 Thomas E. Witte
Thomas E. Witte Nicolas Villeneuve
Nicolas Villeneuve Christopher N. Boddy2*
Christopher N. Boddy2* David P. Overy
David P. Overy
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 23 April 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.664276
This article is part of the Research TopicStrategies for the Discovery of Fungal Natural ProductsView all 5 articles
Accessory chromosomes are strain- or pathotype-specific chromosomes that exist in addition to the core chromosomes of a species and are generally not considered essential to the survival of the organism. Among pathogenic fungal species, accessory chromosomes harbor pathogenicity or virulence factor genes, several of which are known to encode for secondary metabolites that are involved in plant tissue invasion. Accessory chromosomes are of particular interest due to their capacity for horizontal transfer between strains and their dynamic “crosstalk” with core chromosomes. This review focuses exclusively on secondary metabolism (including mycotoxin biosynthesis) associated with accessory chromosomes in filamentous fungi and the role accessory chromosomes play in the evolution of secondary metabolite gene clusters. Untargeted metabolomics profiling in conjunction with genome sequencing provides an effective means of linking secondary metabolite products with their respective biosynthetic gene clusters that reside on accessory chromosomes. While the majority of literature describing accessory chromosome-associated toxin biosynthesis comes from studies of Alternaria pathotypes, the recent discovery of accessory chromosome-associated biosynthetic genes in Fusarium species offer fresh insights into the evolution of biosynthetic enzymes such as non-ribosomal peptide synthetases (NRPSs), polyketide synthases (PKSs) and regulatory mechanisms governing their expression.
Some ascomycete lineages are prolific producers of secondary metabolites (natural products), many of which have associated activity in biological systems. Large scale screening and characterization of fungal extracts has been carried out for over half a century by various industries (including pharmaceutical) and as a result, a considerable breadth of the chemical space associated with in vitro culturing of filamentous ascomycetes has been probed for associated biological activity (Bills et al., 2009). However, with the advent of modern genome sequencing technologies, it has become evident that many secondary metabolite biosynthetic gene clusters are associated with unknown products and are not expressed under axenic conditions (Mao et al., 2018). The fungal natural product community is therefore focused on unlocking and understanding this “cryptic” metabolome and strive to link secondary metabolite products with their respective biosynthetic gene clusters.
In agriculture, fungal secondary metabolites produced by plant pathogens and spoilage fungi can be mycotoxins acting as virulence factors that directly impact crop yields and market suitability. Fungal plant pathogens with biotrophic/necrotrophic and saprophytic lifestyles (such as species from the genera of Fusarium and Alternaria), produce a broad diversity of secondary metabolites. However, secondary metabolism can vary within species populations of some plant pathogens. One reason for observed intra-species variations in secondary metabolism arises from the presence of lineage-specific accessory chromosomes (ACs) within the genome of select strains that encode secondary metabolite biosynthetic gene clusters.
This review focuses exclusively on secondary metabolism (including mycotoxin biosynthesis) associated with ACs in filamentous fungi. We fill in historical gaps and provide a current overview of both the pathways and associated gene clusters involved in the biosynthesis of host specific toxins and other secondary metabolite virulence factors associated with ACs to date. Examples of AC-associated toxin biosynthesis are numerous among Alternaria formae speciales (Alternaria f. sp.) and recent genomic studies of Fusarium species have also detected biosynthetic enzymes on accessory chromosomes. Enzymes of interest include non-ribosomal peptide synthetases (NRPSs), polyketide synthases (PKSs), terpene synthases and the regulatory mechanisms governing their expression. Relationships between ACs and secondary metabolite biosynthesis are discussed, including evidence for horizontal chromosome transfer of ACs between fungi, the relevance of ACs providing a niche for rapid multiplication and diversification of secondary metabolite biosynthesis genes, the implications of regulatory “cross talk” between core chromosomes and ACs, and the role of “repeat-induced point mutation” in detecting and eliminating duplicated genes in the context of AC-associated gene duplications. The utility of long-read sequencing technology to elucidate the structure of genomes through “closed” or “telomere to telomere” chromosomal assemblies is highlighted as essential for the study of ACs, greatly improving analyses of secondary metabolite gene clusters. Finally, a “top down—bottom up” screening strategy coupling genomics based approaches with untargeted metabolomics is presented, where the production of unique secondary metabolite products within species populations can be linked with biosynthetic gene clusters associated with ACs.
The term “accessory chromosome” (or AC) has been adopted by researchers working on a variety of different Ascomycete genera and conceptually encompasses the terms “type B,” “supernumerary,” or “lineage-specific” chromosomes used in the study of plants, animals and fungal species (Covert, 1998; Croll and McDonald, 2012). We have decided to use the term AC in this review as it is consistent with previous fungal research and consensus on the different terms’ proper usage is lacking in the literature. Fungal ACs are generally small, lineage- or pathotype-specific chromosomes that exist in addition to the “core” chromosomes of a species and are not considered essential to the survival of the organism. Among some pathogenic fungi, the concept of an AC is nuanced by the presence of virulence factor genes involved in pathogenicity and/or secondary metabolite production, which play an important role in niche plant invasion and adaptation (Croll and McDonald, 2012). ACs conferring adaptive advantages such as pathogenicity are sometimes described as “conditionally dispensable” chromosomes (popularized by studies of Alternaria species; Tsuge, 2019) or “pathogenicity” chromosomes (Ma et al., 2010; van Dam et al., 2017; Li et al., 2020). ACs are of particular interest in fungal pathogen research due to their capacity for horizontal transfer between strains, which has been experimentally demonstrated to increase pathogen competence in previously deficient strains (He et al., 1998; Akagi et al., 2009; Ma et al., 2010; Shahi et al., 2016; van Dam et al., 2017; Li et al., 2020). Pathogenicity traits associated with ACs have given rise to formae speciales or pathotypes, causal agents of host specific plant diseases.
Fungal ACs display key structural and genetic differences compared to core chromosomes. ACs are distinguished in some species by their low gene density, high level of transposable element (TE) distribution, epigenetic markers, and non-Mendelian inheritance patterns [observed either by spontaneous loss of ACs or more recently by meiotic drive within populations; (Habig et al., 2018)]. ACs vary in size and distribution among individuals of the same species and many do not appear to be under strong selective pressure, exhibiting high levels of sequence polymorphisms or disruption of chromosomal collinearity (for recent reviews of fungal ACs we direct the reader to Bertazzoni et al., 2018 and Yang et al., 2020). Nevertheless, the association of certain ACs with pathogenicity indicates ACs are not trivial to the evolution and survival of fungi outside of the laboratory. ACs contribute to the compartmentalization of genomic regions associated with high repeat content and accelerated evolution of virulence factors as proposed in the “two-speed genome” concept of fungal pathogen adaptation (Raffaele et al., 2010; Croll and McDonald, 2012).
The presence of virulence factors on fungal ACs, and their distribution within populations, is highly relevant to the study of fungal plant pathogens. The best-characterized virulence factors associated with plant pathogen ACs fall into at least three groups: small secreted effector proteins such as the secreted in xylem (SIX) genes in Fusarium oxysporum (Rep et al., 2004; Houterman et al., 2007; Schmidt et al., 2013), detoxification genes such as the pisatin demethylase gene pda6 in the pea-pathogenic strain Fusarium solani f. sp. pisi (Han et al., 2001), and toxin biosynthesis genes such as AM-toxin genes conferring pathogenicity of Alternaria alternata f. sp. mali on apple trees (Malus domestica) (Johnson et al., 2000; Andersen et al., 2006; Harimoto et al., 2007). As more ACs are identified and analyzed, the list of associated genes contributing to pathogenicity expands, as is our understanding of the interplay between core and accessory chromosomes and the role ACs play in coordinating regulatory shifts in their expression.
