
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 15 July 2021
Sec. Microbial Physiology and Metabolism
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.711359
 Ana Nogal1†
Ana Nogal1† Panayiotis Louca1†
Panayiotis Louca1† Xinyuan Zhang1Philippa M. Wells1Claire J. Steves1Tim D. Spector1†
Xinyuan Zhang1Philippa M. Wells1Claire J. Steves1Tim D. Spector1† Mario Falchi1
Mario Falchi1 Ana M. Valdes1,2†
Ana M. Valdes1,2† Cristina Menni1*†
Cristina Menni1*†Background: Acetate is a short-chain fatty acid (SCFA) produced by gut bacteria, which has been implicated in cardio-metabolic health. Here we examine the relationships of circulating acetate levels with gut microbiome composition and diversity and with visceral fat in a large population-based cohort.
Results: Microbiome alpha-diversity was positively correlated with circulating acetate levels (Shannon, Beta [95%CI] = 0.12 [0.06, 0.18], P = 0.002) after adjustment for covariates. Six serum acetate-associated bacterial genera were also identified, including positive correlations with Coprococcus, Barnesiella, Ruminococcus, and Ruminococcaceae NK4A21 and negative correlations were observed with Lachnoclostridium and Bacteroides. We also identified a correlation between visceral fat and serum acetate levels (Beta [95%CI] = −0.07 [−0.11, −0.04], P = 2.8 × 10–4) and between visceral fat and Lachnoclostridium (Beta [95%CI] = 0.076 [0.042, 0.11], P = 1.44 × 10–5). Formal mediation analysis revealed that acetate mediates ∼10% of the total effect of Lachnoclostridium on visceral fat. The taxonomic diversity showed that Lachnoclostridium and Coprococcus comprise at least 18 and 9 species, respectively, including novel bacterial species. By predicting the functional capabilities, we found that Coprococcus spp. present pathways involved in acetate production and metabolism of vitamins B, whereas we identified pathways related to the biosynthesis of trimethylamine (TMA) and CDP-diacylglycerol in Lachnoclostridium spp.
Conclusions: Our data indicates that gut microbiota composition and diversity may influence circulating acetate levels and that acetate might exert benefits on certain cardio-metabolic disease risk by decreasing visceral fat. Coprococcus may play an important role in host health by its production of vitamins B and SCFAs, whereas Lachnoclostridium might have an opposing effect by influencing negatively the circulating levels of acetate and being involved in the biosynthesis of detrimental lipid compounds.
Acetate is a short-chain fatty acid (SCFA) produced by colonic bacteria through the saccharolytic fermentation of fibres (e.g., resistant starch, polysaccharides and simple sugars), which escape digestion and absorption (Topping and Clifton, 2001). The molar ratio of acetate in the colon is three times larger than that of the two other major SCFAs, butyrate and propionate (Cummings et al., 1987). Enteric bacteria, including Ruminococcus spp., Prevotella spp., Bifidobacterium spp., and Akkermansia muciniphila are suggested to be the main acetate-producing bacteria (Rey et al., 2010).
Recently SCFAs have received increasing attention as they have been shown to play an important role in cardio-metabolic diseases (CMD), including obesity, type-2 diabetes (T2D), arterial stiffness and atherosclerosis (Den Besten et al., 2013). Once these bacteria-derived metabolites are synthetised, they have the capacity to reach different systematic tissues, improving the gut barrier integrity, glucose, cholesterol and lipid metabolism, and regulating the immune system and anti-inflammatory response, energy intake, and blood pressure (Martin-Gallausiaux et al., 2020). For instance, acetate was shown to decrease appetite by impacting directly on the hypothalamus (Frost et al., 2014), inhibit endogenous lipolysis (Hron et al., 1978), enhance hepatic uptake of blood cholesterol (Zhao Y. et al., 2017) and reduce hyperglycaemia (Sakakibara et al., 2006). However, to gain further insight into the host-microbial cross-talk involving circulating acetate levels and its implications in cardio-metabolic health (CMH), it is important to integrate different types of data.
In this study, we analyzed the associations between circulating acetate levels, gut microbiome composition and diversity and visceral fat in a cohort of 948 women from TwinsUK. Furthermore, by performing genomic analyses, we have explored the phylogenetic diversity and metabolic complexity of the acetate-associated gut genera.
Study subjects were female twins enrolled in the TwinsUK registry, a national register of adult twins recruited as volunteers without selecting for any particular disease or trait (Moayyeri et al., 2013). In this study, we analyzed data from 948 female twins with concurrent measures of 16S gut microbiome composition, serum acetate levels and visceral fat. The study was approved by NRES Committee London–Westminster, and all twins provided informed written consent. A flowchart of the study design is presented in Figure 1A.
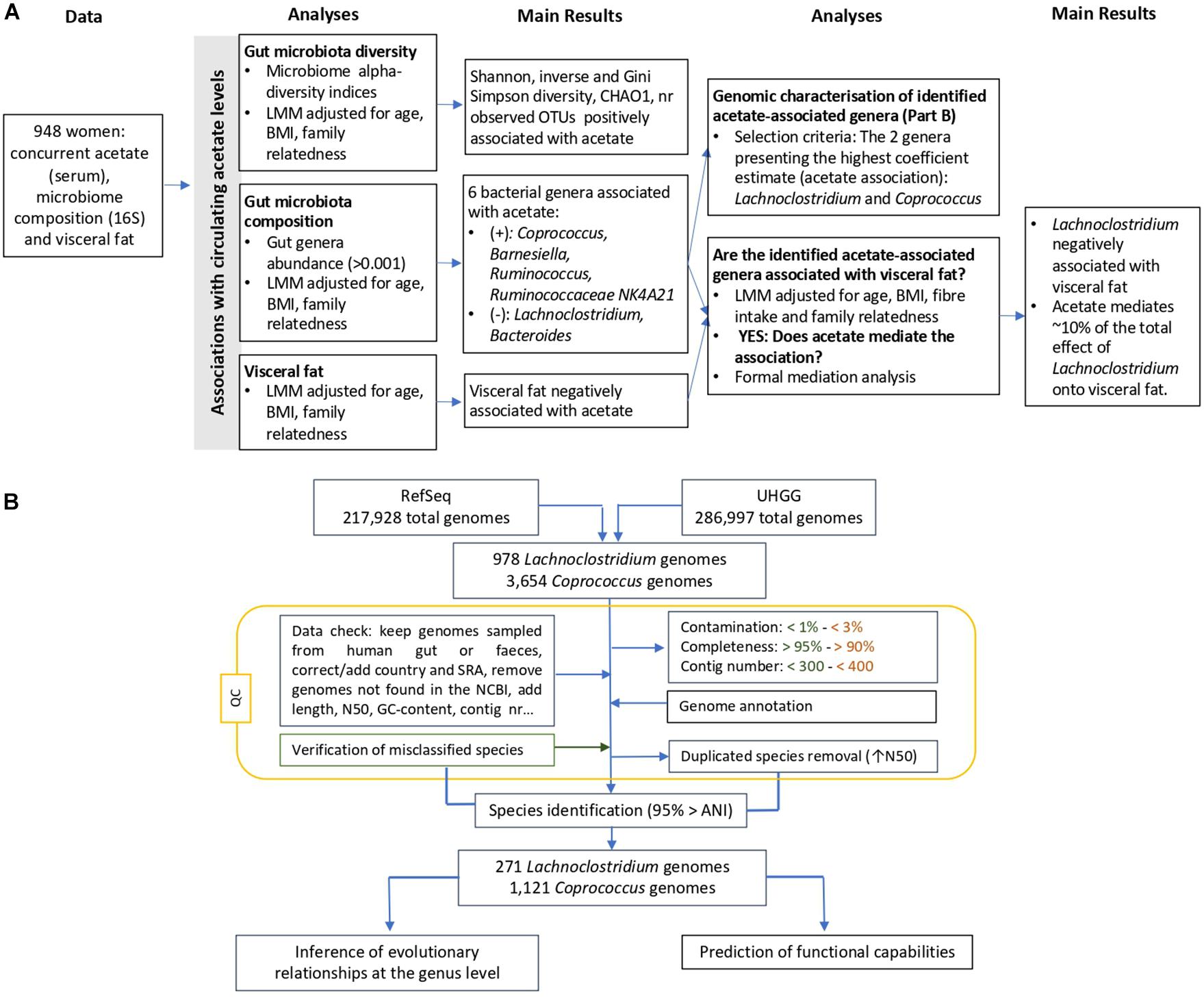
Figure 1. Overview of the flowchart (A) integrating gut microbiota composition and diversity, visceral fat and circulating acetate levels, and (B) showing the applied filters and conducted steps to genomically characterize the Lachnoclostridium spp. and Coprococcus spp. Steps exclusively applied for Lachnoclostridium and Coprococcus are indicated in orange and green, respectively, whereas the rest of steps were conducted in the genomes from both species. ANI, average nucleotide identity; LMM, linear mixed model; UHGG, Unified Human Gastrointestinal Genome; QC, quality control.
