
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 24 June 2021
Sec. Microbiotechnology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.684640
This article is part of the Research Topic Recent Advances in Microbial Biotechnology for the Food Industry View all 6 articles
 Viktor Varga1Vladimír Štefuca2Lenka Mihálová2Zdenko Levarski1,3Eva Struhárňanská1Jaroslav Blaško4Robert Kubinec4
Viktor Varga1Vladimír Štefuca2Lenka Mihálová2Zdenko Levarski1,3Eva Struhárňanská1Jaroslav Blaško4Robert Kubinec4 Pavel Farkaš5Vladimír Sitkey5Ján Turňa1,3Michal Rosenberg2
Pavel Farkaš5Vladimír Sitkey5Ján Turňa1,3Michal Rosenberg2 Stanislav Stuchlík1,3*
Stanislav Stuchlík1,3*The aim of this study was to develop immobilized enzyme systems that reduce carbonyl compounds to their corresponding alcohols. The demand for natural aromas and food additives has been constantly growing in recent years. However, it can no longer be met by extraction and isolation from natural materials. One way to increase the availability of natural aromas is to prepare them by the enzymatic transformation of suitable precursors. Recombinant enzymes are currently being used for this purpose. We investigated trans-2-hexenal bioreduction by recombinant Saccharomyces cerevisiae alcohol dehydrogenase (ScADH1) with simultaneous NADH regeneration by recombinant Candida boidinii formate dehydrogenase (FDH). In a laboratory bioreactor with two immobilized enzymes, 88% of the trans-2-hexenal was transformed to trans-2-hexenol. The initial substrate concentration was 3.7 mM. The aldehyde destabilized ScADH1 by eluting Zn2+ ions from the enzyme. A fed-batch operation was used and the trans-2-hexenal concentration was maintained at a low level to limit the negative effect of Zn2+ ion elution from the immobilized ScADH1. Another immobilized two-enzyme system was used to reduce acetophenone to (S)-1-phenylethanol. To this end, the recombinant alcohol dehydrogenase (RrADH) from Rhodococcus ruber was used. This biocatalytic system converted 61% of the acetophenone to (S)-1-phenylethanol. The initial substrate concentration was 8.3 mM. All enzymes were immobilized by poly-His tag to Ni2+, which formed strong but reversible bonds that enabled carrier reuse after the loss of enzyme activity.
In recent years, there has been an increasing demand for natural aromatic chemicals in the food and cosmetics industries. Natural aromas are produced by isolation from natural materials (Sharmeen et al., 2021), biotransformation of natural compounds, and de novo production by microorganisms (Shaaban et al., 2016; Paulino et al., 2021). Commercially important compounds are derived from green plant parts and are collectively referred to as “green odor.” Eight volatile compounds including C6-aldehydes and C6-alcohols such as the leaf aldehyde trans-2-hexenal and the leaf alcohol cis-3-hexenol contribute to “green odor” (Hatanaka, 1996; Poltronieri et al., 2019). The synthesis of “green odor” compounds starts with free polyunsaturated fatty acids (PUFAs) that are transformed by specific lipoxygenases into hydroperoxy fatty acids subsequently cleaved by specific hydroperoxide lyases into aldehyde and oxoacid moieties (Poltronieri et al., 2019). The aldehydes undergo thermal isomerization or may be reduced by dehydrogenases to other C6 products. Hydroperoxide lyase is inhibited by α-, β-unsaturated aldehydes such as trans-2-hexenal that bind sulfhydryl groups. These aldehydes are generated by isomerization of non-inhibiting primary products such as hexanal or cis-3-hexenal (Suurmeijer et al., 2000; Long et al., 2010). This inhibition is limited by in situ reduction of the aldehydes to their corresponding alcohols via alcohol dehydrogenase (ADH) in pure form or in yeast cells. A two-enzyme system consisting of hydroperoxide lyase and ADH cleaved 13-hydroperoxide linolenic acid faster than hydroperoxide lyase alone (Gargouri et al., 2004).
Alcohol dehydrogenases are also used in natural aroma preparation to reduce ketones to secondary alcohols (Ying et al., 2014; Bakonyi et al., 2020). The secondary alcohol 1-phenylethanol has a mild-floral odor and is used extensively in the cosmetic industry (Hawley and Lewis, 1992). Direct isolation of 1-phenylethanol from plants has a low yield and is not cost-effective. However, an alternative approach to 1-phenylethanol generation is enzymatic acetophenone bioreduction (Zhou et al., 2019).
ADH can be used to reduce aldehydes and ketones. Nevertheless, alcohol dehydrogenases differ in terms of structure and substrate specificity. Therefore, suitable ADH must be found for each application. One widely used type of ADH is secreted by Saccharomyces cerevisiae (ScADH1). This constitutive enzyme reduces acetaldehyde to ethanol during glucose fermentation (Leskovac et al., 1999). It is usually dimeric in higher eukaryotes but homotetrameric in lower eukaryotes such as yeasts. The latter form has a relatively smaller, narrower active site with a comparatively higher affinity for primary carbonyl compounds. In contrast, the alcohol dehydrogenase from the Gram-positive bacterium Rhodococcus ruber (RrADH) reduces ketones and oxidizes secondary alcohols (Hamnevik et al., 2014). RrADH had 5,000-fold higher enzymatic activity on 1-phenylethanol than 2-phenylethanol (Hamnevik et al., 2014). RrADH can bind bulky substrates, as its active site resembles an open cleft. Both proteins are structurally similar NAD(H)-dependent oxidoreductases containing zinc atoms required for their activity. However, RrADH activity is robust at various pH and elevated temperatures. It is also stable in solvents such as acetone used to solubilize hydrophobic ketones and alcohols in the biotransformation. Acetone is also a substrate and co-substrate for certain enzymatic reactions involving nicotinamide cofactor recycling (Karabec et al., 2010; Hamnevik et al., 2014).
Cell-free ADH preparations have high specificity, low product degradation, and superior enzyme activity regulation. However, aldehyde reduction by ADH requires stoichiometric quantities of reduced cofactor. The preparation of certain aromas by aldehyde reduction to alcohols depends on equilibrium reactions. One possible way to improve the efficiency of this process is to shift the reaction equilibrium toward the alcohol product side by increasing the NADH concentration. Therefore, the highest possible NADH concentration should be maintained during the reaction. Nevertheless, these processes are not cost-effective unless reduced cofactors are regenerated. Green aroma chemicals are substantially cheaper than NADH or NADPH. Reduced cofactors may be regenerated enzymatically, electrochemically, or chemically (Weckbecker et al., 2010; Wang et al., 2017). A well-known and highly effective example is the formate dehydrogenase (FDH) reaction using formate as a co-substrate (Shaked and Whitesides, 1980). The equilibrium of the FDH reaction is markedly shifted toward the right because the CO2 product is easily removed from the reaction system (Duman et al., 2020). Co-immobilized amine and formate dehydrogenases were used in the continuous production of chiral amine in a packed bed reactor (Franklin et al., 2021). FDH catalysis served as a model for the development of artificial metalloenzymes reducing nicotinamide cofactors resilient to high temperatures, compatible with solvents, and lacking FDH complexity (Basle et al., 2021).
Only a few FDH-based industrial processes are known. Examples include the production of l-tert-leucine by reductive amination of α-keto acids with an amino acid dehydrogenase (Evonik Industries AG, Germany) (Bommarius et al., 1998), the reduction of 1-phenyl-2-propanone to 1-phenyl-2-propanole (Forschungszentrum Julich GmbH, Germany), the reduction of 2-oxo-4-phenylbutyric acid to 2-hydroxy-4-phenylbutyric acid (CIBA Spezialitatenchemie AG, Switzerland), and the reduction of 5-(1,3-dioxolan-2-yl)-2-oxopentanoic acid to (S)-2-amino-5-(1,3-dioxolan-2-yl)pentanoic acid (Bristol-Myers Squibb, USA) (Liese et al., 2006). FDH is seldom used in industrial biocatalysis because of its high purchase price. A workaround is FDH overexpression by a suitable host and subsequent enzyme immobilization.
