 Mei Peng
Mei Peng Fang Chen4
Fang Chen4 Zhongdao Wu
Zhongdao Wu Jia Shen
Jia Shen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 31 May 2021
Sec. Infectious Agents and Disease
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.670874
This article is part of the Research Topic Needs and Potential Application of One Health Approach in the Control of Vector-borne and Zoonotic Infectious Disease View all 14 articles
Endoplasmic reticulum stress (ER stress) can be induced when cellular protein homeostasis is damaged, and cells can activate the unfolded protein response (UPR) to restore protein homeostasis or induce cell death to facilitate the survival of the whole system. Globally, parasites are a constant threat to human health and are therefore considered a serious public health problem. Parasitic infection can cause ER stress in host cells, and parasites also possess part or all of the UPR under ER stress conditions. In this review, we aim to clarify the role of ER stress pathways and related molecules in parasites for their survival and development, the pathogenesis of parasitosis in hosts, and the artemisinin resistance of Plasmodium, which provides some potential drug design targets to inhibit survival of parasites, relieves pathological damage of parasitosis, and solves the problem of artemisinin resistance.
The endoplasmic reticulum (ER), a vital organelle in eukaryotic cells, is the site of synthesis and processing of membrane and secretory proteins, synthesis of lipids, and storage of Ca2+ (Dolai and Adak, 2014). Therefore, it is important to maintain ER homeostasis. Yet, many factors influence the protein homeostasis of ER, such as plasma cell differentiation (Gass et al., 2002), tunicamycin (Pahl and Baeuerle, 1995), and parasite infection (Galluzzi et al., 2017) which result in accumulated misfolded or unfolded proteins that exceed the folding capacity of ER and trigger endoplasmic reticulum stress (ER stress). Endoplasmic reticulum-associated degradation (ERAD) and unfolded protein response (UPR) are the two major quality control processes of ER stress (Bukau et al., 2006). UPR reduces the synthesis of proteins and eliminates misfolded proteins within the ER by increasing the expression of the ER chaperone proteins.
In mammalian cells, the UPR is mediated by three signaling pathways and activated by three ER-transmembrane proteins: inositol-requiring kinase/endoribonuclease 1 (IRE1), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Figure 1; Hwang and Qi, 2018). Glucose-regulated proteins 78 (GRP78) (Bertolotti et al., 2000; Shen et al., 2002), also called immunoglobulin heavy chain binding protein (Bip) (Bertolotti et al., 2000; Shen et al., 2002), binds to these transmembrane proteins in unstressed cells, while it dissociates from them and binds to unfolded or misfolded proteins in stressed cells (Bertolotti et al., 2000; Shen et al., 2002; Grootjans et al., 2016). After dissociation from Bip, IRE1α will be activated by forming IRE1α homodimers and incise the transcription factor X box-binding protein 1 (XBP1) mRNA into spliced XBP1 (XBP1s) (Bertolotti et al., 2000; Calfon et al., 2002). The function of XBP1s is to maintain the ER function and response to UPR and regulate the expansion of the secretory apparatus (Acosta-Alvear et al., 2007). Activated PERK, induced by oligomerization and autophosphorylation following dissociation from Bip (Bertolotti et al., 2000), phosphorylates the α-subunit of eukaryotic translational initiation factor 2 (eIF2α) and attenuates protein translation, which will reduce the load of newly synthesized proteins within the ER while upregulating the expression of activating transcription factor 4 (ATF4). In addition, the phosphorylation of eIF2α can be dephosphorylated by growth arrest and DNA damage-inducible protein-34 (GADD34). Further, ATF4 is required for the transactivation of GADD34, which will promote the recovery of translation (Novoa et al., 2001; Ma and Hendershot, 2003). Under ER stress, ATF6 translocates from the ER to the Golgi apparatus (Shen et al., 2002). A 90-kDa protein ATF6 (p90ATF6) is converted to a 50-kDa protein ATF6 (p50ATF6, an active and mature form of ATF6) through the cleavage of Golgi-resident proteases—site 1 protease (S1P) and site 2 protease (S2P). P50ATF6 further activates the transcription of ER chaperone genes after entering the nucleus (Haze et al., 1999; Ron and Walter, 2007). Interactions among the three UPR pathways have been found, wherein ATF6 induces the transcription of XBP1 (Yoshida et al., 2001) and PERK-ATF4 upregulates the expression of IRE1α (Tsuru et al., 2016). These interactions will promote UPR to be stronger and more persistent in order to deal with various types of ER stress. Nevertheless, prolonged and severe ER stress can activate various cell death effectors such as BAK, BAX, caspase-12, C/EBP-homologous protein (CHOP), and GADD34 and induce cell death (Ron and Walter, 2007). The mechanism of UPR is evolutionary conservatism across eukaryotes.
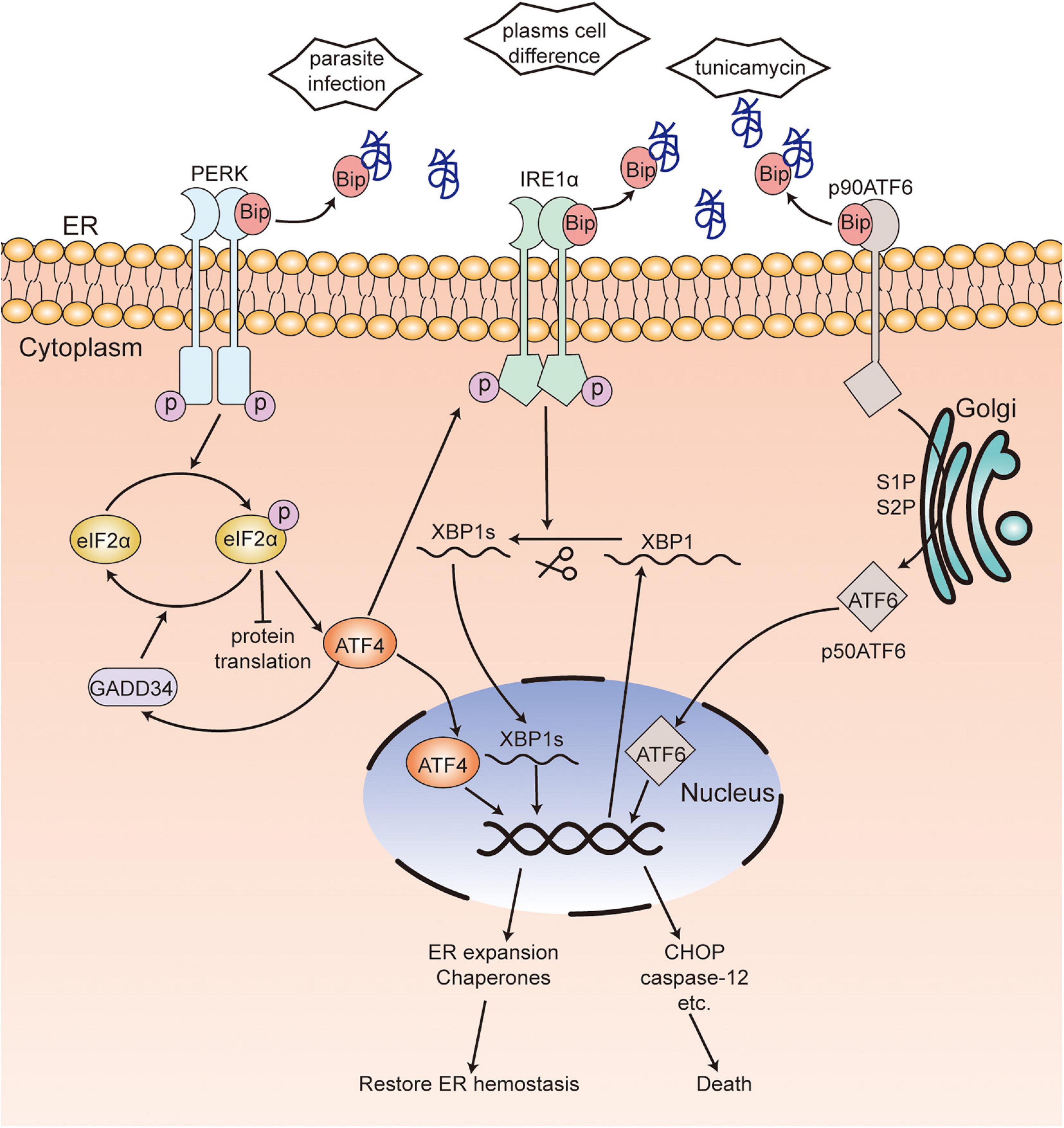
Figure 1. Unfolded protein response in mammalian cells. In unstressed cells, Bip binds to three transmembrane proteins PERK, IRE1α and ATF6. When ER stress occurs, Bip dissociates from these transmembrane proteins and binds to unfolded or misfolded proteins. Activated PERK, induced by oligomerization following dissociation from Bip, phosphorylates eIF2α, which reduces protein translation while upregulating the expression of ATF4. eIF2α phosphorylation can be dephosphorylated by GADD34. After dissociation from Bip, IRE1α is activated by forming IRE1α homodimers and incises XBP1 mRNA into XBP1s. Bip releases from ATF6, which leads to the translocation to Golgi and activation of ATF6. ATF4, XBP1s, and activated ATF6 enter the nucleus and activate the transcription of ER chaperones or various death effectors, which results in the restoration of ER hemostasis or cell death. [ ], unfolded or misfolded proteins; PERK, protein kinase RNA-like ER kinase; IRE1, inositol-requiring kinase/endoribonuclease 1; eIF2α, α-subunit of eukaryotic translational initiation factor 2; ATF6, activating transcription factor 6; ATF4, activating transcription factor 4; XBP1, X box-binding protein 1; CHOP, C/EBP-homologous protein; Bip, immunoglobulin heavy chain binding protein; GADD34, DNA damage-inducible protein-34.