Fecal samples were collected and the composition of the gut microbiome was determined by 16S rRNA gene sequencing carried out as previously described (Goodrich et al., 2016). Briefly, the V4 region of the 16S rRNA gene was amplified and sequenced on Illumina MiSeq. 16S sequences were demultiplexed in QIIME 1 (Caporaso et al., 2010). The following analyses were conducted in RStudio version 1.3.1093. Amplicon sequence variants (ASV) were then generated using the “DADA2” R package following the pipeline described by Wells and colleagues (Wells et al., 2020). The ASVs were grouped into genera and the samples with less than 10,000 reads were discarded. The indices of microbiome alpha-diversity, quantified as Shannon, inverse Simpson, Gini Simpson diversity, CHAO1 and number of observed ASVs were calculated using the “microbiome” package (Lahti and Shetty, 2018).
Circulating levels of acetate were measured from serum by Nightingale Health Ltd. (Helsinki, Finland; previously known as Brainshake Ltd.) using a targeted NMR spectroscopy platform that has been extensively applied for biomarker profiling in epidemiological studies (Würtz et al., 2015) as previously described (Barrios et al., 2018).
Measurements of whole body composition were performed for 948 female twins aged 48 to 87 years using the DXA fan-beam technology (Hologic QDR; Hologic, Inc., Waltham, MA, United States) as was indicated by Menni and colleagues (Menni et al., 2016). This DXA-based measurement has been validated against VF measured by CT scan (Kaul et al., 2012) and shown to be reliable and reproducible.
Briefly, subjects were positioned in a supine position wearing only a gown. The DXA machine was calibrated following the manufacturer’s suggestions. The scans were analyzed using the QDR System Software v12.6. Regions of interest were defined manually by the same operator following the SOP (derived from the manufacturer’s guidelines). The lower and upper horizontal margins were placed just above the iliac crest and at the half of the distance between the acromions and the iliac crest, respectively. The vertical margins were adjusted at the external body borders so that all the soft tissue was included.
A validated 131-item semi-quantitative Food Frequency Questionnaire (FFQ) established for the EPIC (European Prospective Investigations into Cancer and Nutrition)-Norfolk study (Bingham et al., 2001) was used to assess dietary intake. Estimated intakes of fiber (in grams per day) were derived from the UK Nutrient Database (McCance and Widdowson, 2014) and were adjusted for energy intake using the residual method prior to analysis (Willett and Stampfer, 1986).
Statistical analyses were conducted in RStudio version 1.3.1093. We assessed the association between circulating acetate and (i) indices of alpha-diversity (Shannon, inverse Simpson, Gini Simpson, CHAO1, and number of observed OTUs), (ii) gut bacterial genera abundance (genera with abundance >0.001), (iii) visceral fat using linear mixed model adjusting for age, BMI, family relatedness and multiple testing using false discovery rate [Benjamini and Hochberg (Thissen et al., 2002)]. Indices of alpha diversity were also adjusted for sequencing depth. Then, linear mixed models were further employed to investigate the association between visceral fat and any acetate-associated genera. All variables included in the models were Z-score normalized.
Finally, we employed mediation analysis as implemented in the R package “mediation” (Tingley et al., 2014) with 1,000 Monte Carlo draws for a quasi-Bayesian approximation, to test the mediation effects of acetate (indirect effect) on the total effect of Lachnoclostridium on visceral fat adjusting for BMI, age and fiber intake. We constructed a mediation model to quantify both the direct effect Lachnoclostridium on visceral fat and the indirect (mediated) effects mentioned above. The variance accounted for (VAF) score, which represents the ratio of indirect-to-total effect and determines the proportion of the variance explained by the mediation process, was further used to determine the significance of mediation effect.
A flowchart of the steps conducted for the genomic characterisation is presented in Figure 1B.
Genomes belonging to the acetate-associated gut genera (Lachnoclostridium and Coprococcus) and their corresponding metadata were obtained from the UHGG catalog and RefSeq dataset (January, 2021), respectively (Almeida et al., 2020). We removed the RefSeq genomes derived from metagenomes and not sampled from human faeces, stool or the gastrointestinal tract, Inconsistencies related to the variable country were corrected and the missing sample accessions were added. Genomes from sample identifiers not found in the National Center for Biotechnology Information (NCBI) (Sayers et al., 2019) were discarded. The two datasets were merge and we then filtered by completeness, contamination and number of contigs (>90%, <3%, and <400 for Lachnoclostridium and >95%, <1%, and <300 for Coprococcus). The thresholds in Lachnoclostridium were less strict due to the scarcity of genomes presenting higher standards. Duplicated genomes were discarded, keeping the one with the highest N50 value. In total, we downloaded 271 Lachnoclostridium and 1,121 Coprococcus high-quality genomes (Supplementary Table 1). Finally, genomes from uncharacterized species or misclassified species were renamed based on the cluster given by fastANI classification (see section “Materials and Methods”).
Completeness and contamination were estimated with CheckM version 1.1.3 (Parks et al., 2015) using the “lineage_wf” workflow. QUAST version 5.0.2 (Gurevich et al., 2013) was run to retrieve the total length, GC-content, contig number and N50. Genome annotation was performed using Prokka version 1.12 (Seemann, 2014) using the default parameters.
FastANI version 1.32 (Jain et al., 2018) was separately run on Lachnoclostridium and Coprococcus genomes to calculate the average nucleotide identity (ANI) between all pairs of sequences (Supplementary Tables 2, 3, 4). Results were filtered by the alignment fraction (>0.4), and symmetric pairwise ANI dissimilarities (100-95, ANI = 95%) were calculated from the ANI values to construct a dendrogram for each genus using the single linkage hierarchical clustering method [“hclust” R function, stats package (R Core Team and DC, 2019)]. Two networks analyses based on the information given by the dendrograms were conducted using the “layoutwithdrl” layout implemented in the “igraph” R package (Csardi and Nepusz, 2006) with an expansion and simmer attraction of 0, and an innit, liquid and crunch temperature of 100, 50, and 50, respectively.
The inconsistencies in the taxonomic classification were verified using BLASTn. For that, barrnap v0.9 was run to predict the 16S rRNA sequences of genomes from C. eutactus, C. sp. BIOML-A2, C. sp. BIOML-A1, C. sp. NSJ-10, C. sp900066115, and C. sp000154245. These were used as query and subject to perform a BLASTn search. The matches were filtered by 99% of identity and a query cover of 50%.
Evolutionary relationships among the Coprococcus and Lachnoclostridium species were inferred using ezTree version 0.1 (Wu, 2018). For each species, up to three genomes (depending on the number of available genomes) sequenced from isolates were used as input. If genomes sequenced from isolates were not available, then the metagenome-assembled genomes (MAGs) with the highest completeness percentage were selected.
Metabolic Pathway Database (Metacyc) (Caspi et al., 2018) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2014) information for each genome was retrieved using the enzyme commission numbers from the gff files generated by Prokka and MinPath (Minimal set of Pathways) (Ye and Doak, 2009; Supplementary Table 5). For Coprococcus spp, only the KEGG and MetaCyc pathways related to metabolism, and fermentation, biosynthesis and degradation, respectively, were kept. C. sp6 was not included in the analyses due to its scarcity of genomes (n = 1). The retrieved information was utilized to construct heatmaps [“Heatmap” R function implemented in the “ComplexHeatmap” package (Gu et al., 2016)] showing the genome percentage of each species with a given pathway. For KEGG data, only the highly different pathways between species were selected (for a given pathway, at least one species has a percentage <5% and another species has a percentage >80%). Moreover, a principal component analysis (PCA) was performed using the presence/absence matrix with the MetaCyc biosynthesis/degradation pathways using the “prcomp” R function within the “stats” package. For the three major species of Lachnoclostridium (species with >15 genomes), only the MetaCyc pathways related to the lipid metabolism were selected and utilized to construct a heatmap as previously indicated.