In some instances, only a small subsection of a species population can infect a specific host due to the expression of secondary metabolite biosynthetic gene clusters residing on ACs. These secondary metabolites are referred to as host specific toxins (Figure 1). Historically, much research interest has been invested into understanding the production of host specific toxins, including structure determination, genes associated with their biosynthesis and the association of biosynthetic genes with ACs. For example, several unique populations of Alternaria alternata cause disease on niche plant hosts due the production of host specific toxins (described below). The taxonomy of small spored Alternaria species is notoriously convoluted, in part due to strains that are morphologically indistinguishable but produce different host specific toxins and thus have a unique host range. Differing usages of either the term pathotype or formae speciales (or combinations of the two) within the genus Alternaria exist (Akimitsu et al., 2014; Woudenberg et al., 2015; Armitage et al., 2020). The majority of research on host specific toxins in Alternaria was carried out prior to the advent of next generation and long-read sequencing tools. As older studies lack high quality telomere-telomere genomes, localization of host specific toxin biosynthetic gene clusters to ACs was typically based by electrophoretic karyotyping and visualized using a Southern blot probe. To fill in existing gaps in the literature, we illustrate Alternaria biosynthetic pathways and summarize the evidence confirming the localization of their associated biosynthetic genes to ACs.
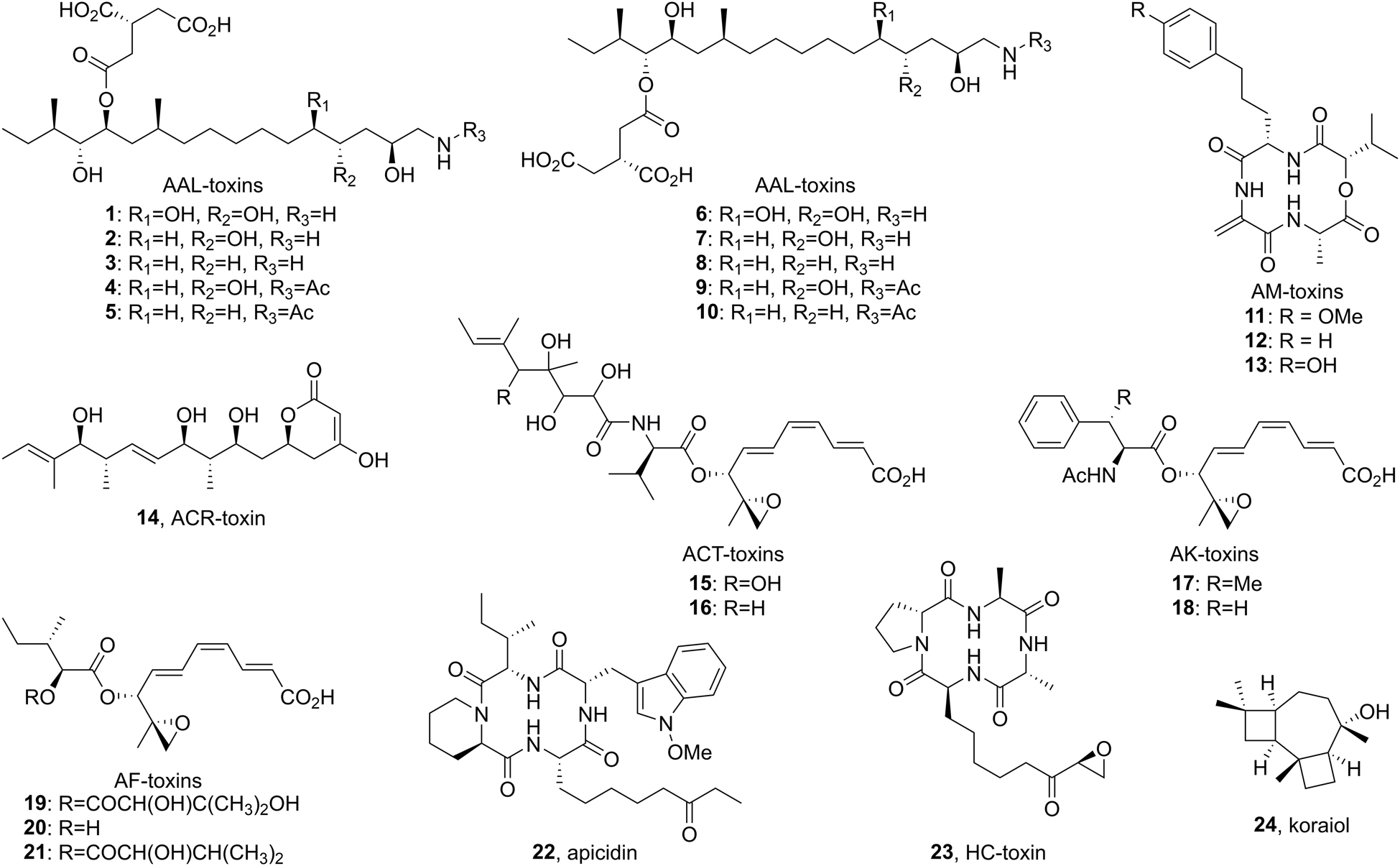
Figure 1. Structure of host specific toxins (1–21) and other secondary metabolites associated with accessory chromosomes (22–24).
The tomato pathotype of A. alternata (Alternaria alternata f. sp. lycopersici), which causes Alternaria stem canker on tomato, produces 5 regioisomeric pairs of AAL-toxins: TA, TB, TC, TD, and TE (Caldas et al., 1998; Figure 1). While direct characterization of AAL-toxin biosynthesis is limited, the well-characterized and highly related fumonisin biosynthetic pathway serves as a reliable guide for AAL-toxin biosynthesis (Figure 2). Homology between the two biosynthetic gene clusters has been confirmed via a complementation experiment where the highly reducing PKS responsible for formation of the polyketide core of AAL-toxin, ALT1, was shown to complement FUM1, the PKS gene linked to the biosynthesis of fumonisin, a sphingolipid inhibitor produced by Fusarium verticillioides (Zhu et al., 2008). Thus, based on the homology to fumonisin biosynthesis, ALT1 can be inferred to generate a 16 carbon long saturated enzyme-linked thioester that is off-loaded from the PKS by the 2-oxoamino synthase ALT4 via a PLP-dependent decarboxylative condensation with glycine (Du et al., 2008). The product is then likely reduced by the NADPH-dependent short chain dehydrogenase ALT6 (Butchko et al., 2003; Yi et al., 2005), in an overall process akin to dihydrosphingosine biosynthesis (Gault et al., 2010). Oxidation at C13 and C14 by the cytochrome p450 enzyme ALT2 and C4 by ALT8 generate an oxidized intermediate, lacking the tricarballyl group and C5 oxidation of the final product (Bojja et al., 2004). The tricarballyl group is generated from aconitate, likely transported from the mitochondria by ALT9, which is converted into the AMP ester via ALT12, and loaded onto the carrier protein of ALT12b. Reduction of the enoyl group by the dehydrogenase ALT3 generates the (R)-configured tricarballyl-enzyme intermediate that is then transferred, via the condensation domain of ALT12b, onto the polyketide chain at either the C14 or C15 alcohol (Lia et al., 2013). The final biosynthetic step is the dioxygenase-catalyzed C5 oxidation by ALT11a (Ding et al., 2004). Among the AAL toxins, TA, TB, TC, and TE all differ in the extent of the oxidation of the polyketide core and the presence or absence of an N-acetate and each of the AAL toxins exist as regioisomeric pairs that differ in the location of the tricarballylate ester, which is on either the C13 (e.g., TA1) or C14 (e.g., TA2) alcohol (Figure 1).