The implementation of enzyme catalysis is often limited by low enzyme thermostability, shear stress sensitivity, and limited use in organic solvents (Chapman et al., 2018). However, the enzymes may be immobilized by binding them to solid supports. The first known attempt was the immobilization of invertase on adsorbent particles. In this manner, the enzyme could be recycled and repeatedly used in catalysis (Nelson and Griffin, 1916). Immobilized enzymes have been extensively investigated and have demonstrated clear advantages over native enzymes. In industrial biocatalysis, enzyme immobilization improves enzyme stability, selectivity, and specificity and reduces inhibition (Mateo et al., 2007; Vasić et al., 2020). This applies to the industrial use of alcohol dehydrogenases which may be facilitated by stabilizing them with suitable immobilization techniques.
The immobilization strategy in this study was based on the use of recombinant poly-His-tag fused enzymes. Several studies showed that this approach enables enzymes to be purified and immobilized on metal chelated supports for subsequent use as biocatalysts (Brena et al., 1994; Beitle and Ataai, 1997; Mateo et al., 2001; Ho et al., 2004). The supports are generally inexpensive and may be prepared from epoxy and iminodiacetic or iminotriacetic acid and subsequent metal cation complexation. This immobilization technique is conducted under gentle conditions compared with direct binding via epoxide groups and eliminates the risk of active site binding to the support. The important advantage of this approach is the possibility to regenerate the biocatalyst by replacing the enzyme at the end of its lifespan. This technique was applied to alcohol dehydrogenases from horse liver (Quaglia et al., 2013), Kluyveromyces polyspora (Zhou et al., 2021), and Aromatoleum aromaticum (Böhmer et al., 2018). His-tagged ADH from Thermus thermophilus was purified and immobilized on a metal support in a single step. In this way, the number of steps required for biocatalyst preparation may be reduced (Kozgus et al., 2014).
This study aimed to develop a biocatalytic redox system based on poly-His-tag fused enzyme immobilization in a flow bioreactor system. Formate dehydrogenase from Candida boidinii and alcohol dehydrogenases from Saccharomyces cerevisiae and Rhodococcus ruber were produced by gene expression in Escherichia coli and immobilization on a nickel carrier. The immobilized enzymes were used in two-enzyme biotransformation systems designed for the reduction of trans-2-hexenal and acetophenone to the corresponding alcohols with simultaneous NADH regeneration.
All chemicals were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA) and Centralchem (Banská Bystrica, Slovakia) unless otherwise stated. D-Trehalose was acquired from Apollo Scientific (Stockford, UK). Acetophenone was obtained from Axxence Slovakia Ltd. (Bratislava, Slovakia). Deionized water (RiOs TM water purification system; EMD Millipore, Billerica, MA, USA) was used in all experiments.
All bacterial strains and plasmids were used in ADH1 production from Saccharomyces cerevisiae (Utekal et al., 2014; Levarski et al., 2018) and in FHD production from Candida boidinii. Gene expressions and enzyme preparations were described in a previous report (Levarski et al., 2018). Bacterial strains and plasmids used in RrADH production are listed in Tables 1, 2.

Table 1. Escherichia coli strains used in this study.

Table 2. Plasmids used in this study.
Rhodococcus ruber sadh was synthesized, codon-optimized for Escherichia coli (GenScript, Piscataway, NJ, USA), and cloned into pET29b via NdeI/XhoI restriction sites. The recombinant plasmid pET29-RrADH was sequenced to confirm the correct sequences and open reading frames (ORFs).
Bacterial strains with chemically induced competence were transformed by the pET29-RrADH plasmid (Hanahan, 1983). Briefly, overnight bacterial cultures were incubated at 37°C in Luria-Bertani (LB) medium without antibiotic until OD600 = 0.5–0.6. The cells were centrifuged for 5 min. at 1,920 × g. The bacterial pellet was resuspended in the buffer according to the protocol and kept on ice for 1.5 h. Glycerol (15% v/v) was added to competent cell aliquots and the suspensions were either used in E. coli transformation or stored at −70°C until the subsequent experiments.
Escherichia coli cells containing the pET-RrADH were grown in 20 mL LB medium (10 g/L peptone, 5 g/L yeast extract, and 5 g/L NaCl), Terrific-Broth medium (TB) (24 g/L yeast extract, 20 g/L peptone, 40 g/L glycerol, 0.017 M KH2PO4, and 0.072 M K2HPO4) and Dynamite medium (24 g/L yeast extract, 20 g/L peptone, 40 g/L glycerol, 1 mM L MgSO4, 0.017 M KH2PO4, and 0.072 M K2HPO4) in Erlenmeyer flasks containing 1% glucose (w/v) and 1 mM kanamycin and incubated overnight. Then 0.5 mL culture was transferred to 50 mL fresh medium supplemented with 1% (w/v) glucose. The culture was shaken at 160 rpm on a Multitron Standard orbital shaker (Infors HT GmBH, Bottmingen, Switzerland) at 20°C, 28°C, or 37°C until exponential growth (OD600 range 0.5–0.8). RrADH was induced by adding isopropyl-β-d-1-thiogalactopyranosid (IPTG) to a final concentration of 1 M. Large-scale expression was performed in 1 L medium plus 0.1% glucose (w/v) inoculated with 100 mL overnight culture in a Biostat B plus-2L MO bioreactor (Sartorius AG, Göttingen, Germany). Bioreactor aeration and initial stirring were set to 0.6 vvm and 200 rpm, respectively. In response to the metabolic activity of the cells, the O2 concentration declined to 2% saturation and was maintained by agitation at 200–2,000 rpm. Samples were taken before and periodically after induction and analyzed by 12% SDS-PAGE electrophoresis (Laemmli, 1970). The cells were incubated for 18 h after induction.
An ultrasonic homogenizer (Sonopuls HD3200; Bandelin Electronic GmbH, Berlin, Germany) was fitted with a KE76 probe and used to disrupt the cells. To increase RrADH solubility during sonication, the effect of adding 0.7 M D-trehalose to the sonication buffer was analyzed. The bacterial pellet was kept on ice, resuspended in 30 mL sonication buffer (0.5 M NaCl, 50 mM Tris HCl (pH 8), and 0.7 M D-trehalose) and disrupted by 10–15 cycles of 30 s sonication and 30 s breaks. The lysate was centrifuged at 7,690 × g for 15 min. A supernatant (soluble fraction) sample was removed for PAGE analysis. The pellet (insoluble fraction) was resuspended in sonication buffer plus 2% SDS (w/v), shaken for 1 h, and centrifuged at 7,690 × g for 1–2 h. The supernatant fractions were pooled and their soluble protein content was quantified. RrADH was purified by IMAC affinity chromatography in an ÄKTA Avant 25 system (GE Healthcare, Little Chalfont, UK) with a 5-mL HisTrap FF column (GE Healthcare, Little Chalfont, UK) equilibrated by Ni2+ ions. The disrupted cell mixture was centrifuged at 7,690 × g and 4°C for 2 × 30 min to remove cell debris. The HisTrap FF column was washed with 2–5 column volumes (CV) water and 2–5 CV equilibration buffer (50 mM Tris HCl (pH 8) and 0.5 M NaCl). The samples were directly loaded onto the column at 0.1–1 CV/min pump speed. The column was washed with 10–15 CV equilibration buffer to remove non-specifically bound material. Then 2–5 CV elution buffer (50 mM Tris HCl (pH 8), 0.5 M NaCl, and 0.5 M imidazole) was used to elute the RrADH. Samples were drawn by an automatic fraction collector and analyzed by SDS-PAGE. GelAnalyzer v. 19.1 (Lazar et al.) was used in densitometric analysis to determine RrADH expression and/or its soluble:insoluble ratio during purification.
The eluted purified protein was mixed with glycerol to a final concentration of 50% (v/v) and stored at −20 °C. The pooled elution fractions were dialyzed against storage buffer (10 mM Tris HCl (pH 8), 100 mM NaCl, and 50% glycerol (v/v) using a 10 kDa MW cutoff membrane. Protein concentration was measured by the Bradford assay (Bradford, 1976).
Enzymes were immobilized according to a previously described method (Mateo et al., 2001). Eupergit-CM (100 mg) was mixed with 2 mL of a solution containing 0.1 M boric acid and 2 M iminodiacetic acid. The solution pH was adjusted to 8.5 with 5 M NaOH. The solution was placed in a sealed dark vial and shaken at 200 rpm on an orbital shaker at room temperature. After 2 h, the support was washed with an excess of deionized water, mixed with 2 mL of 0.05 M sodium phosphate buffer solution (pH 7.0) containing 0.1 M NiCl2 and 1 M NaCl, and agitated at 200 rpm and room temperature for 2 h. The Ni2+ carrier was washed with deionized water and used in enzyme immobilization.