The descriptive characteristics of the study participants are depicted in Table 1. Overall, 948 women were included, aged between 48 and 87 years, with an average BMI of 26.2 km/m2 (SD = 4.9) and concurrent measures of serum acetate levels, 16S microbiome data and visceral fat.
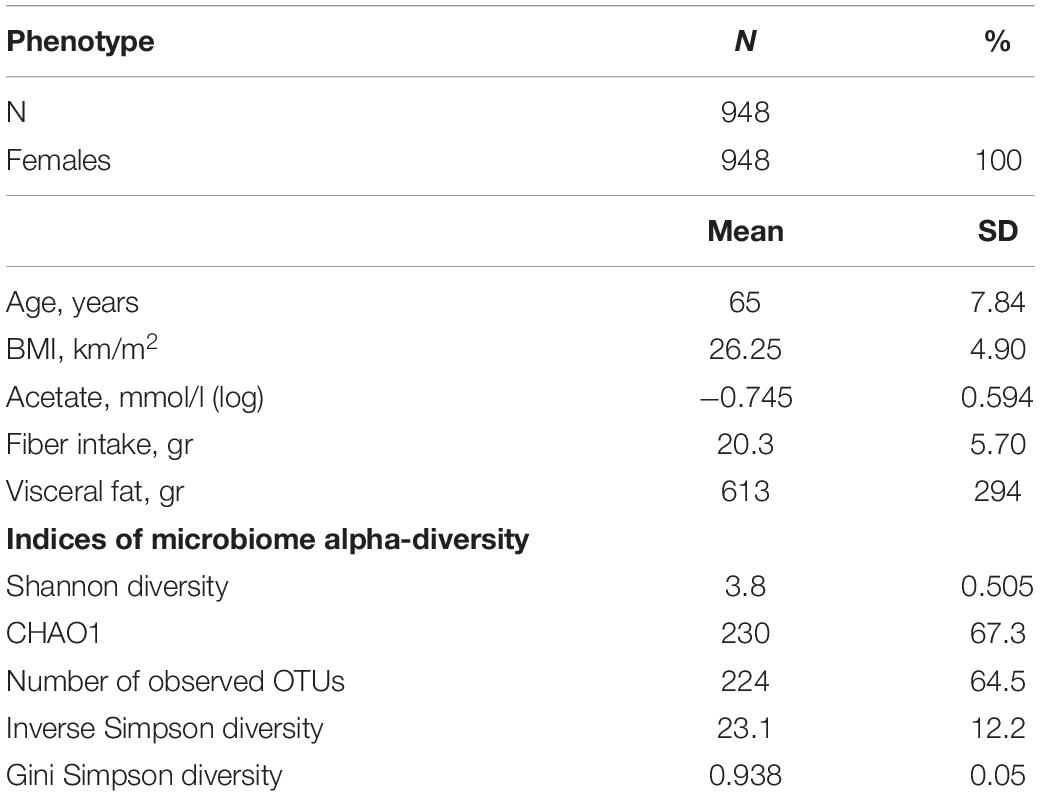
Table 1. Descriptive characteristics of the study population.
As shown in Figure 2, circulating acetate levels were positively correlated with several measures of microbiome alpha-diversity, including Shannon (Beta [95%CI] = 0.12 [0.06, 0.18], P = 0.002), CHAO1 (Beta [95%CI] = 0.14 [0.06, 0.21], P = 0.002), number of observed OTUs (Beta [95%CI] = 0.13 [0.06, 0.21], P = 0.002), inverse Simpson (Beta [95%CI] = 0.095 [0.03, 0.16], P = 0.009) and Gini Simpson (Beta [95%CI] = 0.083 [0.021, 0.15], P = 0.02). We then examined the association between acetate and bacterial genera abundances (genera with abundance >0.001). We identified six genera significantly associated with acetate levels after adjusting for age, BMI, family relatedness and multiple testing using FDR correction (FDR < 0.05) (Figure 2). These include Coprococcus, Barnesiella, Ruminococcus, and Ruminococcaceae NK4A214 positively associated with acetate levels and two genera negatively associated, namely, Lachnoclostridium and Bacteroides. Among them, Lachnoclostridium presented the most robust association (P = 0.006).
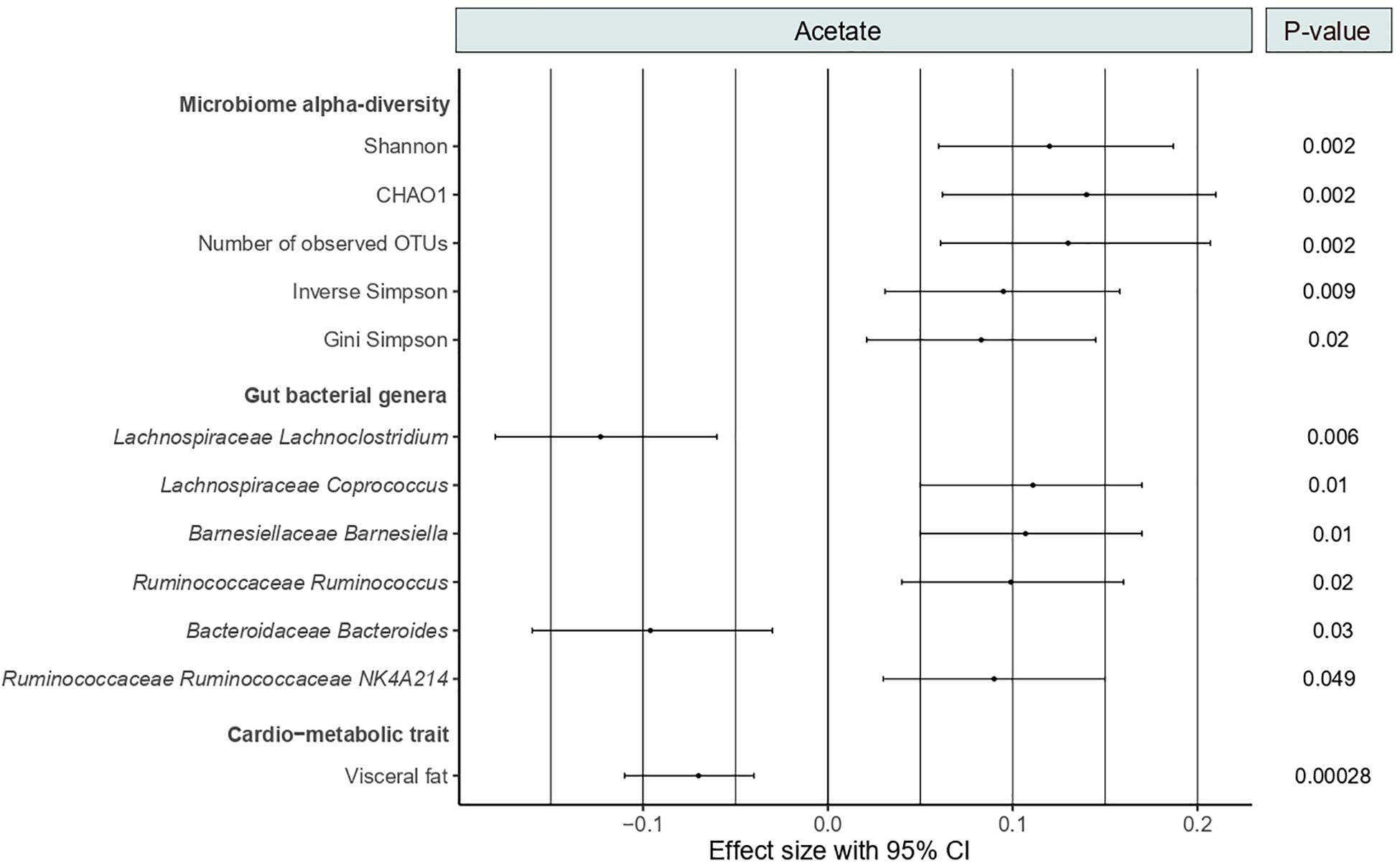
Figure 2. Forest plot showing the significant associations of acetate with microbiome alpha-diversity, gut bacterial genera and visceral fat. P-values are FDR-adjusted.
As SCFAs exert benefits on CMH, we tested the correlation between serum levels of acetate and the cardio-metabolic trait visceral fat. We found a strong negative association between both variables (Beta [95%CI] = −0.07 [−0.11, −0.04], P = 2.8 × 10–4) (Figure 2).
