
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 21 May 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.684092
This article is part of the Research TopicPopulation and Comparative Genomics of Plant Pathogenic BacteriaView all 16 articles
 Ling-Wei Weng1†Yu-Chen Lin2†Chiou-Chu Su3Ching-Ting Huang2Shu-Ting Cho2Ai-Ping Chen2Shu-Jen Chou2Chi-Wei Tsai1*Chih-Horng Kuo2*
Ling-Wei Weng1†Yu-Chen Lin2†Chiou-Chu Su3Ching-Ting Huang2Shu-Ting Cho2Ai-Ping Chen2Shu-Jen Chou2Chi-Wei Tsai1*Chih-Horng Kuo2*The bacterial genus Xylella contains plant pathogens that are major threats to agriculture in America and Europe. Although extensive research was conducted to characterize different subspecies of Xylella fastidiosa (Xf), comparative analysis at above-species levels was lacking due to the unavailability of appropriate data sets. Recently, a bacterium that causes pear leaf scorch (PLS) in Taiwan was described as the second Xylella species (i.e., Xylella taiwanensis; Xt). In this work, we report the complete genome sequence of Xt type strain PLS229T. The genome-scale phylogeny provided strong support that Xf subspecies pauca (Xfp) is the basal lineage of this species and Xylella was derived from the paraphyletic genus Xanthomonas. Quantification of genomic divergence indicated that different Xf subspecies share ∼87–95% of their chromosomal segments, while the two Xylella species share only ∼66–70%. Analysis of overall gene content suggested that Xt is most similar to Xf subspecies sandyi (Xfs). Based on the existing knowledge of Xf virulence genes, the homolog distribution among 28 Xylella representatives was examined. Among the 11 functional categories, those involved in secretion and metabolism are the most conserved ones with no copy number variation. In contrast, several genes related to adhesins, hydrolytic enzymes, and toxin-antitoxin systems are highly variable in their copy numbers. Those virulence genes with high levels of conservation or variation may be promising candidates for future studies. In summary, the new genome sequence and analysis reported in this work contributed to the study of several important pathogens in the family Xanthomonadaceae.
The gammaproteobacterium Xylella fastidiosa (Xf) is an insect-vectored plant pathogen that resides in plant xylem and is fastidious (Wells et al., 1987). To date, at least 563 plant species in 82 families have been reported as hosts for Xf (European Food Safety Authority, 2018). Xf could be classified into at least five subspecies; some of the notable examples include Xf subspecies fastidiosa (Xff) that causes Pierce’s disease (PD) of grapevine, Xf subspecies pauca (Xfp) that causes citrus variegated chlorosis (CVC) and olive quick decline syndrome (OQDS), and Xf subspecies sandyi (Xfs) that causes oleander leaf scorch (OLS). Because of their economic and ecological impacts, substantial resources have been devoted to related research. Notably, several large-scale studies were conducted to investigate the genomic diversity and evolution of Xf (Denancé et al., 2019; Potnis et al., 2019; Vanhove et al., 2019). Based on a comparison of 72 strains, the five Xf subspecies harbor high levels of genetic diversity (Vanhove et al., 2019). With an average gene content of ∼2,150 per strain, the core genome (i.e., genes shared by > 95% of the strains) contains only ∼900 genes, while the pangenome contains ∼10,000 genes. Moreover, although certain patterns of sequence divergence were found among those subspecies (Denancé et al., 2019), extensive recombination occurred at the levels of within- and between-subspecies (Potnis et al., 2019).
In contrast to the extensive genomic research at within-species level, comparative studies of Xf at higher taxonomic levels are lacking. Under the current taxonomy, Xylella belongs to the family Xanthomonadaceae and is most closely related to Xanthomonas (Rodriguez-R et al., 2012; Anderson et al., 2013). However, the genomic divergence between Xylella and Xanthomonas is very high in terms of chromosomal organization, gene content, and sequence variation. Thus, extracting biological insights from such comparisons is difficult. At within-genus level, Xf was largely considered as the only species within this genus since it was formally described in 1987 (Wells et al., 1987), which made between-species comparison infeasible. Intriguingly, a Xylella lineage that causes pear leaf scorch (PLS) in Taiwan was found to exhibit a slightly lower level of 16S rRNA gene sequence identity at 97.8–98.6% when compared to different subspecies of Xf (Su et al., 2012). In 2016, this PLS Xylella was formally reclassified as a novel species Xylella taiwanensis (Xt) based on a polyphasic approach (Su et al., 2016). Although a draft genome sequence of Xt was published earlier (Su et al., 2014), that draft assembly was produced with only ∼20-fold coverage of Roche/454 GS-FLX reads and is highly fragmented (i.e., 85 contigs; N50 = 121 kb). Moreover, no comparative analysis of gene content between Xt and Xf has been conducted.
To fill this gap, we determined the complete genome sequence of the type strain of Xt (i.e., PLS229T) for comparative analysis with its relatives. In addition to providing a genome-level overview of their diversity and evolution, we utilized the existing knowledge of Xff virulence genes and conducted detailed comparisons of virulence gene content among different Xylella lineages.
The strain was acquired from the Bioresource Collection and Research Centre (BCRC) in Taiwan (accession 80915). The procedures for genome sequencing and comparative analysis were based on those described in our previous studies (Lo et al., 2013; Lo et al., 2018; Cho et al., 2020). All bioinformatics tools were used with the default settings unless stated otherwise.
Briefly, the strain was cultivated on PD2 medium as described (Su et al., 2016) for DNA extraction using Wizard Genomic DNA Purification Kit (A1120; Promega, United States). For Illumina sequencing, a paired-end library with a target insert size of 550-bp was prepared using KAPA LTP Library Preparation Kit (KK8232; Roche, Switzerland) without amplification, then sequenced using MiSeq Reagent Nano Kit v2 (MS-103-1003; Illumina, United States) to obtain ∼50X coverage. For Oxford Nanopore Technologies (ONT) sequencing, the library was prepared using ONT Ligation Kit (SQK-LSK109) and sequenced using MinION (FLO-MIN106; R9.4 chemistry and MinKNOW Core v3.6.0) to obtain ∼228X coverage; Guppy v3.4.5 was used for basecalling. The raw reads were combined for de novo assembly by using Unicycler v0.4.8-beta (Wick et al., 2017). For validation, the Illumina and ONT raw reads were mapped to the assembly using BWA v0.7.12 (Li and Durbin, 2009) and Minimap2 v2.15 (Li, 2018), respectively. The results were programmatically checked using SAMtools v1.2 (Li et al., 2009) and manually inspected using IGV v2.3.57 (Robinson et al., 2011). The finalized assembly was submitted to the National Center for Biotechnology Information (NCBI) and annotated using their Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al., 2016).