The Ni2+ carrier was quantitatively transferred from the frit to a vial containing 1 mL alcohol dehydrogenase solution, 10 mM Tris HCl (pH 8), 100 mM NaCl, 50% glycerol (v/v), and 2 mL of 0.1 M phosphate buffer (pH 7.4) and shaken at 200 rpm and room temperature for 24 h. The immobilized enzyme preparation was filtered on a frit, washed with 0.1 M phosphate buffer (pH 7.4), and stored in it. The Bradford assay confirmed that in the case of ScADH1 99% of the protein was bound to the carrier and the protein content on the carrier was 25 mg/g. The activity measurement showed a 20.0% activity yield relative to the total ADH activity in the immobilization solution. The immobilization yield of RrADH was lower due to the lower purity of the enzyme preparation and it was 30 % providing the protein content on the carrier of 10 mg/g and the ADH activity yield of 20% relative to the enzyme activity in the solution used for the immobilization.
The immobilization of format dehydrogenase was carried out in the same way. Two milliliters of the enzyme solution (in 10 mmol/L Tris.HCl pH 8, 100 mmol/L NaCl, 50 % glycerol) were used undiluted. The immobilized enzyme preparation was then washed with 0.1 M phosphate buffer (pH 8) and stored in it. The Bradford assay confirmed that 83% of the protein was bound to the carrier and the protein content on the carrier was 20 mg/g. The activity measurement showed a 21.1% activity yield relative to the total FDH activity in the immobilization solution.
The immobilized enzyme preparations were packed in individual small peek columns (2.1 mm i.d. × 50 mm length; VICI AG International, Schenkon, Switzerland) and used in the subsequent activity and biotransformation measurements.
RrADH and FDH activity levels were determined by continuous spectrophotometry at 340 nm and 25°C. Enzymatic assays were performed in 1 mL reaction mixtures. To determine the oxidation activity of RrADH, isopropanol was the substrate and NAD was the cofactor. The reagent concentrations were 100 mM isopropanol, 100 mM sodium phosphate buffer (pH 7.0–8.0 in 0.2-unit increments), 1.5 mM NAD, and 0.9 μg/mL RrADH. The reduction reaction system consisted of 100 mM Na+ phosphate buffer (pH 7.0–8.0 in 0.2-unit increments), 100 mM acetone, 0.16 mM NADH, and 0.9 μg/mL RrADH. Absorbance at OD340 corresponding to the NADH concentration was monitored for 5 min. One unit (1 U) RrADH activity was defined as the amount of enzyme converting 1 μmol isopropanol to acetone in 1 min in the presence of NAD at pH 7.2 and 25°C.
FDH activity was determined under the same conditions as that for RrADH except the reaction mixture comprised 100 mM Na+ phosphate buffer (pH 8), 100 mM sodium formate, 1.5 mM NAD, and 5.84 μg/mL FDH. One unit (1 U) FDH was defined as the amount of enzyme converting 1 μmol formate to CO2 in 1 min in the presence of NAD at pH 7 and 37°C (Bergmeyer, 1974).
The biotransformation was conducted in a 1 mL reaction volume and the mixture consisted of 100 mM Na+ phosphate buffer (pH 7.2), 100 mM sodium formate, 1.5 mM NAD, 8.33 mM acetophenone, 0.9/9 μg/mL RrADH (in 1 μL and 10 μL solution), and 5.84/58.4 μg/mL FDH (in 1 μL and 10 μL solution). The mixture containing all reagents except RrADH was incubated in a Multitron Standard shaker (Infors HT GmBH, Bottmingen, Switzerland) at 30°C for 30 min allowing the FDH to reduce NAD to NADH. RrADH was then added and the 50 μL samples were regularly removed. The samples were diluted in 200 μL acetone to denature the enzymes and analyzed by gas chromatography as described below.
For testing the ScADH1 activity a substrate solution was used, containing 0.1 mol/L phosphate buffer solution of pH 7.4 with 0.17 mmol/L of NADH and 4 mmol/L of trans-2-hexenal. Alternatively, the substrate solution contained the same buffer and 1.7 mmol/L of NAD and 25 mmol/L of ethanol.
The immobilized enzyme column was placed in a water bath at 30°C and the substrate solution was pumped through the column with a peristaltic pump at a flow rate of 0.9 mL/min. Samples were taken at the column output and NADH concentrations were measured by spectrophotometry at 340 nm. A molar extinction coefficient of 6.22/cm/mM was used to calculate the NADH concentration.
ScADH1-catalyzed bioreduction of trans-2-hexenal to trans-2-hexenol was examined in an infinite recirculation system (Figure 1). The system was fitted with two separate columns containing immobilized ADH and FDH and immersed in a water bath at 30°C. The total void volume of the columns with connection capillaries was 2 mL. The stirred glass-jacketed vessel initially contained 18 mL of 0.1 M phosphate buffer (pH 7.4), 18 mM sodium formate, trans-2-hexenal at various concentrations, or other substances. The solution was pumped by a peristaltic pump through the immobilized enzyme columns at a flow rate of 2 mL/min and returned to the stirred vessel. After flow rate and temperature stabilization, the reaction was initiated by adding 2 mL of 17 mM NAD. During the reaction, samples were withdrawn from the vessel and analyzed for NADH, trans-2-hexenal, and trans-2-hexenol concentrations.
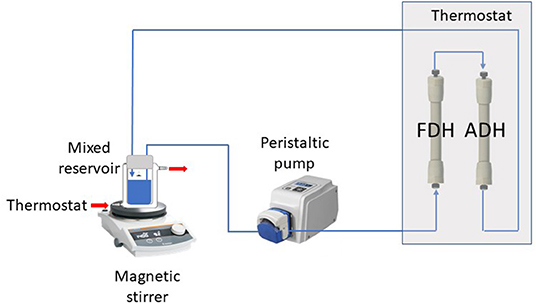
Figure 1. Experimental two-enzyme system with infinite recirculation.
The procedure described previously was applied for acetophenone reduction with RrADH. The initial acetophenone concentration was 8.33 mM or 1 g/L.
Before the analysis, reaction mixture samples were diluted fourfold with acetone (Chromasolv for GC; Sigma-Aldrich Corp., St. Louis, MO, USA). Gas chromatography (GC) measurements were performed on a 6890N gas chromatograph with a flame ionization detector (FID) (Agilent Technologies, Santa Clara, CA, USA). The injection port was maintained at 260°C and a 1-μL sample was injected at a 10:1 split ratio. The samples were separated with a 30 m (L) × 0.25 mm (ID) capillary column coated with a 0.25-μm film (df) of 5% phenylmethylpolysiloxane. The stationary phase was HP-5 (Agilent Technologies, Santa Clara, CA, USA). The column temperature was 80°C for the first min, increased to 130°C at a ramp of 7°C/min, and held at 130°C for 2 min. The carrier gas was He and the flow rate was 1.9 mL/min. The FID temperature was 250°C. Data were processed with Agilent ChemStation v. B.04.03. (Agilent Technologies, Santa Clara, CA, USA).
The enantiomeric purity of the 1-phenylethanol was determined by GC using CYCLOSIL-B (Agilent Technologies, Santa Clara, CA, USA) fitted with a chiral capillary column (30 m (L) × 0.25 mm (ID) × 0.25 μm df). One microliter sample was injected at a 10:1 split ratio. The carrier gas was He and the flow rate was 2.7 mL/min. The column temperature range was 80–240°C and the ramp was 15°C/min. The injector and FID temperatures were 260°C and 250°C, respectively.
To establish Zn2+ cation release from the immobilized ADH, the Zn2+ ion concentration in the column eluent was determined by complexation with dithizone (Song et al., 1976). Fifty milligrams of the carrier with immobilized ADH were placed in two vials containing 1 mL of 0.1 M phosphate buffer (pH 7.4). One vial contained 1.6 mg/mL trans-2-hexenal. The vials were gently agitated for 30 min. The immobilized enzyme particles were removed by filtration and 100 mL of 1 mg/mL dithizone in 0.1 M NaOH was added to each sample. The samples were measured spectrophotometrically at 555 nm against a blank containing phosphate buffer solution mixed with dithizone solution. The zinc concentration was interpolated from a standard curve plotted using various Zn2+ concentrations.
