 Kiem Vu
Kiem Vu Eduardo Blumwald
Eduardo Blumwald Angie Gelli
Angie Gelli
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 05 May 2021
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.673035
One path toward identifying effective and easily accessible antifungals is to repurpose commonly used drugs. Amiloride, a widely used diuretic, inhibits different isoforms of Na+/H+ exchangers, Na+ channels, and Na+/Ca2+ exchangers. Here, we found that amiloride had poor antifungal activity against isolates of Cryptococcus prompting the examination of the amiloride analog, HMA [5-(N,N-hexamethylene)amiloride]. HMA possesses strong activity against Na+/H+ exchangers (NHEs) and little K+-associated toxicity since HMA has only minimal inhibitory effects toward epithelial sodium channels (ENaC), the diuretic and antikaliuretic target of amiloride. Although HMA produced a robust dose-dependent growth inhibition of several fungal isolates, susceptibility assays revealed modest MICs against isolates of Cryptococcus. A checkerboard dilution strategy resulted in fractional inhibitory concentrations (FIC) < 0.5, suggesting that HMA displays synergy with several antifungal azole drugs including posaconazole, voriconazole, and ketoconazole. Itraconazole and ravuconazole showed moderate synergy with HMA across all tested fungal isolates. In combination with HMA, ravuconazole had MICs of 0.004–0.008 μg/ml, a ∼16-fold reduction compared to MICs of ravuconazole when used alone and significantly more effective than the overall MIC90 (0.25 μg/ml) reported for ravuconazole against 541 clinical isolates of Cryptococcus neoformans. In combination with azole drugs, MICs of HMA ranged from 3.2 μM (1 μg/ml) to 26 μM (16 μg/ml), HMA was not cytotoxic at concentrations ≤ 8 μg/ml, but MICs were above the reported HMA Ki of 0.013–2.4 μM for various Na+/H+ exchangers. Our results suggest that HMA has limited potential as a monotherapy and may have additional targets in fungal/yeast cells since strains lacking NHEs remained sensitive to HMA. We determined that the hydrophobic substituent at the 5-amino group of HMA is likely responsible for the observed antifungal activity and synergy with several azoles since derivatives with bulky polar substitutions showed no activity against Cryptococcus, indicating that other 5-substituted HMA derivatives could possess stronger antifungal activity. Moreover, substitution of other positions around the pyrazine core of HMA has not been investigated but could reveal new leads for antifungal drug development.
Invasive fungal infections continue to be a serious threat to human health having claimed 1.6 million deaths annually in recent years (Almeida et al., 2019). Individuals suffering from severe fungal disease have topped one billion but a lack of compulsory surveillance suggests that cases are likely underreported (Almeida et al., 2019). The Cryptococcus spp. complex includes at least seven distinct species that can cause life-threatening disease in immunocompromised and immunocompetent individuals (Kwon-Chung et al., 2017). In regions where HIV infection is prevalent, cryptococcal meningitis is the most common form of adult meningitis (Zuger et al., 1986; Rajasingham et al., 2017).
Serious issues with current antifungal drugs are contributing to the challenges associated with resolving life-threatening fungal infections. The repertoire of antifungal drugs is paltry and the increase in resistance is eliminating their usefulness (Smith et al., 2015; Mpoza et al., 2018). Access to all antifungals is limited and in many cases the countries with the most dire need are unable to acquire the most efficacious drugs because of acquisition costs or counterfeit drugs—an increasingly serious threat to resolving fungal disease in resource-poor nations (Loyse et al., 2013; Africa and Abrantes, 2016).
Azoles are undoubtedly the most common antifungal drugs used in clinical practice. Due to their broad spectrum of activity, azoles are used to treat and prevent a number of different mycoses (Campoy and Adrio, 2017). The antifungal activity of azoles is attributed to their ability to inhibit 14α-lanosterol demethylase, a cytochrome P450-dependent enzyme (CYP51) (Campoy and Adrio, 2017). Encoded by the ERG11 gene, lanosterol demethylase changes lanosterol to ergosterol, but in the presence of azoles ergosterol is depleted from the cell membrane resulting in inhibition of fungal growth and replication (Odds et al., 2003; Campoy and Adrio, 2017). Several new azoles have emerged recently, but fluconazole which has been commercially available since 1988, is by far the most common antifungal used in resource-poor countries due to its efficacy and low acquisition costs (Loyse et al., 2013). However, its efficacy as a monotherapy for cryptococcal meningitis is limited and the development of fluconazole resistance in Candida spp. is further eroding its usefulness (Molloy et al., 2018). A major drawback of fluconazole, like some other azoles, is its mere fungistatic activity, which has prompted a search for other drugs/compounds that can be used in combination with azoles to produce desirable fungicidal effects (Marchetti et al., 2000; Onyewu et al., 2003; Vu and Gelli, 2010; Butts et al., 2017).
One path toward finding effective and easily accessible antifungals is to repurpose commonly used drugs (Kim et al., 2020). Amiloride is a pyrazine compound with a guanidinium-substituent and was initially discovered as an inhibitor of Na+/H+ exchangers (NHEs) (Benos, 1982). NHEs represent a large family of integral membrane proteins that contribute to the acidification of the lumen of intracellular organelles (Nakamura et al., 2005). It is widely known that amiloride inhibits different isoforms of NHEs with an IC50 of 5.3–50 μM and also inhibits epithelial Na+ channels and Na+/Ca2+ exchangers (Cragoe et al., 1967; Li et al., 1985; Palandoken et al., 2005). This lack of specificity prompted the synthesis of several analogs of amiloride by a double substitution on the 5-amino group resulting in HMA [5-(N,N-hexamethylene)amiloride], EIPA [5-(N-ethyl-N-isopropyl)amiloride], and MIA [5-(N-methyl-N-isobutyl)amiloride] (Masereel et al., 2003). Among the analogs, HMA [5-(N,N-hexamethylene)amiloride], is the most active since it specifically inhibits NHEs, and has only minimal inhibitory effects against the epithelial sodium channels (ENaC), the diuretic and antikaliuretic target of amiloride (Cragoe et al., 1967) (Masereel et al., 2003).
From a clinical perspective, amiloride is used as a potassium sparing drug because it blocks Na+ re-absorption by inhibiting epithelial Na+ channels (ENaCs) in the distal tubules of the nephron of kidneys with an IC50 of ∼0.1–0.5 μM (Kleyman and Cragoe, 1988). Normally, sodium reabsorption via ENaCs promotes excretion/loss of K+, but excessive Na+ reabsorption increases loss of K+, resulting in hypokalemia. The potent inhibition of ENaCs by amiloride supports its use as a diuretic because it prevents K+ loss/excretion and decreases water retention, however, this K+-sparing effect can lead to hyperkalemia (Vidt, 1981).
Our goal was to examine whether amiloride could be repurposed for the treatment of fungal infections given that amiloride is widely used in clinical practice, can penetrate the blood-brain barrier, is well-tolerated and accessible. Our impetus for this study was based on the pressing need to develop novel antifungal drugs that can enter the central nervous system, are efficacious and safe. In this study amiloride and its analogs were examined for antifungal activity and their potential use in combination therapy with existing azoles was assessed using a checkerboard dilution strategy to determine fractional inhibitory concentrations (FICs), an in vitro measure of drug synergy.
