 Shukai Zhan
Shukai Zhan Na Li
Na Li Caiguang Liu†
Caiguang Liu† Ren Mao
Ren Mao Xiaojun Zhuang
Xiaojun Zhuang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Microbiol. , 16 July 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.694967
Fibrosis is a complex and difficult to elucidate pathological process with no available therapies. Growing evidence implicates intestinal microbiota in the occurrence and development of fibrosis, and the potential mechanisms involved in different organs have been explored in several studies. In this review, we summarize the causative and preventive effects of gut microbiota on intestinal fibrosis, as well as the relationships between gut microbiota and fibrosis in other organs. Interestingly, several colonized microbes are associated with fibrosis via their structural components and metabolic products. They may also play essential roles in regulating inflammation and fibroblast activation or differentiation, which modulates extracellular matrix formation. While the relationships between intestinal fibrosis and gut microbiota remain unclear, lessons can be drawn from the effects of gut microbiota on hepatic, cardiac, nephritic, and pulmonary fibrosis. Various intestinal microbes alterations have been detected in different fibrotic organs; however, the results were heterogeneous. Mechanisms by which the intestinal microbiota regulate fibrotic processes in other organs, such as novel metabolic products or specific microbes, are also discussed. The specific microbiota associated with fibrosis in other organs could instruct future studies aiming to discover prospective mechanisms regulating intestinal fibrosis.
Fibrosis is a widespread pathological process that affects almost every organ and has poor therapeutic efficacy. It gives rise to end-stage organ failure and aggravates dysfunction, and is a leading cause of mortality worldwide (Liu, 2011; Tsochatzis et al., 2014; Bettenworth and Rieder, 2017; Richeldi et al., 2017; Rosenbloom et al., 2017; Di Ciaula et al., 2020). Fibrosis is the pathological overabundance of extracellular matrix (ECM), which contains several molecular components, including collagens, glycoproteins, and proteoglycans. Physiologically, the ECM maintains a homeostatic balance of synthesis and degradation through complicated regulatory pathways, facilitating wound healing and tissue recovery after injury or inflammation under healthy conditions (Speca et al., 2012; Friedman et al., 2013). However, overstimulation by excessive inflammation or pathologic environmental factors activates mesenchymal cells such as myofibroblasts, smooth muscle cells to continuously proliferate. These cells are characterized by ECM secretion and their abnormal increase will disturb leads to fibrosis (Stallmach et al., 1992; Wynn, 2008). In addition, non-mesenchymal cells, such as fibrocytes, endothelial cells and epithelial cells can transform into fibroblasts when stimulated. This also contributes to ECM accumulation (Kalluri, 2009; Rieder, 2013). The innate and adaptive immune reactions play central roles in bridging causative factors and their effects on fibrosis. The immune system is a highly complex network of various cell types, in which molecules such as cytokines, chemokines, growth factors, angiogenic factors, and reactive oxygen species (ROS), play roles in intercellular communication. Immune system activation by heterogeneous stimuli can activate mesenchymal and non-mesenchymal cells either directly or indirectly, by modulating inflammation (Speca et al., 2012; Rieder, 2013; Tsochatzis et al., 2014).
The gastrointestinal tract is heavily colonized by diverse microbes, including bacteria, fungi, viruses, and parasites (Tremaroli and Bäckhed, 2012; Kamada et al., 2013). Bacteria comprise the majority, up to 100 trillion microbial cells and 1,000 different species (Qin et al., 2010). Recently developed high-throughput sequencing technology and data analysis methods have illustrated the gut microbiome much more precisely than traditional culture-based techniques. This has enabled the study of specific species and even certain microbial structures (Liu et al., 2020). The composition and quantity of this microbial community is relatively homeostatic, depending on various factors such as the dietary patterns, living environment, and health of the host (Dominguez-Bello et al., 2011; Marques et al., 2017). Dysbiosis, which involves compositional alterations in gut microbiota, can increase the risk of disease (Tremaroli and Bäckhed, 2012; Bajaj, 2019).
Gut microbiota, as important environmental factors, are attracting increasing attention in the development of fibrogenesis (Lau et al., 2017; Acharya and Bajaj, 2019; Plata et al., 2019). To date, there have been many studies regarding gut microbial effects on hepatic, cardiac, and nephritic fibrosis; in contrast, the impact on intestinal fibrosis has been only studied sporadically (Speca et al., 2012; Minicis et al., 2014; Yiu et al., 2014; Karbach et al., 2016; Dornas and Lagente, 2019). Unlike other organs, the gastrointestinal tract has a direct and close connection to gut microbiota; therefore, the microbial effects and mechanisms underlying intestinal fibrosis may be unique, and warrant further exploration. Here, we review the latest studies and integrate their findings to summarize the known microbial influences on intestinal fibrosis. Information regarding the effects of intestinal microbiota on fibrosis in other organs is also included.
Intestinal fibrosis occurs in many gastrointestinal tract diseases, including inflammatory bowel disease (IBD), solitary rectal ulcers, radiation enteropathy, and eosinophilic enteropathy, and results in intestinal stenosis and obstruction. Almost all studies investigating relationships between intestinal fibrosis and gut microbiota have focused on IBD (Van Assche et al., 2004; Burke et al., 2007; Wynn, 2008). Encompassing both Crohn’s disease (CD) and ulcerative colitis, IBD is a lifelong relapsing and remitting inflammatory condition of the gastrointestinal tract, which is mainly associated with the activation of intestinal fibroblasts to increases collagen synthesis and facilitate fibrosis (Stallmach et al., 1992). Intestinal fibrosis is detected most frequently in patients with IBD, especially those with CD (Van Assche et al., 2004). Despite the emergence of effective biological therapies for intestinal inflammation, no effective fibrosis treatments exist currently. Fibrosis occurs in more than one-third of patients with CD, causing intestinal obstructions that require surgery in 30–50% of patients within 10 years of disease onset (Cosnes et al., 2002; Pariente et al., 2011). Treatments, such as surgical resection, endoscopic dilation, or section, are all temporary measures, and recurrence rates increase with time (Burke et al., 2007; Rieder et al., 2017).
The close relationship between intestinal microbiota and fibrosis was identified decades ago. After inoculating a fecal suspension sourced from healthy specific pathogen free (SPF) rats into the colonic walls of germ-free mice, researchers noticed obvious increases in collagen accumulation in inoculated regions (Mourelle et al., 1998). Moreover, some patients with CD have circulating antibodies against microbial antigens from Saccharomyces cerevisiae or Pseudomonas fluorescens, which are correlated with the clinical characteristics of intestinal fibrotic stenosis as well as surgical interventions (Mow et al., 2004). These results indicate that gut microbiota can induce fibrosis both directly and indirectly.
All intestinal immune and non-immune cells express pattern recognition receptors (PRRs), such as extracellular toll-like receptors (TLRs) and intracellular nucleotide oligomerization domain-like receptors (NLRs), which recognize pathogen-associated molecular patterns (PAMPs) and transmit intracellular signals. PAMPs are microbe-derived molecules, including components of bacterial cell walls, DNA, and double-stranded RNA (dsRNA) (Liew et al., 2005). When intestinal epithelial barrier integrity is disrupted by dysbiosis and inflammation, gut microbes are continuously exposed to immune or mesenchymal cells and activate intracellular signaling through their corresponding TLRs.
