 Min He1,2†
Min He1,2† Tao Lei2†
Tao Lei2† Fufeng Jiang2,3
Fufeng Jiang2,3 Jumei Zhang2
Jumei Zhang2 Haiyan Zeng2
Haiyan Zeng2 Juan Wang2
Juan Wang2 Moutong Chen2
Moutong Chen2 Liang Xue2
Liang Xue2 Shi Wu2
Shi Wu2 Qinghua Ye2
Qinghua Ye2 Rui Pang2
Rui Pang2 Yu Ding2*
Yu Ding2* Qingping Wu2*
Qingping Wu2*- 1School of Bioscience and Bioengineering, South China University of Technology, Guangzhou, China
- 2Guangdong Provincial Key Laboratory of Microbial Safety and Health, State Key Laboratory of Applied Microbiology Southern China, Institute of Microbiology, Guangdong Academy of Sciences, Guangzhou, China
- 3School of Food and Biological Engineering, Shaanxi University of Science and Technology, Xi’an, China
Vibrio parahaemolyticus is a common foodborne pathogen that causes gastroenteritis worldwide. Determining its prevalence and genetic diversity will minimize the risk of infection and the associated economic burden. Multilocus sequence typing (MLST) is an important tool for molecular epidemiology and population genetic studies of bacteria. Here, we analyzed the genetic and evolutionary relationships of 162 V. parahaemolyticus strains isolated in the Guangdong Province, China, using MLST. In the study, 120 strains were isolated from food samples, and 42 strains were isolated from clinical samples. All strains were categorized into 100 sequence types (STs), of which 58 were novel (48 from the food isolates and 10 from the clinical isolates). ST415 was the most prevalent ST among the food isolates, while ST3 was the most prevalent ST among the clinical isolates. Further, 12 clonal complexes, 14 doublets, and 73 singletons were identified in all ST clusters, indicating high genetic diversity of the analyzed strains. At the concatenated sequence level, non-synonymous sites in both, food and clinical isolates, were associated with purifying selection. Of note, the dN/dS ration was greater than 1 for some housekeeping genes in all isolates. This is the first time that some loci under positive selection were identified. These observations confirm frequent recombination events in V. parahaemolyticus. Recombination was much more important than mutation for genetic heterogeneity of the food isolates, but the probabilities of recombination and mutations were almost equal for the clinical isolates. Based on the phylogenetic analysis, the clinical isolates were concentrated in the maximum-likelihood tree, while the food isolates were heterogeneously distributed. In conclusion, the food and clinical isolates of V. parahaemolyticus from the Guangdong Province are similar, but show different evolutionary trends. This may help prevent large-scale spread of highly virulent strains and provides a genetic basis for the discovery of microevolutionary relationships in V. parahaemolyticus populations.
Introduction
Vibrio parahaemolyticus, a halophilic, Gram-negative bacterium, is commonly found in estuarine and marine environments worldwide. This bacterium is an important pathogen of human. It typically causes acute gastroenteritis, wound infections, and septicemia, with high contamination rates worldwide (Ralph and Currie, 2007; Zhang and Orth, 2013). It is also considered one of the most prevalent foodborne pathogens (Letchumanan et al., 2014).
The main virulence factors of V. parahaemolyticus are thermostable direct hemolysin (TDH), TDH-related hemolysin (TRH), thermolabile hemolysin (TLH), three type 3 secretion systems (T3SS1, T3SS2α, and T3SS2β) and two type 6 secretion systems (T6SS1, T6SS2) (Park et al., 2004; Zhang and Austin, 2005; Okada et al., 2009; Hiyoshi et al., 2010). TDH acts directly on red blood cells and shows hemolytic and enterotoxin activity, cardiotoxicity, and cytotoxicity (Matsuda et al., 2009; Hiyoshi et al., 2010). TRH is composed of 189 amino acids and shares 54.8–68.8% sequence identity with TDH. It is present in a small number of clinical isolates and is encoded by the trh1 and trh2 genes (Kishishita et al., 1992). TLH is an atypical phospholipase that causes hemolysis only in the presence of lecithin (Zhang and Austin, 2005). T3SS1 mainly affects biofilm formation, movement, and cytotoxicity and contributes to the survival of V. parahaemolyticus in the environment. T3SS2 is involved in the negative regulation of cellular inflammatory response, which helps the bacterium to evade the immune response of the host (Calder et al., 2014). Finally, T6SS1 enhances the environmental adaptability of V. parahaemolyticus in the marine environment, while T6SS2 allows V. parahaemolyticus to adhere to host cells (Yu et al., 2012).
Many V. parahaemolyticus serotypes are associated with infection. For instance, the pandemic serotype O3: K6 was the main cause of a global outbreak of V. parahaemolyticus infection (Nair et al., 2007). In fact, many countries have reported outbreaks of V. parahaemolyticus infection, including the US (Shaw et al., 2015), Italy (Caburlotto et al., 2016), Brazil (Leal et al., 2008), Japan (Arakawa et al., 1999), and Bangladesh (Bhuiyan et al., 2002). Consumption of raw or undercooked seafood contaminated with V. parahaemolyticus is the main cause of outbreaks of V. parahaemolyticus infection (McLaughlin et al., 2005; Pal and Das, 2010). In China, where high amounts of seafood are consumed, most foodborne outbreaks and cases of infectious diarrhea are caused by V. parahaemolyticus, especially in the coastal areas (Liu et al., 2018; Paudyal et al., 2018). Further, clinical isolates in China usually represent the pandemic serotype O3: K6 and several of its serovariants (e.g., O1: KUT, O1: K56, and O4: K68) (Yu et al., 2011; Li et al., 2014).
The risk of V. parahaemolyticus infection and the associated economic burden can be minimized by determining the prevalence and genetic diversity of this pathogen at the population level. Molecular biology methods have rapidly advanced in recent years, and many molecular typing techniques are used for molecular epidemiological studies of V. parahaemolyticus (Gonzalez-Escalona et al., 2008; Chen et al., 2012). Multilocus sequence typing (MLST) of V. parahaemolyticus isolates, developed by Gonzalez-Escalona et al. (2008), is an important tool for molecular epidemiology and population genetic studies of this bacterium (Urwin and Maiden, 2003; Turner and Feil, 2007; Gonzalez-Escalona et al., 2008). However, although a number of studies have used MLST to investigate the genetic diversity of V. parahaemolyticus in China, these studies either focused on clinical isolates or food isolates. A comparative analysis of food and clinical isolates is missing.
In the current study, we performed genetic and population analysis of 162 food and clinical isolates of V. parahaemolyticus from the Guangdong Province, China. We compared their genetic diversity, prevalence, and population structure using MLST and investigated possible evolutionary relationships of these isolates by sequence polymorphism analysis. We also determined the differences in the genetic population structure of foodborne and clinical V. parahaemolyticus isolates. The findings provide genetic evidence on the microevolutionary relationships among V. parahaemolyticus isolates from different sources, with the potential to support the development of new strategies for the prevention of infection.
