 Marianoel Pereira-Gómez
Marianoel Pereira-Gómez Lucía Carrau
Lucía Carrau Álvaro Fajardo
Álvaro Fajardo Pilar Moreno
Pilar Moreno Gonzalo Moratorio
Gonzalo Moratorio
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Microbiol., 30 June 2021
Sec. Virology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.676582
This article is part of the Research TopicCodon Usage and Dinucleotide Composition of Virus Genomes: From the Virus-Host Interaction to the Development of VaccinesView all 9 articles
Live-attenuated vaccines have been historically used to successfully prevent numerous diseases caused by a broad variety of RNA viruses due to their ability to elicit strong and perdurable immune-protective responses. In recent years, various strategies have been explored to achieve viral attenuation by rational genetic design rather than using classic and empirical approaches, based on successive passages in cell culture. A deeper understanding of evolutionary implications of distinct viral genomic compositional aspects, as well as substantial advances in synthetic biology technologies, have provided a framework to achieve new viral attenuation strategies. Herein, we will discuss different approaches that are currently applied to modify compositional features of viruses in order to develop novel live-attenuated vaccines.
Recent technological advancements in the field of synthetic biology have enabled the design of synthetic viruses with customized genome. Moreover, affordable DNA synthesis combined with reverse genetics improved the infrastructure to develop live-attenuated vaccines (LAVs). In the near future, synthetic RNA technology will also play an important role in reducing laboratory time and technical steps (Abil et al., 2015; Martínez et al., 2016, 2019; Pardi et al., 2018). The main goal of a LAV is to obtain a virus that does not cause disease but elicits a strong protective immune response against viral diseases. Classically, the generation of attenuated viruses is achieved by passaging the virus in tissue culture under different conditions (e.g., different host species or low temperature), which accumulates mutations supporting viral adaptation to the specific condition (Minor, 2015). As a result, several of these mutations will have a fitness cost in the original host or growth under physiological conditions and thus provide the basis for attenuation. However, the exact mechanisms by which these mutations lead to attenuated phenotypes are usually poorly characterized (Lauring et al., 2010). Despite their great capacity to induce a protective immune response, LAVs have the potential to revert to virulent phenotype by both reversions and/or introduction of compensatory mutations or by recombination with similar viruses (Cann et al., 1984; Kew et al., 2002; Bull et al., 2018). This is of special relevance to RNA viruses, due to their high mutation rates and rapid evolution (Duffy et al., 2008; Sanjuán et al., 2010). In order to overcome the risks presented by traditional LAV development approaches, several methods based on reverse genetics and genome recoding strategies have been developed. For instance, synthetic genome recoding approaches are based on introducing synonymous mutations into a protein coding region without modifying the protein sequence. The goal behind these strategies is that recoded viruses can replicate efficiently in vitro but can have limited or absent virulence in vivo, allowing the host to adopt effective immune responses with minimal risks of disease (Wimmer et al., 2009; Osterrieder and Kunec, 2018). It is worth noting that although numerous point mutations are introduced, the amino acid sequence of the parental virus and, therefore, its antigenic properties are preserved in the genome recoding strategies. In this review, we will focus on the fundamental role that genome composition has on RNA virus biology and evolution to design different attenuation strategies.
Genome composition is a highly variable trait that has a significant impact on the overall biology of organisms. Variability in genome composition can be readily observed by looking into codon usage and codon pair bias. For each species, a preferred set of codons is more frequently used than other synonymous ones that could perform the same role. Also, certain codons tend to be found next to each other more frequently than others, while some codon pair combinations are found infrequently. These results in codon usage or codon pair biases. Compelling evidence suggests that one of the key factors explaining codon usage bias in organisms, including viruses, is their specific nucleotide composition (Knight et al., 2001; Chen et al., 2004; Palidwor et al., 2010; van Hemert et al., 2016; Novoa et al., 2019). In RNA viruses, nucleotide composition is highly influenced by mutational pressure, referring to a bias toward, or away from, certain types of nucleotide substitutions or mutations (Jenkins et al., 2002; Seronello et al., 2011). In fact, studies have shown that mutational pressure or uneven base composition accounts for much of codon usage in RNA viruses (Woelk and Holmes, 2002; Seronello et al., 2011).
Mutational pressure will also determine the genome GC content, referred to as the percentage of guanines and cytosines in a given DNA or RNA sequence, which is also believed to be an important factor shaping codon usage (Sharp et al., 1993; Belalov and Lukashev, 2013). In addition, dinucleotide frequencies – that is, the occurrence of two nucleotides together in a DNA or RNA sequence – can also have significant impact on codon usage or codon pair biases. For instance, CpG dinucleotides are underrepresented in small DNA viruses and most vertebrate RNA viruses, including retroviruses (Karlin et al., 1994; Rima and McFerran, 1997; Shackelton et al., 2006). This CpG restriction is beneficial because it allows the evasion of the host innate immune responses by several pattern recognition receptors. UpA dinucleotide frequency is also strongly underrepresented among both RNA viruses and their corresponding hosts mRNA (Simmonds et al., 2013). This dinucleotide pattern could be explained by the presence of TpA in two out of three stop codons, thus decreasing the probability of generating non-sense mutations (Lobo et al., 2009). Also, some RNA-degrading enzymes involved in RNA stability and protein expression target UpA dinucleotides and present additional explanations for UpA bias in organisms (Beutler et al., 1989; Duan and Antezana, 2003; Simmonds et al., 2013).
Some other factors like RNA secondary structure, viral genomic organization, and life cycle may also contribute to codon bias in RNA viruses: for example, segmented viruses have stronger codon bias than non-segmented ones, and vector-borne viruses have lower codon bias than other viruses, likely due to their need to replicate in disparate hosts (Jenkins and Holmes, 2003).
Other explanations for codon usage bias proposes that as viruses depend on the host cell machinery for their own replication, genome composition and codon biases could reflect their host (Lobo et al., 2009; Simón et al., 2017). For instance, a virus may avoid using less abundant tRNAs present in the host to avoid slower rates of translation (Cattaneo et al., 1988; Hajjar and Linial, 1995). In this sense, it has been observed that some viruses seem to mimic the codon usage of their host (Carbone, 2008; Bahir et al., 2009; Su et al., 2009), but also others show different codon preferences (Gu et al., 2004; Sau et al., 2005, 2006). In addition, some codon pairs deviate significantly from their expected frequency (Moura et al., 2005), which seems to influence elongation rates during translation (Irwin et al., 1995).