Specially, lipopolysaccharide (LPS), a component of the outer membranes of Gram-negative bacteria, is a type of PAMP proven to be fibrogenic. When fibroblasts are exposed to LPS, as mentioned previously herein, this molecule is recognized by corresponding TLR4 in the fibroblast membrane. After that, TLR4 oligomerizes and recruits downstream adaptors to its cytoplasmic toll-interleukin-1 receptor (TIR) domains. The following signaling event can be separated into two pathways, which are dependent and independent of the specific TIR domain-containing adaptor protein, myeloid differentiation primary response gene 88 (MyD88), respectively. The MyD88-dependent pathway leads to phosphorylation and degradation of inhibitory nuclear factor-κB (NF-κB) members, resulting in translocating NF-κB to nucleus and regulating gene transcription (Dauphinee and Karsan, 2006; Lu et al., 2008). Though the underlying mechanism hasn’t been unveiled, it’s known that gene transcription regulation will suppress the expression of SMAD family member 7 (SMAD7), a negative regulator of transforming growth factor β1 (TGFβ1) signaling, leading to enhanced TGFβ1 signaling and increased ECM protein secretion (see Figure 1A; Burke et al., 2010; Frangogiannis, 2020). Furthermore, when human fibrocytes were exposed to LPS, they produced higher amounts of collagen than when exposed to TGFβ1, indicating that LPS can enhance fibrosis independently of inflammatory TGFβ1 stimulation (Sazuka et al., 2014). Another bacterial cell wall polymer, peptidoglycan–polysaccharide, may also increase TGFβ1 expression and collagen accumulation in myofibroblasts through similar preceding mechanisms (van Tol et al., 1999).
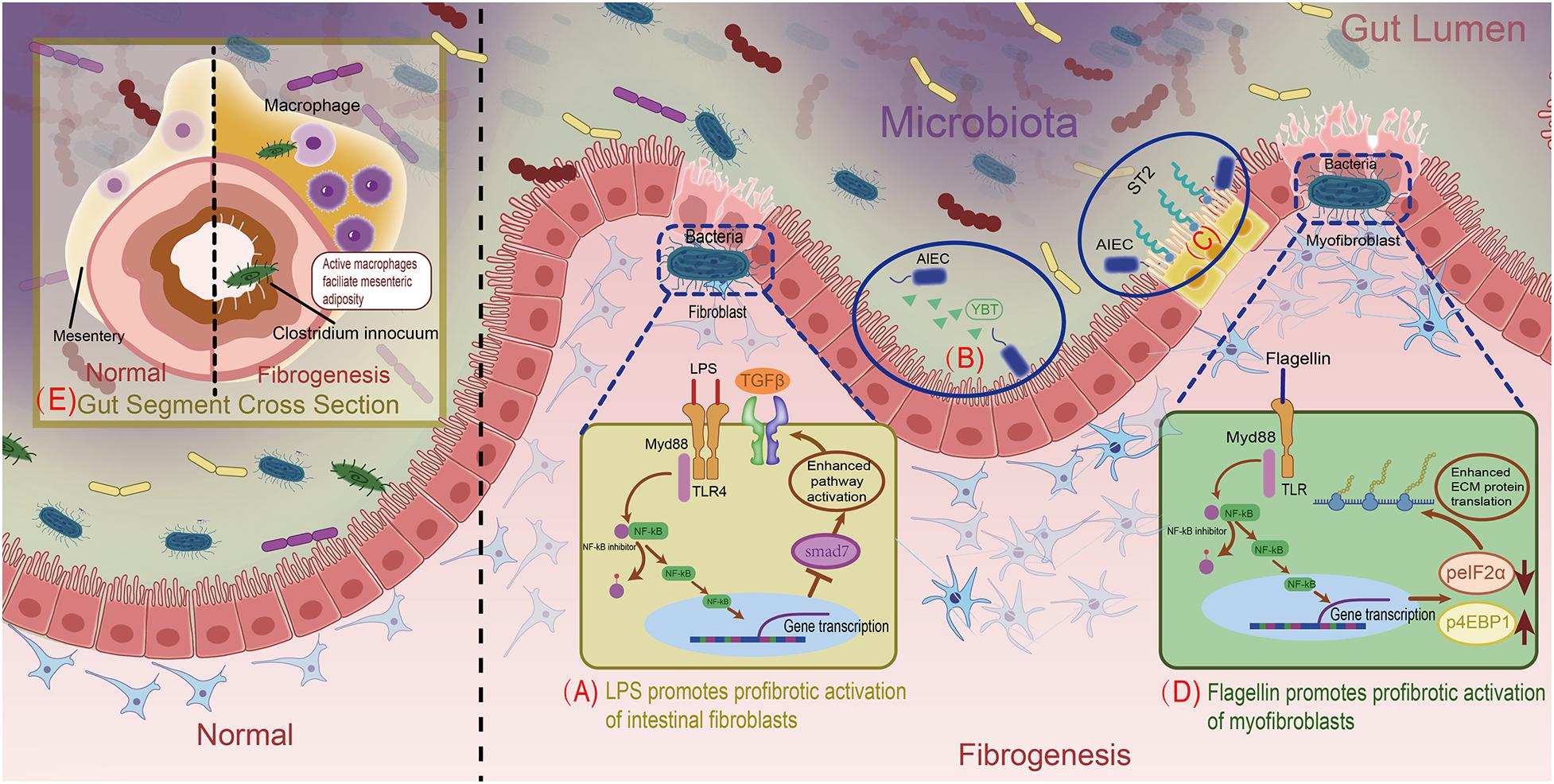
Figure 1. Overview of mechanisms for gut microbiota facilitating intestinal fibrosis. (A) LPS promotes profibrotic activation of intestinal fibroblasts. (B) AIEC’s siderophore Ybt strengthens its intestinal subepithelial localization. (C) AIEC’s flagellin stimulates intestinal epithelial cells to express the IL33 receptor. (D) Bacterial flagellin increases ECM proteins through modulating in the post-transcriptional level. (E) C. innocuum’s translocation to mesenteric adipose drives macrophages activation, resulting in fibrogenesis.
Contrary to traditional research which studied the composition of the gut microbiome as a whole, recent studies use various models to explore the roles of specific microbes in fibrosis. Mucispirillum schaedleri and Ruminococcus in the cecum as well as Streptococcus and Lactobacillus in the ileum were positively correlated with fibrosis in a murine model of transgenic tumor necrosis factor-like cytokine 1A (TL1A) overproduction (Jacob et al., 2018). TL1A is a member of the tumor necrosis factor superfamily that is expressed in many cell types, including immune cells, epithelial cells, and fibroblasts, in response to stimulation by TLR or other molecules such as interleukin (IL)1. Its overexpression in mice leads to spontaneous enteritis and fibrosis (Zheng et al., 2013). Interestingly, gut microbiota are essential for this pathologic process, as they augment fibroblast adhesion, migration, and differentiation into myofibroblasts. However, Oscillospira, Coprococcus, Faecalibacterium prausnitzii, and Bacteroides were negatively correlated with fibrosis (Jacob et al., 2018).
Similarly, Ruminococcus were implicated in stricture development in a multicentric, prospective inception cohort study composed of pediatric patients with newly diagnosed CD (Kugathasan et al., 2017). However, another study discovered that Lactobacillus acidophilus improved inflammation-dependent fibrosis in a dextran sodium sulfate (DSS)-induced chemical enteritis model (Park et al., 2018). The exact mechanisms through which these microbes affect fibrogenesis are unknown.
Adherent/invasive Escherichia coli (AIEC) is an intestinal E. coli subset that is enriched in patients with CD. As AIEC is most frequently isolated from the terminal ileum, which is also the most common site of fibrotic stricture in CD, it is conceivable that AIEC contributes to fibrosis (Rieder et al., 2017; Darfeuille-Michaud et al., 2004). Persistent AIEC infection can augment intestinal inflammation, leading to inflammation-dependent fibrosis (Small et al., 2013). Specifically, the siderophore yersiniabactin (Ybt) of AIEC, which originally sequesters and imports extracellular metals, plays a non-canonical role in fibrosis development (Perry and Fetherston, 2011). Upon its release from AIEC, Ybt strengthens bacterial subepithelial localization to facilitate inflammation-associated fibrogenesis through colonization in the inflammation-prone Il10−/− mouse model (see Figure 1B; Kim et al., 2005; Ellermann et al., 2019). In addition, AIEC flagellin was studied in DSS-induced mice and modified Salmonella enterica serovar Typhimurium-infected mice, in which CD-like colitis was persistent while fibrosis was absent. After AIEC colonization, flagellin stimulation of intestinal epithelial cells significantly increased expression of the IL33 receptor growth stimulation expressed gene 2 (ST2), resulting in the activation of profibrotic NFκB and IL13 (see Figure 1C; Schmitz et al., 2005; Fichtner-Feigl et al., 2007; Imai et al., 2019). In an in vitro intestinal myofibroblast model, bacterial flagellin reduced phosphorylation of eukaryotic initiation factor 2α (eIF2α) and increased phosphorylation of eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4EBP1) via the MyD88-dependent TLR pathway. Phosphorylation of eIF2α blocked pre-initiation mRNA translation complex formation and inhibited translation, while 4EBP1 activation was enhanced (Kimball, 1999; Grewal, 2009), facilitating the translation and accumulation of intracellular ECM proteins (see Figure 1D; Zhao et al., 2020).
