
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 29 June 2021
Sec. Microbial Physiology and Metabolism
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.694824
This article is part of the Research Topic Bacterial Secretion Systems, Volume II View all 17 articles
 Luca Robinson1
Luca Robinson1 Janie Liaw1
Janie Liaw1 Zahra Omole1
Zahra Omole1 Dong Xia2
Dong Xia2 Arnoud H. M. van Vliet3
Arnoud H. M. van Vliet3 Nicolae Corcionivoschi4,5
Nicolae Corcionivoschi4,5 Abderrahman Hachani6†
Abderrahman Hachani6† Ozan Gundogdu1*†
Ozan Gundogdu1*†The Type VI Secretion System (T6SS) has important roles relating to bacterial antagonism, subversion of host cells, and niche colonisation. Campylobacter jejuni is one of the leading bacterial causes of human gastroenteritis worldwide and is a commensal coloniser of birds. Although recently discovered, the T6SS biological functions and identities of its effectors are still poorly defined in C. jejuni. Here, we perform a comprehensive bioinformatic analysis of the C. jejuni T6SS by investigating the prevalence and genetic architecture of the T6SS in 513 publicly available genomes using C. jejuni 488 strain as reference. A unique and conserved T6SS cluster associated with the Campylobacter jejuni Integrated Element 3 (CJIE3) was identified in the genomes of 117 strains. Analyses of the T6SS-positive 488 strain against the T6SS-negative C. jejuni RM1221 strain and the T6SS-positive plasmid pCJDM202 carried by C. jejuni WP2-202 strain defined the “T6SS-containing CJIE3” as a pathogenicity island, thus renamed as Campylobacter jejuni Pathogenicity Island-1 (CJPI-1). Analysis of CJPI-1 revealed two canonical VgrG homologues, CJ488_0978 and CJ488_0998, harbouring distinct C-termini in a genetically variable region downstream of the T6SS operon. CJPI-1 was also found to carry a putative DinJ-YafQ Type II toxin-antitoxin (TA) module, conserved across pCJDM202 and the genomic island CJIE3, as well as several open reading frames functionally predicted to encode for nucleases, lipases, and peptidoglycan hydrolases. This comprehensive in silico study provides a framework for experimental characterisation of T6SS-related effectors and TA modules in C. jejuni.
Bacterial secretion systems, classified from Type I to X according to their genetic and structural organisation and composition, are protein transport machineries enabling niche colonisation, interaction with host cells, and bacterial antagonism (Costa et al., 2015; Palmer et al., 2021). Genes encoding for the Type VI Secretion System (T6SS) are present in more than 25% of Proteobacteria (Bingle et al., 2008; Barret et al., 2013). The injection of a panel of T6SS effectors into competing bacteria promotes the fitness of T6SS-positive strains in polymicrobial environments, including the gut ecosystem (Coulthurst, 2019; Wood et al., 2020). However, the T6SS is not restricted to bacterial antagonism and can mediate host-pathogen interactions. Some T6SS effectors bear anti-eukaryotic activities that subvert the host cell cytoskeleton, evade host defences by countering reactive oxygen species (ROS), and modulate the host inflammatory response (Hachani et al., 2016; Chen et al., 2019).
Despite the multiple roles of T6SSs in complex ecosystems, the genes encoding the T6SS core components are highly conserved into genomic clusters (Coulthurst, 2019). The structure of the T6SS shares features with the bacteriophage T4 contractile apparatus, with structural homology to the phage tail tube and spike proteins (Ho et al., 2014). A fully assembled T6SS apparatus requires a minimal set of 13 core components (Zoued et al., 2014). The machinery is characterised by a puncturing spike (VgrG) that structurally resembles the bacteriophage T4 gp27/gp5 proteins (typically sharpened by a Proline-Alanine-Alanine-Arginine (PAAR) protein), a contractile sheath (formed by the complex TssB and TssC) encasing a needle-like tube (Hcp/TssD) and capped by a core component (TssA) in the cytoplasm. A scaffold formed by a membrane-associated complex (TssJLM) and a cytoplasmic baseplate (TssEFGK) complete the system (Figure 1; Leiman et al., 2009; Zoued et al., 2014; Cianfanelli et al., 2016). Upon contraction, the TssBC sheath propels the VgrG-PAAR complex and associated effectors into target cells or the external milieu (Coulthurst, 2019). The contracted sheath can be depolymerised by the ATPase ClpV/TssH and released TssB and TssC subunits are recycled for assembly (Kapitein et al., 2013; Zoued et al., 2014).
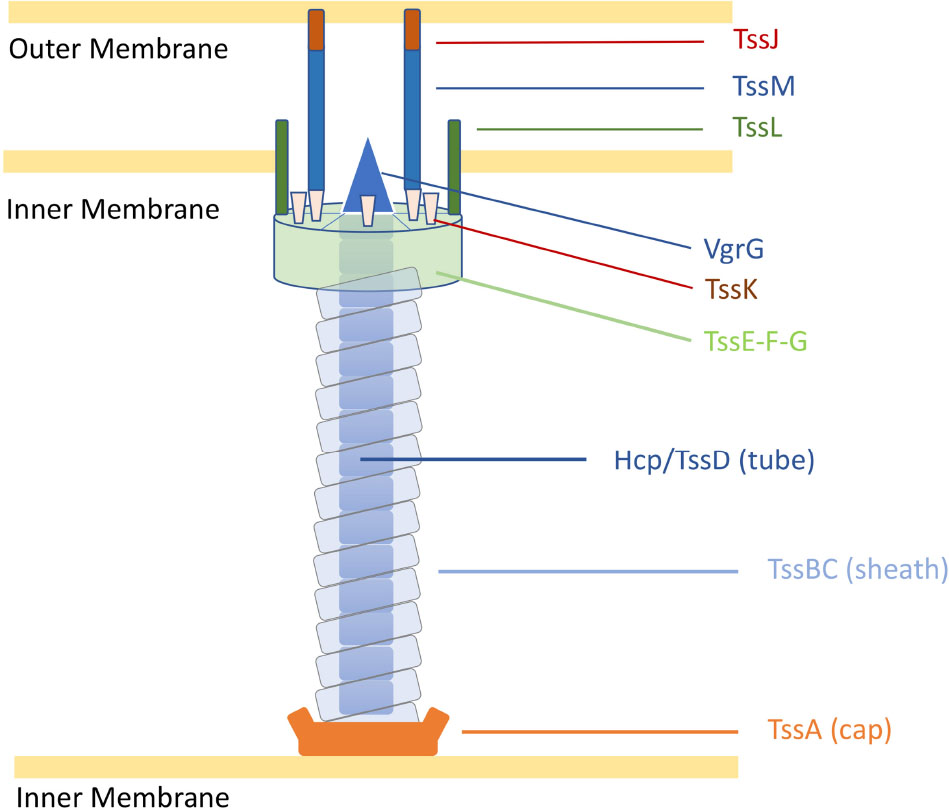
Figure 1. Representation of the C. jejuni T6SS in extended state [adapted from Cherrak et al. (2019)]. Proteins are labelled according to the nomenclature used for C. jejuni T6SS components. Tss, Type six secretion.
Type VI Secretion System effectors exhibit a wide range of functions, mostly anti-bacterial, with some displaying transkingdom activities and few solely targetting eukaryotes (Alteri and Mobley, 2016). To date, T6SS effectors have been shown to harbour functions such as nucleases (Ma et al., 2014; Jana et al., 2019), lipases (Russell et al., 2013; Jiang et al., 2016), peptidoglycan hydrolases (Russell et al., 2011; Whitney et al., 2013), pore-forming activities (English et al., 2012; Fridman et al., 2020), actin cross-linking (Pukatzki et al., 2006), and anti-fungal activities (Trunk et al., 2018, 2019). Translocation of “cargo” effectors is mediated through their non-covalent interaction with Hcp, VgrG, or PAAR proteins, whilst “specialised” effectors consist of catalytic domains covalently fused to Hcp, VgrG, or PAAR proteins (Coulthurst, 2019). Anti-bacterial effector genes are often associated with cognate genes encoding for immunity proteins, producing effector-immunity pairs (Coulthurst, 2019). Immunity proteins are typically located in the cellular compartments targetted by their cognate effectors to prevent self-toxicity (Alcoforado Diniz et al., 2015).
Campylobacter jejuni is a gram-negative microaerophilic bacterium and one of the leading causes of human foodborne gastroenteritis worldwide (Burnham and Hendrixson, 2018). C. jejuni is abundant in the chicken gut, making handling and consumption of contaminated poultry products the primary foodborne transmission route to humans (Ijaz et al., 2018; McKenna et al., 2020). Although considered a commensal of the avian gut, C. jejuni can be an opportunistic pathogen in birds, depending on the genetics of host and bacterial strain (Humphrey et al., 2014; Wigley, 2015). C. jejuni infection in humans can cause bloody diarrhoea, fever, and abdominal pains. In low-resource regions, C. jejuni infections are common in young children and correlate with stunted growth and life-long physical and cognitive deficiencies (Amour et al., 2016). In high-resource regions, it is estimated that 1 in every 100 individuals develop a C. jejuni-related illness each year (Tam et al., 2012). It is still unclear how avian species can tolerate a high presence of C. jejuni without developing overt disease, yet a relatively low infectious dose leads to disease in humans (Gundogdu and Wren, 2020).
In C. jejuni, the T6SS biological functions have been associated with host colonisation, cell adhesion and invasion, survival in bile salts, contact-dependent lysis of erythrocytes, and contributing to oxidative stress (Lertpiriyapong et al., 2012; Bleumink-Pluym et al., 2013; Liaw et al., 2019). So far, Hcp has been suggested as a T6SS effector contributing to C. jejuni host cell adhesion and invasion (Lertpiriyapong et al., 2012; Bleumink-Pluym et al., 2013; Noreen et al., 2018); however, no evidence currently supports the direct contribution to these phenotypes by Hcp in isolation. Whilst few studies have indicated the prevalence of T6SS in C. jejuni (Harrison et al., 2014; Corcionivoschi et al., 2015; Ugarte-Ruiz et al., 2015), a larger comprehensive bioinformatic analysis of T6SS within C. jejuni and the identification of associated effectors are still needed. In this study, we investigated the prevalence and genomic organisation of the T6SS in 513 publicly available C. jejuni genomes through screening of the major T6SS components, previously characterised T6SS effectors, and the integrative element Campylobacter jejuni Integrated Element 3 (CJIE3). Using C. jejuni 488 strain as reference we discovered a novel pathogenicity island (PAI) variant of CJIE3, reclassifying the “T6SS-containing CJIE3” as Campylobacter jejuni Pathogenicity Island-1 (CJPI-1). We also observed that two distinct VgrG proteins are present in the PAI of C. jejuni 488 strain, with a number of other C. jejuni strains also possessing both VgrG proteins. Using bioinformatic analysis, we identified a number of putative T6SS effectors and predicted toxin-antitoxin (TA) modules carried by the CJPI-1 PAI.
Genome sequencing was performed as previously described by Ugarte-Ruiz et al. (2015) and adapted for this study. Briefly, paired-end Fastq data was generated on an Illumina MiSeq and assessed using FastQC (Andrews, 2010). Quality control of the sequencing reads was conducted using Trimmomatic (v0.39) (“leading” and “trailing” setting of 5, a “slidingwindow” setting of 4:20 and a “minlength” of 36 nucleotides) (Bolger et al., 2014). Assembly was performed with VelvetOptimiser (v2.2.6) using n50 optimisation and “kmer” length of 37 nucleotides (Zerbino and Birney, 2008; Gladman and Seemann, 2012). Contigs were ordered against T6SS-positive C. jejuni M129 strain (accession no. CP007749) (Konkel et al., 1992) using ABACAS (v1.3.1) (Assefa et al., 2009). Annotation of the genome was performed with prokka (v.1.14.6) (Seemann, 2014) using C. jejuni NCTC11168 strain (AL111168) (Parkhill et al., 2000). The genome was visualised and manually edited using Artemis and Artemis Comparison Tool (ACT) software (Carver et al., 2005, 2012).
Nucleotide and amino acid sequences of C. jejuni genomes were collected from the NCBI RefSeq genome database release 99 (May 2020) at assembly level “scaffold” or higher (NCBI, 1982b). Reference genomes 108 (JX436460) (Bleumink-Pluym et al., 2013), 43431 (genome sequence from Liaw et al., 2019), and the newly assembled 488 strain were also included into the genome dataset and a local nucleotide and protein database was constructed. Our local database was then filtered to remove genomes that possessed any of the following traits: a total genome size greater than 2 Mb, no assembly protein sequence data available to download from the RefSeq database, a total assembly possessing more than 200 contigs, or the genome possessed an Average Nucleotide Identity (ANI) of less than 95%. ANI was calculated using FastANI with a fragment length of 1000 bp against the reference C. jejuni NCTC11168 (AL111168) strain (Jain et al., 2018). A total of 41 genomes were removed from the dataset. Metadata, including host and sample location, was collected from the NCBI BioSample database (NCBI, 1982a). Genome visualisation was performed in Artemis (Carver et al., 2012).
BLASTP (Altschul et al., 1990) was employed to identify the 13 T6SS components amongst the C. jejuni genomes, using default parameters. The amino acid sequences of the 13 T6SS loci from reference strain 108 (JX36460), which carries a functional T6SS, was aligned against a local protein dataset created for the C. jejuni genomes (Bleumink-Pluym et al., 2013). A similarity percentage was calculated by dividing the bit-score value for each amino acid alignment by two times the specific lengths of the individual query amino acid sequence (Fridman et al., 2020). Protein presence was regarded positive when a minimum of 50% similarity was observed. C. jejuni genomes possessing at least 11 out of the 13 T6SS loci were considered to contain a T6SS (T6SS-positive), and those that possessed fewer than 11 were considered lacking a T6SS (T6SS-negative).
BLASTP (Altschul et al., 1990) was employed to identify the presence of PAAR-motif containing proteins and known T6SS effectors in the local C. jejuni genome database. Representative amino acid sequences from the NCBI-CDD were downloaded for the protein subclasses PAAR1, PAAR2, PAAR3, PAAR4, PAAR5, PAAR-Rhs, PAAR-CT1, and PAAR-CT2 (Marchler-Bauer et al., 2017; Supplementary Data 1). Amino acid sequences characterised as T6SS “cargo” effectors were downloaded from the NCBI Protein database (NCBI, 1982c; Supplementary Table 1). Proteins designated as “cargo” are independent effectors that do not exist as toxin domain-containing extensions of the major T6SS components Hcp, VgrG or PAAR. The amino acid sequences were aligned against a local protein dataset created for the C. jejuni genomes. A minimum threshold expected value of 1e-10 was implemented in the search where a value below this threshold was deemed a positive hit.