A total of 40 genomes, including 27 from Xf, were used for comparative analysis (Table 1). Our taxon sampling mainly focused on the strains that could represent the known Xylella diversity (Vanhove et al., 2019). Two other Xanthomonadaceae genera were also included. For the closely related Xanthomonas, 10 species were selected to represent the key lineages (Parkinson et al., 2009; Rodriguez-R et al., 2012). For the distantly related Pseudoxanthomonas, only two species were sampled.
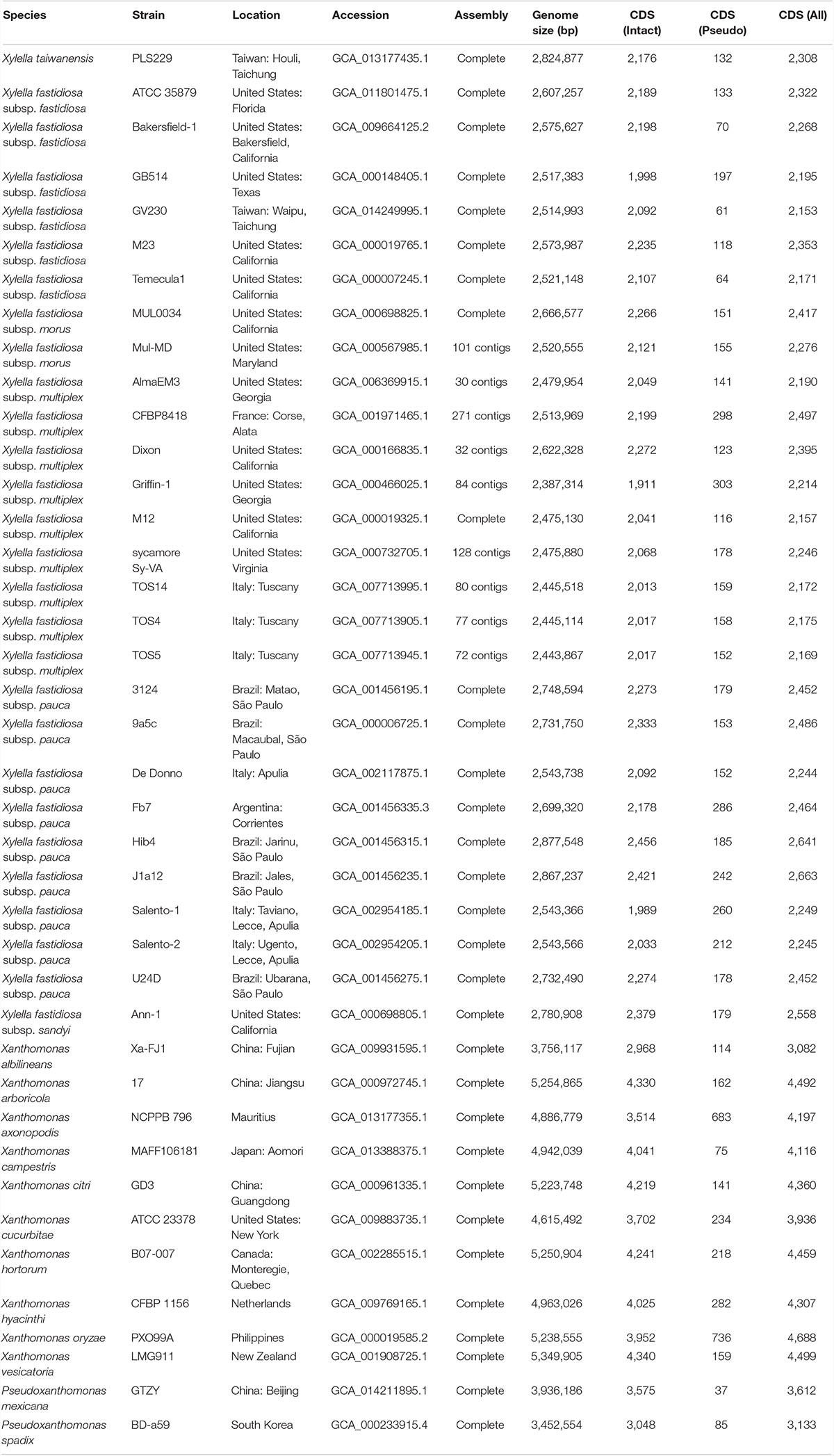
Table 1. List of the genome sequences analyzed.
Chromosomal level comparisons of nucleotide sequences were conducted using fastANI v1.1 (Jain et al., 2018). Homologous gene clusters were identified based on protein sequences using BLASTP v2.10.0 + (Camacho et al., 2009) and OrthoMCL v1.3 (Li et al., 2003). For gene content comparisons, the homolog clustering result was converted into a matrix of genomes by homolog clusters with the value in each cell corresponding to the copy number. This matrix was converted into a Jaccard distance matrix among genomes using VEGAN package v2.5-6 in R, then processed using the principal coordinates analysis function in APE v5.4 (Paradis and Schliep, 2019) and visualized using ggplot2 v3.3.2 (Wickham, 2016). For phylogenetic analysis, homologous sequences were aligned using MUSCLE v3.8.31 (Edgar, 2004). The maximum likelihood inference was performed using PhyML v.3.3.20180214 (Guindon and Gascuel, 2003); the proportion of invariable sites and the gamma distribution parameter were estimated from the data set and the number of substitute rate categories was set to four. The PROTDIST program of PHYLIP v3.697 (Felsenstein, 1989) was used to calculate sequence similarities.
Strain Xt PLS229T has one 2,824,877-bp circular chromosome with 53.3% G + C content; no plasmid was found. The annotation contains two complete sets of 16S-23S-5S rRNA genes, 49 tRNA genes, and 2,176 intact coding sequences (CDSs). This genome size is near the upper range of those Xf representatives (median: 2.54 Mb; range: 2.39–2.88 Mb) and much smaller compared to Xanthomonas spp. (median: 5.09 Mb; range: 3.76–5.35 Mb; Table 1). Among all 40 representative Xanthomonadaceae genomes, the genome sizes and the numbers of intact CDSs have a correlation coefficient of 0.989 (p < 2.2e–16). Compared to those Xf representatives with similar genome sizes (i.e., ∼2.73–2.88 Mb), such as those five Xfp strains from Brazil or the Xfs strain from the United States, the Xt PLS229T genome has fewer intact CDSs (i.e., 2,273–2,456 vs. 2,173) and fewer pseudogenes (i.e., 153–242 vs. 132). It is unclear if these observations were caused by annotation artifacts or have any biological meaning.
