 Garam Choi1,2†
Garam Choi1,2† Sang Ho Choi
Sang Ho Choi- 1National Research Laboratory of Molecular Microbiology and Toxicology, Department of Agricultural Biotechnology, Seoul National University, Seoul, South Korea
- 2Center for Food and Bioconvergence, Seoul National University, Seoul, South Korea
Nitric oxide (NO) is an important antimicrobial effector produced by the host innate immune system to counteract invading pathogens. To survive and establish a successful infection, a fulminating human pathogen Vibrio vulnificus expresses the hmpA gene encoding an NO dioxygenase in an NO-responsive manner. In this study, we identified an Rrf2-family transcriptional regulator NsrR that is predicted to contain the Fe-S cluster coordinated by three cysteine residues. Transcriptome analysis showed that NsrR controls the expression of multiple genes potentially involved in nitrosative stress responses. Particularly, NsrR acts as a strong repressor of hmpA transcription and relieves the repression of hmpA upon exposure to NO. Notably, nsrR and hmpA are transcribed divergently, and their promoter regions overlap with each other. Molecular biological analyses revealed that NsrR directly binds to this overlapping promoter region, which is alleviated by loss of the Fe-S cluster, leading to the subsequent derepression of hmpA under nitrosative stress. We further found that a leucine-responsive regulatory protein (Lrp) negatively regulates hmpA in an NsrR-dependent manner by directly binding to the promoter region, presumably resulting in a DNA conformation change to support the repression by NsrR. Meanwhile, a cyclic AMP receptor protein (CRP) positively regulates hmpA probably through repression of nsrR and lrp by directly binding to each promoter region in a sequential cascade. Altogether, this collaborative regulation of NsrR along with Lrp and CRP enables an elaborate control of hmpA transcription, contributing to survival under host-derived nitrosative stress and thereby the pathogenesis of V. vulnificus.
Introduction
Nitric oxide (NO) is a highly reactive, toxic, and membrane-permeable radical gas. As one of the major components of the host innate immune system, NO is produced by inducible NO synthase (iNOS) which is expressed in phagocytes and epithelial cells under infectious conditions (Fang, 2004; Wang et al., 2010). NO produced by iNOS can subsequently be converted into other toxic reactive nitrogen species (RNS) such as nitrogen dioxide (NO2), peroxynitrite (ONOO−), and dinitrogen trioxide (N2O3) which impose the nitrosative stress on pathogens (Fang, 2004; Stern and Zhu, 2014). Furthermore, intestinal commensals can reduce nitrate () in the diet to nitrite (), which interacts with gastric acid, resulting in RNS that act as antimicrobial barriers against ingested enteric pathogens (Sobko et al., 2005; Tiso and Schechter, 2015). RNS can cause damage to cellular components, including the metal centers of proteins, membrane lipids and nucleotide bases, and thereby inhibit respiration and interfere with the DNA replication of pathogens (Fang, 2004). Therefore, pathogens have evolved sophisticated mechanisms to sense the increased level of RNS and express the proper genes to overcome nitrosative stress in a host (Bang et al., 2006; Stern et al., 2012).
To understand the NO-responsive gene expression in pathogens, numerous transcriptional regulators have been characterized (Spiro, 2007). Among them, two transcriptional regulators, NorR and NsrR, are known to have focused functions on sensing NO in a wide range of bacteria (Stern and Zhu, 2014). NorR directly recognizes NO using its non-heme iron center and controls the expression of genes for NO detoxification: norVW in Escherichia coli and hmpA and nnrS in Vibrio cholerae (D'Autreaux et al., 2005; Stern et al., 2012). Meanwhile, NsrR uses an iron-sulfur (Fe-S) cluster as a cofactor to directly sense NO and regulates a variety of genes involved in NO detoxification and NO damage repair, particularly hmpA in E. coli, Salmonella enterica serovar Typhimurium, and Streptomyces coelicolor (Bang et al., 2006; Spiro, 2007; Tucker et al., 2008). The NO-responsive Fe-S cluster is coordinated to NsrR by three cysteine residues which are widely conserved in various bacterial NsrR (Tucker et al., 2010). A recent study showed that both [2Fe-2S] and [4Fe-4S] clusters can be coordinated to S. coelicolor NsrR (Crack et al., 2015). Upon exposure to NO, the Fe-S cluster is nitrosylated, forming the iron-nitrosyl species such as dinitrosyl iron complex (DNIC), Roussin's Red Ester (RRE), and Roussin's Black Salt (RBS) (Serrano et al., 2016; Crack and Le Brun, 2019). The resulting apo-NsrR lacking an intact Fe-S cluster shows a distinct protein conformation from that of holo-NsrR, leading to loss of DNA-binding activity and the subsequent derepression of its regulons (Crack et al., 2015; Volbeda et al., 2017). NsrR, as a homodimer, binds to the consensus NsrR-binding site consisting of inverted repeats of two 11 bp motifs (AAxATGCATTT; x, any nucleotide) separated by 1 bp spacing (Partridge et al., 2009; Crack et al., 2015).
The opportunistic human pathogen Vibrio vulnificus is a causative agent of foodborne diseases from mild gastroenteritis to primary septicemia (Jones and Oliver, 2009; Baker-Austin and Oliver, 2018). During infection, V. vulnificus exploits various transcriptional regulators to sense host-derived signals and modulate the expression of its virulence genes (Miller et al., 1989; Fang et al., 2016). Particularly, a leucine-responsive regulatory protein (Lrp) and a cyclic AMP receptor protein (CRP) are widely conserved and well-characterized global transcriptional regulators in bacteria (Cho et al., 2008; Manneh-Roussel et al., 2018). Lrp controls diverse cellular functions including amino acid metabolism, stress resistance, and virulence (Jeong et al., 2003; Rhee et al., 2008; Lee et al., 2020). The regulatory activity of Lrp on its regulons can be enhanced, reversed, or unaffected by the binding of a small effector molecule leucine (Cho et al., 2008). CRP is a central regulator of carbon and energy metabolism that forms a complex with cyclic AMP (cAMP) (Kim et al., 2011; Lee et al., 2020). In the absence of glucose, the intracellular cAMP level is increased by adenylate cyclase and the resulting cAMP-CRP complex binds DNA to regulate gene expression (Manneh-Roussel et al., 2018). In this way, Lrp and CRP coordinate the expression of genes involved in metabolism and pathogenesis in response to changing environmental conditions such as nutrient availability.
Like many other enteropathogenic bacteria, V. vulnificus is inevitably exposed to host-derived nitrosative stress in the course of infection. We recently reported that a multidomain NO dioxygenase HmpA is highly expressed in V. vulnificus exposed to NO (Kim et al., 2019). HmpA belongs to the flavohemoglobin family composed of the N-terminal heme-binding globin domain and the C-terminal NAD- and FAD-binding oxidoreductase domain, and detoxifies NO by oxidizing it to a less toxic under aerobic conditions (Bonamore and Boffi, 2008; Forrester and Foster, 2012; Kim et al., 2019). Because the in vitro NO-decomposition activity of V. vulnificus is mostly dependent on HmpA, it has a significant role in the survival and pathogenesis of V. vulnificus under nitrosative stress in a host (Kim et al., 2019). Nevertheless, definitive regulatory mechanisms and transcriptional regulators, by which V. vulnificus senses NO and induces HmpA, have not been yet elucidated in detail. In this study, we newly identified NsrR in V. vulnificus as an NO-responsive transcriptional regulator. The transcriptome analysis of the wild-type and isogenic nsrR-deletion mutant (ΔnsrR) strains revealed that NsrR controls the expression of 47 genes. Notably, hmpA was the most highly induced gene by the nsrR deletion, indicating that NsrR acts as a strong repressor of hmpA. To investigate the exact mechanism by which NsrR regulates hmpA expression, the hmpA transcript levels were compared in the wild-type and ΔnsrR strains under nitrosative stress in vitro and ex vivo. Furthermore, the combined effect of NsrR, Lrp, and CRP on hmpA expression was analyzed at the molecular level. In conclusion, this study suggests that NsrR tightly regulates hmpA transcription in response to nitrosative stress together with Lrp and CRP, contributing to the survival and overall success of V. vulnificus during host infection.
Results
Genome and Transcriptome Analyses Identified NsrR in V. vulnificus
We previously reported that NO-induced HmpA encoded by VVMO6_RS01375 is crucial for survival under host-derived nitrosative stress and pathogenesis of V. vulnificus during infection (Kim et al., 2019). Notably, we further found that the expression of VVMO6_RS01380, which is divergently transcribed from hmpA (Figure 1A), is also induced by NO (Kim et al., 2019). VVMO6_RS01380 encodes an Rrf2-family transcriptional regulator showing an amino acid sequence homology to E. coli NsrR, S. Typhimurium NsrR, and S. coelicolor NsrR (61, 62, and 35% identity, respectively) (Figure 1B). Moreover, the protein encoded by VVMO6_RS01380 contains three conserved cysteine residues, C91, C96, and C102, which are known to be essential for the Fe-S cluster ligation of Rrf2-family transcriptional regulators (Figure 1B) (Volbeda et al., 2017). This observation led us to designate the VVMO6_RS01380 gene product as an Fe-S cluster-containing transcriptional regulator NsrR.