Genome composition also plays a very important role in defining the mutational robustness of organisms, which refers to the capacity to withstand mutations, showing little or no phenotypic variation when a mutation is introduced (de Visser et al., 2003; Wagner, 2005). Mutational robustness is intrinsically linked to codon usage, since synonymous codons will code for the same amino acid, but they can differ in their evolutionary potential or evolutionary trajectories after single mutations, the most common type of mutations in RNA viruses. The evolutionary potential for any codon within a group of synonymous codons will be determined by two main factors: the base composition and the mutation itself. The nucleotide composition for a given codon is fixed and defines its coding capacity. Also, it will define the genetic relation or proximity to other neighboring synonymous or non-synonymous codons. On the other hand, mutation is a random trait that will have different impacts based on the type of point mutation (transition or transversion) and position (first, second, or third position) where the mutation is introduced. As a result, after a point mutation, different synonymous codons will present different likelihoods of remaining synonymous or, on the contrary, mutating into a farther away non-synonymous codon based on these factors. Thus, the genome composition can directly impact mutational robustness.
Strategies to attenuate viruses mostly based on genome recoding have been studied and reported elsewhere (Martínez et al., 2016, 2019; Gonçalves-Carneiro and Bieniasz, 2021). Here, we will summarize reported genome recoding strategies for viral attenuation from an evolutionary standpoint.
As previously stated, vertebrate RNA viruses tend to exhibit underrepresentation or overrepresentation of dinucleotide frequencies (Rima and McFerran, 1997). In particular, UpA and CpG frequencies are consistently underrepresented across RNA viruses. It has been proposed that CpG restriction is beneficial because it allows the evasion of the host innate immune responses via several pattern recognition receptors including retinoic acid-inducible gene-I-like receptors (RIG-I-like receptors), mitochondrial antiviral signaling proteins (MAVS), protein kinase R (PKR), and others (Atkinson et al., 2014; Tulloch et al., 2014). More specifically, CpG underrepresentation has been associated with poor recognition via the zinc finger antiviral protein (ZAP). This protein, which can be ubiquitously expressed or induced the type I interferon (IFN), specifically detects viral RNAs that have a higher frequency of CpG dinucleotides compared to host mRNAs (Meagher et al., 2019) and promotes RNA degradation by endonucleases or RNA exosomes (Gao et al., 2002; Bick et al., 2003; Müller et al., 2007; Takata et al., 2017; Ficarelli et al., 2019; Luo et al., 2020).
Extensive evidence has indicated that enriching viral genomes with CpGs results in a loss of fitness and could therefore provide an alternative strategy to achieve viral attenuation (Figure 1; Burns et al., 2009; Tulloch et al., 2014; Gaunt et al., 2016; Takata et al., 2017). Burns et al. (2009) directly approached this by introducing random sets of synonymous codons into the capsid coding region of the poliovirus genome. In vitro studies with these mutant viruses revealed that only those with increased CpG and UpA frequencies had fitness loss, and this was due to decreased viral infectivity (lower ratio of infectious particle per genome copy number) and not by decreased translation efficiency (Burns et al., 2009). Later, this inverse relationship between dinucleotide frequencies and viral fitness was confirmed in a study on echovirus 7 in which a control virus with decreased CpG and UpA frequencies showed an increase in viral fitness (Atkinson et al., 2014; Tulloch et al., 2014). Also, the authors proposed that the attenuation was mainly driven by enhanced response of the innate immune response. The first studies with recoded viruses in animal models were done with the influenza virus, showing that only the high-CpG virus (but not the high-UpA virus) was attenuated in mice. Moreover, mice infected with high-CpG viruses presented decreased clinical severity, good induction of the innate and adaptive immune responses, and reduced pathology in the lung. Thus, additional evidence to consider these viral constructs as vaccine candidates was provided (Gaunt et al., 2016).
Figure 1. Strategies to rationally attenuate viruses and develop live-attenuated vaccines. (A) Scheme of viral genomes; above is the wild-type (WT) genome, and below is indicated the recoded viral genomes by the main approaches used to attenuate viruses. (B) Viral population growth and (C) survival curves expected from a recoded virus and the WT.
The redundancy of the genetic code allows adjusting the efficiency in the production of proteins at different levels, without modifying the amino acid sequence (Gingold and Pilpel, 2011). Most amino acids are encoded by more than one codon, and the frequency at which these synonymous codons are used defines the codon usage. The preference for different codons has significant effects on mRNA stability (Presnyak et al., 2015), translation efficiency and accuracy (Gingold and Pilpel, 2011), and protein folding (Zhang et al., 2009).
It is assumed that codons that are used to a greater extent by cellular mRNAs correspond to more abundant tRNAs in the host cell (Ikemura, 1985; Plotkin and Kudla, 2011). Consequently, recoding viruses with infrequent host codons should reduce the translation rates and protein yield, leading to its attenuation (Figure 1; Presnyak et al., 2015). This approach was first implemented to recode the capsid protein of polioviruses, leading to reduced amounts of infectious progeny with a sharp decrease in their replicative fitness (Burns et al., 2006). However, the immunogenicity of these engineered viruses was conserved as they continue to elicit robust neutralizing antibody responses (Burns et al., 2006; Mueller et al., 2006). Subsequent studies were performed to rationally recode other picornaviruses (Diaz-San Segundo et al., 2016), as well as different negative stranded RNA viruses (Meng et al., 2014; Nogales et al., 2014; Cheng et al., 2015, 2017; Stobart et al., 2016), giving rise to highly attenuated viruses both in vivo and in vitro, with the exception of rabies virus, in which attenuation could not be achieved (Wirblich and Schnell, 2011).