To identify the presence of CJIE3 in the C. jejuni genome dataset, the gene cje1094 (integrase) from reference strain RM1221 (CP000025) [denoted as a suitable candidate for PCR-identification of CJIE3 by Parker et al. (2006)], and genes cje1105 and cje1153, were used in an in silico identification method. BLASTN (Altschul et al., 1990) was employed to align the nucleotide sequence of the genes against the local nucleotide dataset created for the C. jejuni genomes. A similarity percentage was calculated according to Fridman et al. (2020). To be regarded as positive for CJIE3, a minimum similarity of 50% was required to two of the three genes: cje1094, cje1105, and/or cje1153. To be regarded as possessing a T6SS-harbouing plasmid, a minimum similarity of 50% was required to only gene cje1094 and the presence of at least 11 T6SS loci (T6SS-positive). C. jejuni 108 strain was excluded from this analysis as it does not possess a whole genome sequence, thus reducing this analysis to 512 genomes.
Artemis Comparison Tool and Clinker (Carver et al., 2005; Gilchrist and Chooi, 2021) were used to comparatively align the genome of the re-sequenced and assembled T6SS-positive C. jejuni 488 strain against strain RM1221 (CP000025) and annotated plasmid pCJDM202 (CP014743) (Fouts et al., 2005; Marasini and Fakhr, 2016). Amino acid identity was calculated with BLAST Global Alignment–Protein (Needleman-Wunsch Global Align), using default parameters (Altschul et al., 1990).
Webtools NCBI CDD-BLAST (Marchler-Bauer et al., 2017), SMART (Letunic and Bork, 2018), HmmScan (Potter et al., 2018), Pfam (El-Gebali et al., 2019), SCANPROSITE (de Castro et al., 2006), CDART (Geer et al., 2002), SUPERFAMILY (Gough et al., 2001), InterPro (Mitchell et al., 2019), and MOTIF (GenomeNet, 2015) were used to identify protein domains and characteristic motifs in the CJPI-1 predicted proteins. Signal peptides, transmembrane helices, and subcellular localisation were predicted using Psortb v3.0 (Yu et al., 2010), CELLO v2.5 (Yu et al., 2006), SignalP-5.0 (Armenteros et al., 2019), TMPred (Hofmann and Stoffel, 1993), and TMHMM (Sonnhammer et al., 1998) to assist in protein function prediction. Default parameters were used throughout, with an expected value (E-value) of 0.01 determined as a cut-off and organism group defined as Gram-negative where required. Protein functions were inferred following congruent predictions from at least 5 out of the 9 used webtools (Supplementary Tables 2, 3). Structural homology modelling was performed using the Phyre2 and I-Tasser servers (Zhang, 2008; Kelley et al., 2015).
To assess sequence identity, amino acid sequences for proteins encoded by vgrG1 and vgrG2 in C. jejuni 488 strain were analysed by BLASTP (Altschul et al., 1990) against the NCBI reference protein database, excluding C. jejuni to prevent self-hits. Multiple sequence alignment of vgrG1 and vgrG2 was conducted using Clustal Omega (Madeira et al., 2019).
BLASTN (Altschul et al., 1990) was employed to identify vgrG genes amongst the C. jejuni genomes. The nucleotide sequence of the vgrG gene from the T6SS-positive 108 strain (JX436460) was aligned against a local nucleotide dataset created for the T6SS-positive C. jejuni genomes of assembly level “complete” or higher (Bleumink-Pluym et al., 2013). A cut-off similarity percentage was calculated according to Fridman et al. (2020). To be regarded as positive for a vgrG gene, a minimum similarity of 50% was required. Full length VgrG protein sequences were then obtained from T6SS-positive assembly level genomes “complete” or higher and aligned using MUSCLE (Edgar, 2004) with default parameters. Genomes CJ017CC464, CJ018CCUA, and ZS007 were removed from the analysis due to disrupted open reading frames (ORFs). A phylogenetic tree was constructed from the alignment file using the Maximum-Likelihood method, with JTT modelling, partial deletion (95%), and bootstrapping (n = 500) parameters, conducted in the Molecular Evolutionary Genetics Analysis X (MEGAX) v. 10.1.8 software package (Kumar et al., 2018). The analysis contained 36 amino acid sequences.
The amino acid sequences of the query proteins were searched against the NCBI-CDD (Marchler-Bauer et al., 2017), and the subsequent output alignments corresponding to identified domains were extracted and annotated to identify the conserved catalytic residues described (Zhang et al., 2012; Sun et al., 2015; Tak et al., 2019).
BLASTP (Altschul et al., 1990) was employed to identify the presence and genomic context of the CJPI-1 functionally predicted proteins amongst the C. jejuni genome database. The amino acid sequences were aligned against the local protein dataset created for the C. jejuni genomes. A similarity percentage was calculated according to Fridman et al. (2020). Protein presence regarded as positive required a minimum similarity of 50%. C. jejuni 108 strains was excluded from this analysis as it does not possess a whole genome sequence, therefore this analysis involved 512 genomes.
We have determined the prevalence of the T6SS in publicly available C. jejuni genomes (Supplementary Table 4) by compiling a local dataset of nucleotide and amino acid sequences from isolates with an assembly level “scaffold” or higher from the NCBI RefSeq genome database. This was further populated with C. jejuni 108 and 43431 reference strains and the newly assembled 488 strain, creating a total of 513 genomes. The prevalence study of 13 T6SS core components (TssA-TssM) against our local C. jejuni database classified 136 of the 513 (26.51%) C. jejuni genomes as T6SS-positive and 377 of the 513 (73.49%) as T6SS-negative (Supplementary Tables 5, 6). Interestingly, two T6SS-negative C. jejuni strains, 255 and 10186, were found to possess 10 out of the 13 T6SS genes, with the genes tagH, tssG, and vgrG missing. Furthermore, the genome of C. jejuni OXC6589 strain was identified as the only one without a T6SS complete cluster to present the gene hcp. Our analysis of C. jejuni strains identified a single copy of the T6SS operon, with a conserved set of T6SS core genes sharing synteny with closely related species (Table 1). To date, all T6SS core structural components have been identified in C. jejuni with the exception of TssH (ClpV), the ATPase responsible for disassembly of the contracted sheath components, which is absent from all sequenced C. jejuni T6SS operons. This raises the possibility of an alternative mode of sheath disassembly, or the existence of a ClpV-like ATPase encoded distally from the T6SS cluster (Liaw et al., 2019).
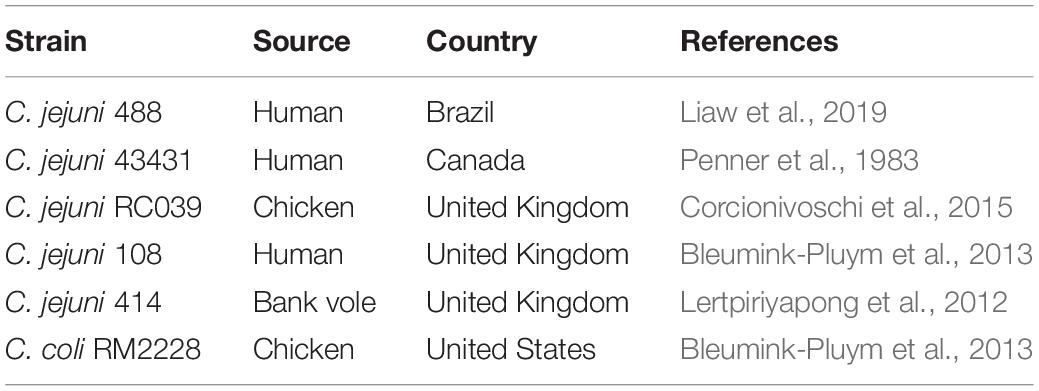
Table 1. C. jejuni and C. coli strains studied to date with a T6SS.
Hitherto, no T6SS-associated effectors have been identified and/or characterised in T6SS-positive C. jejuni (Lertpiriyapong et al., 2012; Bleumink-Pluym et al., 2013; Liaw et al., 2019). Using 40 characterised ‘cargo’ effectors from a range of bacteria including Pseudomonas aeruginosa, Serratia marcescens, Yersinia pseudotuberculosis, and Burkholderia thailandensis (Supplementary Table 1), we performed BLASTP-homology searches for the presence of such effectors within C. jejuni strains. Searches returned no positive matches leading us to conclude that T6SS-containing C. jejuni may possess a subset of unique “cargo” effectors.
The initial study of the T6SS in C. jejuni revealed its integration within the earlier acquired CJIE3; a genomic island displaying a mosaic gene arrangement and present in a number of C. jejuni strains, including RM1221 (a T6SS-negative C. jejuni strain) (Fouts et al., 2005; Bleumink-Pluym et al., 2013). Distribution analyses of CJIE3 in C. jejuni from human and avian isolates report varying prevalence of this integrated element, with only 10% of CJIE3 harbouring a T6SS (Bleumink-Pluym et al., 2013; Kovanen et al., 2019). In this study, we screened the CJIE3 integrase, cje1094, and genes cje1105 and cje1153, using an in silico identification method against a local C. jejuni database as proxies for CJIE3 identification (Supplementary Tables 8–10). Integrase cje1094 possesses strong homology to A0W69_09480 harboured on the T6SS-positive megaplasmid pCJDM202 (Table 2), therefore, we included two further proxies to distinguish between T6SS-containing CJIE3 and T6SS-harbouring plasmids. We observed that 146 of the 512 (28.51%) genomes possessed the CJIE3, of which 117 (80.14%) were T6SS-positive (Supplementary Table 4). Therefore, 117 of the 135 (86.67%) T6SS-positive genomes were identified to possess the CJIE3 and 15 (2.93%) were found harbouring T6SS-positive plasmids (Table 3). Integration of the C. jejuni T6SS was described as occurring between homologues of the genes cje1139 and cje1141/cje1142 from the CJIE3 in RM1221 (Bleumink-Pluym et al., 2013). The genes cje1141 and cje1142 share homology to the major T6SS component tssI/vgrG and possess rhs (rearrangement hotspots) signatures, suggested to have mediated the integration of the T6SS into CJIE3 (Hill, 1999; Jackson et al., 2009; Bleumink-Pluym et al., 2013). Our data supports the findings that the T6SS has been integrated into the CJIE3, as a significant proportion of the T6SS-positive strains in this study also possess the CJIE3. Furthermore, we identified a number of CJIE3-positive genomes that do not possess a complete T6SS cluster, further supporting that integration of the T6SS has occurred subsequently to the acquisition of the integrated element (Supplementary Table 4; Bleumink-Pluym et al., 2013).
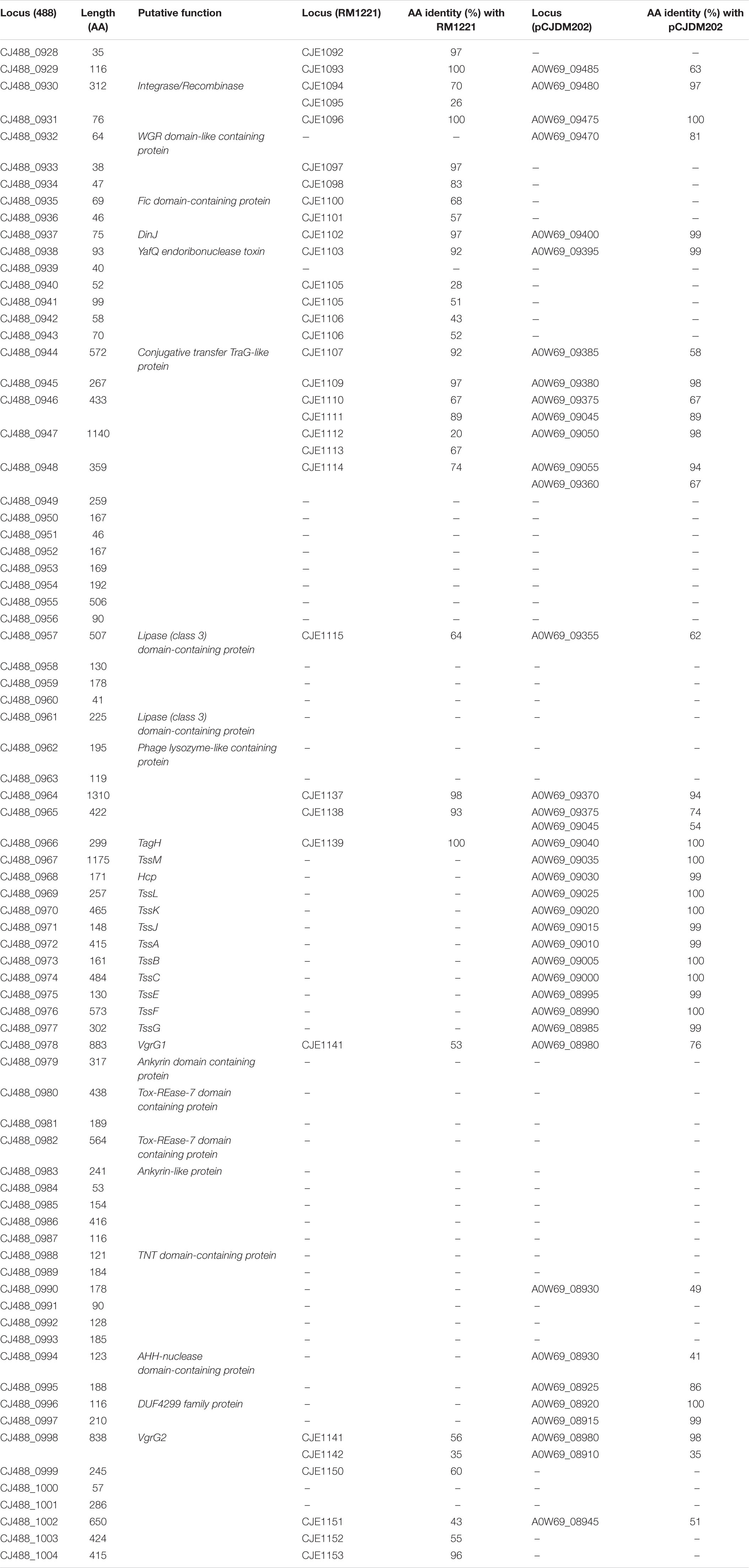
Table 2. Predicted proteins in CJPI-1 of T6SS-positive C. jejuni 488 strain with the respective amino acid length and inferred function. Protein locus tags of homologous proteins found in the CJIE3 of RM1221 and pCJDM202 of WP2-202 are shown with the respective amino acid (aa) identity (%).