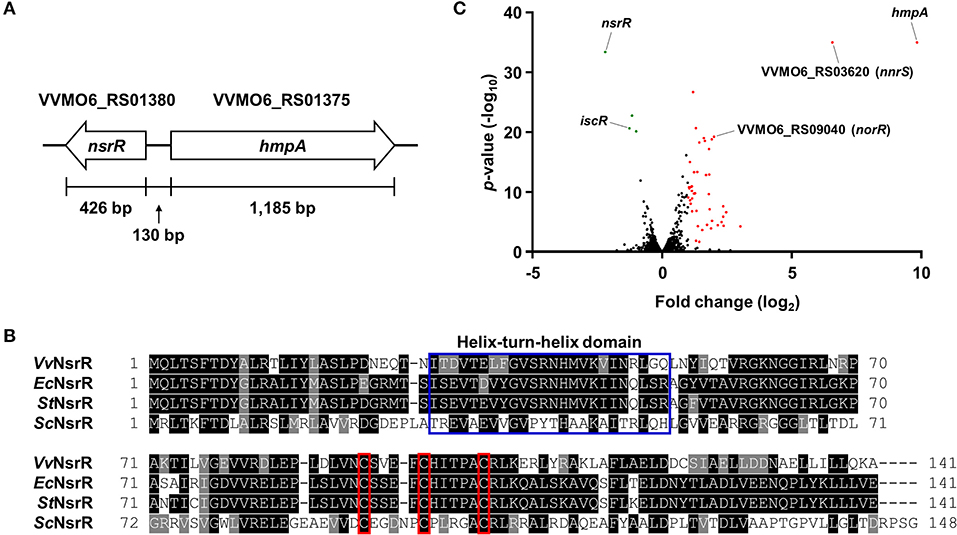
Figure 1. Identification of NsrR and transcriptome analysis of its downstream genes. (A) The physical map of nsrR and hmpA on the V. vulnificus MO6-24/O genome. The open arrows represent the coding regions and transcriptional directions of the genes. (B) The amino acid sequences of various bacterial NsrRs were retrieved from the NCBI protein database and aligned using the T-Coffee alignment program (Notredame et al., 2000). Identical sequences and conserved sequences are shaded in black and gray, respectively. Dashed lines represent missing sequences. Conserved helix-turn-helix DNA-binding motif and three cysteine residues potentially involved in the Fe-S cluster ligation are indicated by a blue open box and red open boxes, respectively. VvNsrR, V. vulnificus NsrR; EcNsrR, E. coli NsrR; StNsrR, S. Typhimurium NsrR; ScNsrR, S. coelicolor NsrR. (C) The volcano plot depicting the genes differentially expressed by the nsrR deletion (fold change ≥ 2 with p < 0.05). The red dots and green dots represent the differentially up-regulated and down-regulated genes, respectively.
For the comprehensive identification of NsrR-regulated genes in V. vulnificus, the transcriptomes of the wild-type and ΔnsrR strains were compared by RNA-seq. The transcriptome analysis revealed that, in total, 47 genes were differentially expressed by the nsrR deletion: 44 genes were up-regulated and 3 genes were down-regulated (Figure 1C, Supplementary Table 1). The overall fold changes of the up-regulated genes were greater than those of the down-regulated genes. This result implies that NsrR serves mainly as a repressor rather than as an activator. Intriguingly, the up-regulated genes included several genes that are predicted to encode proteins involved in the defense against nitrosative stress such as NO dioxygenase HmpA, NO detoxification protein NnrS, reductase large subunit, reductase small subunit, cytochrome c reductase subunit c552 NrfA, and NO reductase transcriptional regulator NorR (Stern et al., 2012; Kim et al., 2019). Among them, hmpA was the most highly up-regulated gene in the ΔnsrR strain (Figure 1C), suggesting that NsrR is a strong repressor of hmpA expression. Meanwhile, the down-regulated genes, iscR, iscS, and iscU, constitute the isc operon (iscRSUA-hscBA-fdx) encoding proteins required for the biogenesis of the Fe-S cluster (Lim and Choi, 2014). Taken together, this result shows that NsrR controls the expression of multiple genes involved in nitrosative stress responses, especially hmpA.
hmpA Transcription Is Derepressed by NsrR in Response to NO
To validate the RNA-seq results and examine whether NsrR mediates the induction of hmpA in response to NO, the hmpA transcript levels in the wild-type and ΔnsrR strains were compared under nitrosative stress in vitro and ex vivo. The hmpA transcript level in the wild-type strain was significantly elevated upon exposure to an in vitro NO donor, NO/PPNPs (NO-releasing poly(lactic-co-glycolic acid)-polyethylenimine nanoparticles) (Figure 2A) (Nurhasni et al., 2015). This result confirms our previous observation that hmpA is induced by NO (Kim et al., 2019). Additionally, the hmpA transcript level was dramatically increased in the ΔnsrR strain compared with that in the wild-type strain even in the absence of NO/PPNPs (Figure 2A), verifying that NsrR negatively regulates hmpA. Strikingly, the hmpA transcript level in the ΔnsrR strain was not affected by the addition of NO/PPNPs (Figure 2A), indicating that NsrR recognizes NO and alleviates the repression of hmpA expression in vitro.
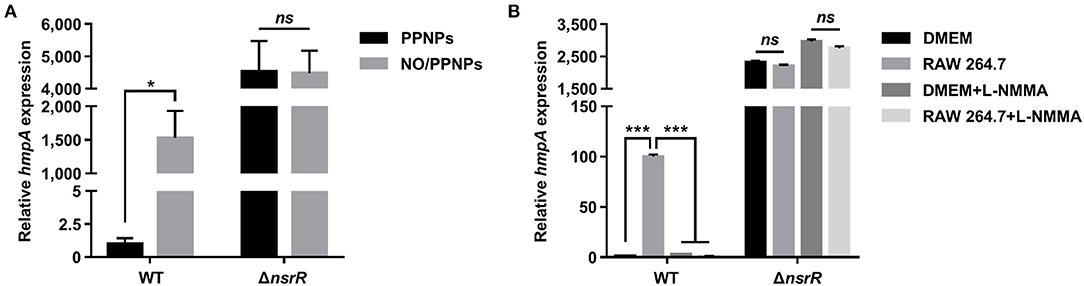
Figure 2. The effect of nitrosative stress and the nsrR mutation on hmpA transcription in vitro and ex vivo. The wild-type and ΔnsrR strains were grown aerobically to an A600 of 0.5, and then exposed to 0.15 mg/ml NO/PPNPs in vitro (A) or NO-producing RAW 264.7 cells ex vivo in the presence or absence of L-NMMA (B) for 10 min. The hmpA transcript levels were determined by qRT-PCR, and the hmpA transcript levels in the wild-type strain exposed to PPNPs (A) or DMEM without L-NMMA (B) were set to 1. Error bars represent the SD. Statistical significance was determined by the Student's t-test (*p < 0.05; ***p < 0.0005; ns, not significant). WT, wild type; ΔnsrR, nsrR-deletion mutant.
The role of NsrR in hmpA expression was further investigated ex vivo using NO-producing murine macrophage RAW 264.7 cells. As shown in Figure 2B, the hmpA transcript level in the wild-type strain exposed to NO-producing RAW 264.7 cells was considerably elevated compared with that exposed to Dulbecco's modified Eagle's medium (DMEM; negative control). The extent of the increase in the hmpA transcript level upon exposure to the RAW 264.7 cells diminished by the addition of the NO synthase inhibitor L-NG-monomethyl arginine citrate (L-NMMA) (Figure 2B). This result suggests that the hmpA induction upon exposure to RAW 264.7 cells is attributable to NO produced by the murine macrophages. In contrast, the highly increased hmpA transcript level in the ΔnsrR strain was not altered by the RAW 264.7 cells and L-NMMA (Figure 2B), confirming that NsrR mediates the derepression of hmpA under nitrosative stress derived from host immune cells. The combined results show that NsrR has a critical role to sense NO and to induce the hmpA expression both in vitro and ex vivo.
Then, we examined whether the introduction of recombinant nsrR can reduce the increased hmpA transcript level in the ΔnsrR strain. Introduction of a nsrR-expressing plasmid significantly decreased the hmpA transcript level, although it was not comparable with that in the wild-type strain (Supplementary Figure 1A). One possible explanation for this lack of complementation is that the recombinant NsrR expressed from the exogenous plasmid is less functional for unknown reasons. On the other hand, ectopic expression of nsrR on the chromosome effectively reduced the hmpA transcript level comparable with that in the wild-type strain (Supplementary Figure 1B). Similarly, the HmpA protein levels in the ΔnsrR strain were highly increased compared with those in the wild-type strain and significantly decreased by complementation (Supplementary Figures 1C,D). Altogether, the results suggest that NsrR is a major transcriptional regulator that recognizes NO and regulates hmpA expression mainly at the transcription level.
Three Conserved Cysteine Residues Are Essential for NsrR to Regulate hmpA and nsrR
As shown in Figure 1B, V. vulnificus NsrR contains three conserved cysteine residues (C91, C96, and C102) that are predicted to act as ligands of the NO-responsive Fe-S cluster (Tucker et al., 2008; Volbeda et al., 2017). To investigate the role of these three cysteine residues, three different strains were constructed: a parent strain GR204 chromosomally encoding 3 × FLAG-tagged NsrR (NsrRFLAG), an isogenic nsrR-deletion mutant, and an isogenic nsrR3CS mutant chromosomally encoding apo-locked NsrRFLAG () (see see Materials and Methods for a detailed description). The hmpA transcript and HmpA protein levels in the ΔnsrR strain were highly elevated compared with those in the parent strain (Figures 3A,B), indicating that NsrRFLAG in the parent strain is still functional as a repressor of hmpA. Notably, the hmpA transcript and HmpA protein levels in the nsrR3CS strain were comparable with those in the ΔnsrR strain (Figures 3A,B). Moreover, a similar effect of the mutation in the three cysteine residues and the nsrR deletion on hmpA expression was observed in the wild-type background (Supplementary Figure 1E). These results that NsrR3CS cannot repress the hmpA transcription reveal that coordination of the Fe-S cluster by the three cysteine residues is essential for the NsrR activity to repress hmpA.
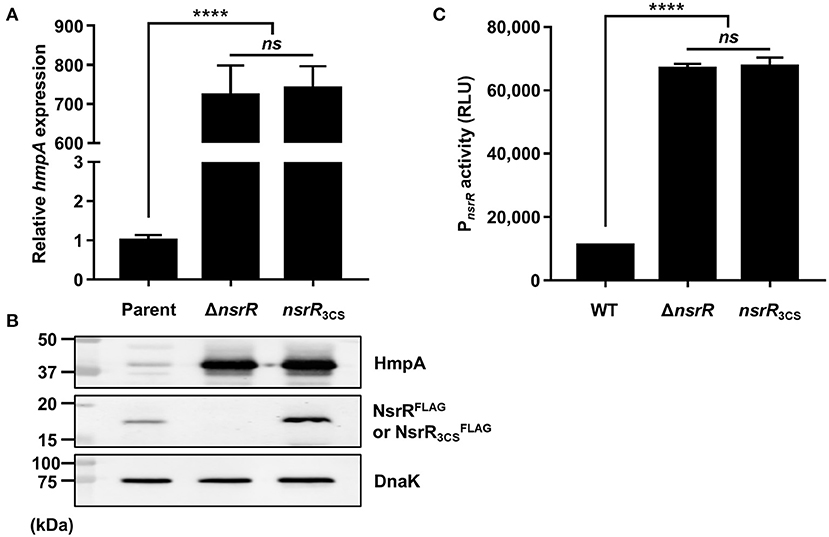
Figure 3. The role of the three cysteine residues in NsrR on hmpA and nsrR transcription. (A,B) Total RNA and proteins were isolated from the parent and mutant strains grown aerobically to an A600 of 0.5. (A) The hmpA transcript levels were determined by qRT-PCR, and the hmpA transcript level in the parent strain was set to 1. (B) The cellular HmpA, NsrRFLAG or , and DnaK (internal control) protein levels were determined by Western blot analysis. Molecular size markers (Bio-Rad) are shown in kDa. Parent, parent strain; ΔnsrR, nsrR-deletion mutant; nsrR3CS, strain expressing apo-locked NsrRFLAG. (C) A PCR fragment carrying the PnsrR was cloned into pBBR-lux to create a reporter plasmid, pGR2025. The wild-type and mutant strains containing pGR2025 were grown aerobically to an A600 of 0.5, and then used to measure the cellular luminescence. Error bars represent the SD. Statistical significance was determined by the Student's t-test (****p < 0.00005; ns, not significant). RLU, relative luminescence unit; WT, wild type; ΔnsrR, nsrR-deletion mutant; nsrR3CS, strain expressing apo-locked NsrR.