], unfolded or misfolded proteins; PERK, protein kinase RNA-like ER kinase; IRE1, inositol-requiring kinase/endoribonuclease 1; eIF2α, α-subunit of eukaryotic translational initiation factor 2; ATF6, activating transcription factor 6; ATF4, activating transcription factor 4; XBP1, X box-binding protein 1; CHOP, C/EBP-homologous protein; Bip, immunoglobulin heavy chain binding protein; GADD34, DNA damage-inducible protein-34.
Parasitosis, caused by parasitic infections, has been harmful to human health and economic development since very long and is still a major global public health problem. As is known, the effective control of parasitic diseases is mainly dependent on the application of parasitic drugs and disruption of the pathogen’s life cycle, such as praziquantel and artemisinin. Unfortunately, drug resistance of parasitic drugs has been reported in recent years (Fallon and Doenhoff, 1994; Dondorp et al., 2009). Therefore, new anti-parasitic drugs including those to alleviate the pathology of the host caused by parasite infection and those to kill parasites should be identified urgently, along with determination of the mechanisms of drug resistance.
When parasites infect the host and obtain nutrients, they will perturb ER homeostasis and induce ER stress and UPR of the host. On the one hand, the induced ER stress of the host is beneficial to the survival and infection of the parasites. For instance, it has been reported that Plasmodium berghei infection induced ER stress of hepatocytes and activated UPR through the XBP1 and cAMP responsive element-binding protein (CREBH, a hepatocyte specific UPR mediator) pathways, which contributed to the infection of Plasmodium by providing phosphatidylcholine and regulating iron level (Inacio et al., 2015). In addition, Leishmania infection induced ER stress of the host to facilitate infection through the PERK-eIF2α-ATF4 and IRE1-XBP1 pathways (Dias-Teixeira et al., 2016, 2017; Galluzzi et al., 2016; Abhishek et al., 2018). And Toxoplasma triggered the UPR in host cells, which affected calcium release from ER, can enhance host cell migration and dissemination of the parasite to host organs (Augusto et al., 2020). However, Poncet et al. (2021) have showed that the IRE1a/XBP1s branch of the UPR was a key regulator of host defense upon Toxoplasma gondii infection, that mice deficient for IRE1a and XBP1 in DCs displayed a severe susceptibility to T. gondii infection, which indicates that the UPR induced by parasites also plays an important role in host immune defense. Anyhow, on the other hand, excessive ER stress and UPR will cause severe pathological damage to the host. Yu et al. (2014) found that the levels of GRP78, CHOP, cleaved caspase-12, and phosphorylated-JNK in the intestine of Trichinella spiralis-infected mice were significantly upregulated, which indicated that the ER stress-induced apoptotic pathway participated in intestinal lesions caused by T. spiralis infection. Thus, inhibition of excessive UPR in the host may be a therapeutic target to alleviate the pathological symptoms.
Additionally, the parasites can sense ER stress and either induce UPR to facilitate their survival when attacked by the host immune system or adapt to the host environment (such as changes in pH and temperature, oxidative stress, nutrient deficiency) (Zuzarte-Luís and Mota, 2018). Therefore, the UPR signaling pathway may be a potential target for inhibiting the survival and development of parasites.
Different parasitic infections result in different pathological damage to different host tissues and organs. Nowadays, increasing reports show that ER stress and UPR play an important role in the development of pathology of parasitosis (Anand and Babu, 2013; Ayyappan et al., 2019).
Plasmodium spp., which are the causative agents of malaria, are obligate intracellular protozoan parasites. Anand and Babu (2013) reported that experimental cerebral malaria (ECM), caused by P. berghei ANKA (PbA) infection, was related to ER stress. They found that PbA infection-induced ER stress could cause the apoptosis of neuronal cells in mice by activating the three branches of UPR—PERK-eIF2α-ATF4/GADD34, IRE1-XBP1s, and ATF6—along with upregulating the levels of CHOP, cleaved caspase-3 and caspase-12 and downregulating the expression of Bip, calreticulin, and calnexin.
Trypanosome cruzi is the causative pathogen of Chagas disease in humans. Reportedly, the trypomastigotes of T. cruzi infection could induce ER stress in the heart of mice, with an increase in the levels of Bip, PERK, eIF2α, ATF4, and CHOP, thereby causing damage to the host. Interestingly, 2-aminopurine (2-APB, an ER stress inhibitor) treatment could alleviate the pathological damage to the heart by decreasing the phosphorylation of eIF2α and its downstream signaling. Therefore, this indicates that inhibition of ER stress may be a therapeutic target for cardiomyopathy in Chagas patients (Ayyappan et al., 2019).
Toxoplasma is an obligate intracellular parasite and opportunistic pathogenic parasite (Sullivan et al., 2004). Toxoplasma encephalitis is the most serious outcome of toxoplasmosis, which may be fatal to immunocompromised individuals. Some studies have found that Toxoplasma encephalitis was related to ER stress. It has been reported that the tachyzoites of T. gondii RH strain and TgCtwh3 (a representative Chinese 1 Toxoplasma strain) induced apoptosis of neural stem cells and neural stem cell line C17.2 by activating CHOP, caspase-12, and JNK (Wang et al., 2014; Zhou et al., 2015). Pretreatment with tauroursodeoxycholic acid (TUDCA, an ER stress inhibitor) and Z-ATAD-FMK (a caspase-12 inhibitor) led to the inhibition of apoptosis (Wang et al., 2014; Zhou et al., 2015), which suggested that neural stem cell apoptosis induced by both TgCtwh3 and RH strain infection was dependent on the ER stress pathway, and ER stress inhibitors could be used to alleviate Toxoplasma encephalitis. In addition, Wan et al. (2015) showed that virulence factor rhoptry protein 18 (ROP18) secreted by T. gondii was involved in nerve cell apoptosis via the ER stress pathway, characterized by an increase in the expression of cleaved caspase-12, CHOP, and cleaved caspase-3. Ran et al. further indicated that ROP18 induced apoptosis of neural cells by phosphorylating reticulon 1-C [RTN1-C, a protein localized in the ER that is preferentially expressed in the neural cells of the central nervous system (CNS) at Ser7/134 and Thr4/8/118], which led to the acetylation of GRP78 and induced ER stress (An et al., 2018). These results suggest that inhibition of ROP18 of T. gondii can be used as a drug target for the treatment of Toxoplasma encephalitis to inhibit the ER stress-induced apoptosis of host cells.
Schistosoma japonicum is the causative agent of schistosomiasis. The pathogenic mechanism of schistosomiasis is primarily attributed to egg-induced hepatic granuloma and fibrosis and cirrhosis (Yu et al., 2016; Duan et al., 2019). Duan et al. (2019) showed that the level of CHOP, a vital factor in the ER stress-mediated apoptosis pathway, was significantly increased in mice at 6 and 10 weeks following infection with S. japonicum. The study indicated that ER stress may be involved in S. japonicum infection-induced hepatic fibrosis. Moreover, Yu et al. (2016) showed that treatment with taurine, an inhibitor of ER stress, significantly suppressed the egg-induced hepatic granuloma and alleviated hepatic fibrosis in mice at 8 weeks post-infection, along with marked reduction of the expression of GRP78. Therefore, ER stress inhibitors may be a therapeutic drug for hepatic fibrosis.
The summary of ER stress in hosts caused by parasitic infection is shown in Figure 2. Therefore, the UPR signaling pathway may be a therapeutic target to alleviate pathological symptoms.