Along with codon usage, codon pairs (or a combination of triplets) have been reported as major players influencing the modulation of translation (Brar, 2016). As mentioned before, in every species, certain codon combinations are observed more frequently than others that are preferably avoided, which is known as codon pair bias (Gutman and Hatfield, 1989). These features can be exploited to reach viral attenuation by recoding viral open reading frames using suboptimal combinations of codon pairs, without affecting codon bias or amino acid profiles (Coleman et al., 2008). This reshuffling approach was successfully implemented to attenuate, both in vitro and in vivo, a broad range of positive (Coleman et al., 2008; Martrus et al., 2013; Shen et al., 2015; Song et al., 2017; Li et al., 2018) and negative stranded RNA viruses (Yang et al., 2013; Broadbent et al., 2016; Le Nouën et al., 2019). Moreover, several vaccines were developed by means of this strategy (Mueller et al., 2010, 2020; Cheng et al., 2017; Tsai et al., 2019), including the recent CDX-005 intranasal COVID-19 vaccine candidate that is currently undergoing phase I trial in the United Kingdom (Lundstrom, 2020; Codagenix starts phase I trial for Covid-19 vaccine in U.K, 2021).
The mechanisms underlying attenuation by deoptimization of codon and codon pairs are still controversial. Although codon choice may have a direct effect on translational efficiency and mRNA stability, codon pair deoptimization strategies will inevitably increase the frequency of usually underrepresented CpGs (Tulloch et al., 2014). This is because, although in the process of shuffling codons the bias among them is maintained, new CpGs emerge as most underrepresented codon pairs contain this dinucleotide at their boundary (Atkinson et al., 2014; Kunec and Osterrieder, 2016). Therefore, many studies have suggested that the attenuation is achieved due to antiviral activity displayed by the recognition of CpG-abundant RNAs by ZAP. However, evidence provided by a recent study suggests that suboptimal codon pairs, rather than CpG increase, are responsible for the attenuation of influenza A virus as a consequence of the reduction in translational efficiency and mRNA stability (Groenke et al., 2020). While opposing, it is likely that attenuation is achieved by a combination of both effects, since usually they go hand in hand, and it is difficult to separate their contributions in experimental settings. Moreover, it is also possible that each has varying impacts on different viral families, and thus, no universal rules can be proposed.
Robustness can be defined as phenotypic conservation in light of genetic variation. One of the most recent developments in the theme of attenuation by codon rearrangements involves decreasing the mutational robustness of RNA viruses (decreasing the capacity to “buffer” mutation effects). By rationally engineering viruses that are less able to “buffer” the burden of mutations, strong attenuation can be readily achieved (Figure 1; Lauring et al., 2012; Moratorio et al., 2017; Carrau et al., 2019). The first study of this kind was done in poliovirus, where synthetic viruses carrying reengineered capsid sequences with hundreds of synonymous mutations versus wild type were compared. The authors found that only the virus with decreased robustness was attenuated in mice and had also decreased in neurovirulence (Lauring et al., 2012). Years after, a study focusing only on synonymous codons with non-sense mutational targets (NSMTs) was proposed (Moratorio et al., 2017). By definition, NSMTs are those codon sites that can produce a non-sense mutation (stop codon) after a single-nucleotide substitution. From the 64 codon triplets, 18 codons containing 19 different non-sense mutation targets can mutate to a stop codon after a single-base substitution (Figure 2). Moreover, single-nucleotide substitutions are the most frequent type of spontaneous mutations in RNA viruses (Sanjuán et al., 2010), and the chance of two or three mutations landing in a single codon is extremely low. Based on these, the authors engineered viral genomes to have more serine and leucine codons with NSMTs, in regions of the Coxsackie virus B3 and influenza A virus, compared with wild-type viruses. The resulting increase in stop codon mutations during replication led to a loss of infectivity in vitro and attenuation in vivo. Moreover, attenuation was even stronger when they coupled it with a low-fidelity polymerase, since more stop codons were being generated during replication. Importantly, in this study, the authors were able to significantly remove the effect of confounding factors such as altering codon pair bias or dinucleotide frequencies and focus solely on the impact of rationally modifying mutational robustness. Next, a study by the same group applied this attenuation method on Chikungunya virus and expanded it by also incorporating codons for arginine and glycine with non-sense mutation targets. The proposed LAV was significantly attenuated in mosquito and mammalian hosts, had significantly reduced dissemination in mice, elicited good antibody responses that protected from challenge and had decreased transmissibility from mosquitoes to mice, thus being proven an efficient design (Carrau et al., 2019).