Furthermore, the protein level in the nsrR3CS strain was significantly elevated compared with the NsrRFLAG protein level in the parent strain (Figure 3B). This observation prompted us to examine the activity of the nsrR promoter (PnsrR, determined in Figures 4B,C) in the wild-type, ΔnsrR, and nsrR3CS strains using the PnsrR-luxCDABE transcriptional fusion reporter. The PnsrR activity in the ΔnsrR strain was higher than that in the wild-type strain (Figure 3C), demonstrating that NsrR represses its own transcription. Additionally, the increased PnsrR activity in the nsrR3CS strain was comparable with that in the ΔnsrR strain (Figure 3C). This result suggests that NsrR relieves the repression of its own transcription by the mutation in the three cysteine residues and the consequent loss of the Fe-S cluster. Combined with the previous data (Figure 2), we propose a model in which holo-NsrR containing the Fe-S cluster represses both hmpA and nsrR transcription, shifts to the clusterless apo-form under nitrosative stress, and then alleviates the repression of hmpA and nsrR transcription.
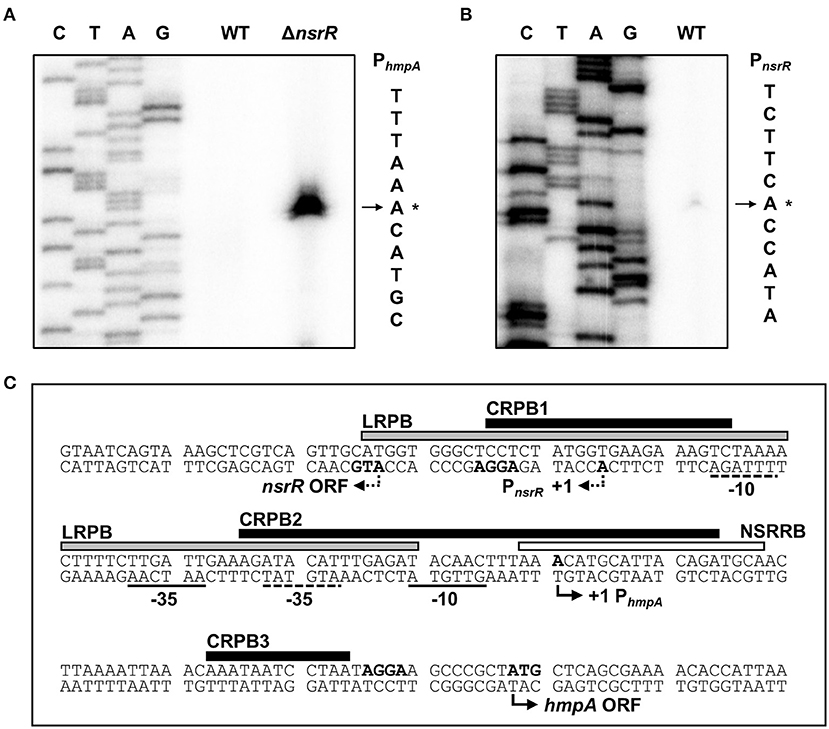
Figure 4. Sequence analysis of the nsrR-hmpA regulatory region. (A,B) The TSSs of hmpA (A) and nsrR (B) were determined by the primer extension of RNA isolated from the wild-type and ΔnsrR strains grown aerobically to an A600 of 0.5. Lanes C, T, A, and G represent the nucleotide sequencing ladders. The asterisks indicate the TSSs. WT, wild type; ΔnsrR, nsrR-deletion mutant. (C) Double-stranded DNA sequence of the nsrR-hmpA regulatory region is shown. The TSS and putative translation start codon of nsrR are indicated by dashed bent arrows, and those of hmpA are indicated by solid bent arrows. The putative −10 and −35 regions are underlined with dashed lines for PnsrR and solid lines for PhmpA. The putative ribosome-binding sites (AGGA) are boldface. The binding sequences of NsrR (NSRRB; a white box), Lrp (LRPB; a gray box), and CRP (CRPB1, CRPB2, CRPB3; black boxes) were determined in the later parts of this study.
PhmpA and PnsrR Overlap Divergently With Each Other
To map the hmpA promoter, the transcription start site (TSS) of hmpA was determined by primer extension analysis. A single reverse transcript was produced from the primer extension of RNA isolated from the ΔnsrR strain grown to an A600 of 0.5 (Figure 4A). This reverse transcript observed in the ΔnsrR strain was not detected in the wild-type strain (Figure 4A), confirming that the hmpA transcription is strongly repressed by NsrR. The 5′-end of hmpA was located 57-bp upstream of the translation start codon of hmpA. Next, the TSS of nsrR was determined in a similar way. A single reverse transcript was produced from the primer extension of RNA isolated from the wild-type strain grown to an A600 of 0.5 (Figure 4B). The 5′-end of nsrR was located 18-bp upstream of the translation start codon of nsrR. The putative promoters constituting the TSSs were named PhmpA and PnsrR to represent the hmpA promoter and the nsrR promoter, respectively. The sequences for putative −10 and −35 regions of each promoter were assigned based on the similarity to the consensus sequences of E. coli σ70 promoters (Figure 4C). Strikingly, these results show that PhmpA and PnsrR overlap with each other. This overlapping promoter region was termed the nsrR-hmpA regulatory region for our further research.
NsrR Directly Binds to the nsrR-hmpA Regulatory Region to Repress hmpA and Its Own Expression
To investigate whether NsrR directly binds to the nsrR-hmpA regulatory region, electrophoretic mobility shift assays (EMSAs) were performed. The addition of NsrR to 6-carboxyfluorescein (6-FAM)-labeled DNA probe encompassing the nsrR-hmpA regulatory region resulted in a single retarded band in an NsrR concentration-dependent manner (Figure 5A). The same unlabeled DNA fragment competed for NsrR binding in a dose-dependent manner (Figure 5A), confirming the specific binding of NsrR. Then, the binding of NsrR3CS to the nsrR-hmpA regulatory region was compared with that of NsrR. The amount of the retarded band of the DNA-NsrR3CS complex was reduced compared with that of the DNA-NsrR complex (Figure 5B). This result implies that the DNA-binding affinity of NsrR3CS is considerably lower than that of NsrR, which leads to the derepression of hmpA and nsrR under nitrosative stress.
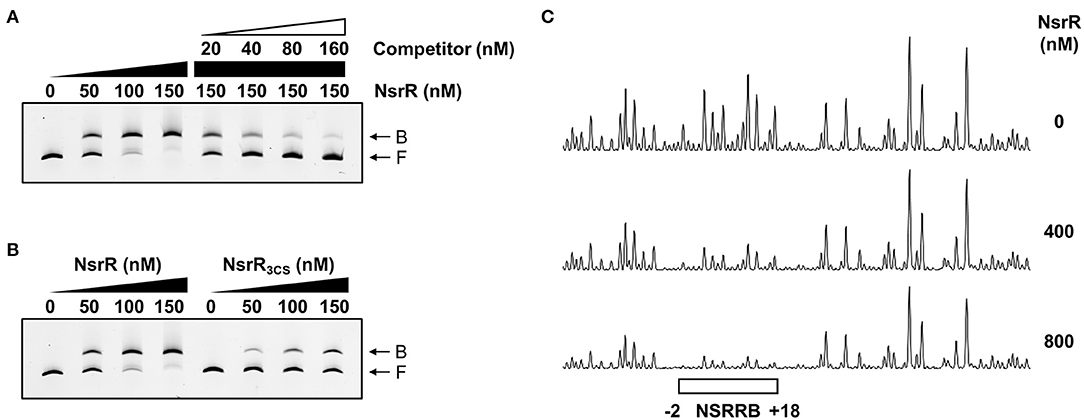
Figure 5. Specific binding of NsrR to the nsrR-hmpA regulatory region. (A,B) A 393-bp DNA fragment of the nsrR-hmpA regulatory region (10 nM) was labeled with 6-FAM, and then incubated with increasing amounts of NsrR (A,B) or NsrR3CS (B) as indicated. For the competition analysis, various amounts of the unlabeled DNA fragment were added as a self-competitor. B, bound DNA; F, free DNA. (C) The same DNA probe (40 nM) was incubated with increasing amounts of NsrR as indicated, and then digested with DNase I. The region protected by NsrR is indicated by a white box (NSRRB). Nucleotide numbers shown are relative to the TSS of hmpA.
To determine the precise location of NsrR-binding site(s) in the nsrR-hmpA regulatory region, DNase I protection assays were performed using the same DNA probe. When NsrR was added to the DNA probe, NsrR protected a single region extending from −2 to +18 (NSRRB, centered at +8.5 from the TSS of hmpA) from DNase I digestion (Figures 4C, 5C). The sequence of NSRRB showed about 87% similarity to the 11-1-11 bp consensus NsrR-binding sequence in E. coli (Bodenmiller and Spiro, 2006; Partridge et al., 2009). Combined with the EMSA data (Figure 5A), these results indicate that NsrR concurrently represses hmpA and its own transcription by directly binding to the single specific sequence in the nsrR-hmpA regulatory region.