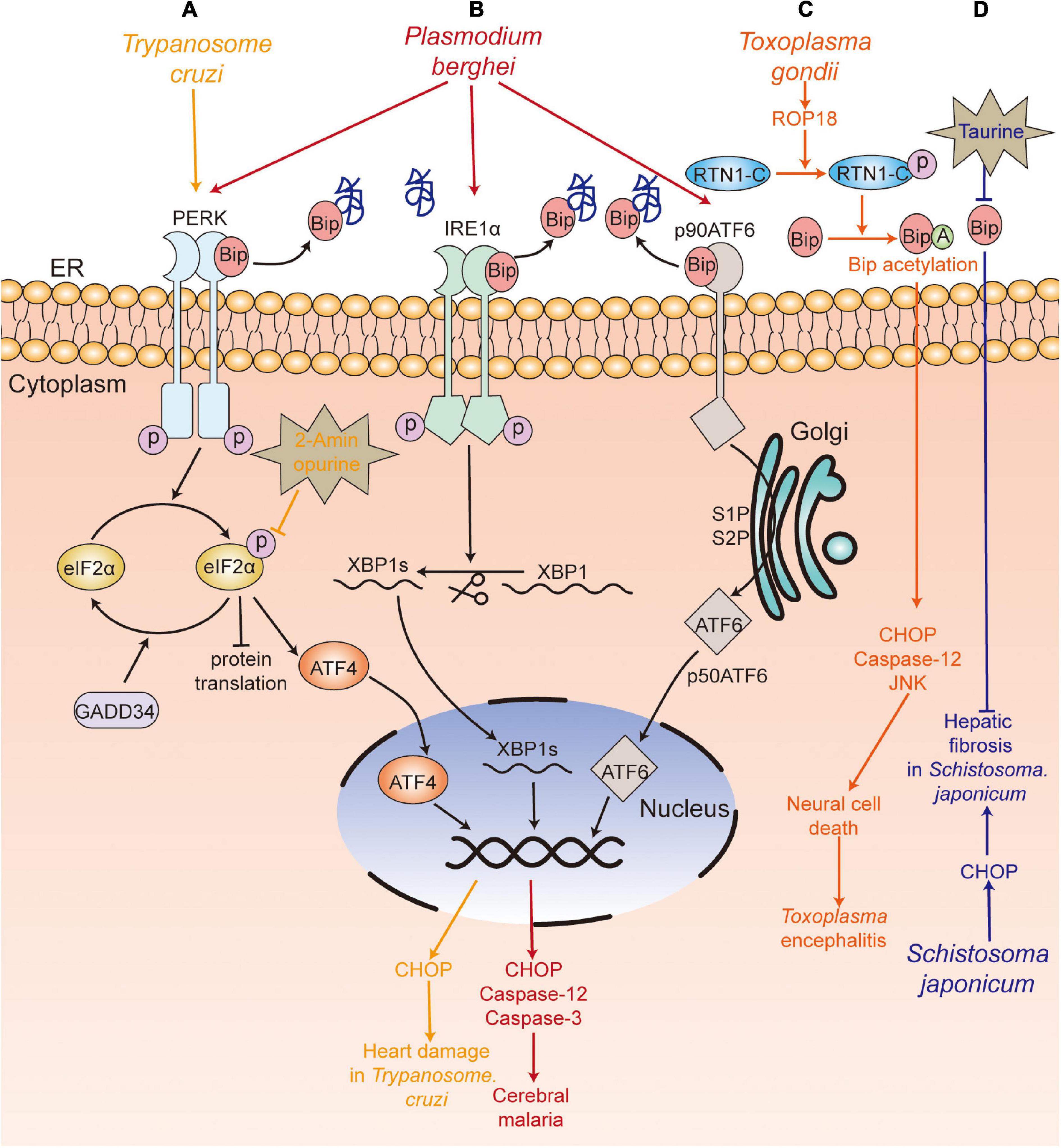
Figure 2. UPR participates in the pathological damage caused by parasite infection. (A) T. Cruzi (yellow) infection caused heart damage, with upregulation of PERK-eIF2α-ATF4-CHOP pathway, and 2-aminopurine treatment alleviated the heart’s pathological damage. (B) P. berghei (red) infection induced neuronal cell death and caused experimental cerebral malaria by activating the three branches of the UPR (PERK-eIF2α-ATF4/GADD34, IRE1-XBP1s, ATF6). (C) Rhoptry protein 18 (ROP18) of T. gondii (orange) phosphorylated reticulon 1-C (RTN1-C), which led to the acetylation of GRP78 and further upregulated the expression of cleaved caspase-12, CHOP, cleaved caspase-3, and induced the apoptosis of neural cells. (D) S. japonicum (blue) infection led to increased levels of CHOP, which was involved in hepatic fibrosis, and the treatment with taurine suppressed the egg-induced hepatic granuloma and fibrosis. PERK, protein kinase RNA-like ER kinase; IRE1, inositol-requiring kinase/endoribonuclease 1; eIF2α, α-subunit of eukaryotic translational initiation factor 2; ATF6, activating transcription factor 6; ATF4, activating transcription factor 4; XBP1, X box-binding protein 1; CHOP, C/EBP-homologous protein; Bip, immunoglobulin heavy chain binding protein;GADD34, DNA damage-inducible protein-34.
Parasites can sense ER stress and induce UPR of themselves to facilitate their survival and development. Different parasites may have different components of ER stress pathway.
Plasmodium has a complicated life cycle, including the merozoite, ring, trophozoite, schizont, and gametophyte stages in humans and the ookinete and sporozoite stages in mosquitoes. Chaubey et al. (2014) showed that Plasmodium falciparum lacked the orthologs of XBP1, IRE1, ATF6, and ATF4, and only retained the PERK-eIF2α pathway to regulate translation under ER stress. Three eIF2α kinases have been identified, namely IK1, IK2, and PK4 (eIF2α kinase of Plasmodium (Möhrle et al., 1997), a PERK homolog of mammals) (Ward et al., 2004). It has been reported that increased phosphorylation of eIF2α leads to reduced levels of protein translation, which is associated with the formation of P. falciparum gametophytes and the conversion of the P. berghei gametophytes into ookinetes when treated with dithiothreitol (DTT) (Chaubey et al., 2014; Duran-Bedolla et al., 2017). In addition, Zhang et al. (2012) have shown that PK4 was involved in the invasion of new red blood cells of merozoite-containing schizonts and the gametocyte infecting Anopheles mosquitoes. The inhibition of PK4 of P. berghei by generating a PK4 conditional mutant (PbPK4cKO) would alleviate the symptoms of malaria and inhibit disease transmission. Another study indicated that treatment of GSK2606414 (a small molecule inhibitor of PERK (Axten et al., 2012), which specifically inhibits PK4 instead of IK1 and IK2 in vitro) could block the transformation of P. falciparum from trophozoites to schizonts (Zhang et al., 2017). The transformation between different forms increased the ability of translational regulation of Plasmodium. In addition, Chen et al. (2018) reported that apoptozole, a novel chemical scaffold, was lethal to the chloroquine-sensitive and chloroquine -resistant P. falciparum parasite strains by inhibiting GRP78 function in vitro. Compared to human GRP78, P. falciparum GRP78 showed a lower affinity to the endogenous ligands, ADP and ATP, which indicated that the competitive inhibitors of GRP78 can be investigated for P. falciparum control (Chen et al., 2018).
According to the above mentioned studies, it appears that the PK4-eIF2α pathway plays an important role in both morphological transformation and host transmission in Plasmodium. Thus, PK4 inhibition would inhibit the development of Plasmodium, which implies that PK4 inhibitors may be a potential target in malaria treatment. However, Bridgford et al. (2018) found that dihydroartemisinin (DHA) increased the toxicity to Plasmodium by prolonging PK4 activation and eIF2α phosphorylation. Therefore, appropriate ER stress is beneficial to the development of Plasmodium, while excessive ER stress would be lethal to the parasites.
Leishmania is the pathogen causing Leishmaniasis and has two forms—promastigote and amastigote. Gosline et al. (2011) proved that Leishmania lacked a transcriptional regulation response to UPR, and only retained the translational regulation in ER stress. They also showed an increased level of phosphorylation of eIF2α in L. donovani after treatment of DTT (Gosline et al., 2011). Moreover, Chow et al. (2011) found that the PERK homolog of Leishmania largely colocalized with Bip in ER, which can phosphorylate eIF2α at threonine 166. They further confirmed that PERK-dependent eIF2α phosphorylation was vital for Leishmania to switch from the promastigote to amastigote form in vitro (Chow et al., 2011). Unlike host macrophages having intact UPR pathway, the mere presence of the PERK pathway in L. donovani promoted the parasite’s susceptibility to DTT-induced ER stress (Gosline et al., 2011), which suggests that inhibition of the PERK pathway and induction of ER stress in Leishmania are both potential targets to kill the parasite. Dolai et al. (2011) proved that tunicamycin treatment induced apoptosis of Leishmania major, with an increase in the level of Bip.
Trypanosome brucei is a protozoan parasite that cycles between the tsetse fly (procyclic form) and mammalian host (blood stream form), which causes African sleeping sickness in humans and nagana in livestock (Zhang et al., 2019). Goldshmidt et al. (2010) reported that the expression of Bip of T. brucei was increased in both procyclic and blood stream forms in DTT-induced ER stress, and irrecoverable ER stress could induce spliced leader RNA silencing pathway (SLS pathway, a unique process in T. brucei), which may accelerate programmed cell death (PCD). Besides, Messias Sandes et al. (2019) showed that both DTT and tunicamycin could induce PCD in T. cruzi.
There were three putative eIF2α kinases (TbeIF2K1-K3) in T. brucei, though its genome lacked the homologs of IRE1/XBP1. It was reported that TbeIF2K2, a transmembrane glycoprotein expressed both in the procyclic and bloodstream forms of Trypanosome (Moraes et al., 2007), shared no similar sequence with known eIF2 kinases of mammals and was localized to the flagellar pocket, where endocytosis and exocytosis occur, and all proteins were transported from the flagellar pocket to the cell membrane (Gull, 2003). Therefore, the localization of TbeIF2K2 indicated that it could sense proteins and regulate protein synthesis near the flagellar pocket of the Trypanosome (Moraes et al., 2007), which suggests that TbeIF2K2 may be a good drug target to destroy T. brucei. In addition, Hope et al. (2014) showed that SEC63 (a factor participating in protein translocation machinery in ER) silence-induced ER stress could activate PK3 (TbeIF2K3) and trigger the release of PK3 from the ER to nucleus in the procyclic form of T. brucei. The deletion of PK3 reduced the death of T. brucei in SEC63 silence-induced ER stress, which suggests that PK3 is required for ER stress-induced PCD. Thus, the results indicate that TbeIF2K2 and TbeIF2K3 could be potential drug targets to eliminate T. brucei.
The PERK-eIF2α pathway is also involved in the form transformation of T. cruzi at different developmental stages. Tonelli et al. (2011) reported that the differentiation of non-infective epimastigotes into infective metacyclic trypomastigotes in T. cruzi requires the phosphorylation of Tc-eIF2α.
In conclusion, the results show that eIF2α phosphorylation plays an important role in the survival and development of Trypanosome, while excessive ER stress induced by DTT or tunicamycin can lead to the death of Trypanosome.
Toxoplasma shows two forms in the human host: tachyzoite (a rapidly growing form) and bradyzoite (a quiescent cyst form) (Black and Boothroyd, 2000; Sullivan et al., 2004). It has been reported that T. gondii lacked the homologs of IRE1 and ATF6 (Joyce et al., 2013), while it possessed four TgIF2α kinases, namely TgIF2K-A, TgIF2K-B, TgIF2K-C, and TgIF2K-D (Narasimhan et al., 2008; Konrad et al., 2014). Narasimhan et al. (2008) showed that only TgIF2K-A was a transmembrane protein localized in the ER and bonded to Bip under unstressed conditions. When ER stress occurred, the binding of Bip to TgIF2K-A was reduced, similar to the binding of BiP to PERK in mammals, which suggests that part of the UPR was conserved in T. gondii (Narasimhan et al., 2008).