A total of 779 single-copy protein-coding genes were found to be shared by the 40 Xanthomonadaceae genomes compared (Table 1). Based on the concatenated alignment of the protein sequences derived from these genes, a robust maximum likelihood phylogeny was inferred (Figure 1). The availability of this Xt genome sequence provided a more appropriate outgroup to root the Xf phylogeny and further supported that Xfp is the basal lineage (Denancé et al., 2019; Potnis et al., 2019; Vanhove et al., 2019).
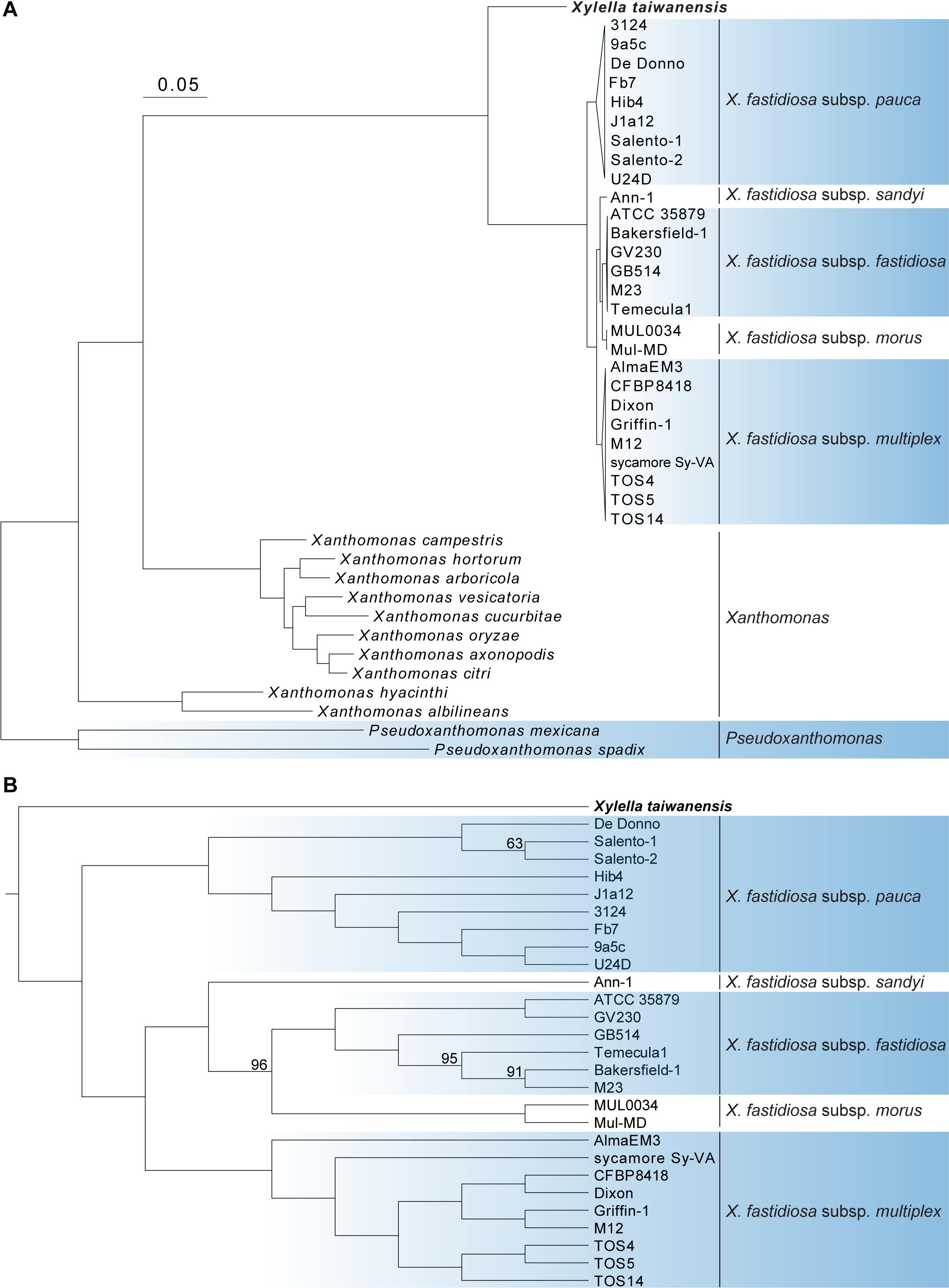
Figure 1. Molecular phylogeny of Xylella and related species in the family Xanthomonadaceae. The maximum likelihood phylogeny was based on 779 shared single-copy genes, the concatenated alignment contains 252,319 aligned amino acid sites. The genus Pseudoxanthomonas was included as the outgroup. (A) A phylogram for illustrating the relationships among all 40 genomes analyzed. Each of the X. fastidiosa subspecies was collapsed into a triangle for simplified visualization. All internal nodes illustrated in this phylogram received > 95% bootstrap support based on 1,000 replicates. (B) A cladogram for illustrating the relationships among those 28 Xylella genomes analyzed. Internal nodes with bootstrap values lower than 100% were labeled.
The genus Xanthomonas was known to be paraphyletic but the relationships of its two major clades (i.e., represented by Xanthomonas albilineans and Xanthomonas campestris, respectively) with Xylella were controversial (Pieretti et al., 2009; Rodriguez-R et al., 2012). With our genome-scale phylogeny, it is clear that Xylella is more closely related to X. campestris (Figure 1) and has experienced genome reduction since their divergence (Table 1).
When the genetic divergence was measured by overall nucleotide sequence conservation using fastANI (Jain et al., 2018), comparisons within each of the five Xf subspecies found that 88.8–99.8% of the chromosomal segments are shared and those segments have 98.5–100% average nucleotide identity (ANI) (Figure 2). For between-subspecies comparisons, 86.6–94.8% of the chromosomal segments are shared and those segments have 96.3–98.8% ANI. When those Xf subspecies were compared to Xt, only 66.4–70.3% of the chromosomal segments are shared and those segments have 82.9–83.4% ANI. These results are consistent with previous findings (Su et al., 2016; Denancé et al., 2019) and provide further support to the current taxonomy based on the 95% ANI threshold recommended for delineating bacterial species (Jain et al., 2018).