Materials and Methods
Bacterial Isolates
In the study, 162 V. parahaemolyticus isolates, including 120 food isolates and 42 clinical isolates, from the Guangdong Province, China, were analyzed (Supplementary Table 1). All isolates were stored and maintained in the authors’ laboratory. Aquatic product and ready-to-eat foods samples were collected in Guangdong province, China, from January to June in 2016. After collection, all samples were placed in sterile sealed plastic bags, transported to the laboratory in a cold box and analyzed immediately. According to National Standards of the People’s Republic of China (GB 4789.7-2013), V. parahaemolyticus were isolated and identified. In brief, each sample (25.0 g) was homogenized in a stomacher bag with 225 mL alkaline peptone water containing 3% (w/v) NaCl. Homogenates were incubated at 37°C for 16–18 h. Samples were streaked onto thiosulfate citrate bile-salt sucrose (TCBS) agar plates and incubated at 37°C for 18–24 h. Presumptive colonies (green or blue green colonies, 2–3 mm in diameter) were streaked onto Chromogenic Vibrio Medium and incubated at 37°C for 24 h. Mauve colonies were selected for further V. parahaemolyticus identification by analysis of oxidase activity, Gram staining, 3.5% NaCl triple sugar iron (TSI) tests, halophilism tests, and API 20E diagnostic strips (Biomerieux Company, France). Thirty-one clinical isolates collected from patients in Guangdong, China were gifted by the Nanshan, Shenzhen Center for Disease Control and Prevention. Eleven clinical isolates were isolated in the authors’ laboratory.
Polymerase Chain Reaction Amplification and Sequencing
Before the analysis, the bacteria were grown in tryptic soy broth (HuanKai Microbial, Guangzhou, China) supplemented with 3% (w/v) NaCl at 37°C overnight. Genomic DNA was extracted from bacteria using a Genomic DNA Extraction kit (Magen Biotech, Guangzhou, China), according to the manufacturer’s instructions. Seven loci (housekeeping genes) were selected for MLST analysis, based on a previously published MLST scheme for V. parahaemolyticus (Gonzalez-Escaloa et al., 2017). These were: recA, encoding RecA protein; dnaE, encoding alpha subunit of DNA polymerase III; gyrB, encoding B subunit of DNA gyrase; dtdS, encoding threonine dehydrogenase; pntA, encoding alpha subunit of transhydrogenase; pyrC, encoding dihydroorotase; and tnaA, encoding tryptophanase. Among them, recA, dnaE, and gyrB are located on chromosome I, and dtdS, pntA, pyrC, and tnaA are located on chromosome II. The genes were amplified following the polymerase chain reaction protocol available on the V. parahaemolyticus PubMLST website1 and using the primer sets listed in Supplementary Table 2. The products of polymerase chain reaction were then subjected to 1.5% (w/v) agarose gel electrophoresis with gold-view staining to purify, and bi-directionally sequenced using an ABI-3700XL instrument (BGI Instruments, Guangdong, China). The raw sequencing data of all strains were in Supplementary Data Sheet 1–8.
MLST Analysis
For MLST analysis, AB1 sequencing files were first imported into Bionumerics v. 7.6 (Applied Maths, Sint-Martens-Latem, Belgium). The sequences were edited and complementary locus fragments from individual isolates were assembled, trimmed and aligned by Bionumerics v. 7.6. Next the allele name and sequence type (ST) of each isolate were determined by querying the gene sequence in the PubMLST database. Whenever novel alleles were identified, their sequences and new allelic profiles were submitted to the PubMLST database, and new numerical identifiers were assigned.
Global optimal eBURST (goeBURST) (Francisco et al., 2009) analysis of related STs was performed using the Phyloviz software,2 and the clonal relationships among isolates were obtained. A single clonal complex (CC) was defined as a complex in which at least six of the seven alleles analyzed were identical among the isolates. Single- and double-locus variants (SLVs and DLVs, respectively) were defined as two STs differing at a single locus or at two loci, respectively. The goeBURST algorithm was also used to take a population snapshot of groups sharing between 0 and 7 loci, to create a full MLST map, where all STs are connected (Francisco et al., 2009).
Phylogenetic Analysis
MUSCLE (Edgar, 2004) was used to perform multiple sequence alignments of each locus. A majority consensus phylogeny of the concatenated loci (3,682 bp) was constructed using the maximum likelihood (ML) method implemented in MEGA X (Kumar et al., 2018). The nodes in the ML tree were evaluated by 1000 bootstrap-based re-sampling.
Population Genetics Analysis
The in-frame sequences were concatenated in the order dnaE-gyrB-recA-dtdS-pntA-pyrC-tnaA to produce a cumulative sequence (3682 bp in length) for each strain. DNASP v. 6 was used to calculate the number of polymorphic sites, nucleotide diversity (π), and the dN/dS ratio for individual loci and the concatenated sequences, to evaluate locus variation in the selected isolates (Rozas et al., 2017). Pi (π) represents the average number of nucleotide differences at each site between two randomly selected strains. The higher the π value, the higher the gene nucleotide polymorphism. The dN/dS ratio is used for the selection and hypothesis tests for neutrality (dN = dS): non-synonymous sites are considered to be under purifying selection (negative selection) if dN/dS < 1; dN/dS > 1 indicates positive selection; and dN/dS = 1 indicates neutrality. In the analysis, dN represents the number of synonymous substitutions per synonymous locus, and dS represents the number of non-synonymous substitutions per non-synonymous locus. Further, recombinant phi test of individual loci in the whole strain collection was performed using SplitsTree v. 4.0 (Huson and Bryant, 2006). For the calculated p-values, p < 0.05 indicated the occurrence of recombination.
The standardized index of association (IAS) was calculated using LIAN v. 3.7 (Haubold and Hudson, 2000) to assess the population structure of V. parahaemolyticus; 10,000 iterations in the Monte Carlo mode were used. For all pairwise comparisons of allelic profiles, IAS was defined as the observed variance of the distribution of allelic mismatches divided by the expected variance of the free recombination population minus 1. Statistics were used to verify the null hypothesis of linkage equilibrium: if alleles are in a linkage equilibrium (i.e., independently distributed at all loci analyzed), recombination occurs frequently and IAS = 0.
LDhat program in RDP4 package (Martin et al., 2017) was used to calculate the mean per-site recombination (rho)/mutation (theta) ratio of concatenated sequences of STs with 1,000,000 MCMC updates. The ratio indicates the probability that a given site is altered by recombination and mutation. Hence, the rho/theta ratio is used to measure the effect of recombination on the diversity of sample relative to a given mutation.
Results
V. parahaemolyticus Isolates From Clinical and Food Sources Show High Levels of Nucleotide and Allelic Diversity
MLST analysis revealed 98 unique STs, 58 (59.18%) of which were novel, as compared with the previous PubMLST database entries. Among the novel STs, 48 (57.14%) were identified among the food isolates and 10 (66.67%) were identified among the clinical isolates (Table 1). Further, 77 unique STs were represented by single isolates, and 21 STs were associated with more than one strain. ST3 was the most frequently detected ST, represented by 23 clinical isolates. ST1820 contained both food and clinical isolates. The observed number of alleles for each locus ranged from 47 (tnaA) to 70 (gyrB). Interestingly, dnaE was the locus showing the most new alleles (n = 10). Accordingly, the nucleotide and allelic diversity of the seven loci were significantly different (Table 2). The nucleotide diversity ranged from 0.00979 to 0.03812 for all isolates, with the greatest diversity observed for tnaA (0.03812). However, pntA was the least polymorphic locus, with the lowest nucleotide diversity per site (π = 0.00979). With regard to the source of the isolates, the values ranged from 0.00604 (dnaE) to 0.0178 (dtdS) for the clinical isolates and from 0.00852 (pntA) to 0.04687(tnaA) for the food isolates (Tables 3, 4). The food isolates showed higher nucleotide diversity than the clinical isolates. Collectively, both, the food and clinical isolates showed high levels of nucleotide and allelic diversity at all loci studied.