Initially we examined whether amiloride possessed antifungal activity. Susceptibility assays revealed an MIC > 278 μM (>64 μg/ml) for amiloride against Cryptococcus neoformans (Table 1). The lack of significant antifungal activity of amiloride led us to examine three analogs that were derived by double substitution of the 5-amino group of amiloride and were known to possess greater specificity for NHEs in plants and animals (Cragoe et al., 1967). We tested MIA [5-(N-methyl-N-isobutyl)amiloride], EIPA [5-(N-ethyl-N-isopropyl)amiloride] and HMA [5-(N,N-hexamethylene)amiloride]. Analog of amiloride, MIA [5-(N-methyl-N-isobutyl)amiloride], EIPA [5-(N-ethyl-N-isopropyl)amiloride] and HMA [5-(N,N-hexamethylene)amiloride], showed a better antifungal activity against cultures of Cryptococcus spp. than amiloride alone, and HMA resulted the best active compound. Based on these results we examined HMA activity further against several fungal isolates via spot sensitivity assays (Figure 1). Of the fungal isolates tested we found that Cryptococcus neoformans (Cn) and Cryptococcus gattii (Cg) displayed growth sensitivity in a dose-dependent manner at concentrations of HMA ≥ 40 μg/ml and were no longer viable at concentrations of 60 μg/ml (Figure 1). Interestingly, the clinical isolates of Cg, specifically JS-110, B-8260, B-8262, and JS5, displayed a more severe growth defect at 60 μg/ml compared to Cn KN99 and Cn H99 isolates (Figure 1). The Cg JS5 and B9322 isolates that were resistant to flucytosine, an antifungal drug, also displayed a severe growth sensitivity to HMA at 60 μg/ml.
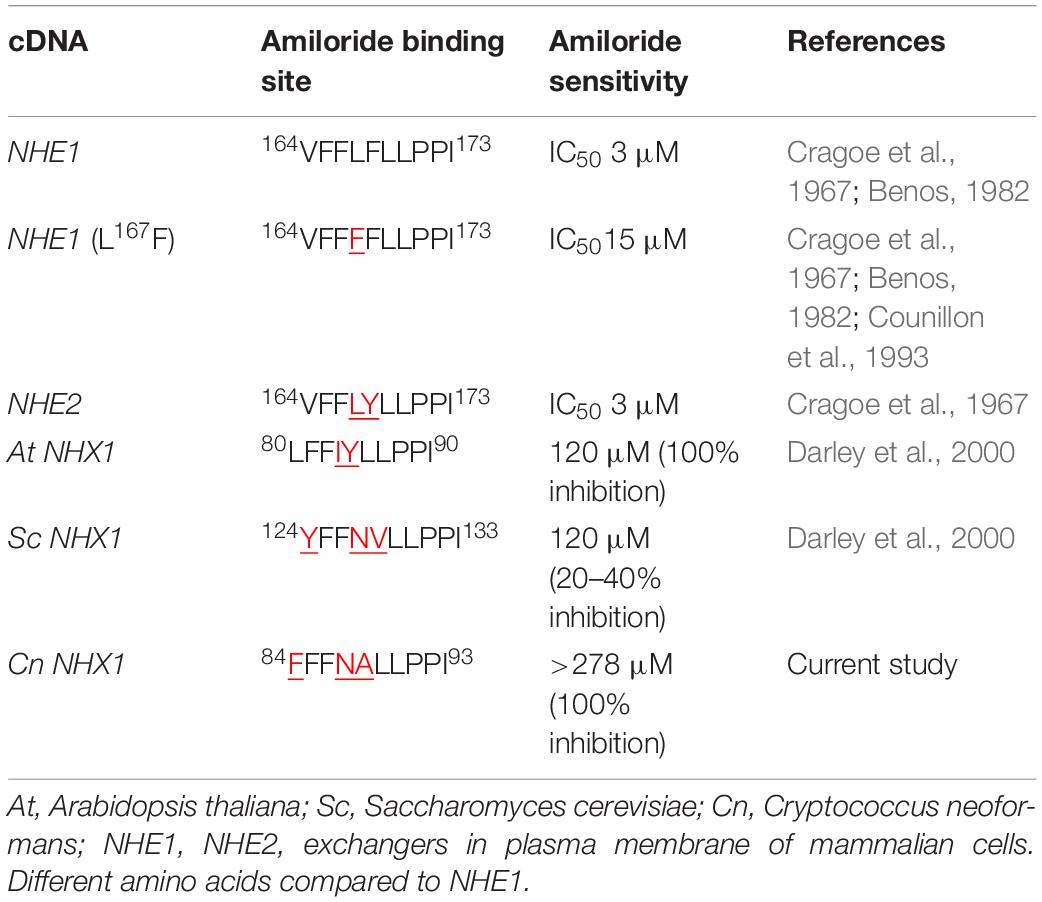
Table 1. Comparison of the amiloride binding site in Na+/H+ exchangers of Cryptococcus neoformans, yeast, plants and mammals.
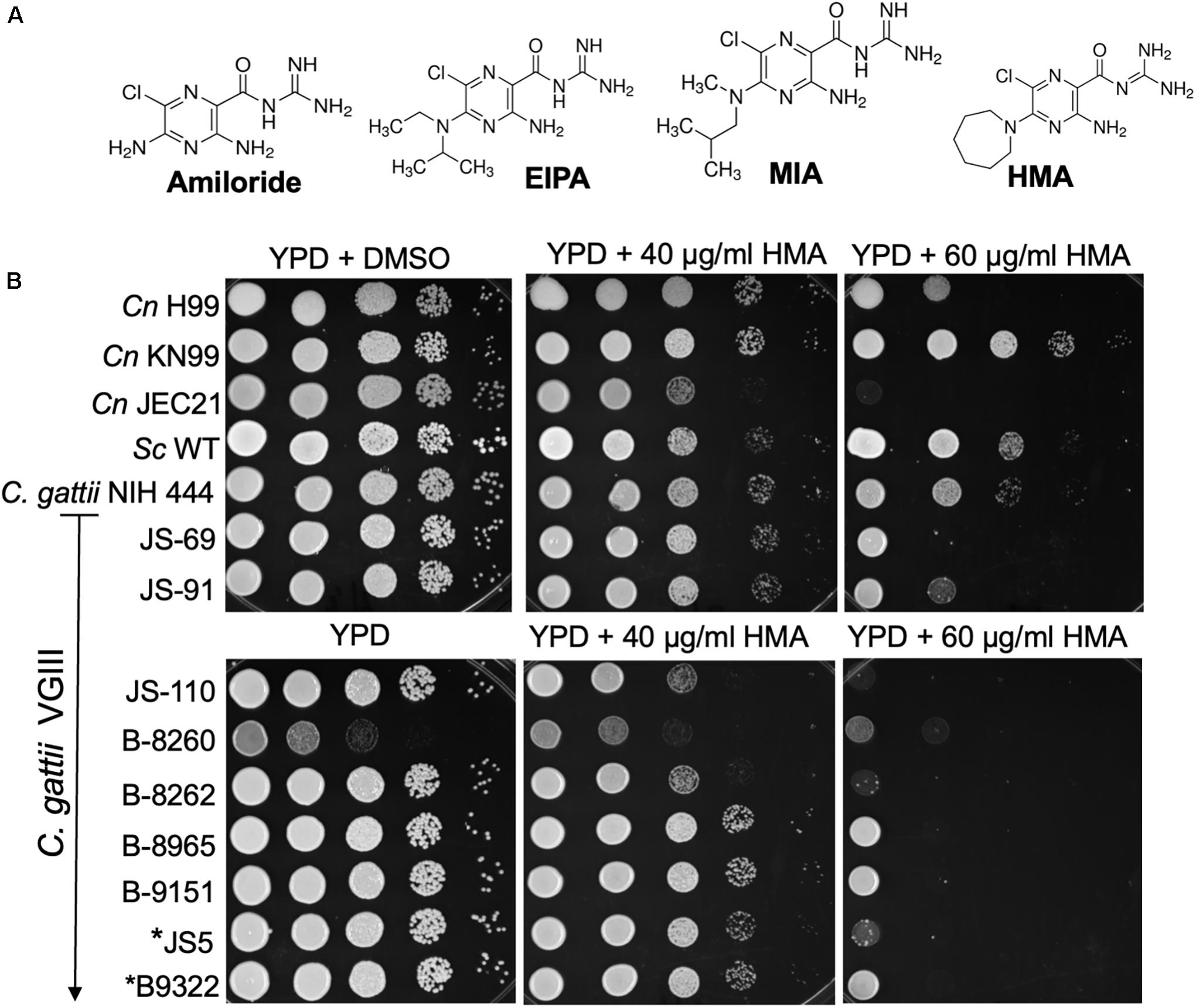
Figure 1. Cryptococcus demonstrates a dose-dependent growth inhibition to HMA. (A) Amiloride and three analogs: EIPA: [5-(N-ethyl-N-isopropyl) amiloride]; MIA: [5-(N-methyl-N-isobutyl)amiloride]; HMA: [5-(N,N-hexamethylene) amiloride]; (B) C. neoformans var. grubii (H99 and KN99), C. neoformans var. neoformans (JEC 21, serotype D), and ten Cryptococcus gattii clinical isolates (ATCC NIH444, JS-69, JS-91, JS-110, B-8260, B-8262, B-8965, B-9151, JS5, and B9322, kindly provided by G. R. Thompson) were examined for their susceptibility to HMA in spot sensitivity assays,. *JS5 and *B9322 were previously identified as flucytosine resistant clinical isolates (Vu et al., 2018). Sc – Saccharomyces cerevisiae, W303a lab strain. Image from three replicates.