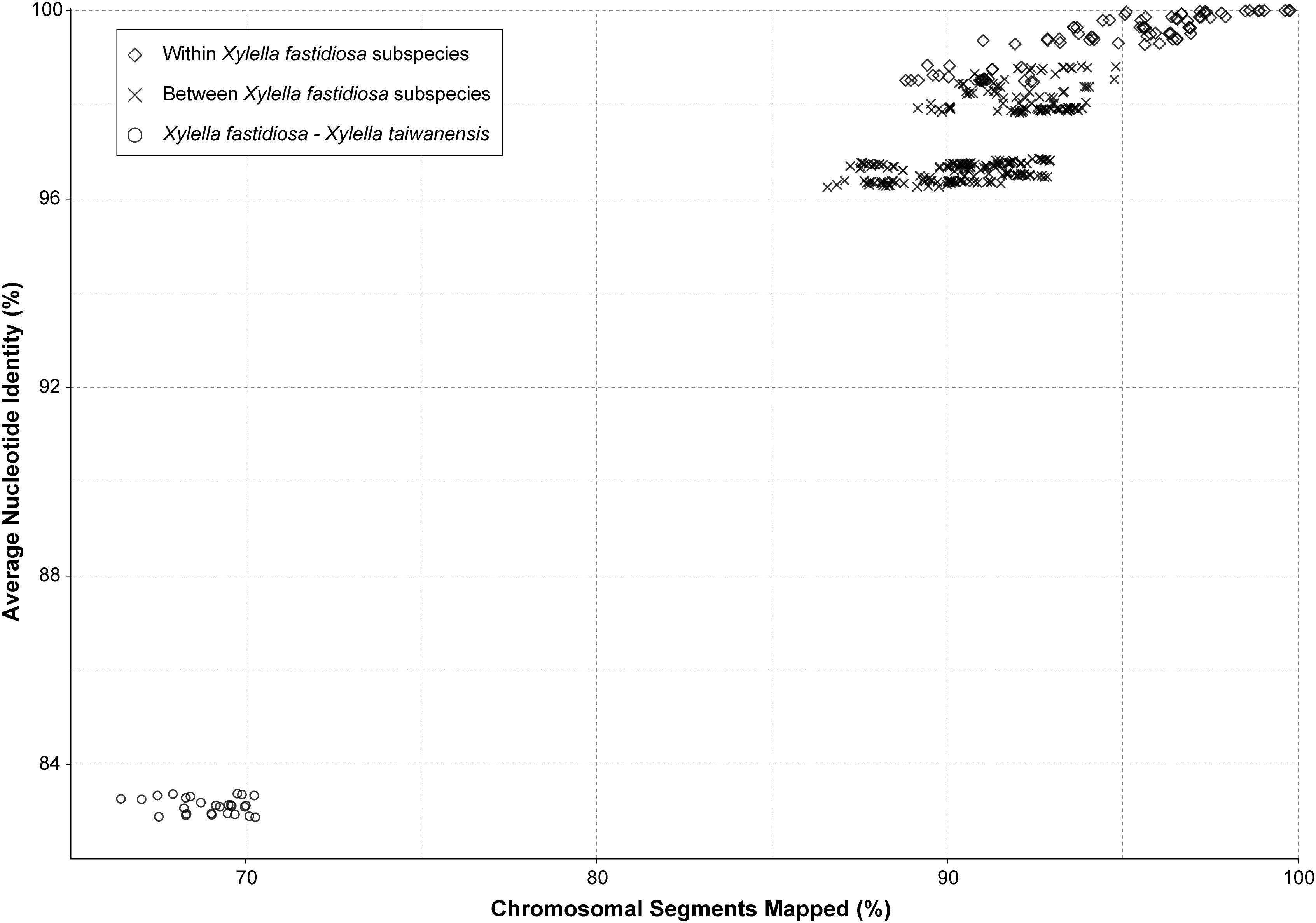
Figure 2. Genome similarity among the representative Xylella strains. The pairwise comparisons were classified into three categories: (1) within the same X. fastidiosa subspecies, (2) between different X. fastidiosa subspecies, and (3) between X. fastidiosa and X. taiwanensis.
Because the ANI approach provides low resolutions when the nucleotide sequence identity drops to ∼80% (Jain et al., 2018) and may not be appropriate for cross-genus comparisons, we also evaluated divergence based on the protein sequences of those 779 Xanthomonadaceae core genes. The two Xylella species have ∼88.8–89.1% protein sequence similarity, which is lower than the values observed in the comparisons among those eight X. campestris clade representatives (median: 93.8%; range: 92.6–97.2%), comparable to the X. albilineans-X. hyacinthi comparison (88.6%), and higher than the Pseudoxanthomonas mexicana-Pseudoxanthomonas spadix comparison (75.4%).
In addition to analysis of sequence divergence based on those 779 core genes (Figure 1) and the entire chromosomes (Figure 2), the divergence in gene content was also examined. The gene content comparisons were based on principal coordinates analysis that examines copy number variation among all homologous gene clusters in the entire pangenome and does not consider sequence divergence within each homologous gene cluster. When all 40 Xanthomonadaceae genomes were compared together based on their 11,455 homologous gene clusters, the grouping patterns (Figure 3A) are consistent with the phylogenetic clades inferred based on sequence divergence of the 779 core genes (Figure 1A). All 27 Xf genomes form a tight cluster (Figure 3A) despite their differences in the number of intact CDSs (range: 1,911–2,456; av. ± std. dev.: 2,156 ± 144; Table 1). In contrast, although the Xt genome has 2,176 intact CDSs, which is close to the average observed among those 27 Xf representatives, it does not fall into the Xf cluster (Figure 3A). This result indicates that the gene content divergence between these two Xylella species is much higher than the divergence among Xf subspecies. For the within-Xylella comparison based on 5,395 homologous gene clusters, the grouping patterns are consistent with the taxonomic assignments and Xt is most similar to Xfs (Figure 3B). It is interesting that Xt and Xfs are similar in having a narrow host range (i.e., Xt is restricted to pear and Xfs is mostly known for oleander infections), while other Xf subspecies can infect a wide range of hosts (Baldi and La Porta, 2017; European Food Safety Authority, 2018; Rapicavoli et al., 2018). However, it is also important to note that the host range information may be limited by sampling and experimental efforts. As more research results become available, this information may be updated. For example, Xfs-related strains have been reported to infect coffee (Jacques et al., 2016) and whether Xt can infect a wider range of plants remains to be investigated.