Lrp Represses hmpA in an NsrR-Dependent Manner by Directly Binding to the nsrR-hmpA Regulatory Region
To determine other factors involved in the hmpA regulation, we further explored various known transcriptional regulators in V. vulnificus. Among them, the role of Lrp in the hmpA regulation was evaluated. The hmpA transcript and HmpA protein levels in the lrp-deletion mutant (Δlrp) were significantly increased compared with those in the parent strain and restored by complementation (Figures 6A,B). The hmpA transcript level in the wild-type strain was not altered by exogenous leucine (Supplementary Figure 2A), suggesting that Lrp negatively affects the hmpA transcription in a leucine-independent manner. To investigate the regulatory relationship between NsrR and Lrp, the lrp-deleted nsrR3CS mutant (nsrR3CSΔlrp), in which both NsrR and Lrp are not functional, was constructed from the parent strain. Interestingly, the hmpA transcript and HmpA protein levels in the nsrR3CSΔlrp strain were comparable with those in the nsrR3CS strain (Figures 6C,D). The observation that Lrp was not able to affect hmpA transcription in the absence of functional NsrR indicates that the negative effect of Lrp on hmpA is mediated by NsrR. This result led us to examine whether Lrp positively regulates the cellular level of NsrR to repress hmpA. However, both the PnsrR activity and NsrRFLAG protein level were not affected by the lrp deletion (Supplementary Figure 2B, Figure 6B).
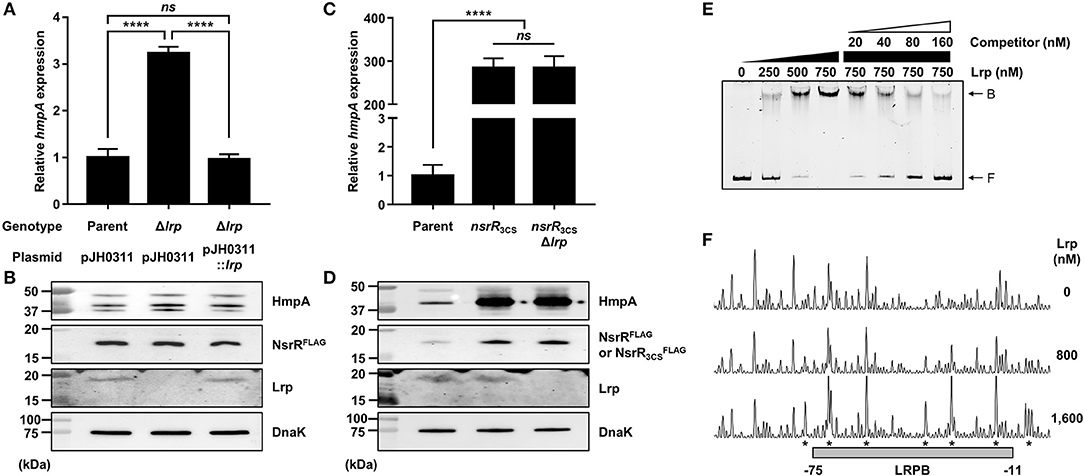
Figure 6. The effect of the lrp mutation on hmpA transcription and the specific binding of Lrp to the nsrR-hmpA regulatory region. (A–D) Total RNA and proteins were isolated from the parent and mutant strains grown aerobically to an A600 of 0.5. (A,C) The hmpA transcript levels were determined by qRT-PCR, and the hmpA transcript levels in the parent strain were set to 1. Error bars represent the SD. Statistical significance was determined by the Student's t-test (****p < 0.00005; ns, not significant). (B,D) The cellular HmpA, NsrRFLAG or , Lrp, and DnaK (internal control) protein levels were determined by Western blot analysis. Molecular size markers (Bio-Rad) are shown in kDa. Parent, parent strain; Δlrp, lrp-deletion mutant; pJH0311, broad-host-range vector; pJH0311::lrp, pJH0311 carrying the lrp gene (pZW1818); nsrR3CS, strain expressing apo-locked NsrRFLAG; nsrR3CSΔlrp, lrp-deletion mutant expressing apo-locked NsrRFLAG. (E) A 393-bp DNA fragment of the nsrR-hmpA regulatory region (10 nM) was labeled with 6-FAM, and then incubated with increasing amounts of Lrp as indicated. For the competition analysis, various amounts of the unlabeled DNA fragment were added as a self-competitor. B, bound DNA; F, free DNA. (F) The same DNA probe (40 nM) was incubated with increasing amounts of Lrp as indicated, and then digested with DNase I. The region protected by Lrp is indicated by a gray box (LRPB). The nucleotides showing enhanced cleavage are indicated by asterisks. Nucleotide numbers shown are relative to the TSS of hmpA.
Next, EMSAs were performed to investigate whether Lrp directly binds to the nsrR-hmpA regulatory region. The addition of Lrp to the DNA probe resulted in a single retarded band in an Lrp concentration-dependent manner (Figure 6E). The same unlabeled DNA fragment showed competition for Lrp binding in a dose-dependent manner (Figure 6E), demonstrating the specific binding of Lrp. DNase I protection assays revealed that Lrp largely protected a single region extending from −75 to −11 (LRPB, centered at −43 from the TSS of hmpA) from DNase I digestion (Figures 4C, 6F). Combined with the EMSA data (Figure 6E), these results indicate that Lrp binds directly and specifically to the nsrR-hmpA regulatory region. Notably, within the region protected by Lrp, a periodic pattern of reduced cleavage followed by short regions of enhanced cleavage was observed (Figure 6F). This pattern known as phased hypersensitivity implies DNA bending by a multimeric Lrp (Pul et al., 2007), suggesting that the Lrp multimer induces a conformation change of the nsrR-hmpA regulatory region. Moreover, EMSA with both NsrR and Lrp showed that NsrR and Lrp simultaneously bind to the nsrR-hmpA regulatory region, rather than displace each other (Supplementary Figure 2C). Altogether, the combined results propose that direct binding of Lrp to the nsrR-hmpA regulatory region does not alter the nsrR transcription but represses hmpA transcription presumably through the modification of the DNA conformation enhancing the hmpA repression by NsrR.
CRP Activates hmpA, but Represses nsrR by Directly Binding to the nsrR-hmpA Regulatory Region
The role of CRP in the hmpA regulation was also explored. The hmpA transcript and HmpA protein levels in the crp-deletion mutant (Δcrp) were considerably decreased compared with those in the parent strain and restored by complementation (Figures 7A,B). In addition, the hmpA transcript level in the wild-type strain was decreased by exogenous glucose while that in the Δcrp strain was not affected (Supplementary Figure 3A). These results indicate that CRP has a positive effect on the hmpA transcription which is relieved in the presence of exogenous glucose. Then, we compared the hmpA transcript and HmpA protein levels in the parent strain, the nsrR3CS strain, and the crp-deleted nsrR3CS mutant (nsrR3CSΔcrp). Similar to Lrp, the hmpA transcript and HmpA protein levels in the nsrR3CSΔcrp strain were comparable with those in the nsrR3CS strain (Figures 7C,D), suggesting that the positive effect of CRP on the hmpA transcription is also mediated by NsrR. Thus, we further examined whether the effect of CRP on hmpA expression results from the increased cellular level of NsrR. Notably, the PnsrR activity and NsrRFLAG protein level were significantly increased by the crp deletion (Supplementary Figure 3B, Figure 7B), showing that CRP acts as a repressor of nsrR transcription. Moreover, the Lrp protein level in the Δcrp strain was elevated compared with that in the parent strain as we observed previously (Figure 7B) (Lee et al., 2020). Accordingly, we hypothesized that CRP indirectly activates hmpA through the repression of both nsrR and lrp in a sequential manner.
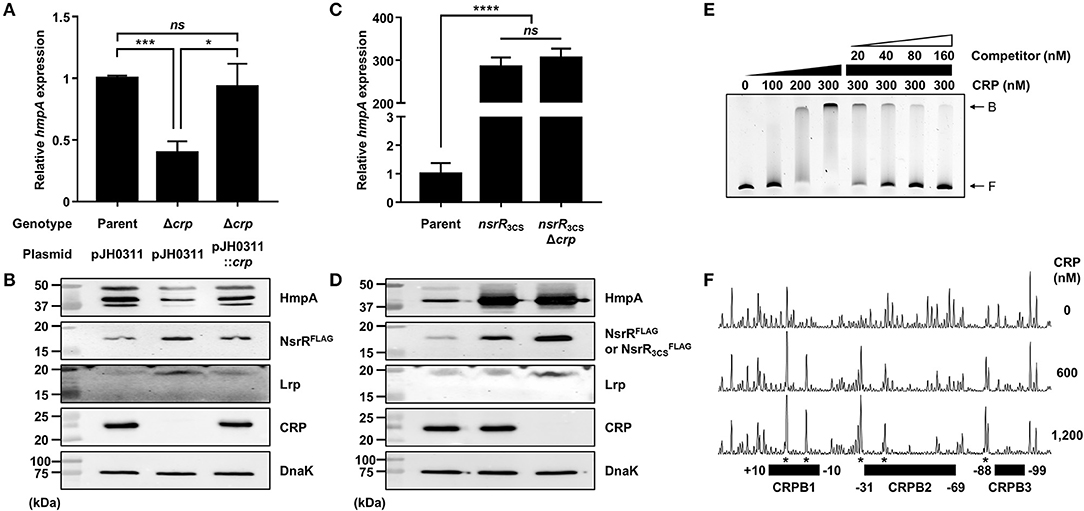
Figure 7. The effect of the crp mutation on hmpA and nsrR transcription, and the specific binding of CRP to the nsrR-hmpA regulatory region. (A–D) Total RNA and proteins were isolated from the parent and mutant strains grown aerobically to an A600 of 0.5. (A,C) The hmpA transcript levels were determined by qRT-PCR, and the hmpA transcript levels in the parent strain were set to 1. Error bars represent the SD. Statistical significance was determined by the Student's t-test (*p < 0.05; ***p < 0.0005; ****p < 0.00005; ns, not significant). (B,D) The cellular HmpA, NsrRFLAG or , Lrp, CRP, and DnaK (internal control) protein levels were determined by Western blot analysis. Molecular size markers (Bio-Rad) are shown in kDa. Parent, parent strain; Δcrp, crp-deletion mutant; pJH0311, broad-host-range vector; pJH0311::crp, pJH0311 carrying the crp gene (pKK1502); nsrR3CS, strain expressing apo-locked NsrRFLAG; nsrR3CSΔcrp, crp-deletion mutant expressing apo-locked NsrRFLAG. (E) A 393-bp DNA fragment of the nsrR-hmpA regulatory region (10 nM) was labeled with 6-FAM, and then incubated with increasing amounts of CRP as indicated. For the competition analysis, various amounts of the unlabeled DNA fragment were added as a self-competitor. B, bound DNA; F, free DNA. (F) The same DNA probe (40 nM) was incubated with increasing amounts of CRP as indicated, and then digested with DNase I. The regions protected by CRP are indicated by block boxes (CRPB1, CRPB2, and CRPB3). The nucleotides showing enhanced cleavage are indicated by asterisks. Nucleotide numbers shown are relative to the TSS of nsrR.