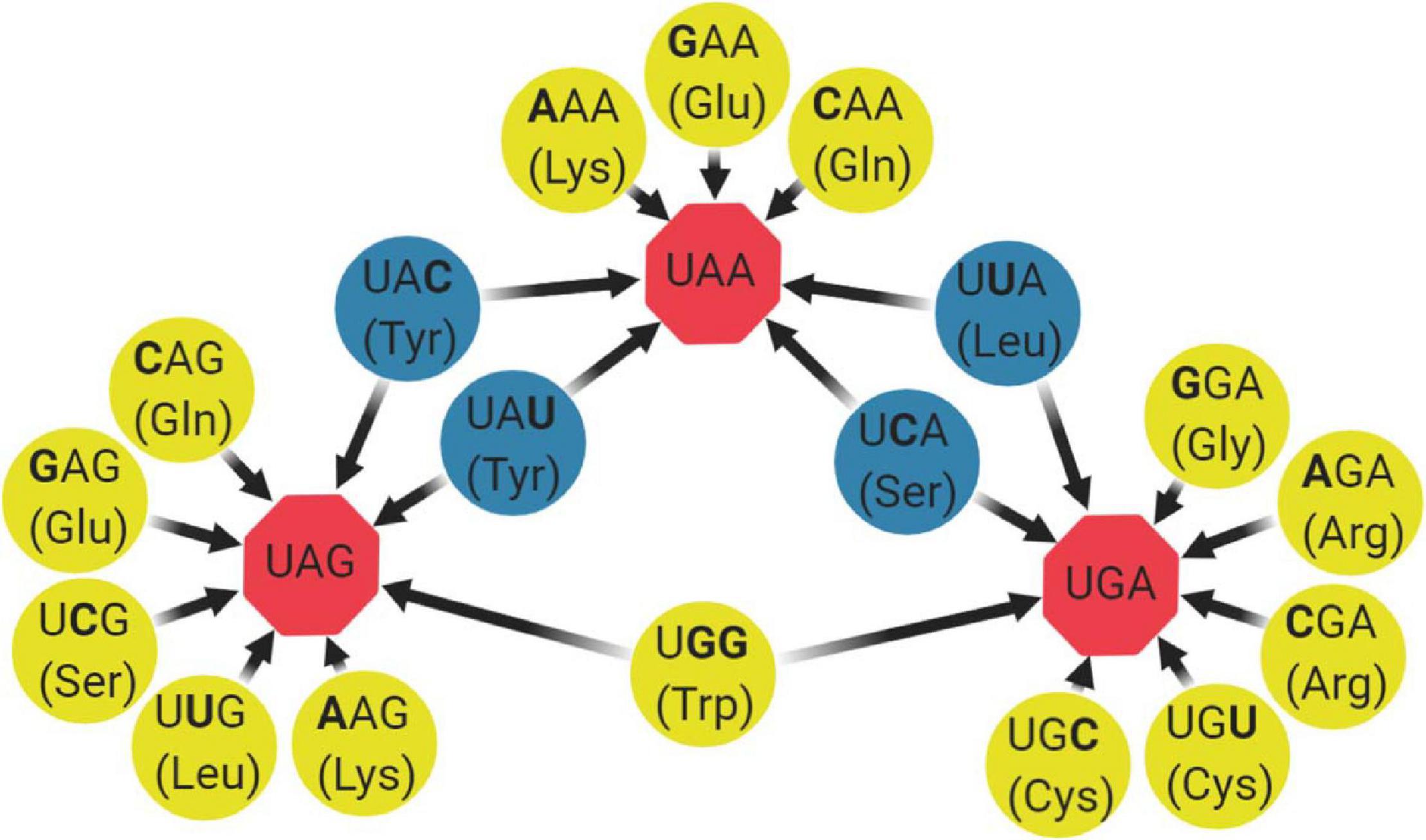
Figure 2. Codons containing non-sense mutation targets. From the 64 codons in the genetic code, 18 codons containing 19 different non-sense mutation targets can mutate to a stop codon after a single-base substitution (boldfaced). In most of the cases, only one of the three possible substitutions produces a stop codon (yellow circles), whereas in some scenarios, two of the three substitutions produce a stop codon (blue circles). Stop codons are represented by red octagons.
Live-attenuated vaccines against viral diseases have been among the most successful medical developments in human history. However, these vaccines, which were generated by classical approaches, have occasionally shown cases of reversion, becoming unsafe and endangering human health. The most paradigmatic case is the oral polio vaccine that showed low genetic stability (Kew, 2012). This is in part because we have not yet fully elucidated the molecular basis of attenuation. In addition, we might underestimate the evolutionary potential of RNA viruses. In this way, the rational design of LAVs, considering the nucleotide composition of viral genomes and their evolution, becomes an important field for future development of LAVs.
Each of the genetic variations on the theme of codon rewiring mentioned in this review has been proposed as new methods for vaccine development or to improve safety of already-existing LAVs. Indeed, they represent good vaccine candidates because they have a strong attenuated phenotype yet exactly similar to wild-type viruses at the protein level, with native antigenicity and complete immunogenicity (Rima and McFerran, 1997; Coleman et al., 2008; Mueller et al., 2010; Cheng et al., 2017; Moratorio et al., 2017; Kaplan et al., 2018). Also, the method can be broadly applied, aided by computational tools that could recode any viral genome (Mueller et al., 2010; Jorge et al., 2015).
The main advantage over other LAV designs is that reversion to pathogenic phenotypes is very unlikely, since no single substitution is responsible for the majority of the attenuated phenotype. Rather, it is presumed that attenuation is the sum of dozens to hundreds of mutations, each imparting minor fitness costs – a strategy aptly described by its creators as “death by a thousand cuts” (Coleman et al., 2008). Nevertheless, if only a subset of codon substitutions has dominant effects, then the risk of reversion to a pathogenic form of virus would be higher than expected. Some in vitro studies have observed slight gain of fitness after extended passaging through reversion or compensatory mutations, but still within the range of the expected evolution for wild-type virus, hence not discouraging the use of these viruses as LAVs (Bull et al., 2012; Nougairede et al., 2013). Interestingly, in a study where the attenuation of a codon-deoptimized human RSV was lost, molecular dynamics simulations identified key positions that could restore the attenuation. When engineered back into the codon-deoptimized genome, a more attenuated virus was rescued with strong immunogenicity and increased stability, advocating for applied rational attenuation (Le Nouën et al., 2017). Even more, in the case of LAVs solely focused on reducing mutational robustness, reversion might seem even more unlikely, given that the introduced mutations do not interfere with replication or translation. If such was the case, selective pressures would be increased in order to restore fitness. Also, compensatory mutations would be less likely, because these viruses will be less able to explore sequence space, restricting the access to beneficial phenotypes.
By further elucidating each mechanism underlying these attenuated phenotypes, a rational evolutionary approach to codon rewiring could take advantage of combining increasing CpG and UpA frequencies to activate host innate immunity (Kumagai et al., 2008), forcing rare codons or codon pair bias to slow down translation (Chevance et al., 2014), and restricting a virus’ mutational robustness and/or detrimental mutational neighborhoods (Lauring et al., 2012; Moratorio et al., 2017; Carrau et al., 2019).
MP-G, LC, and ÁF reviewed literature and performed article conception, design, and writing of the manuscript. MP-G made the figures. PM critically revised the manuscript. GM performed article conception and drafting and critical revision of the manuscript. All authors read and approved the submitted version of the manuscript.
This work was supported by the Agencia Nacional de Investigación e Innovación (ANII FCE_1_2019_1_156157 and FCE_1_2019_1_155930), G4 research group from Institut Pasteur de Montevideo, PEDECIBA, and DARPA PREEMPT administered though DARPA Cooperative Agreement #HR001118S0017-PREEMPT-FP001 (the content of the information does not necessarily reflect the position or the policy of the United States government, and no official endorsement should be inferred).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Diego Simon for critically revising the manuscript.