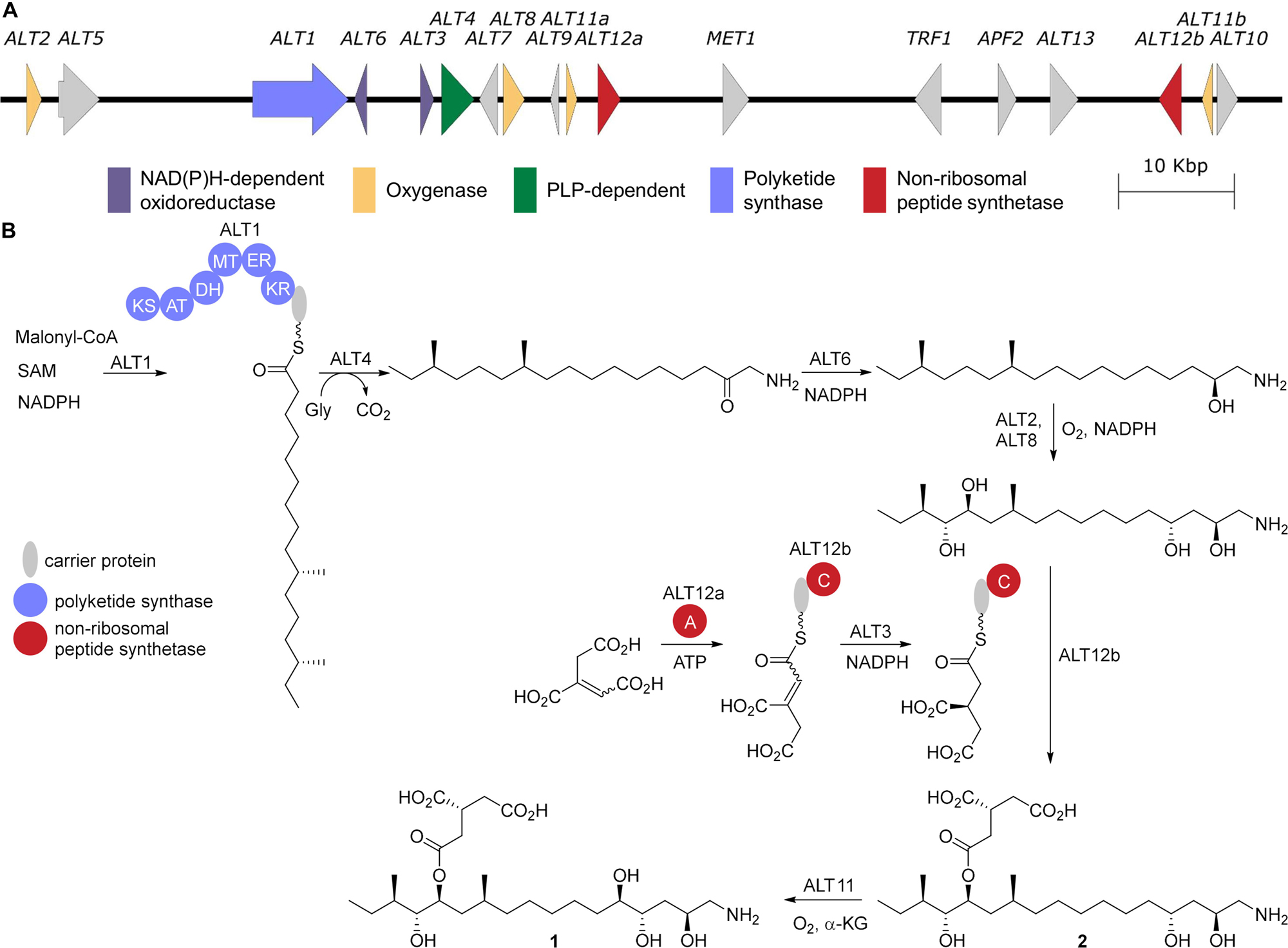
Figure 2. AAL-toxin biosynthesis. (A) The gene cluster for AAL-toxin biosynthesis from A. alternata (AB969680). (B) The proposed biosynthesis for AAL-toxin TA1 based on homology to fumonisin biosynthesis. KS, β-ketosynthase; AT-acyltransferase; KR, β-ketoreductase; DH, dehydratase; ER, enoylreductase; MT methyltransferase; A, adenylation; C, condensation.
Using electrophoretic karyotyping and Southern blot probe labeling experiments, the PKS gene ALT1 was found to be unique to isolates of the tomato pathotype of A. alternata and absent in non-pathogenic strains—ALT1 solely resides on a 1.0 Mb AC in producing strains (Akagi et al., 2009). When produced within the appropriate host plant cell, the AAL-toxins affect the endoplasmic reticulum and mitochondria by preventing the enzyme sphingosine-N-acyltransferase (ceramide synthase) from contributing to the production of ceramide-containing lipids (Gilchrist, 1998). This initiates premature programmed cell death via the accumulation of nitric oxide, reactive oxygen species, and other signaling molecules (Wolpert et al., 2002; Gechev et al., 2004) of the five AAL-toxins isoforms, TA-toxin is the most toxic to tomato and is produced in the highest quantity by producing strains (Spassieva et al., 2002). Interestingly, while the ALT biosynthetic gene cluster encodes an additional copy of ceramide synthase, ALT7, this is not part of a self-resistance mechanism as an ALT7 deletion has no effect on growth rate of the pathogenic strain (Kheder and Akagi, 2012).
The rough lemon pathotype of A. alternata (A. alternata f. sp. citri), is the causal agent of Alternaria leaf spot of rough lemon (Citrus jambhiri Lush.) and rangpur lime (C. limonia Osbeck), and causes the formation of lesions on, and the abscission of, leaves and immature fruit (Peever et al., 2004). The known host range of the A. alternata rough lemon pathotype is rather narrow and attributed to the production of a host specific toxin, ACR-toxin (Masunaka et al., 2005). Within the cell, ACR-toxin affects the mitochondria, which it renders dysfunctional by the uncoupling of oxidative phosphorylation, leakage of NAD + cofactor from the TCA cycle, and a loss of membrane potential (Kohmoto et al., 1984; Akimitsu et al., 1989; Meena et al., 2017).
ACR-toxin is a polyketide consisting of an α-dihydropyrone ring in a 19-carbon polyol (Gardner et al., 1985; Figure 3). The highly reducing type I polyketide synthase encoded by ACRTS2 is likely responsible for its biosynthesis as both RNA silencing of ACRTS2 and homologous recombination-based gene disruption abolishes ACR toxin production (Izumi et al., 2012a, b). A second gene ACRTS1 has also been proposed to be required for ACR-toxin production since disruption of ACRTS1 leads to a decrease in toxin production (Izumi et al., 2012a). While this gene was initially proposed to play a role in generating the dihydropyrone (Izumi et al., 2012a), sequence analysis shows that it encodes a cytochrome p450, which is inconsistent with this proposed function. It thus remains to be seen what, if any, direct role ACRTS1 plays in ACR-toxin biosynthesis. ACRTS2 was confirmed to reside on a 1.5 Mb AC of rough lemon pathotype strains by electrophoretic karyotyping and Southern blot probe labeling experiments and ACRTS2 was shown to be absent in non-producing stains of A. alternata (Masunaka et al., 2005; Izumi et al., 2012b).