Table 1. Properties of each locus and sequence type.
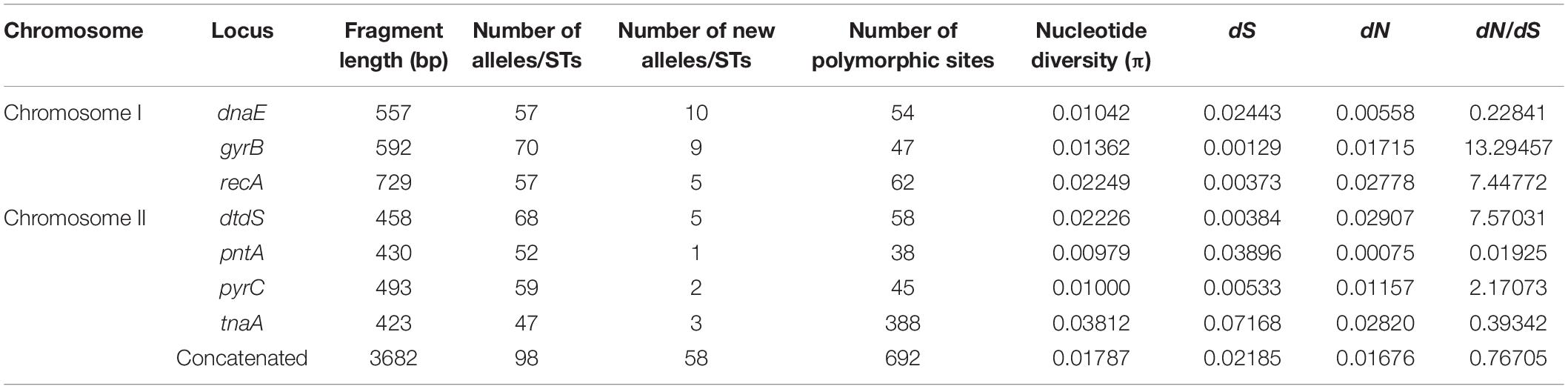
Table 2. Nucleotide and allelic sequences diversity of all isolates.
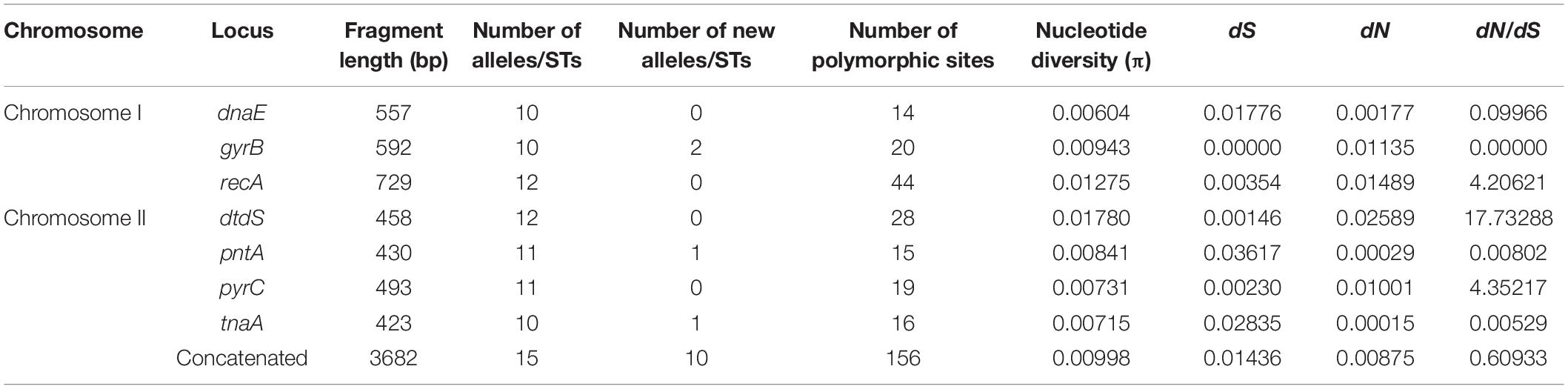
Table 3. Nucleotide and allelic sequences diversity of clinical isolates.
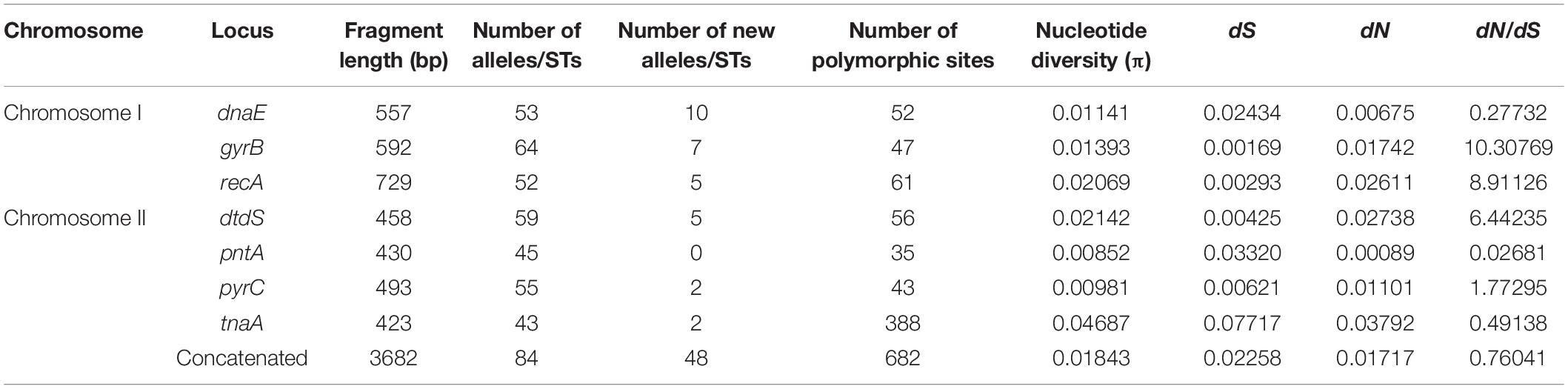
Table 4. Nucleotide and allelic sequences diversity of food isolates.