Next we assessed the activity of HMA by microbroth dilution susceptibility assays to determine the minimum inhibitory concentration (MIC) of HMA. We found that HMA exhibited only minimal fungicidal activity, albeit stronger than amiloride, against three common Cryptococcus isolates (Table 2). The MIC values for HMA against Cn H99, Cn JEC21, and Cg NIH444 were between 32 and 64 μg/ml (Table 2). Since the relatively high MICs of HMA preclude its use as a monotherapy, we examined the possibility of a combination therapy with current azoles. To assess for the presence of synergy between HMA and the azole class of antifungals, the FIC, was used as a measure of drug synergy (Johnson et al., 2004). Several FIC indices were <0.5, suggesting that HMA exhibited moderate synergistic activity with itraconazole, ketoconazole, posaconazole, ravuconazole, and voriconazole (Table 2).
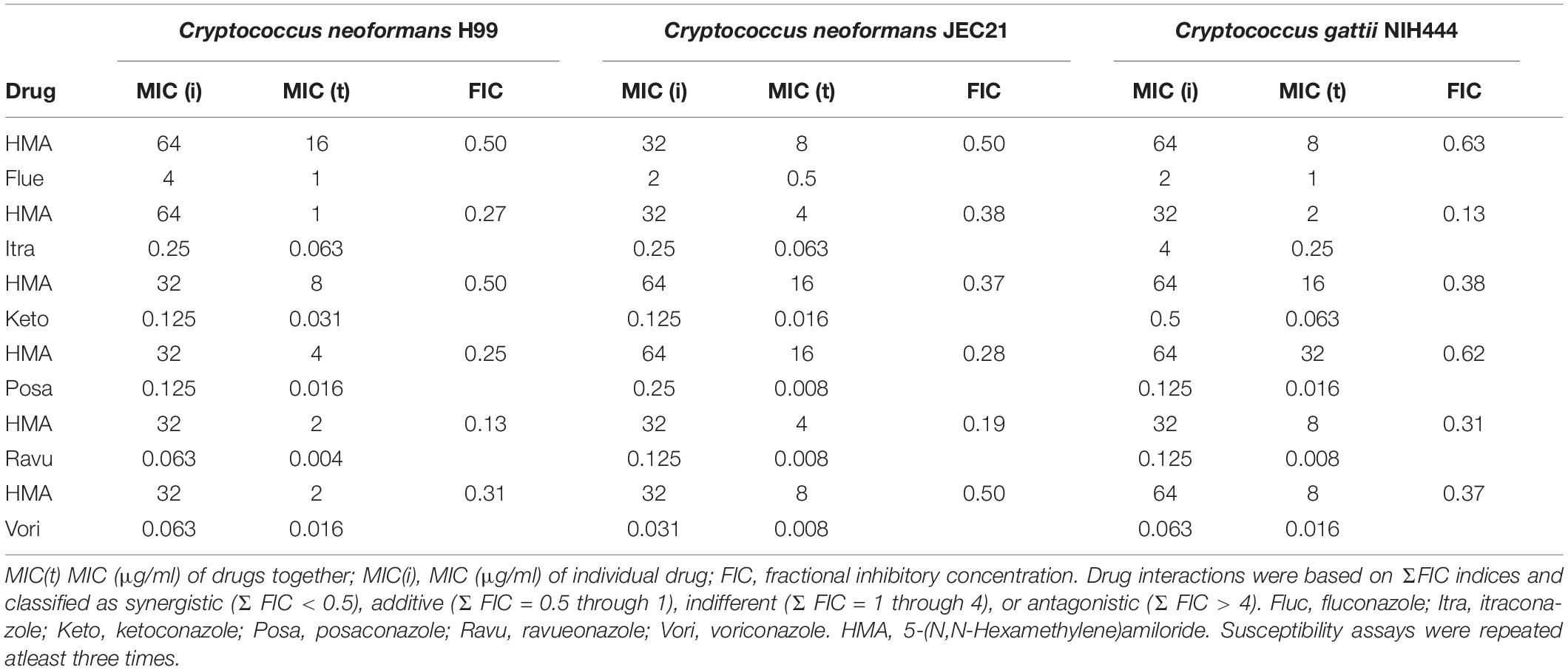
Table 2. Synergy activity of HMA with azole antifungal drugs against strains of Cryptococcus: Cryptococcus neoformans H99, Cryptococcus neoformans JEC21, and Cryptococcus gattii.
Two azole drugs displayed stronger synergistic activity with HMA (FIC index = 0.13, Table 2). The FIC index for HMA plus ravuconazole or itraconazole was 0.13 at 35°C for Cn H99 and Cg, respectively. To illustrate the extent of synergy, the MICs of HMA and ravuconazole given alone were 32 and 0.063 μg/ml, respectively, whereas in combination, the MICs of HMA and ravuconazole decreased to 2 and 0.004 μg/ml, respectively (Table 2). Therefore, in combination, the effective dose of HMA and ravuconazole was ∼16-fold lower. In combination with HMA the MICs of itraconazole, ketoconazole, voriconazole, and posaconazole were reduced between 4- and 32-fold against isolates of Cg and/or Cn (Table 2). The MIC of HMA was reduced between 64-fold and 4- when combined with various azole drugs against Cg and/or Cn (Table 2).
The synergy of HMA with several azole drugs, led us to assess the clinical potential of HMA as an antifungal therapeutic by examining its cytotoxicity profile (Figure 2). HMA was significantly more cytotoxic than amiloride at concentrations ≥ 16 μg/ml (P < 0.01) (Figure 2); however, cytotoxicity was greatly reduced at concentrations of HMA that were synergistic with azoles and not significantly different from that of amiloride (P > 0.05).