To investigate whether CRP directly binds to the nsrR-hmpA regulatory region, EMSAs were performed. As shown in Figure 7E, the addition of CRP to the DNA probe resulted in a single retarded band in a CRP concentration-dependent manner. The same unlabeled DNA fragment competed for CRP binding in a dose-dependent manner (Figure 7E), confirming the specific binding of CRP. DNase I protection assays determined three regions protected by CRP extending from −10 to +10 (CRPB1, centered at −0.5 from the TSS of nsrR), −69 to −31 (CRPB2, centered at −50 from the TSS of nsrR), and −99 to −88 (CRPB3, centered at −93.5 from the TSS of nsrR) from DNase I digestion (Figures 4C, 7F). Combined with the EMSA data showing a single retarded band by CRP (Figure 7E), this result implies that CRP binds to CRPB1, CRPB2, and CRPB3 with similar DNA-binding affinities. Taken together, the combined results propose that CRP directly and specifically binds to the nsrR-hmpA regulatory region to repress nsrR as well as lrp, and consequently induces the hmpA transcription in a sequential cascade. In conclusion, the results in this study suggest that NsrR tightly regulates the hmpA transcription in response to NO, which could be elaborated by Lrp and CRP.
Discussion
In this study, we newly identified and characterized an NO-responsive transcriptional regulator NsrR in V. vulnificus (Figure 1). The transcriptome analysis discovered that 44 genes are negatively regulated and 3 genes are positively regulated by NsrR (Supplementary Table 1). Notably, our previous transcriptome analysis of the wild-type strain revealed that 42 of the 44 genes repressed by NsrR are significantly induced upon exposure to NO (Supplementary Figure 4A) (Kim et al., 2019). Among the 42 genes, we further identified that the induction of nnrS, in addition to hmpA, is mediated by NsrR in response to NO (Figure 2, Supplementary Figure 4B). Accordingly, it is possible to propose that NsrR also regulates the expression of various genes other than hmpA and nnrS by sensing nitrosative stress. Meanwhile, although iscR, iscS, and iscU were positively regulated by NsrR (Supplementary Table 1), NsrR did not directly bind to the promoter region of the isc operon under the conditions tested (Supplementary Figure 5), indicating that NsrR controls the isc operon indirectly. Considering that functional NsrR requires the intact Fe-S cluster, up-regulation of the isc operon would be advantageous for NsrR to control its regulons effectively.
Besides nsrR, we found that the expression of norR, encoding another putative NO-responsive transcriptional regulator NorR, is induced by NO (Kim et al., 2019). In V. cholerae, NorR activates hmpA and nnrS by sensing NO, contributing to NO detoxification and the sustained colonization of host intestines (Stern et al., 2012). However, NorR did not affect the hmpA transcription in V. vulnificus under our experimental conditions (Supplementary Figure 6). Meanwhile, NorR in E. coli is known to activate the norVW genes encoding a flavorubredoxin to detoxify NO (D'Autreaux et al., 2005), but we could not find norVW homologs in the V. vulnificus genome. Although NorR-regulated genes and their role require further studies, NsrR appears to be the major transcriptional regulator for V. vulnificus to respond against nitrosative stress so far.
Figure 8A depicts the regulatory network comprising NsrR, Lrp, and CRP for the hmpA transcription proposed by this study. NsrR relieves the direct repression of hmpA losing its Fe-S cluster and DNA-binding affinity under nitrosative stress (Figures 3, 5). The strong repression of hmpA by NsrR could allow V. vulnificus to prevent unnecessary waste of cellular components such as heme, NAD, and FAD as cofactors of HmpA (Kim et al., 2019). On the other hand, it could facilitate the rapid and strong induction of hmpA when the repression by NsrR is abolished, which may ensure an effective response against nitrosative stress (Alon, 2007). Thus, it is tempting to suppose that NsrR has evolved to regulate hmpA transcription by a derepression mechanism rather than simple activation.
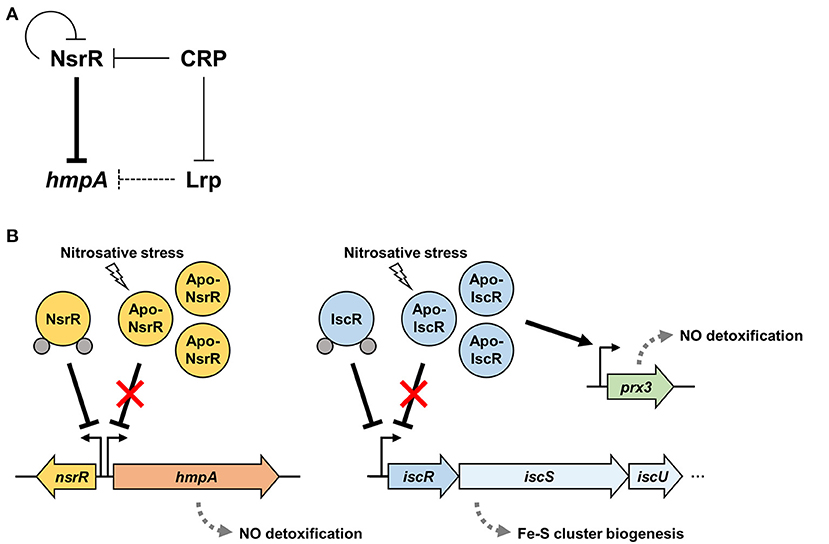
Figure 8. A regulatory network controlling hmpA transcription and nitrosative stress defense systems in V. vulnificus. (A) A regulatory network comprising transcriptional regulators NsrR, Lrp, and CRP controls the hmpA transcription. NsrR directly represses hmpA and nsrR itself. Lrp indirectly represses hmpA presumably by enhancing the repression activity of NsrR through DNA structure remodeling. CRP indirectly activates hmpA possibly through the repression of nsrR and lrp in a sequential cascade. (B) Apo-NsrR alleviates the repression of hmpA and nsrR upon exposure to nitrosative stress. On the other hand, apo-IscR relieves the repression of the isc operon, and the resulting increased apo-IscR directly activates prx3 encoding 1-cysteine peroxiredoxin. The induced NO-decomposition proteins, HmpA and Prx3, would facilitate the survival of V. vulnificus under host-derived nitrosative stress. The gray dots represent the Fe-S cluster.
Furthermore, Lrp and CRP elaborate the hmpA regulation by functional NsrR. Lrp directly binds to the nsrR-hmpA regulatory region but is not able to repress hmpA in the absence of functional NsrR (Figure 6). As one of the bacterial nucleoid-associated proteins, Lrp can modulate gene expression by remodeling the DNA structure (Dillon and Dorman, 2010). Thus, one possible explanation for the NsrR-dependent hmpA repression by Lrp is that the formation of a multicomponent complex containing Lrp multimers and the resulting conformation change of DNA enhance the ability of holo-NsrR to repress hmpA. Meanwhile, this study further demonstrated that CRP acts as a repressor of nsrR by directly binding to the nsrR-hmpA regulatory region (Figure 7, Supplementary Figure 3B). In addition, we confirmed our previous report that CRP directly represses lrp by binding to its promoter (Figure 7B) (Lee et al., 2020). These results led us to propose that CRP activates hmpA by the repression of nsrR and lrp as a sequential cascade.
Particularly, we showed that CRP upregulates the hmpA transcription in response to low levels of glucose (Supplementary Figure 3A). During conditions of intestinal inflammation, NO produced by host cells is rapidly decomposed to less toxic by diverse detoxifying enzymes of enteric pathogens including HmpA. The accumulated in the intestinal lumen can be utilized as an electron acceptor for anaerobic respiration of pathogens in hypoxic environments (Vazquez-Torres and Baumler, 2016; Bueno et al., 2018). In the / respiration, is converted to that is harmful to bacteria. Thus, is subsequently reduced to ammonia (NH3) by an reductase, which can generate NO as a by-product (Spiro, 2007; Tiso and Schechter, 2015). Intriguingly, it has been reported that CRP activates the / respiration under nutrient-poor or low-oxygen conditions in E. coli and Shewanella oneidensis (Stewart et al., 2009; Dong et al., 2012). Accordingly, we could assume that CRP induces hmpA as well as activates / respiration under low-glucose conditions to scavenge the low levels of endogenous NO during / respiration. Since the utilization of host-derived enhances the growth and fitness of pathogens (Vazquez-Torres and Baumler, 2016), NsrR and CRP might coordinate nitrosative stress defense systems and energy production in V. vulnificus for survival during infection. Altogether, the collaborative regulation by NsrR along with Lrp and CRP enables the tight and precise tuning of hmpA transcription by integrating various signals including nitrosative stress and nutrient availability, thereby contributing to the fitness and pathogenesis of V. vulnificus within the host.
Our current understanding of the nitrosative stress defense systems in V. vulnificus is summarized in Figure 8B. In addition to NsrR, we previously demonstrated that V. vulnificus IscR, another Rrf2-family [2Fe-2S] containing transcriptional regulator, also turns to an apo-form lacking the Fe-S cluster under nitrosative stress (Lim and Choi, 2014; Choi et al., 2020). Apo-IscR dissociates from the promoter of the isc operon to express the isc operon and to facilitate the biogenesis of the Fe-S cluster (Lim et al., 2014b). In addition, the resulting increased apo-IscR further activates the expression of prx3 encoding 1-cysteine peroxiredoxin with an NO-decomposition activity by directly binding to the prx3 promoter region (Pprx3) (Lim et al., 2014a; Ahn et al., 2018). The regulatory characteristic of IscR on Pprx3 is distinguishable from that of NsrR on PhmpA in which IscR can bind to Pprx3 in the apo-form, and the increased apo-IscR protein level results in prx3 activation. Taken together, these assorted nitrosative stress defense systems would provide V. vulnificus with the benefit of having inclusive modulation of various NO-detoxifying gene expression and the consequent survival under host-derived nitrosative stress during infection.