Recently, Clostridium innocuum was discovered to translocate from the gut lumen to the mesenteric adipose tissue (MAT) of surgically resected CD samples. This was confirmed using gnotobiotic mice raised with altered Schaedler flora (ASF), a well-defined consortium of eight anaerobic bacterial species (L. acidophilus, Lactobacillus murinus, Bacteroides distasonis, M. schaedleri, Eubacterium plexicaudatum, and two Clostridium species) that promote healthy gut development. After DSS-treated ASF mice were irrigated with C. innocuum, the bacteria were found in the MAT and selectively activated specific profibrotic macrophages in chronically inflamed states. Active macrophages elicited various cytokines or growth factors, leading to mesenteric adiposity and fibrosis. This process is thought to protect the host from the translocation of other microbes or bacterial products into the circulation (see Figure 1E; Schaedler et al., 1965; Ha et al., 2020). Apart from bacteria, which predominate the intestinal microbiome, the genus Anaeroplasma is also positively associated with intestinal fibrosis (Burke et al., 2010).
Hepatic fibrosis, or cirrhosis, is mainly caused by chronic liver damage due to viral infection, alcohol abuse, or non-alcoholic liver disease (Tsochatzis et al., 2014). The liver synthesizes and secretes bile acids and other mediators into the gut lumen through the bile system, contributing to normal intestinal microbiota homeostasis and metabolism (Ridlon et al., 2015). Conversely, homeostatic gut microbiota compose an integral intestinal barrier; as a result, few of their components and metabolites enter the liver through the portal vein, and are cleared by hepatic mononuclear phagocytes (Tripathi et al., 2018).
In chronic liver disease, the intestinal barrier is impaired, leading to dysbiosis or bacteria overgrowth in the small intestine. Alterations in intestinal microbes play a reactive role in increasing intestinal permeability (Wigg et al., 2001). A leaky gut allows many more microbes and microbial components to enter the liver through circulation. Bacteria, LPS, endotoxins, and even bacterial DNA have been found in the blood of patients and various animal models of chronic liver disease (Volynets et al., 2012; Yan and Schnabl, 2012; Hawkesworth et al., 2013; Zhou and Zhong, 2017). These PAMPs are recognized by PRRs on hepatocytes, Kupffer cells, hepatic stellate cells (HSCs), and endothelial cells. Along with the aforementioned LPS–TLR4 signaling axis, hepatic dsRNA-TLR3, flagellin-TLR5, and RNA-NLR1 axes have also been identified (Jäckel et al., 2017; Chen et al., 2019). When HSCs were stimulated directly through PRRs or indirectly via inflammation, ECM synthesis, and deposition increased (Elpek, 2014).
As most studies regarding the relationships between liver fibrogenesis and gut microbiota have focused on PAMPs, the roles of specific microbes in hepatic fibrosis remain mysterious. Moghadamrad et al. (2019) treated ASF-colonized mice and SPF mice with carbon tetrachloride (CCl4) to induce cirrhosis, and discovered that the ASF microbes promoted CCl4-induced liver fibrosis, indicating that gut dysbiosis contributes to cirrhosis through an undefined mechanism.
Several studies have examined gut microbiota changes in cirrhosis. Intestinal dysbiosis varies in cirrhosis, depending on its etiology, the disease period, and the patient’s diet (Bajaj et al., 2018). For example, patients with non-alcoholic steatohepatitis (NASH) without fibrosis exhibited a predominance of Bacteroides, while those with fibrosis had higher relative abundances of Ruminococcus other than Bacteroides (Boursier et al., 2016). Compared with healthy controls, patients with alcoholic cirrhosis had decreased Bacteroides, Parabacteroides, Prevotella, and Clostridium, and increased Lactobacillus and Bifidobacteria (Dubinkina et al., 2017). In another study on cirrhosis, Streptococcus and Veillonella increased while Bacteroidetes and Eubacterium decreased (Qin et al., 2014). A study of patients with viral and alcohol-related cirrhosis revealed reduced Bacteroidetes and increased Proteobacteria and Fusobacteria. More specifically, Enterobacteriaceae, Veillonellaceae, and Staphylococci were abundant (Chen et al., 2011). In patients with alcoholic cirrhosis, Enterobacteriaceae and Halomonadaceae were increased while Lachnospiraceae, Ruminococcaceae, and Clostridiales XIV were decreased compared to abundances in those with non-alcoholic cirrhosis. However, in patients with NASH cirrhosis, the families Bacteroidaceae and Porphyromonadaceae from the phylum Bacteroidetes were enriched and Veillonellaceae was decreased compared to abundances associated with cirrhosis of other etiologies (Bajaj et al., 2014).
Furthermore, intestinal microbiota components differ between compensated and decompensated cirrhosis (Bajaj et al., 2018). Staphylococcae and Lachnospiraceae levels are positively and negatively correlated with the Child–Pugh score, respectively. The microbiota also vary in cirrhosis with and without complications, such as hepatic cell carcinoma, hepatic encephalopathy, and infection (Bajaj et al., 2014; Grąt et al., 2016).
Intestinal microbiota likely fail to directly contact cardiac tissue because of the intestinal barrier, hepatic clearance, and the heterogeneous immune cells in the bloodstream. Therefore, they primarily induce cardiac fibrosis through various molecules, including structural components and metabolites. Cardiac fibrosis results in cardiac insufficiency, which inhibits intestinal venous return, causing overgrowth of anaerobic bacteria and increasing intestinal permeability (Sandek et al., 2007, 2014; Schiattarella et al., 2017).
Several studies have identified intestinal microbiome changes in cardiac insufficiency. One found abnormally high levels of the Proteobacteria genera Campylobacter, Shigella, and Salmonella as well as the Firmicutes genus Lactobacillus in patients with chronic heart failure (Pasini et al., 2016; Kamo et al., 2017). In another study, patients with stable cardiac insufficiency had smaller proportions of Lachnospiraceae compared to controls (Kummen et al., 2018). Decreases in the families Coriobacteriaceae, Erysipelotrichaceae, and Ruminococcaceae and the genera Blautia, Collinsella, uncl. Erysipelotrichaceae, and uncl. Ruminococcaceae were found in patients with heart failure compared to controls (Luedde et al., 2017).
Gut microbiota facilitate cardiac fibrosis, and this was straightly proven with the use of germ-free animal models, though the specific effective factor was not explored at that time (Karbach et al., 2016). Currently, bacterial metabolic products are thought to be contributive. Of these, trimethylamine N-oxide (TMAO) is the most important and well-studied. Dietary phosphatidylcholine, choline, and carnitine are metabolized by intestinal microbiota in the lumen, producing trimethylamine (TMA). TMA is absorbed and travels through the portal vein to the liver, where it is oxidized before entering systemic circulation (Tang et al., 2019). In cardiac fibroblasts, TMAO enhances TGFβ receptor I expression and inhibits the expression of SMAD2, a downstream inhibitor of TGFβ signaling. By facilitating TGFβ signaling, TMAO promotes cardiac fibroblast differentiation into myofibroblasts, causing cytokine secretion, and cardiac fibrosis. This was confirmed in a high choline diet mouse model (Yang et al., 2019). LPS can also aggravate cardiac fibrosis via LPS–TLR4 signaling (Frangogiannis, 2014).