Evidence of Purifying and Positive Selection in V. parahaemolyticus Isolates From Clinical and Food Sources
The dN/dS ratios (Table 2) were < 1 for dnaE, pntA, and tnaA and > 1 for gyrB, recA, dtdS, and pyrC. A similar trend was observed in the food and clinical isolates, except for gyrB, in which no non-synonymous changes were identified in the clinical isolates (Tables 3, 4). To the best of our knowledge, this is the first study reporting dN/dS ratios > 1 for the four housekeeping genes. Since dN/dS < 1 indicates that the evolutionary rate of non-synonymous loci is lower than that of synonymous loci, the dnaE, pntA, and tnaA genes were mainly affected by purifying selection during evolution. However, dN/dS > 1 indicated positive selection for gyrB, recA, dtdS, and pyrC. Of note, the π-value and mean dN/dS ratio of food isolates (0.01843 and 0.76041, respectively) were 1.8- and 1.2-fold higher, respectively, than those of clinical isolates (0.00998 and 0.60933, respectively) (Tables 4, 3). Collectively, these observations suggest that the purifying selection of food and clinical isolates was similar overall, while the genetic evolution of food isolates was faster than that of clinical isolates.
V. parahaemolyticus Isolates From Clinical and Food Sources Show High Level of Recombination
The phi test p-values for the concatenated sequences of all strains were < 0.001, indicating a significant evidence of recombination. This was also true for the clinical and food isolates considered separately, suggesting recombination in these strains. The rho/theta ratio for the concatenated sequences was 8.703, suggesting that recombination was approximately 8.7 times more likely to occur than point mutations during strain evolution. It is consistent with the previous study that rho/theta > 1, indicating that recombination had a more important impact on genetic heterogeneity and diversity than mutation (Dong et al., 2016). The rho/theta values for food and clinical isolates were 14.264 and 1.476, respectively. Both these values were above 1. These observations suggest that recombination occurred more frequently than point mutations in the food isolates, while the probability of recombination was close to that of point mutation in the clinical isolates. Next, the standardized index of association (ISA) was calculated (Table 5). The ISA value for the whole strain collection was 0.6269, significantly different from zero (p < 0.0001). Considering the food and clinical strains separately, the ISA values were 0.4429 and 0.8361, respectively, and were both significantly positive (p < 0.0001). Importantly, the ISA values were consistent with the notion of recombination in the V. parahaemolyticus population. Collectively, these data indicate a tendency of linkage disequilibrium between the alleles, which is consistent with the existing research results (Han et al., 2015; Li et al., 2018).
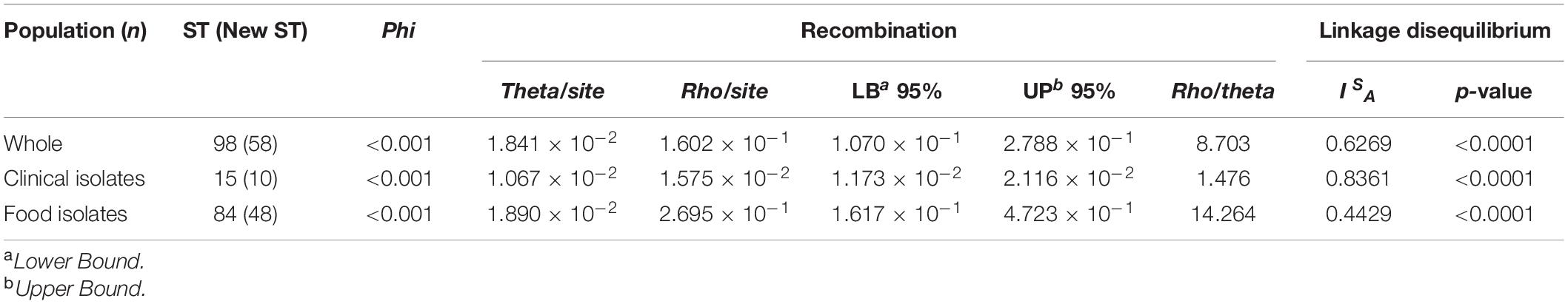
Table 5. Recombination test and estimation.
Identification of CCs
The goeBURST algorithm used in the current study resolved the 98 STs into twelve CCs (CC3, CC8, CC587, CC564, CC114, CC415, CC471, CC515, CC864, CC1236, CC2126, andCC1498) (Figure 1). Among them, except CC415 contains 4 STs, the remaining eleven CCs contain 2 STs. The clinical isolates contained three CCs, CC3, CC8, CC587, and the food isolates contained eight CCs, CC564, CC114, CC415, CC471, CC515, CC864, CC1236, CC2126, respectively. In addition, CC1498 contains both food and clinical isolates. Among the clinical isolates, CC3 was the largest CC, including 24 strains in 2 STs (ST3 and ST2155), of which ST3 contained 23 strains and ST2155 contained 1 strain. Among the food isolates, CC415 was the largest CC, including 11 strains of 4 STs (ST415, ST2129, ST2136, and ST2140), of which ST415 contained 8 strains, and the rest each contained 1 strain. While in food isolates, no ST3 was found in this study, and no STs belonging to the pandemic CC3 were found, which was consistent with the previous study that did not find ST3 and STs belonging to CC3 in food isolates (Dong et al., 2016). CC1498 contained 2 strains from 2 STs (ST1498 and ST2158), of which ST1498 contained one food isolate(Vp266) and ST2158 contained one clinical isolate(r16), indicating that Vp266 and r16 were genetically similar, Vp266 may have potential pathogenicity, and r16 may be derived from food contaminated by highly virulent strains. In addition, fourteen SLVs, five DLVs and 72 singletons were identified from all isolates (Figure 1). In conclusion, the food and clinical isolates contained different CC, suggesting significant evolutionary differences between the food and clinical strains.

Figure 1. Full MLST map of V. parahaemolyticus isolates from food and clinical samples from Guangdong Province, based on nucleotide sequencing. Twelve clonal complexes (CC3, CC8, CC587, CC564, CC114, CC415, CC471, CC515, CC864, CC1236, CC2126, andCC1498), fourteen SLVs, five DLVs and 72 singletons were identified. All connections are shown. STs that are SLVs of each other are connected by black lines. STs that differ in two or more alleles are connected by light gray lines.
Phylogenetic Analysis
ML tree based on the concatenated sequences of the seven housekeeping genes of 162 isolates is shown in Figure 2. Remarkably, the clinical isolates were concentrated in the tree, while the food isolates were heterogeneously distributed. This may be because the food samples were diverse. Specifically, the clinical isolates were separated into three major clusters (Figure 2). Cluster I (n = 23 isolates) was the most homogenous and was composed of one CC (CC3) and only one ST (ST3). Cluster II (n = 6 isolates) was composed of one CC (CC587) and two STs (ST2152 and ST2359). Cluster III (n = 3 isolates) was also composed of one CC (CC8) and two STs (ST8 and ST2151) (Figure 2 and Supplementary Table 1). However, the food isolates were grouped into many small clusters, each composed of different CCs and singletons; 31 small clusters were identified. Of note, a small number of clinical and food isolates appeared in the same cluster, and conversely, single clinical or food isolates formed separate one-isolate clusters.
