. P., Wemheuer, B., Daniel, R., and Meinicke, P. (2015). Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31, 2882–2884. doi: 10.1093/bioinformatics/btv287
Bäckhed, F., Ding, H., Wang, T., Hooper, L. V., Koh, G. Y., Nagy, A., et al. (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U. S. A. 101, 15718–15723. doi: 10.1073/pnas.0407076101
Ben Salem, H., Nefzaoui, A., Makkar, H. P. S., Hochlef, H., Ben Salem, I., and Ben Salem, L. (2005). Effect of early experience and adaptation period on voluntary intake, digestion, and growth in Barbarine lambs given tannin-containing (Acacia cyanophylla Lindl. foliage) or tannin-free (oaten hay) diets. Anim. Feed Sci. Technol. 122, 59–77. doi: 10.1016/j.anifeedsci.2005.04.014
Bi, Y. L., Tu, Y., Zhang, N. F., Wang, S. Q., Zhang, F., Suen, G., et al. (2021). Multiomics analysis reveals the presence of a microbiome in the gut of fetal lambs. Gut 70, 853–864. doi: 10.1136/gutjnl-2020-320951
Calarge, C. A., Murry, D. J., Ziegler, E. E., and Arnold, L. E. (2016). Serum ferritin, weight gain, disruptive behavior, and extrapyramidal symptoms in risperidone-treated youth. J. Child Adolesc. Psychopharmacol. 26, 471–477. doi: 10.1089/cap.2015.0194
Calderón de la Barca, A. M., Castillo-Fimbres, R. S., Mejía-León, M. E., Quihui-Cota, L., Ochoa-Leyva, A., and Aguayo-Patrón, S. V. (2020). Enteric parasitic infection disturbs bacterial structure in Mexican children with autoantibodies for type 1 diabetes and/or celiac disease. Gut Pathog. 12:37. doi: 10.1186/s13099-020-00376-3
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Chambers, E. S., Byrne, C. S., Aspey, K., Chen, Y. J., Khan, S., Morrison, D. J., et al. (2018). Acute oral sodium propionate supplementation raises resting energy expenditure and lipid oxidation in fasted humans. Diabetes Obes. Metab. 20, 1034–1039. doi: 10.1111/dom.13159
Chambers, E. S., Viardot, A., Psichas, A., Morrison, D. J., Murphy, K. G., Zac-Varghese, S. E. K., et al. (2015). Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 64, 1744–1754. doi: 10.1136/gutjnl-2014-307913
Chen, J., Chia, N., Kalari, K. R., Yao, J. Z., Novotna, M., Soldan, M. M. P., et al. (2016). Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 6:28484. doi: 10.1038/srep28484
Chen, S. F., Zhou, Y. Q., Chen, Y. R., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chung, H., Pamp, S. J., Hill, J. A., Surana, N. K., Edelman, S. M., Troy, E. B., et al. (2012). Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149, 1578–1593. doi: 10.1016/j.cell.2012.04.037
David, L. A., Maurice, C. F., Carmody, R. N., Gootenberg, D. B., Button, J. E., Wolfe, B. E., et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. doi: 10.1038/nature12820
Duncan, S. H., Lobley, G. E., Holtrop, G., Ince, J., Johnstone, A. M., Louis, P., et al. (2008). Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. 32, 1720–1724. doi: 10.1038/ijo.2008.155
Earley, Z. M., Akhtar, S., Green, S. J., Naqib, A., Khan, O., Cannon, A. R., et al. (2015). Burn injury alters the intestinal microbiome and increases gut permeability and bacterial translocation. PLoS One 10:e0129996. doi: 10.1371/journal.pone.0129996
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edwards, J. E., Forster, R. J., Callaghan, T. M., Dollhofer, V., Dagar, S. S., Cheng, Y. F., et al. (2017). PCR and omics based techniques to study the diversity, ecology and biology of anaerobic fungi: insights, challenges and opportunities. Front. Microbiol. 8:1657. doi: 10.3389/fmicb.2017.01657
Finegold, S. M., Dowd, S. E., Gontcharova, V., Liu, C. X., Henley, K. E., Wolcott, R. D., et al. (2010). Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 16, 444–453. doi: 10.1016/j.anaerobe.2010.06.008
Forslund, K., Sunagawa, S., Kultima, J. R., Mende, D. R., Arumugam, M., Typas, A., et al. (2013). Country-specific antibiotic use practices impact the human gut resistome. Genome Res. 23, 1163–1169. doi: 10.1101/gr.155465.113
Furusawa, Y., Obata, Y., Fukuda, S., Endo, T. A., Nakato, G., Takahashi, D., et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. doi: 10.1038/nature12721
Giancotti, A., Ambrosio, V. D., De Filippis, A., Aliberti, C., Pasquali, G., Bernardo, S., et al. (2014). Comparison of ultrasound and magnetic resonance imaging in the prenatal diagnosis of Apert syndrome: report of a case. Childs Nerv. Syst. 30, 1445–1448. doi: 10.1007/s00381-014-2377-8
Gill, S. R., Pop, M., Deboy, R. T., Eckburg, P. B., Turnbaugh, P. J., Samuel, B. S., et al. (2006). Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359. doi: 10.1126/science.1124234
Goodrich, J. K., Waters, J. L., Poole, A. C., Sutter, J. L., Koren, O., Blekhman, R., et al. (2014). Human genetics shape the gut microbiome. Cell 159, 789–799. doi: 10.1016/j.cell.2014.09.053