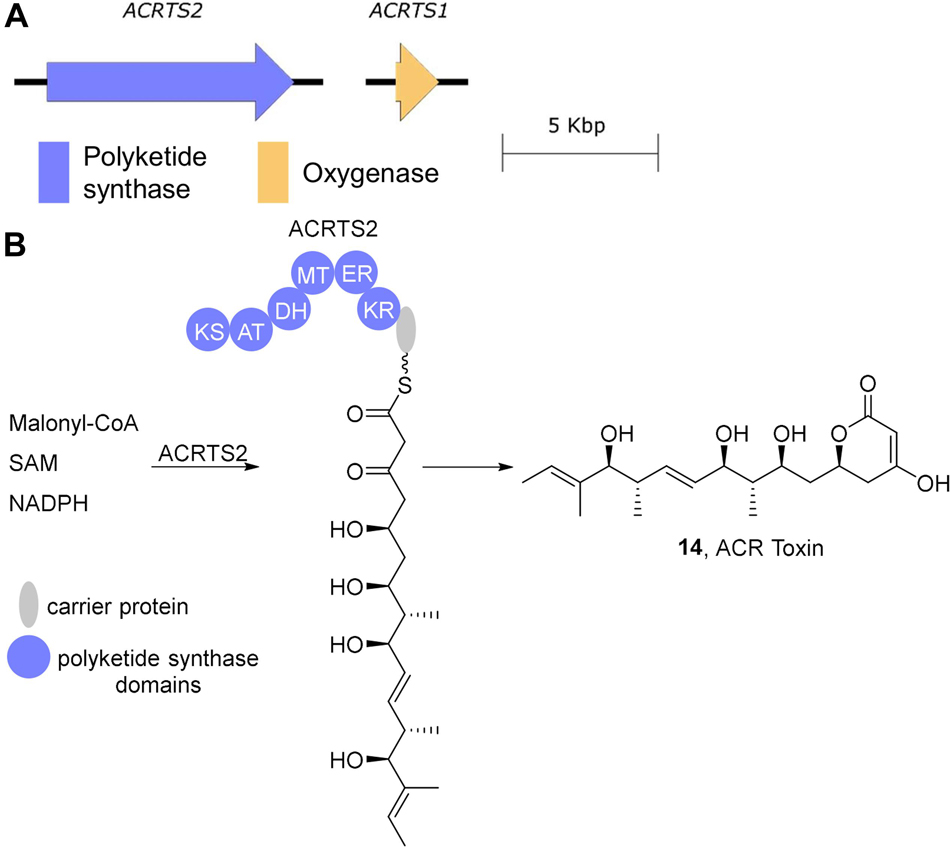
Figure 3. ACR-toxin biosynthesis. (A) The ACRTS1 and ACRTS2 genes involved in ACR-toxin biosynthesis. (B) ACR-toxin is generated from ACRTS2. KS, β-ketosynthase; AT, acyltransferase; KR, β-ketoreductase; DH, dehydratase; ER, enoylreductase; MT, methyltransferase.
ACT-, AF-, and AK-toxins are related toxins with a common core polyketide. ACT-toxin is produced by the tangerine pathotype of A. alternata (A. alternata f. sp. citri) that affects grapefruit, hybrids of grapefruit and tangerine, tangerine and sweet orange (Timmer et al., 2003; Ma et al., 2019). Japanese pear black spot disease is caused by A. gaisen (syn. A. alternata Japanese pear pathotype) due to the production of the host specific AK-toxin (Tsuge et al., 2013). The strawberry pathotype of A. alternata (A. alternata f. sp. fragariae) affects both strawberry and pear due to the production of several host specific toxins (AF-toxins I, II, and III) (Tsuge et al., 2013). ACT-, AF-, and AK-toxins all act to depolarize the cellular plasma membrane resulting in invagination, fragmentation and vesiculation, causing cell necrosis in susceptible leaves (Kohmoto, 1993; Park and Ikeda, 2008).
These toxins all share a 9,10-epoxy-8-hydroxy-9-methyl-decatrienoic acid (EDA) moiety (Kohmoto et al., 1985; Nakatsuka et al., 1986; Kohmoto, 1993; Akimitsu et al., 2003) and differ based on the acyl groups attached to the C8 hydroxyl of the EDA moiety. Biosynthetic gene clusters encoding a PKS have been identified for ACT-, AF-, and AK-toxin biosynthesis and archived in MIBiG, the repository for biosynthetic gene clusters of known function (Tanaka and Tsuge, 2000; Ruswandi et al., 2005; Miyamoto et al., 2010; Takaoka et al., 2014; Kautsar et al., 2020). However, only the PKS from AF- and AK-toxin biosynthesis show significant homology. With a common polyketide core, all three gene clusters would be expected to have at least one homologous PKS. Further examination of the PKS from AF- and AK-toxin biosynthesis shows that the AK-toxin PKS (AFT9h) is pseudogenized. Comparison of the catalytic domains from the AF- and AK-toxin PKSs (AFT9-1 and AFT9h, respectively) show typical highly reducing PKS modules with a C-terminal carnitine O-palmitoyltransferase. Presumably this acyltransferase domain is responsible for off-loading of the completed polyketide, akin to side chain installation in lovastatin biosynthesis (Xie et al., 2009). However, transacylation would produce an ester, inconsistent with the structures of AF-, and AK-toxins that terminate in a carboxylic acid. Potentially this PKS could play a role in side chain installation in AF-toxin biosynthesis, via transacylation of the PKS bound side chain onto the EDA core. In AK-toxin biosynthesis, an aminoacyl chain is added to the C8 hydroxyl group of EDA, thus no transacylating PKS is required, consistent with the pseudogenization of this PKS in the MIBiG AK-toxin biosynthetic gene cluster.
A unique aspect of the EDA core polyketide is its epoxidized isopropyl tail. This unit could arise from either a 2-hydroxyisovalerate or an α,β-unsaturated isovalerate starter unit (Ray and Moore, 2016) for the PKS. Disruption of AFTS1, which encodes a 2-ketoisovalerate reductase and thus should eliminate 2-hydroxyisovalerate formation, did not abolish AF-toxin II biosynthesis (Ito et al., 2004). This strongly suggests that 2-hydroxyisovalerate is not the required starter unit for the core polyketide production.