We then assessed the correlation between the acetate-associated gut genera and visceral fat. We found a strong positive correlation between Lachnoclostridium abundances and visceral fat (Beta [95%CI] = 0.076 [0.042, 0.11], P = 1.44 × 10–5). No significant associations were identified for the remaining five genera. We therefore conducted a formal mediation analysis to determine the indirect effects of acetate on the total effect of Lachnoclostridium on visceral fat. The analysis revealed that acetate acted as a potential partial mediator in the positive association between Lachnoclostridium and visceral fat (VAF = 10.3%, P = 2 × 10–16). These associations remained significant even after adjusting for dietary fiber intake.
Among the bacterial genera identified, we then genomically characterized Lachnoclostridium and Coprococcus because they presented the largest coefficient estimates in the association with acetate (Figure 2).
The dendrograms created from the symmetric pairwise ANI values revealed the grouping of the 271 Lachnoclostridium and 1,121 Coprococcus genomes in 18 and 9 different species, respectively. Among them, most Lachnoclostridium species has been characterized (14 species), whereas Coprococcus presented four novel bacterial species and one has not been formally characterized so far.
In addition, we found that genomes identified as C. sp900066115, C. sp00015424, C. sp. BIOML-A2, C. sp. BIOML-A1, and C. sp. NSJ-10 were assigned to the clusters of C. eutactus, C. sp4 and C. sp5 by the dendrogram. These misclassifications were further verified using their 16S rRNA sequences in a BLASTn search.
We also constructed two networks based on these ANI values, allowing us to study the genomes as members of a connected system (Figure 3). Here, the largest clusters of Lachnoclostridium were shown by C. bolteae (113 genomes, 41% of the total), C. symbiosum (68 genomes, 25% of the total) and C. clostridioforme (37 genomes, 13% of the total), whereas C. eutactus A (549 genomes, 49% of the total), C. sp4 (208 genomes, 19% of the total), C. sp5 (117 genomes, 10% of the total) and C. eutactus (103 genomes, 9% of the total) presented the largest clusters of Coprococcus.
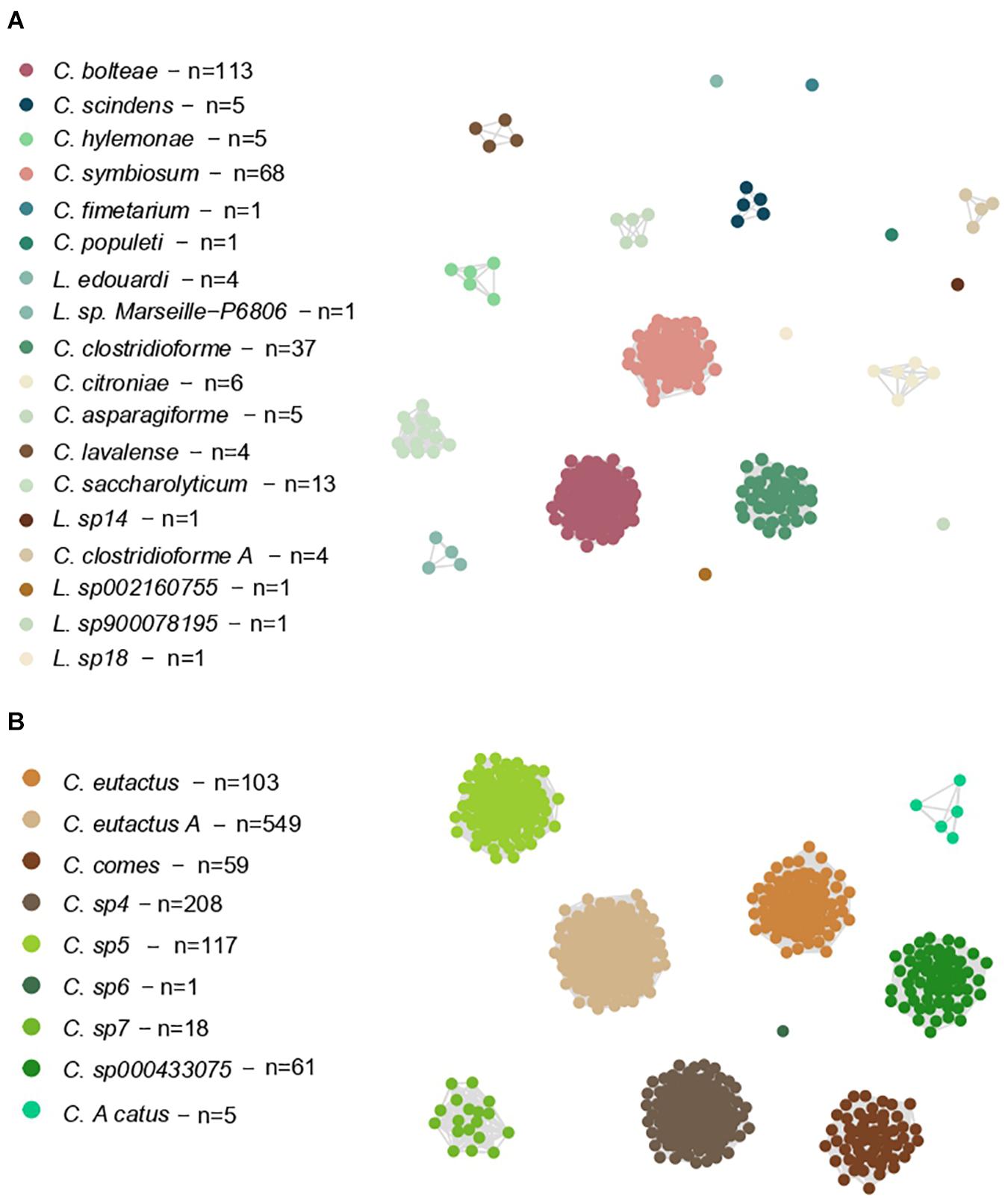
Figure 3. Weighted undirected graph based on symmetric pairwise ANI values of Lachnoclostridium (A) and Coprococcus (B). Each node represents a single genome, and each edge represents a connection between two genomes that share a mean ANI value higher than 95%. Nodes are colored by the belonging Coprococcus and Lachnoclostridium species. The species that have not been formally characterized yet, have been named using “sp” followed by the cluster number given by the dendrogram. “n” indicates the genome number of each species.
Additionally, the computed maximum-likelihood phylogenetic trees show the existing diversity of the gut bacteria Lachnoclostridium and Coprococcus (Figure 4). We observed that all the identified Lachnoclostridium and Coprococcus species represented well-defined independent lineages.
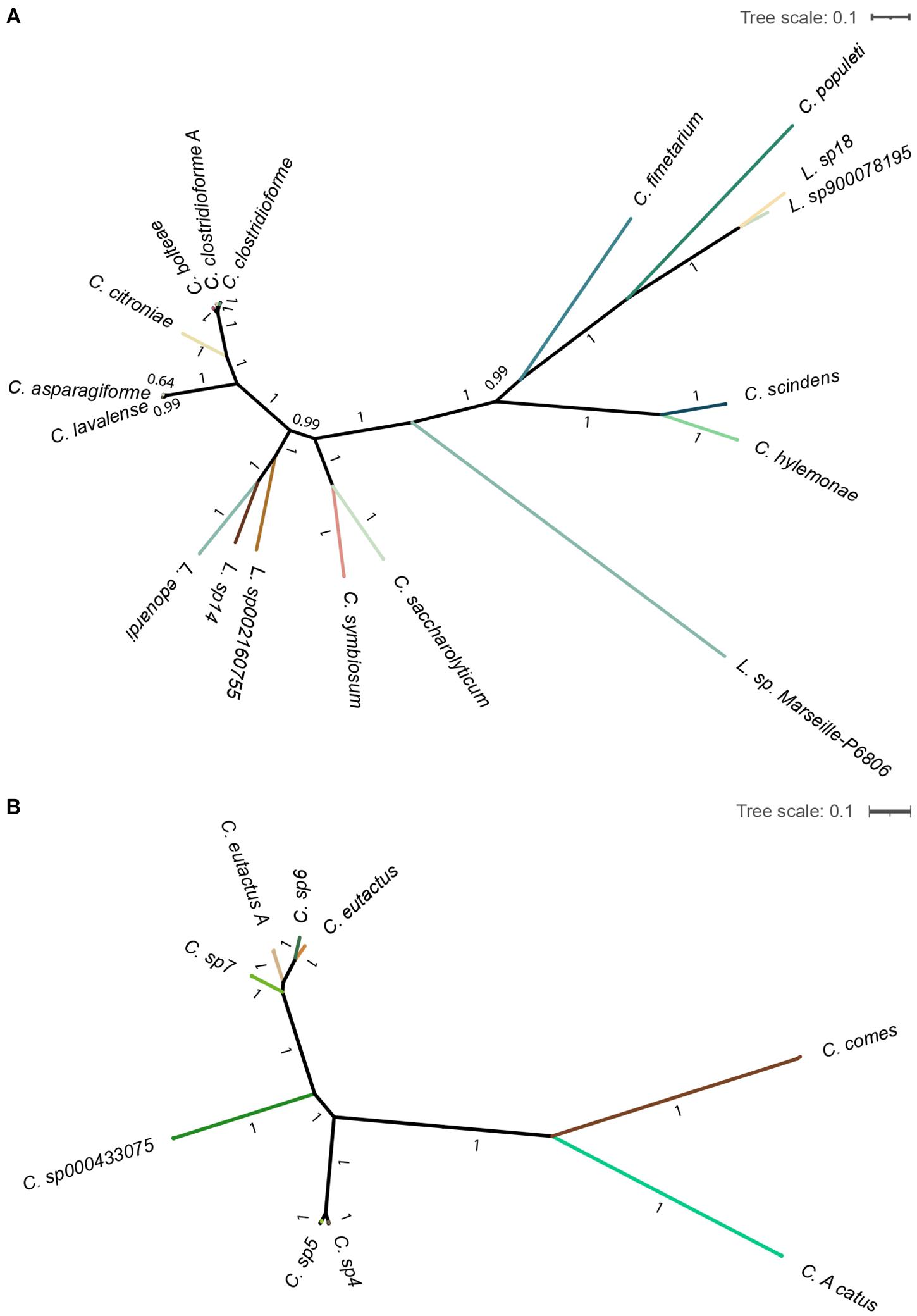
Figure 4. Maximum-likelihood phylogenetic tree of the genera Lachnoclostridium (A) and Coprococcus (B). Tree is collapsed into clades at the species level and colored by species. Bootstrapping values for each branch are shown. The scale bar represents the average number of substitutions per site.