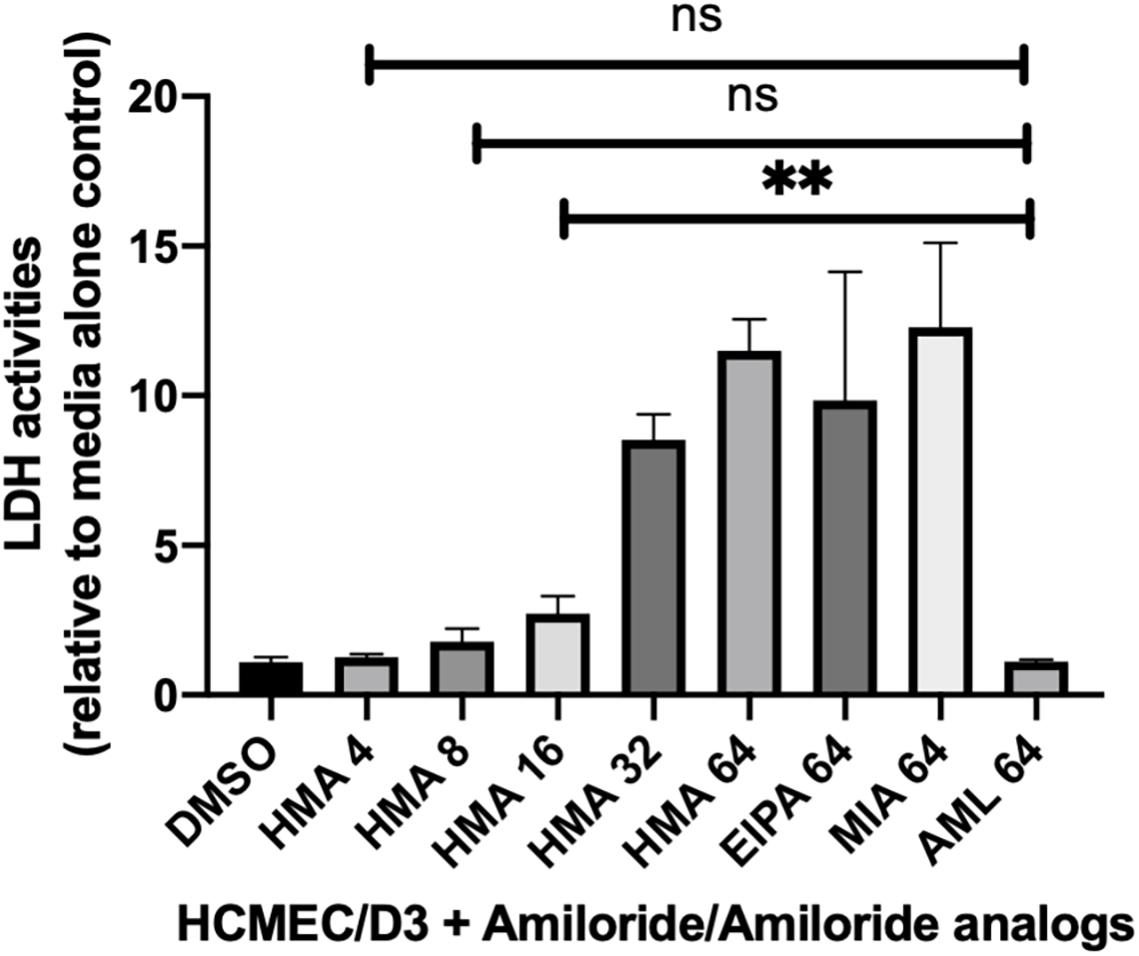
Figure 2. Lactate dehydrogenase (LDH) activity assays in mammalian cells treated with HMA indicate little to no cytotoxicity at concentrations of HMA ≤ 8 μg/ml. The human cerebral microvascular endothelial cell line (HCMEC/D3) was treated with either amiloride (AML) or the amiloride analogs (HMA, EIPA, MIA) at the indicated concentrations (4, 8, 16, 32, or 64 μg/ml) for 8 h prior to measuring LDH activity. The cytotoxicity of HMA at 8 or 4 μg/ml is not significantly different from that of amiloride at 64 μg/ml (P > 0.05). Data shown is from three replicates ± SD; ns, not significant.
To address whether HMA-like analogs might possess more potent antifungal and/or synergistic activity, we examined additional analogs with R-group substitutions at the 5-amino group (Figure 3A). We examined three analogs with bulky polar substitutions at the 5-amino group by disk diffusion assays (Figure 3). We found that analogs UCD74A, UCD38B, and 10-357 (glycinyl-amiloride, five benzyl glycinyl-amiloride, and arylamino amiloride, respectively) had no antifungal activity on their own (Figures 3B,D) and little to no activity in combination with azole drugs based on the absence of a halo for UCD74A and the very minimal halo observed for 10-357 and UCD38B (Figure 3C). We identified several additional analogs in silico with extensive hydrophobic substitutions at the 5-amino group (Supplementary Figure 1) that we intended to examine for antifungal activity, however, these compounds were unavailable (Supplementary Figure 1).
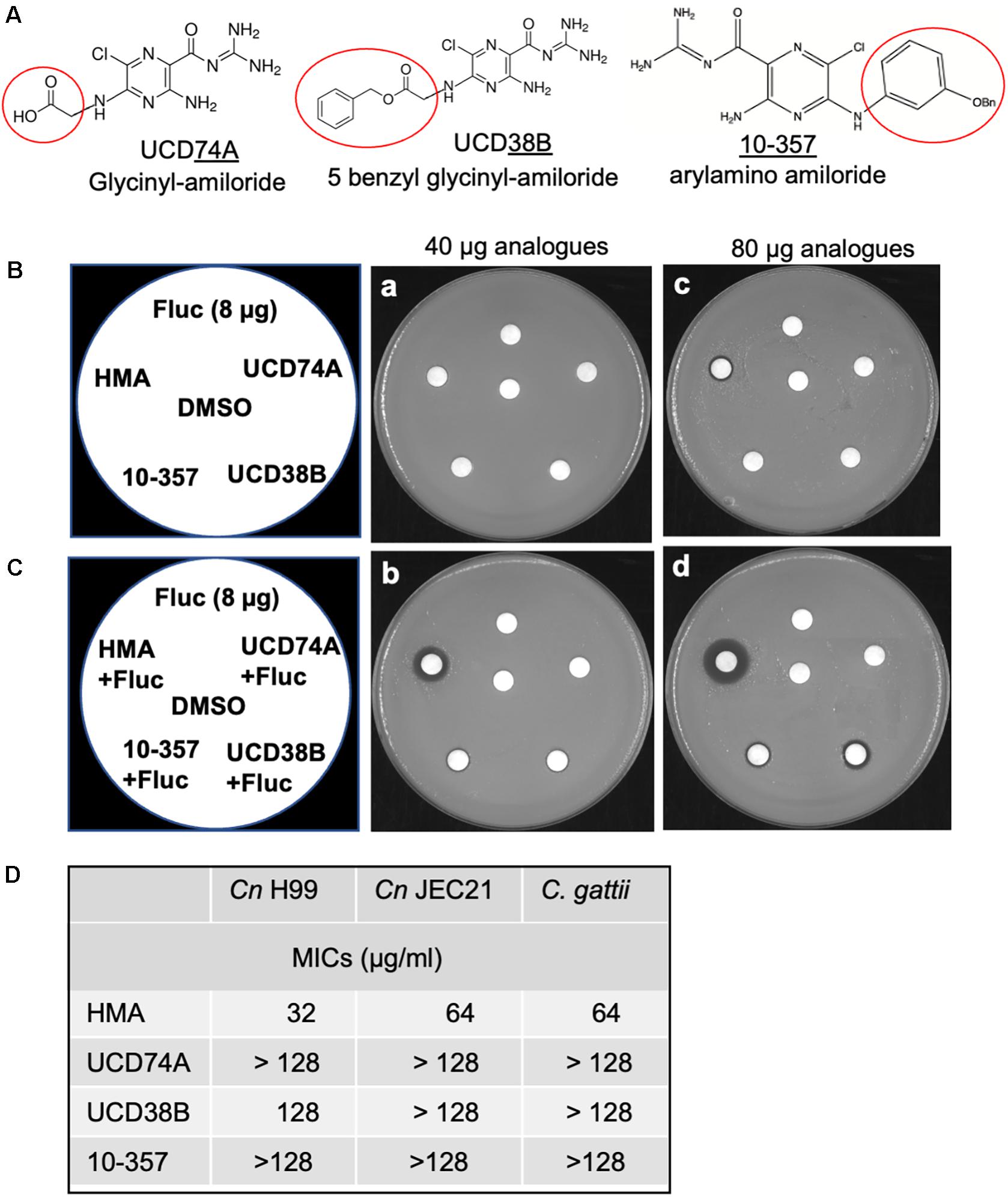
Figure 3. Polar substitutions at the 5-amino group of amiloride eliminate antifungal activity. (A) Three amiloride analogs UCD38B (5-benzyl glycinyl-amiloride), UCD74A (glycinyl-amiloride), and 10-357 (arylamino amiloride, Bn, benzyl group) with polar group substitutions at the 5-amino group of the pyrazine ring (kindly provided by F. Gorin, UC Davis). (B,C) Disk diffusion assays showing susceptibility of the C. neoformans H99 strain to HMA and three other amiloride analogs at 40 and 80 μg and fluconazole (Fluc) at 8 μg either alone (B) or in combination with fluconazole (8 μg) (C). (D) Susceptibility of Cryptococcus neoformans H99, Cryptococcus neoformans JEC21 serotype D, and Cryptococcus gattii, to amiloride analogs HMA, UCD74A, UCD38B, and 10-357 as determined by MICs. Susceptibility assays were performed at least three times.