Materials and Methods
Strains, Plasmids, and Culture Conditions
The strains and plasmids used in this study are listed in Supplementary Table 2. Unless otherwise noted, the V. vulnificus strains were grown aerobically in Luria-Bertani (LB) medium supplemented with 2% (w/v) NaCl (LBS) at 30°C, and their growth was monitored spectrophotometrically at 600 nm (A600). When required, 3 μg/ml chloramphenicol was added to the media. To visualize the cellular NsrR protein levels, V. vulnificus GR204, which carries 3 × FLAG-coding sequence fused to the 3′-end of nsrR ORF on the chromosome, was constructed as a parent strain (Supplementary Table 2). The parent strain and its isogenic mutants were used to quantify the cellular NsrR protein levels. The murine macrophage RAW 264.7 cells were grown in DMEM containing 10% fetal bovine serum (VWR, Radnor, PA) and the antibiotics [100 units/ml penicillin G and 100 μg/ml streptomycin (Gibco-BRL, Gaithersburg, MD)] in air supplemented with 5% CO2 at 37°C. To induce NO production, the RAW 264.7 cells were suspended in fresh DMEM containing 500 ng/ml E. coli O111:B4 lipopolysaccharide (Sigma, St. Louis, MO) and 1 mM L-arginine (Sigma) (Walker et al., 1997; Choi et al., 2020).
Generation and Complementation of the Mutants
For construction of the isogenic deletion mutants, target genes were inactivated in vitro by deletion of each ORF using the PCR-mediated linker-scanning mutation method as described previously (Jang et al., 2016; Choi et al., 2020). Briefly, the deleted ORF fragment was amplified by PCR with appropriate primer pairs (Supplementary Table 3), and the resulting fragment was ligated into SphI-SpeI-digested pDM4 (Milton et al., 1996). E. coli S17-1 λpir (Simon et al., 1983) containing pDM4 with the desired insert was used as a conjugal donor to an appropriate V. vulnificus strain to generate the deletion mutant (Supplementary Table 2). The conjugation and isolation of the transconjugants were conducted using a method described previously (Choi et al., 2020). The lrp-deletion mutant ZW181 and the crp-deletion mutant DI0201 were constructed previously (Choi et al., 2002; Lee et al., 2020).
For construction of the parent strain GR204 encoding NsrRFLAG on the chromosome, the 3 × FLAG-coding sequence was fused to the 3'-end of nsrR ORF by PCR using the primer pairs NSRR01-F and NSRR01F-R, or NSRR02F-F and NSRR02-R (Supplementary Table 3). The amplified fragment was cloned into pDM4, resulting in pGR2008 (Supplementary Table 2). E. coli S17-1 λpir containing pGR2008 was used as a conjugal donor to the ΔnsrR strain as described above to generate GR204 (Supplementary Table 2).
The three cysteine residues in NsrR (C91, C96, and C102) were replaced with serine to examine their regulatory function with the minimal structural change of NsrR. For construction of the nsrR3CS strain DY192, three cysteine residues were substituted with serine in vitro by using the QuikChange® site-directed mutagenesis kit (Agilent Technologies, Loveland, CO) (Bang et al., 2012; Lim et al., 2014a). The complementary mutagenic primers listed in Supplementary Table 3 were used to create pDY1907 carrying the nsrR3CS gene on pDM4 (Supplementary Table 2). E. coli S17-1 λpir containing pDY1907 was used as a conjugal donor to the ΔnsrR strain as described above to generate DY192, and the nsrR3CS mutation in DY192 was confirmed by DNA sequencing. For construction of the 3 × FLAG-tagged nsrR3CS strain GR217 (Supplementary Table 2), a similar method was adopted except using pGR2016 carrying 3 × FLAG-coding sequence fused to the 3′-end of nsrR3CS ORF on pDM4 instead of pDY1907.
To complement the nsrR mutation with a plasmid-based system, the nsrR gene was amplified by PCR using the primer pair NSRRC-F and -R (Supplementary Table 3). The amplified fragment was cloned into the broad-host-range vector pJH0311 (Goo et al., 2006) to create pDY1702 (Supplementary Table 2). To complement the lrp and crp mutation, pZW1818 and pKK1502 carrying the lrp and crp gene on pJH0311, respectively, were used in this study (Supplementary Table 2) (Jang et al., 2017; Lee et al., 2020). The plasmids were transferred into appropriate mutants by conjugation as described above.
To complement the nsrR mutation by ectopic expression of nsrR on the chromosome, the nsrR regulatory region and its ORF was integrated into a cryptic lacZ gene by PCR using specific primer pairs listed in Supplementary Table 3 (Hall, 1999; Chodur et al., 2017). The amplified fragment was cloned into pDM4, resulting in pGR2007 (Supplementary Table 2). E. coli S17-1 λpir containing pGR2007 was used as a conjugal donor to the ΔnsrR strain as described above to generate GR203 (Supplementary Table 2).
RNA-seq and Transcriptome Analysis
To analyze the effect of the nsrR deletion on the V. vulnificus transcriptome, total RNA was isolated from biological duplicates of the wild-type and ΔnsrR strains, grown aerobically to an A600 of 0.5 in M9 minimal media supplemented with 0.4% (w/v) glucose (M9G) and then exposed to PPNPs for 10 min (Nurhasni et al., 2015; Kim et al., 2019). The RNA was further purified by removing DNA using TURBO DNase (Ambion, Austin, TX), and mRNA was selectively enriched by depleting rRNA using a Ribo-Zero rRNA removal kit (Epicenter, Madison, WI) according to the manufacturer's instructions. Strand-specific cDNA libraries were constructed and sequenced using HiSeq 2500 (Illumina, San Diego, CA) as described previously (Lee et al., 2019). The raw sequencing reads were mapped to the V. vulnificus MO6-24/O reference genome (GenBankTM accession numbers: CP002469 and CP002470, www.ncbi.nlm.nih.gov), and the expression level of each gene was calculated as a reads per kilobase of transcript per million mapped sequence reads (RPKM) value using EDGE-pro v1.3.1 (Estimated Degree of Gene Expression in PROkaryots) (Magoc et al., 2013). The RPKM values were normalized and analyzed statistically using DeSeq2 v1.26.0 to identify the differentially expressed genes (fold change ≥ 2 with p < 0.05) (Love et al., 2014). A heat map was generated by the Matplotlib python package using the RPKM-fold change for each gene (Hunter, 2007).
qRT-PCR and Primer Extension Analysis
Relative transcript levels in the total RNA isolated from the V. vulnificus strains grown under various environmental conditions were determined by quantitative RT-PCR (qRT-PCR). In detail, V. vulnificus was grown to an A600 of 0.5 in M9G and then exposed to either 0.15 mg/ml PPNPs (negative control) or NO/PPNPs for 10 min (Nurhasni et al., 2015; Kim et al., 2019). Additionally, V. vulnificus grown to an A600 of 0.5 in LBS was exposed to DMEM (negative control) or RAW 264.7 cells at a multiplicity of infection 10 for 10 min in the presence or absence of 500 μM L-NMMA (Sigma), which is a known NO synthase inhibitor (Nathan and Hibbs, 1991; Choi et al., 2020). When necessary, V. vulnificus was grown to an A600 of 0.5 in LBS with various amounts of L-leucine (Sigma) or 1% glucose (Sigma). Total RNA from the V. vulnificus cells was isolated and quantified using a RNeasy® Mini Kit (Qiagen, Valencia, CA) and a NanoDrop Onec Microvolume UV-Vis Spectrophotometer (Thermo Fisher Scientific, Waltham, MA), respectively. cDNA was synthesized from 500 ng of the total RNA with the iScriptTM cDNA synthesis kit (Bio-Rad, Hercules, CA). Real-time PCR amplification of the cDNA was performed with the Chromo 4 real-time PCR detection system (Bio-Rad) and specific primer pairs (Supplementary Table 3) as described previously (Jang et al., 2017). Relative expression levels were calculated with the 16S rRNA expression level as an internal reference for normalization (Jang et al., 2017).
For primer extension analysis, primers HMPAUP-R and NSRRUP-R (Supplementary Table 3) complementary to the coding region of hmpA and nsrR, respectively, were end-labeled with [γ-32P]-ATP and added to the RNA. The primers were extended with SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA). The cDNA products were purified and resolved on a sequencing gel alongside sequencing ladders generated from pDY1706 and pDY1707 (Supplementary Table 2) with the same primers, respectively. The plasmid pDY1706 was constructed by cloning the 219-bp hmpA upstream region extending from −120 to +99, amplified by PCR using a primer pair HMPAUP-F and -R (Supplementary Table 3), into pGEM-T Easy (Promega, Madison, WI). Similarly, pDY1707 carrying the 198-bp nsrR upstream region extending from −113 to +85 on pGEM-T Easy was constructed using a primer pair NSRRUP-F and -R (Supplementary Table 3). The primer extension product was visualized with the Typhoon FLA 7000 phosphorimager (GE healthcare, Menlo Park, CA).
Protein Purification and Western Blot Analysis
To overexpress NsrR and NsrR3CS, each ORF of nsrR and nsrR3CS was amplified by PCR using specific primer pairs (Supplementary Table 3). The amplified fragments were cloned into pET-28a(+) (Novagen, Madison, WI) to create pEJ1902 and pEJ1903, respectively (Supplementary Table 2). The resulting His6-tagged NsrR and NsrR3CS were expressed in E. coli BL21 (DE3) and purified by affinity chromatography according to the manufacturer's instructions (Qiagen). The buffers used for NsrR and NsrR3CS are as follows: 20 mM Tris-Cl (pH 8.0), 500 mM NaCl, and 5 mM β-mercaptoethanol; additional 10% glycerol for a lysis buffer; additional 20 mM imidazole for a wash buffer; additional 250 mM imidazole for an elution buffer; additional 50% glycerol for a dialysis buffer. To overexpress Lrp and CRP, pZW1903 carrying the lrp gene on pET-28a(+) and pHK0201 carrying the crp gene on pRSET A (Invitrogen) were used in this study (Supplementary Table 2) (Choi et al., 2002; Lee et al., 2020). The His6-tagged Lrp and CRP were purified as described previously (Lee et al., 2020).
For Western blot analysis, V. vulnificus cells were lysed using B-PERTM Bacterial Protein Extraction Reagent with Enzymes (Thermo Fisher Scientific), and residual cell debris was removed by centrifugation to obtain clear cell lysates. The protein levels of HmpA, Lrp, CRP, and DnaK in the clear cell lysates were determined as described previously (Kim et al., 2019; Lee et al., 2020). Similarly, cellular NsrRFLAG protein was detected using Monoclonal ANTI-FLAG® M2 antibody produced in mouse (Sigma).