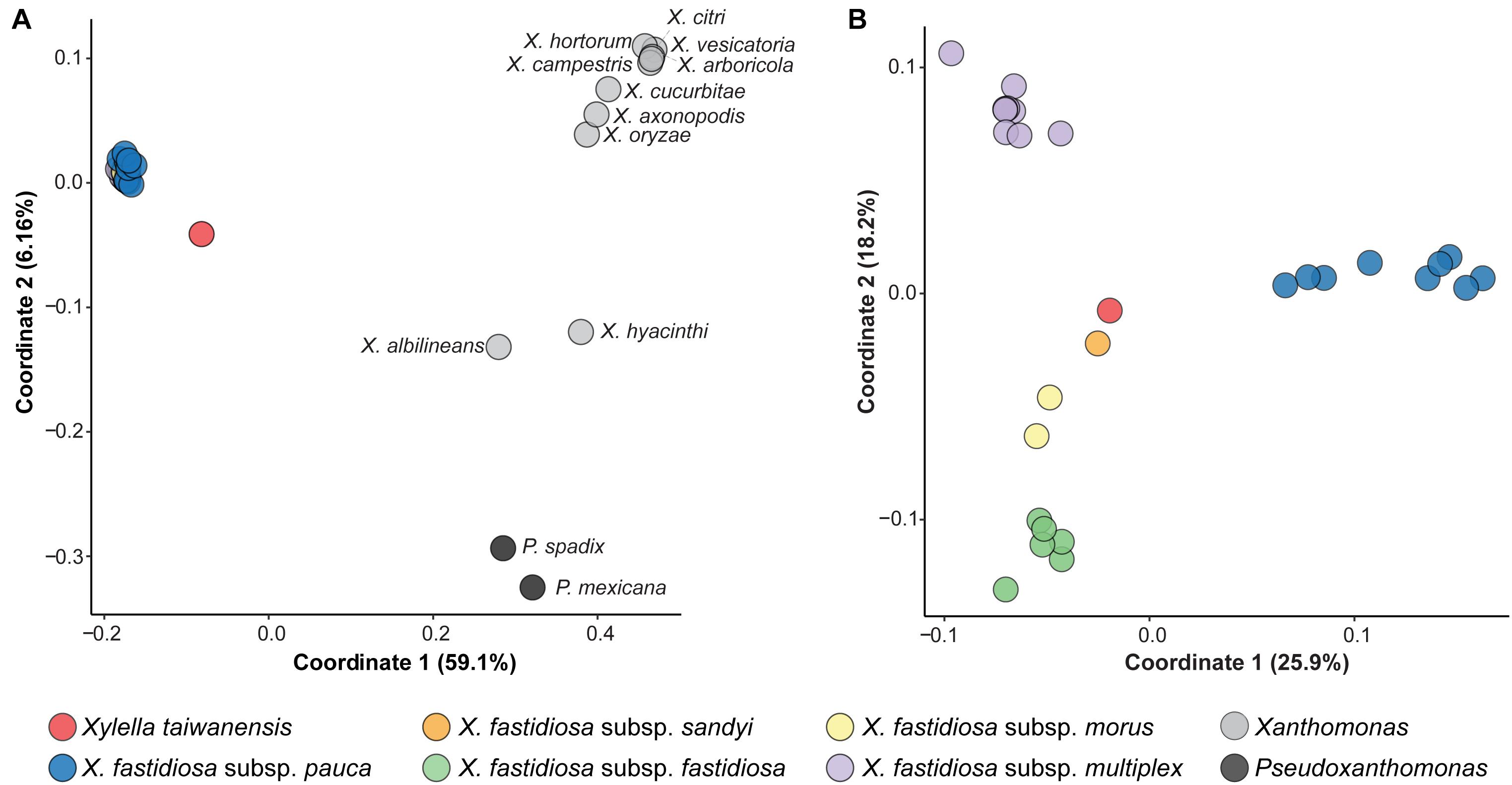
Figure 3. Principal coordinates analysis of gene content dissimilarity. The % variance explained by each coordinate was provided in parentheses. (A) Based on the 11,455 homologous gene clusters found among all 40 Xanthomonadaceae genomes analyzed in this work. (B) Based on the 5,395 homologous gene clusters found among the 28 Xylella genomes analyzed in this work.
Based on the current knowledge of putative virulence genes and pathogenicity factors identified in Xf, type IV pili (T4P) are important for twitching motility and movements within infected plants (Meng et al., 2005; Li et al., 2007; Burdman et al., 2011; Cursino et al., 2011; Hao et al., 2017). The Xylella T4P genes are organized into four major gene clusters with 25 homologs and highly conserved among the 28 representative genomes examined (Figure 4). Among these four T4P gene clusters, cluster II that corresponds to the pil-chp operon (Cursino et al., 2011; Hao et al., 2017) is the most conserved cluster with only two putative gene losses (i.e., chpB in Xfp Salento-1 and chpC in Xt). A notable gene absence is PD1925 in cluster IV, which encodes a hypothetical protein and is absent in all Xfp strains and Xt. The only gene duplication observed involves a tandem duplication of the cluster IV pilA2 homolog in Xt. Intriguingly, Xt PLS229T is the only strain that has two copies of pilA2 and no pilA1. These two type IV pilin paralogs were shown to have different functions in Xff, with pilA1 affecting pilus number and location while pilA2 is required for twitching (Kandel et al., 2018). It is unclear if this pilA2 duplication in Xt PLS229T can complement its lack of pilA1.
