
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Microbiol., 19 August 2021
Sec. Systems Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.709728
 Roel Tans1†
Roel Tans1† Shoumit Dey2†
Shoumit Dey2† Nidhi Sharma Dey2†
Nidhi Sharma Dey2† Grant Calder3Peter O’Toole3
Grant Calder3Peter O’Toole3 Paul M. Kaye2*Ron M. A. Heeren1*
Paul M. Kaye2*Ron M. A. Heeren1*Infectious diseases, including those of viral, bacterial, fungal, and parasitic origin are often characterized by focal inflammation occurring in one or more distinct tissues. Tissue-specific outcomes of infection are also evident in many infectious diseases, suggesting that the local microenvironment may instruct complex and diverse innate and adaptive cellular responses resulting in locally distinct molecular signatures. In turn, these molecular signatures may both drive and be responsive to local metabolic changes in immune as well as non-immune cells, ultimately shaping the outcome of infection. Given the spatial complexity of immune and inflammatory responses during infection, it is evident that understanding the spatial organization of transcripts, proteins, lipids, and metabolites is pivotal to delineating the underlying regulation of local immunity. Molecular imaging techniques like mass spectrometry imaging and spatially resolved, highly multiplexed immunohistochemistry and transcriptomics can define detailed metabolic signatures at the microenvironmental level. Moreover, a successful complementation of these two imaging techniques would allow multi-omics analyses of inflammatory microenvironments to facilitate understanding of disease pathogenesis and identify novel targets for therapeutic intervention. Here, we describe strategies for downstream data analysis of spatially resolved multi-omics data and, using leishmaniasis as an exemplar, describe how such analysis can be applied in a disease-specific context.
Metabolism reflects an intrinsically complex interplay between multiple molecular and cellular networks. These dynamic networks underlie essential biological functions such as inflammatory responses, tissue degeneration, and regeneration. Notably, metabolism has an intimate relationship with immune cell activation and underpins many of the well-defined activation and differentiation states associated with cells of the innate and acquired immune response. For example, studies of macrophage polarization under the influence of LPS and interferon-γ (IFN-γ) or IL-4 [generating M1 and M2 phenotypes, respectively (Murray et al., 2014)] have revealed their strong association with glycolysis (van den Bossche et al., 2012; Tannahill et al., 2013; Palsson-McDermott et al., 2015; Millet et al., 2016; Shirai et al., 2016; Xie et al., 2016) and oxidative phosphorylation (Vats et al., 2006; Van den Bossche et al., 2016), respectively. Integrating in vitro metabolic and transcriptional data on macrophage polarization has revealed how metabolism is affected between polar macrophage activation (Jha et al., 2015). Similarly, differentiation of CD4+ T cell effector function is associated with the regulation of aerobic glycolysis (Chang et al., 2013). Whilst in vitro studies provide clear proof of concept and underscore a link between metabolism and immune function, they may fail to sufficiently account for the more complex and dynamic interactions that occur in vivo in the context of acute and chronic inflammatory microenvironments.
Inflammation reflects the coordinated aggregation of immune cells in response to tissue injury, infection, or other insults that disrupt tissue homeostasis. Inflammatory foci typically contain a wide variety of myeloid and lymphoid cell subsets, with composition varying in a dynamic manner from initiation to resolution. The biology of inflammation has for many years been studied using whole tissue approaches to evaluate gene and protein expression [e.g., whole tissue transcriptomics] or through the use of techniques that require tissue dissociation [e.g., flow cytometry (Leung et al., 1985) or scRNA-seq (Satija and Shalek, 2014)]. Additionally, the advent of dual RNA sequencing of both the host and pathogen has provided further insights into metabolic divergence of different macrophage lineages and pathogen fitness amongst these lineages during infection-associated inflammation (Pisu et al., 2020). Nevertheless, it is becoming increasingly appreciated that a full understanding of the processes governing inflammation requires a spatial context and this is likely to be equally true for studies aiming to delineate and understand the underlying metabolic signatures of immune cells at inflammatory sites.
Analysis of the molecular composition of a tissue is pivotal in understanding the basis of a broad spectrum of disease mechanisms and to determine appropriate diagnostics (Norris and Caprioli, 2013; Aichler and Walch, 2015). In clinical research and diagnostics, medical imaging of tissues is an indispensable technique. Classical hematoxylin and eosin (H&E) staining of tissues reveals tissue histology whereas medical images generated by magnetic resonance imaging (MRI) or computed tomography (CT) reveal tissue morphology and anatomy and enable monitoring of dynamic organ functions in health and disease using high-resolution tomographic images (Porta Siegel et al., 2018).
Technological advances have enabled multi-modal and multi-omics analyses to study the intrinsic interplay of genes, proteins, lipids, and metabolites in single cells which was recognized by Nature as the method of the year 2019 (Single-cell multimodal omics, 2020). Complementing these multi-modal and multi-omics methodologies to investigate immunometabolism is promising, yet they come with a variety of challenges. Two main challenges, which is reviewed elsewhere (Artyomov and van den Bossche, 2020; Lercher et al., 2020; Mazumdar et al., 2020), are to preserve the spatial context of biomolecules and the downstream analysis of the different omes.
Here, we provide a perspective on how multi-modal label-free molecular imaging techniques such as mass spectrometry imaging (MSI) combined with spatially resolved highly multiplexed protein and transcriptome mapping can be used in concert to understand the progression of infectious diseases and delineate inflammatory microenvironments. We discuss strategies for downstream data integration and analysis and describe a case study of research on granulomatous inflammation associated with experimental leishmaniasis as a possible exemplar of how research into inflammatory diseases of infectious (as well as non-infectious) origin can benefit from this approach.
Mass spectrometry (MS) is a technique that measures the mass of atoms and molecules (Chughtai and Heeren, 2010). The specificity of MS allows structural analysis of small and large biomolecules, like metabolites, lipids, or large protein complexes, resulting in unambiguous molecular compound identifications (McDonnell and Heeren, 2007). In addition, rapid advancements of MS-related methodologies have increased its use in a wide variety of (bio)medical sciences, like infectious diseases (Seng et al., 2010; Zheng et al., 2010), oncology (Ifa and Eberlin, 2016; Takats et al., 2017; Mas et al., 2020), pharmacology (Jove et al., 2019), personalized medicine (Chen et al., 2012; Geyer et al., 2016), and more.
Initially described by Caprioli et al. (1997), MSI translates the spatial molecular context of tissues into improved understanding of biological processes underlying health and disease (Chughtai and Heeren, 2011; Porta Siegel et al., 2018). MSI uses an ionization technique that generates charged molecules (ions) from which the mass-to-charge ratios of ions can be determined (Chughtai and Heeren, 2010). Several ionization techniques exist like matrix-assisted laser desorption/ionization (MALDI), desorption electrospray ionization (DESI), and secondary ion mass spectrometry (SIMS) as well as different types of mass analyzers. It is important to recognize that the combination of ionization technique and type of mass analyzer depicts the sensitivity, specificity, and overall spatial resolution of an MSI workflow. In addition, the biomolecule class (i.e., protein, lipid, or metabolite) spatially analyzed in MSI is dependent on sample pre-treatment.
