 Samujjal Bhattacharjee
Samujjal Bhattacharjee Surbhi Kharwar
Surbhi Kharwar Arun Kumar Mishra
Arun Kumar Mishra- Laboratory of Microbial Genetics, Department of Botany, Institute of Science, Banaras Hindu University, Varanasi, India
The functionality of caspase homologs in prokaryotic cell execution has been perceived, yet the dimensions of their metabolic pertinence are still cryptic. Here, a detailed in silico study on putative cyanobacterial caspase homologs, termed orthocaspases, in a sequenced genome of 132 strains was performed. We observed that 473 putative orthocaspases were distributed among 62% cyanobacterial strains subsumed within all the taxonomical orders. However, high diversity among these orthocaspases was also evident as the conventional histidine–cysteine (HC) dyad was present only in 72.03% of orthocaspases (wild-type), whereas the rest 28.18% were pseudo-variants having substituted the catalytic dyad. Besides, the presence of various accessory functional domains with Peptidase C14 probably suggested the multifunctionality of the orthocaspases. Moreover, the early origin and emergence of wild-type orthocaspases were conferred by their presence in Gloeobacter; however, the complex phylogeny displayed by these caspase-homologs perhaps suggested horizontal a gene transfer for their acquisition. However, morpho-physiological advancements and larger genome size favored the acquisition of orthocaspases. Moreover, the conserved caspase hemoglobinase fold not only in the wild-type but also in the pseudo-orthocaspases in Nostoc sp. PCC 7120 ascertained the least effect of catalytic motifs in the protein tertiary structure. Further, the 100-ns molecular dynamic simulation and molecular mechanics/generalized born surface area exhibited stable binding of arginylarginine dipeptide with wild-type orthocaspase of Nostoc sp. PCC 7120, displaying arginine-P1 specificity of wild-type orthocaspases. This study deciphered the distribution, diversity, domain architecture, structure, and basic substrate specificity of putative cyanobacterial orthocaspases, which may aid in functional investigations in the future.
Introduction
Caspases are specialized cysteine-aspartate proteases, recognized to mediate apoptosis in metazoans. These peptidases are synthesized as inactive proenzymes, which upon activation attain dimeric configuration, having p20 and p10 sub-domains in each monomer (Earnshaw et al., 1999; Fuentes-Prior and Salvesen, 2004). The proteolytic activity of caspases is conferred by the p20-subunit that harbors a critical histidine–cysteine (HC) dyad in its active site, which cleaves target proteins after aspartate (D) residue, while the p10 subunit has a regulatory role. Although the caspases are generally harbored by the metazoans, its homologs are abundantly found in lower life forms. Pioneering studies revealed the presence of caspase homologs not only in single-celled eukaryotes (Uren et al., 2000) but also in bacteria (Aravind and Koonin, 2002), portraying the possibility of caspase-homolog-dependent PCD in these life forms.
Caspase homologs are a structurally diverse group of proteases, belonging to the Peptidase C14 family which also subsumed the archetype caspases. This family of proteases is characterized by the presence of a caspase-hemoglobinase fold (CHF) composed of four parallel β-sheets surrounded by three α-helices (Aravind and Koonin, 2002). Further, these homologs are broadly classified into paracaspases (PCAs) (Jaworski and Thome, 2016), metacaspases (MCAs) (Tsiatsiani et al., 2011; Minina et al., 2017), and OCAs (Klemenčič and Funk, 2018), based on their protein architecture. While PCAs and OCAs are devoid of p10-like subunit, the MCAs consist of both the sub-domain homologs. Although the catalytic HC dyad was conserved among caspase homologs, the aspartate P1 cleavage specificity annotated for all the Peptidase C14 members was absent in them. PCAs are arginine (R)-specific Ca2+-independent dimeric proteases (Hachmann et al., 2012), whereas MCAs are Ca2+-dependent monomers which specifically cleave after either R or lysine (K) residues (Hander et al., 2019). Additionally, the OCAs are speculated to have specificity for the basic residues but their detailed structural and functional aspects are yet to be unveiled (Minina et al., 2020). Owning to the significance of PCD in prokaryotes, a metagenomic study in the Baltic Sea exhibited the occurrence of Peptidase C14-containing proteins only among 4% of all the bacterial population (Asplund-Samuelsson et al., 2016). This 4% of bacteria mostly subsumed the members of actinomycetes, α-, β-, and γ-proteobacteria, and cyanobacteria (Koonin and Aravind, 2002; Asplund-Samuelsson et al., 2012). Most of the Peptidase C14-containing proteins in cyanobacteria lacked the p10-like sub-domain (Choi and Berges, 2013) and therefore considered as the OCAs, whereas only 4% accounted for putative MCAs (Klemenčič et al., 2019). An initial study on 33 whole-genome-sequenced cyanobacterial strains depicted the uneven distribution of OCAs, as complex strains with larger genome favored to acquire them (Jiang et al., 2010). Moreover, the substrate specificity of putative OCA, MaOC1, for RR dipeptide and Ca2+-independent autocatalysis was also demonstrated in M. aeruginosa PCC 7806 (Klemenčič et al., 2015). However, the identification of various pseudo-variants of caspase homologs lacking the functional HC dyad increased the obscure underlining of the dimension of their functionalities. In Trypanosoma brucei, the catalytic HC dyad was substituted by the YS (tyrosine–serine) motif in MCAs TbMC1 and TbMC4, probably rendering their proteolytic activity (Proto et al., 2011). The abundance of pseudo-OCAs in cyanobacteria having substituted catalytic motifs may suggest a wider applicability of these proteases (Klemenčič et al., 2019).
This study focuses on the functionality of wild-type OCAs (OCA having an HC catalytic dyad) and their physiological congruity in cyanobacteria. Here a comprehensive bioinformatics study has been conducted to describe the distribution, diversity, abundance, domain configuration, and phylogeny of 473 putative cyanobacterial OCAs. Additionally, the structural analysis of a wild-type OCA from Nostoc sp. PCC 7120 was performed using homology modeling and compared elaborately with their pseudo-counterparts. Further, docking and molecular dynamic (MD) simulation of the protein-substrate complex was performed to ensure the substrate specificity of this OCA. In addition, the binding free energy (ΔGbind) calculated using the molecular mechanics/generalized born surface area (MMGBSA) also assured the stability of the complex. This study will provide an updated and holistic view on the attributes of putative OCAs and may pave new ways to answer the existence of OCA-mediated PCD in cyanobacteria.
Materials and Methods
Retrieving the Sequences of Putative Cyanobacterial OCAs
The protein sequences of putative cyanobacterial OCAs were retrieved from the NCBI database1. The p20-like sub-domain (Peptidase C14)-containing protein sequences in 132 whole-genome-sequenced cyanobacterial strains were identified by performing domain enhanced lookup time accelerated (DELTA) BLAST (Boratyn et al., 2012), using 4AFR (MCA 2) of Trypanosoma brucei (Q585F3.1) as the query. Sequences with incomplete Peptidase C14, either at the N- or C-terminal, or lacking the domain, were manually eliminated. Finally, 473 putative OCA sequences from 82 cyanobacterial strains were further analyzed.
Analyzing the Diversity, Distribution, and Abundance of Putative Cyanobacterial OCAs
All the protein sequences were manually analyzed and subsequently categorized into true (wild-type) and pseudo (mutated) OCAs, based on the catalytic dyad forming the active site. Only those proteins with classical HC dyad were considered as wild-type, whereas proteins having any other amino acid residues in the catalytic dyad were regarded as pseudo-OCAs. Further, the distribution and abundance of all OCA subtypes (true and pseudo OCAs) along the taxonomical order of these 82 cyanobacterial strains were compared against their genome sizes and morphological complexities.
Domain Configuration of Putative Cyanobacterial OCAs
The full-length OCA sequences were subjected to CD search2 (Marchler-Bauer et al., 2017) against the Pfam database (threshold E value = 0.01) (Mistry et al., 2021). CD-search outputs were analyzed, and only specific hits for conserved functional domains were considered since they were top-ranked hits and conferred very high confidence, while superfamily hits were averted. Additionally, transmembrane domains (TM) were identified using TMHMM v2.03 (Krogh et al., 2001). The number of different accessory domains that occurred in 473 putative OCAs was determined; however, successive repeats of domains within the same protein were disregarded. The domain architecture of few OCAs with frequently appearing accessory domains was visualized by Illustrator of Biological Sequence (IBS) v1.0 (Liu et al., 2015). Domain configurations for all the OCAs are enlisted in Supplementary Table 1.
Multiple-Sequence Alignment and Phylogeny
Multiple-sequence alignment (MSA) and phylogeny were performed using only the Peptidase C14 domain of all the putative OCAs. Additional domains associated with the proteases were not considered to avoid any possible uncertainty due to their varied origin and evolutionary history. Four hundred seventy-three Peptidase C14 domains were aligned by MUSCLE (Edgar, 2004), in MEGA v7.0 using default parameters, i.e., gap open penalty = −2.9; gap extended penalty = 0; and hydrophobicity multiplier = 1.2 (Kumar et al., 2016). Subsequently, the features of these sequences were displayed as the Hidden Markov Model (HMM) logo using Skylign4 (Wheeler et al., 2014).
A rooted phylogenetic tree comprising 473 Peptidase C14 sequences was constructed by the neighbor-joining method (Saitou and Nei, 1987) using MEGA v7.0. The MCA-like protein from Thalassiosira pseudonana CCMP1335 (XP_002295354.1) was used as the outgroup, since it is a marine diatom and possesses a closer evolutionary relationship with cyanobacteria (Jiang et al., 2010). The branch lengths were in the same units as those of the evolutionary distances. The evolutionary distances were calculated using the Jones–Taylor–Thornton (JTT) matrix-based method (Jones et al., 1992). The reliability of each branch was tested with 1,000 bootstrap replications. All positions containing gaps and missing data in the MSA file were eliminated during tree construction. Distinct clades were expanded and viewed separately for better inference (Supplementary Figures 1–4). The bootstrap value (above 50) and sequence details for each node were also provided in Supplementary Figures 1–4.
Homology Modeling and Structural Analysis
Phylogenetic analysis displayed YS and YN as the most prominent catalytic dyad substitutions, which otherwise constituted of HC in the wild-type OCAs. Therefore, the theoretical modeling was performed to delineate and compare the structures of wild-type (HC) and mutated (YS and YN) OCAs. Since Nostoc sp. PCC 7120 is a model cyanobacterium and not only harbored the wild-type (HC) OCAs but also consisted of other two abundant forms of pseudo OCAs, i.e., YS and YN types, therefore, three OCAs, i.e., WP_010999143.1 (HC), WP_010999263.1 (YS), and WP_010997820.1 (YN), of Nostoc sp. PCC 7120 were selected. Due to the lack of suitable templates, only the Peptidase C14 domains of WP_010999263.1 (YS) and WP_010997820.1 (YN) were modeled. High-quality homology models were constructed by Swiss-Model5 using default settings (Guex et al., 2009; Benkert et al., 2011; Bertoni et al., 2017; Bienert et al., 2017; Waterhouse et al., 2018; Studer et al., 2020). Further, template-based refinement and energy minimization of models were done using ModRefiner6, for which ideal templates for each model with high resolution, sequence identity, and maximum query coverage were retrieved from Swiss-Model (Supplementary Table 2; Xu and Zhang, 2011). The final models were validated by Ramachandran plot using PROCHECK7 (Laskowski et al., 1993) and ModFOLD88 (Maghrabi and McGuffin, 2017; Supplementary Table 3). Additionally, the folds and topologies of these OCAs were generated by PDBsum9 (Laskowski, 2001). The properties of OCA models were analyzed and visualized by UCSF Chimera (Pettersen et al., 2004).