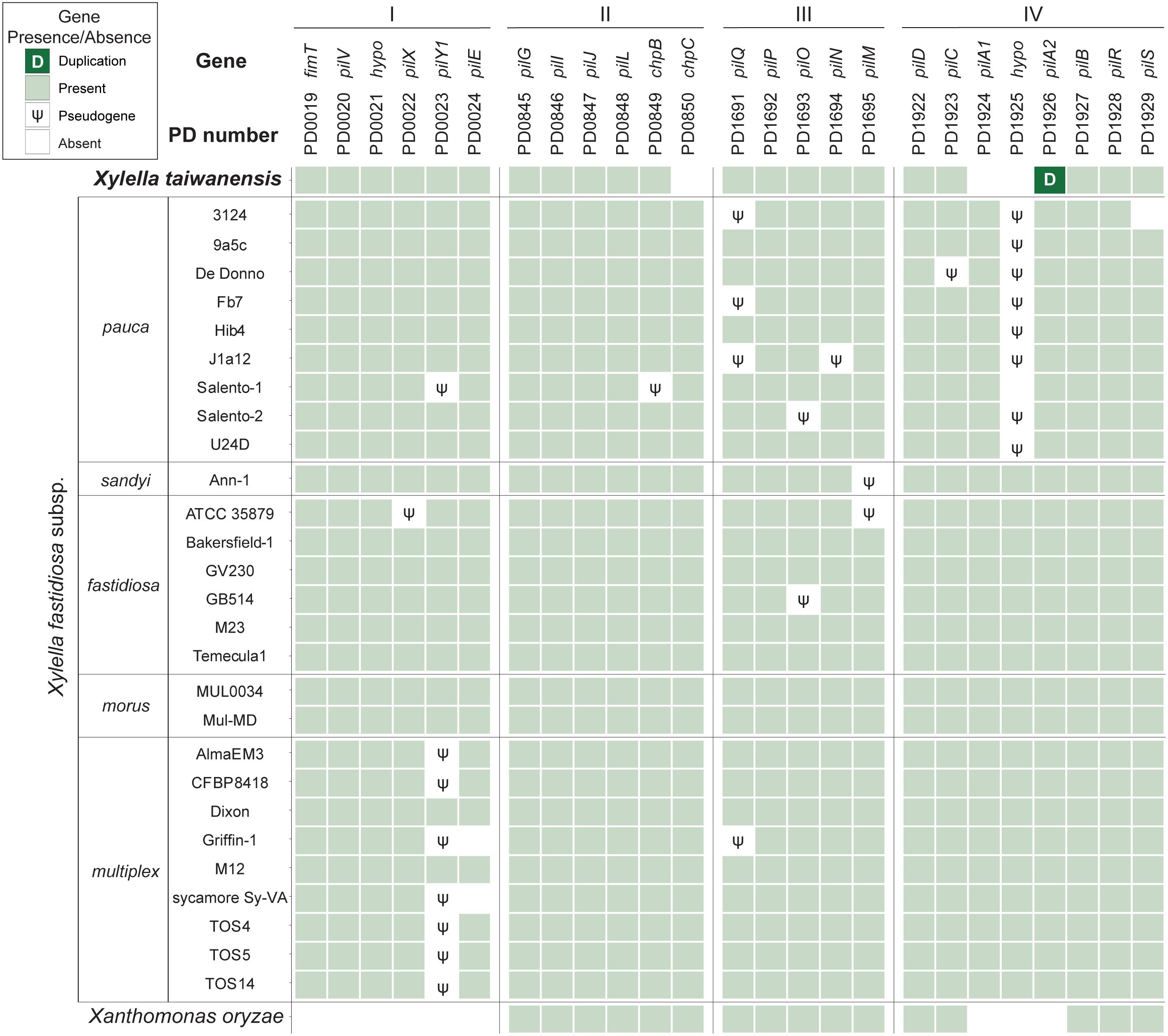
Figure 4. Distribution of the type IV pili genes found among representative Xylella genomes. Xanthomonas oryzae was included as the outgroup. The genes were identified by the PD numbers based on the annotation of Temecula1 genome; those with adjacent PD numbers are located in neighboring regions on the chromosome. A total of four gene clusters (labeled as I–IV) located in syntenic regions were found to be conserved. Gene names were provided when available; “hypo” indicates those annotated as encoding hypothetical proteins. Patterns of gene presence and absence were illustrated in the format of a heatmap. For gene absence, those with identifiable pseudogenes were labeled accordingly. One case of tandem gene duplication was observed for the PD1926 (pilA2) homolog in the Xylella taiwanensis genome.
In addition to the T4P genes, many other Xylella pathogenicity factors have been identified and these additional virulence genes may be classified into 10 major functional categories (Lee et al., 2014; Merfa et al., 2016; Chen et al., 2017; Rapicavoli et al., 2018; Ge et al., 2021). Among these categories, secretion systems and metabolism are the most conserved ones with no variation in gene copy number across all Xylella representatives (Figure 5). Additionally, those genes involved in regulatory systems are also highly conserved. In contrast, several genes related to adhesins, hydrolytic enzymes, and toxin-antitoxin systems are highly variable in copy numbers.

Figure 5. Distribution of putative virulence genes and pathogenicity factors among representative Xylella genomes. Xanthomonas oryzae was included as the outgroup. The homologous gene clusters were identified by the PD numbers based on the annotation of Temecula1 genome; gene names were provided when available. Gene copy numbers were illustrated in the format of a heatmap; values higher than two were labeled with the exact numbers. Two adhesin genes (i.e., PD1792 and PD2118) were assigned to the same homologous gene cluster and were combined for copy number calculation.
For more detailed examination, these putative virulence genes were classified into 38 homologous gene clusters and six are absent in the Xt genome (Figure 5). These include the genes that encode a putative adhesin (PD0986, hemagglutinin-like protein), two hydrolytic enzymes (PD0956, serine protease; PD1485, polygalacturonase), one pair of toxin-antitoxin (PD0370, motility quorum sensing regulator MqsR ribonuclease; PD0371, MqsA antitoxin), and another separate toxin (PD1100, endoribonuclease). Notably, the mqsR-mqsA toxin-antitoxin system genes (Lee et al., 2014; Merfa et al., 2016) are differentially distributed among those Xf subspecies. Homologs of these two genes are entirely conserved in all Xff and Xf subspecies morus strains, present in five out of the nine Xfp strains, and completely absent in Xfs and Xf subspecies multiplex.
Based on previous studies that characterized mutant phenotypes, PD0956 (Gouran et al., 2016) and PD1100 (Burbank and Stenger, 2017) are both antivirulence factors and the loss of either one resulted in hypervirulence of Xff in grapevines. Similarly, PD0370 is another antivirulence factor that reduces the virulence of Xfp against citrus when overexpressed (Merfa et al., 2016). In contrast, both PD0986 and PD1485 are critical for Xff virulence in grapevines. For PD0986, this gene is absent in a Xf biocontrol strain EB92-1 that can infect and persist in grapevines but causes only very slight symptoms. When PD0986 is cloned into EB92-1, the transformant induces significantly increased symptoms that are characteristic of PD (Zhang et al., 2015). For PD1485, the knockout mutant was avirulent due to the loss of ability to systemically colonize grapevines (Roper et al., 2007).
Two gene families appeared to have experienced copy number expansion in the Xt genome. The first family includes homologs of PD1792 and PD2118, which encode hemagglutinins. These adhesins are antivirulence factors that restrict in planta movement by promoting self-aggregation; transposon-insertion mutants of Xff PD1792 and PD2118 both exhibit hypervirulence in grapevines (Guilhabert and Kirkpatrick, 2005). Among the representative Xf and Xff genomes, the median copy numbers of this family are 3 and 8, respectively. In comparison, Xt has 12 copies. It remains to be investigated if the copy number variation is linked to protein expression level and virulence. The second family includes a Zot-like toxin (PD0928). Similar to PD0986 (hemagglutinin-like protein), the biocontrol strain EB92-1 lacks the homolog of PD0928 and the transformant that expresses this gene is virulent (Zhang et al., 2015).