Mass spectrometry imaging defines the molecular composition at the tissue level and even at cellular level, resulting in improved understanding of the tissue microenvironment and can concomitantly identify specific molecular profiles of disease (Ščupáková et al., 2020). Moreover, it facilitates the translation of molecular images to the pathology of tissues (Norris and Caprioli, 2013; Schwamborn et al., 2017). This translation to pathology is enabled by the ability to align histological images, e.g., following H&E staining or immunohistochemistry (IHC), with MSI-generated images. As described earlier (Deutskens et al., 2011), post MALDI-MSI allows classical histological staining and microscopic evaluation of the same tissue section. Such combination of histology and MSI enables the study of molecular patterns associated with specific cell types or tissue structures (Porta Siegel et al., 2018).
A recent development combined MSI with laser microdissection and sensitive liquid chromatography-mass spectrometry (LC-MS) based proteomics. This application uses MSI-guided evaluation of (micro) regions of interest (ROIs) followed by targeted extraction of cellular material and conventional LC-MS/MS proteomics. As a result, MSI-guided spatial omics allows for a more in-depth molecular characterization of a tissue microenvironment (Dewez et al., 2020).
Spatial transcriptomic and multiplexed IHC can provide a transcriptional and functional snapshot of an inflammatory microenvironment. Profiling such in situ microenvironments can be achieved by a variety of techniques. McCaffrey et al. (2020) used multiplexed ion beam imaging by time of flight (MIBI-TOF) to spatially map and comprehensively delineate lung granulomas in patients with tuberculosis into cellular niches. Using individually metal isotope tagged antibodies and imaging based on SIMS, they found that the innermost core of granulomas is rich in myeloid suppressor cells exhibiting high expression of immunomodulatory immune checkpoint molecules IDO-1 and PD-L1 (McCaffrey et al., 2020). The 10× Genomics Visium platform that allows transcriptomic mapping of whole tissue sections using spatially bar-coded capture spots was used in combination with scRNA-seq to delineate granuloma architecture and show that non-immune cells can also produce antimicrobial factors that limit infection in leprosy (Ma et al., 2021). In another study (Carow et al., 2019) in situ mRNA sequencing was used to study murine lung granuloma progression over time. Other platforms like Hyperion that marry CyTOF technology and imaging mass cytometry (IMC) have recently been used to spatially map immune signatures and study lung pathology in COVID-19 (Zhang et al., 2020; Butler et al., 2021; Chevrier et al., 2021). Another key technology CODEX uses iterative cycles of imaging and removal of reporters that bind to complementary barcodes on antibodies used to stain tissue sections. It has been exploited extensively in cancer research (Parra et al., 2019; Schürch et al., 2020) and may be used to probe immune microenvironments.
While the above-named technologies commonly scan an entire tissue section, the Nanostring GeoMx Digital Spatial Profiler (DSP) platform (Merritt et al., 2020) allows more precise selection of user-defined regions of interest by using a unique method of patterned UV light (Merritt et al., 2020). Further, it allows segmentation analysis based on morphology markers for profiling cell types of particular interest. A typical workflow involves staining tissue sections with oligonucleotide tagged antibodies or RNA probes, histological, or fluorescently labeled image guided ROI selection, UV cleavage of tags specifically from the whole ROI or from a segment within an ROI, collection, and finally, barcode counting in the nCounter system (or next gen sequencing). This ability to select biologically relevant regions becomes particularly relevant when DSP is used to assess specific microenvironments, as applied recently in studies of cutaneous leishmaniasis (Dey et al., 2021) and COVID-19 pathology (Margaroli et al., 2021; Rendeiro et al., 2021).
Granulomas are mononuclear cell-rich inflammatory foci formed around “indigestible” materials or pathogens and Leishmania donovani infection provides one of the best studied examples of experimental hepatic granuloma formation (Murray, 2001; Kaye and Beattie, 2016). Further, a transcriptional network enriched in genes for lipid metabolism were identified in uninfected bystander Kupffer cells that are responsible for parasite survival (Beattie et al., 2013). Interestingly, L. brazililensis was shown to affect host cell lipid metabolism (Alves-Ferreira et al., 2020). We show here as an example, a pipeline for the analysis of liver granuloma development following experimental visceral leishmaniasis and their associated lipid profile (using MSI) to help strategize therapeutic approaches. These methods are, however, generally applicable to immune microenvironments (Figure 1).