Analyzing the Substrate-Binding Efficacy of Wild-Type OCA
Two oligopeptides, i.e., arginyl-arginine (RR) and aspartyl-glutamyl-valyl-aspartate (DEVD), were used to study the substrate specificity of wild-type OCA (HC; WP_010999143.1). While DEVD is a classical caspase substrate, RR was previously used as a substrate for cyanobacterial OCA, MaOC1 (Klemenčič et al., 2015). For the docking analysis, the substrate and the protease were treated as ligand and receptor, respectively. Docking was performed with Biovia Discovery Studio 2019 using the CDOCKER program (Wu et al., 2003). CDOCKER is a powerful grid-based molecular docking method that uses Chemistry at Harvard Macromolecular Mechanics (CHARMm). Here, the ligand is allowed to flex while the receptor is held rigid during the refinement. The docked complex was visualized using UCSF Chimera 1.13.1, and LigPlot+ was used to identify the substrate-interacting residues (Laskowski and Swindells, 2011).
Simulation of the HC–RR Complex
The HC–RR complex was subjected to all-atom MD simulation using the Amber16 package (Case et al., 2016). The initial structure was prepared in the tleap module of Amber16, utilizing the ff14SB force field (Maier et al., 2015). The ligand force-field parameters were generated using antechamber (Wang et al., 2006) and gaff (Wang et al., 2004); also, the AM1-bcc method was used to generate charges (Jakalian et al., 2002). The simulation box was solvated by the TIP3P water model with a padding distance of 12 Å (Jorgensen et al., 1983), and NaCl was used as counter-ion in neutralizing the total charge of the system. Joung and Cheatham ion parameters were used for Na+ and Cl– ions (Joung and Cheatham, 2008). The electrostatic interaction was calculated by the particle mesh Ewald (PME) method (Darden et al., 1993). Further, the SHAKE algorithm was used to restrain the binds involving hydrogen (Ryckaert et al., 1977) while the Langevin dynamics (Wu and Brooks, 2003) and Berendsen barostat (Berendsen et al., 1984) were utilized for temperature and pressure control, respectively.
To minimize the energy of the complex, initially, the HC–RR complex was restrained and only the solvent was minimized. Thereafter, the restraint potential was removed from the complex and the entire system was minimized. Following the minimization, gradual heating of the systems was performed at NVT with a 100-kcal restraint potential on the complex, and the temperature was increased to 300K in 300 ps. After heating, each system was subjected to equilibration for 1.8 ns in six cycles of 300 ps in which the restraint potential was subsequently reduced from 100 to 0 kcal/mol (100, 50, 20, 5, 0.5, 0 kcal/mol) at NPT. After equilibration, a production run of 100 ns was performed each at the NPT ensemble. The trajectory was visualized by VMD 1.9.3 and UCSF Chimera 1.13.1.
Calculation of Binding Free Energy
The last 90-ns trajectory was used to calculate the binding free energy of the HC–RR complex (ΔGbind) using the Molecular Mechanics Generalized Born Surface Area (MMGBSA) method implemented in Amber16. The calculation was performed at the dielectric constant of 1 and 80 for solute and solvent, respectively (Ylilauri and Pentikäinen, 2013). The parameters developed by Onfriev et al. was used for GB-calculation (igb=5; α=1.0, β=0.8. γ=4.85) (Onufriev et al., 2004). Additionally, entropic contribution (ΔS) was not taken into account for determining ΔGbind. Other parameters were used as default.
Results
Distribution, Diversity, and Abundance of OCAs in Cyanobacteria
An iterative analysis of 132 whole-genome-sequenced cyanobacteria resulted in the identification of 473 putative OCAs, harbored by 82 strains, accounting for about 62% of the query set (50 cyanobacteria either lacked OCAs or possessed incomplete sequences of Peptidase C14 domain). Out of 473 OCAs, 340 were wild-type (72.03%), having the HC dyad in their active sites, whereas the rest 133 (28.18%) were pseudo-variants. Among the pseudo-OCAs, YS- and YN-substituted catalytic motifs were predominant, accounting for approximately 47% and 31% of all pseudo-variants, respectively. Besides, 16 other pseudo-variants were also identified which comprised YG, YC, YQ, LG, RG, HN, HG, YH, YD, YR, NC, FS, QD, CN, QC, and CS motifs in their active sites (Supplementary Table 4). Among 82 cyanobacteria, few strains, i.e., Gloeobacter kilaueensis JS1, Gloeobacter violaceus PCC 7421, Cyanobacterium aponinum PCC 10605, Cyanobacterium sp. HL-69, Cyanobacterium stanieri PCC 7202, Stanieria sp. NIES-3757, Geitlerinema sp. PCC 7407, and Calothrix sp. PCC 6303, harbored only true OCAs. In contrast, Synechococcus sp. JA-2-3B’a(2-13), Synechococcus sp. PCC 7502, Synechocystis sp. PCC 6714, Synechocystis sp. PCC 6803, Pleurocapsa sp. PCC 7327, Cyanothece sp. PCC 7425, and Nostoc azollae 0708 harbored only pseudo-OCAs (Figure 1). However, at least one copy of both wild-type and pseudo-OCAs was identified in all other strains.
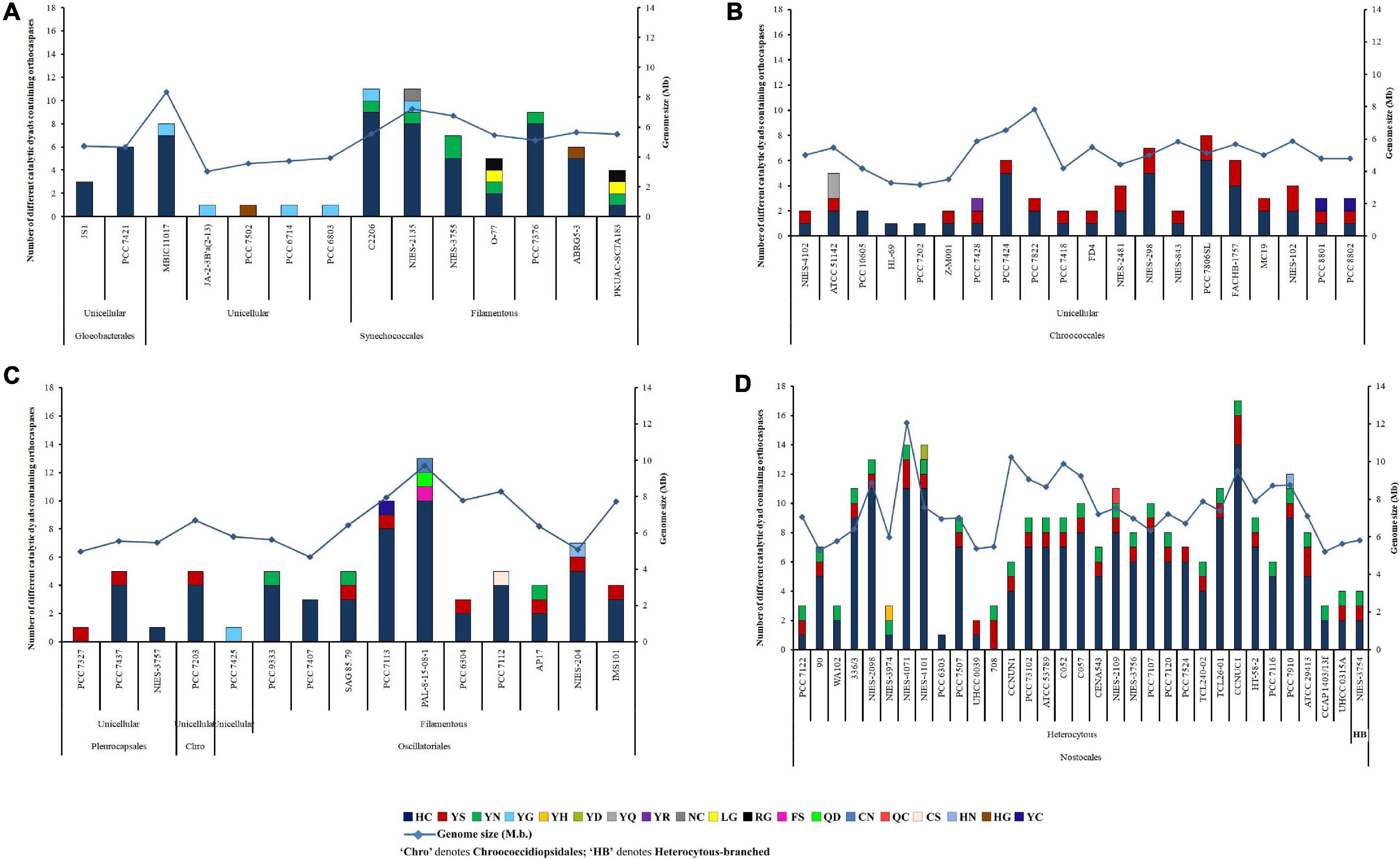
Figure 1. Abundance, distribution, diversity of putative cyanobacterial orthocaspases among (A) Gloeobacteriales, Synechococcales, (B) Chroococcales, (C) Pleurocapsales, Chroococcidiopsidales, Oscillatoriales, and (D) Nostocales. Cyanobacterial strains subsumed within different taxonomical orders are also categorized into unicellular, filamentous, and heterocytous branches based on their morphology. The full forms of the abbreviated strain names are mentioned in Supplementary Table 4.
The abundance of OCA was estimated to be highest in Nostocales and Oscillatoriales with an average occurrence of 0.0921 and 0.0723 true OCA per 100 proteins, respectively. Besides, an abundance of 0.0416 true OCA per 100 proteins was observed in Chroococcales and the lowest abundance of 0.0145 true OCA per 100 proteins was estimated in Synechococcales. The average abundance of pseudo-OCAs was comparable for Nostocales (0.0349/100 proteins), Oscillatoriales (0.0264/100 proteins), and Chroococcales (0.0261/100 proteins), while the lowest abundance was recorded again for Synechococcales (0.0027/100 proteins) (Supplementary Table 4). The actual abundance of OCAs might obscure in Gloeobacterales, Pleurocapsales, and Chroococcidiopsidales due to the lesser number of whole-genome-sequenced cyanobacteria in them, yet OCAs harboring sequenced strains within these orders were examined.