ER stress is also involved in the differentiation of Toxoplasma. Narasimhan et al. (2008) reported that the phosphorylation of TgIF2α induced by tunicamycin treatment resulted in the differentiation of T. gondii from tachyzoite to bradyzoite cysts. Treatment with salubrinal, an inhibitor of eIF2α dephosphorylation, could also induce the differentiation of bradyzoite cysts, which indicated that TgIF2α phosphorylation was involved in the differentiation of bradyzoite cysts (Narasimhan et al., 2008). Cyst formation is a good way to escape from the host’s immune attack. Therefore, the formation of bradyzoite cysts induced by TgIF2α phosphorylation promotes the survival of T. gondii under stressful conditions. Similar results were confirmed by Joyce et al. (2013). Besides, Joyce et al. (2010) reported that the TgIF2α mutant strain of Toxoplasma (i.e., TgIF2α-S71A, which cannot be phosphorylated) showed a lower virulence to the host cell, a lower survival rate and a slower transmitting speed, compared with the control strain of Toxoplasma. Moreover, Augusto et al. (2018) showed that specific inhibition of TgIF2K-A with GSK2606414 could inhibit the lytic cycle of tachyzoites, including attachment/invasion, replication, egress, and differentiation, which prolonged the survival time of mice with acute toxoplasmosis at a lethal dose of 100 RH strain tachyzoites. Interestingly, GSK2606414 did not show apparent detrimental effects on the host cell though with a high concentration in vitro. Therefore, the results suggest that TgIF2K-A and TgIF2α can be used as drug targets to inhibit Toxoplasma survival.
However, DTT treatment and stearoyl-coenzyme A (CoA) desaturase (SCD) accumulation at the ER could trigger ER stress with increasing phosphorylation of TgIF2α and mediated the apoptosis or autophagy of T. gondii (Nguyen et al., 2017; Hao et al., 2019). Therefore, although the TgIF2K-A/TgIF2α pathway plays a protective role in T. gondii under stress conditions, severe disruption of ER homeostasis can lead to the death of T. gondii.
Entamoeba histolytica infection, caused by ingestion of cysts in contaminated water and food, usually induces amoebic dysentery and liver abscesses in humans (Pineda and Perdomo, 2017). Santi-Rocca et al. (2012) found that no genes encoded the orthologs of PERK and ATF6 in E. histolytica amoeba, while the expression of gene encoding eIF2α was upregulated upon treatment with nitric oxide (NO). Hendrick et al. (2016) showed that eIF2α could be phosphorylated at serine-59 in E. histolytica, with a decrease in translation levels during long-term serum starvation, long-term heat shock, and oxidative stress instead of short-term serum starvation, short-term heat shock, and glucose deprivation, and the viability of EheIF2α-S59D (a phosphomimetic variant of eIF2α) was significantly increased during long-term serum starvation. This study suggests that EheIF2α phosphorylation promotes the survival of E. histolytica under stress conditions. DTT treatment can also induce distinct fragmentation of ER and phosphorylation of EheIF2α, while treatment with SNP and DPTA-NON-Oate (NO donors) did not induce phosphorylation of EheIF2α (Walters et al., 2019). Besides, Kumari et al. (2018) identified the ortholog of IRE1 in E. histolytica (EhIre1) and reported that treatment with tunicamycin resulted in the upregulation of EhIre1. In addition, the level of eIF2α phosphorylation was increased during encystation of Entamoeba invadens, but whether eIF2α is necessary for encystation still needs further investigation (Hendrick et al., 2016).
Echinococcus granulosus is the causative cestode of hydatidosis or cystic echinococcosis (CE) and is a worldwide zoonotic infection that affects many organs in human and mammals (Loos et al., 2018). Nicolao et al. (2017) have identified the ortholog of IRE2, XBP1, and ATF6 in the genome of E. granulosus, but the ortholog of PERK/ATF4 was not found. Treatment with bortezomib (a proteasome inhibitor) led to lower viability of E. granulosus in the larval stage in vitro than that in the control group, with an increase of EgGRP78 and EgIRE2/EgXBP1 mRNA levels in protoscoleces; however, no changes were found in the metacestodes (Nicolao et al., 2017). Another study also showed that arsenic trioxide (As2O3) could disturb the intracellular Ca2+ homeostasis and activated ER stress-related apoptosis of protoscoleces in vitro, with an increase in the expression of GRP78, caspase-3, and caspase-12 (Li et al., 2018). These studies show that the induction of ER stress can lead to the apoptosis of protoscoleces in vitro.
In sum, the components of the UPR response such as the PERK-eIF2α pathway of some parasites (Figures 3, 4), including Plasmodium, Leishmania, Trypanosome, Toxoplasma, and E. histolytica, play an important role in their survival and development. However, excessive ER stress could induce the death of parasites such as Plasmodium, Leishmania, Trypanosome, Toxoplasma, and E. granulosus (Figure 4). Considering the toxicity of commonly used ER stress inducers such as DTT and tunicamycin, it is difficult to use them to kill parasites in vivo. For those parasites that are more sensitive to ER stress inducers than their hosts, it is necessary to explore the appropriate concentration of these inducers. TUDCA, a bile salt and chemical chaperone used to treat biliary cirrhosis clinically (Lazaridis et al., 2001), partially inhibits ER stress by lowering the levels of PERK, Bip (Malo et al., 2010; Liu et al., 2015; Li et al., 2019). Thus, TUDCA may be an alternative therapy for parasitosis.
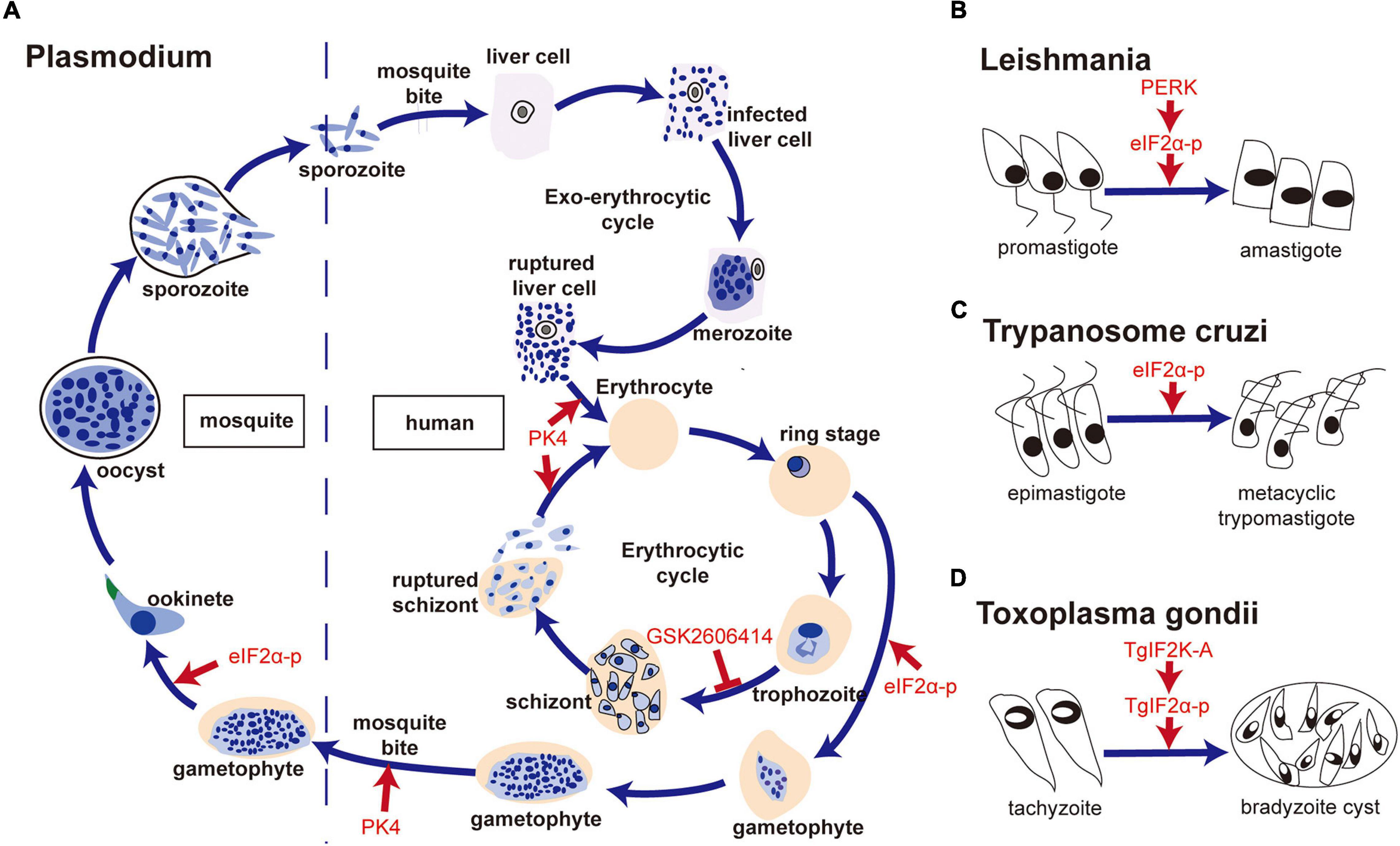
Figure 3. The PERK-eIF2α pathway is involved in forms transformation of parasites. (A) PK4-eIF2α was associated with the formation of gametophytes, conversion of Plasmodium gametophytes into ookinetes, invasion of new red blood cells by merozoite-containing schizonts and the infection of Anopheles mosquitoes by gametocytes; (B) PERK- eIF2α phosphorylation was vital for the switch from promastigote to amastigote in Leishmania; (C) Tc-eIF2α phosphorylation was required for the differentiation of non-infective epimastigotes into infective metacyclic trypomastigotes of T. cruzi; (D) TgIF2K-A/TgIF2α phosphorylation was related to bradyzoite cyst differentiation in T. gondii.