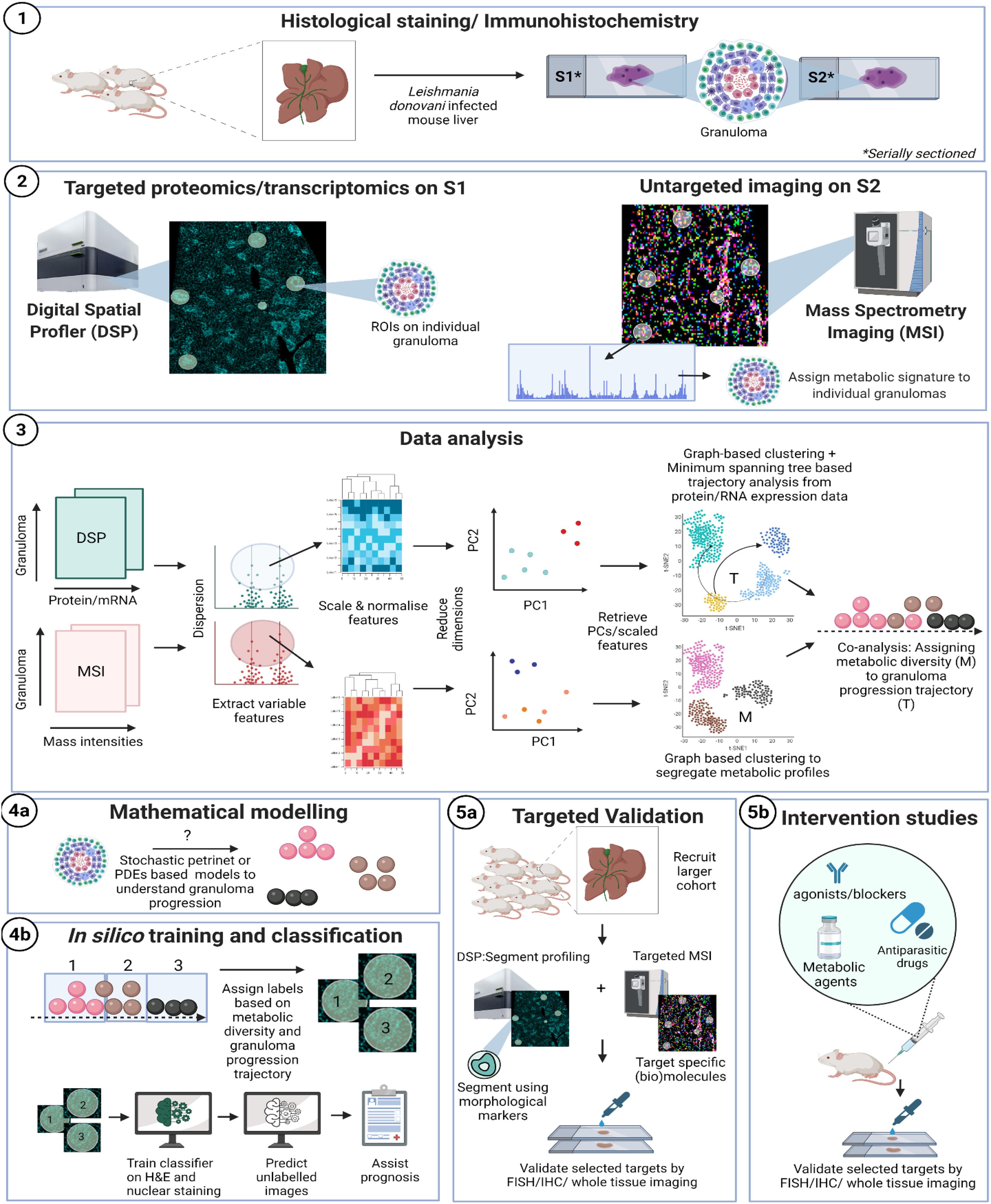
Figure 1. Proposed workflow: (1) classical hematoxylin and eosin (H&E) or nuclear staining of a mouse liver, infected with Leishmania donovani can be used to select granulomatous regions of interest (ROI). (2) Thin serial sections (5 μm) processed in parallel through GeoMx Digital Spatial Profiler (DSP; left) and mass spectroscopy imaging (MSI; right). For DSP: targeted morphology-based selection of ROIs can be probed using antibody-based protein profiling (e.g., mouse Immune Panel, NanoString). In parallel, MSI is used to analyze the whole tissue section at a predefined spatial resolution, typically 10–50 μm pixel size. MSI imaging is used to generate the molecular masses of biomolecules (e.g., of lipids, metabolites, or proteins), depending on the sample’s pretreatment, from each pixel. MSI images can be either co-registered with the corresponding DSP-imaged area based on H&E/nuclear staining. (3) Data obtained from DSP and untargeted MSI imaging are processed in parallel to extract most variable features, which are then scaled, normalized, and finally visualized in reduced dimensions to assess microenvironment (ROI) heterogeneity through PCA. Principal components that explain most of the variance in the data are used to cluster granuloma subtypes based on proteomic/transcriptomic data (DSP) or their metabolic profile (MSI). Progression trajectory inferred from transcriptomic data is overlaid on metabolic signature to identify aberrant lipids as an example. (4a) A mathematical re-construction of granuloma progression and its association to cell types, chemokine/cytokine expression, lipid metabolism (based on 4a,b) can be used to predict heterogeneity in progression dynamics for example associated with different outcomes. (4b) Finally, the knowledge inferred from serial sections using MSI/DSP may be routed back to label granuloma sub-types. Image classifiers can then be used to train on H&E or nuclear stain to learn and predict pathology and function from gross morphology. (5a) Variable features obtained from 1 to 3 are then re-probed on new tissue sections obtained from a larger cohort of subjects and through segmentation analysis or higher resolution targeted MSI. This targeted approach aims at the analytical validation of novel biomarkers discovered during the holistic profiling in 1.1–1.3. (5b) Intervention studies using agonists/blockers/metabolic agents or anti-leishmanial drugs to further establish causality associated with identified targets.
Using GeoMx DSP, ROIs can be created on granulomas using established morphological stains. Next, to identify compositional differences between the granuloma microenvironment and the surrounding parenchymal tissue can be identified using MSI-guided spatial omics. As an exemplar, and in line with our proposed workflow depicted in Figure 1, we show preliminary results on aberrant spatially resolved protein and lipid profiles from granulomas in L. donovani-infected mice using DSP and MSI (Figure 2). High-level (low-plex) exploratory analysis of cellular composition of granulomas using targeted spatial protein profiling and MSI on serial sections can identify heterogeneity in ROI (Figure 2.1) composition and metabolic programming (Figures 1.1, 1.2 for strategy and Figure 2.2 as exemplary data). Such heterogeneity can then be used to explore more mechanistic questions using further iterations of targeted DSP and MSI, e.g., involving cell-specific segmentation (Merritt et al., 2020) in previously characterized ROIs and/or by casting a wider net to delineate heterogeneity at whole transcriptome resolution.