Rhodococcus ruber sadh was expressed in the E. coli expression strains BL21(DE3), C41(DE3), C43 (DE3), Rv308ai, SHuffle T7®, and LEMO21(DE3). The highest sadh expression level ( ≤ 15% of total soluble proteins) was determined for LEMO21(DE3) incubated in TB medium at 28°C according to the SDS-PAGE analysis (Figure 2A) and western blot (Figure 2B). However, most of the protein forming the homotetramer (MW 144.77 kDa) was expressed in inclusion bodies and was, therefore, inactive. After adding D-trehalose to the sonication solution, the soluble fraction markedly increased (Figure 3). Thus, 45–50 mg purified protein was obtained from 1 L culture medium and purity was increased 2.5 × . Subsequent co-expression experiments with chaperone GroEL and 8 M urea during purification did not dramatically improve protein solubility (data not shown).
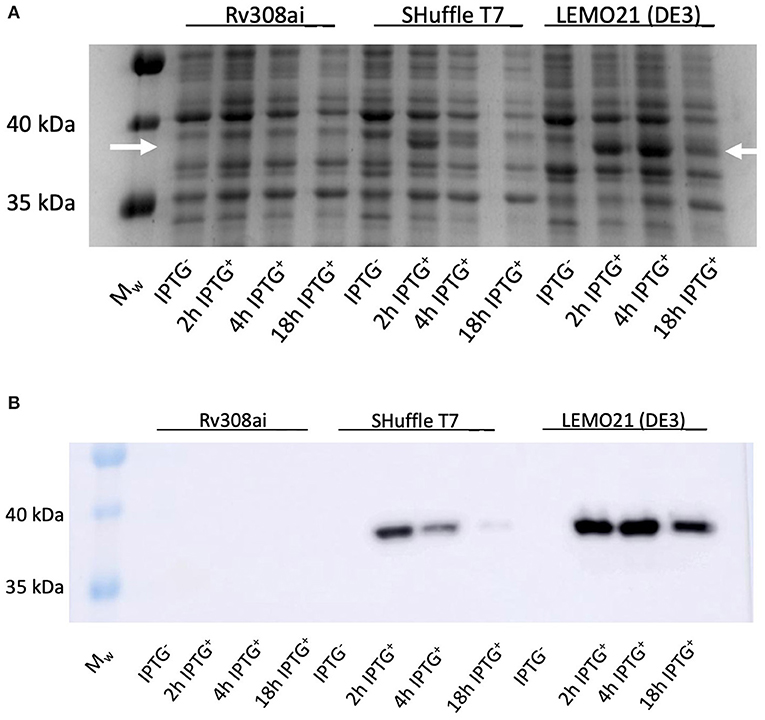
Figure 2. RrADH 50 mL expression in E. coli strains Rv308ai, Shuffle T7, LEMO21 (DE3) in TB medium at 37°C analyzed by SDS-PAGE (A) and Western Blot (B). Mw – molecular weight marker; IPTG − - non-induced expression; 2 h IPTG + - 2 h after induction; 4 h IPTG + - 4 h after induction; 18 h IPTG + - 18 h after induction (arrows indicate the approximate position of RrADH).
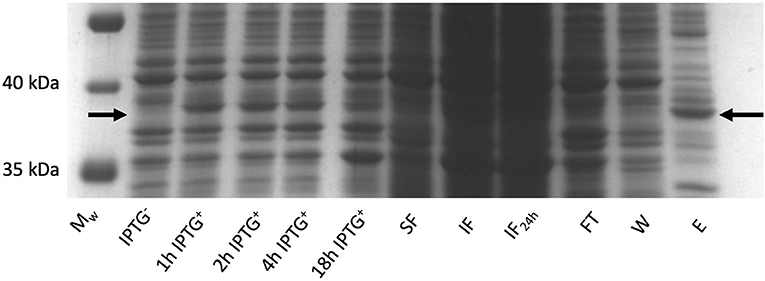
Figure 3. RrADH expression in E. coli LEMO21 (DE3) strain in 1 liter (bioreactor) of TB medium at 28°C analyzed by SDS-PAGE. Mw – protein ladder; IPTG − - before induction with IPTG; 1 h IPTG + - 1 h after induction; 2 h IPTG + - 2 h after induction; 4 h IPTG + - 4 h after induction; 18 h IPTG + - 18 h after induction; SF – soluble fraction; IF – insoluble fraction; IF24 – insoluble fraction incubated in sonication buffer with addition of 2 % SDS (w/v) for 24 h; FT – IMAC flow-through; W – IMAC wash; E – IMAC elution fraction.
The specific activity of RrADH during purification and in the final preparations was determined by measuring isopropanol oxidation. The rate was always in the range of 1–8 U/mg. The final enzyme preparation was characterized in terms of the pH-activity profile that increased in the examined interval of pH values (Figure 4). The pH-activity profile for acetophenone reduction catalyzed by RrADH exhibited the opposite trend with a sight maximum at pH ~7.2. Hence, pH 7.2 was selected for the preliminary trials on acetophenone bioreduction under soluble enzyme conditions.
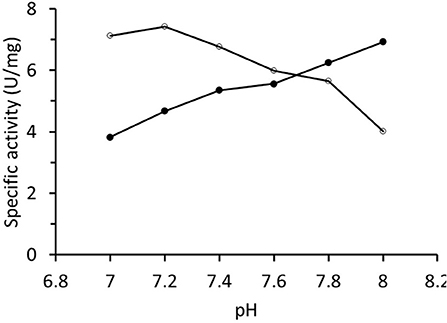
Figure 4. pH-activity profile of recombinant alcohol dehydrogenase from Rhodococcus ruber determined for isopropanol oxidation (∙) and acetone reduction (°).
Freshly prepared RrADH was used for bio-reduction of acetophenone to 1-phenylethanol. The reaction catalyzed by recombinant RrADH was coupled with NADH regeneration by recombinant formate dehydrogenase reaction. Four different enzyme concentrations were tested to identify the conditions required for maximum acetophenone reduction to 1-phenylethanol. According to the results shown in Figure 5, under optimum conditions ACF conversion was >90% whereas it was <10% at suboptimal enzyme concentrations. The reaction kinetics at the optimal enzyme concentrations (9 μg/mL RrADH and 58.4 μg/mL FDH) are shown in Figure 6. The reaction course was followed for 6 days. In the first 3 days, ACF conversion was nearly 90%. RrADH had very high specificity; enantiomeric GC revealed that under all reaction conditions, RrADH produced 100% S-1-phenylethanol.
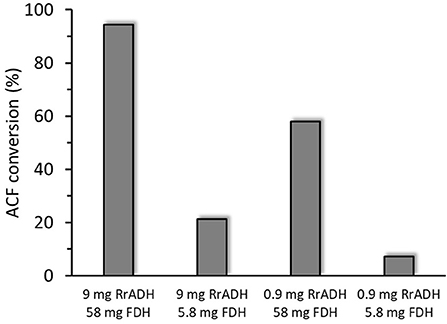
Figure 5. Bioreduction of ACF to 1-phenylethanol in soluble RrADH and FDH system at variable amounts of enzyme in 1 mL of the reaction mixture.
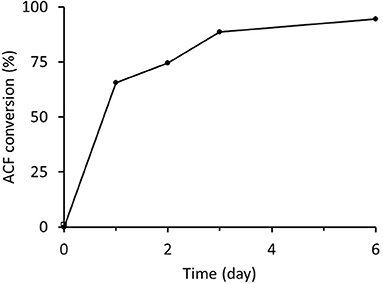
Figure 6. Bioreduction of ACF to 1-phenylethanol in soluble RrADH and FDH system.