Since HMA is a well-known specific inhibitor of NHEs in mammalian cells, we aimed to determine whether Cn Nhx1, the NHE endosomal exchanger in fungi/yeast, was a direct target of HMA. We reasoned that if HMA inhibited Nhx1 activity directly, then there should be little to no effect of HMA on the growth of nhx1Δ deletion strains. To test this, the growth of both Sc nhx1Δ and Cn nhx1Δ strains were examined in the presence of 20–120 μg/ml of HMA (Figure 4). We found that Sc and Cn nhx1Δ strains had a dose-dependent reduction in growth in the presence of HMA in a manner similar to the response of WT cells (Figure 4A). In addition, disk diffusion assay showed a similar response of Cn KN99 and Cn nhx1Δ strains to HMA with or without fluconazole (Figure 4B). Given these results, we questioned whether the plasma membrane Na+/H+ antiporter, Cn Nhe1, was a target of HMA since the mammalian plasma membrane Na+/H+ antiporters, Nhe1 and Nhe2, are known to have the highest sensitivity to amiloride (Figure 4A). Amino acid sequence analysis of Cn Nhe1 indicated that Cn Nhe1 did not appear to contain any obvious amiloride binding domain (ABD) (data not shown). Consistent with Cn Nhe1 lacking a known binding domain for amiloride, the Cn nhe1Δ strain had a similar growth defect as WT cells when exposed to 20–120 μg/ml of HMA (Figure 4A). We determined that the putative (ABD) in Nhx1, had significant sequence similarity among fungi with a common origin (Figure 5); however, the ABD in Sc Nhx1 and Cn Nhx1, which have markedly reduced amiloride sensitivity, differed significantly from that in other NHEs (Table 1; Counillon et al., 1993).
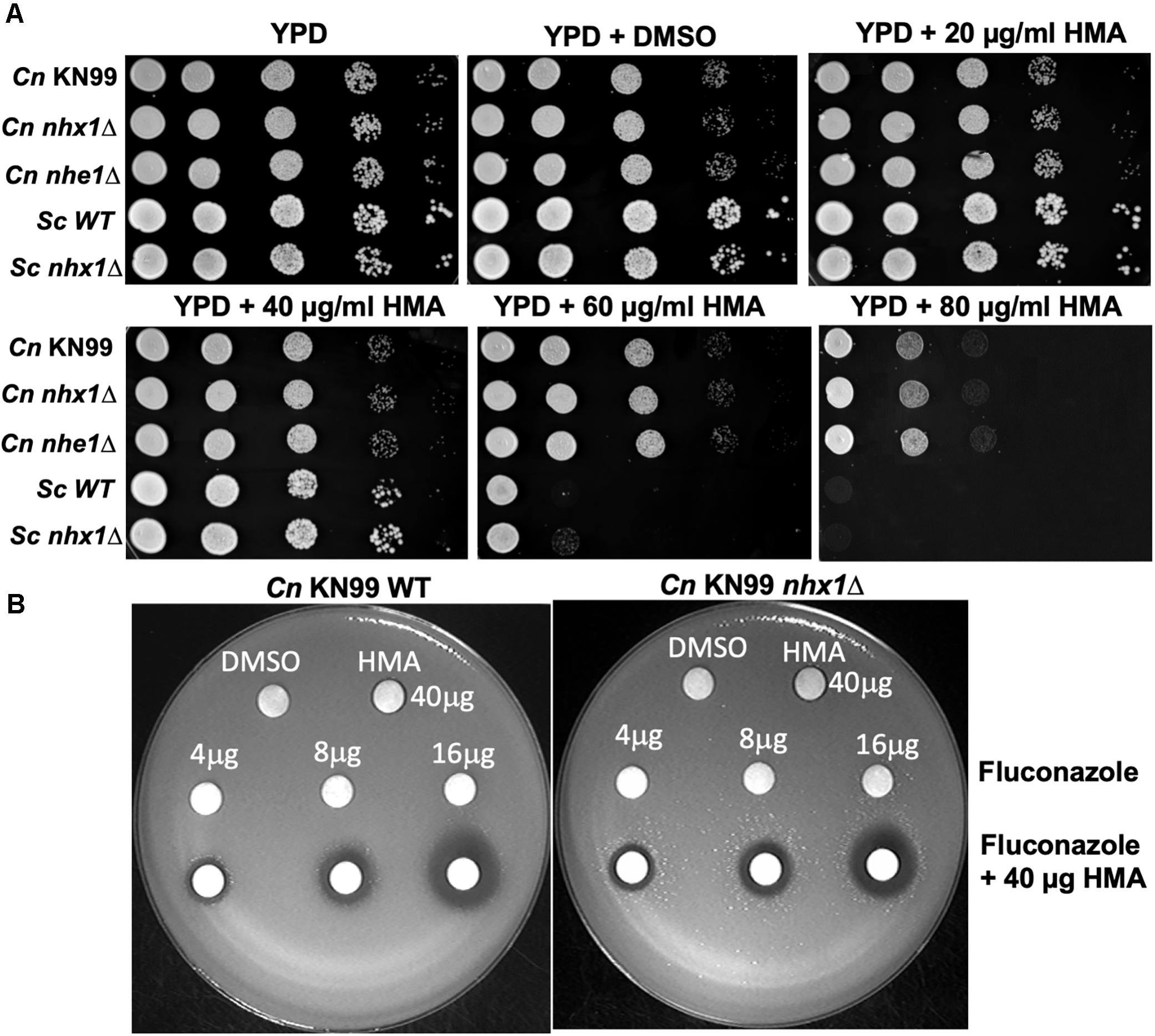
Figure 4. Fungal/yeast cells lacking Na+/H+ exchangers, Nhx1 or Nhe1, are sensitive to HMA. (A,B) Growth sensitivity assays of the C. neoformans KN99 wild type strain, nhx1Δ and nhe1Δ strains demonstrate a similar dose-dependent response to HMA. The background strain of nhx1Δ and nhe1Δ is KN99. Sc – Saccharomyces cerevisiae W303 a strain. Background strain of Sc nhx1Δ is W303 a. (B) Disk diffusion assay shows a similar response of KN99 wild type and nhx1Δ strains to HMA with or without fluconazole. A lawn of KN99 wild type and nhx1Δ strains were tested with either HMA (40 μg), fluconazole (4–16 μg) or fluconazole (4–16 μg) in combination with HMA (40 μg HMA). Images shown are representative of at least three replicates.

Figure 5. The intracellular Na+/H+ exchanger, Nhx1, in fungi. (A) Amiloride binding domain in Nhx1 is highly conserved among fungi. (B) Phylogenetic analysis of Nhx1 protein sequences from 13 fungal species and S. cerevisiae indicates the relative conservation of Nhx1 among these species.
The aim of this study was to determine whether amiloride, a commonly used diuretic, could be repurposed as an antifungal drug. Our studies found that amiloride had little to no antifungal activity at concentrations that would be practical since its activity as a diuretic would supersede any antifungal activity. We questioned whether the noted specificity of HMA, an amiloride analog, for NHEs would promote or improve its antifungal activity and decrease any K+-associated toxicities that might lead to hyperkalemia since HMA is only minimally active toward ENaC. Our results demonstrated that HMA had weak fungicidal activity, although greater than amiloride, as indicated by the MIC values against several isolates of C. neoformans and Cryptococcus gattii. This result along with the LDH activity observed in mammalian cells treated with similar concentrations of HMA and the apparent Ki of HMA for Na+/H+ exchangers would not support the use of HMA as a monotherapy for cryptococcal infections.
We did find, however, that HMA possessed moderate synergy with posaconazole, voriconazole and ketoconazole, although the synergy was strain dependent. Posaconazole and voriconazole appear to be more potent than either itraconazole or fluconazole against published clinical isolates of C. neoformans from Africa and the United States, however, posaconazole has limited penetration of the CNS (Pfaller et al., 1999, 2004). HMA displayed synergistic activity with ravuconazole and itraconazole against all strains tested and our reported MICs were significantly more effective than the overall MIC90 (0.25 μg/ml) reported for ravuconazole against 541 clinical isolates of C. neoformans (Yamazumi et al., 2000). We found that HMA was not cytotoxic at concentrations that promote synergy with the azole antifungals, however, these MICs were above the reported Ki of 0.013–2.4 μM for HMA against various Na+/H+ exchangers (Kleyman and Cragoe, 1988).