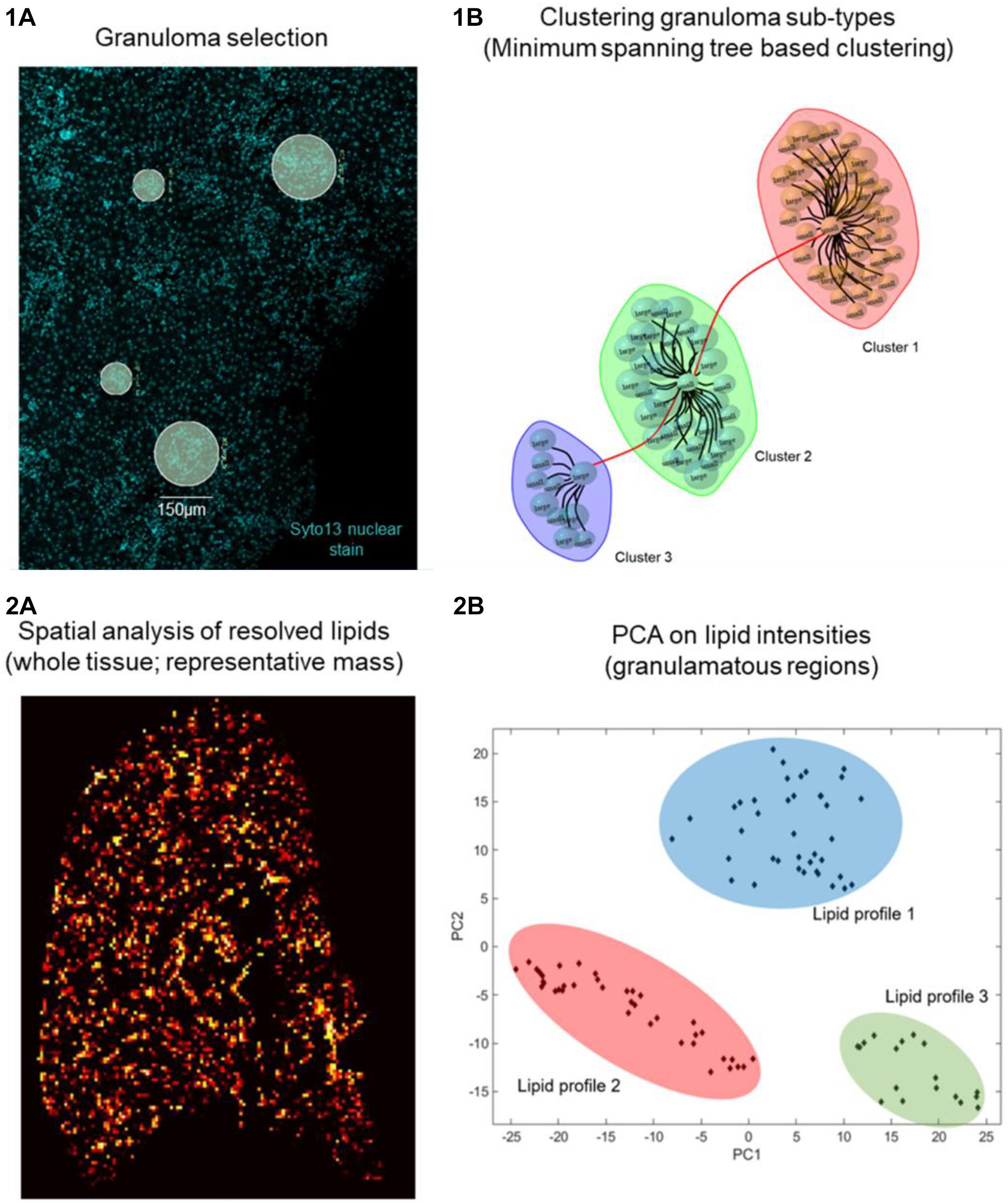
Figure 2. Preliminary data on Leishmania granuloma. Day 28 p.i. L. donovani infected C57BL/6 mice liver sections (n = 5) were 1A stained with SYTO 13 nuclear dye and Nanostring Immune Cell Profiling Core and IO Drug Target Modules (protein panel, 26 oligo labeled antibodies). Eighty granulomas (75 or 150 μ diameter) were selected based on nuclear dye on a Digital Spatial Profiler (DSP, Nanostring). UV light cleaved ROIs from individual granulomas were collected in a 96 well plate, hybridized, and counted on an nCounter system (1B). Distinct granuloma clusters as calculated based on edge betweenness of a minimum spanning tree representation of a granuloma weighted (Pearson’s correlation) graph object. (2A) Day 14 L. donovani infected mouse liver sections (n = 5) were analyzed by MSI. (2B) Principal component analysis (PCA) is used as an unsupervised approach to show variance in spatial distribution of all lipid masses from every pixel across all granuloma regions. The PCA image depicts highly intense pixels, which represent granulomas and contain different lipid profiles. A total of 20 granuloma ROIs per mouse liver section were analyzed. Subsequent PCA shows discriminative lipid profiles, which suggests that granulomas show heterogeneous lipid signatures.
The structure of formatted omics data is often an Xij matrix where the i = r1, r2, r3, …, rm representing the abstraction level of the measured object (e.g., individuals, single-cells, or ROIs) and j = j1, j2, j3, …, jn representing the features of the measured object (which can be single nucleotide polymorphisms, mRNA counts, protein, lipid, or other macromolecular expression intensity). Data generated using DSP and MSI can be easily converted into a rectangular format to describe protein/RNA and metabolite levels as features (column) per ROI (row). Each omics field has developed tools and pipelines for curating and analyzing data, but they provide general ideas for analyzing this type of data (Robinson et al., 2009; de Wit et al., 2012; Butler et al., 2018). We briefly describe methods that can be used to analyze the spatial structure and distribution of multi-modal measurements.
Features with low dispersion in the data are removed to exclude non-informative features (i.e., curse of dimensionality) which can reduce the distance between the data points. Variable features, as selected above, can be normalized to the number of nuclei or area of microenvironment. Features can then be scaled and used as candidates for dimensionality reduction by principal component analysis and this is generally recommended for both small and large datasets (MSI and single-cell RNA) in terms of accuracy and efficiency (Klerk et al., 2007; Sun et al., 2019). It is relevant to assume that heterogeneity in inflammatory microenvironments may be due to cellular activation or composition and that these discrete communities may be classified as discrete clusters. k-means clustering can be used to identify clusters that best model the data by selecting an appropriate “k” using the elbow method (Diday, 1994). Graph-based methods try to detect clusters by identifying nodes that are connected tightly using a distance metric. For example, one approach to this is to calculate the edge betweenness that assigns highest scores for edges that are traversed often to find shortest paths and, therefore, are assumed to connect discrete communities/clusters (Freeman, 1979; Brandes, 2001). Methodologies adopted in high dimensional scRNA sequencing datasets have shown that while there is hardly one clustering method that fits all problems (Lockett, 2020), when it comes to clustering scRNA data graph-based tools such as Seurat (Butler et al., 2018), SCANPY (Wolf et al., 2018), and PhenoGraph (Levine et al., 2015) are able to identify clusters very efficiently (Kiselev et al., 2019). Many algorithms (including non-graph based) have been used for scRNA-seq data [tabulated in Kiselev et al. (2019)] and may be borrowed for analysis of ROI-based high dimensional datasets. An important consideration here is that clustering works well for individual cell types but the signal obtained from a granuloma will be composite and may require additional de-convolution steps (Biancalani et al., 2020; Kleshchevnikov et al., 2020) to aid or inform clustering using publicly available or in-house scRNA sequencing datasets.