In conclusion, this work reported the complete genome sequence of an important plant-pathogenic bacterium that is endemic to Taiwan. In addition to providing the genomic resource that contributes to the study of this pathogen, this species is the only known sister species of Xf, which has extensive genetic variations and devastating effects on agriculture worldwide. The availability of this new Xt genome sequence provides critical genomic information of a key lineage that may improve the study of Xylella evolution and the inference of Xf ancestral states. At above-genus level, our genome-scale phylogenetic inference resolved the relationships between Xylella and Xanthomonas, which are some of the key plant pathogens in the family Xanthomonadaceae.
For gene content analysis, our comparison of the putative virulence genes and pathogenicity factors among representative Xylella strains identified the genes that exhibit high levels of conservation or diversity (Figures 4 and 5). These genes are promising candidates for future functional studies to investigate the molecular mechanisms of Xylella virulence. Previous characterizations of single-gene mutants, particularly those conducted in Xff, have provided a strong foundation (Burdman et al., 2011; Rapicavoli et al., 2018). However, it is important to note that the current knowledge of Xylella virulence genes is mostly derived from those strains that are relatively easy to culture and transform, such that only limited diversity has been investigated in molecular genetics studies. Moreover, infection experiments for the investigation of gene functions were limited to a small number of plant species. For further improvements, experimental studies that examine more diverse Xylella lineages and plant hosts, as well as the combined effects of multiple virulence genes will be critical.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, CP053627, //www.ncbi.nlm.nih.gov/, SRR11805344, https://www.ncbi.nlm.nih.gov/, SRR11805345.
C-WT and C-HK: conceptualization, funding acquisition, project administration, and supervision. L-WW, Y-CL, and C-TH: investigation, validation, and visualization. C-CS, S-TC, A-PC, S-JC, and C-HK: methodology. C-CS, C-WT, and C-HK: resources. L-WW and C-HK: writing—original draft. L-WW, Y-CL, C-CS, C-TH, S-TC, A-PC, S-JC, C-WT, and C-HK: writing—review and editing. All authors contributed to the article and approved the submitted version.
The funding was provided by the Ministry of Science and Technology of Taiwan (MOST 106-2923-B-002-005-MY3) to C-WT and Academia Sinica to C-HK. The funders had no role in study design, data collection and interpretation, and or the decision to submit the work for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Illumina and Oxford Nanopore sequencing library preparation service was provided by the Genomic Technology Core (Institute of Plant and Microbial Biology, Academia Sinica). The Illumina MiSeq sequencing service was provided by the Genomics Core (Institute of Molecular Biology, Academia Sinica).
Anderson, I., Teshima, H., Nolan, M., Lapidus, A., Tice, H., Rio, T. G. D., et al. (2013). Genome sequence of Frateuria aurantia type strain (Kondô 67T), a xanthomonade isolated from Lilium auratium Lindl. Stand. Genomic. Sci. 9, 83–92. doi: 10.4056/sigs.4338002
Baldi, P., and La Porta, N. (2017). Xylella fastidiosa: host range and advance in molecular identification techniques. Front. Plant Sci. 8:944.
Burbank, L. P., and Stenger, D. C. (2017). The DinJ/RelE toxin-antitoxin system suppresses bacterial proliferation and virulence of Xylella fastidiosa in grapevine. Phytopathology 107, 388–394. doi: 10.1094/phyto-10-16-0374-r
Burdman, S., Bahar, O., Parker, J. K., and De La Fuente, L. (2011). Involvement of type IV pili in pathogenicity of plant pathogenic bacteria. Genes 2, 706–735. doi: 10.3390/genes2040706
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2105-10-421
Chen, H., Kandel, P. P., Cruz, L. F., Cobine, P. A., and De La Fuente, L. (2017). The major outer membrane protein MopB is required for twitching movement and affects biofilm formation and virulence in two Xylella fastidiosa strains. Mol. Plant Microbe Interact. 30, 896–905. doi: 10.1094/mpmi-07-17-0161-r
Cho, S.-T., Zwolińska, A., Huang, W., Wouters, R. H. M., Mugford, S. T., Hogenhout, S. A., et al. (2020). Complete genome sequence of “Candidatus Phytoplasma asteris” RP166, a plant pathogen associated with rapeseed phyllody disease in Poland. Microbiol. Resour. Announc. 9:e000760-20.
Cursino, L., Galvani, C. D., Athinuwat, D., Zaini, P. A., Li, Y., De La Fuente, L., et al. (2011). Identification of an operon, Pil-Chp, that controls twitching motility and virulence in Xylella fastidiosa. Mol. Plant Microbe Interact. 24, 1198–1206. doi: 10.1094/mpmi-10-10-0252
Denancé, N., Briand, M., Gaborieau, R., Gaillard, S., and Jacques, M.-A. (2019). Identification of genetic relationships and subspecies signatures in Xylella fastidiosa. BMC Genomics 20:239.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
European Food, and Safety Authority. (2018). Update of the Xylella spp. host plant database. EFSA J. 16:e05408.
Ge, Q., Cobine, P., and De La Fuente, L. (2021). The influence of copper homeostasis genes copA and copB on Xylella fastidiosa virulence is affected by sap copper concentration. Phytopathology doi: 10.1094/PHYTO-12-20-0531-R Online ahead of print.
Gouran, H., Gillespie, H., Nascimento, R., Chakraborty, S., Zaini, P. A., Jacobson, A., et al. (2016). The secreted protease PrtA controls cell growth, biofilm formation and pathogenicity in Xylella fastidiosa. Sci. Rep. 6:31098.
Guilhabert, M. R., and Kirkpatrick, B. C. (2005). Identification of Xylella fastidiosa antivirulence genes: hemagglutinin adhesins contribute to X. fastidiosa biofilm maturation and colonization and attenuate virulence. Mol. Plant Microbe Interact. 18, 856–868. doi: 10.1094/mpmi-18-0856
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Hao, L., Athinuwat, D., Johnson, K., Cursino, L., Burr, T. J., and Mowery, P. (2017). Xylella fastidiosa pil-chp operon is involved in regulating key structural genes of both type I and IV pili. Vitis 56, 55–62.
Jacques, M.-A., Denancé, N., Legendre, B., Morel, E., Briand, M., Mississipi, S., et al. (2016). New coffee plant-infecting Xylella fastidiosa variants derived via homologous recombination. Appl. Environ. Microbiol. 82, 1556–1568. doi: 10.1128/aem.03299-15
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114.