HMA belongs to the amiloride class of pyrazines, in which the two hydrogens at the 5-amino position (adjacent to Cl–) are substituted with a hexamethylene moiety resulting in the formation of an azepane ring (Cragoe et al., 1967). Early work by Cragoe et al. (1967) found that substitutions of the H-atoms at the 5-amino group with hydrophobic R-groups resulted in 3–200-fold increase in the inhibitory potency of the Na+/H+ exchanger activity (Blumwald et al., 1987; Kleyman and Cragoe, 1988). Consistent with this result, we found that polar substitutions of the 5-amino group (i.e., 5-benzyl glycinyl-amiloride, glycinyl-amiloride, or arylamino amiloride) had no antifungal activity indicating that a hydrophobic substituent at the 5-amino group, such as the 5-hexamethylene group in HMA, was likely responsible for the observed antifungal activity. However, we observed that strains lacking Nhx1 or Nhe1, remained sensitive to HMA, suggesting that the Na+/H+ exchangers may not be targets of HMA in fungal/yeast cells, despite the increased potency of HMA for NHEs in higher eukaryotes.
The lack of sequence similarity we noted in the binding domain of amiloride/HMA in NHEs across mammals, plants, and fungi/yeast would further support the presence of other HMA targets in fungi. Mutational analysis of the amino acid at position 4 and 5 of the ABD suggested that a leucine (L) residue at position 4 and either a phenylalanine (F) or tyrosine (Y) residue at position 5 were important for amiloride inhibition (Counillon et al., 1993). In the case of Cn Nhx1, differences in the amino acids at these key positions may have precluded the binding of amioride/HMA. Nevertheless, we could not rule out the possibility that HMA may have indeed inhibited Cn Nhx1 or Cn Nhe1, but other cation/proton exchangers may have compensated (Cagnac et al., 2007). Cagnac et al. (2007) identified a novel type of vacuolar monovalent cation-proton exchanger, Vnx1p, in Saccharomyces cerevisiae with low affinity to Na+ and K+ despite its homology to the CAX (calcium/proton exchangers) family. Similar to Nhx1, the Vnx1 exchanger could be associated with the regulation of ion homeostasis and cellular pH in yeast (Cagnac et al., 2007). A search for VNX1 in the Cryptococcus genome identified an uncharacterized open reading frame listed as a calcium/proton exchanger with 32% amino acid identity to Sc Vnx1. This could be indicative of a Vnx1-like protein in Cn, supporting the notion of a compensatory mechanism active in the absence of Nhx1 activity.
Alternatively, off-target effects of HMA in fungal cells could be possible and would be consistent with other reports that demonstrated HMA activity toward G-protein-coupled receptors, such as the adenosine 2A receptor (Garritsen et al., 1991; Gao and Ijzerman, 2000; Soudijn et al., 2004). The reported anticancer activity of HMA and its analogs stems from their antitumor/metastasis effects due in part to the inhibition of the human urokinase plasminogen protease that functions as a major driver of cell invasiveness (Kleyman and Cragoe, 1988; Aredia et al., 2016; Buckley et al., 2018). These various biological activities of HMA may be indicative of other targets in fungal cells, possibly in addition to Nhx1 or Nhe1.
The goal of our study was to examine the antifungal activity of amiloride and its analogs, and although we originally thought that Nhx1 was targeting by amiloride/HMA, our results suggest otherwise. It should be noted that we previously tested the Cn nhx1 deletion strain in media that was supplemented with various concentrations of NaCl, KCl, CaCl2 and a range of pH values to amplify any effect of amiloride/HMA. In all cases we found no difference in the growth of the Cn nhx1 deletion strain compared to wild type, further supporting the notion that Nhx1 may not be a target of HMA in Cryptococcus. This result is consistent with published data that has clearly shown that HMA has other targets in mammalian cells. There is a possibility, as discussed above, that perhaps HMA does inhibit Cn Nhx1 but other exchangers in Cn may be compensating, in which case it would be difficult to observe the effect of HMA on Nhx1. Clearly, resolving the mechanism of HMA in Cn will require significantly more work and complex transport studies that are beyond the scope of our study.
In conclusion, our study demonstrated that HMA had minimal antifungal activity and moderate synergy with several antifungal drugs. We propose that further derivation of HMA could lead to compounds with significantly greater antifungal activity. The structure-activity relationship trend we observed suggested that the hydrophobic substitution at the 5-amino group of HMA was likely responsible for the antifungal activity and synergy with azoles, indicating that other similar 5-substituted HMA derivatives could possess stronger activity. Moreover, substitution of other positions around the pyrazine core of HMA has not been investigated but could reveal new leads for antifungal drug development.
Cryptococcus neoformans H99 and C. gattii are clinical isolates and were obtained from ATCC (ATCC 208821 and ATTC 32609 (also known as NIH444), respectively). KN99 is a common C. neoformans laboratory strain derived from H99 (Nielsen et al., 2003). The JEC21 strain—a C. neoformans var. neoformans, serotype D—was a gift from Dr. J. Heitman (Duke University) and is also available at ATCC (ATCC® MYA-565). Other C. gattii isolates (JS-69, JS-91, JS-110, B-8260, B-8262, B-8965, B-9151, JS5, and B9322), kindly provided by Dr. G.R. Thompson (UC Davis) included in this study belong to the C. gattii VGIII major molecular type and had previously undergone whole genome sequencing as part of a larger population genomics survey (Vu et al., 2018). Unless noted otherwise, the NIH444 C. gattii strain is used throughout this study. The nhx1Δ and nhe1Δ deletion strains were obtained from the available deletion library1. The S. cerevisiae nhx1Δ deletion mutant and wild-type background strain (W303a) was a gift from Dr. E. Blumwald (UC Davis). All strains were recovered from −80°C frozen stocks, grown at 30°C and maintained on yeast peptone dextrose media (YPD) except when tested in susceptibility assays.
MIA, EIPA, HMA, and DMA amiloride analogs [5-(N-methyl-N-isobutyl)amiloride, 5-(N-ethyl-N-isopropyl) amiloride, 5-(N,N-hexamethylene) amiloride, 5-(N,N-(dimethyl)amiloride, respectively] were purchased from Sigma-Aldrich. Compounds UCD74A, UCD38B, and 10-357 (glycinyl-amiloride, five benzyl glycinyl-amiloride, and arylamino amiloride, respectively) were kindly provided by Dr. F. Gorin (UC Davis). All azole drugs were also purchased from Sigma-Aldrich.
DMSO and HMA were added to freshly prepared YPD agar at the indicated concentrations and the plates were allowed to set for 1 h prior to addition of fungal strains. Inoculum from overnight cultures were used to start new cultures for 8–10 h on the day of the experiment. Inoculum size were determined using a hemocytometer (Bright-LineTM Hemacytometer, Sigma-Aldrich). Cultures were washed three times with 1X phosphate-buffered saline (PBS) and resuspended in 1X PBS. Serial 10-fold dilutions were prepared (101 cells/5 μl to 105 cells/5 μl) on 96-well plates, and 5 μl of each dilution was spotted on assay plates using a multichannel pipet. The plates were incubated at 30°C for 48 h after which images were taken.