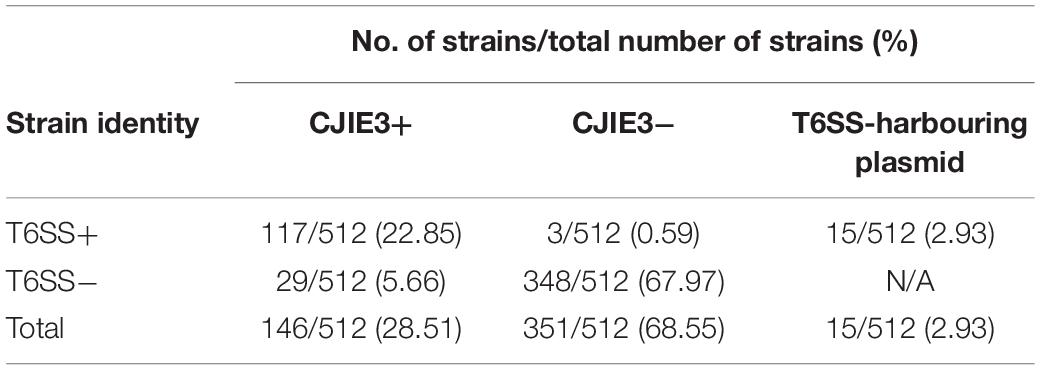
Table 3. C. jejuni strains containing CJIE3 by presence or absence of a T6SS.
C. jejuni Integrated Element 3 belongs to a large family of mobile genetic elements (MGEs) and, as observed in integrative and conjugative elements, could potentially operate horizontal transfer of DNA regions between bacterial species during extended periods of close proximity (Dobrindt et al., 2004; Johnson and Grossman, 2015). MGEs can also exist as PAIs; a large subset of integrative elements (>10 kb) carrying virulence genes, such as secretion systems and their cognate effectors (Jarvis et al., 1995; da Cruz Campos et al., 2020). Members of the order Bacteroidales can display three different “genetic architectures,” with two of these (GA1 and GA2) found on integrative conjugative elements (Coyne et al., 2016). CJIE3 shares sequence homology with proteins encoded on the Campylobacter coli RM2228 megaplasmid and 71-kb pathogenicity island HHGI1 of Helicobacter hepaticus ATCC51449, the latter possessing a T6SS (Fouts et al., 2005; Bartonickova et al., 2013). Interestingly, several C. jejuni megaplasmids also carry T6SS genes (Gunther et al., 2016; Marasini and Fakhr, 2016, 2017). Most recently, megaplasmids pCJDM202 and pCJDM67L from C. jejuni WP2-202 and OD2-67 strains, respectively, were found to harbour the T6SS cluster, along with the tetracycline resistance gene tetO, and T4SS conjugative DNA transfer systems (Marasini et al., 2020). The authors demonstrated that the presence of the T6SS on the megaplasmids contributed to enhanced haemolysis, suggested to support the survival of C. jejuni in retail meats (Marasini et al., 2020).
A newly re-sequenced and assembled genome of the T6SS-positive C. jejuni 488 strain was thus comparatively analysed against the genome of T6SS-negative C. jejuni RM1221 strain and T6SS-positive virulence megaplasmid pCJDM202 to investigate the genomic architecture and integration of the T6SS into the CJIE3 of C. jejuni (Figure 2). We propose to reclassify the T6SS-containing genomic island in the same chromosomal location as CJIE3 (between arginyl-tRNA-3 and cje1156 in RM1221) as a new PAI-variant designated as CJPI-1. We observed that the ∼70 kb PAI is longer than the ∼50 kb CJIE3 of RM1221, containing an integrase/recombinase (CJ488_0930) gene (discussed below) and, like CJIE3, is located immediately adjacent to the chromosomal arginyl-tRNA (Table 4). The G + C% contents of both CJPI-1 and CJIE3 are lower than the average content of the 488 and RM1221 genomes, respectively, confirming the hypothesis that both inserted genetic elements could be considered as independently acquired. Furthermore, a direct repeat sequence designated as the attachment (att) sites, “TCCTCTTGAGCGCACCAT,” flanks both sides of the CJPI-1 and CJIE3 islands. Given the similarities between the integrated islands, CJPI-1 is most likely a derivative of CJIE3 that has undergone multiple recombination and/or genetic exchange events. We also discovered that only 30 proteins encoded in CJPI-1 share homology with those encoded in the CJIE3 of RM1221, highlighting differences in genetic composition (Table 2). Unlike CJIE3, CJPI-1 satisfies criteria commonly used to classify PAIs with the possession of the major T6SS components and putative effectors (discussed below). PAIs in other bacteria have also been found to carry T6SS clusters. Notably, HHGI-1 of H. hepaticus ATCC 51449 possesses a T6SS with a similar gene organisation to C. jejuni (Nano and Schmerk, 2007; Barker et al., 2009; Blondel et al., 2009; Bleumink-Pluym et al., 2013). In all, this data confirms that CJPI-1 can be considered as a bona fide PAI.
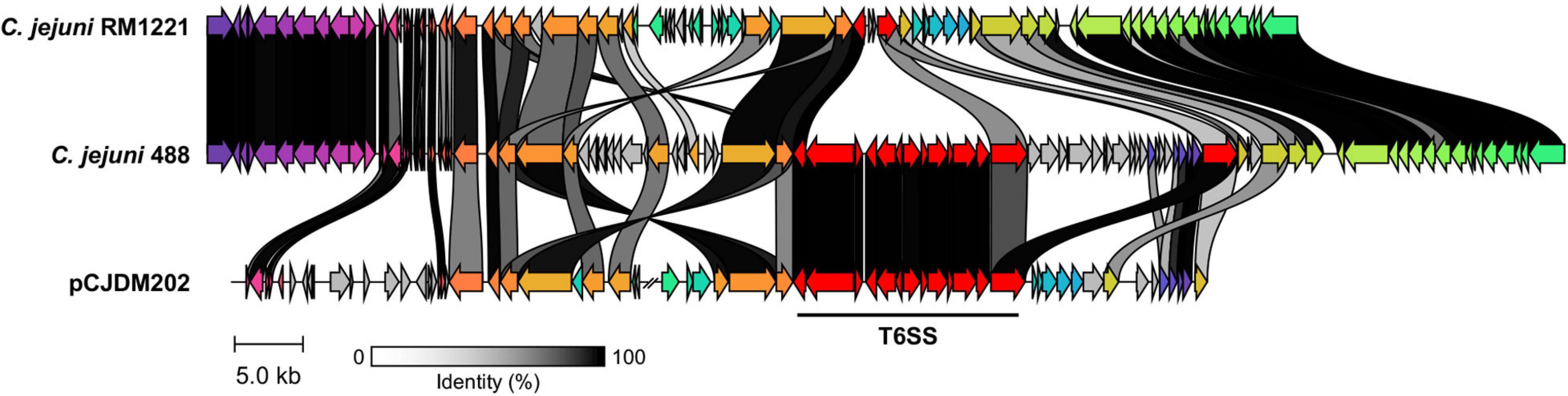
Figure 2. Comparative analyses of CJPI-1 (middle) from T6SS-positive C. jejuni 488 strain to CJIE3 from T6SS-negative strain RM1221 (top) and T6SS-positive megaplasmid pCJDM202 (bottom) from C. jejuni WP2-202 strain. Homologous genes across the three strains share the same arrow colour and are connected by shaded bands corresponding to sequence identity (black = 100%, white = 0%). Red arrows represent major T6SS components. Sequences found in only one strain possess a grey arrow. Only regions of the megaplasmid associated with the T6SS and CJIE3/CJPI-1 regions have been included. The figure was constructed using Clinker.
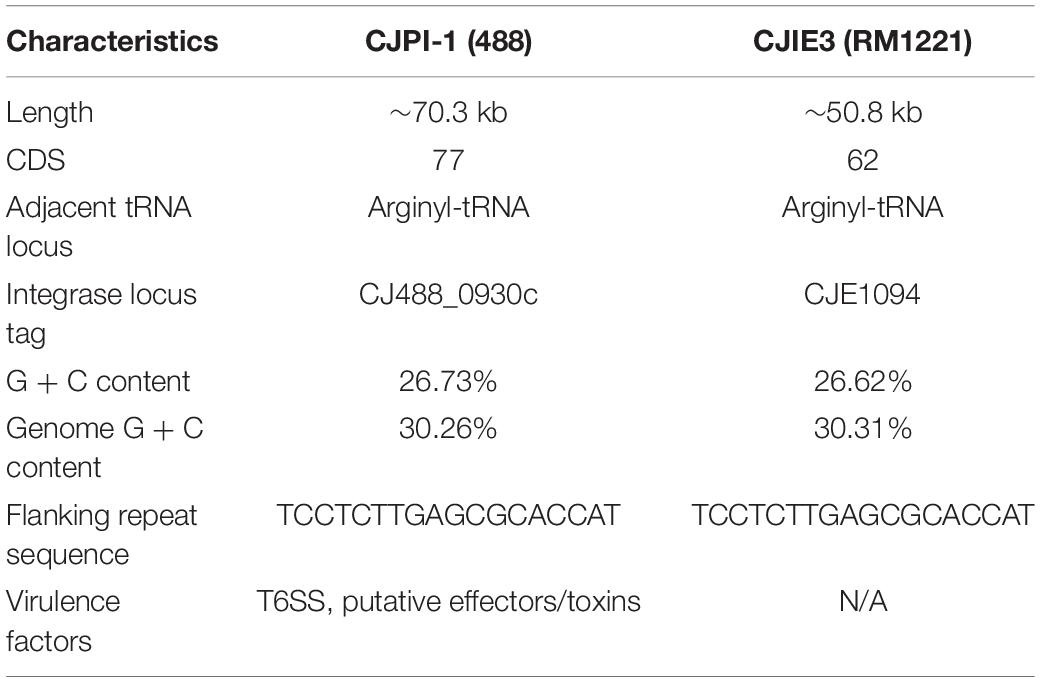
Table 4. Comparative overview of the sequence, gene content, and synteny characteristics of the CJPI-1 from C. jejuni 488 strain and CJIE3 from RM1221.
Comparative analysis of the CJPI-1 to the virulence plasmid pCJDM202 also revealed striking genetic similarities (Figure 2). 35 genes in CJPI-1 matched in pCJDM202, including the T6SS and several genes also found in the CJIE3 (Table 2). Interestingly, the T6SS of CJPI-1 and pCJDM202 share 96% nucleotide similarity across the entire gene cluster. Collectively, this data suggests that CJPI-1 may be resulting from a recombination event of the CJIE3 and a T6SS-containing pCJMD202-like plasmid, leading to the acquisition and integration of the T6SS. However, further analyses are required to understand the genetic events leading to the acquisition of putative effectors (discussed below) which may have occurred through uptake events (mediated by prophages and plasmids conjugation).
Following the identification of this PAI-variant of CJIE3, we set out to bioinformatically investigate the genes encoded in CJPI-1 and assess their genomic context in relation to the PAI and/or T6SS operon. We identified several genes encoding for integrative elements, TA modules, and putative effectors (Table 2 and Figure 3).
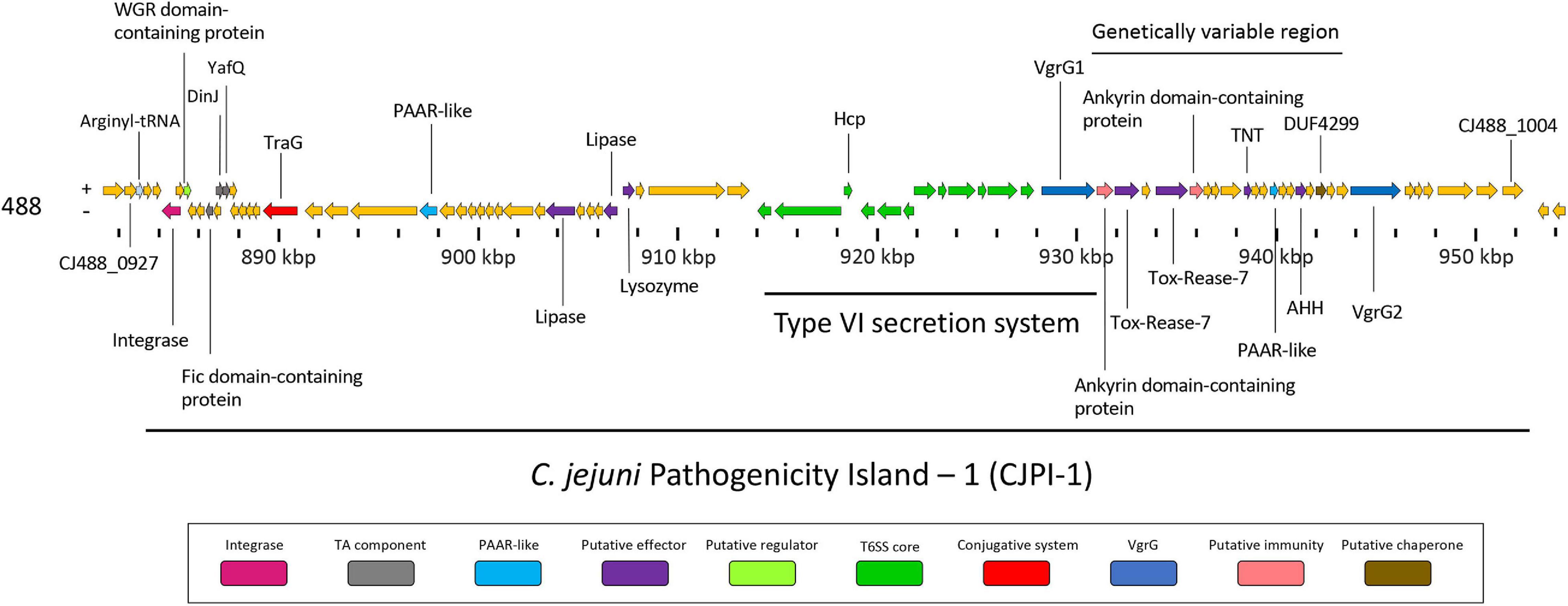
Figure 3. Genomic architecture of the PAI CJPI-1 in T6SS-positive C. jejuni 488 strain. Coloured coding domain sequences (CDS) represent proteins with inferred functions and are labelled. The scale under the CDS represents the nucleotide position [kilobase pair (kbp)] of the pathogenicity island in the genome of C. jejuni 488 strain. The location of the T6SS and genetically variable region are also denoted. The positive (+) symbol on the left-hand side indicates the sense strand in the genome of C. jejuni 488 strain, whilst the negative (–) symbol indicates the antisense strand. Genes sizes are not to scale. Genes are coloured according to predicted function: integrases are shaded magenta, regulators are light green, TA components are grey, conjugative systems are red, putative effectors are purple, PAAR-like proteins are light blue, T6SS components are green, VgrGs are dark blue, putative immunity proteins are pink, and putative chaperone proteins are olive.
A putative integrase/recombinase (CJ488_0930) was identified within CJPI-1, possessing homology to the CJE1094 integrase (70% aa identity) and CJE1095 (26% aa identity) of RM1221, and A0W69_09480 (97% aa identity) from pCJDM202. The CJPI-1 integrase exhibits a Phage_Integrase (PF00589) and a tyrosine recombinase XerD (TIGR02225) domain and is likely a combination of CJE1094 and CJE1095 proteins. Both domains belong to the C-terminal catalytic domains of the DNA breaking-rejoining enzymes superfamily (cl00213). Proteins of this family catalyse recombination of DNA, possessing site-specific integration functions identified in chromosomes, plasmids, and phage genomes (Nash, 1996; Grainge and Jayaram, 1999; Huber and Waldor, 2002). It is predicted that the CJPI-1 and CJIE3 homologous integrases may be responsible for the horizontal transfer and chromosomal integration of the genomic islands to naïve strains at att sites (Santoriello et al., 2020).