In another mouse model of heart failure, researchers found that a high-fiber diet increased the proportion of Bacteroides acidifaciens and ameliorated cardiac fibrosis. Although the underlying mechanism is unclear, decreased synthesis of a master cardiovascular regulator, early growth response protein 1, was suggested (Marques et al., 2017).
Renal fibrosis is a pathological process involved in different types of chronic kidney disease (CKD) that leads to loss of renal function (i.e., end-stage renal disease). Intestinal dysbiosis occurs during CKD, and microbiome changes result in aberrant metabolism and increases in pernicious by-products, most of which are uremic toxins. Similar to that in cardiac disease, the enhanced intestinal permeability in CKD results in increased uremic toxins in the systemic circulation, aggravating nephritic fibrosis (Meijers et al., 2018).
TMAO also causes nephritic fibrosis. It mainly acts on SMAD3, another signaling molecule downstream of TGFβ. When mice were fed a diet supplemented with TMAO or high fat, enhanced SMAD3 phosphorylation, tubulointerstitial fibrosis, and collagen deposition were observed. These results were verified in a CKD cohort (Tang et al., 2015; Sun et al., 2017; El-Deeb et al., 2019).
p-Cresyl sulfate is a uremic toxin produced by aberrant intestinal microbes in nephritic fibrosis. Further, it is a sulfated conjugate of hepatic p-cresol. The increased anaerobic bacteria in the guts of patients with CKD metabolize the dietary amino acids tyrosine and phenylalanine to synthesize p-cresol, which travels though anion transporters and accumulates intracellularly, leading to ROS generation through nicotinamide adenine dinucleotide phosphate oxidase activation. Enhanced oxidative stress activates TGFβ signaling, resulting in collagen accumulation (Miyamoto et al., 2011; Watanabe et al., 2013).
Moreover, a series of tryptophan metabolites, including indole acetic acid, indole lactic acid, and tryptamine, are generated by almost all intestinal microbes. Diverse metabolic products are produced by multiple microbes, forming a complicated network involving heterogeneous pathways that control renal fibrosis by adjusting gene expression. These metabolites were recently reviewed by Liu et al. (2021). In addition, decreased microbial production of short chain fatty acids (SCFAs) like valproic acid or butyrate may contribute to nephritic fibrosis. SCFAs are general histone deacetylase inhibitors that suppress TGFβ signaling, preventing pericyte differentiation into myofibroblasts (Matsumoto et al., 2006; Zhang et al., 2018).
While characterizing the intestinal dysbiosis in nephritic fibrosis, one study comprising 24 stable patients with end-stage renal disease found increases in the bacteria families Alteromonadaceae, Cellulomonadaceae, Clostridiaceae, Dermabacteraceae, Enterobacteriaceae, Halomonadaceae, Methylococcaceae, Micrococcaceae, Moraxellaceae, Polyangiaceae, Pseudomonadaceae, Xanthomonadaceae, and Verrucomicrobiaceae, and decreases in Lactobacillaceae and Prevotellaceae (Wong et al., 2014).
A multitude of causative factors for pulmonary fibrosis have been identified, including autoimmune disorders, silica inhalation, and radiotherapy (Moore and Moore, 2015). However, like in other organs, the relationships between pulmonary fibrosis and gut microbiota are relatively unexplored, and most studies have focused on the roles of airway microbiota (Takahashi et al., 2018; Zhang et al., 2020). Recently, two studies investigated gut microbiota variations in patients and animal models of pulmonary fibrosis. In patients with silica-induced pulmonary fibrosis, species of the genera Megamonas and of the phyla Proteobacteria, Synergistetes, Lentisphaerae, Tenericutes, and Cyanobacteria increased, while Pseudomonas aeruginosa, species of the genera Subdoligranulum, Blautia, Nitrosomonadaceae, and Bifidobacterium, and the phyla Actinobacteria, Acidobacteria, Gemmatimonadetes, Saccharibacteria, Fusobacteria, Aminicenantes, and Verrucomicrobia decreased (Zhou et al., 2019).
Changes in intestinal microbiota and their products will activate various immune and non-immune cells, causing inflammation and stimulating mesenchymal cells to produce ECM. This common mechanism underlies almost all fibrotic processes. Nevertheless, its specific presentation varies dramatically in different organs. In the gut and liver, which are directly exposed to the intestinal microbiota, certain bacteria, or their components have been identified as fibrotic promotors, while in the lung, heart, and kidney, the effects of intestinal microbiota involve their by-products, making it difficult to identify suspected microbes. Multiple microbial products strongly induce fibrosis in these organs (see Table 1).
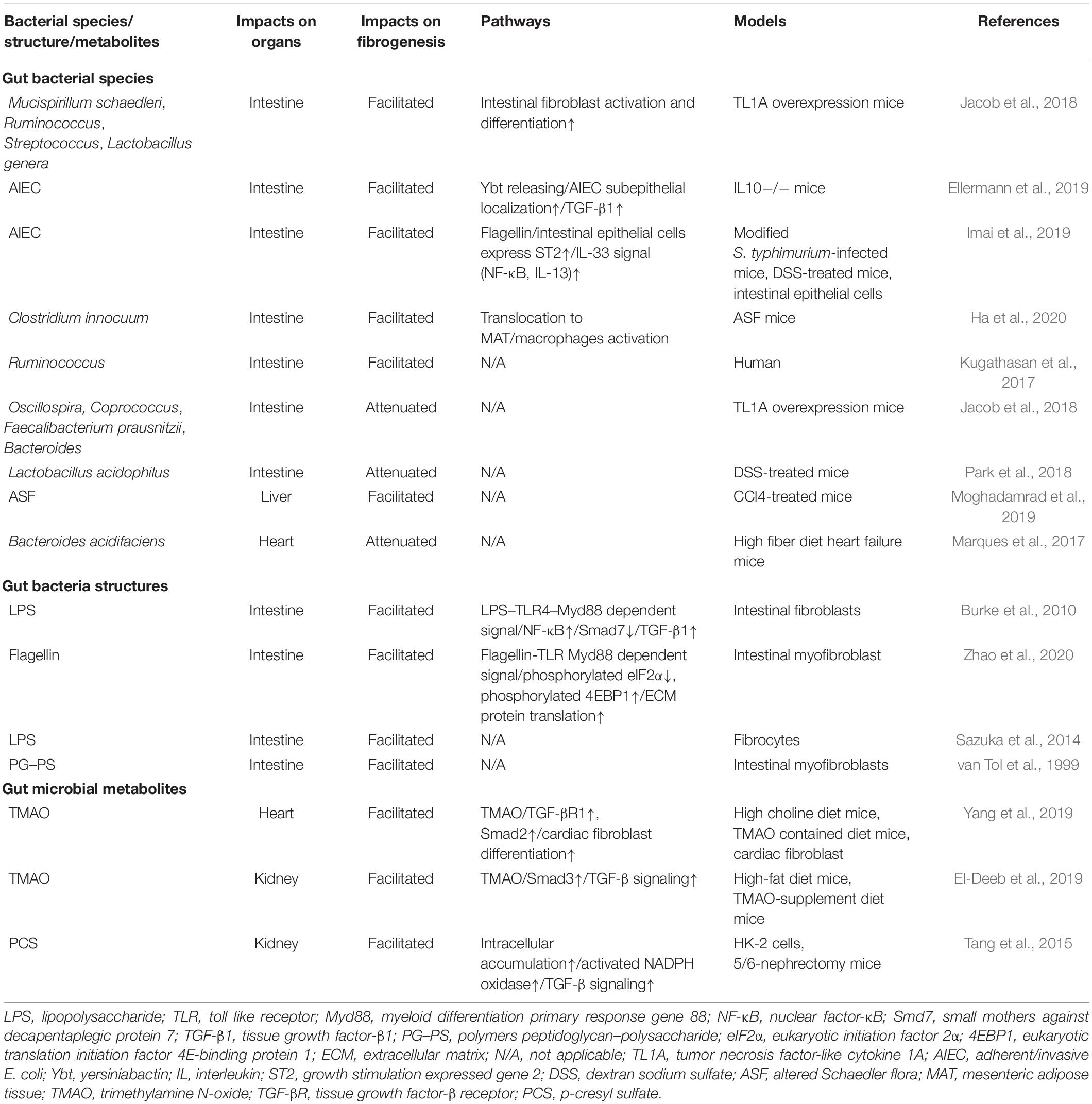
Table 1. Overview of intestinal microbial impacts on fibrogenesis in different organs.