Of note, the shown Clostridium species in Part A of Figures 3, 4 belong to Lachnoclostridium (Yutin and Galperin, 2013).
The percentage of genomes in which a fermentative pathway was predicted in each Coprococcus species is depicted in Figure 5A. Genomes from all the Coprococcus species presented fermentative pathways involved in the acetate formation from pyruvate (range = 90–100% genomes), acetoin biosynthesis (100% genomes), butanediol biosynthesis (100% genomes) and butyrate formation from acetyl-CoA (range = 90–100% genomes). The pyruvate fermentation to acetone and propionate (acrylate pathway) was exclusively present in genomes from C. catus (100% genomes), whereas the production of ethanol from pyruvate was mainly found in genomes from C. comes (80% genomes). Production of lactate from pyruvate was predicted in the latter two species (100% genomes).
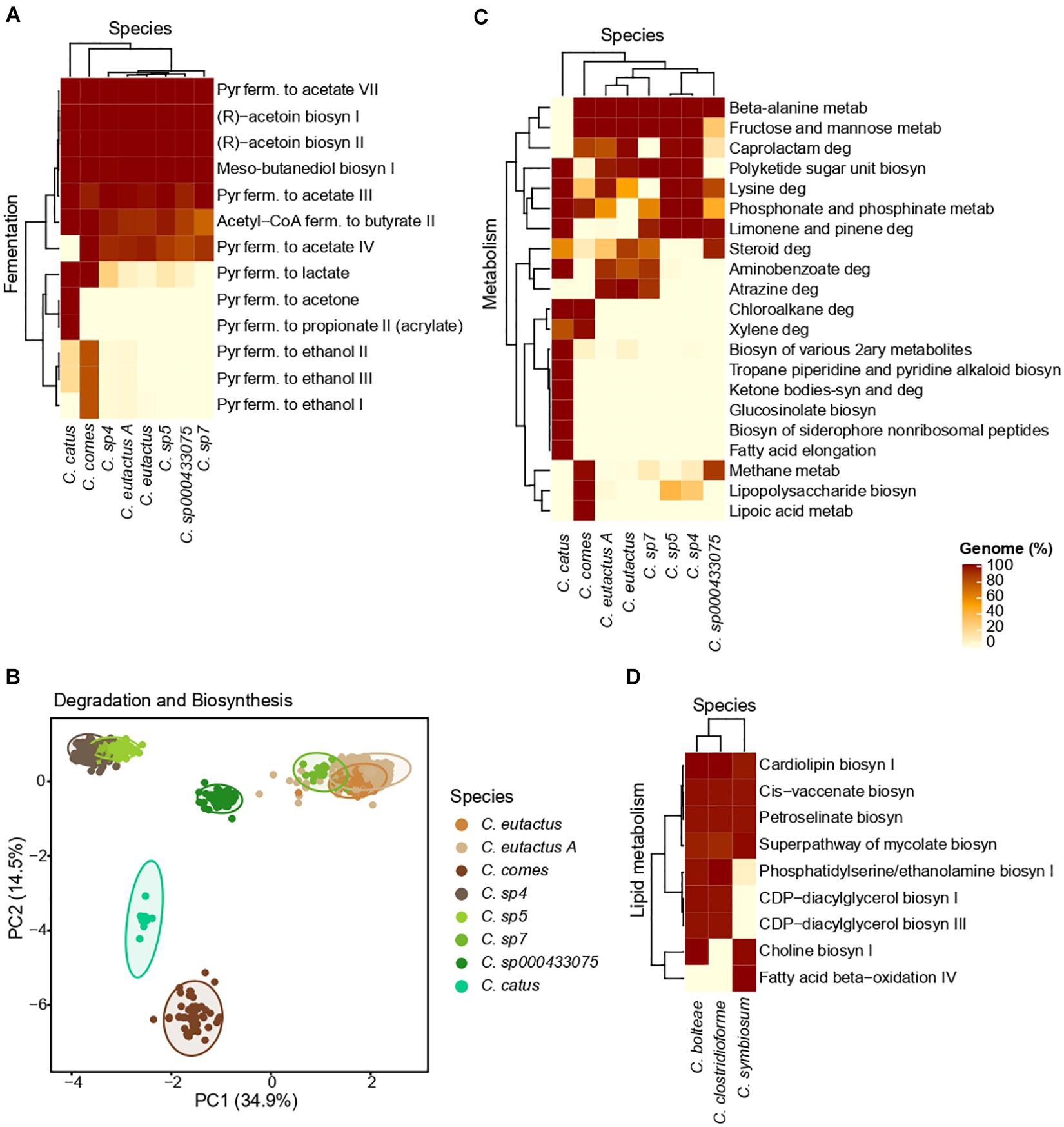
Figure 5. MetaCyc and KEGG results obtained for Coprococcus spp (A–C) and Lachnoclostridium spp (D). (A) Heatmap of the MetaCyc fermentative pathways. (B) PCA clustering based on the MetaCyc data related to the biosynthesis and degradation processes of each species. (C) Heatmap of the KEGG metabolic pathways. Only the pathways highly differential (for a given pathway, at least one species has an abundance value <5% and another species has a value >80%) between species are shown. (D) Heatmap of the MetaCyc pathways related to lipid metabolism in the three major species of Lachnoclostridium (species with >15 genomes). Of note, the shown Clostridium species belong to Lachnoclostridium (Yutin and Galperin, 2013). For all the heatmaps, the pathways and species are hierarchically clustered, and color intensities represent the percentage of genomes of a given species with a specific metabolic pathway. Pyr, pyruvate; ferm., fermentation; metab, metabolism; deg, degradation; biosyn, biosynthesis.
The PCA performed using the presence/absence matrix with the biosynthesis and degradation pathways show each species formed a well-defined cluster, being C. sp4 and C. sp5, and C. eutactus A, C. eutactus, and C. sp7 closely grouped (Figure 5B).
As shown in the heatmap of the KEGG metabolic pathways (Figure 5C), differences in the functional capabilities exit between Coprococcus species. For instance, beta-alanine metabolism was present in all the species (∼100% genomes), except in C. catus, whereas chloroalkene degradation was found only in C. catus and C. comes (100% genomes). On the other hand, some metabolic pathways were present in all the Coprococcu species in a percentage higher than 90% (Supplementary Table 5). The majority of them were related to the metabolism of amino acids such as alanine, aspartate, glutamate, arginine, proline, cysteine, glycine, serine and tyrosine, as well as essential amino acids such histidine, lysine, methionine, threonine, phenylalanine and tryptophan. Additionally, all the species presented the metabolism of several vitamins B, including vitamin B1 (thiamine), B2 (riboflavin), B3 (nicotinate), B6 (pyridoxine), B7 (biotin), and B9 (folate), and pathways involved in the carbohydrate metabolism, such as the pentose phosphate pathway and the starch and sucrose metabolism.