Considerable variations in OCA frequencies were evident within the cyanobacterial orders. About 44% OCA-harboring strains (corresponding to 36 cyanobacteria), belonging to Gloeobacterales, Synechococcales, Chroococcales, Oscillatoriales, and Nostocales, displayed an exclusively high number of wild-type OCAs (≥5); however, the number of pseudo-OCAs ranged from 1 to 3 in 82 cyanobacteria. Nevertheless, the highest was observed in Nostoc sphaeroides CCNUC1 having 14 true and 3 pseudo OCA-encoding genes (Figure 1).
Domain Variability and Configurations
Considerable variability in domain architecture among putative OCAs was observed due to the presence of multiple functional domains. Apart from Peptidase C14, 35 unique Pfam domains were identified in 473 OCAs (Table 1). While the true OCAs were predominantly multidomain proteins, most of the pseudo-OCAs lacked any identifiable domains, except Peptidase C14, showing a lesser domain variability than the wild type. Out of all the OCAs, a single Peptidase C14 domain was present in 34 (7%) and 123 (26%) true and pseudo OCAs, respectively, whereas 306 (65%) true and only 10 (2%) pseudo-OCAs harbored at least two functional domains (including Peptidase C14) (Supplementary Table 1). By disregarding the successive repeats of domains in a single protein, it was observed that the frequency of WD40 (26%) and AAA_16 (19%) was exceptionally high than that of other accessory domains. Moreover, FGE sulfatase (12%), GUN4 (9%), and Pentapeptide repeats (7%) also had a fairly high frequency. In contrast, 11 domains like TPR 19, Pentapeptide-4, AAA_22, GGDEF, CHAT, Ribosomal_L12, PBP-like_2, SAVED, TIR_2, PD40, and Pro-rich were associated with only one OCA each. Additionally, domains such as HEAT_2, TPR 11, HATPase_c, HisKA, DEAD, Helicase_C, TPR 16, TPR 12, TPR 2, Sel1, MAP7, PT, Clp_N, and EAL were also rare. Furthermore, few OCAs also consisted of domains of unknown function (DUF) (Table 1).
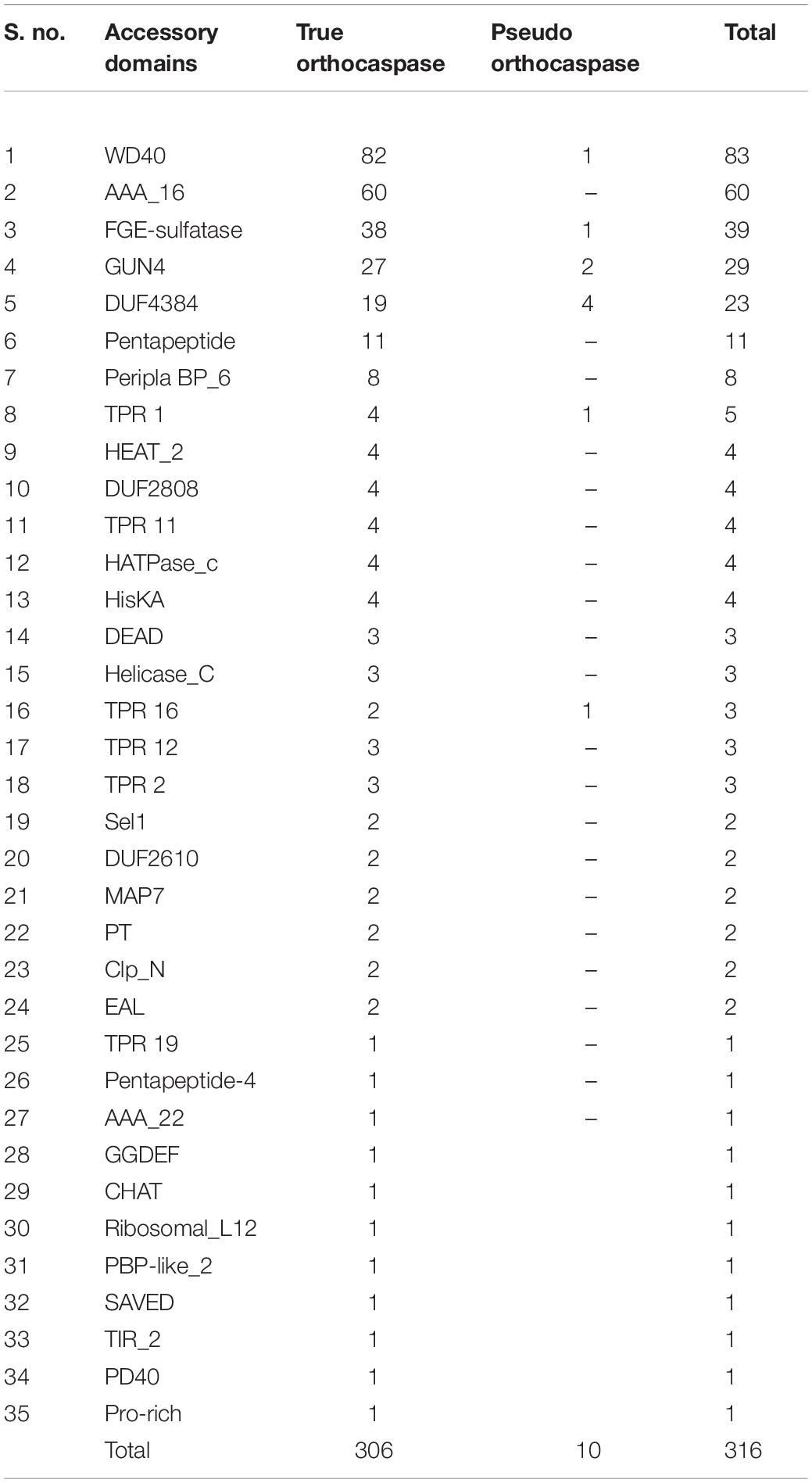
Table 1. Distribution of different accessory domains among putative true and pseudo-orthocaspases in cyanobacteria.
In the 473 putative OCAs, 67 consisted of transmembrane helices (TM), among which 30 and 37 were true and pseudo OCAs, respectively. The presence of one TM was fairly common among OCAs, but WP_015201768.1 (Crinalium epipsammum PCC 9333), WP_158517387.1 (Moorea producens PAL-8-15-08-1), and ABG51878.1 (Trichodesmium erythraeum IMS101) contained two TMs. Although the TM is generally located C-terminally to the Peptidase C14 domain, in AFZ48670.1 (C. stanieri PCC 7202), WP_015220159.1 (C. aponinum PCC 10605), AKV65607.1 (Microcystis panniformis FACHB-1757), WP_125731015.1 (Microcystis viridis NIES 102), WP_083305259.1 (M. producens PAL-8-15-08-1), and WP_167720235.1 (Tolypothrix sp. PCC 7910), TM was identified at the N-terminal with respect to Peptidase C14 (Supplementary Table 3).
Further, domain configuration revealed that the OCAs prevalently acquired a similar architecture. The common domain configurations attained by both wild-type and pseudo OCAs were classified into nine types (Figure 2A). In general, the Peptidase C14 domain was N-terminally located to the accessory domains in multidomain proteins (Figure 2); however, in Type V, the Peptidase C14 was located toward the C-terminal to HEAT_2 domain(s). Such architectures observed were WP_168163431.1 (Calothrix sp. 336/3), BAZ37947.1 (Calothrix sp. NIES 4101), WP_015119030.1 (Rivularia sp. PCC 7116), and WP_011611472.1 (T. erythraeum IMS101). Furthermore, it was observed that the OCAs of strains belonging to Nostocales consisted of all the domain configurations (Figure 2B).

Figure 2. The domain configurations of few putative cyanobacterial orthocaspases with commonly observed accessory domains, and their distribution among taxonomical orders. (A) The domain configurations were categorized into nine different types, and a representative sequence was used to depict the position of domains for each type. Domain architectures obtained CD search using the Pfam database (e-value = 0.01). NCBI accession numbers and strain names are provided for the representative sequence. The figure is drawn to scale with numbers representing protein or domain length (amino acid numbers). Catalytic residues are indicated with a single-letter amino acid code with a position specifier. (B) The distribution of various domain configuration types among cyanobacterial orders is shown. Note that the configuration, i.e., the N- and C-terminal orders of domains, is the same in all the sequences sharing a particular type of domain architecture. However, the position of domain and catalytic residues shown in panel (A) may vary among the sequences.
Multiple-Sequence Alignment and Phylogeny
The sequence alignment of all OCA sequences displayed substantial diversity. The HMM logo exhibited a relatively conserved region surrounding the catalytic dyad (Figure 3). The consensus regions flanking the catalytic residues were (Y/F/H), (F/Y S/A), (G), (H/Y/F), (G/A), and (L/I/M/F), (D), (X), (C/S/N), (H/F/Y/R) (catalytic residues are shown in bold). Besides, G7 and D27 were also conserved in all the OCA subtypes, probably suggesting their essentiality.
