 Terumasa Ikeda
Terumasa Ikeda Yuan Yue1,2
Yuan Yue1,2 Hesham Nasser
Hesham Nasser
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Microbiol. , 15 June 2021
Sec. Virology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.686357
This article is part of the Research Topic Novel Insights into a Functional HIV Cure View all 11 articles
The introduction of combination antiretroviral therapy (cART) has managed to control the replication of human immunodeficiency virus type 1 (HIV-1) in infected patients. However, a complete HIV-1 cure, including a functional cure for or eradication of HIV-1, has yet to be achieved because of the persistence of latent HIV-1 reservoirs in adherent patients. The primary source of these viral reservoirs is integrated proviral DNA in CD4+ T cells and other non-T cells. Although a small fraction of this proviral DNA is replication-competent and contributes to viral rebound after the cessation of cART, >90% of latent viral reservoirs are replication-defective and some contain high rates of G-to-A mutations in proviral DNA. At least in part, these high rates of G-to-A mutations arise from the APOBEC3 (A3) family proteins of cytosine deaminases. A general model has shown that the HIV-1 virus infectivity factor (Vif) degrades A3 family proteins by proteasome-mediated pathways and inactivates their antiviral activities. However, Vif does not fully counteract the HIV-1 restriction activity of A3 family proteins in vivo, as indicated by observations of A3-mediated G-to-A hypermutation in the proviral DNA of HIV-1-infected patients. The frequency of A3-mediated hypermutation potentially contributes to slower HIV-1/AIDS disease progression and virus evolution including the emergence of cytotoxic T lymphocyte escape mutants. Therefore, combined with other strategies, the manipulation of A3-mediated mutagenesis may contribute to an HIV-1 functional cure aimed at cART-free remission. In this mini-review, we discuss the possibility of an HIV-1 functional cure arising from manipulation of A3 mutagenic activity.
Human immunodeficiency virus type 1 (HIV-1) was first discovered in 1983 as the agent that causes acquired immune deficiency syndrome (AIDS) (Barre-Sinoussi et al., 1983). HIV-1 disrupts the immune system by infecting immune cells, such as CD4+ T cells, macrophages, and dendritic cells, which ultimately leads to AIDS and related opportunistic infections. AIDS has become a manageable chronic disease due to the development of combination antiretroviral therapy (cART), which has enabled complete suppression of detectable viremia and controls disease progression in adherent patients. However, cART does not eliminate HIV-1 from these patients because latent reservoirs with the HIV-1 genome integrated into host DNA remain present (Chun et al., 1997; Finzi et al., 1997; Wong et al., 1997) and these latently infected cells clonally expand (Ikeda et al., 2007; Maldarelli et al., 2014; Wagner et al., 2014). When the latent reservoirs harbor replication-competent proviral DNA, they contribute to viral rebound when cART is interrupted (Chun et al., 1997; Finzi et al., 1997; Wong et al., 1997).
Efforts have been made to quantify the size of the latent reservoirs [reviewed by Wang et al. (2018); Lambrechts et al. (2020)]; studies have demonstrated that the majority of proviral DNA in the latent reservoirs is replication-defective and does not contribute to viral rebound when cART ceases. Ideally, complete eradication of HIV-1 from infected patients is the goal of a HIV-1 cure; however, because >90% of latent reservoirs harbor defective viruses, the initial goal should be a functional cure for HIV-1, which would comply with detectable proviral DNA and undetectable (or low-level) plasma viremia without cART. Of particular interest are elite controllers and long-term non-progressors, who represent <1% of all patients living with HIV-1 and are natural models for the functional cure [reviewed by Gonzalo-Gil et al. (2017); Promer and Karris (2018)]; however, little is known about the underlying mechanisms by which the immune systems of such patients control the virus.
APOBEC3 (A3) family proteins are cytosine deaminases that play important roles in mammalian innate immune responses. The human genome encodes seven A3 genes on chromosome 22, which include three single-domain deaminase genes (A3A, A3C, and A3H) and four double-domain deaminase genes (A3B, A3D, A3F, and A3G) [reviewed by Koito and Ikeda (2012); Desimmie et al. (2014); Harris and Dudley (2015); Figure 1A]. In CD4+ T cells, up to five A3 proteins (A3C-Ile188, A3D, A3F, A3G, and A3H) are involved in HIV-1 restriction (Hultquist et al., 2011; Refsland et al., 2012, 2014; Ooms et al., 2013; Wittkopp et al., 2016; Anderson et al., 2018). These A3s are packaged into nascent viral particles and catalyze the deamination of cytosine-to-uracil in reverse transcription cDNA intermediates (Figure 1B) [reviewed by Harris and Dudley (2015); Simon et al. (2015)]. The uracil lesion provides a template for adenine insertion in HIV-1 genomic strands while accumulation of many G-to-A mutations ultimately leads to virus inactivation (Figure 1B). HIV-1 viral infectivity factor (Vif) recruits an E3 ubiquitin ligase complex to promote the degradation of A3 proteins through a proteasome-mediated pathway and counteracts their antiviral activity [reviewed by Harris and Dudley (2015); Salamango and Harris (2020); Figure 1B]. This model is well established and widely accepted. Nevertheless, reports have shown that some fraction of latent reservoirs harbors high rates of G-to-A mutations in proviral DNA (Kieffer et al., 2005; Ho et al., 2013; Imamichi et al., 2014; Bruner et al., 2016, 2019). Importantly, A3-mediated G-to-A hypermutation in proviral DNA is significantly correlated with disease progression in HIV-1-infected patients including elite controllers and long-term non-progressors (Pace et al., 2006; Land et al., 2008; Kourteva et al., 2012; Eyzaguirre et al., 2013; Cuevas et al., 2015). Additionally, CD4+ T cells from HIV-1 controllers with higher A3G expression levels are less susceptible to in vitro HIV-1 infection than are CD4+ T cells from HIV-1 controllers with lower A3G expression levels (Biasin et al., 2007; De Pasquale et al., 2013). Furthermore, A3-mediated mutagenesis seems to influence cytotoxic T lymphocyte (CTL) responses [reviewed by Stavrou and Ross (2015); Figure 2]. Taken together, these observations indicate that the mutagenic activity of A3 family proteins is a factor that determines HIV-1/AIDS disease progression. Therefore, the manipulation of A3-mediated mutagenesis to lethal levels may be a potential target for an HIV-1 functional cure. Here, we discuss the possibility of introducing an HIV-1 functional cure mediated by the mutagenesis of A3 family proteins.