Intestinal microbiome composition changes during fibrosis are well-studied in hepatic, cardiac, and pulmonary fibrosis, and are different and even at times paradoxical between organs (see Figure 2). For example, the genus Ruminococcus, which facilitates intestinal fibrosis and is increased in cirrhosis, is decreased in patients with cardiac insufficiency. Variations in intestinal Firmicutes species are also heterogenous in different fibrotic organs. Conversely, some bacteria are similarly increased in different organs. Streptococcus is thought to induce intestinal fibrosis and is enriched in cirrhosis. Lactobacillus is abundant in patients with hepatic and cardiac fibrosis. Further, Proteobacteria overgrowth is found in patients with cirrhosis and cardiac insufficiency.
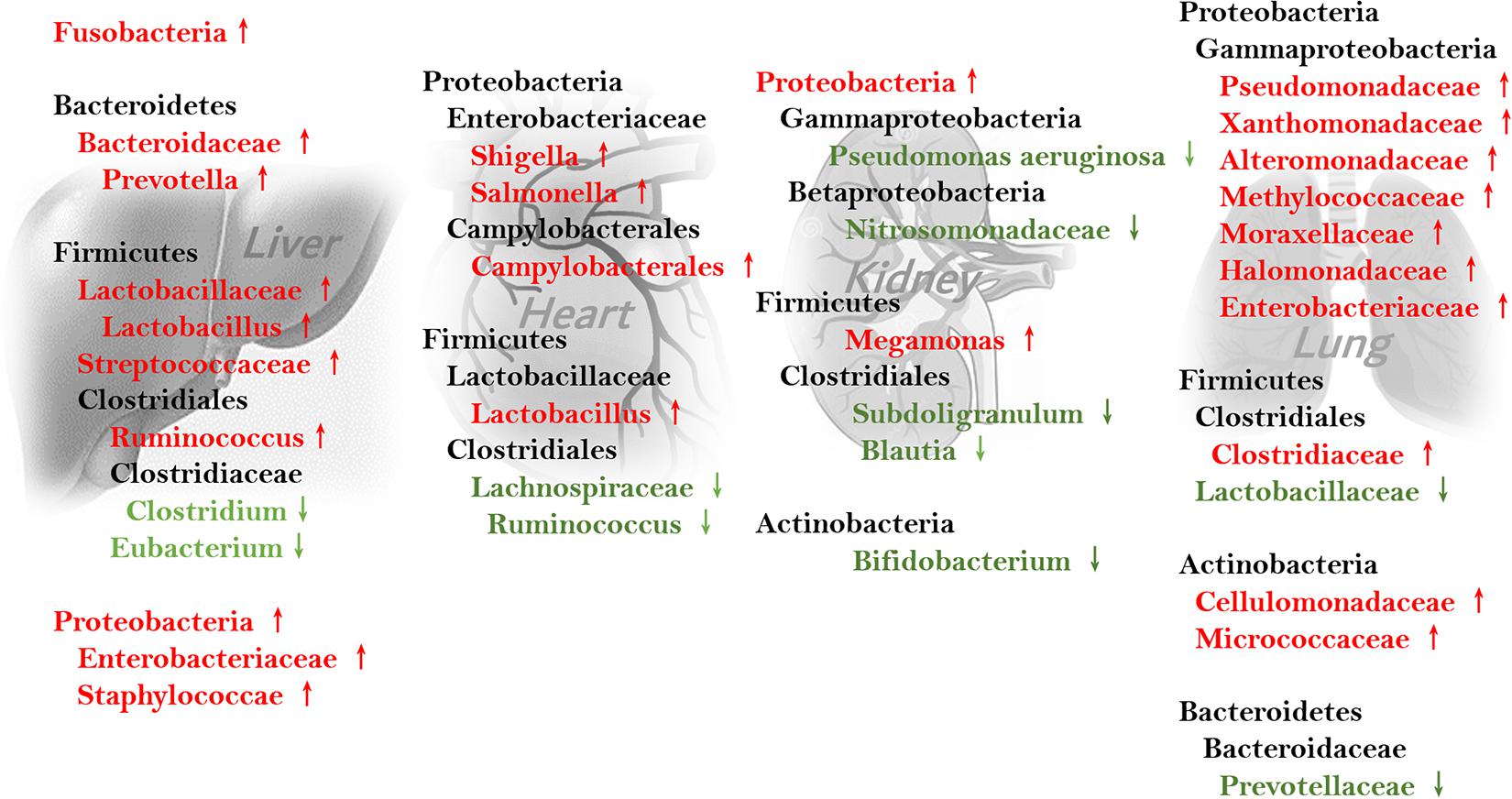
Figure 2. Feces-associated microbiota alterations involved in fibrosis of different organs.
Based on this aforementioned comprehensive analysis, clues from other organs can be gained to further study the issue of gut microbiota and intestinal fibrosis. Certain microbes and microbial metabolites appear to be positively related to fibrosis regarding to other organs, and these have never been previously studied in the intestine. Whether they induce intestinal fibrosis and their underlying mechanism is worthy of further study.
On the other hand, different microbial changes have been identified in patients with cirrhosis with different fibrotic etiologies and cirrhotic disease periods. By analyzing variations in specific intestinal microbes, researchers have constructed models to predict the development of cirrhosis and its complications (Bajaj et al., 2014; Caussy et al., 2019). Recently, a similar microbial prediction model was generated for pulmonary fibrosis (Gong et al., 2021). Therefore, examining the dysbiosis that occurs in intestinal fibrosis and its value in disease prediction may also be worthwhile.
Finally, almost all research has involved bacteria and there are currently limited studies on the impacts of non-bacterial microbes on fibrosis. An examination of these will also expand our understanding of the relationship between gut microbiota and intestinal fibrosis.
XZ and ZZ designed the study. SZ, NL, CL, and RM wrote the manuscript. CL collected the data. DW and TL analyzed the data. MC and ZZ revised the manuscript. All authors approved the final version.