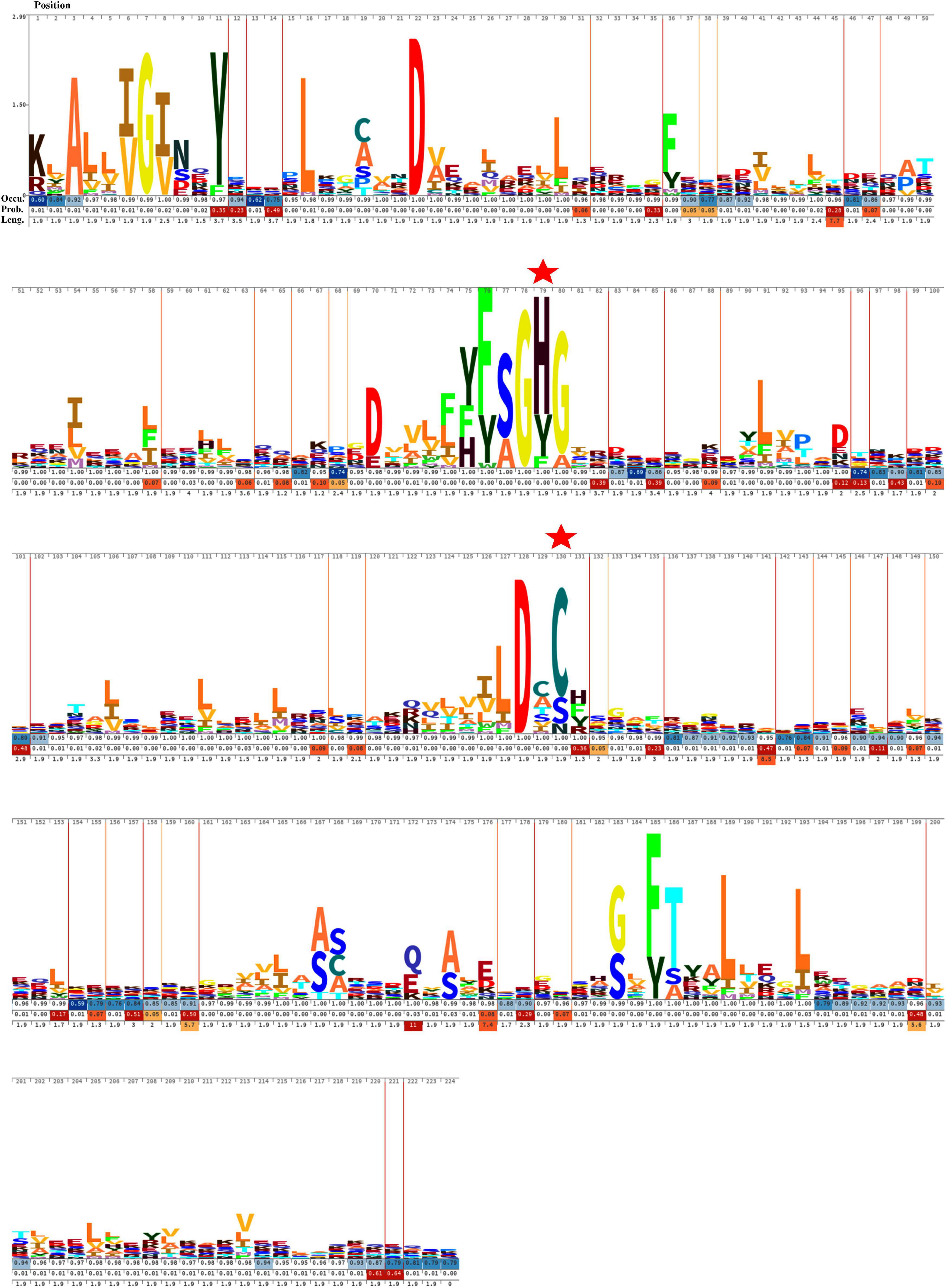
Figure 3. Hidden Markov Model (HMM) logos of the Peptidase C14 domain of putative cyanobacterial orthocaspases. Logos were built with 473 aligned Peptidase C14 domain sequences using Skylign. The values on the Y-axis correspond to the height of each single-letter amino acid code and reflected the probability of its occurrence at that specific position. The larger letter size indicates conserved residues among the protein sequences. The position of each amino acid residue is indicated at the top. The gap parameters, i.e., occupancy (Occu), insert probability (Prob.), and insert length (Leng.), are shown below each residue. The catalytic residues are indicated by “ .”
.”
The phylogenetic relationship between 473 putative cyanobacterial OCAs was determined by an NJ tree constructed using their Peptidase C14 domains. Wild-type Peptidase C14 with HC dyad formed nine clades, whereas YS and YN motifs containing pseudo-variants were accumulated into two distinct clusters. Sequences with any other substituted active site motifs were scattered all along the phylogenetic tree (Figure 4). A thorough assessment of individual clusters revealed that most of the HC clades consisted of sequences from distantly related strains with variable lifestyles and morphologies (unicellular to heterocytous), except HC V and HC VII clades, harboring only multicellular strains of Nostocales and Oscillatoriales (Supplementary Figures 1–3). Evolutionary primitive strains, i.e., G. kilaueensis JS1 and G. violaceus PCC 7421 (Gloeobacteriales) having only wild-type Peptidase C14, were included in HC III and HC VIII clades. Moreover, pseudo-OCAs from Chroococcales, Pleurocapsales, Chroococcidiopsidales, Oscillatoriales, and Nostocales were harbored within the YS clade, whereas the YN clade subsumed only filamentous and heterocytous strains from Synechococcales, Oscillatoriales, and Nostocales (Supplementary Figure 4). Further, OCA-harboring strains of Nostocales showed the widest distribution, as they were present in all the clades.
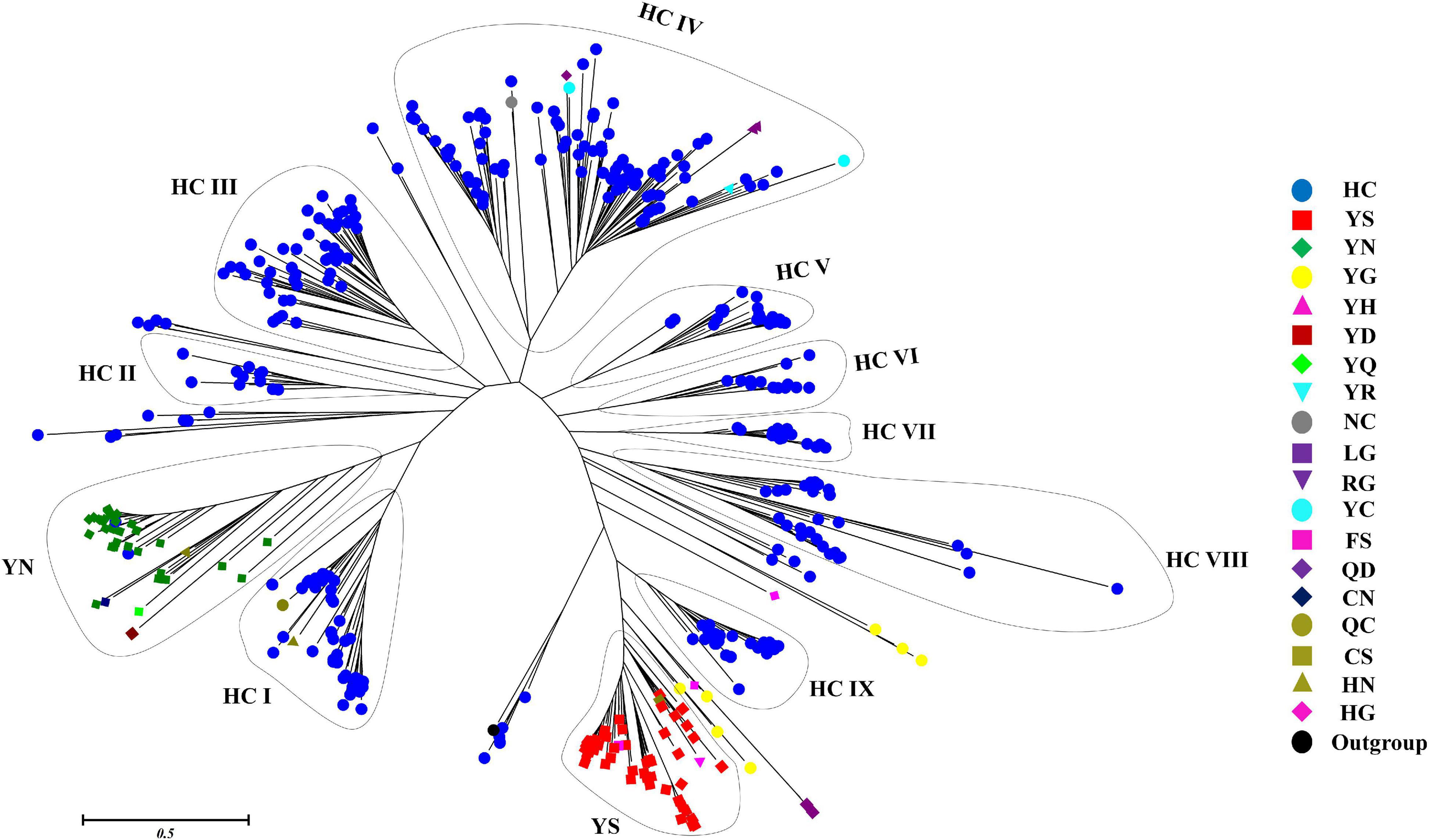
Figure 4. Phylogenetic relationship of putative cyanobacterial orthocaspases (OCAs). The rooted tree of only the Peptidase C14 domain of all the putative OCAs was constructed using the neighbor-joining method with the metacaspase-like protein of Thalassiosira pseudonana CCMP1335 (XP_002295354.1) as the outgroup. The optimal tree has the sum of branch length = 89.299. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances. The evolutionary distances were computed using the JTT matrix-based method and are in the units of the number of amino acid substitutions per site. The analysis involved 474 (473 cyanobacterial Peptidase C14 domain + 1 outgroup) sequences. All positions containing gaps and missing data were eliminated. Distinct clades are assigned based on the active site motifs in majority of the Peptidase C14 sequences. Eleven distinct clades were identified where HC catalytic dyads containing Peptidase C14 sequences formed nine whereas YS and YN catalytic motifs containing Peptidase C14 sequences formed two respective clades. Peptidase sequences with other substitution at catalytic motifs are scattered along the tree.
Structural Analysis of Cyanobacterial OCAs
Homology modeling of WP_010999143.1 (HC), WP_010999263.1 (YS), and WP_010997820.1 (YN) from Nostoc sp. PCC 7120 displayed the presence of typical CHF (Figure 5A). Despite having different catalytic dyads, significant structural alterations among the three proteases were not evident. The core of these proteases comprised four parallel β-sheets, i.e., one N-terminal, two central, and one C-terminal sheets arranged in a 2–1–3–4 fashion, and three α-helices (one each after strands 1, 2, and between 3 and 4) (Figure 5B). However, only in the Peptidase C14 domain of WP_010999143.1 (HC) did the third and fourth β-sheets precede the proximal (H) and distal (C) active sites, respectively. Besides, more hydrophilic residues (blue) were present on the surface of all the three OCAs, conferring them as soluble proteins (Figure 5C).