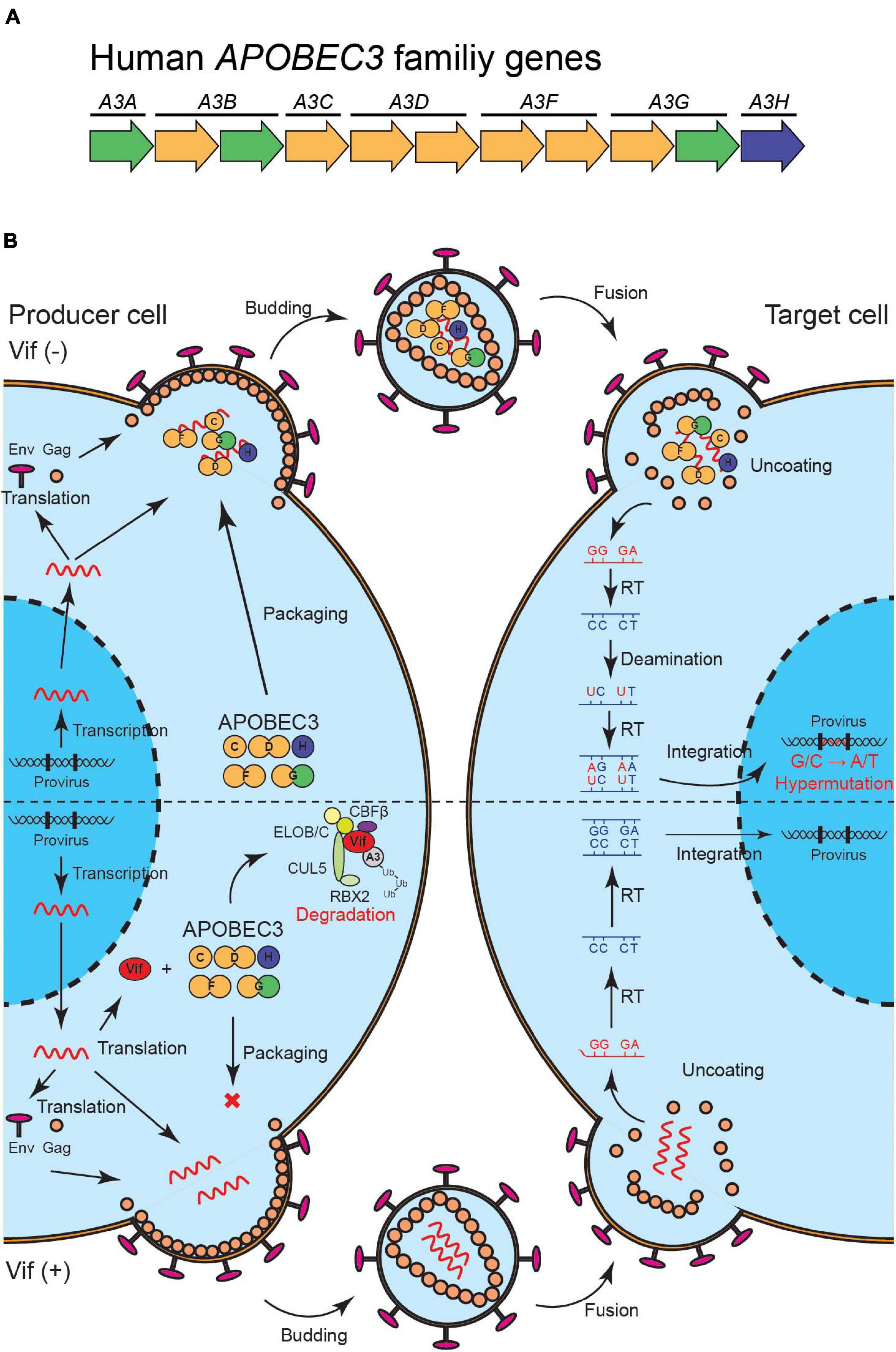
Figure 1. A model of HIV-1 restriction by A3 family proteins and counteraction by HIV-1 Vif. (A) Illustration of human A3 family genes. Human A3 family genes are composed of seven members with one or two zinc-coordinating domains (single of double-domain deaminases); these belong to three phylogenetically different groups, which are shown in green, yellow, and blue. (B) A schematic of HIV-1 restriction by five A3 family proteins and neutralization by HIV-1 Vif. A3C, A3D, A3F, A3G, and A3H are packaged into HIV-1 virions in producer cells and inactivate the virus through cytosine-to-uracil (C-to-U)/guanine-to-adenine (G-to-A) mutations (top). HIV-1 Vif neutralizes the restriction activities of these A3 proteins through proteasome-mediated degradation (bottom). Vif, virus infectivity factor; A3, APOBEC3.

Figure 2. Potential effects of A3-mediated mutagenesis on CTL responses. A3F- and A3G-mediated mutagenesis alters CTL responses through the accumulation of G-to-A mutations on the viral genome, which leads to the modification of epitope sequences and their flanking regions involved in antigen processing/presentation, HLA binding, and TCR recognition. The two examples show that A3-mediated hypermutation on epitope sequences potentially alters HLA binding of the epitopes and TCR recognition. Sublethal A3-mediated mutagenesis is involved in the emergence of CTL escape variants (top), whereas lethal A3-mediated hypermutation likely increases the number of HIV-derived epitopes and consequently enhances HIV-1-specific CTL responses (bottom). A3, APOBEC3; CTL, cytotoxic T lymphocyte; HLA, human leukocyte antigen; TCR, T cell receptor.
The A3H gene is likely a genetic factor that controls the disease progression of HIV-1 with the particular genotype of the vif gene. A3H is the most variable A3 gene in the human population (Figure 1A). Four single-nucleotide polymorphisms and one indel in the A3H gene determine stable and unstable haplotypes of A3H proteins (OhAinle et al., 2008; Wang et al., 2011; Refsland et al., 2014; Ebrahimi et al., 2018). Stable A3H haplotypes are expressed stably and have the ability to inhibit HIV-1, whereas unstable A3H haplotypes are barely detectable or undetectable at the protein level and lack antiviral activity. Interestingly, not all HIV-1 Vif variants from laboratory and natural isolates can degrade stable A3H haplotypes. Several studies have identified amino acid residues at positions 39, 48, and 60–63 that influence the capability of Vif proteins to degrade stable A3H haplotypes but do not affect counteraction activity against A3D, A3F, and A3G (Binka et al., 2012; Ooms et al., 2013; Refsland et al., 2014). Hereafter, we denote Vif proteins that can degrade stable A3H haplotypes and other HIV-1-restrictive A3 proteins (i.e., A3D, A3F, and A3G) as “hyper-functional,” while we term those that fail to degrade stable A3H haplotypes but not other HIV-1 restrictive A3 proteins as “hypo-functional.”