Abil, Z., Xiong, X., and Zhao, H. (2015). Synthetic biology for therapeutic applications. Mol. Pharm. 12, 322–331. doi: 10.1021/mp500392q
Atkinson, N. J., Witteveldt, J., Evans, D. J., and Simmonds, P. (2014). The influence of CpG and UpA dinucleotide frequencies on RNA virus replication and characterization of the innate cellular pathways underlying virus attenuation and enhanced replication. Nucl. Acids Res. 42, 4527–4545. doi: 10.1093/nar/gku075
Bahir, I., Fromer, M., Prat, Y., and Linial, M. (2009). Viral adaptation to host: a proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 5:311. doi: 10.1038/msb.2009.71
Belalov, I. S., and Lukashev, A. N. (2013). Causes and implications of codon usage bias in RNA viruses. PLoS One 8:e56642. doi: 10.1371/journal.pone.0056642
Beutler, E., Gelbart, T., Han, J., Koziol, J. A., and Beutler, B. (1989). Evolution of the genome and the genetic code: selection at the dinucleotide level by methylation and polyribonucleotide cleavage. Proc. Natl. Acad. Sci. U.S.A. 86, 192–196. doi: 10.1073/pnas.86.1.192
Bick, M. J., Carroll, J.-W. N., Gao, G., Goff, S. P., Rice, C. M., and MacDonald, M. R. (2003). Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 77, 11555–11562. doi: 10.1128/jvi.77.21.11555-11562.2003
Brar, G. A. (2016). Beyond the triplet code: context cues transform translation. Cell 167, 1681–1692. doi: 10.1016/j.cell.2016.09.022
Broadbent, A. J., Santos, C. P., Anafu, A., Wimmer, E., Mueller, S., and Subbarao, K. (2016). Evaluation of the attenuation, immunogenicity, and efficacy of a live virus vaccine generated by codon-pair bias de-optimization of the 2009 pandemic H1N1 influenza virus, in ferrets. Vaccine 34, 563–570. doi: 10.1016/j.vaccine.2015.11.054
Bull, J. J., Molineux, I. J., and Wilke, C. O. (2012). Slow fitness recovery in a codon-modified viral genome. Mol. Biol. Evol. 29, 2997–3004. doi: 10.1093/molbev/mss119
Bull, J. J., Smithson, M. W., and Nuismer, S. L. (2018). Transmissible viral vaccines. Trends Microbiol. 26, 6–15. doi: 10.1016/j.tim.2017.09.007
Burns, C. C., Campagnoli, R., Shaw, J., Vincent, A., Jorba, J., and Kew, O. (2009). Genetic inactivation of poliovirus infectivity by increasing the frequencies of CpG and UpA dinucleotides within and across synonymous capsid region codons. J. Virol. 83, 9957–9969. doi: 10.1128/JVI.00508-09
Burns, C. C., Shaw, J., Campagnoli, R., Jorba, J., Vincent, A., Quay, J., et al. (2006). Modulation of poliovirus replicative fitness in HeLa Cells by deoptimization of synonymous codon usage in the capsid region. J. Virol. 80, 3259–3272. doi: 10.1128/JVI.80.7.3259-3272.2006
Cann, A. J., Stanway, G., Hughes, P. J., Minor, P. D., Evans, D. M. A., Schild, G. C., et al. (1984). Reversion to neurovirulence of the live-attenuated sabin type 3 oral pollovirus vaccine. Nucl. Acids Res. 12, 7787–7792. doi: 10.1093/nar/12.20.7787
Carbone, A. (2008). Codon bias is a major factor explaining phage evolution in translationally biased hosts. J. Mol. Evol. 66, 210–223. doi: 10.1007/s00239-008-9068-6
Carrau, L., Rezelj, V. V., Noval, M. G., Levi, L. I., Megrian, D., Blanc, H., et al. (2019). Chikungunya virus vaccine candidates with decreased mutational robustness are attenuated In Vivo and have compromised transmissibility. J. Virol. 93, e00775–e00819. doi: 10.1128/JVI.00775-19
Cattaneo, R., Schmid, A., Eschle, D., Baczko, K., ter Meulen, V., and Billeter, M. A. (1988). Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell 55, 255–265. doi: 10.1016/0092-8674(88)90048-7
Chen, S. L., Lee, W., Hottes, A. K., Shapiro, L., and McAdams, H. H. (2004). Codon usage between genomes is constrained by genome-wide mutational processes. Proc. Natl. Acad. Sci. U.S.A. 101, 3480–3485. doi: 10.1073/pnas.0307827100
Cheng, B. Y. H., Nogales, A., de la Torre, J. C., and Martínez-Sobrido, L. (2017). Development of live-attenuated arenavirus vaccines based on codon deoptimization of the viral glycoprotein. Virology 501, 35–46. doi: 10.1016/j.virol.2016.11.001
Cheng, B. Y. H., Ortiz-Riaño, E., Nogales, A., de la Torre, J. C., and Martínez-Sobrido, L. (2015). Development of live-attenuated arenavirus vaccines based on codon deoptimization. J. Virol. 89, 3523–3533. doi: 10.1128/jvi.03401-14
Chevance, F. F. V., Le Guyon, S., and Hughes, K. T. (2014). The effects of codon context on in vivo translation speed. PLoS Genet. 10:e1004392. doi: 10.1371/journal.pgen.1004392
Codagenix starts phase I trial for Covid-19 vaccine in U.K (2021). Codagenix Starts Phase I trial for COVID-19 Vaccine in U.K. Available online at: https://www.scienceboard.net/index.aspx?sec=ser&sub=def&pag=dis&Item ID=1956 (accessed February 9, 2021).