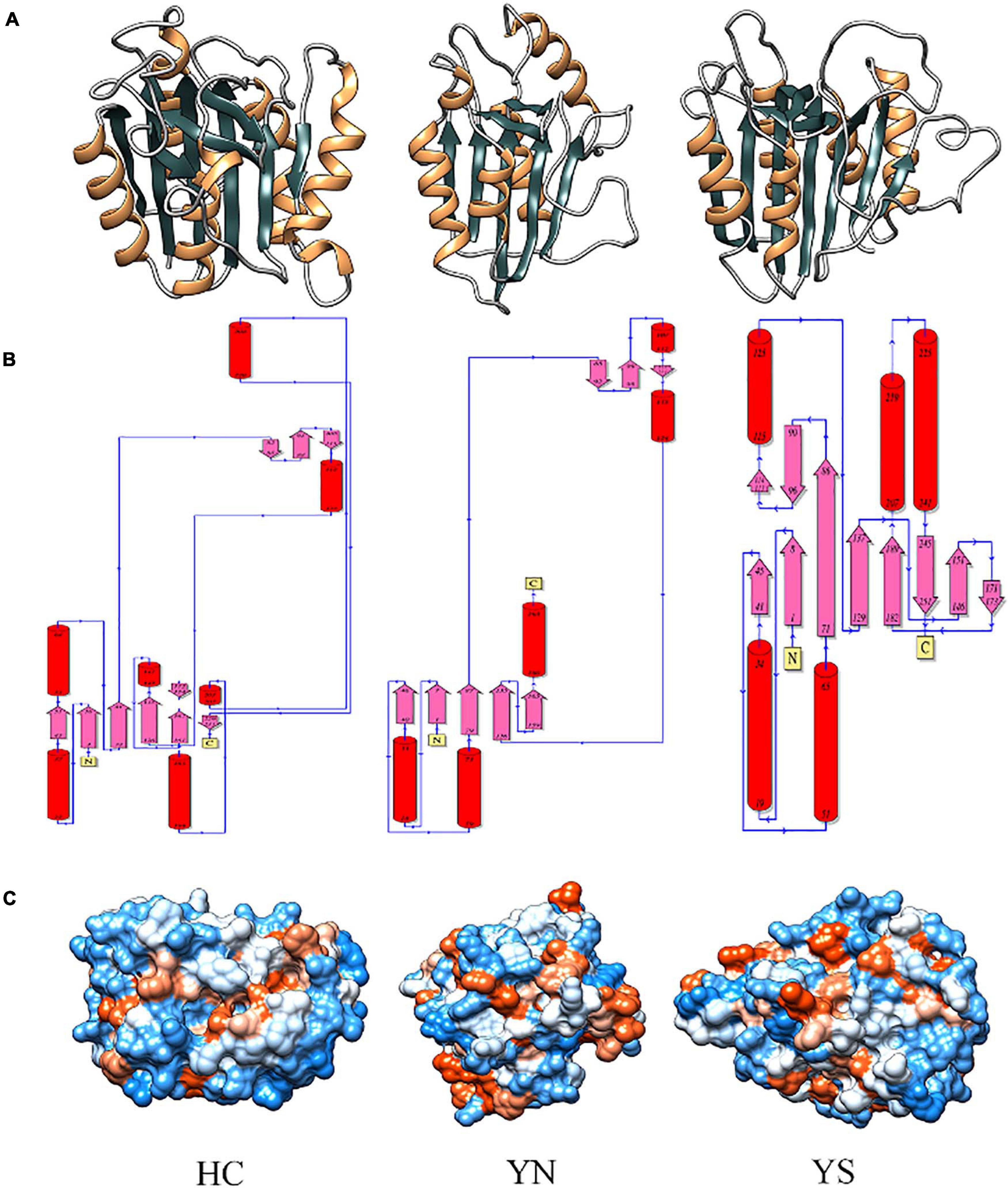
Figure 5. Comparing and analyzing the homology models of WP_010999143.1 (HC), WP_010997820.1 (YN), and WP_010999263.1 (YS) of Nostoc sp. PCC 7120, where HC is the wild-type OCA whereas YN and YS are pseudo orthocaspases. (A) 3D models of putative orthocaspases showing a conserved caspase-hemoglobinase fold (CHF). HC, YN, and YS possessed identical topology (B) due to conserved CHF with four parallel β-sheets and three α-helices. (C) Hydrophobicity surface view of modeled orthocaspases, where blue and orange colors represented hydrophilic and hydrophobic residues, respectively. Note that for WP_010997820.1 (YN) and WP_010999263.1 (YS), only the Peptidase C14 domain has been modeled due to template unavailability, while the full-length homology model was constructed for WP_010999143.1 (HC).
Analyzing the Substrate Specificity of HC
Molecular docking of HC with RR and DEVD revealed the binding of only RR with HC (Figure 6A), whereas no docked complex was formed between HC and DEVD (data not shown). The binding pocket for RR comprised L18, A19, L21, K23, A24, G80, D132, C133, Y173, A174, and E176 along with H81 and C134 (catalytic residues) (Figures 6B,C). Further, 100-ns simulation and MMGBSA illustrated the stability of the HC–RR complex. Residual fluctuation of the HC–RR complex depicted an average RMSF value of 2.45 ± 0.13 Å, among which residues between 18 and 97 and between 107 and 135 recorded lower fluctuations (Figure 7A). Most of the binding pocket residues were subsumed among these. Additionally, for 100-ns simulation the average RMSD of RR was 2.76 ± 0.54 Å, while the HC displayed two distinct trajectory: one from 0 to 50 ns with a lower average RMSD (3.43 ± 0.46 Å) and another from 51 to 100 ns with a higher average RMSD (5.21 ± 0.51 Å) (Figure 7B). However, upon superimposing the average structures, only an RMSD of 0.89 Å was observed, suggesting no major conformational changes in protein topology (Figure 8A). The presence of RR in the binding cavity of HC throughout the 100-ns simulation further conferred the specificity of HC for RR (Figure 8B). Additionally, ΔGbind (= −16.99 ± 5.99) of the HC–RR complex also exhibited high binding efficacy (Table 2).
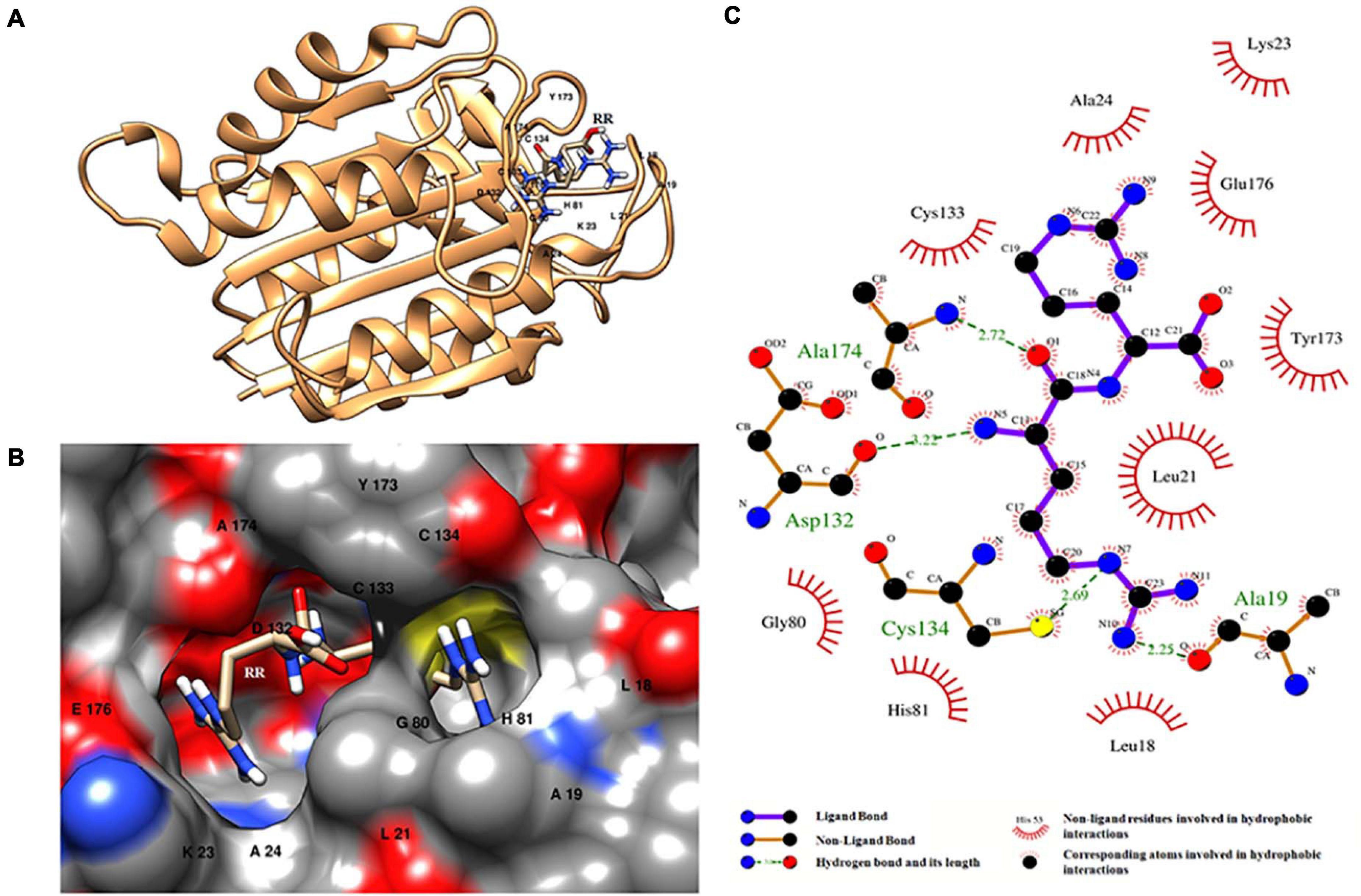
Figure 6. HC–RR complex formed upon docking RR dipeptide with wild-type orthocaspase, WP_010999143.1 (HC), of Nostoc sp. PCC 7120. (A) Binding position of substrate on the protein. (B) Binding pocket accommodating the substrate along with the interacting residues shown with a single-letter amino acid code and position specifier. (C) Interacting amino acid residues and the type of interaction with the substrate; amino acids are shown in a three-letter code with the position specifier.
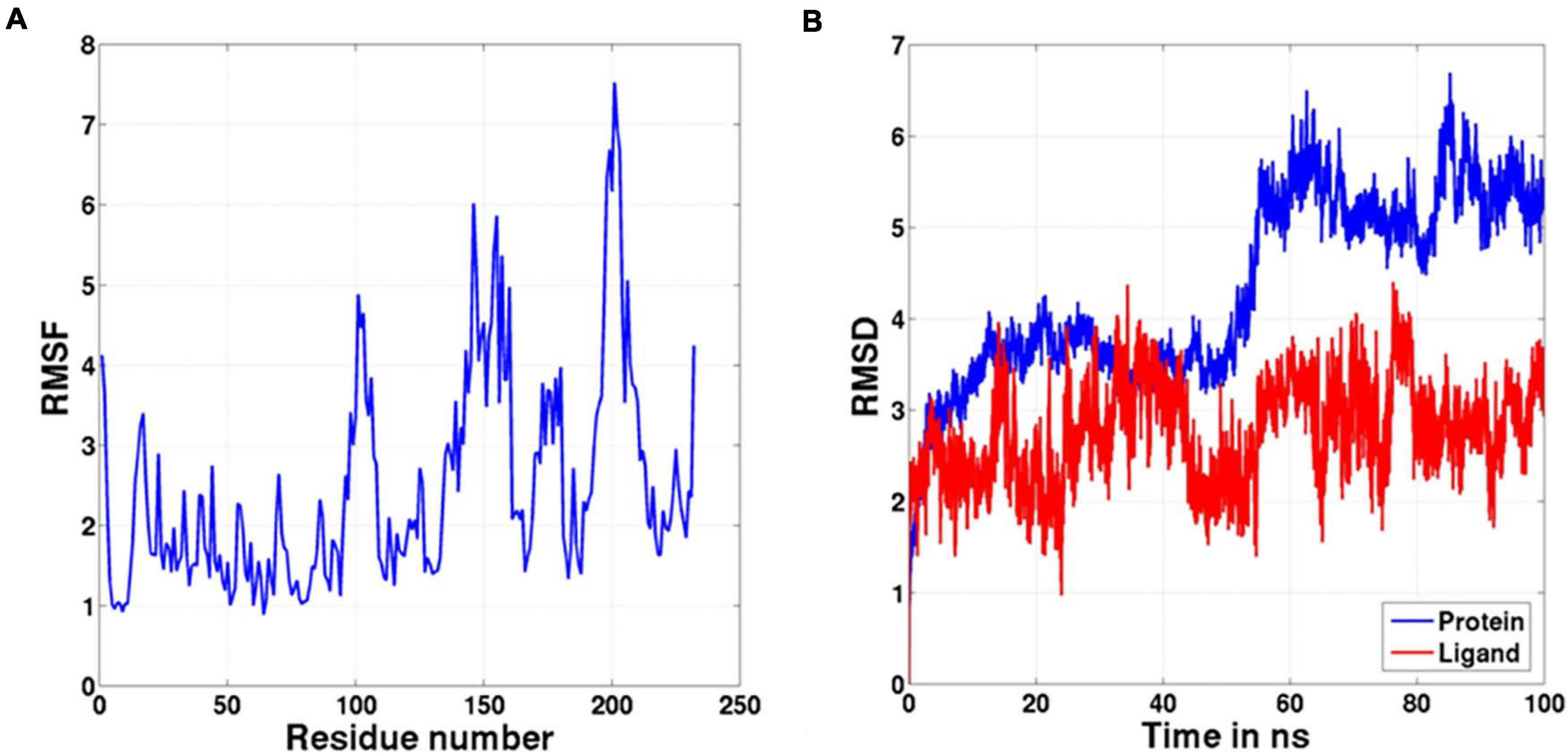
Figure 7. The graph representing (A) RMSF and (B) RMSD values of the complex during the 100-ns molecular dynamic simulation.