Granja-Salcedo, Y. T., Messana, J. D., de Souza, V. C., Dias, A. V. L., Kishi, L. T., Rebelo, L. R., et al. (2017). Effects of partial replacement of maize in the diet with crude glycerin and/or soyabean oil on ruminal fermentation and microbial population in Nellore steers. Br. J. Nutr. 118, 651–660. doi: 10.1017/s0007114517002689
Hampe, J., Cuthbert, A., Croucher, P. J., Mirza, M. M., Mascheretti, S., Fisher, S., et al. (2001). Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet 357, 1925–1928. doi: 10.1016/S0140-6736(00)05063-7
Hilty, M., Burke, C., Pedro, H., Cardenas, P., Bush, A., Bossley, C., et al. (2010). Disordered microbial communities in asthmatic airways. PLoS One 5:e8578. doi: 10.1371/journal.pone.0008578
Hufnagl, K., Pali-Schöll, I., Roth-Walter, F., and Jensen-Jarolim, E. (2020). Dysbiosis of the gut and lung microbiome has a role in asthma. Semin. Immunopathol. 42, 75–93. doi: 10.1007/s00281-019-00775-y
Hummel, G. L., Woodruff, K. L., Austin, K. J., Smith, T. L., and Cunningham-Hollinger, H. C. (2020). Evidence for the amnion-fetal gut-microbial axis in late gestation beef calves. Transl. Anim. Sci. 4, S174–S177. doi: 10.1093/tas/txaa138
Husso, A., Lietaer, L., Pessa-Morikawa, T., Grönthal, T., Govaere, J., Soom, A. V., et al. (2021). The composition of the microbiota in the full-term fetal gut and amniotic fluid: a bovine caesarean section study. Front. Microbiol. 12:626421. doi: 10.3389/fmicb.2021.626421
Jelani, M., Dooley, H. C., Gubas, A., Sheikh, H., Mohamoud, H. S. A., Masood, T., et al. (2019). A mutation in the major autophagy gene, WIPI2, associated with global developmental abnormalities. Brain 142, 1242–1254. doi: 10.1093/brain/awz075
Johansson, M. A., Sjögren, Y. M., Persson, J. O., Nilsson, C., and Sverremark-Ekström, E. (2011). Early colonization with a group of Lactobacilli decreases the risk for allergy at five years of age despite allergic heredity. PLoS One 6:e23031. doi: 10.1371/journal.pone.0023031
Johnson, K. R., Gagnon, L. H., and Chang, B. (2016). A hypomorphic mutation of the gamma-1 adaptin gene (Ap1g1) causes inner ear, retina, thyroid, and testes abnormalities in mice. Mamm. Genome 27, 200–212. doi: 10.1007/s00335-016-9632-0
Joossens, M., Huys, G., Cnockaert, M., Cnockaert, M., De Preter, V., Verbeke, K., et al. (2011). Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 60, 631–637. doi: 10.1136/gut.2010.223263
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2015). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kim, Y. G., Sakamoto, K., Seo, S. U., Pickard, J. M., Gillilland, M. G., Pudlo, N. A., et al. (2017). Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science 356, 315–319. doi: 10.1126/science.aag2029
Le Chatelier, E., Nielsen, T., Qin, J., Prifti, E., Hildebrand, F., Falony, G., et al. (2013). Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546. doi: 10.1038/nature12506
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Lynch, S. V., Goldfarb, K. C., Wild, Y. K., Kong, W. D., De Lisle, R. C., and Brodie, E. L. (2013). Cystic fibrosis transmembrane conductance regulator knockout mice exhibit aberrant gastrointestinal microbiota. Gut Microbes 4, 41–47. doi: 10.4161/gmic.22430
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Morgavi, D. P., Rathahao-Paris, E., Popova, M., Boccard, J., Nielsen, K. F., and Boudra, H. (2015). Rumen microbial communities influence metabolic phenotypes in lambs. Front. Microbiol. 6:1060. doi: 10.3389/fmicb.2015.01060
Murri, M., Leiva, I., Gomez-Zumaquero, J. M., Tinahones, F. J., Cardona, F., Soriguer, F., et al. (2013). Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 11:46. doi: 10.1186/1741-7015-11-46
Murthy, V., Tebaldi, T., Yoshida, T., Erdin, S., Calzonetti, T., Vijayvargia, R., et al. (2019). Hypomorphic mutation of the mouse Huntington’s disease gene orthologue. PLoS Genet. 15:e1007765. doi: 10.1371/journal.pgen.1007765
Nonose, R. W., Lezirovitz, K., de Mello Auricchio, M. T. B., Batissoco, A. C., Yamamoto, G. L., and Mingroni-Netto, R. C. (2018). Mutation analysis of SLC26A4 (Pendrin) gene in a Brazilian sample of hearing-impaired subjects. BMC Med. Genet. 19:73. doi: 10.1186/s12881-018-0585-x
Nylund, L., Satokari, R., Nikkila, J., Rajilić-Stojanović, M., Kalliomäki, M., Isolauri, E., et al. (2013). Microarray analysis reveals marked intestinal microbiota aberrancy in infants having eczema compared to healthy children in at-risk for atopic disease. BMC Microbiol. 13:12. doi: 10.1186/1471-2180-13-12
Ogura, Y., Bonen, D. K., Inohara, N., Nicolae, D. L., Chen, F. F., Ramos, R., et al. (2001). A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606. doi: 10.1038/35079114
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., and Minchin, P. R. (2010). Vegan: Community Ecology Package. R Package Version 1.17-4.