Granulomas may be similar or distinct, as well as asynchronous in their development (Albergante et al., 2013; Moore et al., 2013; Kaye and Beattie, 2016). This suggests that while the selected ROIs may be grouped as discrete clusters, they may also be related to each other in a continuous sense. In 2003, methods to order temporally sampled microarray data correctly by calculating minimum spanning tree of a complete graph were developed (Magwene et al., 2003). These methods have been extended and applied to high-dimensional single cell RNA datasets to infer a pseudotemporal ordering of individual cells. Single-cell transcriptomes show heterogeneity and imply asynchronicity in expression patterns. Thus, cells sequenced at a single time-point can be statistically ordered in pseudotime to reveal lineages (Trapnell et al., 2014). This is not restricted only to single lineages but can indicate the possible branching of trajectories. In the context of inflammatory microenvironments such as granulomas, trajectories can be applied to ascertain progression or maturation. Protein/RNA snapshots at multiple timepoints can further aid computational interpretation of microenvironment progression trajectory. The asynchronicity of granuloma formation and outcome may be mapped on to a pseudo-temporal space. Interpreting such information in the context of underlying spatial metabolism may indicate how the latter affects immune cell composition or activation score in a spatially resolved manner. Further, probabilistic methods of learning temporal trajectory have been described for single cell RNA data that can be applied for multi-modal ROI analysis (Campbell and Yau, 2016a) especially those that can incorporate prior knowledge in trajectory learning (Campbell and Yau, 2016b). At this stage, clustering information of lipid profiles (based on MSI) may be associated [co-analysis (Buescher and Driggers, 2016)] of exact serial sections) with the granuloma progression trajectory as inferred via protein expression data (DSP) to link metabolic profiles to granuloma progression (Figure 1.3).
Further, as both DSP and MSI retain exact spatial information, inferred ROI subtypes can be associated to metadata available from tissue morphology. Correlations found at this step to morphology and pseudotime ordered ROIs can then be applied to probe more mechanistic questions like how proximity to underlying morphology can impact immune cell activation or metabolism. This can be achieved by asking broader fundamental questions by informing mathematical models (Figure 1.4a) of granuloma development in parallel (Albergante et al., 2013; Moore et al., 2013; Siewe et al., 2017; Moyo et al., 2018) or by targeted experiments and interventions, e.g., an ex vivo biomarker validation study in a larger cohort (Figures 1.5a, 1.5b).
Statistically inferred ROI sub-types can be further explored at the single cell level by co-registering MS images with IHC of the entire tissue based on cell types or proteins identified earlier in the process. QuPath (Scupakova et al., 2020), an open-software tool for digital pathology image analysis was successfully aligned with single-cell MALDI-MSI data allowing for automated single-cell morphometrics with their detailed molecular profile (Scupakova et al., 2020). Using other specialized software like Strataquest (TissueGnostics StrataQuest, 2021) and Cell Profiler (Mcquin et al., 2018) it is then possible to identify mechanistic reasons for observed branching in pseudotime. In parallel, classification of ROIs in branched trajectories can be used to label ROIs for training convolutional neural networks to identify such clusters in routinely stained images (Shi et al., 2019). These can be used as a powerful research or diagnostic tool (Figure 1.4b).
Spatially resolved immunometabolism advances our knowledge of different immune lineages and populations central to microbial infections but more specifically can help delineate pathogenesis in important infections such as tuberculosis (Silva Miranda et al., 2012), schistosomiasis (Hams et al., 2013), and leishmaniasis (Kaye and Beattie, 2016) where pathogen induced metabolic re-wiring of host cells has been implicated in the pathological progression of the disease (Wilson et al., 2019; Saunders and McConville, 2020). Recent dual RNA-seq studies evaluating host and pathogen transcriptional response simultaneously show that metabolic interplay between the intracellular pathogen and the host cell can determine microbicidal or permissive property of the microenvironment (Westermann et al., 2016, 2017; Russell et al., 2019). In addition, MSI based reconstruction of host–pathogen lipid metabolic responses has been shown for Francisella infection (Scott et al., 2017) and for specific glycolipids in tuberculosis lesions (Blanc et al., 2018). Further understanding of the heterogeneity in development, function, or progression of these granulomas in relationship to their tissue microenvironment requires further detailed in situ exploration of the relationship between host immune cells, pathogen niches, and local biomolecule (protein, lipid, and transcript) profiles.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
RT, SD, and ND designed and wrote the manuscript. GC and PO’T provided insights and feedback. PMK and RMAH reviewed the manuscript and are project leaders and corresponding authors. All authors contributed to the article and approved the submitted version.
This work was funded by the York-Maastricht Partnership program and supported by a Wellcome Trust Senior Investigator Award to PMK (WT106203). This research was part of the M4I research program and received financial support from the Dutch Province of Limburg under the LINK program. The figures in this article have been created/modified using Biorender.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aichler, M., and Walch, A. (2015). MALDI Imaging mass spectrometry: current frontiers and perspectives in pathology research and practice. Lab. Invest. 95, 422–431. doi: 10.1038/labinvest.2014.156
Albergante, L., Timmis, J., Beattie, L., and Kaye, P. M. (2013). A petri net model of granulomatous inflammation: implications for IL-10 mediated control of leishmania donovani Infection. PLoS Comput. Biol. 9:e1003334. doi: 10.1371/journal.pcbi.1003334
Alves-Ferreira, E. V. C., Ferreira, T. R., Walrad, P., Kaye, P. M., and Cruz, A. K. (2020). Leishmania braziliensis prostaglandin F 2α synthase impacts host infection. Parasit. Vectors 13:9. doi: 10.1186/S13071-020-3883-Z
Artyomov, M. N., and van den Bossche, J. (2020). Immunometabolism in the single-cell era. Cell Metab. 32, 710–725. doi: 10.1016/j.cmet.2020.09.013