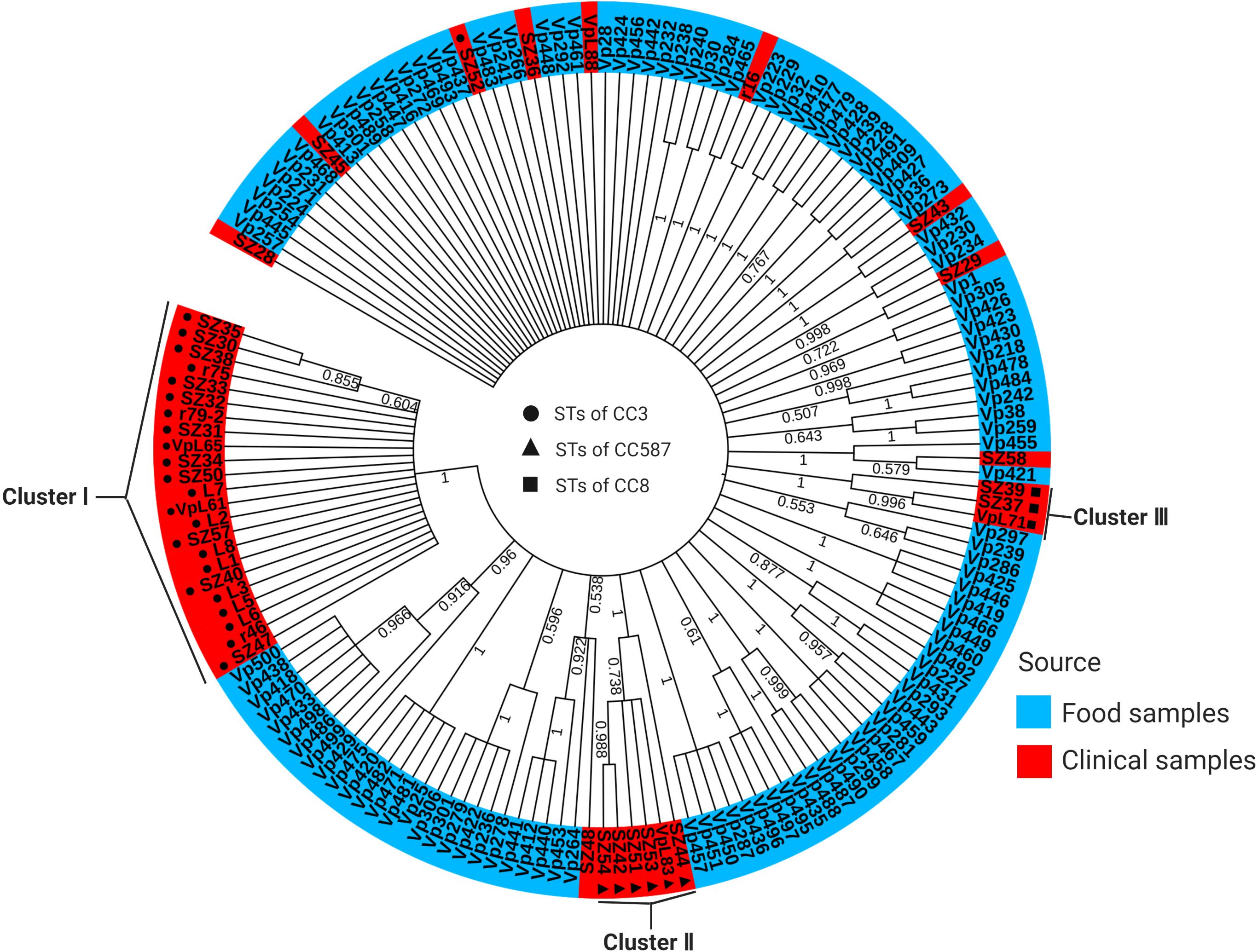
Figure 2. ML tree constructed using concatenated sequences of seven loci (dnaE-gyrB-recA-dtdS-pntA-pyrC-tnaA) of 162 V. parahaemolyticus isolates analyzed in the current study. Food isolates are denoted in blue, and clinical isolates are denoted in red. Circles, triangles, and squares with different shading represent the three CCs of clinical isolates identified by goeBURST. The scale represents the evolutionary distance. The numbers at the nodes represent bootstrap values based on 1000 replications.
Discussion
In the current study, we analyzed the nucleotide diversity of V. parahaemolyticus isolates from food and clinical sources from Guangdong Province, China. We show that the isolates are highly diverse. Overall, 692 polymorphic sites and 100 alleles were identified by concatenated sequence analysis of 162 strains, with 58 novel STs, indicating a high level of genetic diversity of the MLST loci. Interestingly, the clinical isolates (66.67%) harbored a higher proportion of novel STs than the food isolates (55.81%). However, the food strains showed higher nucleotide diversity than the clinical isolates. Further, ST3 was the most common ST, in line with findings of a previous study analyzing samples from various countries (Han et al., 2014).
In the current study, the dN/dS ratio of the concatenated sequences was < 1. However, at the locus level, this was only true for three loci, while the remaining four loci studied showed dN/dS ratios > 1. This indicated that the three loci were subjected to purifying selection, while the remaining four were subjected to positive selection. To the best of our knowledge, this is the first study showing positive selection of housekeeping genes. There are two possible explanations for these findings. The first is related to environmental stressors, such as antibiotic therapy. Extensive antibiotic use is a contributing factor to the increasing incidence of antimicrobial-resistant strains. In fact, according to a recent report, some V. parahaemolyticus isolates are resistant to many commonly used antibiotics, including kanamycin, streptomycin, and ampicillin, because of excessive use of antibiotics in human and aquaculture systems (Park et al., 2018). Of note, all isolates analyzed in this study show various levels of antibiotic resistance (Xu et al., 2016; Xie et al., 2017, 2019). Therefore, to survive under the selective pressure, these V. parahaemolyticus strains had to adapt fast to the external environment, probably by acquiring non-synonymous mutations in some housekeeping genes (rather than acquiring synonymous mutations), in line with what has been reported by others (Yang, 2006). The second explanation relates to the adequateness of using the dN/dS ratio, highlighting a potential limitation of this study. Although this ratio is often calculated for sequences sampled from a single population, it was originally developed for the analysis of distantly divergent sequences. Consequently, this approach ignores the existence of polymorphisms and relies on the indirect assumption that new mutations are instantaneously fixed. This assumption is usually violated and is only reasonable for distantly related species. Of note, violation of the underlying assumption leads to a time-dependence of sequence divergence and biased estimates of the dN/dS ratio, especially for closely related species, where the contribution of ancestral and lineage-specific polymorphisms to sequence divergence is substantial (Mugal et al., 2020). In fact, it has been reported that when a single population under selection is sampled, the dN/dS ratio is relatively insensitive to the selection coefficient. Furthermore, for population samples, the relationship between selection and the dN/dS ratio does not follow a monotonic function; therefore, it may be impossible to infer selection pressures from the dN/dS ratio (Kryazhimskiy and Plotkin, 2008). All of the above point to a need to improve the dN/dS estimation for closely related species by traditional analysis methods. Importantly, the joint usage of polymorphism and divergence data may assist in the inference of selection of closely related species (Mugal et al., 2020). Regardless, high levels of variation generated by purifying and positive selection will ensure a good discriminatory power of MLST and facilitate the subsequent phylogenetic inference.