This research was supported by Guangdong Basic and Applied Basic Research Foundation (2020A1515111087), the L. M. and H. B. Helmsley Charitable Trust Grant (2019PG-CD018), and the National Natural Science Foundation of China (81870384 and 81630018).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acharya, C., and Bajaj, J. S. (2019). Altered microbiome in patients with cirrhosis and complications. Clin. Gastroenterol. Hepatol. 17, 307–321. doi: 10.1016/j.cgh.2018.08.008
Bajaj, J. S. (2019). Altered microbiota in cirrhosis and its relationship to the development of infection. Clin. Liver Dis. 14, 107–111. doi: 10.1002/cld.827
Bajaj, J. S., Heuman, D. M., Hylemon, P. B., Sanyal, A. J., White, M. B., Monteith, P., et al. (2014). Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 60, 940–947. doi: 10.1016/j.jhep.2013.12.019
Bajaj, J. S., Idilman, R., Mabudian, L., Hood, M., Fagan, A., Turan, D., et al. (2018). Diet affects gut microbiota and modulates hospitalization risk differentially in an international cirrhosis cohort. Hepatology 68, 234–247. doi: 10.1002/hep.29791
Bettenworth, D., and Rieder, F. (2017). Pathogenesis of intestinal fibrosis in inflammatory bowel disease and perspectives for therapeutic implication. Dig. Dis. 35, 25–31. doi: 10.1159/000449079
Boursier, J., Mueller, O., Barret, M., Machado, M., Fizanne, L., Araujo-Perezet, F., et al. (2016). The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63, 764–775. doi: 10.1002/hep.28356
Burke, J. P., Cunningham, M. F., Watson, R. W., Docherty, N. G., Coffey, J. C., and O’Connell, P. R. (2010). Bacterial lipopolysaccharide promotes profibrotic activation of intestinal fibroblasts. Br. J. Surg. 97, 1126–1134. doi: 10.1002/bjs.7045
Burke, J. P., Mulsow, J. J., O’Keane, C., Docherty, N. G., Watson, R. W., and O’Connell, P. R. (2007). Fibrogenesis in Crohn’s disease. Am. J. Gastroenterol. 102, 439–448. doi: 10.1111/j.1572-0241.2006.01010.x
Caussy, C., Tripathi, A., Humphrey, G., Bassirian, S., Singh, S., Faulkner, C., et al. (2019). A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 10:1406. doi: 10.1038/s41467-019-09455-9
Chen, D., Le, T. H., Shahidipour, H., Read, S. A., and Ahlenstiel, G. (2019). The role of gut-derived microbial antigens on liver fibrosis initiation and progression. Cells 8:1324. doi: 10.3390/cells8111324
Chen, Y., Yang, F., Lu, H., Wang, B., Chen, Y., Lei, D., et al. (2011). Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 54, 562–572. doi: 10.1002/hep.24423
Cosnes, J., Cattan, S., Blain, A., Beaugerie, L., Carbonnel, F., Parc, R., et al. (2002). Long-term evolution of disease behavior of Crohn’s disease. Inflamm. Bowel Dis. 8, 244–250. doi: 10.1097/00054725-200207000-00002
Darfeuille-Michaud, A., Boudeau, J., Bulois, P., Neut, C., Glasser, A. L., and Barnich, N. (2004). High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127, 412–421. doi: 10.1053/j.gastro.2004.04.061
Dauphinee, S. M., and Karsan, A. (2006). Lipopolysaccharide signaling in endothelial cells. Lab. Invest. 86, 9–22. doi: 10.1038/labinvest.3700366
Di Ciaula, A., Baj, J., Garruti, G., Celano, G., De Angelis, M., Wang, H. H., et al. (2020). Liver steatosis, gut-liver axis, microbiome and environmental factors. A never-ending bidirectional cross-talk. J. Clin. Med. 9:2648. doi: 10.3390/jcm9082648
Dominguez-Bello, M. G., Blaser, M. J., Ley, R. E., and Knight, R. (2011). Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology 140, 1713–1719. doi: 10.1053/j.gastro.2011.02.011
Dornas, W., and Lagente, V. (2019). Intestinally derived bacterial products stimulate development of nonalcoholic steatohepatitis. Pharmacol. Res. 141, 418–428. doi: 10.1016/j.phrs.2019.01.026
Dubinkina, V. B., Tyakht, A. V., Odintsova, V. Y., Yarygin, K. S., Kovarsky, B. A., Pavlenko, A. V., et al. (2017). Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome 5:141. doi: 10.1186/s40168-017-0359-2
El-Deeb, O. S., Atef, M. M., and Hafez, Y. M. (2019). The interplay between microbiota-dependent metabolite trimethylamine N-oxide, transforming growth factor β/SMAD signaling and inflammasome activation in chronic kidney disease patients: a new mechanistic perspective. J. Cell. Biochem. 120, 14476–14485. doi: 10.1002/jcb.28707
Ellermann, M., Gharaibeh, R. Z., Fulbright, L., Dogan, B., Moore, L. N., Broberg, C. A., et al. (2019). Yersiniabactin-producing adherent/invasive Escherichia coli promotes inflammation-associated fibrosis in gnotobiotic Il10(−/−) mice. Infect. Immun. 87:e00587-19. doi: 10.1128/IAI.00587-19
Elpek, G. (2014). Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: an update. World J. Gastroenterol. 20, 7260–7276. doi: 10.3748/wjg.v20.i23.7260
Fichtner-Feigl, S., Fuss, I. J., Young, C. A., Watanabe, T., Geissler, E. K., Schlitt, H. J., et al. (2007). Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J. Immunol. 178, 5859–5870. doi: 10.4049/jimmunol.178.9.5859
Frangogiannis, N. G. (2014). The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 11, 255–265. doi: 10.1038/nrcardio.2014.28
Frangogiannis, N. G. (2020). Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 217:e20190103. doi: 10.1084/jem.20190103
Friedman, S. L., Sheppard, D., Duffield, J. S., and Violette, S. (2013). Therapy for fibrotic diseases: nearing the starting line. Sci. Transl. Med. 5:167sr1. doi: 10.1126/scitranslmed.3004700
Gong, G. C., Song, S. R., and Su, J. (2021). Pulmonary fibrosis alters gut microbiota and associated metabolites in mice: an integrated 16S and metabolomics analysis. Life Sci. 264:118616. doi: 10.1016/j.lfs.2020.118616
Grąt, M., Wronka, K. M., Krasnodȩbski, M., Masior, Ł, Lewandowski, Z., Kosińska, I., et al. (2016). Profile of gut microbiota associated with the presence of hepatocellular cancer in patients with liver cirrhosis. Transplant. Proc. 48, 1687–1691. doi: 10.1016/j.transproceed.2016.01.077
Grewal, S. S. (2009). Insulin/TOR signaling in growth and homeostasis: a view from the fly world. Int. J. Biochem. Cell Biol. 41, 1006–1010. doi: 10.1016/j.biocel.2008.10.010
Ha, C. W. Y., Martin, A., Sepich-Poore, G. D., Shi, B., Wang, Y. Z., Gouin, K., et al. (2020). Translocation of viable gut microbiota to mesenteric adipose drives formation of creeping fat in humans. Cell 183, 666–683. doi: 10.1016/j.cell.2020.09.009
Hawkesworth, S., Moore, S. E., Fulford, A. J., Barclay, G. R., Darboe, A. A., Mark, H., et al. (2013). Evidence for metabolic endotoxemia in obese and diabetic Gambian women. Nutr. Diabetes 3:e83. doi: 10.1038/nutd.2013.24
Imai, J., Kitamoto, S., Sugihara, K., Nagao-Kitamoto, H., Hayashi, A., Morhardt, T. L., et al. (2019). Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. 12, 632–643. doi: 10.1038/s41385-019-0138-4
Jäckel, S., Kiouptsi, K., Lillich, M., Hendrikx, T., Khandagale, A., Kollar, B., et al. (2017). Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2. Blood 130, 542–553. doi: 10.1182/blood-2016-11-754416
Jacob, N., Jacobs, J. P., Kumagai, K., Ha, C. W. Y., Kanazawa, Y., Lagishetty, V., et al. (2018). Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome. Mucosal Immunol. 11, 1466–1476. doi: 10.1038/s41385-018-0055-y
Kalluri, R. (2009). EMT: when epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 119, 1417–1419. doi: 10.1172/JCI39675