Beattie, L., D’El-Rei Hermida, M., Moore, J. W. J., Maroof, A., Brown, N., Lagos, D., et al. (2013). A transcriptomic network identified in uninfected macrophages responding to inflammation controls intracellular pathogen survival. Cell Host Microbe 14, 357–368. doi: 10.1016/j.chom.2013.08.004
Biancalani, T., Scalia, G., Buffoni, L., Avasthi, R., Lu, Z., Sanger, A., et al. (2020). Deep learning and alignment of spatially-resolved whole transcriptomes of single cells in the mouse brain with Tangram. Biorxiv [Preprint] doi: 10.1101/2020.08.29.272831
Blanc, L., Lenaerts, A., Dartois, V., and Prideaux, B. (2018). Visualization of mycobacterial biomarkers and tuberculosis drugs in infected tissue by MALDI-MS imaging. Anal. Chem. 90, 6275–6282. doi: 10.1021/acs.analchem.8b00985
Brandes, U. (2001). A faster algorithm for betweenness centrality. J. Math. Sociol. 25, 163–177. doi: 10.1080/0022250X.2001.9990249
Buescher, J. M., and Driggers, E. M. (2016). Integration of omics: more than the sum of its parts. Cancer Metab. 4:4. doi: 10.1186/s40170-016-0143-y
Butler, A., Hoffman, P., Smibert, P., Papalexi, E., and Satija, R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. doi: 10.1038/nbt.4096
Butler, D., Mozsary, C., Meydan, C., Foox, J., Rosiene, J., Shaiber, A., et al. (2021). Shotgun transcriptome, spatial omics, and isothermal profiling of SARS-CoV-2 infection reveals unique host responses, viral diversification, and drug interactions. Nat. Commun. 12:1660. doi: 10.1038/s41467-021-21361-7
Campbell, K. R., and Yau, C. (2016b). Ouija: incorporating prior knowledge in single-cell trajectory learning using Bayesian nonlinear factor analysis. Biorxiv [Preprint] doi: 10.1101/060442 bioRxiv: 60442,
Campbell, K. R., and Yau, C. (2016a). Order under uncertainty: robust differential expression analysis using probabilistic models for pseudotime inference. PLoS Comput. Biol. 12:e1005212. doi: 10.1371/journal.pcbi.1005212
Caprioli, R. M., Farmer, T. B., and Gile, J. (1997). Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. Anal. Chem. 69, 4751–4760. doi: 10.1021/ac970888i
Carow, B., Hauling, T., Qian, X., Kramnik, I., Nilsson, M., and Rottenberg, M. E. (2019). Spatial and temporal localization of immune transcripts defines hallmarks and diversity in the tuberculosis granuloma. Nat. Commun. 10:1823. doi: 10.1038/s41467-019-09816-4
Chang, C.-H., Curtis, J. D., Maggi, L. B., Faubert, B., Villarino, A. V., O’Sullivan, D., et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. doi: 10.1016/j.cell.2013.05.016
Chen, R., Mias, G. I., Li-Pook-Than, J., Jiang, L., Lam, H. Y. K., Chen, R., et al. (2012). Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148, 1293–1307. doi: 10.1016/j.cell.2012.02.009
Chevrier, S., Zurbuchen, Y., Cervia, C., Adamo, S., Raeber, M. E., de Souza, N., et al. (2021). A distinct innate immune signature marks progression from mild to severe COVID-19. Cell Rep. Med. 2:100166. doi: 10.1016/j.xcrm.2020.100166
Chughtai, K., and Heeren, R. M. (2010). Mass spectrometric imaging for biomedical tissue analysis. Chem. Rev. 110, 3237–3277. doi: 10.1021/cr100012c
Chughtai, K., and Heeren, R. M. A. (2011). Mass spectrometric imaging for biomedical tissue analysis - chemical reviews (ACS publications). Chem. Rev. 110, 3237–3277. doi: 10.11606/T.6.2015.tde-19102015-100242
de Wit, P., Pespeni, M. H., Ladner, J. T., Barshis, D. J., Seneca, F., Jaris, H., et al. (2012). The simple fool’s guide to population genomics via RNA-Seq: an introduction to high-throughput sequencing data analysis. Mol. Ecol. Resourc. 12, 1058–1067. doi: 10.1111/1755-0998.12003
Deutskens, F., Yang, J., and Caprioli, R. M. (2011). High spatial resolution imaging mass spectrometry and classical histology on a single tissue section. J. Mass spectrom. 46, 568–571. doi: 10.1002/jms.1926
Dewez, F., Oejten, J., Henkel, C., Hebeler, R., Neuweger, H., de Pauw, E., et al. (2020). MS Imaging-guided microproteomics for spatial omics on a single instrument. Proteomics 20:e1900369. doi: 10.1002/pmic.201900369
Dey, N. S., Senarathna, S., Somaratne, V., Madarasinghe, N., Seneviratne, B., Forrester, S., et al. (2021). Early reduction in PD-L1 expression predicts faster treatment response in human cutaneous leishmaniasis. Biorxiv [Preprint] doi: 10.1101/2020.02.21.959528
Freeman, L. C. (1979). Centrality in social networks. Soc. Netw. 1, 215–239. doi: 10.1016/0378-8733(78)90021-7
Geyer, P. E., Kulak, N. A., Pichler, G., Holdt, L. M., Teupser, D., and Mann, M. (2016). Plasma proteome profiling to assess human health and disease. Cell Syst. 2, 185–195. doi: 10.1016/j.cels.2016.02.015
Hams, E., Aviello, G., and Fallon, P. G. (2013). The schistosoma granuloma: friend or foe? Front. Immunol. 4:89. doi: 10.3389/fimmu.2013.00089
Ifa, D. R., and Eberlin, L. S. (2016). Ambient ionization mass spectrometry for cancer diagnosis and surgical margin evaluation. Clin. Chem. 62, 111–123. doi: 10.1373/clinchem.2014.237172
Jha, A. K., Huang, S. C. C., Sergushichev, A., Lampropoulou, V., Ivanova, Y., Loginicheva, E., et al. (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430. doi: 10.1016/j.immuni.2015.02.005
Jove, M., Spencer, J., Clench, M., Loadman, P. M., and Twelves, C. (2019). Precision pharmacology: mass spectrometry imaging and pharmacokinetic drug resistance. Crit. Rev. Oncol. Hematol. 141, 153–162. doi: 10.1016/j.critrevonc.2019.06.008
Kaye, P. M., and Beattie, L. (2016). Lessons from other diseases: granulomatous inflammation in leishmaniasis. Semin. Immunopathol. 38, 249–260. doi: 10.1007/s00281-015-0548-7
Kiselev, V. Y., Andrews, T. S., and Hemberg, M. (2019). Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 20, 273–282. doi: 10.1038/s41576-018-0088-9
Klerk, L. A., Broersen, A., Fletcher, I. W., van Liere, R., and Heeren, R. M. A. (2007). Extended data analysis strategies for high resolution imaging MS: new methods to deal with extremely large image hyperspectral datasets. Int. J. Mass Spectrom. 260, 222–236. doi: 10.1016/j.ijms.2006.11.014
Kleshchevnikov, V., Shmatko, A., Dann, E., Aivazidis, A., King, H. W., Li, T., et al. (2020). Comprehensive mapping of tissue cell architecture via integrated single cell and spatial transcriptomics. Biorxiv [Preprint] doi: 10.1101/2020.11.15.378125
Lercher, A., Baazim, H., and Bergthaler, A. (2020). Systemic immunometabolism: challenges and opportunities. Immunity 53, 496–509. doi: 10.1016/J.IMMUNI.2020.08.012
Leung, K.-P., Russell, S. W., LeBlanc, P. A., and Caballero, S. (1985). Heterogeneity among macrophages cultured from mouse bone marrow. Cell Tissue Res. 239, 693–701. doi: 10.1007/BF00219251
Levine, J. H., Simonds, E. F., Bendall, S. C., Davis, K. L., Amir, E. A. D., Tadmor, M. D., et al. (2015). Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197. doi: 10.1016/j.cell.2015.05.047
Lockett, A. J. (2020). No free lunch theorems. Nat. Comput. Series 1, 287–322. doi: 10.1007/978-3-662-62007-6_12