Intriguingly, studies have shown that A3H haplotypes are correlated with the global distribution of HIV-1 Vif alleles (Refsland et al., 2014; Nakano et al., 2017b). For example, around 60% of people have stable A3H haplotypes in Sub-Saharan Africa where hyper-functional Vifs are dominant (Refsland et al., 2014; Nakano et al., 2017b). In contrast, hypo-functional Vifs are prevalent in Asia because most people have unstable A3H haplotypes (Refsland et al., 2014; Nakano et al., 2017b). This biogeographical relationship strongly suggests that HIV-1 with hyper-functional, too Vif has evolved to counteract stable A3H haplotypes but that stable A3H haplotypes remain a potential transmission barrier against HIV-1 with hypo-functional Vif outside Africa. This possibility is supported by previous studies showing that HIV-1-infected patients (including long-term non-progressors) with at least one allele of stable A3H haplotypes have slower disease progression to AIDS (Ooms et al., 2013; Sakurai et al., 2015). Therefore, stable A3H haplotypes may be effective for short-term HIV-1 suppression. However, before stable A3H haplotypes can be exploited for long-term HIV-1 suppression, further studies should ascertain the period over which they can control HIV-1 with the hypo-functional vif gene in the absence of cART and which variants emerge under the selective pressure of stable A3H haplotypes in vivo.
Certain populations of proviral DNA in HIV-1-infected patients comprise defective viruses with A3-mediated G-to-A hypermutation (Kieffer et al., 2005; Ho et al., 2013; Imamichi et al., 2014; Bruner et al., 2016, 2019), suggesting that some of the A3 family proteins may avoid Vif-mediated neutralization in vivo. A simple explanation for this relates to the abundance of A3 family proteins, which can quantitatively exceed the capacity of Vif proteins (Figure 1B). In support of this explanation, CD4+ T cells from HIV-1 controllers with higher A3G expression levels were found to be more resistant to HIV-1 infection in vitro compared with CD4+ T cells from HIV-1 controllers with lower A3G expression levels (Biasin et al., 2007; De Pasquale et al., 2013). Therefore, the upregulation of A3 family proteins seems to increase defective viruses. However, the regulatory mechanisms of A3 family proteins have yet to be fully elucidated, largely because the expression of these proteins is intricately regulated and dependent on multiple determinants such as cell types (Koning et al., 2009, 2011; Refsland et al., 2010; Berger et al., 2011; Burns et al., 2013; Ikeda et al., 2019) and the surrounding environment of development and inflammation, e.g., the interferon (IFN) response (Peng et al., 2006, 2007; Pion et al., 2006; Ellery et al., 2007; Berger et al., 2011; Koning et al., 2011; Land et al., 2013; Mohanram et al., 2013).
Interestingly, most A3 family proteins are incorporated into ribonucleoprotein complexes in human cells (e.g., CD4+ T cells). In these cells, A3G forms enzymatically inactive high molecular mass (HMM) complexes immediately after translation, which are composed of A3G-binding proteins, A3G-binding RNAs, and RNA-binding proteins (Chiu et al., 2006; Soros et al., 2007; Stopak et al., 2007). These complexes are shifted to enzymatically active low molecular mass (LMM) complexes by RNase treatment (Soros et al., 2007). Hence, the formation of A3G HMM complexes in cells is one factor that determines permissiveness to HIV-1 infection (Pion et al., 2006; Ellery et al., 2007), being markedly altered by cytokines such as IFNs and chemokines (Kreisberg et al., 2006; Stopak et al., 2007). Similarly, the formation of HMM and LMM complexes in cells is conserved among other HIV-1 restrictive A3s (at least in A3F and A3H) (Niewiadomska et al., 2007; Ito et al., 2018). Although >100 A3G interactors have been identified (Chiu et al., 2006; Kozak et al., 2006; Gallois-Montbrun et al., 2007; Shirakawa et al., 2008; Sugiyama et al., 2011), bona fide interactors that regulate the restrictive capacity of A3s against HIV-1 are yet to be clearly defined. Accordingly, identifying novel A3 interactors that modulate the mutagenic activity of A3 enzymes against HIV-1 and resolving the molecular mechanisms that control these regulatory factors will be important for developing an HIV-1 functional cure that functions via A3-mediated mutagenesis.
APOBEC3 proteins are examples of host factors that restrict cross-species transmission of lentiviruses [reviewed by Nakano et al. (2017a); Uriu et al. (2021)]. Interactions between mammalian A3 proteins and lentiviral Vifs are largely specific to viruses in their hosts (Mariani et al., 2003; Compton et al., 2012; Etienne et al., 2015; Yamada et al., 2016; Zhang et al., 2016; Nakano et al., 2020). As examples, HIV-1 Vif can degrade A3G protein from humans but not from African green monkeys or rhesus macaques (Bogerd et al., 2004; Schrofelbauer et al., 2004; Xu et al., 2004). Three laboratories have demonstrated that a single amino acid residue at position 128 of the A3G protein determines this interaction; human A3G D128K proteins, therefore, become resistant to HIV-1 Vif (Bogerd et al., 2004; Schrofelbauer et al., 2004; Xu et al., 2004). One potential strategy for controlling HIV-1 under cART-free conditions would be the use of Vif-resistant A3 proteins such as human A3G D128K proteins. Recently, Delviks-Frankenberry et al. (2019) took a novel approach to control HIV-1 using self-activating lentiviral vectors that deliver the human A3G D128K gene to target cells. T cell lines in which the human A3G D128K gene was transduced by this system were shown to potently inhibit HIV-1 replication and suppress the emergence of resistant viruses against human A3G D128K proteins for >3.5 months. The delivery system was also used to transduce primary CD4+ T cells as well as CD34+ hematopoietic stem and progenitor cells with transduction efficiencies of around 15 and 30%, respectively. Thus, developments in the efficiency of gene-of-interest delivery may improve the feasibility of gene therapy involving Vif-resistant A3 proteins as an HIV-1 functional cure.
Another interesting challenge is the creation of a “super restriction factor” to function as an inhibitor more potent than the original protein (McDonnell et al., 2020). Single-domain deaminase A3C proteins have weak HIV-1 restriction activity compared with that of double-domain deaminases, e.g., A3D, A3F, and A3G proteins (Bishop et al., 2004; Hultquist et al., 2011; Wittkopp et al., 2016; Anderson et al., 2018). Synthetic tandem double deaminase A3C proteins represent one attempt to create HIV-1 restrictive A3C proteins that are more potent than the native proteins (McDonnell et al., 2020). The novel double-domain A3C proteins showed improved HIV-1 restriction activity and were largely resistant to Vif-mediated degradation. Although an efficient delivery system to HIV-1 target cells must be established, a strategy by which single deaminases are genetically connected could generate deaminases that are more potent and Vif-resistant; such proteins could be exploited to develop an HIV-1 functional cure.