Intriguingly, a larger gene cluster spanning 90 kb of the ACT-toxin producing tangerine pathotype A. alternata Z7 genome was proposed to encode the ACT-gene cluster (Wang et al., 2016; Figure 4). Of particular note in this cluster is the presence of a HMG-CoA synthase and an enoyl-CoA hydratase, that are suggestive of a potential mechanism for installing an α,β-unsaturated isovalerate via a β-branching type mechanism (Walker et al., 2020). Results from isotope labeling experiments with 13C labeled acetate are consistent with this hypothesis (Nakatsuka et al., 1990). Additionally, disruption of the two copies of enoyl-CoA hydratase encoding ACTT6 in A. alternata SH20 eliminates ACT-toxin production (Miyamoto et al., 2009). Lastly, while an HMG-CoA synthase has not yet been found in the AF-toxin biosynthetic gene cluster, one is associated with AK-toxin biosynthesis, AKT4 (Takaoka et al., 2014). Based on these data, the PKS AALTg11757 in combination with the HMG-CoA synthase AALTg11755 (previously identified as ACTT3; Miyamoto et al., 2008) and the cytochrome p450 AALTg11750 are responsible for assembling the common core of ACT-, AF-, and AK-toxin (Figure 4). Presumably, homologs of all these genes will be identified in AF- and AK-toxin producers as well. PKS AALTg11750 (ACTTS3) (Miyamoto et al., 2010) in combination with the NRPS AALTg11749 generate the side chain, which is added to the core via the condensation domain AALTg11738. Lastly epoxidation of the side chain by the Flavin monooxygenase AALTg11736 followed by epoxide hydrolysis via AALTg11743 (ACTT2) (Miyamoto et al., 2008) gives ACT-toxin I, 15, as shown.
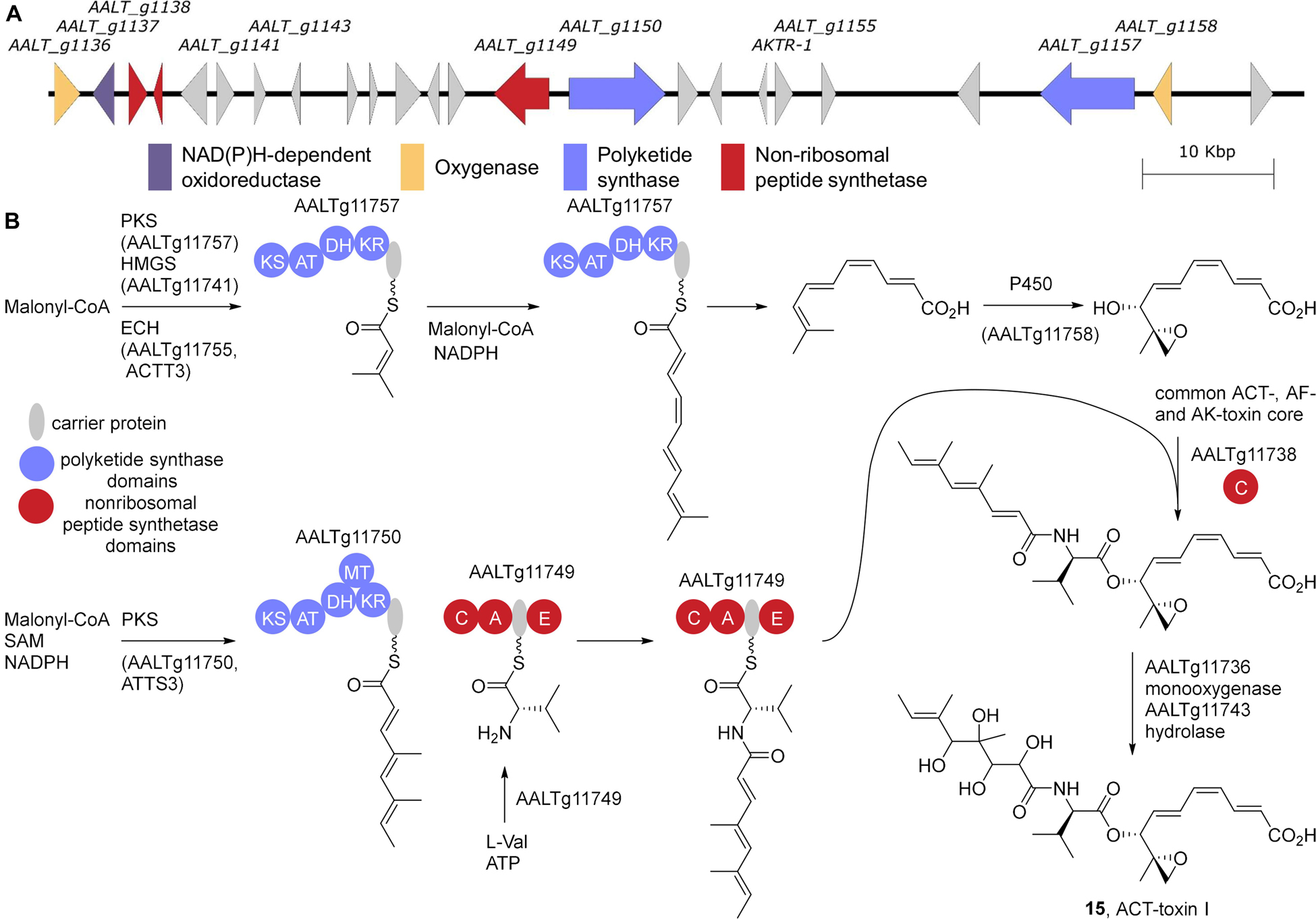
Figure 4. ACT-toxin biosynthesis. (A) The gene cluster from A. alternata Z7 (LPVP01000048). (B) The proposed biosynthesis of the ACT-, AF-, and AK-toxin core and the addition of the ACT-toxin I side chain. KS, β-ketosynthase; AT, acyltransferase; KR, β-ketoreductase; DH, dehydratase; MT, methyltransferase; A, adenylation; C, condensation; E, epimerization.
While the proposed biosyntheses of these toxins are yet to be fully characterized, strong evidence shows that their biosynthetic gene clusters are all present on ACs. For example, ACT-toxin associated genes ACTT1 and ACTT2 were shown by pulsed-field gel electrophoresis and Southern hybridization to be present only on the 1.9 Mb chromosome in the tangerine pathotype A. alternata SH20 (Masunaka et al., 2005). Similarly, a 32kb cosmid clone from the 1.05 Mb AC of the strawberry pathotype A. alternata NAF8 encoded the AF-toxin associated genes AFT1, AFTR and AFT3 (Hatta et al., 2002). Lastly probes for AK-toxin genes AKT1, AKT2, AKTR, and AKT3 were shown to hybridize to the 4.1 Mb AC of the pear pathotype A. alternata A15 (Tanaka and Tsuge, 2000).
AM-toxin is a four-residue cyclic depsipeptide produced by a pathotype of A. alternata (Alternata f. sp. mali) causing Alternaria blotch on a narrow range of apple cultivars world-wide (Ueno et al., 1975; Kohmoto et al., 1977; Filajdic and Sutton, 1991). AM-toxin affects both the chloroplast and plasma membrane of leaf cells, where the toxin inhibits photosynthetic CO2 fixation and induces electrolyte loss, resulting in tissue damage of susceptible leaves (Kohmoto et al., 1983; Shimomura et al., 1991; Zhang et al., 2015).