As we found a positive association between visceral fat and Lachnoclostridium, we focused on the metabolic pathways related to lipid metabolism. As depicted in Figure 5D, a high homogeneity in the functional capabilities related to lipid metabolism is presented in the three major species of Lachnoclostridium, above all between C. bolteae and C. clostridioforme. Furthermore, all the predicted pathways belong to the higher category of lipid biosynthesis, such as biosynthesis of choline I, CDP-diacylglycerol I and III, with the exception of the fatty acid beta-oxidation IV pathway, which belongs to the lipid degradation category. Moreover, genomes from the three major species presented pathways involved in the production of trimethylamine (TMA), including the biosynthesis of choline (∼100% genomes of C. bolteae and C. symbiosum) and phosphatidylethanolamine (∼100% genomes of C. bolteae and C. clostridioforme).
In what is to our knowledge the largest study to date investigating the associations of circulating acetate levels with gut microbiome composition and diversity and visceral fat, we report that circulating acetate levels are positively associated with microbiome alpha-diversity, while different gut bacterial genera are associated with either higher or lower acetate levels, and higher serum levels of acetate are correlated with lower visceral fat. We have also shown for the first time that the identified acetate-associated genus Lachnoclostridium has a strong positive correlation with visceral fat, and such association is partially mediated by acetate. Moreover, this is the first study genomically characterizing the acetate-associated gut genera Lachnoclostridium and Coprococcus, specifically, presenting their diversity and evolution at the genus level and annotating the functional capabilities of their species.
The identified positive associations between acetate and Barnesiella and Ruminococcus are consistent with the fact they contain genes involved in acetate production (Rey et al., 2010; Lustgarten, 2019). We also found a negative correlation between acetate levels and Bacteroides. Strikingly, Bacteroides spp. are acetate producers (Miller, 1978; Robert et al., 2007). We speculate that a plausible reason why Bacteroides present a negative correlation is the co-presence of other bacteria that might utilize the acetate produced by Bacteroides to generate other metabolites.
Among the bacterial genera identified, we genomically characterized Lachnoclostridium and Coprococcus as they presented the largest coefficient estimates in the association with acetate, as well as the positive association between visceral fat and Lachnoclostridium. In addition, these two genera presented opposing effects on acetate levels, even though they are within the same family, and thus, their genomic characterisation can provide a more holistic perspective of the influence of the gut bacteria on human health.
The dendrogram of Coprococcus revealed that half of the identified species remained uncharacterized, indicating that Coprococcus is still a poorly known genus, whereas most Lachnoclostridium species presented less than six genomes suggesting that its members are very rare (low prevalence) or that may be present in the human gut at such extremely low abundances that are difficult to detect.
In addition, the dendrogram allowed us to identify misclassified genomes, emphasizing the importance of performing quality controls and taxonomic classification. We could further confirm that the groups of species obtained using an ANI threshold of 95% were correct, since all the identified species formed completely independent lineages. It is important to note that the bacterial species delineation was not affected by the high proportion of MAGs used (92% of Coprococcus genomes and 71% of Lachnoclostridium genomes from the total number). Therefore, the genomic methods proposed here can be generalizable to genomes from other bacterial species, independently of the genome type (reconstructed from metagenomes or sequenced from isolates).
Furthermore, to the best of our knowledge, the phylogenetic results represent the most complete overview of the phylogenetic relationships of species from the genera Coprococcus and Lachnoclostridium so far, as it includes non-characterized species.
The annotation of the fermentative pathways confirmed that the identified Coprococcus species present genes involved in the formation of acetate, explaining the found positive association between this genus and acetate. Moreover, Coprococcus species are known as butyrate producers (Pryde et al., 2002), supporting with our results, which show that the formation of butyrate was predicted in all species. Our results are also in line with the fact that C. catus can produce propionate via the acrylate pathway (Reichardt et al., 2014). C. catus and C. comes present fermentative pathways (e.g., ethanol and acetone production) which are not found in other species. Interestingly, these species clustered in a different clade in the phylogenetic tree at the genus level. Additionally, both might produce lactate. It is known that C. comes can also produce lactate and C. catus can produce propionate from this compound (Reichardt et al., 2014), however, C. catus is not recognized as a lactate producer. We hypothesize that the produced lactate in C. catus might be used to generate propionate or that this fermentative pathway is not active, as this genomic approach facilitates the prediction of the functional capabilities of this genus, but unable to infer active pathways.
When we analyzed the diversity of the functional capabilities related to the biosynthesis and degradation of compounds using a PCA, we observed a considerable functional diversity among species. Of note, C. sp5 and C. sp6, and C. eutactus A, C. eutactus and C. sp7 were closely clustered, again, these are closely related according to the phylogenetic tree, and thus, the lack of differences might be due to their evolutionary closeness. These results suggest that different species might be distinguished by their metabolic functional capabilities.
We also noted differences in several KEGG metabolic pathways between species. Some of these pathways have been associated with CMH. For instance, a higher aminobenzoate degradation has been associated with a body weight decrease (Pataky et al., 2016). Our results show that genomes from C. catus, C. eutactus A, C. eutactus and C. sp7 might degrade aminobenzoate, and thus, positively influencing body weight.
Regarding the shared KEGG metabolic pathways, all the genomes presented starch and sucrose metabolism and pentose phosphate pathway, which are necessary to produce SCFAs (Topping and Clifton, 2001; Basen and Kurrer, 2020), and metabolism of essential amino acids, which can be absorbed meeting the amino acids requirements (Fuller and Tomé, 2005). Furthermore, all the species might be able to metabolize several vitamins/nutrients; including vitamins B, which has been associated with protective pathways involved in CMH; folate levels, which have been correlated with a lower metabolic syndrome score, plasma fasting glucose and a higher plasma HDL cholesterol (Navarrete-Muñoz et al., 2020); biotin, which has been shown to be involved in the glucose and lipid homeostasis (Fernandez-Mejia, 2005); thiamine, which may attenuate hypertension (Alaei-Shahmiri et al., 2015); and pyridoxine, might decrease triglyceride levels (Mottaghian et al., 2020).
Finally, we examined the lipid metabolism of the three major species of Lachnoclostridium as we found a positive association with visceral fat, as well as several studies have reported it to be related to diet-induced obesity (Zhao L. et al., 2017; Li et al., 2019; Sun et al., 2020), total cholesterol and LDL-C (Wang et al., 2020). Additionally, the mechanisms by through Lachnoclostridium impacts obesity remain unknown. Our results suggest that Lachnoclostridium spp. might negatively impact obesity and T2D. For instance, Lachnoclostridium spp might biosynthesize choline and phosphatidylethanolamine. Phosphatidylethanolamine can be methylated producing choline (Li and Vance, 2008), which can be subsequently used to produce TMA, and then trimethylamine-N-oxide (TMAO) in the liver (Zhu et al., 2018). This is in line with the fact that Lachnoclostridium has been suggested to be a TMA-producing bacteria (Jameson et al., 2016). Likewise, TMAO pathway has been associated with CMD in humans such as obesity and T2D (Dambrova et al., 2016; Schugar et al., 2017). Moreover, we identified in C. bolteae and C. clostridioforme two pathways involved in the biosynthesis of CDP-diacylglycerol, which might be a potential mediator of insulin resistance (Petersen and Shulman, 2018).
We are aware of some limitations in this study. The study sample includes only woman, and thus, our results might not be generalisable to men or different ranges of age. Only measures of acetate were available in this study, and therefore, we could not assess the associations between other relevant SCFAs, such as butyrate and propionate, and gut microbiota and visceral fat. These measures were performed using NMR, which provides different levels as compared to the gold standard LC-MS methodology. Furthermore, the association study was performed using 16S rRNA gene sequencing data. Our findings would have benefited from metagenomic sequencing analyses and an independent dataset to replicate our results or in vitro demonstrations.
Notwithstanding the above limitations, we have shown for the first time that higher abundances of Lachnoclostridium lead to lower circulating levels of acetate, resulting in increasing visceral fat. In addition, Coprococcus may play an important role in host health by its production of vitamins B and SCFAs, whereas Lachnoclostridium might have a negative impact on CMH by influencing negatively the circulating levels of acetate and being involved in the biosynthesis of harmful lipid compounds, such as TMA and CDP-diacylglycerol. We have also presented a dataset that compiles 271 and 1,121 high-quality genomes of Lachnoclostridium and Coprococcus, respectively, which can be very useful for scientists working in this area.