CJ488_0935, encoded in CJPI-1, was predicted to belong to the globular Fic/Doc (PF02661) family, harbouring the conserved Fic-motif H-x-F-x-[DE]-[AG]-N-[GK]-R, including the catalytic histidine residue which contributes to AMPylation activity (Sprenger et al., 2017; Veyron et al., 2018). Within our local database, 61 out of the 512 (11.91%) genomes were found to possess the protein CJ488_0935, of which 37 were T6SS-positive (60.66%) and 24 were T6SS-negative (39.34%). Of the T6SS-negative genomes, 12 out of the 24 were found to contain the CJIE3 (Supplementary Table 11). The protein CJE1100 from RM1221 also shares homology to CJ488_0935 (68% aa identity). Identified in a number of bacterial virulence factors (including Type III and IV secreted effectors), the AMPylation activity of Fic proteins have been demonstrated to catalyse the post-translational modifications of host proteins. Such activity leads to cytotoxicity of targetted host cells, as demonstrated in VopS from Vibrio parahaemolyticus and IbpA from Haemophilus somnus (Schmid et al., 2006; Worby et al., 2009; Yarbrough et al., 2009; Engel et al., 2012).
Fic domains can also be found as part of Type II TA toxin modules, as recently discovered in Campylobacter fetus (Goepfert et al., 2013; Sprenger et al., 2017). TA modules consist of a pair of antagonistic genes that encode for a stable toxin and adjacent, unstable antitoxin (Page and Peti, 2016). Many bacterial and archaeal chromosomes bear TA modules, with roles ranging from plasmid inheritance, MGE stability, growth arrest, and control to stress responses (Leplae et al., 2011; Page and Peti, 2016; Fraikin et al., 2020). Type II TA modules are the most widely studied TA systems in bacteria and have been identified to maintain and stabilise integrative elements, as well as involved to increase colonisation and virulence in pathogenic bacteria (Wozniak and Waldor, 2009; Leplae et al., 2011; Norton and Mulvey, 2012). TA modules have been characterised in C. jejuni, as observed in the pVir plasmids of 81-176 and IA3902 strains, and more recently in the YH002 strain isolated from retail beef liver (Shen et al., 2016; Ghatak et al., 2020).
Here, bioinformatic analysis inferred CJ488_0937 and CJ488_0938 as a putative Type II TA module, with the latter found to contain a predicted YafQ_toxin (PF15738) domain. YafQ toxins exhibit endoribonuclease activity and acts as the toxin component, with its activity inhibited by the cognate antitoxin DinJ (Motiejunaite et al., 2007; Prysak et al., 2009). Homologous proteins to CJ488_0938 were also found in RM1221 and pCJDM202, sharing 92 and 99% amino acid identity to CJE1103 and A0W69_09395, respectively. Within our local C. jejuni database, 95 out of the 512 (18.55%) genomes were found to possess the protein CJ488_0938, of which 64 were T6SS-positive (67.37%) and 31 were T6SS-negative (32.63%). Of the T6SS-negative genomes, 13 out of the 31 were found to contain the CJIE3 (Supplementary Table 11). The protein encoded by the upstream gene CJ488_0937 (Figure 3) also shared an amino acid identity of 97% and 99% to proteins CJE1102 and A0W69_09400, respectively. CJ488_0937 does not possess any identifiable domains, therefore, we performed structural homology modelling of CJ488_0937 in the Phyre2 and I-Tasser servers using the amino acid sequence as a template (Zhang, 2008; Kelley et al., 2015). I-Tasser confidently identified the Escherichia coli DinJ-YafQ Type II TA complex (PDB: 4Q2U, Z-score: 1.60) as the most suitable candidate template for modelling, with an exclusive alignment to the DinJ antitoxin (PDB: 4Q2U_1) amino acid sequence. Further, both servers generated models with two predicted helix-turn-helix motifs suggesting, a DNA-binding function commonly identified in type II antitoxins (Aravind et al., 2005; Page and Peti, 2016). This is consistent with the DNA-binding ability and subsequent transcriptional autorepression activity of DinJ, the YafQ antitoxin (Ruangprasert et al., 2014). These elements suggest CJ488_0937 may be acting as the cognate DinJ antitoxin, with experimental confirmation warranted to validate these roles. To our knowledge, this is the first report of a DinJ-YafQ TA module in Campylobacter spp.
The protein encoded by the gene CJ488_0944 was predicted to contain a TraG N-terminal region (PF07916) domain and shares homology to proteins CJE1107 (92% aa identity) and A0W69_09385 (58% aa identity) from RM1221 and pCJDM202, respectively, with the latter annotated as a conjugation transfer protein TraG (Marasini and Fakhr, 2016). Within our local C. jejuni database, 93 out of the 512 (18.16%) genomes were found to possess the protein CJ488_0944 (average per genome = 1.04), of which 33 were T6SS-positive (35.48%) and 60 were T6SS-negative (64.52%). Of the T6SS-negative genomes, 5 out of the 60 were found to contain CJIE3 (Supplementary Table 11). The N-terminus of TraG is required for F pilus assembly; a long filament mediating the conjugative transfer of genetic material (Frost et al., 1994). Homologues of TraG and transfer coupling protein VirD4, a component of the Agrobacterium tumefaciens Type IVa secretion system (T4SSa), have been previously identified in the chromosomes of C. jejuni 81-176 and ATCC 43431, as well as in plasmids pCC31 and pTet (Batchelor et al., 2004; Poly et al., 2005; Chandran Darbari and Waksman, 2015). T4SS DNA conjugation systems were recently found harboured on megaplasmids in C. jejuni (Grohmann et al., 2018; Marasini et al., 2020). The T6SS-positive and TraG-containing megaplasmid pCJDM202 from C. jejuni WP2-202 strain was successfully transferred by conjugation to a T6SS-negative mutant NCTC11168 Nal+ recipient cell, with the hcp gene observed in the transconjugants, thus demonstrating a role in the transfer of T6SS-containing megaplasmids to transconjugants (Marasini et al., 2020).
CJ488_0932 was predicted to contain the nucleic acid binding domain WGR (smart00773), belonging to the WGR superfamily (cl01581), and shares homology to the protein A0W69_09470 (81% aa identity) from pCJDM202. Within our local C. jejuni database, 63 out of the 512 (12.30%) genomes were found to possess the protein CJ488_0932, of which 51 were T6SS-positive (80.95%) and 12 were T6SS-negative (19.05%) (Supplementary Table 11). WGR domains have been identified in poly(ADP-ribose) polymerases of eukaryotes, as well as in a molybdate metabolism regulator in E. coli and a number of predicted proteins (Citarelli et al., 2010); however, the precise function of the domain remains unclear.
A number of T6SS lipase effectors have been previously described to target bacterial and eukaryotic membranes (Miyata et al., 2011; Russell et al., 2013; Jiang et al., 2014). In CJPI-1, two predicted lipase (Lipase class 3) domain-containing proteins (PF01764), CJ488_0957 and CJ488_0961, were identified upstream of the T6SS operon (Figure 3), with the former sharing homology to the proteins CJE1115 (64% aa identity) and A0W69_09355 (62% aa identity) from RM1221 and pCJDM202, respectively. Within our local C. jejuni database, 108 out of the 512 (21.09%) genomes were found to possess the protein CJ488_0957 (average per genome = 1.06), of which 98 were T6SS-positive (90.74%) and 10 were T6SS-negative (9.26%). Furthermore, 49 out of the 512 (9.57%) genomes were found to possess the protein CJ488_0961 (average per genome = 1.00), of which 45 were T6SS-positive (91.84%) and four were T6SS-negative (8.16%) (Supplementary Table 11). Previous PF01764 domain-containing proteins have been predicted as T4SS effectors in several bacteria targetting prokaryotic membranes, suggesting these two lipase-domain containing proteins belong to a larger family of hydrolysing effectors that can be delivered by several effector delivery systems (Sgro et al., 2019).
A phage lysozyme-like protein, CJ488_0962 was also inferred in CJPI-1 and predicted to contain a Phage_lysozyme (PF00959) and autolysin/endolysin family (cd00737) domain. Within our local C. jejuni database, 96 out of the 512 (18.75%) genomes were found to possess the protein CJ488_0962 (average per genome = 1.02), of which 91 were T6SS-positive (94.79%) and five were T6SS-negative (5.21%) (Supplementary Table 11). PF00959 is described as a glycoside hydrolase, associated with bacteriophage enzymes that degrade bacterial peptidoglycan in the cell wall (El-Gebali et al., 2019). Several characterised bacteriophage endolysins have been demonstrated to exhibit lytic antibacterial activity, containing both domains predicted in CJ488_0962 (Li et al., 2016; Ding et al., 2020). Interestingly, a prevalence study identifying endolysins in phage genomes identified PF00959 as the most frequently detected domain amongst analysed phage endolysins predicted to target Proteobacteria (Fernandez-Ruiz et al., 2018).
The protein VgrG is an essential component of the T6SS with roles including the promotion of the T6SS machinery assembly, the puncturing of target cells, and the delivery of effectors via their C-terminal domains (Pukatzki et al., 2007). Orphan vgrG genes can be located distally from their cognate T6SS structural operons; however, to date only one VgrG protein has been described in T6SS-containing C. jejuni (De Maayer et al., 2011; Bleumink-Pluym et al., 2013; Lopez et al., 2020). Here, we have identified two vgrG genes in the CJPI-1 of C. jejuni 488 (Figure 3), hereafter referred to as, vgrG1 (CJ488_0978) and vgrG2 (CJ488_0998) (Table 2 and Supplementary Table 2). Sequence alignment and identification of conserved and additional domains revealed a conserved N-terminal region possessing the VgrG domain (COG3501) and a region matching the superfamily VI_Rhs_Vgr (TIGR03361) (Figure 4). As observed in other bacteria, VgrG1 and VgrG2 differ in length, attributed to divergent C-terminal regions and associated domains (Hachani et al., 2011). The C-terminal region of VgrG1 (aa 564-883) shares an amino acid identity of 35% to the C-terminal region of VgrG2 (aa 564-838). Bioinformatic analysis revealed a Jag domain (COG1847, E-value: 0.008) in VgrG1, following the domain COG3501, which is potentially linked to RNA-binding. However, a structural homology search of the VgrG1 C-terminal region (aa 564-883) using Phyre2 matched to the C-terminal domain of the phage tail-lysozyme protein Gp5 (PDB: 1K28, Confidence: 100%). VgrG2 was found to contain a five superfamily/Gp5_C domain (PHA02596/PF06715) in its C-terminal region. The Gp5 C-terminal domain is commonly found in the bacteriophage T4 tail lysozyme protein Gp5 and VgrG proteins of bacteria, forming the membrane-puncturing β-helix structure of the spike proteins (Kanamaru et al., 2002; Pukatzki et al., 2007; El-Gebali et al., 2019). The C-termini Gp5 regions of some VgrG proteins may be also extended with catalytic domains (Hachani et al., 2011; Wettstadt et al., 2020). Furthermore, these extensions may also contribute to the recruitment of additional effectors (Flaugnatti et al., 2020).

Figure 4. Full amino acid sequence alignment of the two VgrG homologues present in T6SS-positive C. jejuni 488 strain using ClustalO. Conserved residues found in both sequences correspond to letters in uppercase below the aligned sequences. Protein locus tag and amino acid position are provided for each respective sequence. The N-terminal VgrG domain (COG3501) in both sequences is highlighted black. The Jag domain (COG1847) is highlighted light grey for VgrG1, while letters in bold represent the Phyre2 structural homology match to the C-terminal lysozyme domain of Gp5 (PDB: 1K28). The Gp5_C domain (PF06715) is highlighted dark grey for VgrG2.
A BLASTP-homology search was then conducted to assess the sequence identity of VgrG1 and VgrG2 against the NCBI Reference Protein database. The amino acid sequence of VgrG1 shares homology with the VgrG proteins of C. coli (WP_070241948.1–98.87% aa identity) and to Epsilonproteobacteria Helicobacter sp. MIT 11-5569 (WP_138109445.1–69.88% aa identity). Similarly, the amino acid sequence of VgrG2 shares homology with the VgrG proteins of C. coli (WP_072231509.1–99.76% aa identity) and to Helicobacter sp. MIT 11-5569 (WP_181881862.1–79.01% aa identity). The highly homologous matches suggest that these species too possess more than one VgrG protein with similar, if not identical domain architectures to VgrG1 and VgrG2, respectively. Further exploration is needed to assess whether both VgrG proteins exist in T6SS-containing C. coli and/or Helicobacter spp. genomes.
Genomic analyses of T6SS-positive C. jejuni strains showed that a number of putative vgrG genes were located downstream of T6SS gene operons. A BLASTN search for vgrG detected 41 homologous genes in the T6SS-positive C. jejuni genomes (with assembly level “complete” or higher) (Supplementary Table 12). Interestingly, 1 of the 24 T6SS-positive “complete” C. jejuni genomes, IF1100, does not encode any vgrG gene, whereas 13 out of 24 encoded two or more. Phylogenetic analysis classified the VgrG proteins into two distinct clades, grouped with either VgrG1 or VgrG2 from C. jejuni 488 strain (Figure 5) (five VgrG amino acid sequences from genomes CJ017CC464, CJ018CCUA, and ZS007 were removed due to fragmented ORFs). A domain search using NCBI-CDD revealed that 28 of the 36 identified VgrG protein sequences contain an additional domain in the divergent C-terminus. Of which, 16 possessed the Jag (COG1847) domain, 12 possessed the five superfamily (PHA02596) domain, and eight possessed no identifiable domains (Supplementary Table 13). Collectively, this data suggests that two distinct VgrG proteins exist within the T6SS-positive C. jejuni isolates, with some strains bearing multiple VgrG proteins. It is predicted that isolates may exploit these different VgrG proteins in an interchangeable puncturing role in the spike complex, translocating specific effectors via interaction with the distinct C-terminal regions (Hachani et al., 2014; Jana and Salomon, 2019).