AM-toxin is a non-ribosomal peptide and the NRPS associated with its biosynthesis, AMT1, has been identified (Johnson et al., 2000). In addition, a 2-ketoisovalerate reductase (AMT2), a cytochrome p450 (AMT3), and a thioesterase (AMT4) were associated with AM-toxin biosynthesis (Ito et al., 2004; Harimoto et al., 2007). Sequencing of cDNA clones that hybridized with the chromosome containing AMT1 and AMT2 identified a number of key metabolic genes required for the complete biosynthesis of AM-toxin (Harimoto et al., 2007), all of which could be found on the sequenced BAC clone AM-BAC-14 (accession number AB525198, Figure 5A). Based on these genes, it is clear that AM-toxin is produced by a NRPS using 2-hydroxyisovalerate, 2-amino-p-hydroxyphenylvalerate (Ahv), and two alanines (Figure 5B). The 2-hydroxyisovalerate is generated from 2-ketoisovalerate via the reductase AMT2. Ahv is generated from tyrosine following two cycles of one carbon homologation, akin to leucine biosynthesis from valine (Kohlhaw, 2003). Specially, the transaminase AMT5 converts tyrosine into the corresponding α-ketoacid. AMT7, a 2-isopropylmalate synthase, adds acetyl-CoA to the 2-keto acid, and the product is isomerized by AMT8, an aconitase, to generate the precursor for AMT6, a 3-isopropyl malate dehydrogenase, which effects oxidative decarboxylation generating the α-ketoacid of homotyrosine (Yue et al., 2015). Reiteration of this one-carbon homologation catalyzed by AMT7, AMT8, and AMT6 generates the 2-keto-p-hydroxyphenylvalerate, which is converted into the α-amino acid via AMT5 producing Ahv. These enzymatic reactions provide access to the key building blocks required for AM-toxin biosynthesis.
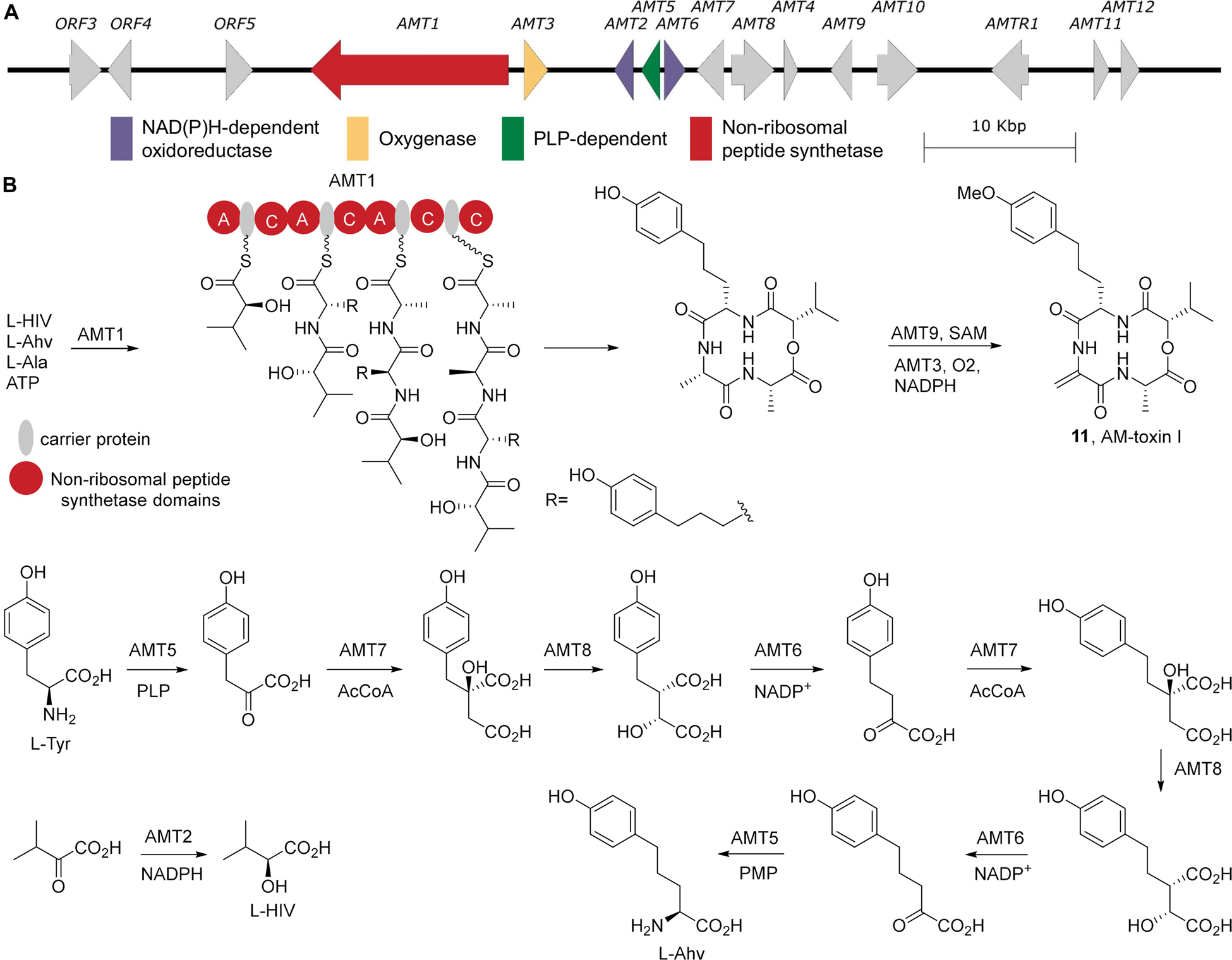
Figure 5. AM-toxin Biosynthesis. (A) The biosynthetic gene cluster from A. alternata strain NBRC 8984 (AB525198) (B) The biosynthetic pathway for AM-toxin I and its non-proteinogenic building blocks. A, adenylation; C, condensation.
As AM-toxin is a cyclic tetrapeptide, the NRPS responsible for its production, AMT1, must incorporate four amino acids, however examination of its domain architecture shows it lacks an adenylation domain in the final elongation module. Homologous to basidiomycete type VI siderophore synthetases (Brandenburger et al., 2017), AMT1 possesses an A3-T3-C4-T4-Ct architecture. Based on this similarity, A3 is predicted to amino acylate both T3 and T4 with alanine. Thus A1 is selective for 2-hydroxyisovalerate, A2 for Ahv, and A3 for alanine. The completed tetrapeptide linked to T4 is then cyclized by the offloading Ct domain (Gao et al., 2012). The cyclic tetrapeptide is subsequently O-methylated by AMT9 and presumably oxidized by the cytochrome p450 to generate the dehdyroalanine residues. This late stage installation of the dehydroalanine residue through oxidation is consistent with the AM-toxin NRPS and supported by the observation that the fusaristatin biosynthetic gene cluster also possess a cytochrome p450 (FGSG_08207), which may convert an alanine residue into a dehydroalanine on the macrocycle (Sørensen et al., 2014). However, biochemical confirmation of this biosynthesis is still required.
The AM-toxin biosynthetic gene cluster has clearly been localized to an AC. The AMT1 gene was shown by electrophoretic karyotyping and Southern blot probe labeling experiments to localize to ACs of 1.1–1.7 Mb in size (where AC size was dependent on the A. alternata apple pathotype strain examined). Furthermore, the AMT1 gene is absent in non-pathogenic strains (Akamatsu et al., 1999; Johnson et al., 2001). Loss of the associated ACs coincided with the loss of AMT1 gene expression, resulting in an observed AM-toxin-minus phenotype in a leaf necrosis bioassay (Johnson et al., 2001).