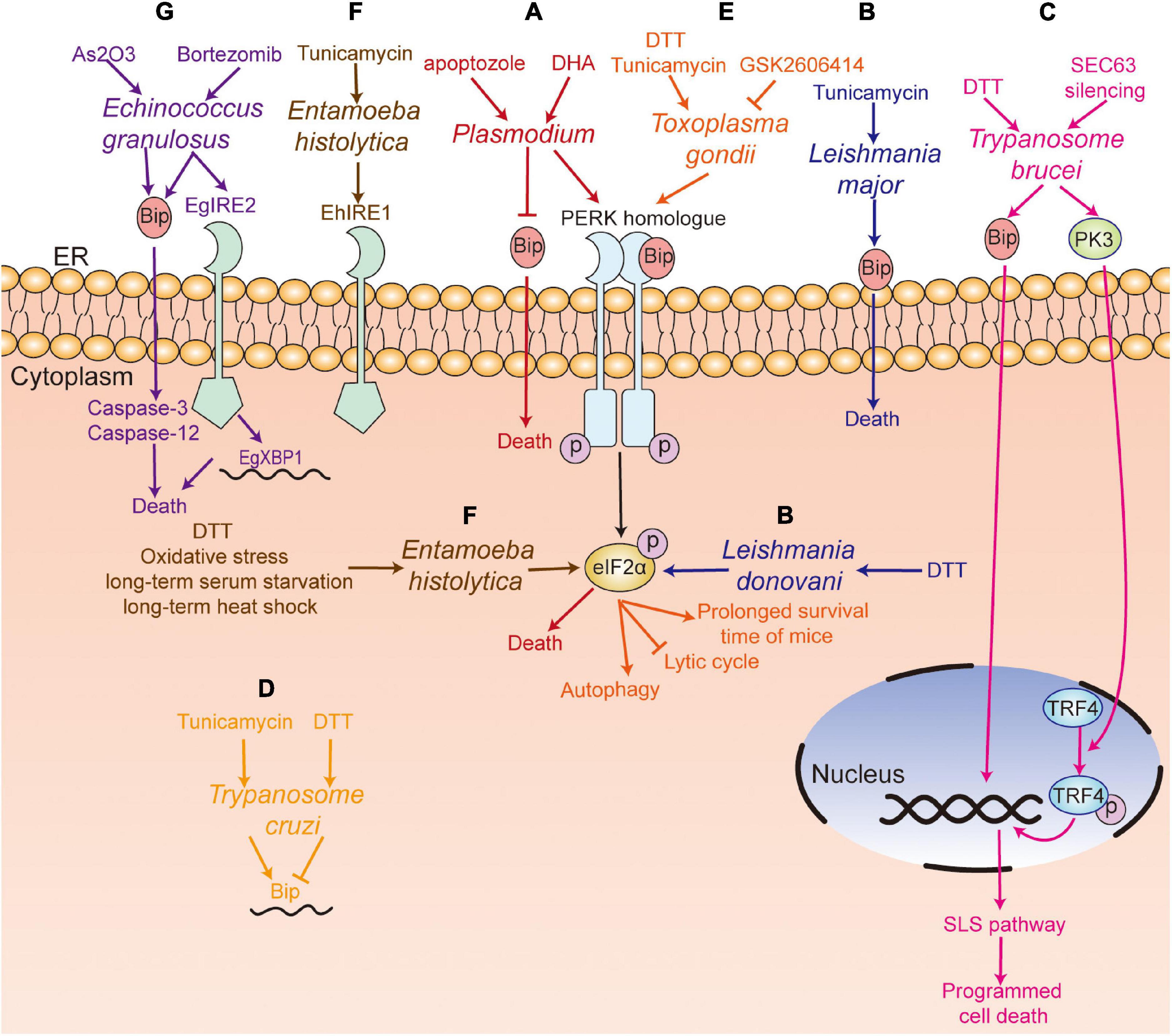
Figure 4. UPR pathway in parasites. (A) Plasmodium spp. (red) with the PERK homolog; apoptozole was lethal to the chloroquine-sensitive and chloroquine-resistant P. falciparum parasite strains by inhibiting GRP78 function in vitro, and DHA treatment could induce the death of P. falciparum through the PERK-eIF2α pathway; (B) Tunicamycin treatment induced the death of L. major (blue) with an increased level of Bip, and DTT treatment upregulated the phosphorylation of eIF2α in L. donovani; (C) DTT treatment induced programmed cell death. SEC63 silencing activated PK3, which further induced programmed cell death by phosphorylating TRF4 and inducing the SLS pathway in T. brucei (pink); (D) Tunicamycin treatment increased the level of Bip mRNA, while DTT treatment decreased the level of Bip in T. Cruzi (yellow); (E) GSK2606414 inhibited the lytic cycle of tachyzoites, including attachment/invasion, replication, egress, and prolonged the survival time of infected mice. DTT treatment mediated the apoptosis or autophagy of T. gondii (orange) by increasing the phosphorylation of TgIF2α; (F) DTT treatment, long-term serum starvation, long-term heat shock and oxidative stress induced the phosphorylation of eIF2α, and the ortholog of IRE1 in E. histolytica (EhIre1) (gray) was identified; (G) Bortezomib treatment induced the death of E. granulosus (purple) with an increase of EgGRP78 and EgIRE2/EgXBP1 mRNA. Arsenic trioxide treatment induced the death of E. granulosus (purple) with upregulation of the expression of GRP78, caspase-3, and caspase-12 in protoscoleces. PERK, protein kinase RNA-like ER kinase; IRE1, inositol-requiring kinase/endoribonuclease 1; XBP1, X box-binding protein 1; Bip, immunoglobulin heavy chain binding protein; eIF2α, α-subunit of eukaryotic translational initiation factor 2; TRF4, TBP-related factor 4; SLS pathway, spliced leader RNA silencing pathway; DTT, dithiothreitol; DHA, dihydroartemisinin.
Endoplasmic reticulum-associated degradation is another way for maintaining ER homeostasis, which can degrade misfolded protein (Hwang and Qi, 2018). And ERAD also exists in parasites, such as trypanosomes (Tiengwe et al., 2016). In addition, some apicomplexan parasites, including P. falciparum, T. gondii and cryptosporidium, harbor an apicoplast, which is important for parasite survival (Agrawal et al., 2013). Reportedly, ERAD components were associated with importing the apicoplast protein, and lose of ERAD components would lead to the death of parasites (Agrawal et al., 2009; Spork et al., 2009). Thus, apicoplast is a potential anti-parasitic drug target. Ubiquitin-dependent ERAD is essential for the survival of Plasmodium (Chung et al., 2012). Harbut et al. found that the inhibitors of signal peptide peptidase (SPP, a protein of ERAD) was lethal to P. falciparum (Harbut et al., 2012). It was reported that NITD731, a SPP inhibitor, was effective against T. cruzi and T. gondii, and it showed no toxicity to human cell lines (Harbut et al., 2012). Above studies further showed that parasites were much more sensitive to the disruption of protein homeostasis. Thus, inhibition of the two key quality-control mechanisms, UPR and ERAD, may be a potential way for parasites control.
As is well known, UPR can restore ER homeostasis. Therefore, when parasites are exposed to external risk factors such as drugs, they are capable of restoring their own homeostasis by inducing ER stress and activating UPR; hence, it is not surprising that ER stress and UPR are involved in the mechanism of drug resistance.
Artemisinin-based combination therapies (ACTs) are efficient frontline drugs to treat malaria. However, since artemisinin resistance was first discovered in vivo in western Cambodia (Dondorp et al., 2009), it gradually become a great challenge in malaria treatment. In recent years, many researches have focused on the mechanism of artemisinin resistance, and some of them have suggested that UPR is an important mechanism for artemisinin resistance. By analyzing 1,043 P. falciparum samples isolated from the peripheral blood of patients with acute malaria, Mok et al. (2015) found that artemisinin-resistant parasites exhibited decelerated development in the early ring stage and the expression of two molecular chaperone complexes of UPR were upregulated, such as Plasmodium reactive oxidative stress complex (PROSC, BiP belonging to the family) (Haldar et al., 2018) and TCP-1 ring complex (TRiC) (Mok et al., 2015). Thus, they speculated that the decelerated development of artemisinin-resistant P. falciparum may be associated with the upregulation of their UPR, which as a proteostatic mechanism that can repair the artemisinin induced impaired protein and reduce artemisinin-induced toxic proteopathy (Mok et al., 2015). Souvik et al. further clarified that the amplified phosphatidylinositol-3-phosphate (PI3P) tubules/vesicles in the parasite’s ER in infected red cells extensively spread the proteostatic capacity of UPR, which may neutralize artemisinin’s toxic proteopathy and participate in artemisinin resistance (Bhattacharjee et al., 2018). Therefore, ER stress inhibitors or PI3P tubules/vesicles inhibitors may be used in patients with artemisinin resistance.
Zhang et al. (2017) studied the relationship between PK4-eIF2α pathway and recrudescence of Plasmodium and found that treatment of ARTs could activate the phosphorylation of PK4-eIF2α and promote latency in the ring stage. Treatment with salubrinal significantly increased the recrudescence rate, while the PK4 inhibitor GSK2606414 abolished recrudescence after ARTs treatment in P. berghei-infected mice. Furthermore, they also showed that eIF2α phosphorylation was only observed in the young ring stage of Dd2C580Y but not in Dd2, an ART-sensitive and chloroquine-resistant Plasmodium line. This study indicated that the recrudescence of Plasmodium was related to the activation of PK4 and phosphorylation of eIF2α following ART treatment. The results show that artemisinin can be combined with PK4 inhibitor to prevent the recurrence of Plasmodium.