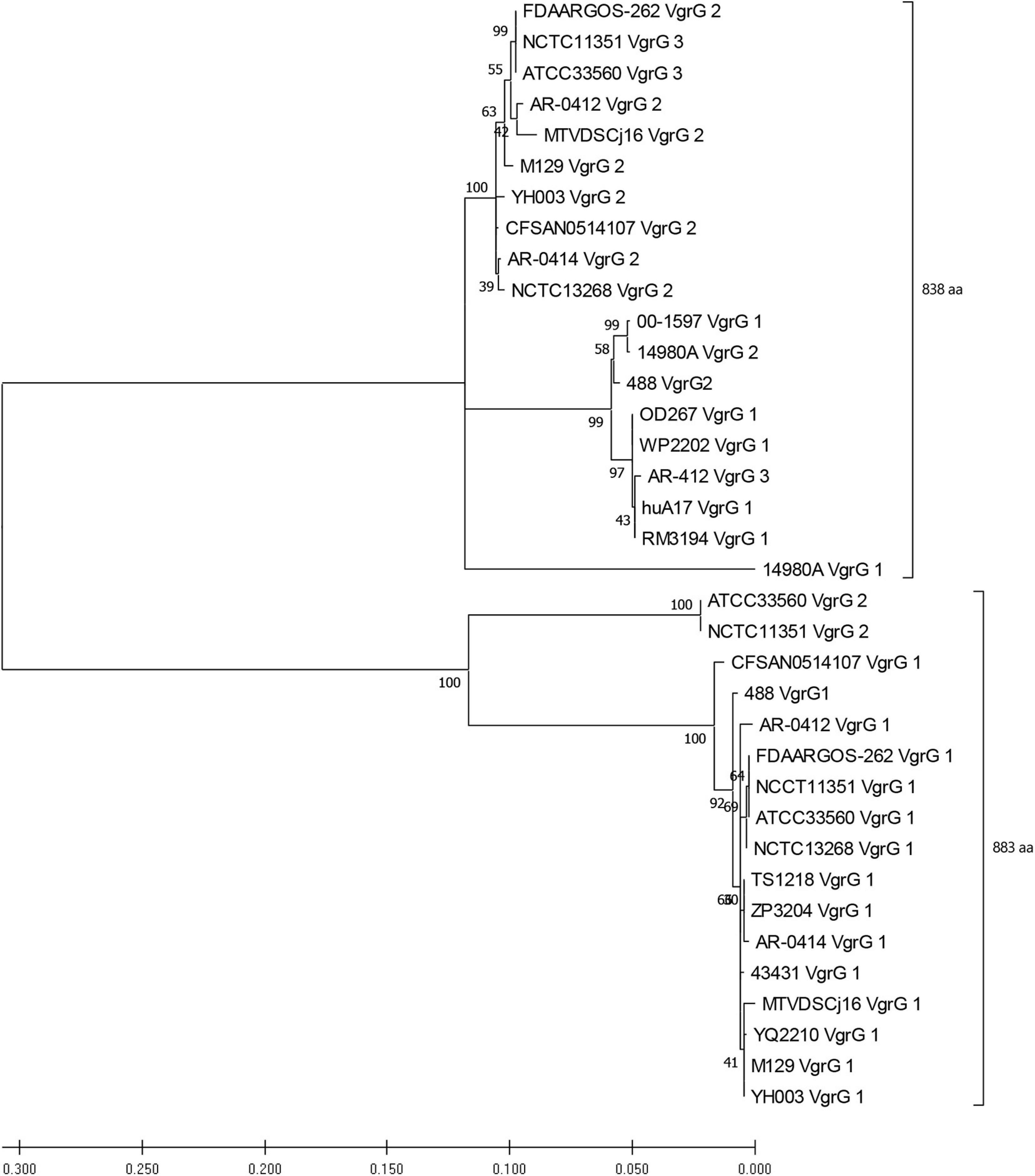
Figure 5. Phylogenetic tree derived from a Maximum likelihood analysis on a MUSCLE alignment of VgrG amino acid sequences from T6SS-positive C. jejuni of assembly level “complete” or higher (bootstrap n = 500, partial deletion). The value of each node is given. Top bracket = 838 amino acids (aa), bottom bracket = 883 aa.
The T6SS puncturing structure is composed by a VgrG trimer further sharpened with a capping PAAR domain-containing protein tip (Wettstadt et al., 2020). To assess the prevalence of these “effector markers,” the amino acid sequences of representative proteins belonging to all classes of PAAR subgroups found in the superfamily CL21497 were aligned against a local protein database of C. jejuni genomes (Supplementary Data 1). We identified only PAAR4 (representative protein AGP36489.1 of Sorangium cellulosum So0157-2) which matched positively to 500 genomes. However, the results were considered not significant as the alignment exclusively occurred at the C-terminal S41-peptidase domain (PF03572) of the representative protein and not the N-terminal PAAR-motif. We hypothesised that compared to other bacteria, significant sequence divergence in C. jejuni PAAR genes might exist and thus homology-based searches may not identify PAAR proteins. Further iterative based analysis (data not shown) was able to predict a novel PAAR-like domain (∼ 125 amino acids) present in two proteins within CJPI-1, CJ488_0948, and CJ488_0990, with the latter found in the genetically variable region downstream of the T6SS operon (Figure 3). Multiple sequence alignments uncovered the novel domain possesses a series of conserved cysteine and histidine residues similar to the PAAR-like domain DUF4280 (Rigard et al., 2016) and PAAR-containing proteins (Shneider et al., 2013) and is present in a wide range of bacterial families. A number of the PAAR-like domain-containing proteins possess N- or C-terminal extensions harbouring characterised toxin domains (Figure 6), conferring a toxin translocation function to PAAR in addition its sharpening role (Shneider et al., 2013). The predicted novel PAAR-like domain-containing proteins in strain 488 possessed no other identifiable domains, confining a sole sharpening role to these proteins in this strain (Shneider et al., 2013). An alignment using the amino acid sequences of representative proteins for MIX clans I–V was also conducted; however returned no positive matches (Supplementary Table 7; Salomon et al., 2014).
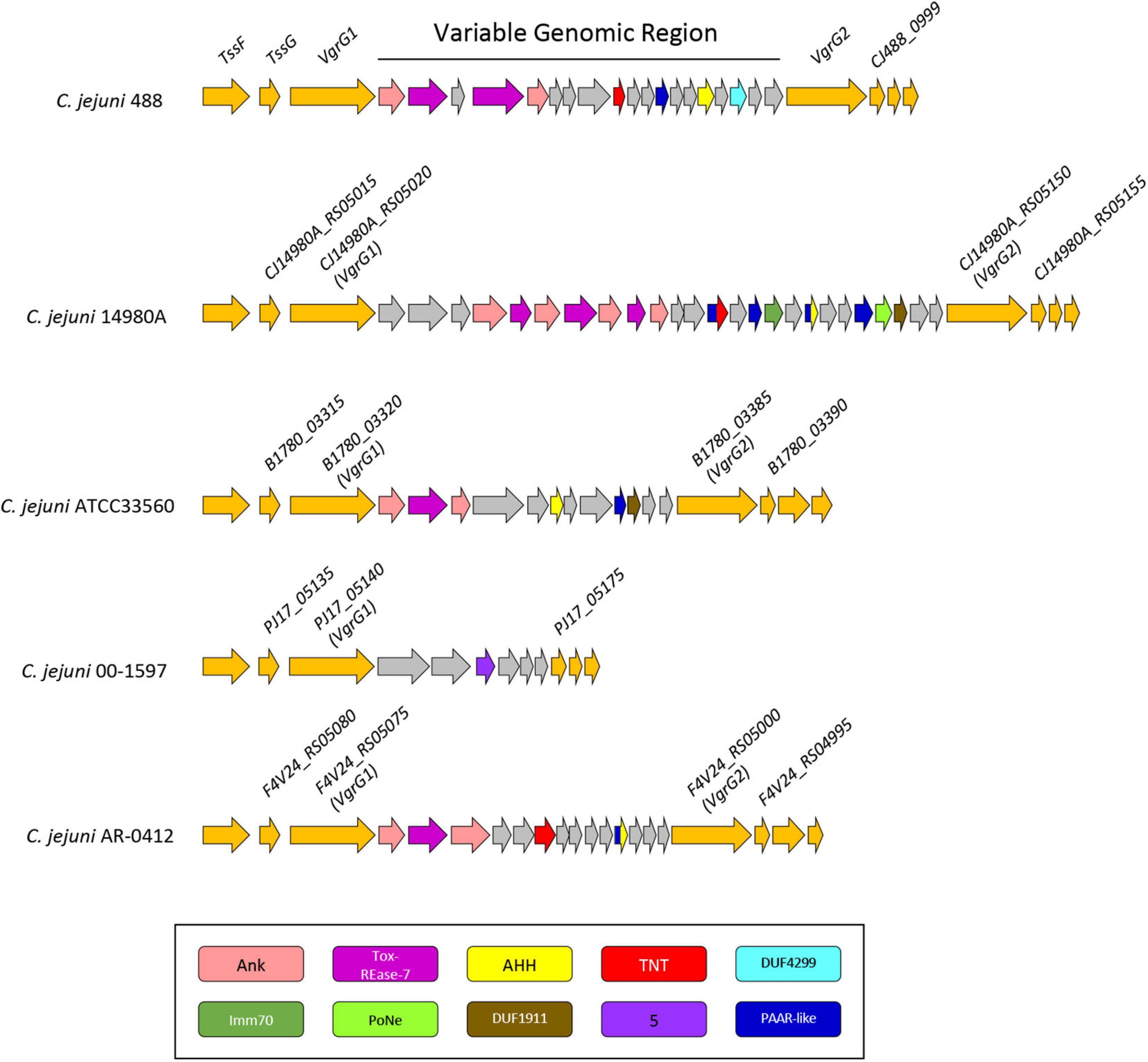
Figure 6. Genomic architecture of the genetically variable region downstream of the T6SS operon in five T6SS-positive C. jejuni strains; 488, 14980A, ATCC33560, 00-1597, and AR-0412. Genes found within the variable region possessing a domain by NCBI CDD (Marchler-Bauer et al., 2017) are coloured according to the domains identified: Ankyrin domains are pink, Tox-REase-7 are purple, AHH are yellow, TNT are red, DUF4299 are sky blue, Imm70 are dark green, PoNe are light green, DUF1911 are brown, five superfamily domain are lavender, and PAAR-like are dark blue. Variable region genes with no predicted function or identifiable domains are shaded grey and genes neighbouring the variable region are orange. Gene sizes are not to scale. The corresponding locus tags for selected genes is labelled above accordingly.
Genomic regions neighbouring vgrG genes often encode a number of hypothetical proteins, with many determined as T6SS effector and immunity proteins (De Maayer et al., 2011; Lopez et al., 2020). Analyses of T6SS-positive C. jejuni completely assembled genomes revealed a genetically variable region immediately downstream of the T6SS operon that did not share sequence identity to any ORFs found in the CJIE3 of strain RM1221. This region ranged from six genes in the 00-1597 strain, to 25 genes in the 14980A strain, located downstream of the main cluster gene vgrG and was commonly located upstream of the second orphan vgrG gene (Figure 6). Within this genetically variable region in 488, a number of ORFs between vgrG1 and vgrG2 were analysed using predictive programes (Figure 3). This resulted in the predicted identification of four putative effectors (CJ488_0980, CJ488_0982, CJ488_0988, and CJ488_0994), two ankyrin repeat domain-containing proteins (CJ488_0979 and CJ488_0983), and a DUF4299 family protein (CJ488_0996).
Among the putative effectors, proteins CJ488_0980 and CJ488_0982 contain predicted domains belonging to the restriction endonuclease family Tox-REase-7 (PF15649), whilst CJ488_0994 possesses a predicted domain belonging to the Tox-AHH HNH/ENDO VII superfamily nuclease (PF14412). Both domains belong to large toxin superfamilies with predicted functions as DNases/nucleases and are secreted by a wide range of polymorphic effector delivery systems, including the T6SS (Zhang et al., 2011, 2012). CJ488_0994 shares weak homology to the protein A0W69_08930 (41% aa identity) from pCJDM202, also predicted to possess a Tox-AHH domain. Interestingly, none of the other inferred putative effectors were found on pCJDM202 or CJIE3, suggesting separate genetic transfer events have mediated the acquisition of these putative T6SS-associated effectors.
Both Tox-REase-7 domain-containing proteins, CJ488_0980 and CJ488_0982, contain the conserved catalytic residues IxD, ExK, and Q, and metal chelating site signature D-[EQ]xK, characteristic of REase-fold toxins, specifically Tox-REase-7 (Figure 7A). The AHH domain-containing protein CJ488_0994 also possesses the conserved catalytic residues HH, N, H, H, and Y, specific to the Tox-AHH fold/motif (Figure 7B; Zhang et al., 2012). Previous studies have successfully identified AHH domain-containing proteins as T6SS-associated effectors in both human and plant pathogenic bacterium, notably VP1415 from V. parahaemolyticus, therefore strengthening the prediction of CJ488_0994 as a T6SS effector (Figure 7B; Salomon et al., 2014; Santos et al., 2019). Within our local C. jejuni database, 88 out of the 512 (17.18%) genomes were identified to possess the protein CJ488_0980 (average per genome = 1.05), of which 87 were T6SS-positive (98.86%) and 1 was T6SS-negative (1.14%). The T6SS-negative genome was found to contain the CJIE3. Further, 100 out of 512 (19.53%) genomes were found to possess the protein CJ488_0994 (average per genome = 1.24), of which 95 were T6SS-positive (95%) and 5 were T6SS-negative (5%). Of the T6SS-negative genomes, 4 out of 5 were found to contain the CJIE3. Within the T6SS-positive population, 87 out of the 135 (64.44%) T6SS-positive genomes possessed the protein CJ488_0980 and 95 out of the 135 (70.37%) T6SS-positive genomes possessed the protein CJ488_0994 (Supplementary Table 11).

Figure 7. Partial amino acid sequence alignment of Tox-REase-7 and AHH domain-containing representative amino acids against putative effectors downstream of the T6SS from C. jejuni 488 strain. (A) Alignment of CJ488_0980 and CJ488_0982 against Tox-REase-7 representative amino acids. (B) Alignment of CJ488_0994 against AHH representative amino acids. Catalytic residues for each domain are highlighted in yellow, and the hash (#) indicates the position of the residue in the C. jejuni 488 proteins. Red letters indicate amino acids with greater than 90% homology across all sequences for the alignment. Protein locus tag, organism name, and amino acid position are to the left and either side of the respective sequences.
The final putative effector, CJ488_0988, was predicted to contain the C-terminal domain tuberculosis necrotising toxin (TNT) (PF14021) from the Mycobacterium tuberculosis protein CpnT (Sun et al., 2015). Within our local C. jejuni database, 83 out of the 512 (16.21%) genomes were found to possess the protein CJ488_0988 (average per genome = 1.12), of which 80 were T6SS-positive (96.39%) and three were T6SS-negative (3.61%). All of the T6SS-negative genomes were found to contain the CJIE3. Within the T6SS-positive population, 80 out of the 135 (59.26%) T6SS-positive genomes possessed the protein CJ488_0994 (Supplementary Table 11). TNT, characterised as a NAD+- and NAD(P)+-glycohydrolase, is transported to the cell surface of M. tuberculosis by CpnT and released by proteolytic cleavage, where inside the eukaryotic target cell it depletes cellular NAD+ inducing host-cell death (Danilchanka et al., 2014; Sun et al., 2015; Tak et al., 2019). The TNT domain of CJ488_0988 possesses the conserved residues Arg5, Arg29, and Gln101 (Arg757, Arg780, and Gln822 of TNT in M. tuberculosis), identified as the putative NAD+ binding pocket essential for NAD+ hydrolysis (Sun et al., 2015; Tak et al., 2019). The third tyrosine (Y) residue, Tyr765 in TNT of CpnT, was found to be replaced by a phenylalanine (F) residue, Phe13, in the TNT domain of CJ488_0988. Mutations of the Tyr765 residue were demonstrated to significantly reduce the catalytic activity of TNT yet did not eradicate its cytotoxic activity (Sun et al., 2015; Tak et al., 2019). Structural homology of CJ488_0988 using the Phyre2 server confidently identified the domain TNT (PDB: 4QLP/c4qlpB, Confidence: 99.6%) as the most suitable template for modelling with a sequence coverage of 98%, indicating a high sequence and structural similarity (Kelley et al., 2015). Several interbacterial Type VI effectors have also been characterised to exhibit NAD(P)+-glycohydrolase activity, inducing bacteriostasis in target cells (Whitney et al., 2015; Tang et al., 2018).