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385
Pasqualetti, M., Neun, R., Davenne, M., and Rijli, F. M. (2001). Retinoic acid rescues inner ear defects in Hoxa1 deficient mice. Nat. Genet. 29, 34–39. doi: 10.1038/ng702
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J. X., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Qiao, R. M., He, Y. Y., Pan, B., Xiao, S. J., Zhang, X. F., Li, J., et al. (2015). Understanding the molecular mechanisms of human microtia via a pig model of HOXA1 syndrome. Dis. Model Mech. 8, 611–622. doi: 10.1242/dmm.018291
Sfera, A., Osorio, C., Diaz, E. L., Maguire, G., and Cummings, M. (2020). The other obesity epidemic-of drugs and bugs. Front. Endocrinol. 11:488. doi: 10.3389/fendo.2020.00488
Stinson, L. F., Boyce, M. C., Payne, M. S., and Keelan, J. A. (2019). The not-so-sterile womb: evidence that the human fetus is exposed to bacteria prior to birth. Front. Microbiol. 10:1124. doi: 10.3389/fmicb.2019.01124
Strati, F., Cavalieri, D., Albanese, D., De Felice, C., Donati, C., Hayek, J., et al. (2016). Altered gut microbiota in Rett syndrome. Microbiome 4:41. doi: 10.1186/s40168-016-0185-y
Tougaard, P., Skov, S., Pedersen, A. E., Krych, L., Nielsen, D. S., Bahl, M. I., et al. (2015). TL1A regulates TCRγδ+ intraepithelial lymphocytes and gut microbial composition. Eur. J. Immunol. 45, 865–875. doi: 10.1002/eji.201444528
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007). The human microbiome project. Nature 449, 804–810. doi: 10.1038/nature06244
Vitetta, L., Coulson, S., Thomsen, M., Nguyen, T., and Hall, S. (2017). Probiotics, D-Lactic acidosis, oxidative stress and strain specificity. Gut Microbes 8, 311–322. doi: 10.1080/19490976.2017.1279379
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microb. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wood, K. M., Palmer, S. I., Steele, M. A., Metcalf, J. A., and Penner, G. B. (2015). The influence of age and weaning on permeability of the gastrointestinal tract in Holstein bull calves. J. Dairy Sci. 98, 7226–7237. doi: 10.3168/jds.2015-9393
Wu, C. H., Ko, J. L., Liao, J. M., Huang, S. S., Lin, M. Y., Lee, L. H., et al. (2019). D-methionine alleviates cisplatin-induced mucositis by restoring the gut microbiota structure and improving intestinal inflammation. Ther. Adv. Med. Oncol. 11, 1–18. doi: 10.1177/1758835918821021
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. doi: 10.1038/nature11053
Zhou, H. M., Chen, Y. X., Hu, Y. Q., Gao, S., Lu, W., and He, Y. Y. (2021). Administration of all-Trans retinoic acid to pregnant sows improves the developmental defects of Hoxa1–/– fetal pigs. Front. Vet. Sci. 7:618660. doi: 10.3389/fvets.2020.618660
Zhuang, X., Xiong, L., Li, L., Li, M. Y., and Chen, M. H. (2017). Alterations of gut microbiota in patients with irritable bowel syndrome: a systematic review and meta-analysis. J.Gastroenterol. Hepatol. 32, 28–38. doi: 10.1111/jgh.13471
Keywords: Hoxa1 mutation, all-trans retinoic acid, pregnant sows, gut bacterial community, neonatal piglets
Citation: Zhou H, Wu H, Chen Y, Zou W, Lu W and He Y (2021) Administration of All-Trans Retinoic Acid to Pregnant Sows Alters Gut Bacterial Community of Neonatal Piglets With Different Hoxa1 Genotypes. Front. Microbiol. 12:712212. doi: 10.3389/fmicb.2021.712212
Received: 20 May 2021; Accepted: 29 June 2021;
Published: 26 July 2021.
Edited by:
Peng Huang, Hunan Agricultural University, ChinaReviewed by:
Xiangfeng Kong, Institute of Subtropical Agriculture, Chinese Academy of Sciences, ChinaCopyright © 2021 Zhou, Wu, Chen, Zou, Lu and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuyong He, d2xraDEwMTJAMTYzLmNvbQ==; Wei Lu, bHcyMDAzMDUwOEAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.