The principal aim of this study was to prepare and immobilize recombinant enzymes to construct two-enzyme biocatalytic systems that reduce aliphatic and aromatic carbonyl compounds to their corresponding alcohols. The first system was based on ADH from Saccharomyces cerevisiae known as ScADH1 that is suitable for aliphatic aldehyde reduction. It is a homotetramer with a total molecular weight of 149.54 kDa. Each monomer is 36.84 kDa. Experiments on immobilized ScADH1 showed that operational stability was a critical point in trans-2-hexenal reduction (Figure 7A). A breakthrough in NADH concentration was measured at the output of the small column packed with immobilized ScADH1 after switching from buffer to reaction mixture containing trans-2-hexenal and NADH. The concentration of the latter was measured up to quasi-steady-state conditions. Nevertheless, the actual steady state was never reached. The output NADH concentration progressively increased as its consumption rate declined in response to the loss of enzyme activity. In contrast, when ScADH1 was used to catalyze ethanol oxidation, steady-state output NADH concentration was reached (Figure 7B).
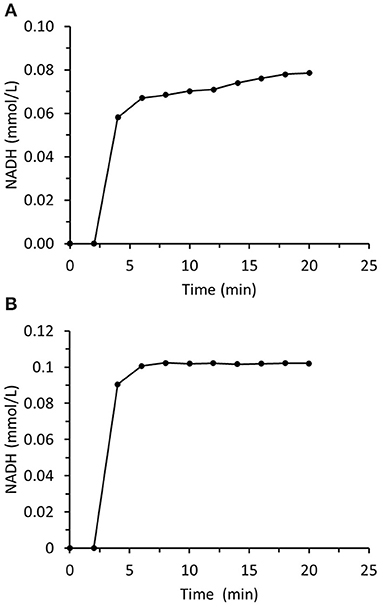
Figure 7. Time dependence of NADH concentration in the output of the column packed with immobilized ScADH1. The column was fed with the substrate solution containing (A) −0.17 mmol/L of NADH and 4 mmol/L of trans-2-hexenal; (B) −1.7 mmol/L of NAD and 25 mmol/L of ethanol.
Figure 8 summarizes data for experiments wherein the inlet trans-2-hexenal concentration was varied. The rate of loss of enzyme activity increased with trans-2-hexenal concentration. An analogous experiment involving trans-2-hexenol oxidation presented no enzyme activity loss (Figure 9).
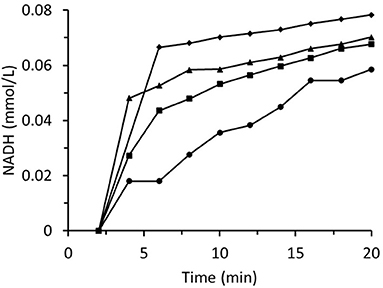
Figure 8. Time dependences of output NADH concentration for the column packed with immobilized ScADH1 and fed with 0.17 mmol/L of NADH solution containing variable inlet trans-2-hexenal concentrations (mg/mL): ♦ - 0.2; ▴ - 0.4; ■ - 0.8; ∙ - 1.6.
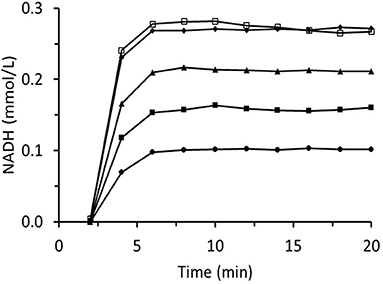
Figure 9. Time dependences of output NADH concentration for the column packed with immobilized ScADH1 and fed with 0.17 mmol/L of NADH solution containing variable inlet trans-2-hexenol concentrations (mg/mL): ♦ - 0.2; ▴ - 0.4; ■ - 0.8; ∙ - 1.6; □ - 3.2.
These results could be explained by the role of Zn2+ cations in the enzyme structure (Hao and Maret, 2006). The reactions between the enzyme Cys residues and the aldehyde groups broke the bonds between the enzyme and zinc cations. As Zn2+ is a part of the active site of ScADH1, the reactions would automatically inactivate the enzyme. This hypothesis was tested by incubating immobilized ScADH1 in 1.6 mg/mL trans-2-hexenal. Then, the release of Zn2+ ions into the surrounding solution was analyzed. The zinc concentration was 2.4 mg/mL which corresponded to 67% of the amount of zinc originally bound to the enzyme molecules. Therefore, the aldehyde concentration in the reaction system must be minimized during biocatalysis.
The immobilized two-enzyme biocatalytic system for trans-2-hexenal reduction is illustrated in Figure 10. The NADH concentration was constantly low, which means that the limiting factor was FDH activity. In an experiment using sixfold greater FDH activity, higher NADH concentrations were reached and the trans-2-hexenol production rate was enhanced (Figure 11A). In this case, 88% conversion and a final 3.4 mM trans-2-hexenol concentration were achieved after 8 h. A fed-batch operation was also tested. The trans-2-hexenal was added incrementally to keep its concentration low and limit the effect of zinc cation elution from the immobilized ScADH1. A maximum trans-2-hexenol concentration of 5.5 mM (0.55 mg/mL) was attained by sequential addition of trans-2-hexenal (Figure 11B).
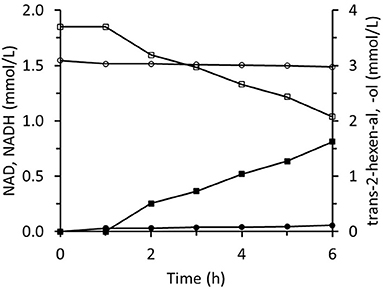
Figure 10. Reaction course in the infinite recycling system with FDA and ADH: ■ - trans-2-hexenol; □ - trans-2-hexenal; ∙ - NADH; ° - NAD.
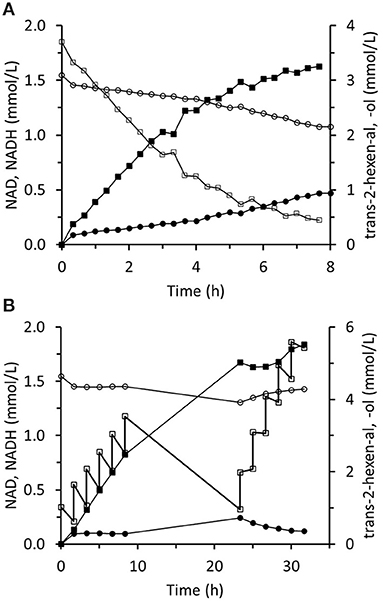
Figure 11. Reaction course in the infinite recycling system with increased FDH amount under batch operation (A) and fed-batch operation (B). ■ - trans-2-hexenol; □ - trans-2-hexenal; ∙ - NADH; ° - NAD.
The method of ScADH1-catalyzed trans-2-hexenal bioreduction was applied to acetophenone bioreduction using immobilized RrADH. The required NADH was regenerated during biotransformation via coupled formate oxidation catalyzed by immobilized FDH. Biotransformation in the infinite recirculation system during the first 8 h of reaction is depicted in Figure 12A. The 1-phenylethanol concentration reached 5.1 mM and corresponded to an ACF conversion of 61% and remained constant during the following 18 h. The experiment was repeated using the same immobilized enzymes and virtually the same results were obtained. This shows that the immobilized enzymes were stable, and the reaction was stopped by reaching equilibrium conditions. The immobilized enzyme preparations were regenerated by replacing the spent enzymes with fresh ones (Figure 12B). Immobilized RrADH and FDH were desorbed by pumping 1 M imidazole through packed columns. Enzyme solutions of the foregoing compositions and concentrations were immobilized on the carriers by recirculation through the columns for 24 h. The reaction course was tested, however, in this case with a three-fold reduced volume of the reaction mixture in a stirred tank to accelerate bioreduction. Figure 12B shows that this adjustment considerably increased the reaction rate, but the resulting ACF conversion remained at 61%.
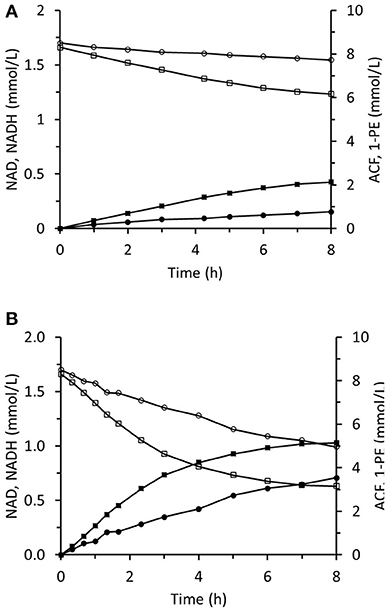
Figure 12. Reaction course in the infinite recycling system with FDA and RrADH: ■ - 1-phenylethanol; □ - acetophenone; ∙ - NADH; ° - NAD. The volume of reaction solution in mixed reservoir was 18 mL (A) and 6 mL (B).