Kamada, N., Seo, S. U., and Núñez, G. (2013). Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 13, 321–335. doi: 10.1038/nri3430
Kamo, T., Akazawa, H., Suda, W., Saga-Kamo, A., Shimizu, Y., Yagi, H., et al. (2017). Dysbiosis and compositional alterations with aging in the gut microbiota of patients with heart failure. PLoS One 12:e0174099. doi: 10.1371/journal.pone.0174099
Karbach, S. H., Schönfelder, T., Brandão, I., Wilms, E., Hörmann, N., Jäckel, S., et al. (2016). Gut microbiota promote angiotensin II-induced arterial hypertension and vascular dysfunction. J. Am. Heart Assoc. 5:e003698. doi: 10.1161/JAHA.116.003698
Kim, S. C., Tonkonogy, S. L., Albright, C. A., Tsang, J., Balish, E. J., Braun, J., et al. (2005). Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 128, 891–906. doi: 10.1053/j.gastro.2005.02.009
Kimball, S. R. (1999). Eukaryotic initiation factor eIF2. Int. J. Biochem. Cell Biol. 31, 25–29. doi: 10.1016/s1357-2725(98)00128-9
Kugathasan, S., Denson, L. A., Walters, T. D., Kim, M. O., Marigorta, U. M., Schirmer, M., et al. (2017). Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. Lancet 389, 1710–1718. doi: 10.1016/S0140-6736(17)30317-3
Kummen, M., Mayerhofer, C. C. K., Vestad, B., Broch, K., Awoyemi, A., Storm-Larsen, C., et al. (2018). Gut microbiota signature in heart failure defined from profiling of 2 independent cohorts. J. Am. Coll. Cardiol. 71, 1184–1186. doi: 10.1016/j.jacc.2017.12.057
Lau, K., Srivatsav, V., Rizwan, A., Nashed, A., Liu, R., Shen, R., et al. (2017). Bridging the gap between gut microbial dysbiosis and cardiovascular diseases. Nutrients 9:859. doi: 10.3390/nu9080859
Liew, F. Y., Xu, D., Brint, E. K., and O’Neill, L. A. (2005). Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 5, 446–458. doi: 10.1038/nri1630
Liu, J. R., Miao, H., Deng, D. Q., Vaziri, N. D., Li, P., and Zhao, Y. Y. (2021). Gut microbiota-derived tryptophan metabolism mediates renal fibrosis by aryl hydrocarbon receptor signaling activation. Cell. Mol. Life Sci. 78, 909–922. doi: 10.1007/s00018-020-03645-1
Liu, Y. (2011). Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 7, 684–696. doi: 10.1038/nrneph.2011.149
Liu, Y. X., Qin, Y., Chen, T., Lu, M., Qian, X., Guo, X., et al. (2020). A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 12, 315–330. doi: 10.1007/s13238-020-00724-8
Lu, Y. C., Yeh, W. C., and Ohashi, P. S. (2008). LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151. doi: 10.1016/j.cyto.2008.01.006
Luedde, M., Winkler, T., Heinsen, F. A., Rühlemann, M. C., Spehlmann, M. E., Bajrovic, A., et al. (2017). Heart failure is associated with depletion of core intestinal microbiota. ESC Heart Failure 4, 282–290. doi: 10.1002/ehf2.12155
Marques, F. Z., Nelson, E., Chu, P. Y., Horlock, D., Fiedler, A., Ziemann, M., et al. (2017). High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation 135, 964–977. doi: 10.1161/CIRCULATIONAHA.116.024545
Matsumoto, N., Riley, S., Fraser, D., Al-Assaf, S., Ishimura, E., Wolever, T., et al. (2006). Butyrate modulates TGF-beta1 generation and function: potential renal benefit for Acacia(sen) SUPERGUM (gum arabic)? Kidney Int. 69, 257–265. doi: 10.1038/sj.ki.5000028
Meijers, B., Farré, R., Dejongh, S., Vicario, M., and Evenepoel, P. (2018). Intestinal barrier function in chronic kidney disease. Toxins 10:298. doi: 10.3390/toxins10070298
Minicis, S. D., Rychlicki, C., Agostinelli, L., Saccomanno, S., Candelaresi, C., Trozzi, L., et al. (2014). Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 59, 1738–1749. doi: 10.1002/hep.26695
Miyamoto, Y., Watanabe, H., Noguchi, T., Kotani, S., Nakajima, M., Kadowaki, D., et al. (2011). Organic anion transporters play an important role in the uptake of p-cresyl sulfate, a uremic toxin, in the kidney. Nephrol. Dial. Transplant. 26, 2498–2502. doi: 10.1093/ndt/gfq785
Moghadamrad, S., Hassan, M., McCoy, K. D., Kirundi, J., Kellmann, P., and Gottardi, A. D. (2019). Attenuated fibrosis in specific pathogen-free microbiota in experimental cholestasis- and toxin-induced liver injury. FASEB J. 33, 12464–12476. doi: 10.1096/fj.201901113R
Moore, B. B., and Moore, T. A. (2015). Viruses in idiopathic pulmonary fibrosis: etiology and exacerbation. Ann. Am. Thorac. Soc. 12(Suppl. 2) S186–S192. doi: 10.1513/AnnalsATS.201502-088AW
Mourelle, M., Salas, A., Guarner, F., García-Lafuente, A., and Malagelada, J. R. (1998). Stimulation of transforming growth factor beta1 by enteric bacteria in the pathogenesis of rat intestinal fibrosis. Gastroenterology 114, 519–526. doi: 10.1016/s0016-5085(98)70535-9
Mow, W. S., Vasiliauskas, E. A., Lin, Y. C., Fleshner, P. R., Papadakis, K. A., Taylor, K. D., et al. (2004). Association of antibody responses to microbial antigens and complications of small bowel Crohn’s disease. Gastroenterology 126, 414–424. doi: 10.1053/j.gastro.2003.11.015
Pariente, B., Cosnes, J., Danese, S., Sandborn, W. J., Lewin, M., Fletcher, J. G., et al. (2011). Development of the Crohn’s disease digestive damage score, the Lémann score. Inflamm. Bowel Dis. 17, 1415–1422. doi: 10.1002/ibd.21506
Park, J. S., Choi, J. W., Jhun, J., Kwon, J. Y., Lee, B. I., Yang, C. W., et al. (2018). Lactobacillus acidophilus improves intestinal inflammation in an acute colitis mouse model by regulation of Th17 and Treg cell balance and fibrosis development. J. Med. Food 21, 215–224. doi: 10.1089/jmf.2017.3990
Pasini, E., Aquilani, R., Testa, C., Baiardi, P., Angioletti, S., Boschi, F., et al. (2016). Pathogenic gut flora in patients with chronic heart failure. JACC Heart Failure 4, 220–227. doi: 10.1016/j.jchf.2015.10.009
Perry, R. D., and Fetherston, J. D. (2011). Yersiniabactin iron uptake: mechanisms and role in Yersinia pestis pathogenesis. Microbes Infect. 13, 808–817. doi: 10.1016/j.micinf.2011.04.008
Plata, C., Cruz, C., Cervantes, L. G., and Ramírez, V. (2019). The gut microbiota and its relationship with chronic kidney disease. Int. Urol. Nephrol. 51, 2209–2226. doi: 10.1007/s11255-019-02291-2
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
Qin, N., Yang, F., Li, A., Prifti, E., Chen, Y., Shao, L., et al. (2014). Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64. doi: 10.1038/nature13568
Richeldi, L., Collard, H. R., and Jones, M. G. (2017). Idiopathic pulmonary fibrosis. Lancet 389, 1941–1952. doi: 10.1016/S0140-6736(17)30866-8
Ridlon, J. M., Kang, D. J., Hylemon, P. B., and Bajaj, J. S. (2015). Gut microbiota, cirrhosis, and alcohol regulate bile acid metabolism in the gut. Dig. Dis. 33, 338–345. doi: 10.3748/wjg.v21.i41.11597
Rieder, F. (2013). The gut microbiome in intestinal fibrosis: environmental protector or provocateur? Sci. Transl. Med. 5:190s10. doi: 10.1126/scitranslmed.3004731
Rieder, F., Fiocchi, C., and Rogler, G. (2017). Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology 152, 340–350. doi: 10.1053/j.gastro.2016.09.047
Rosenbloom, J., Macarak, E., Piera-Velazquez, S., and Jimenez, S. A. (2017). Human fibrotic diseases: current challenges in fibrosis research. Methods Mol. Biol. 1627, 1–23. doi: 10.1007/978-1-4939-7113-8