Ma, F., Hughes, T. K., Teles, R. M. B., Andrade, P. R., Silva, B. J., de, A., et al. (2021). The cellular architecture of the antimicrobial response network in human leprosy granulomas. Nat. Immunol. 22, 839–850. doi: 10.1038/s41590-021-00956-8
Magwene, P. M., Lizardi, P., and Kim, J. (2003). Reconstructing the temporal ordering of biological samples using microarray data. Bioinformatics 19, 842–850. doi: 10.1093/bioinformatics/btg081
Margaroli, C., Benson, P., Sharma, N. S., Madison, M. C., Robison, S. W., Arora, N., et al. (2021). Spatial mapping of SARS-CoV-2 and H1N1 lung injury identifies differential transcriptional signatures. Cell Rep. Med. 2:100242. doi: 10.1016/j.xcrm.2021.100242
Mas, S., Torro, A., Fernández, L., Bec, N., Gongora, C., Larroque, C., et al. (2020). MALDI imaging mass spectrometry and chemometric tools to discriminate highly similar colorectal cancer tissues. Talanta 208:120455. doi: 10.1016/j.talanta.2019.120455
Mazumdar, C., Driggers, E. M., and Turka, L. A. (2020). The untapped opportunity and challenge of immunometabolism: a new paradigm for drug discovery. Cell Metab. 31, 26–34. doi: 10.1016/J.CMET.2019.11.014
McCaffrey, E. F., Donato, M., Keren, L., Chen, Z., Fitzpatrick, M., Jojic, V., et al. (2020). Multiplexed imaging of human tuberculosis granulomas uncovers immunoregulatory features conserved across tissue and blood. Biorxiv [Preprint] doi: 10.1101/2020.06.08.140426
McDonnell, L. A., and Heeren, R. M. (2007). Imaging mass spectrometry. Mass Spectrom. Rev. 26, 606–643. doi: 10.1002/mas.20124
Mcquin, C., Goodman, A., Chernyshev, V., Kamentsky, L., Cimini, A., Karhohs, K. W., et al. (2018). CellProfiler 3.0: next-generation image processing for biology. PLoS Biol. 16:e2005970. doi: 10.1371/journal.pbio.2005970
Merritt, C. R., Ong, G. T., Church, S. E., Barker, K., Danaher, P., Geiss, G., et al. (2020). Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 38, 586–599. doi: 10.1038/s41587-020-0472-9
Millet, P., Vachharajani, V., McPhail, L., Yoza, B., and McCall, C. E. (2016). GAPDH binding to TNF-α mRNA contributes to posttranscriptional repression in monocytes: a novel mechanism of communication between inflammation and metabolism. J. Immunol. 196, 2541–2551. doi: 10.4049/jimmunol.1501345
Moore, J. W. J., Moyo, D., Beattie, L., Andrews, P. S., Timmis, J., and Kaye, P. M. (2013). Functional complexity of the Leishmania granuloma and the potential of in silico modeling. Front. Immunol. 4:35. doi: 10.3389/fimmu.2013.00035
Moyo, D., Beattie, L., Andrews, P. S., Moore, J. W. J., Timmis, J., Sawtell, A., et al. (2018). Macrophage transactivation for chemokine production identified as a negative regulator of granulomatous inflammation using agent-based modeling. Front. Immunol. 9:637. doi: 10.3389/fimmu.2018.00637
Murray, H. W. (2001). Tissue granuloma structure-function in experimental visceral leishmaniasis. Int. J. Exp. Pathol. 82, 249–267. doi: 10.1046/j.1365-2613.2001.00199.x
Murray, P. J., Allen, J. E., Biswas, S. K., Fisher, E. A., Gilroy, D. W., Goerdt, S., et al. (2014). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20. doi: 10.1016/j.immuni.2014.06.008
Norris, J. L., and Caprioli, R. M. (2013). Analysis of tissue specimens by matrix-assisted laser desorption/ionization imaging mass spectrometry in biological and clinical research. Chem. Rev. 113, 2309–2342. doi: 10.1021/cr3004295
Palsson-McDermott, E. M., Curtis, A. M., Goel, G., Lauterbach, M. A. R., Sheedy, F. J., Gleeson, L. E., et al. (2015). Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 21, 65–80. doi: 10.1016/j.cmet.2014.12.005
Parra, E. R., Francisco-Cruz, A., and Wistuba, I. I. (2019). State-of-the-art of profiling immune contexture in the era of multiplexed staining and digital analysis to study paraffin tumor tissues. Cancers 11:247. doi: 10.3390/cancers11020247
Pisu, D., Huang, L., Grenier, J. K., and Russell, D. G. (2020). Dual RNA-Seq of Mtb-infected macrophages in vivo reveals ontologically distinct host-pathogen interactions. Cell Rep. 30, 335–350. doi: 10.1016/j.celrep.2019.12.033
Porta Siegel, T., Hamm, G., Bunch, J., Cappell, J., Fletcher, J. S., and Schwamborn, K. (2018). Mass spectrometry imaging and integration with other imaging modalities for greater molecular understanding of biological tissues. Mol. Imaging Biol. 20, 888–901. doi: 10.1007/s11307-018-1267-y
Rendeiro, A. F., Ravichandran, H., Bram, Y., Chandar, V., Kim, J., Meydan, C., et al. (2021). The spatial landscape of lung pathology during COVID-19 progression. Nature 593, 564–569. doi: 10.1038/s41586-021-03475-6
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2009). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Russell, D. G., Huang, L., and VanderVen, B. C. (2019). Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 19, 291–304. doi: 10.1038/s41577-019-0124-9
Satija, R., and Shalek, A. K. (2014). Heterogeneity in immune responses: from populations to single cells. Trends Immunol. 35, 219–229. doi: 10.1016/j.it.2014.03.004
Saunders, E. C., and McConville, M. J. (2020). Immunometabolism of leishmania granulomas. Immunol. Cell Biol. 98, 832–844. doi: 10.1111/imcb.12394
Schürch, C. M., Bhate, S. S., Barlow, G. L., Phillips, D. J., Noti, L., Zlobec, I., et al. (2020). Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive. Front. Cell 182:1341–1359. doi: 10.1016/j.cell.2020.07.005
Schwamborn, K., Kriegsmann, M., and Weichert, W. (2017). MALDI imaging mass spectrometry–from bench to bedside. Biochim. Biophys. Acta Proteins Proteom. 1865, 776–783. doi: 10.1016/j.bbapap.2016.10.014
Scott, A. J., Post, J. M., Lerner, R., Ellis, S. R., Lieberman, J., Shirey, K. A., et al. (2017). Host-based lipid inflammation drives pathogenesis in Francisella infection. Proc. Natl. Acad. Sci. U.S.A. 114, 12596–12601. doi: 10.1073/pnas.1712887114
Ščupáková, K., Balluff, B., Tressler, C., Adelaja, T., Heeren, R. M. A., Glunde, K., et al. (2020). Cellular resolution in clinical MALDI mass spectrometry imaging: the latest advancements and current challenges. Clin. Chem. Lab. Med. 58, 914–929. doi: 10.1515/cclm-2019-0858
Scupakova, K., Dewez, F., Walch, A. K., Heeren, R. M. A., and Balluff, B. (2020). Morphometric cell classification for single-cell MALDI-mass spectrometry imaging. Angew. Chem. Int. Ed. Engl. 59, 17447–17450. doi: 10.1002/anie.202007315
Seng, P., Rolain, J.-M., Fournier, P. E., La Scola, B., Drancourt, M., and Raoult, D. (2010). MALDI-TOF-mass spectrometry applications in clinical microbiology. Future Microbiol. 5, 1733–1754. doi: 10.2217/fmb.10.127
Shi, F., Chen, G., Wang, Y., Yang, N., Chen, Y., Dey, N., et al. (2019). “Texture features based microscopic image classification of liver cellular granuloma using artificial neural networks,” in Proceedings of the 2019 IEEE 8th Joint International Information Technology and Artificial Intelligence Conference (ITAIC), (Chongqing: Institute of Electrical and Electronics Engineers), 432–439.