Coleman, J. R., Papamichail, D., Skiena, S., Futcher, B., Wimmer, E., and Mueller, S. (2008). Virus attenuation by genome-scale changes in codon pair bias. Science (80). 320, 1784–1787. doi: 10.1126/science.1155761
de Visser, J. A. G. M., Hermisson, J., Wagner, G. P., Ancel Meyers, L., Bagheri-Chaichian, H., Blanchard, J. L., et al. (2003). Perspective: evolution and detection of genetic robustness. Evolution 57, 1959–1972. doi: 10.1111/j.0014-3820.2003.tb00377.x
Diaz-San Segundo, F., Medina, G. N., Ramirez-Medina, E., Velazquez-Salinas, L., Koster, M., Grubman, M. J., et al. (2016). Synonymous deoptimization of foot-and-mouth disease virus causes attenuation in vivo while inducing a strong neutralizing antibody response. J. Virol. 90, 1298–1310. doi: 10.1128/jvi.02167-15
Duan, J., and Antezana, M. A. (2003). Mammalian mutation pressure, synonymous codon choice, and mRNA degradation. J. Mol. Evol. 57, 694–701. doi: 10.1007/s00239-003-2519-1
Duffy, S., Shackelton, L. A., and Holmes, E. C. (2008). Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 9, 267–276. doi: 10.1038/nrg2323
Ficarelli, M., Wilson, H., Galão, R. P., Mazzon, M., Antzin-Anduetza, I., Marsh, M., et al. (2019). KHNYN is essential for the zinc finger antiviral protein (ZAP) to restrict HIV-1 containing clustered CpG dinucleotides. eLife 8:e46767. doi: 10.7554/eLife.46767
Gao, G., Guo, X., and Goff, S. P. (2002). Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science (80) 297, 1703–1706. doi: 10.1126/science.1074276
Gaunt, E., Wise, H. M., Zhang, H., Lee, L. N., Atkinson, N. J., Nicol, M. Q., et al. (2016). Elevation of CpG frequencies in influenza a genome attenuates pathogenicity but enhances host response to infection. Elife 5:e12735. doi: 10.7554/eLife.12735
Gingold, H., and Pilpel, Y. (2011). Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 7:481. doi: 10.1038/msb.2011.14
Gonçalves-Carneiro, D., and Bieniasz, P. D. (2021). Mechanisms of attenuation by genetic recoding of viruses. MBio 12, 1–14. doi: 10.1128/mBio.02238-20
Groenke, N., Trimpert, J., Merz, S., Conradie, A. M., Wyler, E., Zhang, H., et al. (2020). Mechanism of virus attenuation by codon pair deoptimization. Cell Rep. 31:107586. doi: 10.1016/j.celrep.2020.107586
Gu, W., Zhou, T., Ma, J., Sun, X., and Lu, Z. (2004). Analysis of synonymous codon usage in SARS Coronavirus and other viruses in the Nidovirales. Virus Res. 101, 155–161. doi: 10.1016/j.virusres.2004.01.006
Gutman, G. A., and Hatfield, G. W. (1989). Nonrandom utilization of codon pairs in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 86, 3699–3703. doi: 10.1073/pnas.86.10.3699
Hajjar, A. M., and Linial, M. L. (1995). Modification of retroviral RNA by double-stranded RNA adenosine deaminase. J. Virol. 69, 5878–5882. doi: 10.1128/jvi.69.9.5878-5882.1995
Ikemura, T. (1985). Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 2, 13–34. doi: 10.1093/oxfordjournals.molbev.a040335
Irwin, B., Heck, J. D., and Hatfield, G. W. (1995). Codon pair utilization biases influence translational elongation step times. J. Biol. Chem. 270, 22801–22806. doi: 10.1074/jbc.270.39.22801
Jenkins, G. M., and Holmes, E. C. (2003). The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 92, 1–7. doi: 10.1016/s0168-1702(02)00309-x
Jenkins, G. M., Rambaut, A., Pybus, O. G., and Holmes, E. C. (2002). Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J. Mol. Evol. 54, 156–165. doi: 10.1007/s00239-001-0064-3
Jorge, D. M., de, M., Mills, R. E., and Lauring, A. S. (2015). CodonShuffle: a tool for generating and analyzing synonymously mutated sequences. Virus Evol. 1:vev012. doi: 10.1093/ve/vev012
Kaplan, B. S., Souza, C. K., Gauger, P. C., Stauft, C. B., Robert Coleman, J., Mueller, S., et al. (2018). Vaccination of pigs with a codon-pair bias de-optimized live attenuated influenza vaccine protects from homologous challenge. Vaccine 36, 1101–1107. doi: 10.1016/j.vaccine.2018.01.027
Karlin, S., Doerfler, W., and Cardon, L. R. (1994). Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J. Virol. 68, 2889–2897. doi: 10.1128/jvi.68.5.2889-2897.1994
Kew, O. (2012). Reaching the last one per cent: progress and challenges in global polio eradication. Curr. Opin. Virol. 2, 188–198. doi: 10.1016/j.coviro.2012.02.006
Kew, O., Morris-Glasgow, V., Landaverde, M., Burns, C., Shaw, J., Garib, Z., et al. (2002). Outbreak of poliomyelitis in hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science (80) 296, 356–359. doi: 10.1126/science.1068284
Knight, R. D., Freeland, S. J., and Landweber, L. F. (2001). A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2, esearch0010.1. doi: 10.1186/gb-2001-2-4-research0010
Kumagai, Y., Takeuchi, O., and Akira, S. (2008). TLR9 as a key receptor for the recognition of DNA. Adv. Drug Deliv. Rev. 60, 795–804. doi: 10.1016/j.addr.2007.12.004
Kunec, D., and Osterrieder, N. (2016). Codon pair bias is a direct consequence of dinucleotide bias. Cell Rep. 14, 55–67. doi: 10.1016/j.celrep.2015.12.011
Lauring, A. S., Acevedo, A., Cooper, S. B., and Andino, R. (2012). Codon usage determines the mutational robustness, evolutionary capacity, and virulence of an RNA virus. Cell Host Microbe 12, 623–632. doi: 10.1016/j.chom.2012.10.008
Lauring, A. S., Jones, J. O., and Andino, R. (2010). Rationalizing the development of live attenuated virus vaccines. Nat. Biotechnol. 28, 573–579. doi: 10.1038/nbt.1635
Le Nouën, C., Collins, P. L., and Buchholz, U. J. (2019). Attenuation of human respiratory viruses by synonymous genome recoding. Front. Immunol. 10:1250. doi: 10.3389/fimmu.2019.01250