In bacteria, ankyrin repeat-containing proteins have been characterised to act as immunity proteins (ImmAnk) against decrosslinking enzymes and a wide range of T6SS-associated toxin domains, including Tox-AHH (Zhang et al., 2012; Lambert et al., 2015). Both predicted ankyrin repeat-containing proteins, CJ488_0979 and CJ488_0983, were found adjacent to the putative Tox-REase-7 effectors, CJ488_0980 and CJ488_0982, and are predicted to encode the cognate immunity proteins to the respective effectors, presenting two identical effector-immunity pairs. Immunity proteins for the remaining predicted effectors could not be determined using our in silico methods, suggesting they may play an anti-eukaryotic role and are not involved in interbacterial competition.
CJ488_0996 was predicted to contain the domain DUF4299 (PF14132) and shares homology to the protein A0W69_08920 (100%) from pCJDM2020. However, no further functional data was available. Within our local C. jejuni database, 127 out of the 512 (24.80%) genomes were found to possess the protein CJ488_0996 (average per genome = 1.06), of which 122 were T6SS-positive (96.06%) and five were T6SS-negative (3.94%) (Supplementary Table 11). The significantly high proportion of T6SS-positive genomes identified to possess CJ488_0996 suggests there is unknown link ensuring a strong conservation of both the T6SS and this DUF4299 domain-containing protein together. We speculate that the DUF4299 domain-containing protein may be playing an adaptor/chaperone protein role for the T6SS, as no identifiable toxin or effector domains could be determined (Liang et al., 2015; Bondage et al., 2016).
The roles of T6SSs have been associated with interbacterial competition, host colonisation and virulence, as well as environmental survival. We have conducted in this study a comprehensive bioinformatic analysis to understand the genotypic T6SS organisation and its functional roles in C. jejuni. Using more than 500 publicly available genomes, we have identified co-occurrence of the T6SS and the integrative element CJIE3, confirming their association. Interestingly, genetic recombination with a T6SS-harbouring pCJDM202-like plasmid gives the potential for chromosomal integration. The analysis of our newly re-sequenced and assembled T6SS-positive 488 strain shows poor homology between the “T6SS-containing genomic island” and CJIE3, thus endorsing the reclassification of the former as a PAI termed CJPI-1. To note, Clark et al. (2018) make a similar observation when comparing the genomes and proteomes of four C. jejuni strains; however, here we present a comprehensive bioinformatics overview of the dynamic C. jejuni genome, its respective T6SS, and prediction of associated effectors (Clark et al., 2018). Two canonical VgrG proteins were identified within T6SS-positive C. jejuni genomes, as well as a wide range of predicted T6SS effectors and toxins, with some found in a genetically variable region downstream of the T6SS. Furthermore, we uncovered a putative DinJ-YafQ Type II TA module with predicted links to the stability of MGEs within C. jejuni. However, we cannot exclude from our analyses the possibility of these toxins to be recognised by the T6SS and exploited as effectors (Yadav et al., 2021). Collectively, these observations emphasise the diversity of genetic elements within C. jejuni strains, highlighting their contributions to bacterial survival in a wide range of hosts (i.e., chickens and humans) and in mediating competition in polymicrobial environments via multiple virulence factors.
Our data predicts the first T6SS-associated effectors in C. jejuni and identifies their putative functions as nucleases and a NAD+-glycohydrolase, based on their close proximity, high prevalence, and genomic context to the T6SS. Furthermore, the presence of putative anti-eukaryotic and anti-prokaryotic effectors suggests that C. jejuni encodes a multifunctional T6SS, as observed in other bacteria, that may have evolved during its evolutionary adaptation to host gastrointestinal tracts amongst polymicrobial communities (Miyata et al., 2013). Encoding a diverse repertoire of effectors, in close proximity to the T6SS operon, may allow for C. jejuni to secrete several effectors into prey cells and the surrounding milieu, overcoming bacterial competition and host defences to support its fitness.
This study also highlights that the acquisition of the T6SS and its related effectors into CJIE3 may have not occurred as a single event but rather upon multiple and independent genetic uptakes. Indeed, a small number of CJIE3-positive genomes were identified to possess some of the putative effectors in absence of a T6SS operon. This raises questions whether CJIE3-containing genomes once possessed a T6SS but was consequently lost through unknown events whilst retaining the putative effectors. Conversely, the effectors may have been associated to a number of pre-existing genes, some possibly as part of TA modules, subsequently repurposed as effectors upon successful acquisition of the T6SS into the genome via a plasmid, as previously mentioned. Interestingly, several T6SS-positive genomes were also identified to possess none of the putative effectors characterised in C. jejuni 488, suggesting they may instead harbour effector subsets that are yet to be discovered.
The original raw data used in this study are publicly available. This data can be found here: PRJEB41135. Publicly available datasets were analysed in this study. This data can be found here: https://www.ncbi.nlm. nih.gov.
OG and LR conceived the study. LR conducted the bioinformatic analysis and analysed the data. OG managed the study. LR, AH, and OG drafted the manuscript with contributions from JL, ZO, DX, AHMVV, and NC. All authors contributed to data interpretation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Geunhye Hong and Cadi Davies for helpful discussions. We also thank Cadi Davies for her version of the 488 Fastq files. Data included in this study previously appeared in the online thesis of Robinson (2020).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.694824/full#supplementary-material
Supplementary Table 1 | List of characterised T6SS effectors screened against the local C. jejuni protein database.
Supplementary Table 2 | Domain and motif hits of predicted proteins in CJPI-1 using NCBI CDD-BLAST, Pfam, SMART, HmmScan, PROSITE, CDART, SUPERFAMILY, MOTIF, and InterPro.
Supplementary Table 3 | Results of subcellular localisation, signal prediction and transmembrane helices prediction of CJPI-1 predicted proteins with inferred functions using Psortb, CELLO, SignalP, TMPred, and TMHMM.
Supplementary Table 4 | Table of C. jejuni genomes “scaffold” level or higher under study with T6SS and CJIE3 analysis.
Supplementary Table 5 | BLASTP-homology search for T6SS proteins in the local Campylobacter protein database.
Supplementary Table 6 | Core T6SS proteins identified in the C. jejuni genomes under study.
Supplementary Table 7 | List of MIX proteins screened against the local C. jejuni protein database.
Supplementary Table 8 | BLASTN-homology search and analysis for cje1094 in the local Campylobacter nucleotide database.
Supplementary Table 9 | BLASTN-homology search and analysis for cje1105 in the local Campylobacter nucleotide database.
Supplementary Table 10 | BLASTN-homology search and analysis for cje1153 in the local Campylobacter nucleotide database.
Supplementary Table 11 | Analysis of the BLASTP-homology searches for CJPI-1 functionally predicted proteins in the local C. jejuni protein database.
Supplementary Table 12 | BLASTN-homology search and analysis for vgrG genes in a local database of T6SS-positive C. jejuni genomes of assembly level “complete” or higher.
Supplementary Table 13 | NCBI-CDD webtool results for VgrG proteins from T6SS-positive C. jejuni genomes of assembly level “complete” or higher.
Supplementary Data 1 | PAAR amino acid sequences screened against the local C. jejuni protein database.
Supplementary Data 2 | C. jejuni 488 strain assembled GenBank (.gbk) file.
Alcoforado Diniz, J., Liu, Y. C., and Coulthurst, S. J. (2015). Molecular weaponry: diverse effectors delivered by the Type VI secretion system. Cell Microbiol. 17, 1742–1751. doi: 10.1111/cmi.12532
Alteri, C. J., and Mobley, H. L. T. (2016). The versatile Type VI secretion system. Microbiol. Spectr. 4:10.1128/microbiolsec.VMBF-0026-2015. doi: 10.1128/microbiolspec.VMBF-0026-2015
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Amour, C., Gratz, J., Mduma, E., Svensen, E., Rogawski, E. T., Mcgrath, M., et al. (2016). Epidemiology and Impact of campylobacter infection in children in 8 low-resource settings: results from the MAL-ED study. Clin. Infect. Dis. 63, 1171–1179.
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed October 5, 2020).
Aravind, L., Anantharaman, V., Balaji, S., Babu, M. M., and Iyer, L. M. (2005). The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol. Rev. 29, 231–262. doi: 10.1016/j.femsre.2004.12.008
Armenteros, J. J. A., Tsirigos, K. D., Sonderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/s41587-019-0036-z
Assefa, S., Keane, T. M., Otto, T. D., Newbold, C., and Berriman, M. (2009). ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25, 1968–1969. doi: 10.1093/bioinformatics/btp347
Barker, J. R., Chong, A., Wehrly, T. D., Yu, J. J., Rodriguez, S. A., Liu, J., et al. (2009). The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol. Microbiol. 74, 1459–1470. doi: 10.1111/j.1365-2958.2009.06947.x
Barret, M., Egan, F., and O’gara, F. (2013). Distribution and diversity of bacterial secretion systems across metagenomic datasets. Environ. Microbiol. Rep. 5, 117–126. doi: 10.1111/j.1758-2229.2012.00394.x
Bartonickova, L., Sterzenbach, T., Nell, S., Kops, F., Schulze, J., Venzke, A., et al. (2013). Hcp and VgrG 1 are secreted components of the H elicobacter hepaticus type VI secretion system and VgrG 1 increases the bacterial colitogenic potential. Cell. Microbiol. 15, 992–1011. doi: 10.1111/cmi.12094
Batchelor, R. A., Pearson, B. M., Friis, L. M., Guerry, P., and Wells, J. M. (2004). Nucleotide sequences and comparison of two large conjugative plasmids from different Campylobacter species. Microbiology 150, 3507–3517. doi: 10.1099/mic.0.27112-0
Bingle, L. E., Bailey, C. M., and Pallen, M. J. (2008). Type VI secretion: a beginner’s guide. Curr. Opin. Microbiol. 11, 3–8.
Bleumink-Pluym, N. M., Van Alphen, L. B., Bouwman, L. I., Wosten, M. M., and Van Putten, J. P. (2013). Identification of a functional type VI secretion system in Campylobacter jejuni conferring capsule polysaccharide sensitive cytotoxicity. PLoS Pathog. 9:e1003393. doi: 10.1371/journal.ppat.1003393
Blondel, C. J., Jimenez, J. C., Contreras, I., and Santiviago, C. A. (2009). Comparative genomic analysis uncovers 3 novel loci encoding type six secretion systems differentially distributed in Salmonella serotypes. BMC Genom. 10:354. doi: 10.1186/1471-2164-10-354
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bondage, D. D., Lin, J. S., Ma, L. S., Kuo, C. H., and Lai, E. M. (2016). VgrG C terminus confers the type VI effector transport specificity and is required for binding with PAAR and adaptor-effector complex. Proc. Natl. Acad. Sci. U.S.A. 113, E3931–E3940.
Burnham, P. M., and Hendrixson, D. R. (2018). Campylobacter jejuni: collective components promoting a successful enteric lifestyle. Nat. Rev. Microbiol. 16, 551–565. doi: 10.1038/s41579-018-0037-9
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and Mcquillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
Carver, T. J., Rutherford, K. M., Berriman, M., Rajandream, M. A., Barrell, B. G., and Parkhill, J. (2005). ACT: the artemis comparison tool. Bioinformatics 21, 3422–3423. doi: 10.1093/bioinformatics/bti553
Chandran Darbari, V., and Waksman, G. (2015). Structural biology of bacterial Type IV secretion systems. Annu. Rev. Biochem. 84, 603–629. doi: 10.1146/annurev-biochem-062911-102821
Chen, C., Yang, X., and Shen, X. (2019). Confirmed and potential roles of bacterial T6SSs in the intestinal ecosystem. Front. Microbiol. 10:1484. doi: 10.3389/fmicb.2019.01484
Cherrak, Y., Flaugnatti, N., Durand, E., Journet, L., and Cascales, E. (2019). Structure and activity of the Type VI secretion system. Microbiol. Spectr. 7, 329–342.
Cianfanelli, F. R., Monlezun, L., and Coulthurst, S. J. (2016). Aim, load, fire: the Type VI secretion system, a bacterial Nanoweapon. Trends Microbiol. 24, 51–62. doi: 10.1016/j.tim.2015.10.005
Citarelli, M., Teotia, S., and Lamb, R. S. (2010). Evolutionary history of the poly(ADP-ribose) polymerase gene family in eukaryotes. BMC Evol. Biol. 10:308. doi: 10.1186/1471-2148-10-308
Clark, C. G., Chen, C. Y., Berry, C., Walker, M., Mccorrister, S. J., Chong, P. M., et al. (2018). Comparison of genomes and proteomes of four whole genome-sequenced Campylobacter jejuni from different phylogenetic backgrounds. PLoS One 13:e0190836. doi: 10.1371/journal.pone.0190836
Corcionivoschi, N., Gundogdu, O., Moran, L., Kelly, C., Scates, P., Stef, L., et al. (2015). Virulence characteristics of hcp (+) Campylobacter jejuni and Campylobacter coli isolates from retail chicken. Gut Pathog. 7:20.
Costa, T. R., Felisberto-Rodrigues, C., Meir, A., Prevost, M. S., Redzej, A., Trokter, M., et al. (2015). Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat. Rev. Microbiol. 13, 343–359. doi: 10.1038/nrmicro3456
Coulthurst, S. (2019). The Type VI secretion system: a versatile bacterial weapon. Microbiology 165, 503–515. doi: 10.1099/mic.0.000789
Coyne, M. J., Roelofs, K. G., and Comstock, L. E. (2016). Type VI secretion systems of human gut Bacteroidales segregate into three genetic architectures, two of which are contained on mobile genetic elements. BMC Genom. 17:58. doi: 10.1186/s12864-016-2377-z
da Cruz Campos, A. C., Couto, N., Lucas Da Silva Andrade, N., Friedrich, A. W., De Paula Rosa, A. C., Vieira Damasco, P., et al. (2020). Virulence and resistance properties of E. coli isolated from urine samples of hospitalized patients in Rio de Janeiro, Brazil - The role of mobile genetic elements. Int. J. Med. Microbiol. 310:151453. doi: 10.1016/j.ijmm.2020.151453
Danilchanka, O., Sun, J., Pavlenok, M., Maueroder, C., Speer, A., Siroy, A., et al. (2014). An outer membrane channel protein of Mycobacterium tuberculosis with exotoxin activity. Proc. Natl. Acad. Sci. U.S.A. 111, 6750–6755. doi: 10.1073/pnas.1400136111
de Castro, E., Sigrist, C. J., Gattiker, A., Bulliard, V., Langendijk-Genevaux, P. S., Gasteiger, E., et al. (2006). ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365.