16S sequencing data used for this study is deposited in the European Nucleotide Archive (ERP015317). All other TwinsUK data are available upon request on the department website (http://www.twinsuk.ac.uk/dataaccess/accessmanagement/). All the metagenome data generated during the current study are included in the Supplementary Material.
Twins provided informed written consent and the study was approved by St. Thomas’ Hospital Research Ethics Committee (REC Ref: EC04/015).
CM and AMV conceived and designed the experiments. AN analyzed the data. PL, XZ, PMW, CJS, TDS, and MF contributed reagents/materials/analysis tools. AN drafted the first version of the manuscript. AMV and CM edited and revised the manuscript. All authors have readthe final manuscript and approved it for publication.
The Department of Twin Research receives support from grants from the Wellcome Trust (212904/Z/18/Z), the Medical Research Council (MRC)/British Heart Foundation (BHF) Ancestry and Biological Informative Markers for Stratification of Hypertension (AIM-HY; MR/M016560/1), the European Union, the Chronic Disease Research Foundation (CDRF), Zoe Global Ltd., and the NIHR Clinical Research Facility and Biomedical Research Centre (based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London). CM is funded by the Chronic Disease Research Foundation and by the MRC AIM-HY project grant. PL and AN are funded by the Chronic Disease Research Foundation. AMV is supported by the National Institute for Health Research Nottingham Biomedical Research Centre.
TDS is co-founder of Zoe Global Ltd. AMV is a consultant for Zoe Global Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all the participants of TwinsUK for contributing and supporting our research.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.711359/full#supplementary-material
Supplementary Table 1 | The created dataset containing 1,121 ultra high-quality genomes belonging to Coprococcus and 271 high-quality genomes belonging to Lachnoclostridium and their respective metadata.
Supplementary Tables 2, 3, 4 | Average nucleotide identity (ANI) values obtained for each pair of genomes belonging to Lachnoclostridium and Coprococcus using FastANI. Previously, genomes were filtered by alignment fraction (>0.4).
Supplementary Table 5 | KEGG and MetaCyc pathways obtained for Coprococcus and Lachnoclostridium genomes along with the percentage of genomes of each species presenting a given pathway.
ANI, average nucleotide identity; ASV, amplicon sequence variants; CMD, cardio-metabolic diseases; CMH, cardio-metabolic health; CVD, cardiovascular diseases; KEGG, Kyoto Encyclopedia of Genes and Genomes; MAG, metagenome-assembled genomes; MetaCyc, Metabolic Pathway Database; NCBI, National Center for Biotechnology Information; SCFA, short-chain fatty acids; T2D, type-2 diabetes; TMA, trimethylamine; TMAO, trimethylamine-N-oxide; UHGG, Unified Human Gastrointestinal Genome.
Alaei-Shahmiri, F., Soares, M., Zhao, Y., and Sherriff, J. (2015). The impact of thiamine supplementation on blood pressure, serum lipids and C-reactive protein in individuals with hyperglycemia: a randomised, double-blind cross-over trial. Diabetes Metab. Syndrome: Clin. Res. Rev. 9, 213–217. doi: 10.1016/j.dsx.2015.04.014
Almeida, A., Nayfach, S., Boland, M., Strozzi, F., Beracochea, M., Shi, Z. J., et al. (2020). A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114. doi: 10.1038/s41587-020-0603-3
Barrios, C., Zierer, J., Würtz, P., Haller, T., Metspalu, A., Gieger, C., et al. (2018). Circulating metabolic biomarkers of renal function in diabetic and non-diabetic populations. Sci. Rep. 8;15249.
Basen, M., and Kurrer, S. E. (2020). A close look at pentose metabolism of gut bacteria. FEBS J. 288, 1804–1808. doi: 10.1111/febs.15575
Bingham, S. A., Welch, A. A., McTaggart, A., Mulligan, A. A., Runswick, S. A., Luben, R., et al. (2001). Nutritional methods in the European prospective investigation of cancer in Norfolk. Public Health Nutr. 4, 847–858. doi: 10.1079/phn2000102
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7, 335–336.
Caspi, R., Billington, R., Fulcher, C. A., Keseler, I. M., Kothari, A., Krummenacker, M., et al. (2018). The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 46, D633–D639.
Csardi, G., and Nepusz, T. (2006). The igraph software package for complex network research. InterJ. Complex Syst. 1695, 1–9.
Cummings, J., Pomare, E., Branch, W., Naylor, C., and Macfarlane, G. (1987). Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28, 1221–1227. doi: 10.1136/gut.28.10.1221
Dambrova, M., Latkovskis, G., Kuka, J., Strele, I., Konrade, I., Grinberga, S., et al. (2016). Diabetes is associated with higher trimethylamine N-oxide plasma levels. Exp. Clin. Endocrinol. Diabetes 124, 251–256. doi: 10.1055/s-0035-1569330
Den Besten, G., Van Eunen, K., Groen, A. K., Venema, K., Reijngoud, D.-J., and Bakker, B. M. (2013). The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 54, 2325–2340. doi: 10.1194/jlr.r036012
Fernandez-Mejia, C. (2005). Pharmacological effects of biotin. J. Nutr. Biochem. 16, 424–427. doi: 10.1016/j.jnutbio.2005.03.018
Frost, G., Sleeth, M. L., Sahuri-Arisoylu, M., Lizarbe, B., Cerdan, S., Brody, L., et al. (2014). The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 5, 1–11.
Fuller, M. F., and Tomé, D. (2005). In vivo determination of amino acid bioavailability in humans and model animals. J. AOAC Int. 88, 923–934. doi: 10.1093/jaoac/88.3.923
Goodrich, J. K., Davenport, E. R., Beaumont, M., Jackson, M. A., Knight, R., Ober, C., et al. (2016). Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19, 731–743. doi: 10.1016/j.chom.2016.04.017
Gu, Z., Eils, R., and Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. doi: 10.1093/bioinformatics/btw313
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hron, W., Menahan, L., and Lech, J. (1978). Inhibition of hormonal stimulation of lipolysis in perfused rat heart by ketone bodies. J. Mol. Cell. Cardiol. 10, 161–174. doi: 10.1016/0022-2828(78)90040-8
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 1–8.
Jameson, E., Doxey, A. C., Airs, R., Purdy, K. J., Murrell, J. C., and Chen, Y. (2016). Metagenomic data-mining reveals contrasting microbial populations responsible for trimethylamine formation in human gut and marine ecosystems. Microb. Genomics 2:e000080.
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205.
Kaul, S., Rothney, M. P., Peters, D. M., Wacker, W. K., Davis, C. E., Shapiro, M. D., et al. (2012). Dual-energy X-ray absorptiometry for quantification of visceral fat. Obesity 20, 1313–1318. doi: 10.1038/oby.2011.393
Li, S., Li, J., Mao, G., Yan, L., Hu, Y., Ye, X., et al. (2019). Effect of the sulfation pattern of sea cucumber-derived fucoidan oligosaccharides on modulating metabolic syndromes and gut microbiota dysbiosis caused by HFD in mice. J. Funct. Foods 55, 193–210. doi: 10.1016/j.jff.2019.02.001
Li, Z., and Vance, D. E. (2008). Thematic review series: glycerolipids. Phosphatidylcholine and choline homeostasis. J. Lipid Res. 49, 1187–1194. doi: 10.1194/jlr.r700019-jlr200
Lustgarten, M. S. (2019). The role of the gut microbiome on skeletal muscle mass and physical function: 2019 update. Front. Physiol. 10:1435. doi: 10.3389/fphys.2019.01435
Martin-Gallausiaux, C., Marinelli, L., Blottière, H. M., Larraufie, P., and Lapaque, N. (2020). SCFA: mechanisms and functional importance in the gut. Proc. Nutr. Soc. 80, 37–49. doi: 10.1017/s0029665120006916
McCance, R. A., and Widdowson, E. M. (2014). McCance and Widdowson’s the Composition of Foods. Royal Society of Chemistry.