Various mechanisms of recombination operate in bacteria, such as transformation, phage transduction, and conjugation, which promote the replacement of DNA regions of core genes with homologous sequences from strains of the same or closely related species (Thomas and Nielsen, 2005; Didelot and Maiden, 2010). The recombination rate varies greatly between different bacterial species, and high levels of recombination have been detected in V. parahaemolyticus (Cui et al., 2015). Of note, homologous recombination is of great significance for the survival and evolution of bacterial populations. Relative to spontaneous mutations, homologous recombination is associated with faster mutation speed than spontaneous mutation, which is important for a quick increase of the genetic diversity and the environmental adaptability potential of a population (Vos, 2009). Newly acquired mutated loci affect gene function or regulation, leading to new phenotypes of the corresponding strains, such as evading host immune response, developing multi-drug resistance, and increasing pathogenicity (Hacker and Carniel, 2001; Arnold et al., 2008). Interestingly, the MLST data generated in this study indicated that recombination replacement contributed 8.7 times more than point mutations to the genetic variations among all isolates. Therefore, recombination has a much greater effect than mutation on the generation and maintenance of genetic diversity in V. parahaemolyticus. In fact, the frequency of recombination and mutation in the clinical isolates was almost the same. On the one hand, that may be because the maintenance of some virulence-related functions requires gene stability and hence, the frequency of recombination is reduced. On the other hand, mutations can make the gene change at the level of function or regulation, leading to new phenotypes in corresponding strains, such as the virulence is enhanced, so the mutation frequency in clinical strains may increase to increase the virulence. In clinical strains, the frequency of recombination decreases and that of mutation increases, and ultimately, the two become close. However, the frequency of recombination in the food isolates was approximately 14 times that of mutation, indicating that recombination plays a greater role in maintaining genetic diversity of these isolates. Food isolates were obtained from different types of food sampled at different places and inhabit complex and diverse environments. Recombination might occur more frequently than mutation in these isolates to allow better adaptation to these environments.
Clonal, epidemic, and panmictic are three models of population structure that are used to distinguish bacterial pathogens. Clonal population contains strains derived from a common ancestor that diversified predominantly by mutation rather than recombination within linkages (Smith et al., 1993). In epidemic population, well-adapted clones become widely distributed, with recombination occurring much more frequently than mutation (Smith et al., 2000). Finally, in a panmictic population, all strains are potential partners without genetic mating restrictions, recombination occurs freely, and polymorphic sites are all at linkage equilibrium (Suerbaum et al., 1998).
V. parahaemolyticus bacteria are widely distributed in estuarine, marine, and coastal environments, suggesting they are able to rapidly adapt to various environments. Consistently, high levels of genotypic diversity were observed for V. parahaemolyticus isolates in this study and numerous singletons were detected in the population snapshot. Further, the ISA values were significantly different from zero for all the analyzed strain sets, indicating a tendency of linkage disequilibrium between the alleles. Of note, in the clonal population, recombination does not occur freely, and there is no random distribution of alleles in general; however, recombination can occur within different subpopulations (Gonzalez-Escalona et al., 2008). Consequently, the data suggest that V. parahaemolyticus populations follow the epidemic model of clonal expansion (Chao et al., 2011; Yan et al., 2011).
In the current study, when the eBURST algorithm and the ML tree were used to assess phylogenetic relationships, the results obtained were different. These two analysis tools have different advantages. eBURST analysis can be used to quickly identify clonal complexes and their ancestral STs, while the ML trees are better at revealing evolutionary relationships among different STs. The high degree of allelic diversity among the analyzed strains decreased the ability of goeBURST to identify related genotypes; evolutionary relationships are reliable only for identical or closely related strains (SLVs to TLVs) (Francisco et al., 2009). Therefore, simultaneous application of the two models would lead to more reliable conclusions than using either model alone.
Phylogenetic analysis revealed that the clinical isolates were more concentrated in the ML tree, while the food isolates were more dispersed, and few food isolates and clinical isolates were in the same cluster. A possible explanation is that the maintenance of some virulence-related functions of the clinical isolates requires gene stability, and the frequency of recombination is relatively reduced, while the food isolates frequently recombine to adapt to various environments. High-frequency recombination disrupts the vertical evolutionary relationship. The phylogeny is presented as a radial structure, which cannot reflect the sequence of evolution and the distance between species (Cui et al., 2015). Food isolates in the same cluster as clinical isolates may be potentially pathogenic strains that have contaminated the food but did not cause infection, resulting in outbreaks of foodborne diseases. Therefore, these strains should become key strains for the assessment of V. parahaemolyticus contamination. In addition, no ST3 and STs belonging to the pandemic CC3 were found in the food isolates, but ST3 was the most prevalent ST in the clinical isolates, if ST3 or STs belonging to CC3 is present in food isolates, these bacteria will be a priority for monitoring This observation can provide a new reference strategy for tracing the sources of clinical isolates and preventing the outbreaks of V. parahaemolyticus infection.
Conclusion
The study revealed high genetic diversity of the analyzed V. parahaemolyticus strains, with many novel alleles and STs identified. Importantly, in both, food and clinical isolates, the housekeeping genes were mainly affected by purifying selection during the evolutionary process. We also showed that recombination plays a much greater role than mutation in the generation of genetic heterogeneity, particularly in the food isolates. The ISA values were significantly different from zero for all the analyzed strain sets, indicating that the alleles are in linkage disequilibrium in both food and clinical isolates. The clinical isolates showed a concentrated distribution in the ML tree, while the food isolates were more distinct. Of note, CC415 was the most prevalent CC among the food isolates, while CC3 was the most prevalent CC among the clinical isolates. Collectively, these observations suggest that while the food and clinical isolates of V. parahaemolyticus from Guangdong Province are similar, they show different evolutionary trends. This may help prevent large-scale spread of highly virulent strains.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
QW and YD conceived the study. TL and HZ designed the experiments. MH and FJ conducted the experiments. MH wrote the manuscript. MC, RP, and LX contributed to data analysis. JZ supervised the experiment. JW, SW, and QY reviewed and edited the manuscript. All authors agreed to accountable for the content of the work.
Funding
This study was funded by the National Natural Science Foundation of China (NSFC) (Grant No. 31730070), Guangdong Provincial Key Laboratory of Microbial Safety and Health (Grant No. 2020B121201009), and GDAS’ Special Project of Science and Technology Development (Grant No. 2019GDASYL-0201001). The fund providers had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Xiaoke Xu, Kui Wu, and Tengfei Xie for the isolation and purification of V. parahaemolyticus from food and clinical samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.708795/full#supplementary-material
Footnotes
References
Arakawa, E., Murase, T., Shimada, T., Okitsu, T., Yamai, S., and Watanabe, H. (1999). Emergence and prevalence of a novel Vibrio parahaemolyticus O3: K6 clone in Japan. Jpn. J. Infect. Dis. 52, 246–247.