De Maayer, P., Venter, S. N., Kamber, T., Duffy, B., Coutinho, T. A., and Smits, T. H. (2011). Comparative genomics of the Type VI secretion systems of Pantoea and Erwinia species reveals the presence of putative effector islands that may be translocated by the VgrG and Hcp proteins. BMC Genom. 12:576. doi: 10.1186/1471-2164-12-576
Ding, Y., Zhang, Y., Huang, C., Wang, J., and Wang, X. (2020). An endolysin LysSE24 by bacteriophage LPSE1 confers specific bactericidal activity against multidrug-resistant Salmonella strains. Microorganisms 8:737. doi: 10.3390/microorganisms8050737
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424. doi: 10.1038/nrmicro884
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432.
Engel, P., Goepfert, A., Stanger, F. V., Harms, A., Schmidt, A., Schirmer, T., et al. (2012). Adenylylation control by intra- or intermolecular active-site obstruction in Fic proteins. Nature 482, 107–110. doi: 10.1038/nature10729
English, G., Trunk, K., Rao, V. A., Srikannathasan, V., Hunter, W. N., and Coulthurst, S. J. (2012). New secreted toxins and immunity proteins encoded within the Type VI secretion system gene cluster of Serratia marcescens. Mol. Microbiol. 86, 921–936. doi: 10.1111/mmi.12028
Fernandez-Ruiz, I., Coutinho, F. H., and Rodriguez-Valera, F. (2018). Thousands of novel endolysins discovered in uncultured phage genomes. Front. Microbiol. 9:1033. doi: 10.3389/fmicb.2018.01033
Flaugnatti, N., Rapisarda, C., Rey, M., Beauvois, S. G., Nguyen, V. A., Canaan, S., et al. (2020). Structural basis for loading and inhibition of a bacterial T6SS phospholipase effector by the VgrG spike. EMBO J. 39:e104129.
Fouts, D. E., Mongodin, E. F., Mandrell, R. E., Miller, W. G., Rasko, D. A., Ravel, J., et al. (2005). Major structural differences and novel potential virulence mechanisms from the genomes of multiple Campylobacter species. PLoS Biol. 3:e15. doi: 10.1371/journal.pbio.0030015
Fraikin, N., Goormaghtigh, F., and Van Melderen, L. (2020). Type II Toxin-antitoxin systems: evolution and revolutions. J. Bacteriol. 202:e00763-19.
Fridman, C. M., Keppel, K., Gerlic, M., Bosis, E., and Salomon, D. (2020). A comparative genomics methodology reveals a widespread family of membrane-disrupting T6SS effectors. Nat. Commun. 11:1085.
Frost, L. S., Ippen-Ihler, K., and Skurray, R. A. (1994). Analysis of the sequence and gene products of the transfer region of the F sex factor. Microbiol. Rev. 58, 162–210. doi: 10.1128/mr.58.2.162-210.1994
Geer, L. Y., Domrachev, M., Lipman, D. J., and Bryant, S. H. (2002). CDART: protein homology by domain architecture. Genome Res. 12, 1619–1623. doi: 10.1101/gr.278202
GenomeNet (2015). MOTIF: Searching Protein Sequence Motifs. Available online at: https://www.genome.jp/tools/motif/ (accessed July 1, 2020).
Ghatak, S., He, Y., Reed, S., and Irwin, P. (2020). Comparative genomic analysis of a multidrug-resistant Campylobacter jejuni strain YH002 isolated from Retail Beef Liver. Foodborne Pathog. Dis. 17, 576–584. doi: 10.1089/fpd.2019.2770
Gilchrist, C. L. M., and Chooi, Y. H. (2021). Clinker & clustermap.js: automatic generation of gene cluster comparison figures. Bioinformatics 2021:btab007.
Gladman, S., and Seemann, T. (2012). ”VelvetOptimiser”. 2.2.5 ed.). Available online at: https://github.com/tseemann/VelvetOptimiser (accessed October 5, 2020).
Goepfert, A., Harms, A., Schirmer, T., and Dehio, C. (2013). “Type II toxin-antitoxin loci: The fic family,” in Prokaryotic Toxin-Antitoxins, ed. G. Kenn (Heidelberg: Springer), 177–187. doi: 10.1007/978-3-642-33253-1_10
Gough, J., Karplus, K., Hughey, R., and Chothia, C. (2001). Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J. Mol. Biol. 313, 903–919. doi: 10.1006/jmbi.2001.5080
Grainge, I., and Jayaram, M. (1999). The integrase family of recombinase: organization and function of the active site. Mol. Microbiol. 33, 449–456. doi: 10.1046/j.1365-2958.1999.01493.x
Grohmann, E., Christie, P. J., Waksman, G., and Backert, S. (2018). Type IV secretion in Gram-negative and Gram-positive bacteria. Mol. Microbiol. 107, 455–471. doi: 10.1111/mmi.13896
Gundogdu, O., and Wren, B. W. (2020). Microbe profile: Campylobacter jejuni–survival instincts. Microbiology 166, 230–232. doi: 10.1099/mic.0.000906
Gunther, N. W., Reichenberger, E. R., and Bono, J. L. (2016). Complete genome sequence of UV-resistant Campylobacter jejuni RM3194, including an 81.08-kilobase plasmid. Genome Announc. 4:e0305-16.
Hachani, A., Allsopp, L. P., Oduko, Y., and Filloux, A. (2014). The VgrG proteins are “a la carte” delivery systems for bacterial type VI effectors. J. Biol. Chem. 289, 17872–17884. doi: 10.1074/jbc.m114.563429
Hachani, A., Lossi, N. S., Hamilton, A., Jones, C., Bleves, S., Albesa-Jove, D., et al. (2011). Type VI secretion system in Pseudomonas aeruginosa: secretion and multimerization of VgrG proteins. J. Biol. Chem. 286, 12317–12327.
Hachani, A., Wood, T. E., and Filloux, A. (2016). Type VI secretion and anti-host effectors. Curr. Opin. Microbiol. 29, 81–93. doi: 10.1016/j.mib.2015.11.006
Harrison, J. W., Dung, T. T., Siddiqui, F., Korbrisate, S., Bukhari, H., Tra, M. P., et al. (2014). Identification of possible virulence marker from Campylobacter jejuni isolates. Emerg. Infect. Dis. 20, 1026–1029.
Hill, C. W. (1999). Large genomic sequence repetitions in bacteria: lessons from rRNA operons and Rhs elements. Res. Microbiol. 150, 665–674. doi: 10.1016/s0923-2508(99)00125-4
Ho, B. T., Dong, T. G., and Mekalanos, J. J. (2014). A view to a kill: the bacterial type VI secretion system. Cell Host Microb. 15, 9–21. doi: 10.1016/j.chom.2013.11.008
Hofmann, K., and Stoffel, W. (1993). TMbase—a database of membrane spanning protein segments. Biol. Chem. Hoppe Seyler 374:166.
Huber, K. E., and Waldor, M. K. (2002). Filamentous phage integration requires the host recombinases XerC and XerD. Nature 417, 656–659. doi: 10.1038/nature00782
Humphrey, S., Chaloner, G., Kemmett, K., Davidson, N., Williams, N., Kipar, A., et al. (2014). Campylobacter jejuni is not merely a commensal in commercial broiler chickens and affects bird welfare. mBio 5:e01364-14.
Ijaz, U. Z., Sivaloganathan, L., Mckenna, A., Richmond, A., Kelly, C., Linton, M., et al. (2018). Comprehensive longitudinal microbiome analysis of the chicken cecum reveals a shift from competitive to environmental drivers and a window of opportunity for Campylobacter. Front. Microbiol. 9:2452. doi: 10.3389/fmicb.2018.02452
Jackson, A. P., Thomas, G. H., Parkhill, J., and Thomson, N. R. (2009). Evolutionary diversification of an ancient gene family (rhs) through C-terminal displacement. BMC Genom. 10:584. doi: 10.1186/1471-2164-10-584
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114.
Jana, B., Fridman, C. M., Bosis, E., and Salomon, D. (2019). A modular effector with a DNase domain and a marker for T6SS substrates. Nat. Commun. 10:3595.
Jana, B., and Salomon, D. (2019). Type VI secretion system: a modular toolkit for bacterial dominance. Future Microbiol. 14, 1451–1463. doi: 10.2217/fmb-2019-0194
Jarvis, K. G., Giron, J. A., Jerse, A. E., Mcdaniel, T. K., Donnenberg, M. S., and Kaper, J. B. (1995). Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc. Natl. Acad. Sci. U.S.A. 92, 7996–8000. doi: 10.1073/pnas.92.17.7996
Jiang, F., Wang, X., Wang, B., Chen, L., Zhao, Z., Waterfield, N. R., et al. (2016). The Pseudomonas aeruginosa Type VI secretion PGAP1-like effector induces host autophagy by activating endoplasmic reticulum stress. Cell Rep. 16, 1502–1509. doi: 10.1016/j.celrep.2016.07.012
Jiang, F., Waterfield, N. R., Yang, J., Yang, G., and Jin, Q. (2014). A Pseudomonas aeruginosa type VI secretion phospholipase D effector targets both prokaryotic and eukaryotic cells. Cell Host Microb. 15, 600–610. doi: 10.1016/j.chom.2014.04.010
Johnson, C. M., and Grossman, A. D. (2015). Integrative and Conjugative Elements (ICEs): what they do and how they work. Annu. Rev. Genet. 49, 577–601. doi: 10.1146/annurev-genet-112414-055018
Kanamaru, S., Leiman, P. G., Kostyuchenko, V. A., Chipman, P. R., Mesyanzhinov, V. V., Arisaka, F., et al. (2002). Structure of the cell-puncturing device of bacteriophage T4. Nature 415, 553–557. doi: 10.1038/415553a
Kapitein, N., Bonemann, G., Pietrosiuk, A., Seyffer, F., Hausser, I., Locker, J. K., et al. (2013). ClpV recycles VipA/VipB tubules and prevents non-productive tubule formation to ensure efficient type VI protein secretion. Mol. Microbiol. 87, 1013–1028. doi: 10.1111/mmi.12147
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Konkel, M. E., Corwin, M. D., Joens, L. A., and Cieplak, W. (1992). Factors that influence the interaction of Campylobacter jejuni with cultured mammalian cells. J. Med. Microbiol. 37, 30–37. doi: 10.1099/00222615-37-1-30
Kovanen, S., Rossi, M., Pohja-Mykra, M., Nieminen, T., Raunio-Saarnisto, M., Sauvala, M., et al. (2019). Population genetics and characterization of Campylobacter jejuni isolates from Western Jackdaws and game birds in Finland. Appl. Environ. Microbiol. 85:e02365-18.
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lambert, C., Cadby, I. T., Till, R., Bui, N. K., Lerner, T. R., Hughes, W. S., et al. (2015). Ankyrin-mediated self-protection during cell invasion by the bacterial predator Bdellovibrio bacteriovorus. Nat. Commun. 6:8884.
Leiman, P. G., Basler, M., Ramagopal, U. A., Bonanno, J. B., Sauder, J. M., Pukatzki, S., et al. (2009). Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc. Natl. Acad. Sci. U.S.A. 106, 4154–4159. doi: 10.1073/pnas.0813360106
Leplae, R., Geeraerts, D., Hallez, R., Guglielmini, J., Dreze, P., and Van Melderen, L. (2011). Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res. 39, 5513–5525. doi: 10.1093/nar/gkr131
Lertpiriyapong, K., Gamazon, E. R., Feng, Y., Park, D. S., Pang, J., Botka, G., et al. (2012). Campylobacter jejuni type VI secretion system: roles in adaptation to deoxycholic acid, host cell adherence, invasion, and in vivo colonization. PLoS One 7:e42842. doi: 10.1371/journal.pone.0042842
Letunic, I., and Bork, P. (2018). 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496.
Li, M., Li, M., Lin, H., Wang, J., Jin, Y., and Han, F. (2016). Characterization of the novel T4-like Salmonella enterica bacteriophage STP4-a and its endolysin. Arch. Virol. 161, 377–384. doi: 10.1007/s00705-015-2647-0
Liang, X., Moore, R., Wilton, M., Wong, M. J., Lam, L., and Dong, T. G. (2015). Identification of divergent type VI secretion effectors using a conserved chaperone domain. Proc. Natl. Acad. Sci. U.S.A. 112, 9106–9111. doi: 10.1073/pnas.1505317112
Liaw, J., Hong, G., Davies, C., Elmi, A., Sima, F., Stratakos, A., et al. (2019). The Campylobacter jejuni Type VI secretion system enhances the oxidative stress response and host colonization. Front. Microbiol. 10:2864. doi: 10.3389/fmicb.2019.02864
Lopez, J., Ly, P. M., and Feldman, M. F. (2020). The tip of the VgrG spike is essential to functional Type VI secretion system assembly in Acinetobacter baumannii. mBio 11:e02761-19.
Ma, L. S., Hachani, A., Lin, J. S., Filloux, A., and Lai, E. M. (2014). Agrobacterium tumefaciens deploys a superfamily of type VI secretion DNase effectors as weapons for interbacterial competition in planta. Cell Host Microb. 16, 94–104. doi: 10.1016/j.chom.2014.06.002
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641.
Marasini, D., and Fakhr, M. K. (2016). Complete genome sequences of Campylobacter jejuni strains OD267 and WP2202 isolated from retail chicken livers and gizzards reveal the presence of novel 116-Kilobase and 119-kilobase megaplasmids with Type VI secretion systems. Genome Announc. 4:e01060-16.
Marasini, D., and Fakhr, M. K. (2017). Complete genome sequences of plasmid-bearing multidrug-resistant Campylobacter jejuni and Campylobacter coli strains with type VI secretion systems, isolated from retail turkey and pork. Genome Announc. 5:e01360-17.
Marasini, D., Karki, A. B., Bryant, J. M., Sheaff, R. J., and Fakhr, M. K. (2020). Molecular characterization of megaplasmids encoding the type VI secretion system in Campylobacter jejuni isolated from chicken livers and gizzards. Sci. Rep. 10, 1–10.
Marchler-Bauer, A., Bo, Y., Han, L., He, J., Lanczycki, C. J., Lu, S., et al. (2017). CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203.
McKenna, A., Ijaz, U. Z., Kelly, C., Linton, M., Sloan, W. T., Green, B. D., et al. (2020). Impact of industrial production system parameters on chicken microbiomes: mechanisms to improve performance and reduce Campylobacter. Microbiome 8:128.