The routine accessibility of long-read or single-molecule sequencing platforms has catalyzed an explosion of highly contiguous fungal genomes in recent years. Platforms developed by Oxford Nanopore Technologies and Pacific Biosciences (PacBio) can effectively resolve genomic regions of low complexity and/or highly repetitive content, producing “closed” or “telomere-to-telomere” assemblies. The quality of fungal genome assemblies is often improved by inclusion of massively parallelized, high-fidelity shorter read sequences (such as those derived from Illumina platforms) in assembly pipelines. The resulting closed genomes are highly desirable for the study of rearrangements, regulation of clustered genes, gene duplications, transposable element proliferation and other forms of gene migration between genome compartments such as ACs, accessory regions and core chromosomes (Thomma et al., 2016).
To explore relationships between secondary metabolite biosynthesis and ACs, we compiled 15 recently published fungal genomes containing sequences identified as ACs and scanned them using the fungal version of antiSMASH (Blin et al., 2019) to detect secondary metabolite biosynthetic gene clusters (Table 1). Most of the genomes included in the analysis were produced using long-read sequencing; however, we included three genomes obtained by short-read sequencing platforms where chromosomes were identified as “lineage-specific chromosomes” or “minichromosomes” by optical mapping, electrophoretic karyotyping and/or strain- or species-specific genome comparisons (Ma et al., 2010; O’Connell et al., 2012; Wiemann et al., 2013). When available, gene predictions from the literature were used and in cases where no predictions were available, genes were predicted using antiSMASH default fungal parameters (see Supplementary Table S1 for predicted gene locations). Fungal taxa investigated included representatives from the genera Alternaria, Colletotrichum, Fusarium, Magnaporthe, and Zymoseptoria.
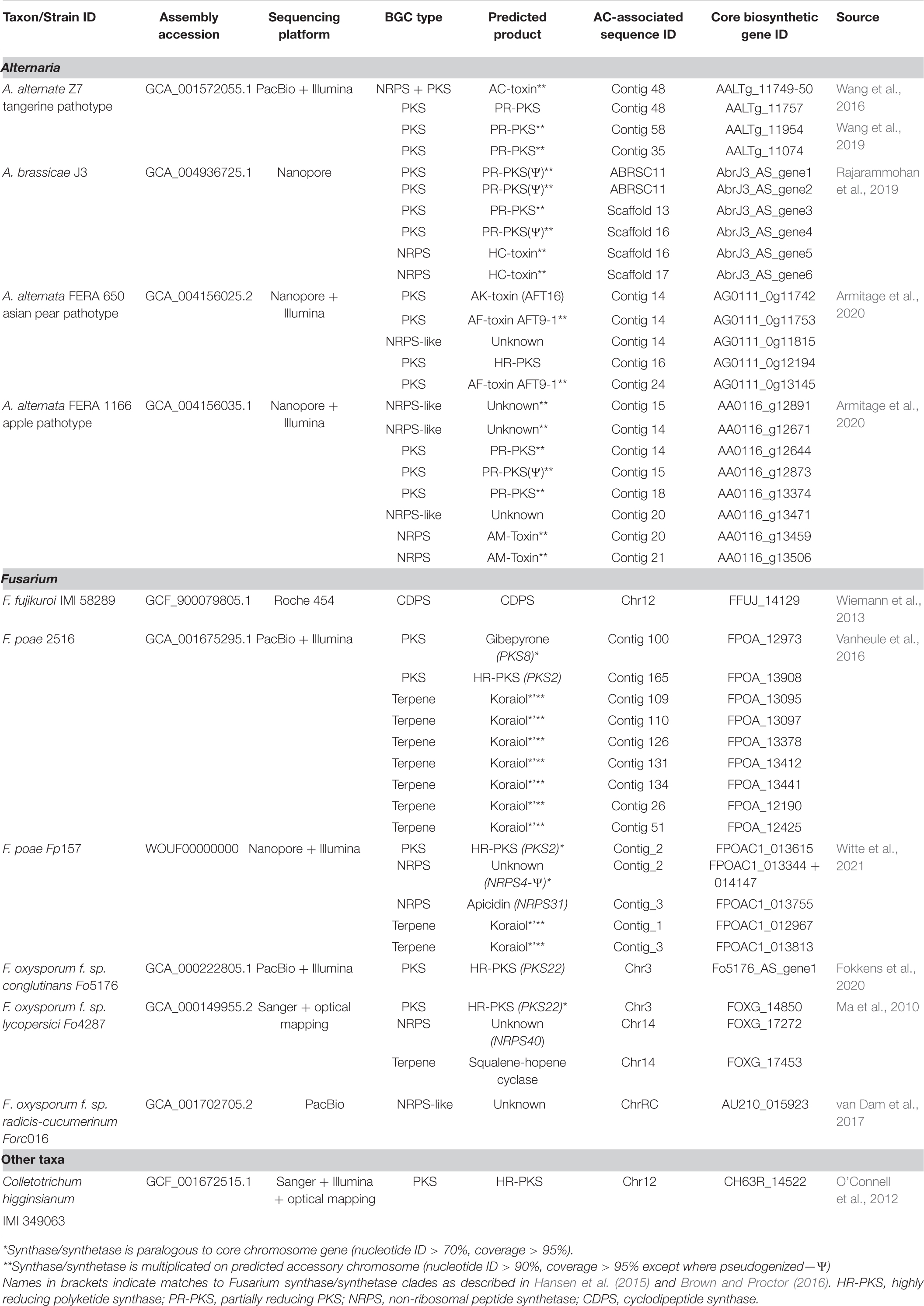
Table 1. Secondary metabolite biosynthetic gene clusters (BGCs) predicted from recently published fungal accessory chromosome (AC) sequences.
Most of the biosynthetic gene clusters associated with ACs in this subset of fungi are encoded by isolates of Fusarium and Alternaria, an unsurprising fact as these are genera associated with diverse secondary metabolism products and are likely over-represented in available databases of high-quality genomes. Published genomes derived from isolates of Zymoseptoria tritici, Magnaporthe oryzae, Colletotrichum graminicola, Fusarium oxysporum f. sp. lini and Fusarium solani (anamorph of Nectria haematococca) all lacked AC-associated secondary metabolite biosynthetic gene clusters, whereas Alternaria and Fusarium genomes contained numerous secondary metabolite biosynthetic gene multiplications in AC-associated sequences. The gene multiplication exhibited on ACs suggests that ACs are fertile grounds for secondary metabolite biosynthetic gene amplification and neofunctionalization (discussed below).
Our analysis underlines the utility of long-read sequencing in associating biosynthetic gene clusters to AC sequences in fungi. This marks an important shift in the study of AC-associated secondary metabolism, which can now be inferred and characterized without prior study of phenotypic or biological association. Three fungal secondary metabolites were recently attributed to biosynthetic gene clusters on AC-associated sequences thanks to long-read sequencing efforts. HC-toxin and apicidin are two structurally related cyclic tetrapeptides encoded by AC-associated biosynthetic gene clusters in A. brassicae strain J3 and F. poae strain Fp157, respectively (Rajarammohan et al., 2019; Witte et al., 2021), and koraiol is the product of a terpene synthase/cyclase detected in numerous Fusarium AC sequences (Table 1). The functional role of these secondary metabolites as virulence factors in the context of Alternaria and Fusarium host pathogenicity is currently unknown. Both HC-toxin and apicidin are known histone deacetylase inhibitors associated with potent bioactivity and their associated presence with ACs is likely not trivial to the evolution of fungal plant pathogenicity.