Kandel, P. P., Chen, H., and De La Fuente, L. (2018). A short protocol for gene knockout and complementation in Xylella fastidiosa shows that one of the type IV pilin paralogs (PD1926) is needed for twitching while another (PD1924) affects pilus number and location. Appl. Environ. Microbiol. 84:e01167-18.
Lee, M. W., Tan, C. C., Rogers, E. E., and Stenger, D. C. (2014). Toxin-antitoxin systems mqsR/ygiT and dinJ/relE of Xylella fastidiosa. Physiol. Mol. Plant Pathol. 87, 59–68. doi: 10.1016/j.pmpp.2014.07.001
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, Y., Hao, G., Galvani, C. D., Meng, Y., De La Fuente, L., Hoch, H. C., et al. (2007). Type I and type IV pili of Xylella fastidiosa affect twitching motility, biofilm formation and cell–cell aggregation. Microbiology 153, 719–726. doi: 10.1099/mic.0.2006/002311-0
Lo, W.-S., Chen, L.-L., Chung, W.-C., Gasparich, G. E., and Kuo, C.-H. (2013). Comparative genome analysis of Spiroplasma melliferum IPMB4A, a honeybee-associated bacterium. BMC Genom. 14:22. doi: 10.1186/1471-2164-14-22
Lo, W.-S., Gasparich, G. E., and Kuo, C.-H. (2018). Convergent evolution among ruminant-pathogenic Mycoplasma involved extensive gene content changes. Genome Biol. Evol. 10, 2130–2139. doi: 10.1093/gbe/evy172
Meng, Y., Li, Y., Galvani, C. D., Hao, G., Turner, J. N., Burr, T. J., et al. (2005). Upstream migration of Xylella fastidiosa via pilus-driven twitching motility. J. Bacteriol. 187, 5560–5567. doi: 10.1128/jb.187.16.5560-5567.2005
Merfa, M. V., Niza, B., Takita, M. A., and De Souza, A. A. (2016). The MqsRA toxin-antitoxin system from Xylella fastidiosa plays a key role in bacterial fitness, pathogenicity, and persister cell formation. Front. Microbiol. 7:904.
Paradis, E., and Schliep, K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Parkinson, N., Cowie, C., Heeney, J., and Stead, D. (2009). Phylogenetic structure of Xanthomonas determined by comparison of gyrB sequences. Int. J. Syst. Evol. Microbiol. 59, 264–274. doi: 10.1099/ijs.0.65825-0
Pieretti, I., Royer, M., Barbe, V., Carrere, S., Koebnik, R., Cociancich, S., et al. (2009). The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. BMC Genomics 10:616. doi: 10.1186/1471-2164-10-616
Potnis, N., Kandel, P. P., Merfa, M. V., Retchless, A. C., Parker, J. K., Stenger, D. C., et al. (2019). Patterns of inter- and intrasubspecific homologous recombination inform eco-evolutionary dynamics of Xylella fastidiosa. ISME J. 13, 2319–2333. doi: 10.1038/s41396-019-0423-y
Rapicavoli, J., Ingel, B., Blanco-Ulate, B., Cantu, D., and Roper, C. (2018). Xylella fastidiosa: an examination of a re-emerging plant pathogen. Mol. Plant Pathol. 19, 786–800. doi: 10.1111/mpp.12585
Robinson, J. T., Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotech. 29, 24–26. doi: 10.1038/nbt.1754
Rodriguez-R, L. M., Grajales, A., Arrieta-Ortiz, M. L., Salazar, C., Restrepo, S., and Bernal, A. (2012). Genomes-based phylogeny of the genus Xanthomonas. BMC Microbiol. 12:43. doi: 10.1186/1471-2180-12-43
Roper, M. C., Greve, L. C., Warren, J. G., Labavitch, J. M., and Kirkpatrick, B. C. (2007). Xylella fastidiosa requires polygalacturonase for colonization and pathogenicity in Vitis vinifera grapevines. Mol. Plant Microbe Interact. 20, 411–419. doi: 10.1094/mpmi-20-4-0411
Su, C.-C., Chang, C.-J., Yang, W.-J., Hsu, S.-T., Tzeng, K.-C., Jan, F.-J., et al. (2012). Specific characters of 16S rRNA gene and 16S–23S rRNA internal transcribed spacer sequences of Xylella fastidiosa pear leaf scorch strains. Eur. J. Plant Pathol. 132, 203–216. doi: 10.1007/s10658-011-9863-6
Su, C.-C., Deng, W.-L., Jan, F.-J., Chang, C.-J., Huang, H., and Chen, J. (2014). Draft genome sequence of Xylella fastidiosa pear leaf scorch strain in Taiwan. Genome Announc. 2:e00166-14.
Su, C.-C., Deng, W.-L., Jan, F.-J., Chang, C.-J., Huang, H., Shih, H.-T., et al. (2016). Xylella taiwanensis sp. nov., causing pear leaf scorch disease. Int. J. Syst. Evol. Microbiol. 66, 4766–4771. doi: 10.1099/ijsem.0.001426
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Vanhove, M., Retchless, A. C., Sicard, A., Rieux, A., Coletta-Filho, H. D., De La Fuente, L., et al. (2019). Genomic diversity and recombination among Xylella fastidiosa subspecies. Appl. Environ. Microbiol. 85:e02972-18.
Wells, J. M., Raju, B. C., Hung, H.-Y., Weisburg, W. G., Mandelco-Paul, L., and Brenner, D. J. (1987). Xylella fastidiosa gen. nov., sp. nov: gram-negative, xylem-limited, fastidious plant bacteria related to Xanthomonas spp. Int. J. Syst. Bacteriol. 37, 136–143. doi: 10.1099/00207713-37-2-136
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Keywords: Xylella, Xanthomonadaceae, plant pathogens, pear leaf scorch, genome, virulence
Citation: Weng L-W, Lin Y-C, Su C-C, Huang C-T, Cho S-T, Chen A-P, Chou S-J, Tsai C-W and Kuo C-H (2021) Complete Genome Sequence of Xylella taiwanensis and Comparative Analysis of Virulence Gene Content With Xylella fastidiosa. Front. Microbiol. 12:684092. doi: 10.3389/fmicb.2021.684092
Received: 22 March 2021; Accepted: 27 April 2021;
Published: 21 May 2021.
Edited by:
Jeffrey Jones, University of Florida, United StatesReviewed by:
Prem P. Kandel, Pennsylvania State University (PSU), United StatesCopyright © 2021 Weng, Lin, Su, Huang, Cho, Chen, Chou, Tsai and Kuo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chi-Wei Tsai, chiwei@ntu.edu.tw; Chih-Horng Kuo, chk@gate.sinica.edu.tw
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.