Arnold, M. L., Sapir, Y., and Martin, N. H. (2008). Genetic exchange and the origin of adaptations: prokaryotes to primates. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 2813–2820. doi: 10.1098/rstb.2008.0021
Bhuiyan, N. A., Ansaruzzaman, M., Kamruzzaman, M., Alam, K., Chowdhury, N. R., Nishibuchi, M., et al. (2002). Prevalence of the pandemic genotype of Vibrio parahaemolyticus in Dhaka, Bangladesh, and significance of its distribution across different serotypes. J. Clin. Microbiol. 40, 284–286. doi: 10.1128/jcm.40.1.284-286.2002
Caburlotto, G., Suffredini, E., Toson, M., Fasolato, L., Antonetti, P., Zambon, M., et al. (2016). Occurrence and molecular characterisation of Vibrio parahaemolyticus in crustaceans commercialised in Venice area, Italy. Int. J. Food Microbiol. 220, 39–49. doi: 10.1016/j.ijfoodmicro.2015.12.007
Calder, T., Santos, M.d.S., Attah, V., Klimko, J., Fernandez, J., Salomon, D., et al. (2014). Structural and regulatory mutations in Vibrio parahaemolyticus type III secretion systems display variable effects on virulence. FEMS Microbiol. Lett. 361, 107–114. doi: 10.1111/1574-6968.12619
Chao, G., Wang, F., Zhou, X., Jiao, X., Huang, J., Pan, Z., et al. (2011). Origin of Vibrio parahaemolyticus O3:K6 pandemic clone. Int. J. Food Microbiol. 145, 459–463. doi: 10.1016/j.ijfoodmicro.2011.01.022
Chen, W., Xie, Y., Xu, J., Wang, Q., Gu, M., Yang, J., et al. (2012). Molecular typing of Vibrio parahaemolyticus isolates from the middle-east coastline of China. Int. J. Food Microbiol. 153, 402–412. doi: 10.1016/j.ijfoodmicro.2011.12.001
Cui, Y., Yang, X., Didelot, X., Guo, C., Li, D., Yan, Y., et al. (2015). Epidemic clones, oceanic gene pools, and eco-ld in the free living marine pathogen Vibrio parahaemolyticus. Mol. Biol. Evol. 32, 1396–1410. doi: 10.1093/molbev/msv009
Didelot, X., and Maiden, M. C. J. (2010). Impact of recombination on bacterial evolution. Trends Microbiol. 18, 315–322. doi: 10.1016/j.tim.2010.04.002
Dong, X., Li, Z., Wang, X., Zhou, M., Lin, L., Zhou, Y., et al. (2016). Characteristics of Vibrio parahaemolyticus isolates obtained from crayfish (Procambarus clarkii) in freshwater. Int. J. Food Microbiol. 238, 132–138. doi: 10.1016/j.ijfoodmicro.2016.09.004
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Francisco, A. P., Bugalho, M., Ramirez, M., and Carrico, J. A. (2009). Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics 10:152. doi: 10.1186/1471-2105-10-152
Gonzalez-Escaloa, N., Jolley, K. A., Reed, E., and Martinez-Urtaza, J. (2017). Defining a core genome multilocus sequence typing scheme for the global epidemiology of Vibrio parahaemolyticus. J. Clin. Microbiol. 55, 1682–1697. doi: 10.1128/jcm.00227-17
Gonzalez-Escalona, N., Martinez-Urtaza, J., Romero, J., Espejo, R. T., Jaykus, L.-A., and DePaola, A. (2008). Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J. Bacteriol. 190, 2831–2840. doi: 10.1128/jb.01808-07
Hacker, J., and Carniel, E. (2001). Ecological fitness, genomic islands and bacterial pathogenicity–a darwinian view of the evolution of microbes. EMBO Rep. 2, 376–381. doi: 10.1093/embo-reports/kve097
Han, D., Tang, H., Lu, J., Wang, G., Zhou, L., Min, L., et al. (2014). Population structure of clinical Vibrio parahaemolyticus from 17 coastal countries, determined through multilocus sequence analysis. Plos One 9:e107371. doi: 10.1371/journal.pone.0107371
Han, D., Tang, H., Ren, C., Wang, G., Zhou, L., and Han, C. (2015). Prevalence and genetic diversity of clinical Vibrio parahaemolyticus isolates from China, revealed by multilocus sequence typing scheme. Front. Microbiol. 6:291. doi: 10.3389/fmicb.2015.00291
Haubold, B., and Hudson, R. R. (2000). LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics 16, 847–848. doi: 10.1093/bioinformatics/16.9.847
Hiyoshi, H., Kodama, T., Iida, T., and Honda, T. (2010). Contribution of Vibrio parahaemolyticus virulence factors to cytotoxicity, enterotoxicity, and lethality in mice. Infect. Immun. 78, 1772–1780. doi: 10.1128/iai.01051-09
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Kishishita, M., Matsuoka, N., Kumagai, K., Yamasaki, S., Takeda, Y., and Nishibuchi, M. (1992). Sequence variation in the thermostable direct hemolysin-related hemolysin (trh) gene of Vibrio parahaemolyticus. Appl. Environ. Microbiol. 58, 2449–2457. doi: 10.1128/aem.58.8.2449-2457.1992
Kryazhimskiy, S., and Plotkin, J. B. (2008). The population genetics of dN/dS. PLoS Genet. 4:e1000304. doi: 10.1371/journal.pgen.1000304
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Leal, N. C., da Silva, S. C., Cavalcanti, V. O., Figueiroa, A.C.T.d.A., Nunes, V. V. F., Miralles, I. S., et al. (2008). Vibrio parahaemolyticus serovar O3: K6 gastroenteritis in northeast Brazil. J. Appl. Microbiol. 105, 691–697. doi: 10.1111/j.1365-2672.2008.03782.x
Letchumanan, V., Chan, K.-G., and Lee, L.-H. (2014). Vibrio parahaemolyticus: a review on the pathogenesis, prevalence, and advance molecular identification techniques. Front. Microbiol. 5:705. doi: 10.3389/fmicb.2014.00705
Li, P., Xin, W., Kang, L., Chen, Z., Guo, C., Gao, S., et al. (2018). Genetic and population analyses of Vibrio parahaemolyticus isolates from three major coastal regions in China. Future Microbiol. 13, 1261–1269. doi: 10.2217/fmb-2018-0060
Li, Y., Xie, X., Shi, X., Lin, Y., Qiu, Y., Mou, J., et al. (2014). Vibrio parahaemolyticus, southern coastal region of China, 2007-2012. Emerg. Infect. Dis. 20, 685–688. doi: 10.3201/eid2004.130744
Liu, J., Bai, L., Li, W., Han, H., Fu, P., Ma, X., et al. (2018). Trends of foodborne diseases in China: lessons from laboratory-based surveillance since 2011. Front. Med. 12:48–57. doi: 10.1007/s11684-017-0608-6
Martin, D. P., Murrell, B., Khoosal, A., and Muhire, B. (2017). Detecting and analyzing genetic recombination using RDP4. Methods Mol. Biol. 1525, 433–460.
Matsuda, S., Kodama, T., Okada, N., Okayama, K., Honda, T., Iida, T. J. I., et al. (2009). Association of Vibrio parahaemolyticus thermostable direct hemolysin with lipid rafts is essential for cytotoxicity but not hemolytic activity. Infect. Immun. 78, 603–610.