Sandek, A., Bauditz, J., Swidsinski, A., Buhner, S., Weber-Eibel, J., Haehling, S. V., et al. (2007). Altered intestinal function in patients with chronic heart failure. J. Am. Coll. Cardiol. 50, 1561–1569. doi: 10.1016/j.jacc.2007.07.016
Sandek, A., Swidsinski, A., Schroedl, W., Watson, A., Valentova, M., Herrmann, R., et al. (2014). Intestinal blood flow in patients with chronic heart failure: a link with bacterial growth, gastrointestinal symptoms, and cachexia. J. Am. Coll. Cardiol. 64, 1092–1102. doi: 10.1016/j.jacc.2014.06.1179
Sazuka, S., Katsuno, T., Nakagawa, T., Saito, M., Saito, K., Maruoka, D., et al. (2014). Fibrocytes are involved in inflammation as well as fibrosis in the pathogenesis of Crohn’s disease. Dig. Dis. Sci. 59, 760–768. doi: 10.1007/s10620-013-2813-8
Schaedler, R. W., Dubos, R., and Costello, R. (1965). The development of the bacterial flora in the gastrointestinal tract of mice. J. Exp. Med. 122, 59–66. doi: 10.1084/jem.122.1.59
Schiattarella, G. G., Sannino, A., Toscano, E., Giugliano, G., Gargiulo, G., Franzone, A., et al. (2017). Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: a systematic review and dose-response meta-analysis. Eur. Heart J. 38, 2948–2956. doi: 10.1093/eurheartj/ehx342
Schmitz, J., Owyang, A., Oldham, E., Song, Y., Murphy, E., McClanahan, T. K., et al. (2005). IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490. doi: 10.1016/j.immuni.2005.09.015
Small, C. L., Reid-Yu, S. A., McPhee, J. B., and Coombes, B. K. (2013). Persistent infection with Crohn’s disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat. Commun. 4:1957. doi: 10.1038/ncomms2957
Speca, S., Giusti, I., Rieder, F., and Latella, G. (2012). Cellular and molecular mechanisms of intestinal fibrosis. World J. Gastroenterol. 18, 3635–3661. doi: 10.3748/wjg.v18.i28.3635
Stallmach, A., Schuppan, D., Riese, H. H., Matthes, H., and Riecken, E. O. (1992). Increased collagen type III synthesis by fibroblasts isolated from strictures of patients with Crohn’s disease. Gastroenterology 102, 1920–1929. doi: 10.1016/0016-5085(92)90314-o
Sun, G., Yin, Z., Liu, N., Bian, X., Yu, R., Su, X., et al. (2017). Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem. Biophys. Res. Commun. 493, 964–970. doi: 10.1016/j.bbrc.2017.09.108
Takahashi, Y., Saito, A., Chiba, H., Kuronuma, K., Ikeda, K., Kobayashi, T., et al. (2018). Impaired diversity of the lung microbiome predicts progression of idiopathic pulmonary fibrosis. Respir. Res. 19:34. doi: 10.1186/s12931-018-0736-9
Tang, W. H. W., Li, D. Y., and Hazen, S. L. (2019). Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 16, 137–154. doi: 10.1038/s41569-018-0108-7
Tang, W. H., Wang, Z., Kennedy, D. J., Wu, Y., Buffa, J. A., Agatisa-Boyle, B., et al. (2015). Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 116, 448–455. doi: 10.1161/CIRCRESAHA.116.305360
Tremaroli, V., and Bäckhed, F. (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249. doi: 10.1038/nature11552
Tripathi, A., Debelius, J., Brenner, D. A., Karin, M., Loomba, R., Schnabl, B., et al. (2018). The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 15, 397–411. doi: 10.1038/s41575-018-0011-z
Tsochatzis, E. A., Bosch, J., and Burroughs, A. K. (2014). Liver cirrhosis. Lancet 383, 1749–1761. doi: 10.1016/S0140-6736(14)60121-5
Van Assche, G., Geboes, K., and Rutgeerts, P. (2004). Medical therapy for Crohn’s disease strictures. Inflamm. Bowel Dis. 10, 55–60. doi: 10.1097/00054725-200401000-00009
van Tol, E. A., Holt, L., Li, F. L., Rippe, R., Yamauchi, M., Pucilowska, J., et al. (1999). Bacterial cell wall polymers promote intestinal fibrosis by direct stimulation of myofibroblasts. Am. J. Physiol. 277, G245–G255. doi: 10.1152/ajpgi.1999.277.1.G245
Volynets, V., Küper, M. A., Strahl, S., Maier, I. B., Spruss, A., Wagnerberger, S., et al. (2012). Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig. Dis. Sci. 57, 1932–1941. doi: 10.1007/s10620-012-2112-9
Watanabe, H., Miyamoto, Y., Honda, D., Tanaka, H., Wu, Q., Endo, M., et al. (2013). p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 83, 582–592. doi: 10.1038/ki.2012.448
Wigg, A. J., Roberts-Thomson, I. C., Dymock, R. B., McCarthy, P. J., Grose, R. H., and Cummins, A. G. (2001). The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 48, 206–211. doi: 10.1136/gut.48.2.206
Wong, J., Piceno, Y. M., DeSantis, T. Z., Pahl, M., Andersen, G. L., and Vaziri, N. D. (2014). Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 39, 230–237. doi: 10.1159/000360010
Wynn, T. A. (2008). Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210. doi: 10.1002/path.2277
Yan, A. W., and Schnabl, B. (2012). Bacterial translocation and changes in the intestinal microbiome associated with alcoholic liver disease. World J. Hepatol. 4, 110–118. doi: 10.4254/wjh.v4.i4.110
Yang, W., Zhang, S., Zhu, J., Jiang, H., Jia, D., Ou, T., et al. (2019). Gut microbe-derived metabolite trimethylamine N-oxide accelerates fibroblast-myofibroblast differentiation and induces cardiac fibrosis. J. Mol. Cell. Cardiol. 134, 119–130. doi: 10.1016/j.yjmcc.2019.07.004
Yiu, W. H., Lin, M., and Tang, S. C. W. (2014). Toll-like receptor activation: from renal inflammation to fibrosis. Kidney Int. Suppl. 4, 20–25. doi: 10.1038/kisup.2014.5
Zhang, D., Li, S., Wang, N., Tan, H. Y., Zhang, Z., and Feng, Y. (2020). The cross-talk between gut microbiota and lungs in common lung diseases. Front. Microbiol. 11:301. doi: 10.3389/fmicb.2020.00301
Zhang, Y., Gao, F., Tang, Y., Xiao, J., Li, C., Ouyang, Y., et al. (2018). Valproic acid regulates Ang II-induced pericyte-myofibroblast trans-differentiation via MAPK/ERK pathway. Am. J. Transl. Res. 10, 1976–1989.
Zhao, S., Dejanovic, D., Yao, P., Bhilocha, S., Sadler, T., Schirbel, A., et al. (2020). Selective deletion of MyD88 signaling in α-SMA positive cells ameliorates experimental intestinal fibrosis via post-transcriptional regulation. Mucosal Immunol. 13, 665–678. doi: 10.1038/s41385-020-0259-9
Zheng, L., Zhang, X., Chen, J., Ichikawa, R., Wallace, K., Pothoulakis, C., et al. (2013). Sustained TL1A (TNFSF15) expression on both lymphoid and myeloid cells leads to mild spontaneous intestinal inflammation and fibrosis. Eur. J. Microbiol. Immunol. 3, 11–20. doi: 10.1556/EuJMI.3.2013.1.2
Zhou, Y., Chen, L., Sun, G., Li, Y., and Huang, R. (2019). Alterations in the gut microbiota of patients with silica-induced pulmonary fibrosis. J. Occup. Med. Toxicol. 14:5. doi: 10.1186/s12995-019-0225-1
Keywords: intestinal fibrosis, fibrogenesis, gut microbiota, microbiota alteration, metabolites
Citation: Zhan S, Li N, Liu C, Mao R, Wu D, Li T, Chen M, Zhuang X and Zeng Z (2021) Intestinal Fibrosis and Gut Microbiota: Clues From Other Organs. Front. Microbiol. 12:694967. doi: 10.3389/fmicb.2021.694967
Received: 14 April 2021; Accepted: 24 June 2021;
Published: 16 July 2021.
Edited by:
Paul Laszlo Bollyky, Stanford University, United StatesReviewed by:
Christoph Reinhardt, Johannes Gutenberg University Mainz, GermanyCopyright © 2021 Zhan, Li, Liu, Mao, Wu, Li, Chen, Zhuang and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojun Zhuang, emh1YW5neGo5QG1haWwuc3lzdS5lZHUuY24=; Zhirong Zeng, emVuZ3poaXJvbmdAbWFpbC5zeXN1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.