Construction of PnsrR-luxCDABE Transcriptional Fusion
A 393-bp nsrR-hmpA regulatory region (−262 to +131 from the TSS of nsrR) was amplified with the primer PnsrR-F carrying a SacI restriction site and PnsrR-R carrying a SpeI restriction site (Supplementary Table 3). The resulting DNA fragment was cloned into the SacI-SpeI-digested pBBR-lux carrying the promoterless luxCDABE genes to create pGR2025 (Supplementary Table 2) (Lenz et al., 2004). pGR2025 was transferred into the V. vulnificus strains by conjugation as described above. The cellular luminescence and growth (A600) of each strain grown to an A600 of 0.5 in LBS were measured using a microplate reader (InfiniteTM microplate reader, Tecan, Männedorf, Switzerland), and RLUs were calculated by dividing the luminescence with the A600 (Lee et al., 2019).
EMSA and DNase I Protection Assay
For the EMSAs, a 393-bp nsrR-hmpA regulatory region (−186 to +207 from the TSS of hmpA, equivalent to −262 to +131 from the TSS of nsrR) was amplified by PCR using 6-FAM-labeled PnsrRhmpA-F and -R as primers (Supplementary Table 3). Similarly, a 321-bp isc operon regulatory region [−194 to +127 from the TSS of isc operon (Lim et al., 2014b)] was amplified by PCR using 6-FAM-labeled Pisc-F and -R as primers (Supplementary Table 3). The 6-FAM-labeled DNA probe (10 nM) was then incubated with purified NsrR or CRP for 30 min at 30°C in a 20-μl reaction mixture containing 1 × NsrR binding buffer (10 mM Tris-Cl (pH 8.0), 10 mM KCl, 1 mM DTT, and 100 μg BSA; additional 1 mM cAMP only for CRP) and 0.1 μg of poly(dI-dC) (Sigma) as a non-specific competitor. Similarly, the DNA probe was incubated with purified Lrp or both NsrR and Lrp for 30 min at 30°C in a 20-μl reaction mixture containing 1 × Lrp binding buffer (50 mM Tris-Cl (pH 8.0), 20 mM KCl, 1 mM DTT, and 100 μg BSA, and 10% glycerol) and 0.1 μg of poly(dI-dC) (Sigma) as a non-specific competitor. For the competition analysis, various concentrations of unlabeled DNA fragment were added as a self-competitor to the reaction mixture before incubation. Electrophoretic analysis of the DNA-protein complexes was performed as described previously (Lee et al., 2020).
The same 393-bp nsrR-hmpA regulatory region was amplified by PCR using unlabeled PnsrRhmpA-F and 6-FAM-labeled PnsrRhmpA-R as primers for the DNase I protection assays (Supplementary Table 3). The binding of NsrR, Lrp, and CRP to the DNA probe (40 nM) was performed as described above, and DNase I digestion of the DNA-protein complexes followed the procedures described previously (Jang et al., 2017). The digested DNA products were precipitated with ethanol and eluted in sterilized H2O, and then analyzed using an ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA) with Peak ScannerTM Software v1.0 (Applied Biosystems) (Hwang et al., 2019).
Data Analysis
Average and standard deviation (SD) values were calculated from at least three independent experiments. Statistical analysis was performed by the Student's t-test using GraphPad Prism 7.0 (GraphPad Software, San Diego, CA).
Data Availability Statement
The raw data of the RNA-seq analysis can be found in the NCBI BioProject database–PRJNA704465; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA704465.
Author Contributions
GC, DK, and SC designed the research. GC and DK performed the experiments. GC and SC wrote the manuscript. All authors analyzed, interpreted the data, reviewed the results, and approved the final version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea, funded by the Ministry of Science, ICT, and Future Planning (2017R1E1A1A01074639) to SC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Eun Jung Na for kindly providing the NsrR- and NsrR3CS-expressing plasmids pEJ1902 and pEJ1903. Also, we are grateful to all the members of Choi laboratory for their valuable discussion and technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.681196/full#supplementary-material
References
Ahn, J., Jang, K. K., Jo, I., Nurhasni, H., Lim, J. G., Yoo, J. W., et al. (2018). Crystal structure of peroxiredoxin 3 from Vibrio vulnificus and its implications for scavenging peroxides and nitric oxide. IUCrJ 5, 82–92. doi: 10.1107/S205225251701750X
Alon, U. (2007). Network motifs: theory and experimental approaches. Nat. Rev. Genet. 8, 450–461. doi: 10.1038/nrg2102
Baker-Austin, C., and Oliver, J. D. (2018). Vibrio vulnificus: new insights into a deadly opportunistic pathogen. Environ. Microbiol. 20, 423–430. doi: 10.1111/1462-2920.13955
Bang, I. S., Liu, L., Vazquez-Torres, A., Crouch, M. L., Stamler, J. S., and Fang, F. C. (2006). Maintenance of nitric oxide and redox homeostasis by the Salmonella flavohemoglobin Hmp. J. Biol. Chem. 281, 28039–28047. doi: 10.1074/jbc.M605174200
Bang, Y. J., Oh, M. H., and Choi, S. H. (2012). Distinct characteristics of two 2-Cys peroxiredoxins of Vibrio vulnificus suggesting differential roles in detoxifying oxidative stress. J. Biol. Chem. 287, 42516–42524. doi: 10.1074/jbc.M112.421214
Bodenmiller, D. M., and Spiro, S. (2006). The yjeB (nsrR) gene of Escherichia coli encodes a nitric oxide-sensitive transcriptional regulator. J. Bacteriol. 188, 874–881. doi: 10.1128/JB.188.3.874-881.2006
Bonamore, A., and Boffi, A. (2008). Flavohemoglobin: structure and reactivity. IUBMB Life 60, 19–28. doi: 10.1002/iub.9
Bueno, E., Sit, B., Waldor, M. K., and Cava, F. (2018). Anaerobic nitrate reduction divergently governs population expansion of the enteropathogen Vibrio cholerae. Nat. Microbiol. 3, 1346–1353. doi: 10.1038/s41564-018-0253-0
Cho, B. K., Barrett, C. L., Knight, E. M., Park, Y. S., and Palsson, B. O. (2008). Genome-scale reconstruction of the Lrp regulatory network in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 105, 19462–19467. doi: 10.1073/pnas.0807227105
Chodur, D. M., Guo, L., Pu, M., Bruger, E., Fernandez, N., Waters, C., and Rowe-Magnus, D. A. (2017). The proline variant of the W[F/L/M][T/S]R Cyclic Di-gmp binding motif suppresses dependence on signal association for regulator function. J. Bacteriol. 199:e00344–17. doi: 10.1128/JB.00344-17
Choi, G., Jang, K. K., Lim, J. G., Lee, Z. W., Im, H., and Choi, S. H. (2020). The transcriptional regulator IscR integrates host-derived nitrosative stress and iron starvation in activation of the vvhBA operon in Vibrio vulnificus. J. Biol. Chem. 295, 5350–5361. doi: 10.1074/jbc.RA120.012724
Choi, H. K., Park, N. Y., Kim, D. I., Chung, H. J., Ryu, S., and Choi, S. H. (2002). Promoter analysis and regulatory characteristics of vvhBA encoding cytolytic hemolysin of Vibrio vulnificus. J. Biol. Chem. 277, 47292–47299. doi: 10.1074/jbc.M206893200
Crack, J. C., and Le Brun, N. E. (2019). Mass Spectrometric Identification of [4Fe-4S](NO)x intermediates of nitric oxide sensing by regulatory iron-sulfur cluster proteins. Chemistry 25, 3675–3684. doi: 10.1002/chem.201806113
Crack, J. C., Munnoch, J., Dodd, E. L., Knowles, F., Al Bassam, M. M., Kamali, S., et al. (2015). NsrR from Streptomyces coelicolor is a nitric oxide-sensing [4Fe-4S] cluster protein with a specialized regulatory function. J. Biol. Chem. 290, 12689–12704. doi: 10.1074/jbc.M115.643072
D'Autreaux, B., Tucker, N. P., Dixon, R., and Spiro, S. (2005). A non-haem iron centre in the transcription factor NorR senses nitric oxide. Nature 437, 769–772. doi: 10.1038/nature03953
Dillon, S. C., and Dorman, C. J. (2010). Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 8, 185–195. doi: 10.1038/nrmicro2261
Dong, Y., Wang, J., Fu, H., Zhou, G., Shi, M., and Gao, H. (2012). A Crp-dependent two-component system regulates nitrate and nitrite respiration in Shewanella oneidensis. PLoS ONE 7:e51643. doi: 10.1371/journal.pone.0051643
Fang, F. C. (2004). Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2, 820–832. doi: 10.1038/nrmicro1004
Fang, F. C., Frawley, E. R., Tapscott, T., and Vazquez-Torres, A. (2016). Bacterial stress responses during host infection. Cell Host Microbe 20, 133–143. doi: 10.1016/j.chom.2016.07.009
Forrester, M. T., and Foster, M. W. (2012). Protection from nitrosative stress: a central role for microbial flavohemoglobin. Free Radic. Biol. Med. 52, 1620–1633. doi: 10.1016/j.freeradbiomed.2012.01.028
Goo, S. Y., Lee, H. J., Kim, W. H., Han, K. L., Park, D. K., Lee, H. J., et al. (2006). Identification of OmpU of Vibrio vulnificus as a fibronectin-binding protein and its role in bacterial pathogenesis. Infect. Immun. 74, 5586–5594. doi: 10.1128/IAI.00171-06
Hall, B. G. (1999). Experimental evolution of Ebg enzyme provides clues about the evolution of catalysis and to evolutionary potential. FEMS. Microbiol. Lett. 174, 1–8. doi: 10.1111/j.1574-6968.1999.tb13542.x
Hunter, J. D. (2007). Matplotlib: a 2D graphics environment. Comp. Sci. Eng. 9, 90–95. doi: 10.1109/MCSE.2007.55
Hwang, S. H., Park, J. H., Lee, B., and Choi, S. H. (2019). A regulatory network controls cabABC expression leading to biofilm and rugose colony development in Vibrio vulnificus. Front. Microbiol. 10:3063. doi: 10.3389/fmicb.2019.03063
Jang, K. K., Gil, S. Y., Lim, J. G., and Choi, S. H. (2016). Regulatory characteristics of Vibrio vulnificus gbpA gene encoding a mucin-binding protein essential for pathogenesis. J. Biol. Chem. 291, 5774–5787. doi: 10.1074/jbc.M115.685321