Checkerboard titrations were performed in order to assess drug interactions according to the Clinical and Laboratory Standards Institute (CLSI). In vitro testing was carried out in RPMI 1640 medium containing L-glutamine, without sodium bicarbonate and buffered to pH 7.0 with MOPS in 96-well plates (96-well cell culture cluster, flat-bottom, Costar). Inoculum of C. neoformans (100 μl) was prepared in accordance with the CLSI standard (M27-A3), added to the 96-well plates and incubated for 48 h at 35°C without shaking. Readings were taken by visual inspection of the opacity of the wells. The MIC of drugs alone or in combination was defined as the lowest drug concentration in a well at which 100% reduction in optical density was observed compared to the no-drug control well. FICs and FIC indices (Σ FIC) were determined as previously described (Johnson et al., 2004). The FIC index is defined as the sum of the FICs for each of the drugs and the FICs are defined as the MIC of each drug when used in combination divided by the MIC of the drug when used alone (Johnson et al., 2004). Drug interactions were based on Σ FIC indices and classified as synergistic (Σ FIC < 0.5), additive (Σ FIC = 0.5 through 1), indifferent (Σ FIC = 1 through 4), or antagonistic (Σ FIC > 4) (Johnson et al., 2004). Determination of MICs of amiloride and amiloride analogs were repeated at least three times. The checkerboard dilutions used to calculate FIC were done twice with similar results.
Toxicity of HMA was measured via lactate dehydrogenase (LDH) activity using the LDH assay kit (Sigma-Aldrich). Briefly, LDH activity, which is a marker of cell viability, was measured following 8 h co- incubation of the human brain microvascular endothelial cell line with 4–64 μg/ml of HMA. Absorbance was measured at 490 nm for 30 min at 37°C, taking the highest time-point reading before absorbance exceeded the maximum tested standard. LDH activity was calculated using an NADH standard set provided by the kit.
Strains were inoculated into liquid medium and grown overnight at 30°C. Approximately 2 × 107 cells were inoculated into 8 ml of top agar pre-warmed at 42°C and subsequently spread onto YPD plates. All drugs and the solvent control were applied to 6-mm BBL disks (Becton Dickinson). These disks were placed on the solidified top agar surface of the YPD plates as indicated and the strains were grown at 30°C for 48 h.
Nhx1 protein sequence from C. neoformans var. grubii (H99 serotype A strain) was used in a BLAST search for homologous proteins in a number of fungal species. The top ‘‘hits’’ were collated and analyzed using the freely available Clustal X 2.1 program2. Clustal alignment results were viewed and annotated using Jalview3. Phylogenetic trees were obtained by entering Clustal alignment data into FigTree version 1.4.04. Accession numbers are provided in Supplementary Figure 2.
A standard unpaired t test was used for comparison of two groups and a one-way analysis of variance (ANOVA) was used when comparing more than two groups. Statistical significance was established at P < 0.05 (∗∗P < 0.01). Statistical analysis was performed with commercially available software (Prism GraphPad version 8.4.3).
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
KV carried out the experiments. AG and EB developed the idea presented, and supervised the study. AG wrote the manuscript. KV and EB assisted with the edits. All authors analyzed and discussed the results and contributed to the final manuscript.
This study was supported by the Will W. Lester Endowment of the University of California awarded to EB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to F. Gorin for providing UCD38B and UCD74A small molecules and to J. Heitman and G. R. Thompson for providing fungal strains/isolates.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.673035/full#supplementary-material
Supplementary Figure 1 | Candidate amiloride analogs with hydrophobic substitutions at the 5-amino group of the pyrazine ring. (A) 3-Amino-N-(aminoiminomethyl)-5-azacyclotridec-1-yl-6-chloro-2-pyrazinecarboxamide; (B) 3-amino-N-(aminoiminomethyl)-6-chloro-5-(3,4-dihydro-2(1H)-isoquinolinyl)-2Pyrazinecarboxamide; (C) 3-amino-N-(aminoiminomethyl)-6-chloro-5-(2,3-dihydro-1H-indol-1-yl)-2-Pyrazinecarboxamide; (D) 3-amino-N-(aminoiminomethyl)-6-chloro-5-(methylphenylamino)-2-Pyrazinecarboxamide; (E) 3-amino-N-(aminoiminomethyl)-6-chloro-5-(cyclohexylmethylamino)-2-Pyrazinecarboxamide; (F) 3-amino-N-(aminoiminomethyl)-6-chloro-5-[(cyclohexylmethyl)amino]-2-Pyrazinecarboxamide; (G) 3-amino-N-(aminoiminomethyl)-6-chloro-5-(dipropylamino)-2- Pyrazinecarboxamide.
Supplementary Figure 2 | Accession numbers for fungal amino acid sequences of Nhx1. Shown are the corresponding accession numbers for Nhx1 for each species of fungi used in the Clustal alignment.
Africa, C. W., and Abrantes, P. M. (2016). Candida antifungal drug resistance in sub-Saharan African populations: a systematic review. F1000Res 5:2832. doi: 10.12688/f1000research.10327.2
Almeida, F., Rodrigues, M. L., and Coelho, C. (2019). The still underestimated problem of fungal diseases worldwide. Front. Microbiol. 10:214. doi: 10.3389/fmicb.2019.00214
Aredia, F., Czaplinski, S., Fulda, S., and Scovassi, A. I. (2016). Molecular features of the cytotoxicity of an NHE inhibitor: evidence of mitochondrial alterations, ROS overproduction and DNA damage. BMC Cancer 16:851. doi: 10.1186/s12885-016-2878-9
Benos, D. J. (1982). Amiloride: a molecular probe of sodium transport in tissues and cells. Am. J. Physiol. 242, C131–C145. doi: 10.1152/ajpcell.1982.242.3.C131
Blumwald, E., Cragoe, E. J., and Poole, R. J. (1987). Inhibition of Na+/H+ antiport activity in sugar beet tonoplast by analogs of amiloride. Plant Physiol. 85, 30–33. doi: 10.1104/pp.85.1.30
Buckley, B. J., Aboelela, A., Minaei, E., Jiang, L. X., Xu, Z., Ali, U., et al. (2018). 6-Substituted Hexamethylene Amiloride (HMA) derivatives as potent and selective inhibitors of the human urokinase plasminogen activator for use in cancer. J. Med. Chem. 61, 8299–8320. doi: 10.1021/acs.jmedchem.8b00838
Butts, A., Palmer, G. E., and Rogers, P. D. (2017). Antifungal adjuvants: preserving and extending the antifungal arsenal. Virulence 8, 198–210. doi: 10.1080/21505594.2016.1216283
Cagnac, O., Leterrier, M., Yeager, M., and Blumwald, E. (2007). Identification and characterization of Vnx1p, a novel type of vacuolar monovalent cation/H+ antiporter of Saccharomyces cerevisiae. J. Biol. Chem. 282, 24284–24293. doi: 10.1074/jbc.M703116200
Campoy, S., and Adrio, J. L. (2017). Antifungals. Biochem. Pharmacol. 133, 86–96. doi: 10.1016/j.bcp.2016.11.019
Counillon, L., Franchi, A., and Pouyssegur, J. (1993). A point mutation of the Na+/H+ exchanger gene (NHE1) and ampl1ification of the mutated allele confer amiloride resistance upon chronic acidosis. Proc. Natl. Acad. Sci. U.S.A. 90, 4508–4512. doi: 10.1073/pnas.90.10.4508
Cragoe, E. J. Jr., Woltersdorf, O. W. Jr., Bicking, J. B., Kwong, S. F., and Jones, J. H. (1967). Pyrazine diuretics. II. N-amidino-3-amino-5-substituted 6-halopyrazinecarboxamides. J. Med. Chem. 10, 66–75. doi: 10.1021/jm00313a014