Parasitic infection-induced pathological damage in hosts largely depends on ER stress. Therefore, inhibition of ER stress in hosts can be an effective treatment approach for parasitic diseases. In addition, considering that ER stress of parasites participates in their survival, development, and infection, the components or molecules of ER stress of parasites may be used as drug targets to kill or inhibit the development of parasites. The ER stress components or molecules that may be potential targets for the treatment of parasitic diseases are summarized in Table 1. Chemical chaperones TUDCA, trehalose, and 4-phenylbutyrate (4-PBA) have been used to reduce ER stress (Hetz et al., 2013), and they may be available for clinical application. Besides, clinical trials have shown that TUDCA therapy improved the sensitivity of insulin in the liver and muscle of insulin-resistant obese patients by affecting ER stress (Kars et al., 2010). Low dose of naltrexone treatment improved the function of epithelial barrier in IBD patients by reducing ER stress (Lie et al., 2018). These studies further demonstrate the feasibility of ER stress inhibitors as a treatment for parasitic diseases. In addition, the selectively target molecules of ER stress are more likely to be used, such as the inhibitors of ER stress molecules that play a key role in the survival and development of parasites (e.g., GSK2606414, PERK inhibitor) (Zhang et al., 2017), or the molecules of ER stress of parasites that are different from the host genes (e.g., GRP78 of P. falciparum) (Chen et al., 2018), or the parasites that are more sensitive to ER stress inducers than the host (e.g., DTT-induced ER stress in Leishmania) (Gosline et al., 2011). Moreover, because ER stress is involved in drug resistance, the inhibitors of ER stress molecules can be used in combination with anti-parasite drugs, such as ER stress inhibitors or PI3P tubules/vesicles inhibitors that may be used in patients with artemisinin resistance (Mok et al., 2015; Bhattacharjee et al., 2018).
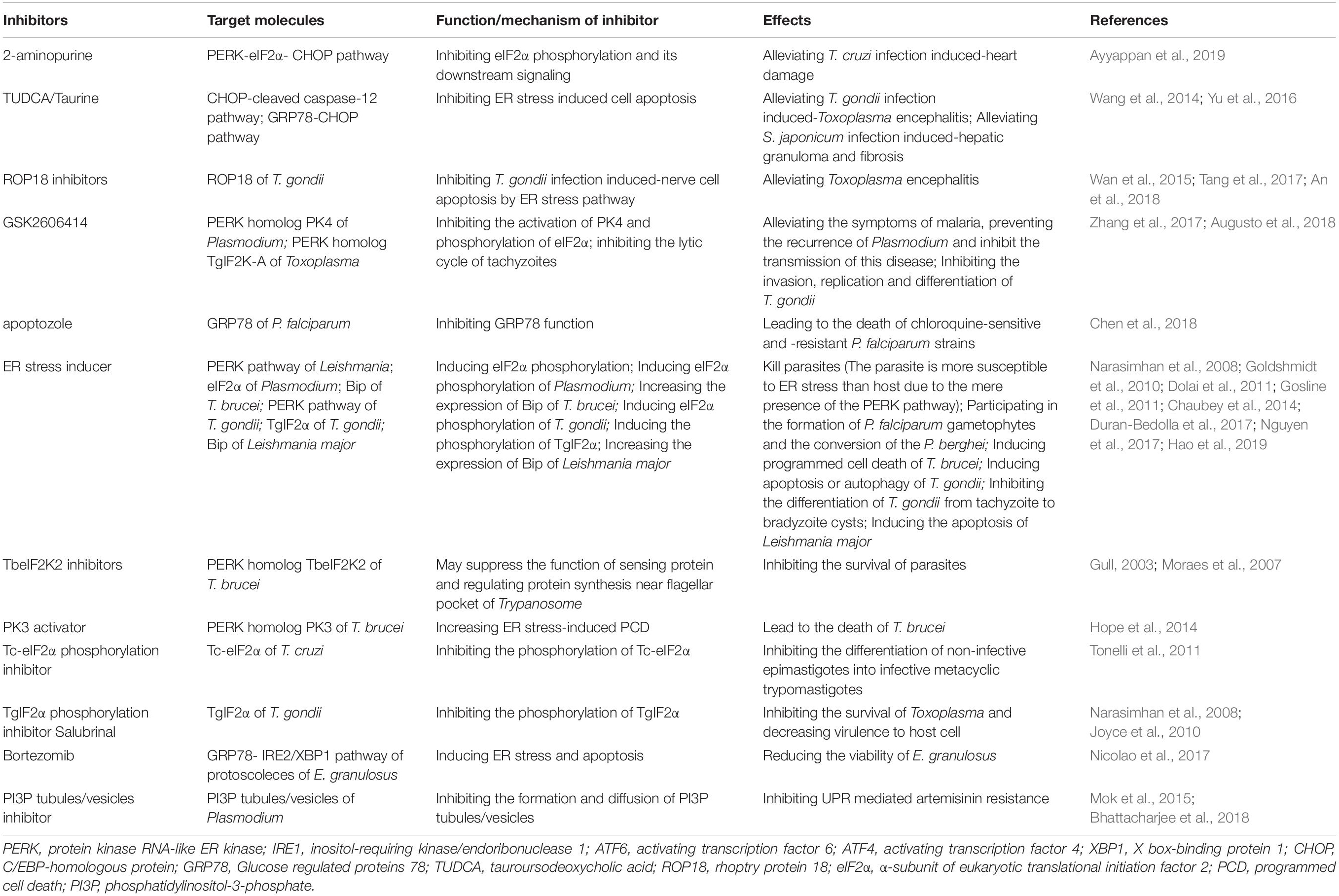
Table 1. Potential drug targets of UPR for treatment of parasitosis.
JS advocated writing this review, reviewed, edited, and approved its final version. MP collected literature and wrote the manuscript. FC collected and reviewed literature. ZW provided some suggestions for this review. All authors contributed to the article and approved the submitted version.
This work was supported by grants from National Natural Science Foundation of China (grant nos. 81802036 and 81871682), the Natural Science Foundation of Guangdong Province, China (2020A1515010896), the China Postdoctoral Science Foundation (nos. 2018M631027 and 2019T120770), and the Fundamental Research Funds for the Central Universities (no. 19ykpy165).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abhishek, K., Das, S., Kumar, A., Kumar, A., Kumar, V., Saini, S., et al. (2018). Leishmania donovani induced unfolded protein response delays host cell apoptosis in PERK dependent manner. PLoS Neglect. Trop. Dis. 12:e0006646. doi: 10.1371/journal.pntd.0006646
Acosta-Alvear, D., Zhou, Y., Blais, A., Tsikitis, M., Lents, N. H., Arias, C., et al. (2007). XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66.
Agrawal, S., Chung, D. W. D., Ponts, N., van Dooren, G. G., Prudhomme, J., Brooks, C. F., et al. (2013). An apicoplast localized Ubiquitylation system is required for the import of nuclear-encoded plastid proteins. PLoS Pathog. 9:e1003426. doi: 10.1371/journal.ppat.1003426
Agrawal, S., van Dooren, G. G., Beatty, W. L., and Striepen, B. (2009). Genetic evidence that an endosymbiont-derived endoplasmic reticulum-associated protein degradation (ERAD) system functions in import of apicoplast proteins. J. Biol. Chem. 284, 33683–33691. doi: 10.1074/jbc.M109.044024
An, R., Tang, Y., Chen, L., Cai, H., Lai, D.-H., Liu, K., et al. (2018). Encephalitis is mediated by ROP18 of, a severe pathogen in AIDS patients. Proc.Natl. Acad. Sci. U.S.A. 115, E5344–E5352. doi: 10.1073/pnas.1801118115
Anand, S. S., and Babu, P. P. (2013). Endoplasmic reticulum stress and neurodegeneration in experimental cerebral malaria. Neuro Signals 21:79. doi: 10.1159/000336970
Augusto, L., Martynowicz, J., Amin, P. H., Alakhras, N. S., Kaplan, M. H., Wek, R. C., et al. (2020). Toxoplasma gondii Co-opts the unfolded protein response to enhance migration and dissemination of infected host cells. mBio 11:e00915-20. doi: 10.1128/mBio.00915-20
Augusto, L., Martynowicz, J., Staschke, K. A., Wek, R. C., and Sullivan, W. J. (2018). Effects of PERK eIF2α kinase inhibitor against Toxoplasma gondii. Antimicrob. Agents Chemother. 62:e001442-18. doi: 10.1128/AAC.01442-18
Axten, J. M., Medina, J. R., Feng, Y., Shu, A., Romeril, S. P., Grant, S. W., et al. (2012). Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 55, 7193–7207. doi: 10.1021/jm300713s
Ayyappan, J. P., Lizardo, K., Wang, S., Yurkow, E., and Nagajyothi, J. F. (2019). Inhibition of ER stress by 2-aminopurine treatment modulates cardiomyopathy in a murine chronic chagas disease model. Biomol. Ther. 27, 386–394. doi: 10.4062/biomolther.2018.193
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332.
Bhattacharjee, S., Coppens, I., Mbengue, A., Suresh, N., Ghorbal, M., Slouka, Z., et al. (2018). Remodeling of the malaria parasite and host human red cell by vesicle amplification that induces artemisinin resistance. Blood 131, 1234–1247. doi: 10.1182/blood-2017-11-814665
Black, M. W., and Boothroyd, J. C. (2000). Lytic cycle of Toxoplasma gondii. Microbiol. Mol. Biol. Rev. 64, 607–623.
Bridgford, J. L., Xie, S. C., Cobbold, S. A., Pasaje, C. F. A., Herrmann, S., Yang, T., et al. (2018). Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat. Commun. 9:3801. doi: 10.1038/s41467-018-06221-1
Bukau, B., Weissman, J., and Horwich, A. (2006). Molecular chaperones and protein quality control. Cell 125, 443–451.
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96.