Clearly, A3 family proteins contribute to innate immunity against retroviruses including HIV-1. In addition, there is evidence that A3-mediated mutagenesis is involved in adaptive immunity, such as CTL responses, through changes to proviral sequences (Figure 2). This can be validated because A3 family protein expression in antigen-presenting cells, e.g., macrophages, varies upon HIV-1 infection or cytokine stimulation with IFN (Peng et al., 2006, 2007; Pion et al., 2006; Biasin et al., 2007; Stopak et al., 2007; Land et al., 2008; Koning et al., 2009, 2011; Berger et al., 2011; Hultquist et al., 2011; Mohanram et al., 2013). Moreover, this process provides an additional layer to a potential HIV-1 functional cure by A3-mediated mutagenesis, i.e., via innate and adaptive immune responses.
Human immunodeficiency virus type 1-specific CTL responses and their human leukocyte antigen (HLA) restrictions are likely to be determinants of viral replication control in HIV-1-infected individuals [reviewed by Macatangay and Rinaldo (2015)]. However, mutations accumulate on the HIV-1 genome during infection and escape variants that can avoid HIV-1-specific CTL responses subsequently emerge. HIV-1 sequencing data from patients has indicated that A3-mediated G-to-A mutations (mainly A3F and A3G) are embedded within the predicted CTL epitopes and flanking regions, suggesting that sublethal levels of A3-mediated mutagenesis are involved in the emergence of CTL escape variants [reviewed by Grant and Larijani (2017); Venkatesan et al. (2018); Figure 2]. In contrast, one study in which the Vif-null virus was used revealed that A3G-induced mutations enhance HIV-1-specific CTL responses (Casartelli et al., 2010). Interestingly, engineered defective viruses with premature termination codons caused by A3G can enhance the abundance of HIV-1-derived epitopes, resulting in the activation of HIV-1-specific CTLs (Casartelli et al., 2010; Figure 2). The degradation of polypeptides from defective mRNAs is efficiently presented on major histocompatibility complex class I molecules (Trentini et al., 2020). Indeed, defective HIV-1 proviruses retain the ability to transcribe mRNAs and produce proteins that can be recognized by HIV-1-specific CTLs (Imamichi et al., 2016, 2020; Pollack et al., 2017). Therefore, lethal A3-mediated hypermutation may contribute to the generation of HIV-1 peptides supplied to the pool of antigens presented on major histocompatibility complex class I molecules and to the enhancement of CTL responses (Figure 2). Although effects on CTL responses seem to differ depending on lethal or sublethal levels of A3-mediated mutagenesis, A3-mediated mutagenesis undoubtedly contributes to the quantity and quality of HIV-specific CTL responses. For successful HIV-1 control without cART, A3 family proteins may be required to function alongside factors associated with adaptive immunity such as CTLs. Understanding the potential role of A3 proteins in innate and adaptive immunity would create new avenues of possibility for an HIV-1 functional cure.
Proteasomal degradation of HIV-1-restrictive A3s by Vif limits the packaging of these A3 enzymes into viral particles and thereby prevents A3-mediated hypermutation in the subsequent viral infection (Figure 1B). Nevertheless, evidence increasingly suggests that sublethal levels of A3-mediated mutagenesis contribute to virus evolution including drug resistance and immune escape [reviewed by Grant and Larijani (2017); Venkatesan et al. (2018)]. One possible therapeutic strategy would involve restoration of the lethal mutagenic activity of A3 family proteins by direct inhibition of Vif. Indeed, this viral protein is an attractive therapeutic target because it is not known to have mammalian homologs. Moreover, it is difficult for Vif-null HIV-1 to adapt to cells expressing full HIV-1 restrictive A3 proteins simultaneously (Haché et al., 2008; Albin et al., 2010; Ikeda et al., 2018).
Many attempts have been made to discover small molecules that target Vif proteins. Of these small molecules, RN-18 and its analogs are particularly well studied. RN-18 was discovered by screening 30,000 small molecules for the effects of Vif on fluorescence-tagged A3G signals (Nathans et al., 2008). RN-18 has been shown to exhibit A3G-dependent anti-HIV-1 activity in three non-permissive T cell lines (IC50 = 6 μM in H9); RN-18 treatment reduces the expression of cellular Vif and increases A3G expression levels, leading to the packaging of more A3G in viral particles. In addition, structure–activity relationship studies have revealed RN-18 analogs with improved potency in their antiviral activity relative to that of RN-18 (Ali et al., 2012; Mohammed et al., 2012, 2016; Zhou et al., 2017; Sharkey et al., 2019). For example, the compound 12c enhances the antiviral activity of A3G by >150-fold compared with the effect of RN-18 (Zhou et al., 2017). Furthermore, a recent cell culture study reported the emergence of a resistant virus against the RN-18 analog IMC15 and proposed a docking model of IMC15 bound to the Vif-A3G-E3 ligase complex (Sharkey et al., 2019). Along with RN-18 and its analogs, IMB-26/35, MM-1/2, and VEC-5 have been identified as lead compounds [reviewed by Olson et al. (2018)]. Overall, the development of small molecules that restore the mutagenic activity of A3 enzymes remains at an early stage. However, structural findings, such as the elucidation of the A3F–Vif interface (Hu et al., 2019), help to develop Vif antagonists that are more potent and can improve strategies for an HIV-1 functional cure.
HIV-1 has adapted to human cells such as CD4+ T cells; consequently, restriction factors, including A3 family proteins, are less effective against the virus. However, certain populations of HIV-1 are still exposed to lethal or sublethal A3-mediated mutagenesis, suggesting that the manipulation of this process is an attractive target through which to develop a functional cure for HIV-1. Therefore, in combination with several other strategies or therapies, A3-mediated mutagenesis could potentially be applied to functionally cure HIV-1.
TI drafted the manuscript. All authors edited, contributed to the manuscript, and approved the submitted version.
This work was supported in part by the JSPS Leading Initiative for Excellent Young Researchers (LEADER), AMED Research Program on Emerging and Re-emerging Infectious Diseases 20fk0108413, JST A-STEP JPMJTM20SL, Takeda Science Foundation, Mitsubishi Foundation, Shin-Nihon Foundation of Advanced Medical Research, an intramural grant from Kumamoto University COVID-19 Research Projects (AMABIE), and Intercontinental Research and Educational Platform Aiming for Eradication of HIV/AIDS (to TI).