Darley, C. P., van Wuytswinkel, O. C., van der Woude, K., Mager, W. H., and de Boer, A. H. (2000). Arabidopsis thaliana and Saccharomyces cerevisiae NHX1 genes encode amiloride sensitive electroneutral Na+/H+ exchangers. Biochem. J. 351(Pt 1), 241–249. doi: 10.1042/0264-6021:3510241
Gao, Z. G., and Ijzerman, A. P. (2000). Allosteric modulation of A(2A) adenosine receptors by amiloride analogues and sodium ions. Biochem Pharmacol 60, 669–676. doi: 10.1016/s0006-2952(00)00360-9
Garritsen, A., Ijzerman, A. P., Tulp, M. T., Cragoe, E. J. Jr., and Soudijn, W. (1991). Receptor binding profiles of amiloride analogues provide no evidence for a link between receptors and the Na+/H+ exchanger, but indicate a common structure on receptor proteins. J. Recept. Res. 11, 891–907. doi: 10.3109/10799899109064686
Johnson, M. D., MacDougall, C., Ostrosky-Zeichner, L., Perfect, J. R., and Rex, J. H. (2004). Combination antifungal therapy. Antimicrob. Agents Chemother. 48, 693–715. doi: 10.1128/aac.48.3.693-715.2004
Kim, J. H., Cheng, L. W., Chan, K. L., Tam, C. C., Mahoney, N., Friedman, M., et al. (2020). Antifungal drug repurposing. Antibiotics (Basel) 9:812. doi: 10.3390/antibiotics9110812
Kleyman, T. R., and Cragoe, E. J. Jr. (1988). Amiloride and its analogs as tools in the study of ion transport. J. Membr. Biol. 105, 1–21. doi: 10.1007/BF01871102
Kwon-Chung, K. J., Bennett, J. E., Wickes, B. L., Meyer, W., Cuomo, C. A., Wollenburg, K. R., et al. (2017). The case for adopting the “Species Complex” nomenclature for the etiologic agents of Cryptococcosis. mSphere 2, e00357-e00416. doi: 10.1128/mSphere.00357-16
Li, J. H., Cragoe, E. J. Jr., and Lindemann, B. (1985). Structure-activity relationship of amiloride analogs as blockers of epithelial Na+ channels: I. Pyrazine-ring modifications. J. Membr. Biol. 83, 45–56. doi: 10.1007/BF01868737
Loyse, A., Thangaraj, H., Easterbrook, P., Ford, N., Roy, M., Chiller, T., et al. (2013). Cryptococcal meningitis: improving access to essential antifungal medicines in resource-poor countries. Lancet Infect. Dis. 13, 629–637. doi: 10.1016/S1473-3099(13)70078-1
Marchetti, O., Entenza, J. M., Sanglard, D., Bille, J., Glauser, M. P., and Moreillon, P. (2000). Fluconazole plus cyclosporine: a fungicidal combination effective against experimental endocarditis due to Candida albicans. Antimicrob. Agents Chemother. 44, 2932–2938. doi: 10.1128/aac.44.11.2932-2938.2000
Masereel, B., Pochet, L., and Laeckmann, D. (2003). An overview of inhibitors of Na+/H+ exchanger. Eur. J. Med. Chem. 38, 547–554. doi: 10.1016/s0223-5234(03)00100-4
Molloy, S. F., Kanyama, C., Heyderman, R. S., Loyse, A., Kouanfack, C., Chanda, D., et al. (2018). Antifungal combinations for treatment of Cryptococcal Meningitis in Africa. N. Engl. J. Med. 378, 1004–1017. doi: 10.1056/NEJMoa1710922
Mpoza, E., Rhein, J., and Abassi, M. (2018). Emerging fluconazole resistance: implications for the management of cryptococcal meningitis. Med. Mycol. Case Rep. 19, 30–32. doi: 10.1016/j.mmcr.2017.11.004
Nakamura, N., Tanaka, S., Teko, Y., Mitsui, K., and Kanazawa, H. (2005). Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J. Biol. Chem. 280, 1561–1572. doi: 10.1074/jbc.M410041200
Nielsen, K., Cox, G. M., Wang, P., Toffaletti, D. L., Perfect, J. R., and Heitman, J. (2003). Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and alpha isolates. Infect. Immun. 71, 4831–4841. doi: 10.1128/iai.71.9.4831-4841.2003
Odds, F. C., Brown, A. J., and Gow, N. A. (2003). Antifungal agents: mechanisms of action. Trends Microbiol. 11, 272–279. doi: 10.1016/s0966-842x(03)00117-3
Onyewu, C., Blankenship, J. R., Del Poeta, M., and Heitman, J. (2003). Ergosterol biosynthesis inhibitors become fungicidal when combined with calcineurin inhibitors against Candida albicans, Candida glabrata, and Candida krusei. Antimicrob. Agents Chemother. 47, 956–964. doi: 10.1128/aac.47.3.956-964.2003
Palandoken, H., By, K., Hegde, M., Harley, W. R., Gorin, F. A., and Nantz, M. H. (2005). Amiloride peptide conjugates: prodrugs for sodium-proton exchange inhibition. J. Pharmacol. Exp. Ther. 312, 961–967. doi: 10.1124/jpet.104.076984
Pfaller, M. A., Messer, S. A., Boyken, L., Hollis, R. J., Rice, C., Tendolkar, S., et al. (2004). In vitro activities of voriconazole, posaconazole, and fluconazole against 4,169 clinical isolates of Candida spp. and Cryptococcus neoformans collected during 2001 and 2002 in the ARTEMIS global antifungal surveillance program. Diagn. Microbiol. Infect. Dis. 48, 201–205. doi: 10.1016/j.diagmicrobio.2003.09.008
Pfaller, M. A., Zhang, J., Messer, S. A., Brandt, M. E., Hajjeh, R. A., Jessup, C. J., et al. (1999). In vitro activities of voriconazole, fluconazole, and itraconazole against 566 clinical isolates of Cryptococcus neoformans from the United States and Africa. Antimicrob. Agents Chemother. 43, 169–171. doi: 10.1093/oxfordjournals.jac.a020873
Rajasingham, R., Smith, R. M., Park, B. J., Jarvis, J. N., Govender, N. P., Chiller, T. M., et al. (2017). Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect. Dis. 17, 873–881. doi: 10.1016/S1473-3099(17)30243-8
Smith, K. D., Achan, B., Hullsiek, K. H., McDonald, T. R., Okagaki, L. H., Alhadab, A. A., et al. (2015). Increased antifungal drug resistance in clinical isolates of Cryptococcus neoformans in Uganda. Antimicrob. Agents Chemother. 59, 7197–7204. doi: 10.1128/AAC.01299-15
Soudijn, W., Van Wijngaarden, I., and Ap, I. J. (2004). Allosteric modulation of G protein-coupled receptors: perspectives and recent developments. Drug Discov. Today 9, 752–758. doi: 10.1016/S1359-6446(04)03220-9
Vidt, D. G. (1981). Mechanism of action, pharmacokinetics, adverse effects, and therapeutic uses of amiloride hydrochloride, a new potassium-sparing diuretic. Pharmacotherapy 1, 179–187. doi: 10.1002/j.1875-9114.1981.tb02539.x
Vu, K., and Gelli, A. (2010). Astemizole and an analogue promote fungicidal activity of fluconazole against Cryptococcus neoformans var. grubii and Cryptococcus gattii. Med. Mycol. 48, 255–262. doi: 10.1080/13693780903081968
Vu, K., Thompson, G. R. III, Roe, C. C., Sykes, J. E., Dreibe, E. M., Lockhart, S. R., et al. (2018). Flucytosine resistance in Cryptococcus gattii is indirectly mediated by the FCY2-FCY1-FUR1 pathway. Med. Mycol. 56, 857–867. doi: 10.1093/mmy/myx135
Yamazumi, T., Pfaller, M. A., Messer, S. A., Houston, A., Hollis, R. J., and Jones, R. N. (2000). In vitro activities of ravuconazole (BMS-207147) against 541 clinical isolates of Cryptococcus neoformans. Antimicrob. Agents Chemother. 44, 2883–2886. doi: 10.1128/aac.44.10.2883-2886.2000
Keywords: Cryptococcus, antifungal, amiloride, HMA, synergy, Nhx1, azoles, Na+/H+ exchanger
Citation: Vu K, Blumwald E and Gelli A (2021) The Antifungal Activity of HMA, an Amiloride Analog and Inhibitor of Na+/H+ Exchangers. Front. Microbiol. 12:673035. doi: 10.3389/fmicb.2021.673035
Received: 26 February 2021; Accepted: 12 April 2021;
Published: 05 May 2021.
Edited by:
Miguel Cacho Teixeira, University of Lisbon, PortugalReviewed by:
Claudia Cafarchia, University of Bari Aldo Moro, ItalyCopyright © 2021 Vu, Blumwald and Gelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angie Gelli, YWNnZWxsaUB1Y2RhdmlzLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.