Numerous enzymes of plant, microbial, and animal origin are used to prepare natural flavors by biocatalysis. The enzymes are classified according to the types of reactions they catalyze. Pertinent examples include oxidations and reductions of oxygen-bearing functional groups catalyzed by alcohol dehydrogenases (Paulino et al., 2021). There is a wide diversity of natural aromas bearing functional groups with different reactivities and they are used in industry. Bioreduction and bio-oxidation often require various types of alcohol dehydrogenases. However, alcohol dehydrogenase application in industrial production may be limited by high costs and low stability (Hummel, 1997). Hence, this study aimed to identify suitable alcohol dehydrogenases and economically efficient ways to prepare them by recombinant techniques. Here, we successfully prepared recombinant enzymes and applied them in two important bioreduction reactions. We used them to reduce the aliphatic unsaturated aldehyde trans-2-hexenal and the aromatic ketone acetophenone.
The main goal of expressing a biologically active protein is to achieve a high product yield. The E. coli system is usually the first choice in this objective as it can be easily manipulated, inexpensively cultured, and rapidly propagated. However, most E. coli strains only reach their highest production efficiency after several optimization steps. Therefore, a combination of maximum cell density and soluble protein production is the best way to obtain a high protein yield.
Alcohol dehydrogenase from Rhodococcus ruber was expressed in six different E. coli strains cultured in Luria-Bertani medium (LB), Terrific broth (TB), or Dynamite medium and at 20°C, 28°C, or 37°C. The LB medium differed from the other two in that it lacked glycerol and had a unique salt content. Ideal results were obtained with the BL21(DE3) and LEMO21(DE3) strains grown in TB medium at 28°C as previously described (Utekal et al., 2014; Levarski et al., 2018). Maximum RrADH expression with respect to soluble fraction content was detected 18 h after induction in LEMO21(DE3) (Figure 2). The addition of 0.7 M D-trehalose during cell homogenization improved protein solubility in vivo as previously described (Leibly et al., 2012). The yield of purified RrADH was 0.9 mg/mL after one-step IMAC chromatography and was determined by Bradford assay and SDS-PAGE densitometric analysis. Thus, enough enzymes could be prepared for the biotransformation experiments.
A kinetic analysis of native purified RrADH with isopropanol substrate indicated low specific activity (maximum 8 U/mg). Comparison of the pH-activity profiles of opposing reactions (oxidation/reduction) showed substantially different pH dependencies, namely, increasing for isopropanol oxidation and decreasing for acetone reduction (Figure 4). This observation was consistent with the pH-activity profiles for (S)-1-phenylethanol oxidation and acetophenone reduction reported for wild type ADH from Rhodococcus ruber (Hamnevik et al., 2014) and recombinant ADH from Rhodococcus erythropolis (Kasprzak et al., 2016). This behavior is not exceptional in ADH-catalyzed reactions. Markedly different pH profiles were observed for oxidation and reduction reactions catalyzed by Zn-dependent ADH from Chloroflexus aurantiacus (Loderer et al., 2018) and Aeropyrum pernix (Hirakawa et al., 2004).
The enzymes prepared herein were successfully tested in a one-pot reaction system for the bioreduction of acetophenone to 1-phenyletanol. There was nearly 95% acetophenone conversion to 1-phenylethanol in 6 days (Figure 5) and 100% of the product was the desired S-1-phenylethanol. Hence, this finding lays the foundation for the design and optimization of immobilized enzymatic biocatalysis systems.
The bioreduction capacity of an immobilized two-enzyme system comprising recombinant poly-His tagged Saccharomyces cerevisiae ADH1 and Candida boidinii FDH was tested using trans-2-hexenal as a substrate in a flow system with infinite recirculation (Figure 1). A similar experimental configuration was designed and tested for bioreduction and simultaneous cofactor regeneration (Zhou et al., 2021) using two enzymes co-immobilized on a packed bed column. The results of the present study demonstrated that co-immobilization is suboptimal when one of the enzymes is substantially less stable and must be replaced more frequently than the other. The aldehyde in the reaction medium diminished ScADH1 activity possibly because of the elution of Zn2+ ions vital to the enzyme (Yang and Zhou, 2001). The foregoing observations and the low stability of yeast ADH (Magonet et al., 1992; Leskovac et al., 2002; Bolivar et al., 2012) indicate that immobilized ADH must be regenerated more frequently than immobilized FDH. The use of two enzymes in separate columns enabled optimization of the biotransformation system by increasing the amount of immobilized FDH that limited overall bioreduction because of slow NADH regeneration. According to an earlier study (Zhou et al., 2021), adding an adsorption column for in situ product removal (ISPR) could efficiently shift the reaction equilibrium and increase trans-2-hexenol production. Yeast alcohol dehydrogenases effectively catalyze transformations of short-chain primary aliphatic alcohols and aldehydes (Dickinson and Monger, 1973; Green et al., 1993). However, they are relatively less effective on secondary alcohols and ketones when they have neighboring alkyl groups larger than the methyl group and are, therefore, sterically hindered (Dickinson and Dalziel, 1967). Considering this substrate specificity, the recombinant poly-His tagged ADH from Rhodococcus ruber was chosen instead of yeast ADH for the bioreduction of acetophenone to (S)-1-phenylethanol. Tests on both systems showed that the final bioreduction process did not fully consume the substrate and the conversion was <100%. To ameliorate system performance, future work must investigate the equilibrium and product inhibition phenomena that limit biotransformation. It is also necessary to identify the various pH optima for the oxidation and reduction reactions. Our studies on alcohol dehydrogenase from Rhodococcus (Figure 4) corroborate findings reported earlier (Hamnevik et al., 2014; Kasprzak et al., 2016). Bioreduction process improvement may be achieved by implementing hybrid systems combining reaction with in situ product removal, as they are promising ways to intensify biocatalytic processes (Schügerl and Hubbuch, 2005) (Woodley, 2017).
In terms of practical applications, there is a need for immobilization methods that reuse carriers after enzyme inactivation (Fraas and Franzreb, 2017). His-tagged enzymes combined with metal chelating supports offer the possibility to integrate enzyme purification and immobilization in a single step (Mateo et al., 2006). her advantage is the possibility to reuse the immobilization carrier after the enzyme inactivation (Fraas and Franzreb, 2017). In the present study, immobilized RrADH was desorbed and washed from the column with imidazole solution, replaced with fresh enzyme, and repeatedly used in subsequent acetophenone bioreduction batches.
In this study, an effective dual biotransformation system by combining recombinant alcohol dehydrogenase and formate dehydrogenase was created and used for aldehyde and ketone bio-reduction. The main strategy was based on the use of poly-His tagged enzymes that can be reversibly immobilized on poly-histidine on metal-chelated carriers. Separate enzyme immobilization instead of co-immobilization on the same support enables the regeneration of an individual immobilized enzyme by replacement after inactivation. The protocol designed and tested herein serves as a model for the development of alcohol dehydrogenase-based biocatalytic systems with NADH regeneration.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
SS, VŠ, JT, PF, VS, and MR: conceived and designed the experiments. VV, LM, ZL, ES, JB, and RK: performed the experiments. SS and VŠ: performed the data analyses. All authors contributed to the article and approved the submitted version.
This research was supported by Slovak Research and Development Agency grants (Nos. APVV-15-0466 and APVV-17-0333), ERDF projects: SmartFarm (No. ITMS2014: 313011V465), and ACCORD (No. ITMS: 313021X329).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Prof. Remus T. Dame of Leiden University for critical manuscript reading.