Chaubey, S., Grover, M., and Tatu, U. (2014). Endoplasmic reticulum stress triggers gametocytogenesis in the malaria parasite. J. Biol. Chem. 289, 16662–16674. doi: 10.1074/jbc.M114.551549
Chen, Y., Murillo-Solano, C., Kirkpatrick, M. G., Antoshchenko, T., Park, H. W., and Pizarro, J. C. (2018). Repurposing drugs to target the malaria parasite unfolding protein response. Sci. Rep. 8:10333. doi: 10.1038/s41598-018-28608-2
Chow, C., Cloutier, S., Dumas, C., Chou, M.-N., and Papadopoulou, B. (2011). Promastigote to amastigote differentiation of Leishmania is markedly delayed in the absence of PERK eIF2alpha kinase-dependent eIF2alpha phosphorylation. Cell. Microbiol. 13, 1059–1077. doi: 10.1111/j.1462-5822.2011.01602.x
Chung, D.-W. D., Ponts, N., Prudhomme, J., Rodrigues, E. M., and Le Roch, K. G. (2012). Characterization of the ubiquitylating components of the human malaria parasite’s protein degradation pathway. PLoS One 7:e43477. doi: 10.1371/journal.pone.0043477
Dias-Teixeira, K. L., Calegari-Silva, T. C., dos Santos, G. R. R. M., Vitorino Dos Santos, J., Lima, C., Medina, J. M., et al. (2016). The integrated endoplasmic reticulum stress response in Leishmania amazonensis macrophage infection: the role of X-box binding protein 1 transcription factor. FASEB J. 30, 1557–1565. doi: 10.1096/fj.15-281550
Dias-Teixeira, K. L., Calegari-Silva, T. C., Medina, J. M., Vivarini, A. C., Cavalcanti, A., Teteo, N., et al. (2017). Emerging role for the PERK/eIF2alpha/ATF4 in human Cutaneous Leishmaniasis. Sci. Rep. 7:17074. doi: 10.1038/s41598-017-17252-x
Dolai, S., and Adak, S. (2014). Endoplasmic reticulum stress responses in Leishmania. Mol. Biochem. Parasitol. 197, 1–8. doi: 10.1016/j.molbiopara.2014.09.002
Dolai, S., Pal, S., Yadav, R. K., and Adak, S. (2011). Endoplasmic reticulum stress-induced apoptosis in Leishmania through Ca2+-dependent and caspase-independent mechanism. J. Biol. Chem. 286, 13638–13646. doi: 10.1074/jbc.M110.201889
Dondorp, A. M., Nosten, F., Yi, P., Das, D., Phyo, A. P., Tarning, J., et al. (2009). Artemisinin resistance in Plasmodium falciparum malaria. New Engl. J. Med. 361, 455–467. doi: 10.1056/NEJMoa0808859
Duan, M., Yang, Y., Peng, S., Liu, X., Zhong, J., Guo, Y., et al. (2019). C/EBP homologous protein (CHOP) activates macrophages and promotes liver fibrosis in Schistosoma japonicum-infected mice. J. Immunol. Res. 2019:5148575. doi: 10.1155/2019/5148575
Duran-Bedolla, J., Tellez-Sosa, J., Valdovinos-Torres, H., Pavon, N., Buelna-Chontal, M., Tello-Lopez, A. T., et al. (2017). Cellular stress associated with the differentiation of Plasmodium berghei ookinetes. Biochem. Cell Biol. 95, 310–317. doi: 10.1139/bcb-2016-0028
Fallon, P. G., and Doenhoff, M. J. (1994). Drug-resistant schistosomiasis: resistance to praziquantel and oxamniquine induced in Schistosoma mansoni in mice is drug specific. Am. J. Trop. Med. Hyg. 51, 83–88.
Galluzzi, L., Diotallevi, A., De Santi, M., Ceccarelli, M., Vitale, F., Brandi, G., et al. (2016). Leishmania infantum induces mild unfolded protein response in infected macrophages. PLoS One 11:e0168339. doi: 10.1371/journal.pone.0168339
Galluzzi, L., Diotallevi, A., and Magnani, M. (2017). Endoplasmic reticulum stress and unfolded protein response in infection by intracellular parasites. Future Sci. OA 3:FSO198. doi: 10.4155/fsoa-2017-0020
Gass, J. N., Gifford, N. M., and Brewer, J. W. (2002). Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J. Biol. Chem. 277, 49047–49054.
Goldshmidt, H., Matas, D., Kabi, A., Carmi, S., Hope, R., and Michaeli, S. (2010). Persistent ER stress induces the spliced leader RNA silencing pathway (SLS), leading to programmed cell death in Trypanosoma brucei. PLoS Pathog. 6:e1000731. doi: 10.1371/journal.ppat.1000731
Gosline, S. J. C., Nascimento, M., McCall, L.-I., Zilberstein, D., Thomas, D. Y., Matlashewski, G., et al. (2011). Intracellular eukaryotic parasites have a distinct unfolded protein response. PLoS One 6:e19118. doi: 10.1371/journal.pone.0019118
Grootjans, J., Kaser, A., Kaufman, R. J., and Blumberg, R. S. (2016). The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 16, 469–484. doi: 10.1038/nri.2016.62
Gull, K. (2003). Host-parasite interactions and trypanosome morphogenesis: a flagellar pocketful of goodies. Curr. Opin. Microbiol. 6, 365–370.
Haldar, K., Bhattacharjee, S., and Safeukui, I. (2018). Drug resistance in Plasmodium. Nat. Rev. Microbiol. 16, 156–170. doi: 10.1038/nrmicro.2017.161
Hao, P., Cui, X., Liu, J., Li, M., Fu, Y., and Liu, Q. (2019). Identification and characterization of stearoyl-CoA desaturase in Toxoplasma gondii. Acta Biochim. Biophys. Sinica 51, 615–626. doi: 10.1093/abbs/gmz040
Harbut, M. B., Patel, B. A., Yeung, B. K., McNamara, C. W., Bright, A. T., Ballard, J., et al. (2012). Targeting the ERAD pathway via inhibition of signal peptide peptidase for antiparasitic therapeutic design. Proc. Natl. Acad. Sci. U.S.A. 109, 21486–21491. doi: 10.1073/pnas.1216016110
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799.
Hendrick, H. M., Welter, B. H., Hapstack, M. A., Sykes, S. E., Sullivan, W. J., and Temesvari, L. A. (2016). Phosphorylation of eukaryotic initiation factor-2α during Stress and encystation in Entamoeba Species. PLoS Pathog. 12:e1006085. doi: 10.1371/journal.ppat.1006085
Hetz, C., Chevet, E., and Harding, H. P. (2013). Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719. doi: 10.1038/nrd3976
Hope, R., Ben-Mayor, E., Friedman, N., Voloshin, K., Biswas, D., Matas, D., et al. (2014). Phosphorylation of the TATA-binding protein activates the spliced leader silencing pathway in Trypanosoma brucei. Sci. Signal. 7:ra85. doi: 10.1126/scisignal.2005234
Hwang, J., and Qi, L. (2018). Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 43, 593–605. doi: 10.1016/j.tibs.2018.06.005
Inacio, P., Zuzarte-Luis, V., Ruivo, M. T., Falkard, B., Nagaraj, N., Rooijers, K., et al. (2015). Parasite-induced ER stress response in hepatocytes facilitates Plasmodium liver stage infection. EMBO Rep. 16, 955–964. doi: 10.15252/embr.201439979
Joyce, B. R., Queener, S. F., Wek, R. C., and Sullivan, W. J. (2010). Phosphorylation of eukaryotic initiation factor-2{alpha} promotes the extracellular survival of obligate intracellular parasite Toxoplasma gondii. Proc. Natl. Acad. Sci. U.S.A. 107, 17200–17205. doi: 10.1073/pnas.1007610107
Joyce, B. R., Tampaki, Z., Kim, K., Wek, R. C., and Sullivan, W. J. (2013). The unfolded protein response in the protozoan parasite Toxoplasma gondii features translational and transcriptional control. Eukaryot. Cell 12, 979–989. doi: 10.1128/EC.00021-13
Kars, M., Yang, L., Gregor, M. F., Mohammed, B. S., Pietka, T. A., Finck, B. N., et al. (2010). Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 59, 1899–1905. doi: 10.2337/db10-0308
Konrad, C., Wek, R. C., and Sullivan, W. J. (2014). GCN2-like eIF2α kinase manages the amino acid starvation response in Toxoplasma gondii. Intern. J. Parasitol. 44, 139–146. doi: 10.1016/j.ijpara.2013.08.005
Kumari, R., Gupta, P., and Tiwari, S. (2018). Ubc7/Ube2g2 ortholog in Entamoeba histolytica: connection with the plasma membrane and phagocytosis. Parasitol. Res. 117, 1599–1611. doi: 10.1007/s00436-018-5842-6
Lazaridis, K. N., Gores, G. J., and Lindor, K. D. (2001). Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders’. J. Hepatol. 35, 134–146.
Li, J., Tang, G., Qin, W., Yang, R., Ma, R., Ma, B., et al. (2018). Toxic effects of arsenic trioxide on Echinococcus granulosus protoscoleces through ROS production, and Ca2+-ER stress-dependent apoptosis. Acta Biochimi. Biophys. Sinica 50, 579–585. doi: 10.1093/abbs/gmy041
Li, P., Fu, D., Sheng, Q., Yu, S., Bao, X., and Lv, Z. (2019). TUDCA attenuates intestinal injury and inhibits endoplasmic reticulum stress-mediated intestinal cell apoptosis in necrotizing enterocolitis. Intern. Immunopharmacol. 74:105665. doi: 10.1016/j.intimp.2019.05.050
Lie, M. R. K. L., van der Giessen, J., Fuhler, G. M., de Lima, A., Peppelenbosch, M. P., van der Ent, C., et al. (2018). Low dose Naltrexone for induction of remission in inflammatory bowel disease patients. J. Transl. Med. 16:55. doi: 10.1186/s12967-018-1427-5
Liu, F., Cui, Y., Ge, P., Luan, J., Zhou, X., and Han, J. (2015). Tauroursodeoxycholic acid attenuates inorganic phosphate-induced osteoblastic differentiation and mineralization in NIH3T3 fibroblasts by inhibiting the ER stress response PERK-eIF2α-ATF4 pathway. Drug Discov. Therap. 9, 38–44.