Jang, K. K., Lee, Z. W., Kim, B., Jung, Y. H., Han, H. J., Kim, M. H., et al. (2017). Identification and characterization of Vibrio vulnificus plpA encoding a phospholipase A2 essential for pathogenesis. J. Biol. Chem. 292, 17129–17143. doi: 10.1074/jbc.M117.791657
Jeong, H. S., Rhee, J. E., Lee, J. H., Choi, H. K., Kim, D., Lee, M. H., et al. (2003). Identification of Vibrio vulnificus lrp and its influence on survival under various stresses. J. Microbiol. Biotechnol. 13, 159–163. Available online at: https://www.koreascience.or.kr/article/JAKO200311921574129.page
Jones, M. K., and Oliver, J. D. (2009). Vibrio vulnificus: disease and pathogenesis. Infect. Immun. 77, 1723–1733. doi: 10.1128/IAI.01046-08
Kim, B. S., Hwang, J., Kim, M. H., and Choi, S. H. (2011). Cooperative regulation of the Vibrio vulnificus nan gene cluster by NanR protein, cAMP receptor protein, and N-acetylmannosamine 6-phosphate. J. Biol. Chem. 286, 40889–40899. doi: 10.1074/jbc.M111.300988
Kim, D., Na, E. J., Kim, S., Kim, J. S., Jung, Y. H., Cao, J., et al. (2019). Transcriptomic identification and biochemical characterization of HmpA, a nitric oxide dioxygenase, essential for pathogenesis of Vibrio vulnificus. Front. Microbiol. 10:2208. doi: 10.3389/fmicb.2019.02208
Lee, Z. W., Kim, B. S., Jang, K. K., Bang, Y. J., Kim, S., Ha, N. C., et al. (2019). Small-molecule inhibitor of HlyU attenuates virulence of Vibrio species. Sci. Rep. 9:4346. doi: 10.1038/s41598-019-39554-y
Lee, Z. W., Hwang, S. H., Choi, G., Jang, K. K., Lee, T. H., Chung, K. M., et al. (2020). A MARTX toxin rtxA gene is controlled by host environmental signals through a CRP-coordinated regulatory network in Vibrio vulnificus. mBio 11:e00723-20. doi: 10.1128/mBio.00723-20
Lenz, D. H., Mok, K. C., Lilley, B. N., Kulkarni, R. V., Wingreen, N. S., and Bassler, B. L. (2004). The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 118, 69–82. doi: 10.1016/j.cell.2004.06.009
Lim, J. G., Bang, Y. J., and Choi, S. H. (2014a). Characterization of the Vibrio vulnificus 1-Cys peroxiredoxin Prx3 and regulation of its expression by the Fe-S cluster regulator IscR in response to oxidative stress and iron starvation. J. Biol. Chem. 289, 36263–36274. doi: 10.1074/jbc.M114.611020
Lim, J. G., and Choi, S. H. (2014). IscR is a global regulator essential for pathogenesis of Vibrio vulnificus and induced by host cells. Infect. Immun. 82, 569–578. doi: 10.1128/IAI.01141-13
Lim, J. G., Park, J. H., and Choi, S. H. (2014b). Low cell density regulator AphA upregulates the expression of Vibrio vulnificus iscR gene encoding the Fe-S cluster regulator IscR. J. Microbiol. 52, 413–421. doi: 10.1007/s12275-014-3592-4
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Magoc, T., Wood, D., and Salzberg, S. L. (2013). EDGE-pro: estimated degree of gene expression in prokaryotic genomes. Evol. Bioinform Online 9, 127–136. doi: 10.4137/EBO.S11250
Manneh-Roussel, J., Haycocks, J. R. J., Magan, A., Perez-Soto, N., Voelz, K., Camilli, A., et al. (2018). cAMP receptor protein controls Vibrio cholerae gene expression in response to host colonization. mBio 9:e00966-18. doi: 10.1128/mBio.00966-18
Miller, J. F., Mekalanos, J. J., and Falkow, S. (1989). Coordinate regulation and sensory transduction in the control of bacterial virulence. Science 243, 916–922. doi: 10.1126/science.2537530
Milton, D. L., O'toole, R., Horstedt, P., and Wolf-Watz, H. (1996). Flagellin A is essential for the virulence of Vibrio anguillarum. J. Bacteriol. 178, 1310–1319. doi: 10.1128/JB.178.5.1310-1319.1996
Nathan, C. F., and Hibbs, J. B. Jr. (1991). Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr. Opin. Immunol. 3, 65–70. doi: 10.1016/0952-7915(91)90079-G
Notredame, C., Higgins, D. G., and Heringa, J. (2000). T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217. doi: 10.1006/jmbi.2000.4042
Nurhasni, H., Cao, J., Choi, M., Kim, I., Lee, B. L., Jung, Y., et al. (2015). Nitric oxide-releasing poly(lactic-co-glycolic acid)-polyethylenimine nanoparticles for prolonged nitric oxide release, antibacterial efficacy, and in vivo wound healing activity. Int.J. Nanomed. 10, 3065–3080. doi: 10.2147/IJN.S82199
Partridge, J. D., Bodenmiller, D. M., Humphrys, M. S., and Spiro, S. (2009). NsrR targets in the Escherichia coli genome: new insights into DNA sequence requirements for binding and a role for NsrR in the regulation of motility. Mol. Microbiol. 73, 680–694. doi: 10.1111/j.1365-2958.2009.06799.x
Pul, U., Wurm, R., and Wagner, R. (2007). The role of LRP and H-NS in transcription regulation: involvement of synergism, allostery and macromolecular crowding. J. Mol. Biol. 366, 900–915. doi: 10.1016/j.jmb.2006.11.067
Rhee, J. E., Kim, K. S., and Choi, S. H. (2008). Activation of the Vibrio vulnificus cadBA Operon by Leucine-responsive regulatory protein is mediated by CadC. J. Microbiol. Biotechnol. 18, 1755–1761. doi: 10.4014/jmb.0800.121
Serrano, P. N., Wang, H., Crack, J. C., Prior, C., Hutchings, M. I., Thomson, A. J., et al. (2016). Nitrosylation of nitric-oxide-sensing regulatory proteins containing [4Fe-4S] clusters gives rise to multiple iron-nitrosyl complexes. Angew. Chem. Int. Ed. Engl. 55, 14575–14579. doi: 10.1002/anie.201607033
Simon, R., Priefer, U., and Puhler, A. (1983). A broad host range mobilization system for in vivo genetic-engineering - transposon mutagenesis in Gram-negative bacteria. Bio-Technology 1, 784–791. doi: 10.1038/nbt1183-784
Sobko, T., Reinders, C. I., Jansson, E., Norin, E., Midtvedt, T., and Lundberg, J. O. (2005). Gastrointestinal bacteria generate nitric oxide from nitrate and nitrite. Nitric Oxide 13, 272–278. doi: 10.1016/j.niox.2005.08.002
Spiro, S. (2007). Regulators of bacterial responses to nitric oxide. FEMS Microbiol. Rev. 31, 193–211. doi: 10.1111/j.1574-6976.2006.00061.x
Stern, A. M., Hay, A. J., Liu, Z., Desland, F. A., Zhang, J., Zhong, Z., et al. (2012). The NorR regulon is critical for Vibrio cholerae resistance to nitric oxide and sustained colonization of the intestines. mBio 3, e00013–e00012. doi: 10.1128/mBio.00013-12
Stern, A. M., and Zhu, J. (2014). An introduction to nitric oxide sensing and response in bacteria. Adv. Appl. Microbiol. 87, 187–220. doi: 10.1016/B978-0-12-800261-2.00005-0
Stewart, V., Bledsoe, P. J., Chen, L. L., and Cai, A. (2009). Catabolite repression control of napF (periplasmic nitrate reductase) operon expression in Escherichia coli K-12. J. Bacteriol. 191, 996–1005. doi: 10.1128/JB.00873-08
Tiso, M., and Schechter, A. N. (2015). Correction: nitrate reduction to nitrite, nitric oxide and ammonia by gut bacteria under physiological conditions. PLoS ONE 10:e0127490. doi: 10.1371/journal.pone.0127490
Tucker, N. P., Hicks, M. G., Clarke, T. A., Crack, J. C., Chandra, G., Le Brun, N. E., et al. (2008). The transcriptional repressor protein NsrR senses nitric oxide directly via a [2Fe-2S] cluster. PLoS ONE 3:e3623. doi: 10.1371/journal.pone.0003623
Tucker, N. P., Le Brun, N. E., Dixon, R., and Hutchings, M. I. (2010). There's NO stopping NsrR, a global regulator of the bacterial NO stress response. Trends Microbiol. 18, 149–156. doi: 10.1016/j.tim.2009.12.009
Vazquez-Torres, A., and Baumler, A. J. (2016). Nitrate, nitrite and nitric oxide reductases: from the last universal common ancestor to modern bacterial pathogens. Curr. Opin. Microbiol. 29, 1–8. doi: 10.1016/j.mib.2015.09.002
Volbeda, A., Dodd, E. L., Darnault, C., Crack, J. C., Renoux, O., Hutchings, M. I., et al. (2017). Crystal structures of the NO sensor NsrR reveal how its iron-sulfur cluster modulates DNA binding. Nat. Commun. 8:15052. doi: 10.1038/ncomms15052
Walker, G., Pfeilschifter, J., and Kunz, D. (1997). Mechanisms of suppression of inducible nitric-oxide synthase (iNOS) expression in interferon (IFN)-gamma-stimulated RAW 264.7 cells by dexamethasone. Evidence for glucocorticoid-induced degradation of iNOS protein by calpain as a key step in post-transcriptional regulation. J. Biol. Chem. 272, 16679–16687. doi: 10.1074/jbc.272.26.16679
Keywords: Vibrio vulnificus, gene regulation, transcriptional regulator, nitric oxide dioxygenase, nitric oxide, nitrosative stress, stress response
Citation: Choi G, Kim D, Im H and Choi SH (2021) A Nitric Oxide-Responsive Transcriptional Regulator NsrR Cooperates With Lrp and CRP to Tightly Control the hmpA Gene in Vibrio vulnificus. Front. Microbiol. 12:681196. doi: 10.3389/fmicb.2021.681196
Received: 16 March 2021; Accepted: 23 April 2021;
Published: 21 May 2021.
Edited by:
Jun Lin, The University of Tennessee, Knoxville, United StatesReviewed by:
Ryo Nasuno, Nara Institute of Science and Technology (NAIST), JapanKambiz Morabbi Heravi, University of Hohenheim, Germany
Copyright © 2021 Choi, Kim, Im and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sang Ho Choi, Y2hvaXNoQHNudS5hYy5rcg==
†These authors have contributed equally to this work
‡Present address: Dukyun Kim, Department of Environmental Health Sciences, Graduate School of Public Health, and Bio-MAX/N-Bio, Seoul National University, Seoul, South Korea