The reviewer DS declared a past co-authorship with the author TI to the handling editor. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Drs. C. Motozono and Y. Satou for the productive comments.
Albin, J. S., Haché, G., Hultquist, J. F., Brown, W. L., and Harris, R. S. (2010). Long-term restriction by APOBEC3F selects human immunodeficiency virus type 1 variants with restored Vif function. J. Virol. 84, 10209–10219. doi: 10.1128/JVI.00632-10
Ali, A., Wang, J., Nathans, R. S., Cao, H., Sharova, N., Stevenson, M., et al. (2012). Synthesis and structure-activity relationship studies of HIV-1 virion infectivity factor (Vif) inhibitors that block viral replication. ChemMedChem 7, 1217–1229. doi: 10.1002/cmdc.201200079
Anderson, B. D., Ikeda, T., Moghadasi, S. A., Martin, A. S., Brown, W. L., and Harris, R. S. (2018). Natural APOBEC3C variants can elicit differential HIV-1 restriction activity. Retrovirology 15:78. doi: 10.1186/s12977-018-0459-5
Barre-Sinoussi, F., Chermann, J. C., Rey, F., Nugeyre, M. T., Chamaret, S., Gruest, J., et al. (1983). Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220, 868–871. doi: 10.1126/science.6189183
Berger, G., Durand, S., Fargier, G., Nguyen, X. N., Cordeil, S., Bouaziz, S., et al. (2011). APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog 7:e1002221. doi: 10.1371/journal.ppat.1002221
Biasin, M., Piacentini, L., Lo Caputo, S., Kanari, Y., Magri, G., Trabattoni, D., et al. (2007). Apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G: a possible role in the resistance to HIV of HIV-exposed seronegative individuals. J. Infect. Dis. 195, 960–964. doi: 10.1086/511988
Binka, M., Ooms, M., Steward, M., and Simon, V. (2012). The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G. APOBEC3F, and APOBEC3H. J. Virol. 86, 49–59. doi: 10.1128/JVI.06082-11
Bishop, K. N., Holmes, R. K., Sheehy, A. M., Davidson, N. O., Cho, S. J., and Malim, M. H. (2004). Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14, 1392–1396. doi: 10.1016/j.cub.2004.06.057
Bogerd, H. P., Doehle, B. P., Wiegand, H. L., and Cullen, B. R. (2004). A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc. Natl. Acad. Sci. U S A. 101, 3770–3774. doi: 10.1073/pnas.0307713101
Bruner, K. M., Murray, A. J., Pollack, R. A., Soliman, M. G., Laskey, S. B., Capoferri, A. A., et al. (2016). Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 22, 1043–1049. doi: 10.1038/nm.4156
Bruner, K. M., Wang, Z., Simonetti, F. R., Bender, A. M., Kwon, K. J., Sengupta, S., et al. (2019). A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 566, 120–125. doi: 10.1038/s41586-019-0898-8
Burns, M. B., Lackey, L., Carpenter, M. A., Rathore, A., Land, A. M., Leonard, B., et al. (2013). APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370. doi: 10.1038/nature11881
Casartelli, N., Guivel-Benhassine, F., Bouziat, R., Brandler, S., Schwartz, O., and Moris, A. (2010). The antiviral factor APOBEC3G improves CTL recognition of cultured HIV-infected T cells. J. Exp. Med. 207, 39–49. doi: 10.1084/jem.20091933
Chiu, Y. L., Witkowska, H. E., Hall, S. C., Santiago, M., Soros, V. B., Esnault, C., et al. (2006). High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. U S A. 103, 15588–15593. doi: 10.1073/pnas.0604524103
Chun, T. W., Stuyver, L., Mizell, S. B., Ehler, L. A., Mican, J. A., Baseler, M., et al. (1997). Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U S A. 94, 13193–13197. doi: 10.1073/pnas.94.24.13193
Compton, A. A., Hirsch, V. M., and Emerman, M. (2012). The host restriction factor APOBEC3G and retroviral Vif protein coevolve due to ongoing genetic conflict. Cell Host Microbe 11, 91–98. doi: 10.1016/j.chom.2011.11.010
Cuevas, J. M., Geller, R., Garijo, R., Lopez-Aldeguer, J., and Sanjuan, R. (2015). Extremely high mutation rate of HIV-1 in vivo. PLoS Biol. 13:e1002251. doi: 10.1371/journal.pbio.1002251
De Pasquale, M., Kourteva, Y., Allos, T., and D’aquila, R. T. (2013). Lower HIV provirus levels are associated with more APOBEC3G protein in blood resting memory CD4+ T lymphocytes of controllers in vivo. PLoS One 8:e76002. doi: 10.1371/journal.pone.0076002
Delviks-Frankenberry, K. A., Ackerman, D., Timberlake, N. D., Hamscher, M., Nikolaitchik, O. A., Hu, W. S., et al. (2019). Development of lentiviral vectors for HIV-1 gene therapy with Vif-resistant APOBEC3G. Mol. Ther. Nucleic Acids 18, 1023–1038. doi: 10.1016/j.omtn.2019.10.024
Desimmie, B. A., Delviks-Frankenberrry, K. A., Burdick, R. C., Qi, D., Izumi, T., and Pathak, V. K. (2014). Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 426, 1220–1245. doi: 10.1016/j.jmb.2013.10.033
Ebrahimi, D., Richards, C. M., Carpenter, M. A., Wang, J., Ikeda, T., Becker, J. T., et al. (2018). Genetic and mechanistic basis for APOBEC3H alternative splicing, retrovirus restriction, and counteraction by HIV-1 protease. Nat. Commun. 9:4137. doi: 10.1038/s41467-018-06594-3
Ellery, P. J., Tippett, E., Chiu, Y. L., Paukovics, G., Cameron, P. U., Solomon, A., et al. (2007). The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J. Immunol. 178, 6581–6589.