Le Nouën, C., McCarty, T., Brown, M., Smith, M. L., Lleras, R., Dolan, M. A., et al. (2017). Genetic stability of genome-scale deoptimized RNA virus vaccine candidates under selective pressure. Proc. Natl. Acad. Sci. U.S.A. 114, E386–E395. doi: 10.1073/pnas.1619242114
Li, P., Ke, X., Wang, T., Tan, Z., Luo, D., Miao, Y., et al. (2018). Zika virus attenuation by codon pair deoptimization induces sterilizing immunity in mouse models downloaded from. Am. Soc. Microbiol. 92, 701–719. doi: 10.1128/JVI.00701-18
Lobo, F. P., Mota, B. E. F., Pena, S. D. J., Azevedo, V., Macedo, A. M., Tauch, A., et al. (2009). Virus-host coevolution: common patterns of nucleotide motif usage in flaviviridae and their hosts. PLoS One 4:e6282. doi: 10.1371/journal.pone.0006282
Lundstrom, K. (2020). The current status of COVID-19 vaccines. Front. Genome Ed. 2:579297. doi: 10.3389/fgeed.2020.579297
Luo, X., Wang, X., Gao, Y., Zhu, J., Liu, S., Gao, G., et al. (2020). Molecular mechanism of RNA recognition by zinc-finger antiviral protein. Cell Rep. 30, 46–52. doi: 10.1016/j.celrep.2019.11.116
Martínez, M. A., Jordan-Paiz, A., Franco, S., and Nevot, M. (2016). Synonymous virus genome recoding as a tool to impact viral fitness. Trends Microbiol. 24, 134–147. doi: 10.1016/j.tim.2015.11.002
Martínez, M. A., Jordan-Paiz, A., Franco, S., and Nevot, M. (2019). Synonymous genome recoding: a tool to explore microbial biology and new therapeutic strategies. Nucl. Acids Res. 47, 10506–10519. doi: 10.1093/nar/gkz831
Martrus, G., Nevot, M., Andres, C., Clotet, B., and Martinez, M. A. (2013). Changes in codon-pair bias of human immunodeficiency virus type 1 have profound effects on virus replication in cell culture. Retrovirology 10:78. doi: 10.1186/1742-4690-10-78
Meagher, J. L., Takata, M., Gonçalves-Carneiro, D., Keane, S. C., Rebendenne, A., Ong, H., et al. (2019). Structure of the zinc-finger antiviral protein in complex with RNA reveals a mechanism for selective targeting of CG-rich viral sequences. Proc. Natl. Acad. Sci. U.S.A. 116, 24303–24309. doi: 10.1073/pnas.1913232116
Meng, J., Lee, S., Hotard, A. L., and Moore, M. L. (2014). Refining the balance of attenuation and immunogenicity of respiratory syncytial virus by Targeted codon deoptimization of virulence genes. MBio 5, e01704–e01714. doi: 10.1128/mBio.01704-14
Minor, P. D. (2015). Live attenuated vaccines: historical successes and current challenges. Virology 47, 379–392. doi: 10.1016/j.virol.2015.03.032
Moratorio, G., Henningsson, R., Barbezange, C., Carrau, L., Bordería, A. V., Blanc, H., et al. (2017). Attenuation of RNA viruses by redirecting their evolution in sequence space. Nat. Microbiol. 2:17088. doi: 10.1038/nmicrobiol.2017.88
Moura, G., Pinheiro, M., Silva, R., Miranda, I., Afreixo, V., Dias, G., et al. (2005). Comparative context analysis of codon pairs on an ORFeome scale. Genome Biol. 6:R28. doi: 10.1186/gb-2005-6-3-r28
Mueller, S., Coleman, J. R., Papamichail, D., Ward, C. B., Nimnual, A., Futcher, B., et al. (2010). Live attenuated influenza virus vaccines by computer-aided rational design. Nat. Biotechnol. 28, 723–726. doi: 10.1038/nbt.1636
Mueller, S., Papamichail, D., Coleman, J. R., Skiena, S., and Wimmer, E. (2006). Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 80, 9687–9696. doi: 10.1128/jvi.00738-06
Mueller, S., Stauft, C. B., Kalkeri, R., Koidei, F., Kushnir, A., Tasker, S., et al. (2020). A codon-pair deoptimized live-attenuated vaccine against respiratory syncytial virus is immunogenic and efficacious in non-human primates. Vaccine 38, 2943–2948. doi: 10.1016/j.vaccine.2020.02.056
Müller, S., Möller, P., Bick, M. J., Wurr, S., Becker, S., Günther, S., et al. (2007). Inhibition of filovirus replication by the zinc finger antiviral protein. J. Virol. 81, 2391–2400. doi: 10.1128/jvi.01601-06
Nogales, A., Baker, S. F., Ortiz-Riano, E., Dewhurst, S., Topham, D. J., and Martinez-Sobrido, L. (2014). Influenza a virus attenuation by codon deoptimization of the NS gene for vaccine development. J. Virol. 88, 10525–10540. doi: 10.1128/jvi.01565-14
Nougairede, A., De Fabritus, L., Aubry, F., Gould, E. A., Holmes, E. C., and de Lamballerie, X. (2013). Random codon re-encoding induces stable reduction of replicative fitness of chikungunya virus in primate and mosquito cells. PLoS Pathog. 9:e1003172. doi: 10.1371/journal.ppat.1003172
Novoa, E. M., Jungreis, I., Jaillon, O., Kellis, M., and Leitner, T. (2019). Elucidation of codon usage signatures across the domains of life. Mol. Biol. Evol. 36, 2328–2339. doi: 10.1093/molbev/msz124
Osterrieder, N., and Kunec, D. (2018). Attenuation of viruses by large-scale recoding of their genomes: the selection is always biased. Curr. Clin. Microbiol. Rep. 5, 66–72. doi: 10.1007/s40588-018-0080-3
Palidwor, G. A., Perkins, T. J., and Xia, X. (2010). A general model of Codon bias due to GC mutational bias. PLoS One 5:e13431. doi: 10.1371/journal.pone.0013431
Pardi, N., Hogan, M. J., Porter, F. W., and Weissman, D. (2018). mRNA vaccines-a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279. doi: 10.1038/nrd.2017.243
Plotkin, J. B., and Kudla, G. (2011). Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42. doi: 10.1038/nrg2899
Presnyak, V., Alhusaini, N., Chen, Y. H., Martin, S., Morris, N., Kline, N., et al. (2015). Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124. doi: 10.1016/j.cell.2015.02.029
Rima, B. K., and McFerran, N. V. (1997). Dinucleotide and stop codon frequencies in single-stranded RNA viruses. J. Gen. Virol. 78(Pt 11), 2859–2870. doi: 10.1099/0022-1317-78-11-2859