McLaughlin, J. B., DePaola, A., Bopp, C. A., Martinek, K. A., Napolilli, N. P., Allison, C. G., et al. (2005). Outbreak of Vibrio parahaemolyticus gastroenteritis associated with Alaskan oysters. N. Engl. J. Med. 353, 1463–1470. doi: 10.1056/NEJMoa051594
Mugal, C. F., Kutschera, V. E., Botero-Castro, F., Wolf, J. B. W., and Kaj, I. (2020). Polymorphism data assist estimation of the nonsynonymous over synonymous fixation rate ratio ω for closely related species. Mol. Biol. Evol. 37, 260–279. doi: 10.1093/molbev/msz203
Nair, G. B., Ramamurthy, T., Bhattacharya, S. K., Dutta, B., Takeda, Y., and Sack, D. A. (2007). Global dissemination of Vibrio parahaemolyticus serotype O3: K6 and its serovariants. Clin. Microbiol. Rev. 20, 39–48. doi: 10.1128/cmr.00025-06
Okada, N., Iida, T., Park, K.-S., Goto, N., Yasunaga, T., Hiyoshi, H., et al. (2009). Identification and characterization of a novel type III secretion system in trh-positive Vibrio parahaemolyticus strain TH3996 reveal genetic lineage and diversity of pathogenic machinery beyond the species level. Infect. Immun. 77, 904–913. doi: 10.1128/iai.01184-08
Pal, D., and Das, N. (2010). Isolation, identification and molecular characterization of Vibrio parahaemolyticus from fish samples in Kolkata. Eur. Rev. Med. Pharmacol. Sci. 14, 545–549.
Park, K., Mok, J. S., Kwon, J. Y., Ryu, A., Kim, S. H., and Lee, H. J. (2018). Food-borne outbreaks, distributions, virulence, and antibiotic resistance profiles of Vibrio parahaemolyticus in Korea from 2003 to 2016: a review. Fish. Aquatic Sci. 21, 1–10.
Park, K. S., Ono, T., Rokuda, M., Jang, M. H., Okada, K., Idia, T., et al. (2004). Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect. Immun. 72, 6659–6665. doi: 10.1128/iai.72.11.6659-6665.2004
Paudyal, N., Pan, H., Liao, X., Zhang, X., Li, X., Fang, W., et al. (2018). A meta-analysis of major foodborne pathogens in chinese food commodities between 2006 and 2016. Foodborne Pathog. Dis. 15, 187–197. doi: 10.1089/fpd.2017.2417
Ralph, A., and Currie, B. J. (2007). Vibrio vulnificus and V. parahaemolyticus necrotising fasciitis in fishermen visiting an estuarine tropical northern Australian location. J. Infect. 54, e111–e114. doi: 10.1016/j.jinf.2006.06.015
Rozas, J., Ferrer-Mata, A., Carlos Sanchez-DelBarrio, J., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Shaw, K. S., Sapkota, A. R., Jacobs, J. M., He, X., and Crump, B. C. (2015). Recreational swimmers’ exposure to Vibrio vulnificus and Vibrio parahaemolyticus in the Chesapeake Bay, Maryland, USA. Environ. Int. 74, 99–105. doi: 10.1016/j.envint.2014.09.016
Smith, J. M., Feil, E. J., and Smith, N. H. (2000). Population structure and evolutionary dynamics of pathogenic bacteria. Bioessays 22, 1115–1122. doi: 10.1002/1521-1878(200012)22:12<1115::Aid-bies9<3.0.Co;2-r
Smith, J. M., Smith, N. H., Orourke, M., and Spratt, B. G. (1993). How clonal are bacteria. Proc. Natl. Acad. Sci. U.S.A. 90, 4384–4388. doi: 10.1073/pnas.90.10.4384
Suerbaum, S., Smith, J. M., Bapumia, K., Morelli, G., Smith, N. H., Kunstmann, E., et al. (1998). Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 95, 12619–12624. doi: 10.1073/pnas.95.21.12619
Thomas, C. M., and Nielsen, K. M. (2005). Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 3, 711–721. doi: 10.1038/nrmicro1234
Turner, K. M. E., and Feil, E. J. (2007). The secret life of the multilocus sequence type. Int. J. Antimicrob. Agents 29, 129–135. doi: 10.1016/j.ijantimicag.2006.11.002
Urwin, R., and Maiden, M. C. J. (2003). Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 11, 479–487. doi: 10.1016/j.tim.2003.08.006
Vos, M. (2009). Why do bacteria engage in homologous recombination? Trends Microbiol. 17, 226–232. doi: 10.1016/j.tim.2009.03.001
Xie, T., Wu, G., He, X., Lai, Z., Zhang, H., and Zhao, J. (2019). Prevalence and genetic diversity of Vibrio parahaemolyticus strains from salmon in Chinese markets. FEMS Microbiol. Lett. 366:fnz103. doi: 10.1093/femsle/fnz103
Xie, T., Wu, Q., Zhang, J., Xu, X., and Cheng, J. (2017). Comparison of Vibrio parahaemolyticus isolates from aquatic products and clinical by antibiotic susceptibility, virulence, and molecular characterisation. Food Control 71, 315–321. doi: 10.1016/j.foodcont.2016.06.046
Xu, X., Cheng, J., Wu, Q., Zhang, J., and Xie, T. (2016). Prevalence, characterization, and antibiotic susceptibility of Vibrio parahaemolyticus isolated from retail aquatic products in North China. BMC Microbiol. 16:32. doi: 10.1186/s12866-016-0650-6
Yan, Y., Cui, Y., Han, H., Xiao, X., Wong, H.-c, Tan, Y., et al. (2011). Extended MLST-based population genetics and phylogeny of Vibrio parahaemolyticus with high levels of recombination. Int. J. Food Microbiol. 145, 106–112. doi: 10.1016/j.ijfoodmicro.2010.11.038
Yu, Y., Hu, W., Wu, B., Zhang, P., Chen, J., Wang, S., et al. (2011). Vibrio parahaemolyticus isolates from southeastern Chinese coast are genetically diverse with circulation of clonal complex 3 strains since 2002. Foodborne Pathog. Dis. 8, 1169–1176. doi: 10.1089/fpd.2011.0865
Yu, Y., Yang, H., Li, J., Zhang, P., Wu, B., Zhu, B., et al. (2012). Putative type VI secretion systems of Vibrio parahaemolyticus contribute to adhesion to cultured cell monolayers. Arch. Microbiol. 194, 827–835. doi: 10.1007/s00203-012-0816-z
Zhang, L., and Orth, K. (2013). Virulence determinants for Vibrio parahaemolyticus infection. Curr. Opin. Microbiol. 16, 70–77. doi: 10.1016/j.mib.2013.02.002
Keywords: Vibrio parahaemolyticus, genetic diversity, population structure, clinical isolates, food isolates.
Citation: He M, Lei T, Jiang F, Zhang J, Zeng H, Wang J, Chen M, Xue L, Wu S, Ye Q, Pang R, Ding y and Wu Q (2021) Genetic Diversity and Population Structure of Vibrio parahaemolyticus Isolated From Clinical and Food Sources. Front. Microbiol. 12:708795. doi: 10.3389/fmicb.2021.708795
Received: 12 May 2021; Accepted: 02 July 2021;
Published: 27 July 2021.
Edited by:
Lin Lin, Jiangsu University, ChinaReviewed by:
Jinquan Li, Huazhong Agricultural University, ChinaAnshul Sharma, Gachon University, South Korea
Copyright © 2021 He, Lei, Jiang, Zhang, Zeng, Wang, Chen, Xue, Wu, Ye, Pang, Ding and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Ding, dingyu@jnu.edu.cn; Qingping Wu, wuqp203@163.com
†These authors have contributed equally to this work