Etienne, L., Bibollet-Ruche, F., Sudmant, P. H., Wu, L. I., Hahn, B. H., and Emerman, M. (2015). The role of the antiviral APOBEC3 gene family in protecting chimpanzees against lentiviruses from monkeys. PLoS Pathog 11:e1005149. doi: 10.1371/journal.ppat.1005149
Eyzaguirre, L. M., Charurat, M., Redfield, R. R., Blattner, W. A., Carr, J. K., and Sajadi, M. M. (2013). Elevated hypermutation levels in HIV-1 natural viral suppressors. Virology 443, 306–312. doi: 10.1016/j.virol.2013.05.019
Finzi, D., Hermankova, M., Pierson, T., Carruth, L. M., Buck, C., Chaisson, R. E., et al. (1997). Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300. doi: 10.1126/science.278.5341.1295
Gallois-Montbrun, S., Kramer, B., Swanson, C. M., Byers, H., Lynham, S., Ward, M., et al. (2007). Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 81, 2165–2178. doi: 10.1128/JVI.02287-06
Gonzalo-Gil, E., Ikediobi, U., and Sutton, R. E. (2017). Mechanisms of virologic control and clinical characteristics of HIV+ elite/viremic controllers. Yale J. Biol. Med. 90, 245–259.
Grant, M., and Larijani, M. (2017). Evasion of adaptive immunity by HIV through the action of host APOBEC3G/F enzymes. AIDS Res. Ther. 14:44. doi: 10.1186/s12981-017-0173-8
Haché, G., Shindo, K., Albin, J. S., and Harris, R. S. (2008). Evolution of HIV-1 isolates that use a novel Vif-independent mechanism to resist restriction by human APOBEC3G. Curr. Biol. 18, 819–824. doi: 10.1016/j.cub.2008.04.073
Harris, R. S., and Dudley, J. P. (2015). APOBECs and virus restriction. Virology 479-480C, 131–145. doi: 10.1016/j.virol.2015.03.012
Ho, Y. C., Shan, L., Hosmane, N. N., Wang, J., Laskey, S. B., Rosenbloom, D. I., et al. (2013). Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155, 540–551. doi: 10.1016/j.cell.2013.09.020
Hu, Y., Desimmie, B. A., Nguyen, H. C., Ziegler, S. J., Cheng, T. C., Chen, J., et al. (2019). Structural basis of antagonism of human APOBEC3F by HIV-1 Vif. Nat. Struct. Mol. Biol. 26, 1176–1183. doi: 10.1038/s41594-019-0343-6
Hultquist, J. F., Lengyel, J. A., Refsland, E. W., Larue, R. S., Lackey, L., Brown, W. L., et al. (2011). Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85, 11220–11234. doi: 10.1128/JVI.05238-11
Ikeda, T., Molan, A. M., Jarvis, M. C., Carpenter, M. A., Salamango, D. J., Brown, W. L., et al. (2019). HIV-1 restriction by endogenous APOBEC3G in the myeloid cell line THP-1. J. Gen. Virol. 100, 1140–1152. doi: 10.1099/jgv.0.001276
Ikeda, T., Shibata, J., Yoshimura, K., Koito, A., and Matsushita, S. (2007). Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. J. Infect. Dis. 195, 716–725. doi: 10.1086/510915
Ikeda, T., Symeonides, M., Albin, J. S., Li, M., Thali, M., and Harris, R. S. (2018). HIV-1 adaptation studies reveal a novel Env-mediated homeostasis mechanism for evading lethal hypermutation by APOBEC3G. PLoS Pathog 14:e1007010. doi: 10.1371/journal.ppat.1007010
Imamichi, H., Dewar, R. L., Adelsberger, J. W., Rehm, C. A., O’doherty, U., Paxinos, E. E., et al. (2016). Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. U S A. 113, 8783–8788. doi: 10.1073/pnas.1609057113
Imamichi, H., Natarajan, V., Adelsberger, J. W., Rehm, C. A., Lempicki, R. A., Das, B., et al. (2014). Lifespan of effector memory CD4+ T cells determined by replication-incompetent integrated HIV-1 provirus. AIDS 28, 1091–1099. doi: 10.1097/QAD.0000000000000223
Imamichi, H., Smith, M., Adelsberger, J. W., Izumi, T., Scrimieri, F., Sherman, B. T., et al. (2020). Defective HIV-1 proviruses produce viral proteins. Proc. Natl. Acad. Sci. U S A. 117, 3704–3710. doi: 10.1073/pnas.1917876117
Ito, F., Yang, H., Xiao, X., Li, S. X., Wolfe, A., Zirkle, B., et al. (2018). Understanding the structure, multimerization, subcellular localization and mC selectivity of a genomic mutator and anti-HIV factor APOBEC3H. Sci. Rep. 8:3763. doi: 10.1038/s41598-018-21955-0
Kieffer, T. L., Kwon, P., Nettles, R. E., Han, Y., Ray, S. C., and Siliciano, R. F. (2005). G–>A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J. Virol. 79, 1975–1980. doi: 10.1128/JVI.79.3.1975-1980.2005
Koito, A., and Ikeda, T. (2012). Apolipoprotein B mRNA-editing, catalytic polypeptide cytidine deaminases and retroviral restriction. Wiley Interdiscip Rev. RNA 3, 529–541. doi: 10.1002/wrna.1117
Koning, F. A., Goujon, C., Bauby, H., and Malim, M. H. (2011). Target cell-mediated editing of HIV-1 cDNA by APOBEC3 proteins in human macrophages. J. Virol. 85, 13448–13452. doi: 10.1128/JVI.00775-11
Koning, F. A., Newman, E. N., Kim, E. Y., Kunstman, K. J., Wolinsky, S. M., and Malim, M. H. (2009). Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 83, 9474–9485. doi: 10.1128/JVI.01089-09
Kourteva, Y., De Pasquale, M., Allos, T., Mcmunn, C., and D’aquila, R. T. (2012). APOBEC3G expression and hypermutation are inversely associated with human immunodeficiency virus type 1 (HIV-1) burden in vivo. Virology 430, 1–9. doi: 10.1016/j.virol.2012.03.018
Kozak, S. L., Marin, M., Rose, K. M., Bystrom, C., and Kabat, D. (2006). The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J. Biol. Chem. 281, 29105–29119. doi: 10.1074/jbc.M601901200
Kreisberg, J. F., Yonemoto, W., and Greene, W. C. (2006). Endogenous factors enhance HIV infection of tissue naive CD4 T cells by stimulating high molecular mass APOBEC3G complex formation. J. Exp. Med. 203, 865–870. doi: 10.1084/jem.20051856
Lambrechts, L., Cole, B., Rutsaert, S., Trypsteen, W., and Vandekerckhove, L. (2020). Emerging PCR-based techniques to study HIV-1 reservoir persistence. Viruses 12:149. doi: 10.3390/v12020149