Sanjuán, R., Nebot, M. R., Chirico, N., Mansky, L. M., and Belshaw, R. (2010). Viral mutation rates. J. Virol. 84, 9733–9748. doi: 10.1128/JVI.00694-10
Sau, K., Gupta, S. K., Sau, S., Mandal, S. C., and Ghosh, T. C. (2006). Factors influencing synonymous codon and amino acid usage biases in Mimivirus. BioSystems 85, 107–113. doi: 10.1016/j.biosystems.2005.12.004
Sau, K., Sau, S., Mandal, S. C., and Ghosh, T. C. (2005). Factors influencing the synonymous codon and amino acid usage bias in AT-rich Pseudomonas aeruginosa phage PhiKZ. Acta Biochim. Biophys. Sin. (Shanghai). 37, 625–633. doi: 10.1111/j.1745-7270.2005.00089.x
Seronello, S., Montanez, J., Presleigh, K., Barlow, M., Park, S. B., and Choi, J. (2011). Ethanol and reactive species increase basal sequence heterogeneity of hepatitis C virus and produce variants with reduced susceptibility to antivirals. PLoS One 6:e27436. doi: 10.1371/journal.pone.0027436
Shackelton, L. A., Parrish, C. R., and Holmes, E. C. (2006). Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J. Mol. Evol. 62, 551–563. doi: 10.1007/s00239-005-0221-1
Sharp, P. M., Stenico, M., Peden, J. F., and Lloyd, A. T. (1993). Codon usage: mutational bias, translational selection, or both? Biochem. Soc. Trans. 21, 835–841. doi: 10.1042/bst0210835
Shen, S. H., Stauft, C. B., Gorbatsevych, O., Song, Y., Ward, C. B., Yurovsky, A., et al. (2015). Large-scale recoding of an arbovirus genome to rebalance its insect versus mammalian preference. Proc. Natl. Acad. Sci. U.S.A. 112, 4749–4754. doi: 10.1073/pnas.1502864112
Simmonds, P., Xia, W., Baillie, J. K., and McKinnon, K. (2013). Modelling mutational and selection pressures on dinucleotides in eukaryotic phyla -selection against CpG and UpA in cytoplasmically expressed RNA and in RNA viruses. BMC Genomics 14:610. doi: 10.1186/1471-2164-14-610
Simón, D., Fajardo, A., Sóñora, M., Delfraro, A., and Musto, H. (2017). Host influence in the genomic composition of flaviviruses: A multivariate approach. Biochem. Biophys. Res. Commun. 492, 572–578. doi: 10.1016/j.bbrc.2017.06.088
Song, Y., Gorbatsevych, O., Liu, Y., Mugavero, J. A., Shen, S. H., Ward, C. B., et al. (2017). Limits of variation, specific infectivity, and genome packaging of massively recoded poliovirus genomes. Proc. Natl. Acad. Sci. U.S.A. 114, E8731–E8740. doi: 10.1073/pnas.1714385114
Stobart, C. C., Rostad, C. A., Ke, Z., Dillard, R. S., Hampton, C. M., Strauss, J. D., et al. (2016). A live RSV vaccine with engineered thermostability is immunogenic in cotton rats despite high attenuation. Nat. Commun. 7:13916. doi: 10.1038/ncomms13916
Su, M.-W., Lin, H.-M., Yuan, H. S., and Chu, W.-C. (2009). Categorizing host-dependent RNA viruses by principal component analysis of their codon usage preferences. J. Comput. Biol. 16, 1539–1547. doi: 10.1089/cmb.2009.0046
Takata, M. A., Gonçalves-Carneiro, D., Zang, T. M., Soll, S. J., York, A., Blanco-Melo, D., et al. (2017). CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 550, 124–127. doi: 10.1038/nature24039
Tsai, Y.-H., Huang, S.-W., Hsieh, W.-S., Cheng, C.-K., Chang, C.-F., Wang, Y.-F., et al. (2019). Enterovirus A71 containing codon-deoptimized VP1 and high-fidelity polymerase as next-generation vaccine candidate. Downloaded from. jvi.asm.org 1. J. Virol. 93, 2308–2326. doi: 10.1128/JVI
Tulloch, F., Atkinson, N. J., Evans, D. J., Ryan, M. D., and Simmonds, P. (2014). RNA virus attenuation by codon pair deoptimisation is an artefact of increases in CpG/UpA dinucleotide frequencies. Elife 3:e04531. doi: 10.7554/eLife.04531
van Hemert, F., van der Kuyl, A. C., and Berkhout, B. (2016). Impact of the biased nucleotide composition of viral RNA genomes on RNA structure and codon usage. J. Gen. Virol. 97, 2608–2619. doi: 10.1099/jgv.0.000579
Wagner, A. (2005). Robustness, evolvability, and neutrality. FEBS Lett. 579, 1772–1778. doi: 10.1016/j.febslet.2005.01.063
Wimmer, E., Mueller, S., Tumpey, T. M., and Taubenberger, J. K. (2009). Synthetic viruses: a new opportunity to understand and prevent viral disease. Nat. Biotechnol. 27, 1163–1172. doi: 10.1038/nbt.1593
Wirblich, C., and Schnell, M. J. (2011). Rabies Virus (RV) glycoprotein expression levels are not critical for pathogenicity of RV. J. Virol. 85, 697–704. doi: 10.1128/jvi.01309-10
Woelk, C. H., and Holmes, E. C. (2002). Reduced positive selection in vector-borne RNA viruses. Mol. Biol. Evol. 19, 2333–2336. doi: 10.1093/oxfordjournals.molbev.a004059
Yang, C., Skiena, S., Futcher, B., Mueller, S., and Wimmer, E. (2013). Deliberate reduction of hemagglutinin and neuraminidase expression of influenza virus leads to an ultraprotective live vaccine in mice. Proc. Natl. Acad. Sci. U.S.A. 110, 9481–9486. doi: 10.1073/pnas.1307473110
Keywords: RNA viruses, genome composition, codon usage, codon pair bias, mutational robustness, attenuation, vaccines
Citation: Pereira-Gómez M, Carrau L, Fajardo Á, Moreno P and Moratorio G (2021) Altering Compositional Properties of Viral Genomes to Design Live-Attenuated Vaccines. Front. Microbiol. 12:676582. doi: 10.3389/fmicb.2021.676582
Received: 05 March 2021; Accepted: 01 June 2021;
Published: 30 June 2021.
Edited by:
Akio Adachi, Kansai Medical University, JapanReviewed by:
Ron Geller, University of Valencia, SpainCopyright © 2021 Pereira-Gómez, Carrau, Fajardo, Moreno and Moratorio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gonzalo Moratorio, bW9yYXRvcmlvQHBhc3RldXIuZWR1LnV5
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.