Bakonyi, D., Toelzer, C., Stricker, M., Hummel, W., Niefind, K., and Gröger, H. (2020). Expanding the application range of microbial oxidoreductases by an alcohol dehydrogenase from Comamonas testosteroni with a broad substrate spectrum and pH profile. Catalysts 10:1281. doi: 10.3390/catal10111281
Basle, M., Padley, H. A. W., Martins, F. L., Winkler, G. S., Jäger, C. M., and Pordea, A. (2021). Design of artificial metalloenzymes for the reduction of nicotinamide cofactors. J. Inorg. Biochem. 220:111446. doi: 10.1016/j.jinorgbio.2021.111446
Beitle, R. R., and Ataai, M. M. (1997). “Use of divalent metal ions chelated to agarose derivatives for reversible immobilization of proteins,” in Immobilization of Enzymes and Cells, ed. G. F. Bickerstaff (Totowa, NJ: Humana Press), 339–343. doi: 10.1385/0-89603-386-4:339
Böhmer, W., Kraus, T., and Mutti, F. (2018). Hydrogen-borrowing alcohol bioamination with coimmobilized dehydrogenases. ChemCatChem. 10, 731–735. doi: 10.1002/cctc.201701366
Bolivar, J. M., Rocha-Martín, J., Mateo, C., and Guisan, J. M. (2012). Stabilization of a highly active but unstable alcohol dehydrogenase from yeast using immobilization and post-immobilization techniques. Process Biochem. 47, 679–686. doi: 10.1016/j.procbio.2012.01.012
Bommarius, A. S., Schwarm, M., and Drauz, K. (1998). Biocatalysis to amino acid-based chiral pharmaceuticals – examples and perspectives. J. Mol. Cat. B Enzym. 5, 1–11. doi: 10.1016/S1381-1177(98)00009-5
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1006/abio.1976.9999
Brena, B. M., Rydén, L. G., and Porath, J. (1994). Immobilization of beta-galactosidase on metal-chelate-substituted gels. Biotechnol. Appl. Biochem. 19, 217–231.
Chapman, J., Ismail, A. E., and Dinu, C. Z. (2018). Industrial applications of enzymes: recent advances, techniques, and outlooks. Catalysts 8:238. doi: 10.3390/catal8060238
Dickinson, F. M., and Dalziel, K. (1967). Substrate specificity and stereospecificity of alcohol dehydrogenases. Nature 214, 31–33. doi: 10.1038/214031a0
Dickinson, F. M., and Monger, G. P. (1973). A study of the kinetics and mechanism of yeast alcohol dehydrogenase with a variety of substrates. Biochem. J. 131, 261–270. doi: 10.1042/bj1310261
Duman, Z. E., Duraksoy, B. B., Aktaş, F., Woodley, J. M., and Binay, B. (2020). High-level heterologous expression of active chaetomium thermophilum FDH in pichia pastoris. Enzyme Microb. Technol. 137:109552. doi: 10.1016/j.enzmictec.2020.109552
Fraas, R., and Franzreb, M. (2017). Reversible covalent enzyme immobilization methods for reuse of carriers. Biocatal. Biotransform. 35, 337–348. doi: 10.1080/10242422.2017.1344229
Franklin, R. D., Whitley, J. A., Caparco, A. A., Bommarius, B. R., Champion, J. A., and Bommarius, A. S. (2021). Continuous production of a chiral amine in a packed bed reactor with co-immobilized amine dehydrogenase and formate dehydrogenase. Chem. Eng. J. 407:127065. doi: 10.1016/j.cej.2020.127065
Gargouri, M., Akacha, N. B., and Legoy, M. D. (2004). Coupled hydroperoxide lyase and alcohol dehydrogenase for selective synthesis of aldehyde or alcohol. Appl. Biochem. Biotechnol. 119, 171–180. doi: 10.1385/ABAB:119:2:171
Green, D. W., Sun, H. W., and Plapp, B. V. (1993). Inversion of the substrate specificity of yeast alcohol dehydrogenase. J. Biol. Chem. 268, 7792–7798. doi: 10.1016/S0021-9258(18)53028-3
Hamnevik, E., Blikstad, C., Norrehed, S., and Widersten, M. (2014). Kinetic characterization of Rhodococcus ruber DSM 44541 alcohol dehydrogenase A. J. Mol. Cat. B Enzym. 99, 68–78. doi: 10.1016/j.molcatb.2013.10.023
Hanahan, D. (1983). Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557–580. doi: 10.1016/S0022-2836(83)80284-8
Hao, Q., and Maret, W. (2006). Aldehydes release zinc from proteins. a pathway from oxidative stress/lipid peroxidation to cellular functions of zinc. FEBS J. 273, 4300–4310. doi: 10.1111/j.1742-4658.2006.05428.x
Hatanaka, A. (1996). The fresh green odor emitted by plants. Food Rev. Int. 12, 303–350. doi: 10.1080/87559129609541083
Hawley, G. G., and Lewis, R. J. (1992). Hawley's Condensed Chemical Dictionary (New York: Van: Nostrand-Reinhold).
Hirakawa, H., Kamiya, N., Kawarabayashi, Y., and Nagamune, T. (2004). Properties of an alcohol dehydrogenase from the hyperthermophilic archaeon Aeropyrum pernix K1. J. Biosci. Bioeng. 97, 202–206. doi: 10.1016/S1389-1723(04)70191-7
Ho, L. F., Li, S. Y., Lin, S. C., and Hsu, W. H. (2004). Integrated enzyme purification and immobilization processes with immobilized metal affinity adsorbents. Process Biochem. 39, 1573–1581. doi: 10.1016/S0032-9592(03)00288-7
Hummel, W. (1997). “New alcohol dehydrogenases for the synthesis of chiral compounds,” in New Enzymes Org. Synth. Screen. Supply Eng. (Berlin, Heidelberg: Springer). 145–184. doi: 10.1007/BFb0103304
Karabec, M., Łyskowski, A., Tauber, K. C., Steinkellner, G., Kroutil, W., Grogan, G., et al. (2010). Structural insights into substrate specificity and solvent tolerance in alcohol dehydrogenase ADH-‘A' from Rhodococcus ruber DSM 44541. Chem. Commun. 46, 6314–6316. doi: 10.1039/c0cc00929f
Kasprzak, J., Bischoff, F., Rauter, M., Becker, K., Baronian, K., Bode, R., et al. (2016). Synthesis of 1-(S)-phenylethanol and ethyl (R)-4-chloro-3-hydroxybutanoate using recombinant Rhodococcus erythropolis alcohol dehydrogenase produced by two yeast species. Biochem. Eng. J. 106, 107–117. doi: 10.1016/j.bej.2015.11.007
Kozgus, O. G., Ece, S., Evran, S., Medine, E. I., Demirkol, D. O., Unak, P., et al. (2014). Isolation and immobilization of his-tagged alcohol dehydrogenase on magnetic nanoparticles in one step: application as biosensor platform. J. Macromol. Sci. A. 51, 699–705. doi: 10.1080/10601325.2014.936236
Krahulec, J., Hyrsov,á, M., Pepeliaev, S., Jílková, J., Cerný, Z., and Machálková, J. (2010). High level expression and purification of antimicrobial human cathelicidin LL-37 in Escherichia coli. Appl. Microbiol. Biotechnol. 88, 167–175. doi: 10.1007/s00253-010-2736-7
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. doi: 10.1038/227680a0
Leibly, D. J., Nguyen, T. N., Kao, L. T., Hewitt, S. N., Barrett, L. K., and Van Voorhis, W. C. (2012). Stabilizing additives added during cell lysis aid in the solubilization of recombinant proteins. PLoS ONE 7:e52482. doi: 10.1371/journal.pone.0052482
Leskovac, V., Trivic, S., and Anderson, B. M. (1999). Comparison of the chemical mechanisms of action of yeast and equine liver alcohol dehydrogenase. Eur. J. Biochem. 264, 840–847. doi: 10.1046/j.1432-1327.1999.00675.x
Leskovac, V., Trivić, S., and Peričin, D. (2002). The three zinc-containing alcohol dehydrogenases from baker's yeast, Saccharomyces cerevisiae. FEMS Yeast Res. 2, 481–494. doi: 10.1111/j.1567-1364.2002.tb00116.x
Levarski, Z., Blasko, J., Stuchlík, S., Frano, M., Bírová, S., Struharnanska, E., et al. (2018). Small scale biotransformation of food additive trans-2-hexenal to trans-2-hexenol by recombinant alcohol dehydrogenase and formate dehydrogenase produced in Escherichia coli. J. Food Nutr. Res. 57, 201–207.