Land, A. M., Ball, T. B., Luo, M., Pilon, R., Sandstrom, P., Embree, J. E., et al. (2008). Human immunodeficiency virus (HIV) type 1 proviral hypermutation correlates with CD4 count in HIV-infected women from Kenya. J. Virol. 82, 8172–8182. doi: 10.1128/JVI.01115-08
Land, A. M., Law, E. K., Carpenter, M. A., Lackey, L., Brown, W. L., and Harris, R. S. (2013). Endogenous APOBEC3A DNA cytosine deaminase is cytoplasmic and nongenotoxic. J. Biol. Chem. 288, 17253–17260. doi: 10.1074/jbc.M113.458661
Macatangay, B. J., and Rinaldo, C. R. (2015). Preserving HIV-specific T cell responses: does timing of antiretroviral therapy help? Curr. Opin. HIV AIDS 10, 55–60. doi: 10.1097/COH.0000000000000124
Maldarelli, F., Wu, X., Su, L., Simonetti, F. R., Shao, W., Hill, S., et al. (2014). HIV latency. specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345, 179–183. doi: 10.1126/science.1254194
Mariani, R., Chen, D., Schrofelbauer, B., Navarro, F., Konig, R., Bollman, B., et al. (2003). Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114, 21–31. doi: 10.1016/s0092-8674(03)00515-4
McDonnell, M. M., Crawford, K. H. D., Dingens, A. S., Bloom, J. D., and Emerman, M. (2020). APOBEC3C tandem domain proteins create super restriction factors against HIV-1. mBio 11:e00737-20. doi: 10.1128/mBio.00737-20
Mohammed, I., Kummetha, I. R., Singh, G., Sharova, N., Lichinchi, G., Dang, J., et al. (2016). 1,2,3-Triazoles as amide bioisosteres: discovery of a new class of potent HIV-1 Vif antagonists. J. Med. Chem. 59, 7677–7682. doi: 10.1021/acs.jmedchem.6b00247
Mohammed, I., Parai, M. K., Jiang, X., Sharova, N., Singh, G., Stevenson, M., et al. (2012). SAR and lead optimization of an HIV-1 Vif-APOBEC3G axis inhibitor. ACS Med. Chem. Lett. 3, 465–469. doi: 10.1021/ml300037k
Mohanram, V., Skold, A. E., Bachle, S. M., Pathak, S. K., and Spetz, A. L. (2013). IFN-alpha induces APOBEC3G, F, and A in immature dendritic cells and limits HIV-1 spread to CD4+ T cells. J. Immunol. 190, 3346–3353. doi: 10.4049/jimmunol.1201184
Nakano, Y., Aso, H., Soper, A., Yamada, E., Moriwaki, M., Juarez-Fernandez, G., et al. (2017a). A conflict of interest: the evolutionary arms race between mammalian APOBEC3 and lentiviral Vif. Retrovirology 14:31. doi: 10.1186/s12977-017-0355-4
Nakano, Y., Misawa, N., Juarez-Fernandez, G., Moriwaki, M., Nakaoka, S., Funo, T., et al. (2017b). HIV-1 competition experiments in humanized mice show that APOBEC3H imposes selective pressure and promotes virus adaptation. PLoS Pathog 13:e1006348. doi: 10.1371/journal.ppat.1006348
Nakano, Y., Yamamoto, K., Ueda, M. T., Soper, A., Konno, Y., Kimura, I., et al. (2020). A role for gorilla APOBEC3G in shaping lentivirus evolution including transmission to humans. PLoS Pathog 16:e1008812. doi: 10.1371/journal.ppat.1008812
Nathans, R., Cao, H., Sharova, N., Ali, A., Sharkey, M., Stranska, R., et al. (2008). Small-molecule inhibition of HIV-1 Vif. Nat. Biotechnol. 26, 1187–1192. doi: 10.1038/nbt.1496
Niewiadomska, A. M., Tian, C., Tan, L., Wang, T., Sarkis, P. T., and Yu, X. F. (2007). Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J. Virol. 81, 9577–9583. doi: 10.1128/JVI.02800-06
OhAinle, M., Kerns, J. A., Li, M. M., Malik, H. S., and Emerman, M. (2008). Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 4, 249–259. doi: 10.1016/j.chom.2008.07.005
Olson, M. E., Harris, R. S., and Harki, D. A. (2018). APOBEC enzymes as targets for virus and cancer therapy. Cell Chem. Biol. 25, 36–49. doi: 10.1016/j.chembiol.2017.10.007
Ooms, M., Brayton, B., Letko, M., Maio, S. M., Pilcher, C. D., Hecht, F. M., et al. (2013). HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe 14, 411–421. doi: 10.1016/j.chom.2013.09.006
Pace, C., Keller, J., Nolan, D., James, I., Gaudieri, S., Moore, C., et al. (2006). Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. J. Virol. 80, 9259–9269. doi: 10.1128/JVI.00888-06
Peng, G., Greenwell-Wild, T., Nares, S., Jin, W., Lei, K. J., Rangel, Z. G., et al. (2007). Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 110, 393–400. doi: 10.1182/blood-2006-10-051763
Peng, G., Lei, K. J., Jin, W., Greenwell-Wild, T., and Wahl, S. M. (2006). Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J. Exp. Med. 203, 41–46. doi: 10.1084/jem.20051512
Pion, M., Granelli-Piperno, A., Mangeat, B., Stalder, R., Correa, R., Steinman, R. M., et al. (2006). APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J. Exp. Med. 203, 2887–2893. doi: 10.1084/jem.20061519
Pollack, R. A., Jones, R. B., Pertea, M., Bruner, K. M., Martin, A. R., Thomas, A. S., et al. (2017). Defective HIV-1 proviruses are expressed and can be recognized by cytotoxic T lymphocytes, which shape the proviral landscape. Cell Host Microbe 21, 494–506.e4. doi: 10.1016/j.chom.2017.03.008
Promer, K., and Karris, M. Y. (2018). Current treatment options for HIV elite controllers: a review. Curr. Treat Options Infect. Dis. 10, 302–309. doi: 10.1007/s40506-018-0158-8
Refsland, E. W., Hultquist, J. F., and Harris, R. S. (2012). Endogenous origins of HIV-1 G to A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog 8:e1002800. doi: 10.1371/journal.ppat.1002800
Refsland, E. W., Hultquist, J. F., Luengas, E. M., Ikeda, T., Shaban, N. M., Law, E. K., et al. (2014). Natural polymorphisms in human APOBEC3H and HIV-1 Vif combine in primary T lymphocytes to affect viral G-to-A mutation levels and infectivity. PLoS Genet 10:e1004761. doi: 10.1371/journal.pgen.1004761