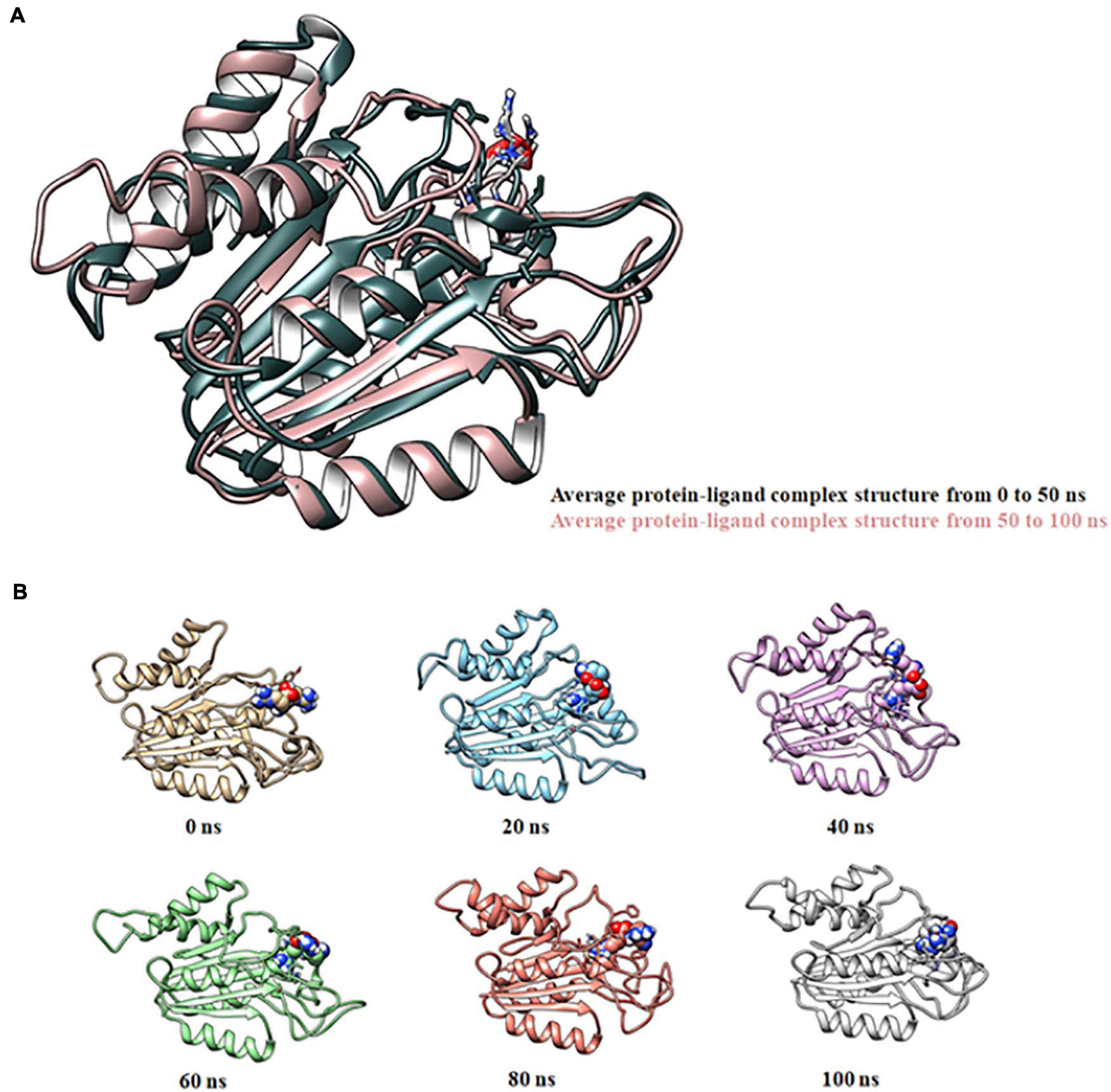
Figure 8. Molecular dynamic simulation of the HC–RR complex for 100 ns displaying its stability. (A) Superimposition of average complex structures between 0 and 50 ns and between 51 and 100 ns for determining the RMSD. RMSD was found to be 0.89 Å, suggesting minor deviation. (B) Snapshot of the HC-RR complex at 0, 20, 40, 60, 80, and 100 ns displaying stable incorporation of RR in the HC throughout the simulation time frame.
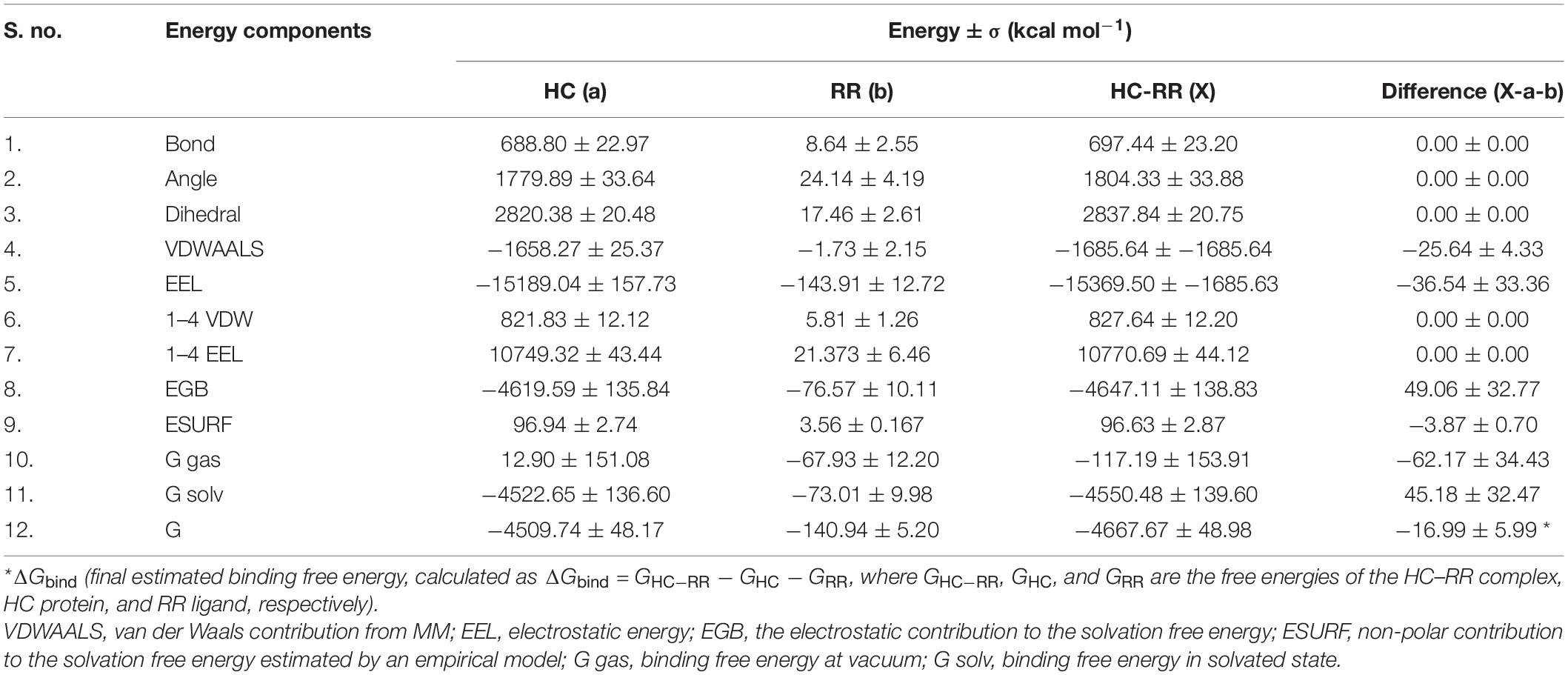
Table 2. Molecular mechanical energies combined with the generalized born and surface area continuum salvation (MMGBSA) calculation for the HC–RR complex.
Discussion
Rich Pool of Cyanobacterial OCAs Having Considerable Diversity and Sharing a Complex Phylogenetic Relationship
The distribution of putative OCAs among cyanobacteria exhibited a clear relation with larger genome size and morphological complexities (Kharwar et al., 2021), since the Oscillatoriales and Nostocales abundantly harbored them. This assumption was also supported by the absence of OCAs in the majority of the unicellular members of Synechococcales and their presence in the filamentous ones. However, the occurrence of considerable putative OCA genes among the Chroococcales, Pleurocapsales, and Chroococcidiopsidales suggested the existence of at least one more factor apart from genome size and morphology, favoring the prevalence of these genes. For example, the probable existence of OCA activity in colonies of Microcystis may confer advantages upon stress through altruistic adaptation, thereby enhancing inclusive fitness of the population (Durand et al., 2016). Further, the early origin of OCAs among cyanobacteria was depicted by the presence of these genes in the primitive cyanobacterium, Gloeobacter (Asplund-Samuelsson et al., 2012). Therefore, the loss of OCAs largely in the monophyletic clades of unicellular marine cyanobacteria Prochlorococcus, Synechococcus, and Synechocystis might be a cumulative consequence of extensive genome streamlining and solitary lifestyle (Ran et al., 2010; Larsson et al., 2011). On the other hand, the higher abundance of OCAs in the marine cyanobacterium Trichodesmium erythraeum IMS101 not only indicated the varied lifestyle preferences among cyanobacteria despite sharing the same habitat but also suggested lesser contribution of habitat in favoring the acquisition of these proteases. The occurrence of more OCAs in the mesophilic Leptolyngbya as compared to the thermophilic counterpart (Thermoleptolyngbya sp. PKUAC-SCTA183), further strengthened the previous assumption.