Liese, A., Seelbach, K., and Wandrey, C. (2006). Industrial Biotransformations (Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA). doi: 10.1002/3527608184
Loderer, C., Wagner, D., Morgenstern, F., Spieß, A., and Ansorge-Schumacher, M. B. (2018). Discovery of a novel thermostable Zn2+-dependent alcohol dehydrogenase from Chloroflexus aurantiacus through conserved domains mining. J. Appl. Microbiol. 124, 480–490. doi: 10.1111/jam.13664
Long, Z., Kong, X., Zhang, C., Jiang, B., and Hua, Y. (2010). Purification and characterization of hydroperoxide lyase from amaranth tricolor (Amaranthus mangostanus L.) leaves. Eur. Food Res. Technol. 231, 865–871. doi: 10.1007/s00217-010-1337-0
Magonet, E., Hayen, P., Delforge, D., Delaive, E., and Remacle, J. (1992). Importance of the structural zinc atom for the stability of yeast alcohol dehydrogenase. Biochem. J. 287, 361–365. doi: 10.1042/bj2870361
Mateo, C., Fernández-Lorente, G., Cortés, E., Garcia, J. L., Fernández-Lafuente, R., and Guisan, J. M. (2001). One-step purification, covalent immobilization, and additional stabilization of poly-His-tagged proteins using novel heterofunctional chelate-epoxy supports. Biotechnol. Bioeng. 76, 269–276. doi: 10.1002/bit.10019
Mateo, C., Palomo, J. M., Fernandez-Lorente, G., Guisan, J. M., and Fernandez-Lafuente, R. (2007). Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol. 40, 1451–1463. doi: 10.1016/j.enzmictec.2007.01.018
Mateo, C., Pessela, B. C. C., Grazu, V., Torres, R., López-Gallego, F., Guisan, J. M., et al. (2006). “One-step purification, immobilization, and stabilization of poly-histidine-tagged enzymes using metal chelate-epoxy supports,” in Immobilization of Enzymes and Cells, ed. J. M. Guisan (Totowa, NJ: Humana Press), 117–128. doi: 10.1007/978-1-59745-053-9_11
Nelson, J. M., and Griffin, E. G. (1916). Adsorption of invertase. J. Am. Chem. Soc. 38, 1109–1115. doi: 10.1021/ja02262a018
Paulino, B. N., Sales, A., Felipe, L., Pastore, G. M., Molina, G., and Bicas, J. L. (2021). Recent advances in the microbial and enzymatic production of aroma compounds. Curr. Opin. Food Sci. 37, 98–106. doi: 10.1016/j.cofs.2020.09.010
Poltronieri, P., De Domenico, S., Bonsegna, S., and Santino, A. (2019). “Chapter 32 - oxylipins and green leaf volatiles: pplication of enzymes from plant origin to produce flavors and antifungal aldehydes,” in Enzymes in Food Biotechnology, ed. M. Kuddus (Cambridge, MA: Academic press), 551–567. doi: 10.1016/B978-0-12-813280-7.00032-3
Quaglia, D., Pori, M., Galletti, P., Emer, E., Paradisi, F., and Giacomini, D. (2013). His-tagged Horse Liver Alcohol Dehydrogenase: immobilization and application in the bio-based enantioselective synthesis of (S)-arylpropanols. Process Biochem. 48, 810–818. doi: 10.1016/j.procbio.2013.03.016
Schügerl, K., and Hubbuch, J. (2005). Integrated bioprocesses. Curr. Opin. Microbiol. 8, 294–300. doi: 10.1016/j.mib.2005.01.002
Shaaban, H., Mahmoud, K., Amin, A., and Banna, H. (2016). Application of biotechnology to the production of natural flavor and fragrance chemicals. Res. J. Pharm. Biol. Chem. Sci. 7, 2670–2717.
Shaked, Z. E., and Whitesides, G. M. (1980). Enzyme-catalyzed organic synthesis: NADH regeneration by using formate dehydrogenase. J. Am. Chem. Soc. 102, 7104–7105. doi: 10.1021/ja00543a038
Sharmeen, J. B., Mahomoodally, F. M., Zengin, G., and Maggi, F. (2021). Essential oils as natural sources of fragrance compounds for cosmetics and cosmeceuticals. Molecules 26:666. doi: 10.3390/molecules26030666
Song, M. K., Adham, N. F., and Rinderknecht, H. (1976). A simple, highly sensitive colorimetric method for the determination of zinc in serum. Am. J. Clin. Pathol. 65, 229–233. doi: 10.1093/ajcp/65.2.229
Suurmeijer, C. N. S. P., Pérez-Gilabert, M., an Unen, D. J., van der Hijden, H. T. W. M., Veldink, G. A., and Vliegenthart, J. F. G. (2000). Purification, stabilization and characterization of tomato fatty acid hydroperoxide lyase. Phytochemistry 53, 177–185. doi: 10.1016/S0031-9422(99)00504-X
Utekal, P., Tóth, C., Illésová, A., Koiš, P., Bocánová, L., Turna, J., et al. (2014). Expression of soluble Saccharomyces cerevisiae alcohol dehydrogenase in Escherichia coli applicable to oxido-reduction bioconversions. Biologia 69, 722–726. doi: 10.2478/s11756-014-0376-6
Vasić, K., Knez, Ž., and Leitgeb, M. (2020). Immobilization of alcohol dehydrogenase from Saccharomyces cerevisiae onto carboxymethyl dextran-coated magnetic nanoparticles: a novel route for biocatalyst improvement via epoxy activation. Sci. Rep. 10:19478. doi: 10.1038/s41598-020-76463-x
Wang, X., Saba, T., Yiu, H. H. P., Howe, R. F., Anderson, J. A., and Shi, J. (2017). Cofactor NAD(P)H regeneration inspired by heterogeneous pathways. Chem 2, 621–654. doi: 10.1016/j.chempr.2017.04.009
Weckbecker, A., Gröger, H., and Hummel, W. (2010). Regeneration of nicotinamide coenzymes: principles and applications for the synthesis of chiral compounds. Adv. Biochem. Eng. Biotechnol. 120, 195–242. doi: 10.1007/10_2009_55
Woodley, J. M. (2017). Bioprocess intensification for the effective production of chemical products. Comput. Chem. Eng. 105, 297–307. doi: 10.1016/j.compchemeng.2017.01.015
Yang, Y., and Zhou, H. M. (2001). Effect of zinc ions on conformational stability of yeast alcohol dehydrogenase. Biochemistry (Mosc) 66, 47–54. doi: 10.1023/A:1002829628836
Ying, X., Wang, Y., Xiong, B., Wu, T., Xie, L., Yu, M., et al. (2014). Characterization of an allylic/benzyl alcohol dehydrogenase from Yokenella sp. strain WZY002, an organism potentially useful for the synthesis of α,β-unsaturated alcohols from allylic aldehydes and ketones. Appl. Environ. Microbiol. 80, 2399–2409. doi: 10.1128/AEM.03980-13
Zhou, J., Wu, Y., Zhang, Q., Xu, G., and Ni, Y. (2021). Co-immobilized alcohol dehydrogenase and glucose dehydrogenase with resin extraction for continuous production of chiral diaryl alcohol. Appl. Biochem. Biotechnol. doi: 10.1007/s12010-021-03561-5
Keywords: biotransformation, green notes, recombinant protein production and purification, alcohol dehydrogenase, formate dehydrogenase, acetophenone reduction, immobilized enzyme regeneration
Citation: Varga V, Štefuca V, Mihálová L, Levarski Z, Struhárňanská E, Blaško J, Kubinec R, Farkaš P, Sitkey V, Turňa J, Rosenberg M and Stuchlík S (2021) Recombinant Enzymatic Redox Systems for Preparation of Aroma Compounds by Biotransformation. Front. Microbiol. 12:684640. doi: 10.3389/fmicb.2021.684640
Received: 25 March 2021; Accepted: 03 May 2021;
Published: 24 June 2021.
Edited by:
Laurent Dufossé, Université de la Réunion, FranceReviewed by:
Deniz Yildirim, Çukurova University, TurkeyCopyright © 2021 Varga, Štefuca, Mihálová, Levarski, Struhárňanská, Blaško, Kubinec, Farkaš, Sitkey, Turňa, Rosenberg and Stuchlík. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stanislav Stuchlík, c3RhbmlzbGF2LnN0dWNobGlrQHVuaWJhLnNr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.