Shirai, T., Nazarewicz, R. R., Wallis, B. B., Yanes, R. E., Watanabe, R., Hilhorst, M., et al. (2016). The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 213, 337–354. doi: 10.1084/jem.20150900
Siewe, N., Yakubu, A. A., Satoskar, A. R., and Friedman, A. (2017). Granuloma formation in leishmaniasis: a mathematical model. J. Theor. Biol. 412, 48–60. doi: 10.1016/j.jtbi.2016.10.004
Silva Miranda, M., Breiman, A., Allain, S., Deknuydt, F., and Altare, F. (2012). The tuberculous granuloma: an unsuccessful host defence mechanism providing a safety shelter for the bacteria? Clin. Dev. Immunol. 2012:139127. doi: 10.1155/2012/139127
Single-cell multimodal omics (2020). Method of the year 2019: single-cell multimodal omics. Nat. Methods 17:1. doi: 10.1038/s41592-019-0703-5
Sun, S., Zhu, J., Ma, Y., and Zhou, X. (2019). Accuracy, robustness and scalability of dimensionality reduction methods for single-cell RNA-seq analysis. Genome Biol. 20:269. doi: 10.1186/s13059-019-1898-6
Takats, Z., Strittmatter, N., and McKenzie, J. S. (2017). “Chapter nine - ambient mass spectrometry in cancer research,” in Advances in Cancer Research, eds R. R. Drake and L. A. McDonnell (Cambridge, MA: Academic Press), 231–256. doi: 10.1016/bs.acr.2016.11.011
Tannahill, G. M., Curtis, A. M., Adamik, J., Mcgettrick, A. F., Goel, G., Frezza, C., et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242. doi: 10.1038/nature11986
TissueGnostics StrataQuest (2021). TissueGnostics StrataQuest–Meyer Instruments. Available online at: https://www.meyerinst.com/brand/tissuegnostics/tissuegnostics-strataquest/ (accessed April 28, 2021).
Trapnell, C., Cacchiarelli, D., Grimsby, J., Pokharel, P., Li, S., Morse, M., et al. (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. doi: 10.1038/nbt.2859
Van den Bossche, J., Baardman, J., Otto, N. A., van der Velden, S., Neele, A. E., van den Berg, S. M., et al. (2016). Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 17, 684–696. doi: 10.1016/j.celrep.2016.09.008
van den Bossche, J., Lamers, W. H., Koehler, E. S., Geuns, J. M. C., Alhonen, L., Uimari, A., et al. (2012). Pivotal advance: arginase-1-independent polyamine production stimulates the expression of IL-4-induced alternatively activated macrophage markers while inhibiting LPS-induced expression of inflammatory genes. J. Leukoc. Biol. 91, 685–699. doi: 10.1189/jlb.0911453
Vats, D., Mukundan, L., Odegaard, J. I., Zhang, L., Smith, K. L., Morel, C. R., et al. (2006). Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 4, 13–24. doi: 10.1016/j.cmet.2006.05.011
Westermann, A. J., Barquist, L., and Vogel, J. (2017). Resolving host–pathogen interactions by dual RNA-seq. PLoS Pathog. 13:e1006033. doi: 10.1371/journal.ppat.1006033
Westermann, A. J., Förstner, K. U., Amman, F., Barquist, L., Chao, Y., Schulte, L. N., et al. (2016). Dual RNA-seq unveils noncoding RNA functions in host-pathogen interactions. Nature 529, 496–501. doi: 10.1038/nature16547
Wilson, J. L., Mayr, H. K., and Weichhart, T. (2019). Metabolic programming of macrophages: implications in the pathogenesis of granulomatous disease. Front. Immunol. 10:2265. doi: 10.3389/fimmu.2019.02265
Wolf, A. F., Angerer, P., and Theis, F. J. (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19:15. doi: 10.1111/1462-2920.13787
Xie, M., Yu, Y., Kang, R., Zhu, S., Yang, L., Zeng, L., et al. (2016). PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 7:13280. doi: 10.1038/ncomms13280
Zhang, Y., Gao, Y., Qiao, L., Wang, W., and Chen, D. (2020). Inflammatory response cells during acute respiratory distress syndrome in patients with coronavirus disease 2019 (COVID-19). Ann. Intern. Med. 173, 402–404. doi: 10.7326/L20-0227
Zheng, L., T’Kind, R., Decuypere, S., von Freyend, S. J., Coombs, G. H., and Watson, D. G. (2010). Profiling of lipids in Leishmania donovani using hydrophilic interaction chromatography in combination with Fourier transform mass spectrometry. Rapid Commun. Mass Spectrom. 24, 2074–2082. doi: 10.1002/rcm.4618
Keywords: immunometabolism, infectious disease, inflammation, granulomas, mass spectrometry imaging, spatial transcriptomics, multi-omics, leishmaniasis
Citation: Tans R, Dey S, Dey NS, Calder G, O’Toole P, Kaye PM and Heeren RMA (2021) Spatially Resolved Immunometabolism to Understand Infectious Disease Progression. Front. Microbiol. 12:709728. doi: 10.3389/fmicb.2021.709728
Received: 17 May 2021; Accepted: 30 July 2021;
Published: 19 August 2021.
Edited by:
George Tsiamis, University of Patras, GreeceReviewed by:
Nupur Biswas, Indian Institute of Chemical Biology (CSIR), IndiaCopyright © 2021 Tans, Dey, Dey, Calder, O’Toole, Kaye and Heeren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul M. Kaye, cGF1bC5rYXllQHlvcmsuYWMudWs=; orcid.org/0000-0002-8796-4755; Ron M. A. Heeren, ci5oZWVyZW5AbWFhc3RyaWNodHVuaXZlcnNpdHkubmw=; orcid.org/0000-0002-6533-7179
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.