Refsland, E. W., Stenglein, M. D., Shindo, K., Albin, J. S., Brown, W. L., and Harris, R. S. (2010). Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38, 4274–4284. doi: 10.1093/nar/gkq174
Sakurai, D., Iwatani, Y., Ohtani, H., Naruse, T. K., Terunuma, H., Sugiura, W., et al. (2015). APOBEC3H polymorphisms associated with the susceptibility to HIV-1 infection and AIDS progression in Japanese. Immunogenetics 67, 253–257. doi: 10.1007/s00251-015-0829-2
Salamango, D. J., and Harris, R. S. (2020). Dual functionality of HIV-1 Vif in APOBEC3 counteraction and cell cycle arrest. Front. Microbiol. 11:622012. doi: 10.3389/fmicb.2020.622012
Schrofelbauer, B., Chen, D., and Landau, N. R. (2004). A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc. Natl. Acad. Sci. U S A. 101, 3927–3932. doi: 10.1073/pnas.0307132101
Sharkey, M., Sharova, N., Mohammed, I., Huff, S. E., Kummetha, I. R., Singh, G., et al. (2019). HIV-1 escape from small-molecule antagonism of Vif. mBio 10:e00144-19. doi: 10.1128/mBio.00144-19
Shirakawa, K., Takaori-Kondo, A., Yokoyama, M., Izumi, T., Matsui, M., Io, K., et al. (2008). Phosphorylation of APOBEC3G by protein kinase A regulates its interaction with HIV-1 Vif. Nat. Struct. Mol. Biol. 15, 1184–1191. doi: 10.1038/nsmb.1497
Simon, V., Bloch, N., and Landau, N. R. (2015). Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat. Immunol. 16, 546–553. doi: 10.1038/ni.3156
Soros, V. B., Yonemoto, W., and Greene, W. C. (2007). Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog 3:e15. doi: 10.1371/journal.ppat.0030015
Stavrou, S., and Ross, S. R. (2015). APOBEC3 Proteins in Viral Immunity. J. Immunol. 195, 4565–4570. doi: 10.4049/jimmunol.1501504
Stopak, K. S., Chiu, Y. L., Kropp, J., Grant, R. M., and Greene, W. C. (2007). Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J. Biol. Chem. 282, 3539–3546. doi: 10.1074/jbc.M610138200
Sugiyama, R., Nishitsuji, H., Furukawa, A., Katahira, M., Habu, Y., Takeuchi, H., et al. (2011). Heat shock protein 70 inhibits HIV-1 Vif-mediated ubiquitination and degradation of APOBEC3G. J. Biol. Chem. 286, 10051–10057. doi: 10.1074/jbc.M110.166108
Trentini, D. B., Pecoraro, M., Tiwary, S., Cox, J., Mann, M., Hipp, M. S., et al. (2020). Role for ribosome-associated quality control in sampling proteins for MHC class I-mediated antigen presentation. Proc. Natl. Acad. Sci. U S A. 117, 4099–4108. doi: 10.1073/pnas.1914401117
Uriu, K., Kosugi, Y., Ito, J., and Sato, K. (2021). The battle between retroviruses and APOBEC3 genes: its past and present. Viruses 13:124. doi: 10.3390/v13010124
Venkatesan, S., Rosenthal, R., Kanu, N., Mcgranahan, N., Bartek, J., Quezada, S. A., et al. (2018). Perspective: APOBEC mutagenesis in drug resistance and immune escape in HIV and cancer evolution. Annals Oncol. 29, 563–572. doi: 10.1093/annonc/mdy003
Wagner, T. A., Mclaughlin, S., Garg, K., Cheung, C. Y., Larsen, B. B., Styrchak, S., et al. (2014). HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 345, 570–573. doi: 10.1126/science.1256304
Wang, X., Abudu, A., Son, S., Dang, Y., Venta, P. J., and Zheng, Y. H. (2011). Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J. Virol. 85, 3142–3152. doi: 10.1128/JVI.02049-10
Wang, Z., Simonetti, F. R., Siliciano, R. F., and Laird, G. M. (2018). Measuring replication competent HIV-1: advances and challenges in defining the latent reservoir. Retrovirology 15:21. doi: 10.1186/s12977-018-0404-7
Wittkopp, C. J., Adolph, M. B., Wu, L. I., Chelico, L., and Emerman, M. (2016). A single nucleotide polymorphism in human APOBEC3C enhances restriction of lentiviruses. PLoS Pathog 12:e1005865. doi: 10.1371/journal.ppat.1005865
Wong, J. K., Hezareh, M., Gunthard, H. F., Havlir, D. V., Ignacio, C. C., Spina, C. A., et al. (1997). Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278, 1291–1295. doi: 10.1126/science.278.5341.1291
Xu, H., Svarovskaia, E. S., Barr, R., Zhang, Y., Khan, M. A., Strebel, K., et al. (2004). A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc. Natl. Acad. Sci. U S A. 101, 5652–5657. doi: 10.1073/pnas.0400830101
Yamada, E., Yoshikawa, R., Nakano, Y., Misawa, N., Kobayashi, T., Ren, F., et al. (2016). A naturally occurring bovine APOBEC3 confers resistance to bovine lentiviruses: implication for the co-evolution of bovids and their lentiviruses. Sci. Rep. 6:33988. doi: 10.1038/srep33988
Zhang, Z., Gu, Q., Jaguva Vasudevan, A. A., Hain, A., Kloke, B. P., Hasheminasab, S., et al. (2016). Determinants of FIV and HIV Vif sensitivity of feline APOBEC3 restriction factors. Retrovirology 13:46. doi: 10.1186/s12977-016-0274-9
Zhou, M., Luo, R. H., Hou, X. Y., Wang, R. R., Yan, G. Y., Chen, H., et al. (2017). Synthesis, biological evaluation and molecular docking study of N-(2-methoxyphenyl)-6-((4-nitrophenyl)sulfonyl)benzamide derivatives as potent HIV-1 Vif antagonists. Eur. J. Med. Chem. 129, 310–324. doi: 10.1016/j.ejmech.2017.01.010
Keywords: APOBEC3-mediated mutagenesis, genetic factors, A3 expression, A3-interacting proteins, adaptive immunity, Vif inhibitors
Citation: Ikeda T, Yue Y, Shimizu R and Nasser H (2021) Potential Utilization of APOBEC3-Mediated Mutagenesis for an HIV-1 Functional Cure. Front. Microbiol. 12:686357. doi: 10.3389/fmicb.2021.686357
Received: 26 March 2021; Accepted: 10 May 2021;
Published: 15 June 2021.
Edited by:
Taisuke Izumi, American Type Culture Collection, United StatesReviewed by:
Daniel Salamango, Stony Brook University, United StatesCopyright © 2021 Ikeda, Yue, Shimizu and Nasser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Terumasa Ikeda, aWtlZGF0QGt1bWFtb3RvLXUuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.