Loos, J. A., Nicolao, M. C., and Cumino, A. C. (2018). Metformin promotes autophagy in Echinococcus granulosus larval stage. Mol. Biochem. Parasitol. 224, 61–70. doi: 10.1016/j.molbiopara.2018.07.003
Ma, Y., and Hendershot, L. M. (2003). Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 278, 34864–34873.
Malo, A., Krüger, B., Seyhun, E., Schäfer, C., Hoffmann, R. T., Göke, B., et al. (2010). Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, trypsin activation, and acinar cell apoptosis while increasing secretion in rat pancreatic acini. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G877–G886. doi: 10.1152/ajpgi.00423.2009
Messias Sandes, J., Nascimento Moura, D. M., Divina da Silva Santiago, M., Barbosa de Lima, G., Cabral Filho, P. E., da Cunha Goncalves de Albuquerque, S., et al. (2019). The effects of endoplasmic reticulum stressors, tunicamycin and dithiothreitol on Trypanosoma cruzi. Exp. Cell Res. 383:111560. doi: 10.1016/j.yexcr.2019.111560
Möhrle, J. J., Zhao, Y., Wernli, B., Franklin, R. M., and Kappes, B. (1997). Molecular cloning, characterization and localization of PfPK4, an eIF-2alpha kinase-related enzyme from the malarial parasite Plasmodium falciparum. Biochem. J. 328(Pt 2), 677–687.
Mok, S., Ashley, E. A., Ferreira, P. E., Zhu, L., Lin, Z., Yeo, T., et al. (2015). Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 347, 431–435. doi: 10.1126/science.1260403
Moraes, M. C. S., Jesus, T. C. L., Hashimoto, N. N., Dey, M., Schwartz, K. J., Alves, V. S., et al. (2007). Novel membrane-bound eIF2alpha kinase in the flagellar pocket of Trypanosoma brucei. Eukaryot. Cell 6, 1979–1991.
Narasimhan, J., Joyce, B. R., Naguleswaran, A., Smith, A. T., Livingston, M. R., Dixon, S. E., et al. (2008). Translation regulation by eukaryotic initiation factor-2 kinases in the development of latent cysts in Toxoplasma gondii. J. Biol. Chem. 283, 16591–16601. doi: 10.1074/jbc.M800681200
Nguyen, H. M., Berry, L., Sullivan, W. J., and Besteiro, S. (2017). Autophagy participates in the unfolded protein response in Toxoplasma gondii. FEMS Microbiol. Lett. 364:fnx153. doi: 10.1093/femsle/fnx153
Nicolao, M. C., Loos, J. A., Rodriguez Rodrigues, C., Beas, V., and Cumino, A. C. (2017). Bortezomib initiates endoplasmic reticulum stress, elicits autophagy and death in Echinococcus granulosus larval stage. PLoS One 12:e0181528. doi: 10.1371/journal.pone.0181528
Novoa, I., Zeng, H., Harding, H. P., and Ron, D. (2001). Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 153, 1011–1022.
Pahl, H. L., and Baeuerle, P. A. (1995). A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-kappa B. EMBO J. 14, 2580–2588.
Pineda, E., and Perdomo, D. (2017). Entamoeba histolytica under oxidative stress: what countermeasure mechanisms are in place? Cells 6:44. doi: 10.3390/cells6040044
Poncet, A. F., Bosteels, V., Hoffmann, E., Chehade, S., Rennen, S., Huot, L., et al. (2021). The UPR sensor IRE1α promotes dendritic cell responses to control Toxoplasma gondii infection. EMBO Rep. 22:e49617. doi: 10.15252/embr.201949617
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529.
Santi-Rocca, J., Smith, S., Weber, C., Pineda, E., Hon, C.-C., Saavedra, E., et al. (2012). Endoplasmic reticulum stress-sensing mechanism is activated in Entamoeba histolytica upon treatment with nitric oxide. PLoS One 7:e31777. doi: 10.1371/journal.pone.0031777
Shen, J., Chen, X., Hendershot, L., and Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111.
Spork, S., Hiss, J. A., Mandel, K., Sommer, M., Kooij, T. W. A., Chu, T., et al. (2009). An unusual ERAD-Like complex is targeted to the apicoplast of Plasmodium falciparum. Eukaryot. Cell 8, 1134–1145. doi: 10.1128/Ec.00083-09
Sullivan, W. J., Narasimhan, J., Bhatti, M. M., and Wek, R. C. (2004). Parasite-specific eIF2 (eukaryotic initiation factor-2) kinase required for stress-induced translation control. Biochem. J. 380(Pt 2), 523–531.
Tang, Y. W., Zheng, M. J., An, R., Chen, L. J., Gong, L. L., Cai, H. J., et al. (2017). Proteasomal degradation of T. gondii ROP18 requires Derlin2. Acta Trop. 174, 106–113. doi: 10.1016/j.actatropica.2017.06.027
Tiengwe, C., Muratore, K. A., and Bangs, J. D. (2016). Surface proteins, ERAD and antigenic variation in Trypanosoma brucei. Cell. Microbiol. 18, 1673–1688. doi: 10.1111/cmi.12605
Tonelli, R. R., Augusto Lda, S., Castilho, B. A., and Schenkman, S. (2011). Protein synthesis attenuation by phosphorylation of eIF2alpha is required for the differentiation of Trypanosoma cruzi into infective forms. PLoS One 6:e27904. doi: 10.1371/journal.pone.0027904
Tsuru, A., Imai, Y., Saito, M., and Kohno, K. (2016). Novel mechanism of enhancing IRE1α-XBP1 signalling via the PERK-ATF4 pathway. Sci. Rep. 6:24217. doi: 10.1038/srep24217
Walters, H. A., Welter, B. H., Sullivan, W. J., and Temesvari, L. A. (2019). Phosphorylation of eukaryotic initiation factor-2α in response to endoplasmic reticulum and nitrosative stress in the human protozoan parasite, Entamoeba histolytica. Mol. Biochem. Parasitol. 234:111223. doi: 10.1016/j.molbiopara.2019.111223
Wan, L., Gong, L., Wang, W., An, R., Zheng, M., Jiang, Z., et al. (2015). T. gondii rhoptry protein ROP18 induces apoptosis of neural cells via endoplasmic reticulum stress pathway. Parasit. Vect. 8:554. doi: 10.1186/s13071-015-1103-z
Wang, T., Zhou, J., Gan, X., Wang, H., Ding, X., Chen, L., et al. (2014). Toxoplasma gondii induce apoptosis of neural stem cells via endoplasmic reticulum stress pathway. Parasitology 141, 988–995. doi: 10.1017/S0031182014000183
Ward, P., Equinet, L., Packer, J., and Doerig, C. (2004). Protein kinases of the human malaria parasite Plasmodium falciparum: the kinome of a divergent eukaryote. BMC Genom. 5:79. doi: 10.1186/1471-2164-5-79
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891.
Yu, Y.-R., Deng, M.-J., Lu, W.-W., Zhang, J.-S., Jia, M.-Z., Huang, J., et al. (2014). Endoplasmic reticulum stress-mediated apoptosis is activated in intestines of mice with Trichinella spiralis infection. Exper. Parasitol. 145, 1–6. doi: 10.1016/j.exppara.2014.06.017
Yu, Y.-R., Ni, X.-Q., Huang, J., Zhu, Y.-H., and Qi, Y.-F. (2016). Taurine drinking ameliorates hepatic granuloma and fibrosis in mice infected with Schistosoma japonicum. Intern. J. Parasitol. Drugs Drug Resist. 6, 35–43. doi: 10.1016/j.ijpddr.2016.01.003
Zhang, M., Gallego-Delgado, J., Fernandez-Arias, C., Waters, N. C., Rodriguez, A., Tsuji, M., et al. (2017). Inhibiting the Plasmodium eIF2alpha Kinase PK4 prevents artemisinin-induced latency. Cell Host Microb. 22, 766–776.e764. doi: 10.1016/j.chom.2017.11.005
Zhang, M., Mishra, S., Sakthivel, R., Rojas, M., Ranjan, R., Sullivan, W. J., et al. (2012). PK4, a eukaryotic initiation factor 2 (eIF2) kinase, is essential for the development of the erythrocytic cycle of Plasmodium. Proc. Natl. Acad. Sci. U.S.A. 109, 3956–3961. doi: 10.1073/pnas.1121567109
Zhang, X., An, T., Pham, K. T. M., Lun, Z.-R., and Li, Z. (2019). Functional analyses of cytokinesis regulators in bloodstream stage Trypanosoma brucei parasites identify functions and regulations specific to the life cycle stage. mSphere 4:e0199-19. doi: 10.1128/mSphere.00199-19
Zhou, J., Gan, X., Wang, Y., Zhang, X., Ding, X., Chen, L., et al. (2015). Toxoplasma gondii prevalent in China induce weaker apoptosis of neural stem cells C17.2 via endoplasmic reticulum stress (ERS) signaling pathways. Parasit. Vect. 8:73. doi: 10.1186/s13071-015-0670-3
Keywords: endoplasmic reticulum stress, drug targets, parasite, parasitosis, drug resistance
Citation: Peng M, Chen F, Wu Z and Shen J (2021) Endoplasmic Reticulum Stress, a Target for Drug Design and Drug Resistance in Parasitosis. Front. Microbiol. 12:670874. doi: 10.3389/fmicb.2021.670874
Received: 22 February 2021; Accepted: 03 May 2021;
Published: 31 May 2021.
Edited by:
Karl Kuchler, Medical University of Vienna, AustriaCopyright © 2021 Peng, Chen, Wu and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Shen, c2hlbmoyOUBtYWlsLnN5c3UuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.