Menni, C., Migaud, M., Glastonbury, C. A., Beaumont, M., Nikolaou, A., Small, K. S., et al. (2016). Metabolomic profiling to dissect the role of visceral fat in cardiometabolic health. Obesity (Silver Spring) 24, 1380–1388. doi: 10.1002/oby.21488
Miller, T. L. (1978). The pathway of formation of acetate and succinate from pyruvate by Bacteroides succinogenes. Arch. Microbiol. 117, 145–152. doi: 10.1007/bf00402302
Moayyeri, A., Hammond, C. J., Valdes, A. M., and Spector, T. D. (2013). Cohort profile: TwinsUK and healthy ageing twin study. Int. J. Epidemiol. 42, 76–85. doi: 10.1093/ije/dyr207
Mottaghian, M., Salehi, P., Teymoori, F., Mirmiran, P., Hosseini-Esfahani, F., and Azizi, F. (2020). Nutrient patterns and cardiometabolic risk factors among Iranian adults: tehran lipid and glucose study. BMC Public Health 20:653. doi: 10.1186/s12889-020-08767-6
Navarrete-Muñoz, E.-M., Vioque, J., Toledo, E., Oncina-Canovas, A., Martínez-González, M. A., and Salas-Salvado, J. (2020). Dietary folate intake and metabolic syndrome in participants of PREDIMED-Plus study: a cross-sectional study. Eur. J. Nutr. 60, 1125–1136.
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pataky, Z., Genton, L., Spahr, L., Lazarevic, V., Terraz, S., Gaïa, N., et al. (2016). Impact of hypocaloric hyperproteic diet on gut microbiota in overweight or obese patients with nonalcoholic fatty liver disease: a pilot study. Digest. Dis. Sci. 61, 2721–2731. doi: 10.1007/s10620-016-4179-1
Petersen, M. C., and Shulman, G. I. (2018). Mechanisms of insulin action and insulin resistance. Physiol. Rev. 98, 2133–2223. doi: 10.1152/physrev.00063.2017
Pryde, S. E., Duncan, S. H., Hold, G. L., Stewart, C. S., and Flint, H. J. (2002). The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 217, 133–139. doi: 10.1111/j.1574-6968.2002.tb11467.x
R Core Team and DC R. (2019). A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing.
Reichardt, N., Duncan, S. H., Young, P., Belenguer, A., Leitch, C. M., Scott, K. P., et al. (2014). Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 8, 1323–1335. doi: 10.1038/ismej.2014.14
Rey, F. E., Faith, J. J., Bain, J., Muehlbauer, M. J., Stevens, R. D., Newgard, C. B., et al. (2010). Dissecting the in vivo metabolic potential of two human gut acetogens. J. Biol. Chem. 285, 22082–22090. doi: 10.1074/jbc.m110.117713
Robert, C., Chassard, C., Lawson, P. A., and Bernalier-Donadille, A. (2007). Bacteroides cellulosilyticus sp. nov., a cellulolytic bacterium from the human gut microbial community. Int. J. Syst. Evol. Microbiol. 57, 1516–1520. doi: 10.1099/ijs.0.64998-0
Sakakibara, S., Yamauchi, T., Oshima, Y., Tsukamoto, Y., and Kadowaki, T. (2006). Acetic acid activates hepatic AMPK and reduces hyperglycemia in diabetic KK-A (y) mice. Biochem. Biophys. Res. Commun. 344, 597–604. doi: 10.1016/j.bbrc.2006.03.176
Sayers, E. W., Agarwala, R., Bolton, E. E., Brister, J. R., Canese, K., Clark, K., et al. (2019). Database resources of the national center for biotechnology information. Nucleic Acids Res. 47:D23.
Schugar, R. C., Shih, D. M., Warrier, M., Helsley, R. N., Burrows, A., Ferguson, D., et al. (2017). The TMAO-producing enzyme flavin-containing monooxygenase 3 regulates obesity and the beiging of white adipose tissue. Cell Rep. 19, 2451–2461. doi: 10.1016/j.celrep.2017.05.077
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sun, X., Zhao, H., Liu, Z., Sun, X., Zhang, D., Wang, S., et al. (2020). Modulation of gut microbiota by fucoxanthin during alleviation of obesity in high-fat diet-fed mice. J. Agric. Food Chem. 68, 5118–5128. doi: 10.1021/acs.jafc.0c01467
Thissen, D., Steinberg, L., and Kuang, D. (2002). Quick and easy implementation of the Benjamini-Hochberg procedure for controlling the false positive rate in multiple comparisons. J. Educ. Behav. Stat. 27, 77–83. doi: 10.3102/10769986027001077
Tingley, D., Yamamoto, T., Hirose, K., Keele, L., and Imai, K. (2014). Mediation: R package for Causal Mediation Analysis.
Topping, D. L., and Clifton, P. M. (2001). Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 81, 1031–1064. doi: 10.1152/physrev.2001.81.3.1031
Wang, Y., Ye, X., Ding, D., and Lu, Y. (2020). Characteristics of the intestinal flora in patients with peripheral neuropathy associated with type 2 diabetes. J. Int. Med. Res. 48:0300060520936806.
Wells, P. M., Adebayo, A. S., Bowyer, R. C. E., Freidin, M. B., Finckh, A., Strowig, T., et al. (2020). Associations between gut microbiota and genetic risk for rheumatoid arthritis in the absence of disease: a cross-sectional study. Lancet Rheumatol. 2, e418–e427.
Willett, W., and Stampfer, M. J. (1986). Total energy intake: implications for epidemiologic analyses. Am. J. Epidemiol. 124, 17–27. doi: 10.1093/oxfordjournals.aje.a114366
Wu, Y.-W. (2018). ezTree: an automated pipeline for identifying phylogenetic marker genes and inferring evolutionary relationships among uncultivated prokaryotic draft genomes. BMC Genomics 19:921. doi: 10.1186/s12864-017-4327-9
Würtz, P., Havulinna, A. S., Soininen, P., Tynkkynen, T., Prieto-Merino, D., Tillin, T., et al. (2015). Metabolite profiling and cardiovascular event risk: a prospective study of 3 population-based cohorts. Circulation 131, 774–785. doi: 10.1161/circulationaha.114.013116
Ye, Y., and Doak, T. G. (2009). A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 5:e1000465. doi: 10.1371/journal.pcbi.1000465
Yutin, N., and Galperin, M. Y. (2013). A genomic update on clostridial phylogeny: g ram-negative spore formers and other misplaced clostridia. Environ. Microbiol. 15, 2631–2641.
Zhao, L., Zhang, Q., Ma, W., Tian, F., Shen, H., and Zhou, M. (2017). A combination of quercetin and resveratrol reduces obesity in high-fat diet-fed rats by modulation of gut microbiota. Food Funct. 8, 4644–4656. doi: 10.1039/c7fo01383c
Zhao, Y., Liu, J., Hao, W., Zhu, H., Liang, N., He, Z., et al. (2017). Structure-specific effects of short-chain fatty acids on plasma cholesterol concentration in male syrian hamsters. J. Agric. Food Chem. 65, 10984–10992. doi: 10.1021/acs.jafc.7b04666
Zhu, W., Buffa, J., Wang, Z., Warrier, M., Schugar, R., Shih, D., et al. (2018). Flavin monooxygenase 3, the host hepatic enzyme in the metaorganismal trimethylamine N-oxide-generating pathway, modulates platelet responsiveness and thrombosis risk. J. Thrombosis Haemostasis 16, 1857–1872. doi: 10.1111/jth.14234
Keywords: acetate, Lachnoclostridium, Coprococcus, human gut microbiota, visceral fat
Citation: Nogal A, Louca P, Zhang X, Wells PM, Steves CJ, Spector TD, Falchi M, Valdes AM and Menni C (2021) Circulating Levels of the Short-Chain Fatty Acid Acetate Mediate the Effect of the Gut Microbiome on Visceral Fat. Front. Microbiol. 12:711359. doi: 10.3389/fmicb.2021.711359
Received: 18 May 2021; Accepted: 21 June 2021;
Published: 15 July 2021.
Edited by:
Hui Wu, East China University of Science and Technology, ChinaReviewed by:
Congying Chen, Jiangxi Agricultural University, ChinaCopyright © 2021 Nogal, Louca, Zhang, Wells, Steves, Spector, Falchi, Valdes and Menni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Menni, Y3Jpc3RpbmEubWVubmlAa2NsLmFjLnVr
†ORCID: Ana Nogal, orcid.org/0000-0002-0973-4313; Panayiotis Louca, orcid.org/0000-0001-5956-1433; Tim D. Spector, orcid.org/0000-0002-9795-0365; Ana M. Valdes, orcid.org/0000-0003-1141-4471; Cristina Menni, orcid.org/0000-0001-9790-0571
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.