The rich pool of putative OCAs in cyanobacteria also displayed significant diversity based on the active site motif. While ∼72% accounted for wild-type OCAs (having conventional HC dyad) the rest ∼28% was pseudo-OCAs. In general, these pseudo-OCAs in cyanobacteria are considered as catalytically inert due to lack of functional HC motif in the active site, but in Trypanosoma brucei the role of TbMC4 (MCA having YS catalytic dyad) in the cell cycle progression and virulence warranted their activity in cellular processes (Proto et al., 2011). A recent in silico study also proposed the putative role of these pseudo-enzymes in photosynthesis (Klemenčič et al., 2019). Additionally, the presence of only true OCAs in Gloeobacter possibly indicated the origin of these pseudo-OCAs from their wild-type relatives in cyanobacteria (Klemenčič et al., 2019).
Being a rich repertoire of functional domains, apart from Peptidase C14, cyanobacterial OCAs presumably display multifunctionality, since these domains possess diverse catalytic capacities like protein–protein interactions (WD40), protein modification (FGE sulfatase), scaffold (HEAT_2), and signaling (GUN4). Mostly, these accessory domains are located C-terminally to Peptidase C14 but in some OCAs the C-terminally positioned Peptidase C14 domain was also observed, although these alterations in domain architecture may not have any functional amendments. In addition, the distribution of accessory domains was also uneven among cyanobacteria, since the OCAs from heterocytous strains with broader physiological abilities exhibited the presence of diverse accessory domains. The underlying reason is possibly the higher abundance of true OCAs in heterocytous strains. Additionally, the employment of these multidomain OCAs may benefit the physiologically robust life strategies. Furthermore, the presence of TM infers membrane localization of some OCAs, while others devoid of TM are probably cytosolic. The presence of putative cytosolic and membrane-bound true OCAs depicted the possibility of both intrinsic and extrinsic PCD in cyanobacteria (Bhattacharjee and Mishra, 2020), yet they are poorly appreciated in prokaryotes and require extensive research to validate.
MSA and phylogeny revealed significant sequence diversity and intermixing of cyanobacterial OCAs from different taxonomical orders. The wild-type OCAs were subsumed within nine distinct clades, whereas YS and YN motifs containing pseudo-variants formed two distinct clades, respectively; however, all other pseudo-OCAs were scattered throughout the tree. Mostly, the clades bracketed cyanobacterial strains with different morphological features and/or lifestyle preferences, suggesting the possibility of horizontal gene transfer (HGT) events for acquiring these genes (Bidle and Falkowski, 2004). Additionally, the appearance of several OCAs from the same strain in different clades may also indicate multiple HGT events (Bhattacharjee and Mishra, 2020). Interestingly, the YN pseudo-variant was only harbored by nitrogen-fixing multicellular cyanobacteria, whereas the YS variant was harbored by unicellular to multicellular strains, thus insinuating the possible physiological advantages of the YN variant in multicellular nitrogen-fixing strains. Besides, the distinct clades for YS and YN perhaps imply some functional similarities of these pseudo-proteases, while the scattered distribution of other pseudo-OCAs presumably implied their independent acquisition in cyanobacteria (Klemenčič et al., 2019). Nonetheless, in the future with more sequences of the putative cyanobacterial OCAs, a clearer phylogenetic relationship could be unveiled.
Docking and MD Simulation Simultaneously Revealed Basic Substrate Specificity for Wild-Type OCA
The structural homology of three putative OCA variants with the HC, YS, and YN catalytic dyads is conferred by conserved CHF in their Peptidase C14 domain. Therefore, the substitution of catalytic motifs does not considerably amend the 3D conformation of these proteins (Klemenčič et al., 2019); however, the catalytic capabilities or substrate specificity will presumably alter. True OCAs preferably display substrate specificity for basic substrates due to critical negatively charged amino acid residues in their specificity pocket, as earlier observed in MaOC1 (Klemenčič et al., 2015). However, preference for basic substrates is common for all the characterized caspase homologs, till date. Expectedly, in our study, the putative wild-type OCA, WP_010999143.1 (HC), displayed high substrate affinity for the RR dipeptide, whereas no binding complex was formed with DEVD. Furthermore, the structural stability of the HC-RR complex was validated by analyzing the trajectory for the 100-ns MD simulation run. The negative binding energy calculated using MMGBSA also confirmed the binding efficacy of the RR dipeptide with the protease. The accommodation for the RR dipeptide was possibly determined by the two acidic residues, i.e., D132 and E176, in the binding pocket, thus providing Arg-P1 cleavage specificity to the protease. Similarly, in TbMC2 (MCA of T. brucei) D95 and D211 exhibited their significance in binding basic substrates (Bidle and Falkowski, 2004). Additionally, D353, E500, and D266 were previously identified as significant contributors for substrate-binding pockets in NtMC1 (MCA of Nicotiana tabacum L.), PCA MALT-1, and AtMC9 (MCA of Arabidopsis thaliana), respectively (Jong et al., 2011; Acosta-Maspons et al., 2014).
Conclusion
The identification of caspase homologs in prokaryotes advocated for the possibility of a protease-based cell death mechanism among these life forms. However, the involvement of all these caspase homologs in cell execution pathways is still uncertain. The present study exclusively describes the distribution, diversity, abundance, domain architectures, and phylogenetic relationship of putative cyanobacterial OCAs. Further, this work unraveled the conservation of CHF and Arg-P1 specificity of wild-type OCA in Nostoc sp. PCC 7120 through detailed structural analysis, molecular docking, and MD simulation. Although the wild-type cyanobacterial OCAs possessed conserved fold and catalytic dyad, the protease activity and substrate specificity cannot be corroborated without experimental validations. However, delineating the functionality of the OCAs in cyanobacteria, progenitor of oxygenic photosynthesis, is crucial for understanding not only the evolution and acquisition of caspase homologs in the plant kingdom but also the physiological pertinence of these proteases in PCD.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
SB conceptualized the study and performed the computational analysis. SB and SK retrieved sequences from the database and performed the curation and analysis. AKM conceived and supervised the work. SB wrote the manuscript with contribution and thorough revision from SK and AKM. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Marina Klemenčič, University of Ljubljana, Ljubljana, Slovenia, for her generous guidance during this work. We are also grateful to Dr. Vinay Singh, Information Officer, School of Biotechnology, Banaras Hindu University, Varanasi, India, for providing molecular docking facility by Biovia Discovery Studio 2019. We sincerely acknowledge the support from Institutions of Eminence (IoE-6031), Banaras Hindu University. We also acknowledge Dr. Atul Jaiswal Research Associate, Department of Biochemical Engineering and Biotechnology, IIT, Delhi, India, and Quickiscool.org for performing MD simulation. SB is thankful to Mr. S. Keertic Aswin, Pigmentation Cell Biology Laboratory, CSIR-IGIB, New Delhi, India, for his constant support and guidance. SB and SK are also grateful to the Council of Scientific and Industrial Research (CSIR) and University Grants Commission (UGC), New Delhi, for the award of a Junior Research Fellowship (JRF) and Senior Research Fellowship (SRF), respectively.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.682306/full#supplementary-material
Supplementary Figure 1 | Neighbor-joining tree defining phylogenetic relationship within distinct (A) HC I, (B) HC II and (C) HC III clades.
Supplementary Figure 2 | Neighbor-joining tree defining phylogenetic relationship within distinct HC IV clades.
Supplementary Figure 3 | Neighbor-joining tree defining phylogenetic relationship within distinct (A) HC V, (B) HC VI, (C) HC VII, (D) HC VIII and (E) HC IX clades.
Supplementary Figure 4 | Neighbor-joining tree defining phylogenetic relationship within distinct (A) YS and (B) YN clades.
Supplementary Table 1 | Domain positioning and configuration of all the putative cyanobacterial orthocaspases.
Supplementary Table 2 | Detail of templates used for model refinement and energy minimization for three homology models of putative orthocaspases of Nostoc sp. PCC 7120.
Supplementary Table 3 | Structure quality assessment using Ramachandran plot (PROCHECK) and ModFold8.
Supplementary Table 4 | Detailed description of cyanobacterial strains and the number of diverse putative orthocaspases harbored by them.
Abbreviations
DELTA-BLAST, domain enhanced lookup time accelerated-BLAST; DEVD, aspartyl-glutamyl-valyl-aspartate; MMGBSA, molecular mechanics/generalized born surface area; OCA, orthocaspase; PCD, programmed cell death; RMSD, root mean square deviation; RMSF, root mean square fluctuations; RR, arginylarginine.
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/
- ^ https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi
- ^ http://www.cbs.dtu.dk/services/TMHMM-2.0/
- ^ http://skylign.org/
- ^ https://swissmodel.expasy.org/interactive
- ^ https://zhanglab.ccmb.med.umich.edu/ModRefiner/
- ^ https://servicesn.mbi.ucla.edu/PROCHECK/
- ^ https://www.reading.ac.uk/bioinf/ModFOLD/
- ^ https://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=index.html
References
Acosta-Maspons, A., Sepúlveda-García, E., Sánchez-Baldoquín, L., Marrero-Gutiérrez, J., Pons, T., Rocha-Sosa, M., et al. (2014). Two aspartate residues at the putative p10 subunit of a type II metacaspase from Nicotiana tabacum L. may contribute to the substrate-binding pocket. Planta 239, 147–160. doi: 10.1007/s00425-013-1975-0
Aravind, L., and Koonin, E. V. (2002). Classification of the caspase–hemoglobinase fold: detection of new families and implications for the origin of the eukaryotic separins. Proteins 46, 355–367. doi: 10.1002/prot.10060
Asplund-Samuelsson, J., Bergman, B., and Larsson, J. (2012). Prokaryotic caspase homologs: phylogenetic patterns and functional characteristics reveal considerable diversity. PLoS One 7:e49888. doi: 10.1371/journal.pone.0049888
Asplund-Samuelsson, J., Sundh, J., Dupont, C. L., Allen, A. E., McCrow, J. P., Celepli, N. A., et al. (2016). Diversity and expression of bacterial metacaspases in an aquatic ecosystem. Front. Microbiol. 7:1043. doi: 10.3389/fmicb.2016.01043
Benkert, P., Biasini, M., and Schwede, T. (2011). Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–350. doi: 10.1093/bioinformatics/btq662
Berendsen, H. J., Postma, J. V., van Gunsteren, W. F., DiNola, A. R. H. J., and Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. doi: 10.1063/1.448118
Bertoni, M., Kiefer, F., Biasini, M., Bordoli, L., and Schwede, T. (2017). Modeling protein quaternary structure of homo-and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 7:10480.