Mitchell, A. L., Attwood, T. K., Babbitt, P. C., Blum, M., Bork, P., Bridge, A., et al. (2019). InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360.
Miyata, S. T., Bachmann, V., and Pukatzki, S. (2013). Type VI secretion system regulation as a consequence of evolutionary pressure. J. Med. Microbiol. 62, 663–676. doi: 10.1099/jmm.0.053983-0
Miyata, S. T., Kitaoka, M., Brooks, T. M., Mcauley, S. B., and Pukatzki, S. (2011). Vibrio cholerae requires the type VI secretion system virulence factor VasX to kill Dictyostelium discoideum. Infect Immun. 79, 2941–2949. doi: 10.1128/iai.01266-10
Motiejunaite, R., Armalyte, J., Markuckas, A., and Suziedeliene, E. (2007). Escherichia coli dinJ-yafQ genes act as a toxin-antitoxin module. FEMS Microbiol. Lett. 268, 112–119. doi: 10.1111/j.1574-6968.2006.00563.x
Nano, F. E., and Schmerk, C. (2007). The Francisella pathogenicity island. Ann. N.Y. Acad. Sci. 1105, 122–137. doi: 10.1196/annals.1409.000
Nash, H. A. (1996). “Site-specific recombination: integration, excision, resolution, and inversion of defined DNA segments,” in Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd Edn, Vol. 2, ed. F. C. Neidhart (Washington, DC: American Society for Microbiology), 2363–2376.
NCBI (1982a). BioSample. Available online at: https://www.ncbi.nlm.nih.gov/biosample (accessed May 25, 2020).
NCBI (1982b). Genome List - Genome. Available online at: https://www.ncbi.nlm.nih.gov/genome/browse#!/overview/ (accessed May 25, 2020).
NCBI (1982c). Protein. Available online at: https://www.ncbi.nlm.nih.gov/protein/?term= (accessed May 25, 2020).
Noreen, Z., Jobichen, C., Abbasi, R., Seetharaman, J., Sivaraman, J., and Bokhari, H. (2018). Structural basis for the pathogenesis of Campylobacter jejuni Hcp1, a structural and effector protein of the Type VI secretion system. FEBS J. 285, 4060–4070. doi: 10.1111/febs.14650
Norton, J. P., and Mulvey, M. A. (2012). Toxin-antitoxin systems are important for niche-specific colonization and stress resistance of uropathogenic Escherichia coli. PLoS Pathog. 8:e1002954. doi: 10.1371/journal.ppat.1002954
Page, R., and Peti, W. (2016). Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 12, 208–214. doi: 10.1038/nchembio.2044
Palmer, T., Finney, A. J., Saha, C. K., Atkinson, G. C., and Sargent, F. (2021). A holin/peptidoglycan hydrolase-dependent protein secretion system. Mol. Microbiol. 115, 345–355. doi: 10.1111/mmi.14599
Parker, C. T., Quinones, B., Miller, W. G., Horn, S. T., and Mandrell, R. E. (2006). Comparative genomic analysis of Campylobacter jejuni strains reveals diversity due to genomic elements similar to those present in C. jejuni strain RM1221. J. Clin. Microbiol. 44, 4125–4135. doi: 10.1128/jcm.01231-06
Parkhill, J., Wren, B. W., Mungall, K., Ketley, J. M., Churcher, C., Basham, D., et al. (2000). The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403, 665–668. doi: 10.1038/35001088
Penner, J. L., Hennessy, J. N., and Congi, R. V. (1983). Serotyping ofCampylobacter jejuni andCampylobacter coli on the basis of thermostable antigens. Eur. J. Clin. Microbiol. 2, 378–383.
Poly, F., Threadgill, D., and Stintzi, A. (2005). Genomic diversity in Campylobacter jejuni: identification of C. jejuni 81-176-specific genes. J. Clin. Microbiol. 43, 2330–2338. doi: 10.1128/jcm.43.5.2330-2338.2005
Potter, S. C., Luciani, A., Eddy, S. R., Park, Y., Lopez, R., and Finn, R. D. (2018). HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204. doi: 10.1093/nar/gky448
Prysak, M. H., Mozdzierz, C. J., Cook, A. M., Zhu, L., Zhang, Y., Inouye, M., et al. (2009). Bacterial toxin YafQ is an endoribonuclease that associates with the ribosome and blocks translation elongation through sequence-specific and frame-dependent mRNA cleavage. Mol. Microbiol. 71, 1071–1087. doi: 10.1111/j.1365-2958.2008.06572.x
Pukatzki, S., Ma, A. T., Revel, A. T., Sturtevant, D., and Mekalanos, J. J. (2007). Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc. Natl. Acad. Sci. U.S.A. 104, 15508–15513. doi: 10.1073/pnas.0706532104
Pukatzki, S., Ma, A. T., Sturtevant, D., Krastins, B., Sarracino, D., Nelson, W. C., et al. (2006). Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc. Natl. Acad. Sci. U.S.A. 103, 1528–1533. doi: 10.1073/pnas.0510322103
Rigard, M., Broms, J. E., Mosnier, A., Hologne, M., Martin, A., Lindgren, L., et al. (2016). Francisella tularensis IglG belongs to a novel family of PAAR-Like T6SS proteins and harbors a unique n-terminal extension required for virulence. PLoS Pathog. 12:e1005821. doi: 10.1371/journal.ppat.1005821
Robinson, L. (2020). Bioinformatic Analysis of the Campylobacter jejuni Type VI Secretion System, Masters Thesis. London: LSHTM.
Ruangprasert, A., Maehigashi, T., Miles, S. J., Giridharan, N., Liu, J. X., and Dunham, C. M. (2014). Mechanisms of toxin inhibition and transcriptional repression by Escherichia coli DinJ-YafQ. J. Biol. Chem. 289, 20559–20569. doi: 10.1074/jbc.m114.573006
Russell, A. B., Hood, R. D., Bui, N. K., Leroux, M., Vollmer, W., and Mougous, J. D. (2011). Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475, 343–347.
Russell, A. B., Leroux, M., Hathazi, K., Agnello, D. M., Ishikawa, T., Wiggins, P. A., et al. (2013). Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature 496, 508–512. doi: 10.1038/nature12074
Salomon, D., Kinch, L. N., Trudgian, D. C., Guo, X., Klimko, J. A., Grishin, N. V., et al. (2014). Marker for type VI secretion system effectors. Proc. Natl. Acad. Sci. U.S.A. 111, 9271–9276.
Santoriello, F. J., Michel, L., Unterweger, D., and Pukatzki, S. (2020). Pandemic Vibrio cholerae shuts down site-specific recombination to retain an interbacterial defence mechanism. Nat. Commun. 11:6246.
Santos, M. N. M., Cho, S. T., Wu, C. F., Chang, C. J., Kuo, C. H., and Lai, E. M. (2019). Redundancy and specificity of Type VI secretion vgrG loci in antibacterial activity of Agrobacterium tumefaciens 1D1609 strain. Front. Microbiol. 10:3004. doi: 10.3389/fmicb.2019.03004
Schmid, M. C., Scheidegger, F., Dehio, M., Balmelle-Devaux, N., Schulein, R., Guye, P., et al. (2006). A translocated bacterial protein protects vascular endothelial cells from apoptosis. PLoS Pathog. 2:e115. doi: 10.1371/journal.ppat.0020115
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sgro, G. G., Oka, G. U., Souza, D. P., Cenens, W., Bayer-Santos, E., Matsuyama, B. Y., et al. (2019). Bacteria-Killing Type IV secretion systems. Front. Microbiol. 10:1078. doi: 10.3389/fmicb.2019.01078
Shen, Z., Patil, R. D., Sahin, O., Wu, Z., Pu, X. Y., Dai, L., et al. (2016). Identification and functional analysis of two toxin–antitoxin systems in Campylobacter jejuni. Mol. Microbiol. 101, 909–923. doi: 10.1111/mmi.13431
Shneider, M. M., Buth, S. A., Ho, B. T., Basler, M., Mekalanos, J. J., and Leiman, P. G. (2013). PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature 500, 350–353. doi: 10.1038/nature12453
Sonnhammer, E. L., Von Heijne, G., and Krogh, A. (1998). A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182.
Sprenger, H., Kienesberger, S., Pertschy, B., Poltl, L., Konrad, B., Bhutada, P., et al. (2017). Fic proteins of Campylobacter fetus subsp. venerealis form a network of functional toxin-antitoxin systems. Front. Microbiol. 8:1965. doi: 10.3389/fmicb.2017.01965
Sun, J., Siroy, A., Lokareddy, R. K., Speer, A., Doornbos, K. S., Cingolani, G., et al. (2015). The tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nat. Struct. Mol. Biol. 22, 672–678. doi: 10.1038/nsmb.3064
Tak, U., Vlach, J., Garza-Garcia, A., William, D., Danilchanka, O., De Carvalho, L. P. S., et al. (2019). The tuberculosis necrotizing toxin is an NAD(+) and NADP(+) glycohydrolase with distinct enzymatic properties. J. Biol. Chem. 294, 3024–3036. doi: 10.1074/jbc.ra118.005832
Tam, C. C., Rodrigues, L. C., Viviani, L., Dodds, J. P., Evans, M. R., Hunter, P. R., et al. (2012). Longitudinal study of infectious intestinal disease in the UK (IID2 study): incidence in the community and presenting to general practice. Gut 61, 69–77. doi: 10.1136/gut.2011.238386
Tang, J. Y., Bullen, N. P., Ahmad, S., and Whitney, J. C. (2018). Diverse NADase effector families mediate interbacterial antagonism via the type VI secretion system. J. Biol. Chem. 293, 1504–1514. doi: 10.1074/jbc.ra117.000178
Trunk, K., Coulthurst, S. J., and Quinn, J. (2019). A New Front in Microbial Warfare-Delivery of Antifungal Effectors by the Type VI Secretion System. J. Fungi 5:50. doi: 10.3390/jof5020050
Trunk, K., Peltier, J., Liu, Y. C., Dill, B. D., Walker, L., Gow, N. A. R., et al. (2018). The type VI secretion system deploys antifungal effectors against microbial competitors. Nat. Microbiol. 3, 920–931. doi: 10.1038/s41564-018-0191-x
Ugarte-Ruiz, M., Stabler, R. A., Dominguez, L., Porrero, M. C., Wren, B. W., Dorrell, N., et al. (2015). Prevalence of Type VI Secretion System in Spanish Campylobacter jejuni Isolates. Zoonoses Public Health 62, 497–500. doi: 10.1111/zph.12176
Veyron, S., Peyroche, G., and Cherfils, J. (2018). FIC proteins: from bacteria to humans and back again. Pathog. Dis. 76:fty012.
Wettstadt, S., Lai, E. M., and Filloux, A. (2020). Solving the Puzzle: connecting a Heterologous Agrobacterium tumefaciens T6SS effector to a Pseudomonas aeruginosa spike complex. Front. Cell Infect. Microbiol. 10:291. doi: 10.3389/fcimb.2020.00291
Whitney, J. C., Chou, S., Russell, A. B., Biboy, J., Gardiner, T. E., Ferrin, M. A., et al. (2013). Identification, structure, and function of a novel type VI secretion peptidoglycan glycoside hydrolase effector-immunity pair. J. Biol. Chem. 288, 26616–26624. doi: 10.1074/jbc.m113.488320
Whitney, J. C., Quentin, D., Sawai, S., Leroux, M., Harding, B. N., Ledvina, H. E., et al. (2015). An interbacterial NAD(P)(+) glycohydrolase toxin requires elongation factor Tu for delivery to target cells. Cell 163, 607–619. doi: 10.1016/j.cell.2015.09.027
Wigley, P. (2015). Blurred lines: pathogens, commensals, and the healthy gut. Front. Vet. Sci. 2:40. doi: 10.3389/fvets.2015.00040
Wood, T. E., Aksoy, E., and Hachani, A. (2020). From welfare to warfare: the arbitration of host-microbiota interplay by the Type VI secretion system. Front. Cell Infect. Microbiol. 10:587948. doi: 10.3389/fcimb.2020.587948
Worby, C. A., Mattoo, S., Kruger, R. P., Corbeil, L. B., Koller, A., Mendez, J. C., et al. (2009). The fic domain: regulation of cell signaling by adenylylation. Mol. Cell 34, 93–103. doi: 10.1016/j.molcel.2009.03.008
Wozniak, R. A., and Waldor, M. K. (2009). A toxin-antitoxin system promotes the maintenance of an integrative conjugative element. PLoS Genet. 5:e1000439. doi: 10.1371/journal.pgen.1000439
Yadav, S. K., Magotra, A., Ghosh, S., Krishnan, A., Pradhan, A., Kumar, R., et al. (2021). Immunity proteins of dual nuclease T6SS effectors function as transcriptional repressors. EMBO Rep. 22:e51857.
Yarbrough, M. L., Li, Y., Kinch, L. N., Grishin, N. V., Ball, H. L., and Orth, K. (2009). AMPylation of Rho GTPases by Vibrio VopS disrupts effector binding and downstream signaling. Science 323, 269–272. doi: 10.1126/science.1166382
Yu, C. S., Chen, Y. C., Lu, C. H., and Hwang, J. K. (2006). Prediction of protein subcellular localization. Proteins 64, 643–651.
Yu, N. Y., Wagner, J. R., Laird, M. R., Melli, G., Rey, S., Lo, R., et al. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615. doi: 10.1093/bioinformatics/btq249
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Zhang, D., De Souza, R. F., Anantharaman, V., Iyer, L. M., and Aravind, L. (2012). Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol. Direct. 7:18. doi: 10.1186/1745-6150-7-18
Zhang, D., Iyer, L. M., and Aravind, L. (2011). A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res. 39, 4532–4552. doi: 10.1093/nar/gkr036
Zhang, Y. (2008). I-TASSER server for protein 3D structure prediction. BMC Bioinform. 9:40. doi: 10.1186/1471-2105-9-40
Keywords: Campylobacter jejuni, Type VI Secretion System, T6SS effectors, T6SS immunity proteins, toxin-antitoxin, pathogenicity island
Citation: Robinson L, Liaw J, Omole Z, Xia D, van Vliet AHM, Corcionivoschi N, Hachani A and Gundogdu O (2021) Bioinformatic Analysis of the Campylobacter jejuni Type VI Secretion System and Effector Prediction. Front. Microbiol. 12:694824. doi: 10.3389/fmicb.2021.694824
Received: 13 April 2021; Accepted: 07 June 2021;
Published: 29 June 2021.
Edited by:
Eric Cascales, Aix-Marseille Université, FranceReviewed by:
Alessia Levante, University of Parma, ItalyCopyright © 2021 Robinson, Liaw, Omole, Xia, van Vliet, Corcionivoschi, Hachani and Gundogdu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ozan Gundogdu, b3phbi5ndW5kb2dkdUBsc2h0bS5hYy51aw==
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.