Bhattacharjee, S., and Mishra, A. K. (2020). The tale of caspase homologues and their evolutionary outlook: deciphering programmed cell death in cyanobacteria. J. Exp. Bot. 71, 4639–4657. doi: 10.1093/jxb/eraa213
Bidle, K. D., and Falkowski, P. G. (2004). Cell death in planktonic, photosynthetic microorganisms. Nat. Rev. Microbiol. 2, 643–655. doi: 10.1038/nrmicro956
Bienert, S., Waterhouse, A., de Beer, T. A., Tauriello, G., Studer, G., Bordoli, L., et al. (2017). The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 45, D313–D319.
Boratyn, G. M., Schäffer, A. A., Agarwala, R., Altschul, S. F., Lipman, D. J., and Madden, T. L. (2012). Domain enhanced lookup time accelerated BLAST. Biol. Direct. 7:12. doi: 10.1186/1745-6150-7-12
Case, D. A., Betz, R. M., Cerutti, D. S., Cheatham, T., Darden, T., Duke, R., et al. (2016). Amber 2016. San Francisco, CA: University of California.
Choi, C. J., and Berges, J. A. (2013). New types of metacaspases in phytoplankton reveal diverse origins of cell death proteases. Cell Death Dis. 4:e490. doi: 10.1038/cddis.2013.21
Darden>Darden, T., York, D., and Pedersen, L. (1993). Particle mesh Ewald: an N⋅ log (N) method for Ewald sums in large systems. Int. J. Chem. Phys. 98, 10089–10092. doi: 10.1063/1.464397
Durand, P. M., Sym, S., and Michod, R. E. (2016). Programmed cell death and complexity in microbial systems. Curr. Biol. 26, R587–R593.
Earnshaw, W. C., Martins, L. M., and Kaufmann, S. H. (1999). Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68, 383–424. doi: 10.1146/annurev.biochem.68.1.383
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fuentes-Prior, P., and Salvesen, G. S. (2004). The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 384, 201–232. doi: 10.1042/bj20041142
Guex, N., Peitsch, M. C., and Schwede, T. (2009). Automated comparative protein structure modeling with SWISS-MODEL and Swiss-Pdb Viewer: a historical perspective. Electrophoresis 30, S162–S173.
Hachmann, J., Snipas, S. J., van Raam, B. J., Cancino, E. M., Houlihan, E. J., Poreba, M., et al. (2012). Mechanism and specificity of the human paracaspase MALT1. Biochem. J. 443, 287–295. doi: 10.1042/bj20120035
Hander, T., Fernández-Fernández, ÁD., Kumpf, R. P., Willems, P., Schatowitz, H., Rombaut, D., et al. (2019). Damage on plants activates Ca2+-dependent metacaspases for release of immune modulatory peptides. Science 363:eaar7486. doi: 10.1126/science.aar7486
Jakalian, A., Jack, D. B., and Bayly, C. I. (2002). Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. parameterization and validation. J. Comput. Chem. 23, 1623–1641. doi: 10.1002/jcc.10128
Jaworski, M., and Thome, M. (2016). The paracaspase MALT1: biological function and potential for therapeutic inhibition. Cell. Mol. Life Sci. 73, 459–473. doi: 10.1007/s00018-015-2059-z
Jiang, Q., Qin, S., and Wu, Q. Y. (2010). Genome-wide comparative analysis of metacaspases in unicellular and filamentous cyanobacteria. BMC Genomics 11:198. doi: 10.1186/1471-2164-11-198
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Jong, W. Y., Jeffrey, P. D., Ha, J. Y., Yang, X., and Shi, Y. (2011). Crystal structure of the mucosa-associated lymphoid tissue lymphoma translocation 1 (MALT1) paracaspase region. Proc. Natl. Acad. Sci. U.S.A. 108, 21004–21009. doi: 10.1073/pnas.1111708108
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. Int. J. Chem. Phys. 79, 926–935. doi: 10.1063/1.445869
Joung, I. S., and Cheatham, T. E. (2008). Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B. 112, 9020–9041. doi: 10.1021/jp8001614
Kharwar, S., Bhattacharjee, S., and Mishra, A. K. (2021). Disentangling the impact of sulfur limitation on exopolysaccharide and functionality of Alr2882 by in silico approaches in Anabaena sp. PCC 7120. Appl. Biochem. Biotechnol. 193, 1447–1468. doi: 10.1007/s12010-021-03501-3
Klemenèiè, M., and Funk, C. (2018). Structural and functional diversity of caspase homologues in non-metazoan organisms. Protoplasma 255, 387–397. doi: 10.1007/s00709-017-1145-5
Klemenčič, M., Asplund-Samuelsson, J., Dolinar, M., and Funk, C. (2019). Phylogenetic distribution and diversity of bacterial pseudo-orthocaspases underline their putative role in photosynthesis. Front. Plant Sci. 10:293. doi: 10.3389/fpls.2019.00293
Klemenčič, M., Novinec, M., and Dolinar, M. (2015). Orthocaspases are proteolytically active prokaryotic caspase homologues: the case of Microcystis aeruginosa. Mol. Microbiol. 98, 142–150. doi: 10.1111/mmi.13110
Koonin, E. V., and Aravind, L. (2002). Origin and evolution of eukaryotic apoptosis: the bacterial connection. Cell Death Differ. 9, 394–404. doi: 10.1038/sj.cdd.4400991
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Larsson, J., Nylander, J. A., and Bergman, B. (2011). Genome fluctuations in cyanobacteria reflect evolutionary, developmental and adaptive traits. BMC Evol. Biol. 11:187. doi: 10.1186/1471-2148-11-187
Laskowski, R. A. (2001). PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res. 29, 221–222. doi: 10.1093/nar/29.1.221
Laskowski, R. A., and Swindells, M. B. (2011). LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786. doi: 10.1021/ci200227u
Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. doi: 10.1107/s0021889892009944
Liu, W., Xie, Y., Ma, J., Luo, X., Nie, P., Zuo, Z., et al. (2015). IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics 31, 3359–3361. doi: 10.1093/bioinformatics/btv362
Maghrabi, A. H. A., and McGuffin, L. J. (2017). ModFOLD6: an accurate web server for the global and local quality estimation of 3D models of proteins. Nucleic Acids Res. 45, W416–W421.
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015). ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713. doi: 10.1021/acs.jctc.5b00255
Marchler-Bauer, A., Bo, Y., Han, L., He, J., Lanczycki, C. J., Lu, S., et al. (2017). CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203.
Minina, E. A., Coll, N. S., Tuominen, H., and Bozhkov, P. V. (2017). Metacaspases versus caspases in development and cell fate regulation. Cell Death Differ. 24, 1314–1325. doi: 10.1038/cdd.2017.18
Minina, E. A., Staal, J., Alvarez, V. E., Berges, J. A., Berman-Frank, I., Beyaert, R., et al. (2020). Classification and nomenclature of metacaspases and paracaspases: no more confusion with caspases. Mol. Cell. 77, 927–929.
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L., et al. (2021). Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419.
Onufriev, A., Bashford, D., and Case, D. A. (2004). Exploring protein native states and large−scale conformational changes with a modified generalized born model. Proteins 55, 383–394. doi: 10.1002/prot.20033
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Proto, W. R., Castanys-Munoz, E., Black, A., Tetley, L., Moss, C. X., and Juliano, L. (2011). Trypanosoma brucei metacaspase 4 is a pseudopeptidase and a virulence factor. J. Biol. Chem. 286, 39914–39925. doi: 10.1074/jbc.m111.292334
Ran, L., Larsson, J., Vigil-Stenman, T., Nylander, J. A., Ininbergs, K., Zheng, W. W., et al. (2010). Genome erosion in a nitrogen-fixing vertically transmitted endosymbiotic multicellular cyanobacterium. PLoS One 5:e11486. doi: 10.1371/journal.pone.0011486
Ryckaert, J. P., Ciccotti, G., and Berendsen, H. J. (1977). Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. doi: 10.1016/0021-9991(77)90098-5
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Studer, G., Rempfer, C., Waterhouse, A. M., Gumienny, R., Haas, J., and Schwede, T. (2020). QMEANDis Co-distance constraints applied on model quality estimation. Bioinformatics 36, 1765–1771. doi: 10.1093/bioinformatics/btz828
Tsiatsiani, L., van Breusegem, F., Gallois, P., Zavialov, A., Lam, E., and Bozhkov, P. V. (2011). Metacaspases. Cell Death Differ. 18, 1279–1288.
Uren, A. G., O’Rourke, K., Aravind, L. A., Pisabarro, M. T., Seshagiri, S., Koonin, E. V., et al. (2000). Identification of paracaspases and metacaspases: two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol. Cell. 6, 961–967. doi: 10.1016/s1097-2765(00)00094-0
Wang, J., Wang, W., Kollman, P. A., and Case, D. A. (2006). Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 25, 247–260. doi: 10.1016/j.jmgm.2005.12.005
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., and Case, D. A. (2004). Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174. doi: 10.1002/jcc.20035
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303.
Wheeler, T. J., Clements, J., and Finn, R. D. (2014). Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinformatics 15:7. doi: 10.1186/1471-2105-15-7
Wu, G., Robertson, D. H., Brooks, C. L., and Vieth, M. (2003). Detailed analysis of grid-based molecular docking: a case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 24, 1549–1562. doi: 10.1002/jcc.10306
Wu, X., and Brooks, B. R. (2003). Self-guided Langevin dynamics simulation method. Chem. Phys. Lett. 381, 512–518. doi: 10.1016/j.cplett.2003.10.013
Xu, D., and Zhang, Y. (2011). Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 101, 2525–2534. doi: 10.1016/j.bpj.2011.10.024
Keywords: cyanobacteria, horizontal gene transfer, orthocaspases, phylogeny, programmed cell death
Citation: Bhattacharjee S, Kharwar S and Mishra AK (2021) Insights Into the Phylogenetic Distribution, Diversity, Structural Attributes, and Substrate Specificity of Putative Cyanobacterial Orthocaspases. Front. Microbiol. 12:682306. doi: 10.3389/fmicb.2021.682306
Received: 18 March 2021; Accepted: 02 June 2021;
Published: 02 July 2021.
Edited by:
Leda Giannuzzi, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Daniela Sueldo, Norwegian University of Science and Technology, NorwayJens Staal, Ghent University, Belgium
Gerard Abraham, Indian Agricultural Research Institute (ICAR), India
Copyright © 2021 Bhattacharjee, Kharwar and Mishra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arun Kumar Mishra, YWttaXNocmFhdUBob3RtYWlsLmNvbQ==
†ORCID: Samujjal Bhattacharjee, orcid.org/0000-0002-9802-1461; Surbhi Kharwar, orcid.org/0000-0003-1737-3068; Arun Kumar Mishra, orcid.org/0000-0001-6096-8097