The apicidin family of non-ribosomal peptides are produced by various Fusarium species, including F. semitectum, F. sambucinum, and F. fujikuroi (Darkin-Rattray et al., 1996; Singh et al., 1996, 2001, 2002; Park et al., 1999; Jin et al., 2008, 2010; Von Bargen et al., 2013; Niehaus et al., 2014). The apicidins are potent, reversible histone deactylase inhibitors (Darkin-Rattray et al., 1996) that show antiprotozoal activity against Plasmodium berghei and P. falciparum (Darkin-Rattray et al., 1996) and induce apoptosis in human leukemic HL-60 cells (Kwon et al., 2002). Histone deacetylase inhibition can lead to hyper-acetylation of histones, impacting an organism’s ability to regulate genetic transcription. From in planta experiments, apicidins were observed to negatively impact the development of roots in seedlings, resulting in chlorosis and necrosis on leaves, distorted leaf shape in developing leaves, and stunted plant growth (Jin et al., 2008). Exposure of duckweed to apicidins caused an inhibition of cell division, cellular leakage, and an impairment of chlorophyl synthesis due to chloroplast deterioration (Abbas et al., 2001). Apicidins are structurally similar to HC-toxin, another cyclic tetrapeptide histone deacetylase-inhibitor with a well-documented role as a virulence factor during infection by the fungal plant pathogen Cochliobolus carbonum.
The parent compound apicidin B is a cyclic tetrapeptide comprised of a L-pipecolic acid, an L-isoleucine, a N-methoxy-L-tryptophan, and a L-8-keto-2-aminodecanoic acid residue (Figure 6). The biosynthetic gene clusters responsible for apicidin production in F. semitectum and apicidin F production in F. fujikuroi have been identified (Jin et al., 2010; Niehaus et al., 2014) and both clusters show high nucleotide identity over the coding regions of the assigned biosynthetic genes (>60%). Bioinformatic based comparisons show that the gene clusters are also present in the genomes of F. incarnatum and F. scirpi (Villani et al., 2019). The building blocks for the NRPS-mediated biosynthesis of apicidin are the proteinogenic amino acids L-isoleucine and L-tryptophan. The non-proteinogenic amino acid L-pipecolic acid (L-Pip) and L-2-aminodecanoic acid (L-Adc) must be generated specifically for apicidin biosynthesis. APS3 encodes a reductase responsible for converting 2-aminoapidate semialdehyde into L-Pip. Consistent with this, deletion of APS3 leads to the production of apicidin B, an analog where the L-Pip is replaced with L-proline (Jin et al., 2010). The genes encoding L-Adc formation are not all present in the apicidin biosynthetic gene cluster. A fatty acid synthase α-subunit, APS5 is present and is required for apicidin production, however the corresponding β-subunit is absent in the biosynthetic gene cluster and is likely located elsewhere in the genome. These synthases likely generate decanoate, which is further elaborated via α-oxidation by an as yet uncharacterized set of enzymes into 2-ketodecanoate. The aminotransferase APS4 then coverts this into L-Adc. APS1, the NRPS then epimerizes and condenses the L-Pip to L-Adc, L-tryptophan, and L-isoleucine, followed by cyclization via its C-terminal domain (Gao et al., 2012). The tryptophan and Adc side chains are then oxidized, though the order of oxidation is unclear. C8 oxidation of Adc via the monooxygenase APS9, generates an S-configured alcohol, which is followed by the oxidation to the ketone by the cytochrome p450 APS7 (Jin et al., 2010). N-oxidation of the tryptophan by APS8 (Niehaus et al., 2014) followed by O-methylation via APS6 (Jin et al., 2010) completes the biosynthesis.
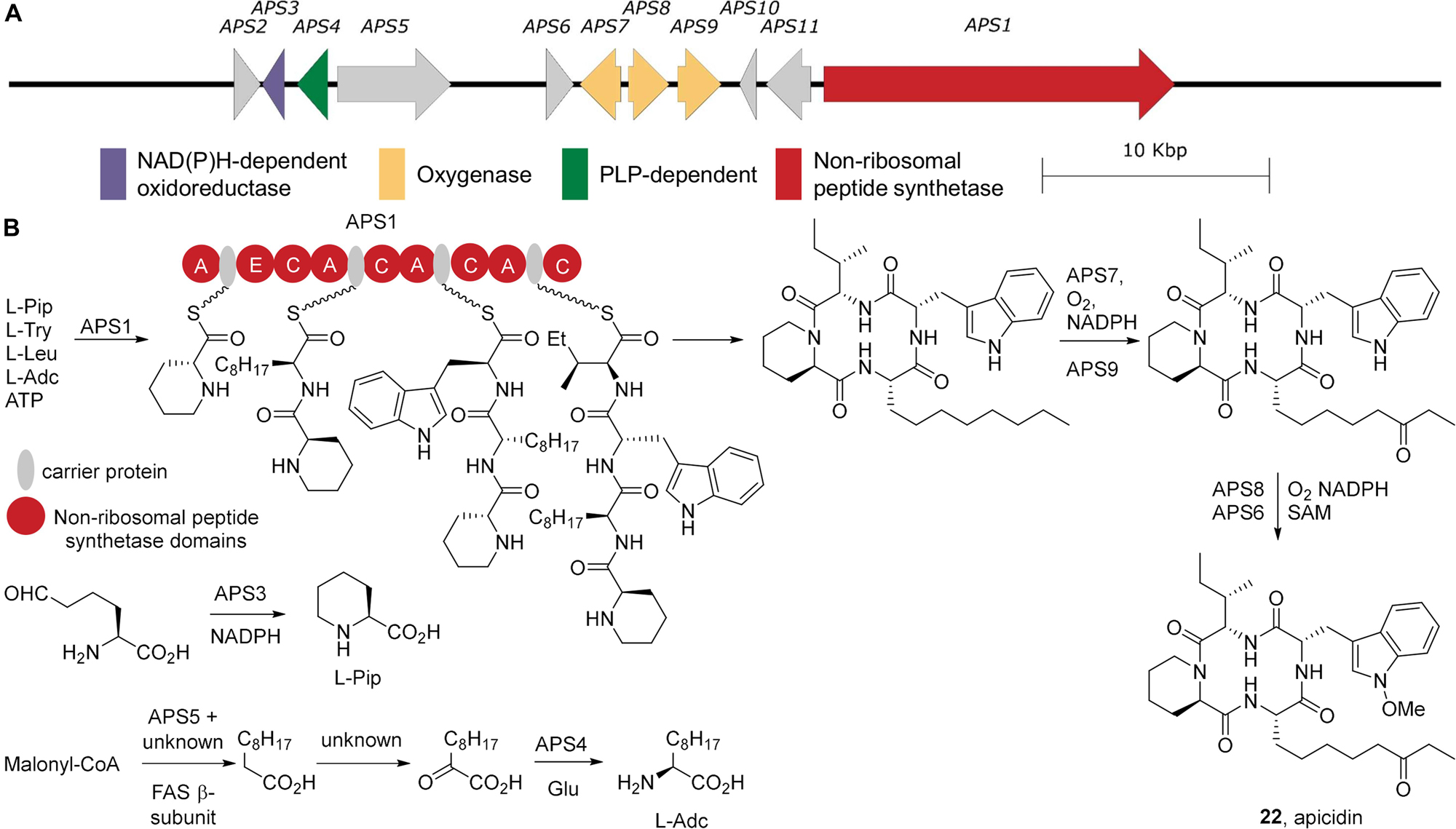
Figure 6. Apicidin biosynthesis. (A) The apicidin biosynthetic gene cluster from Fusarium incarnatum. (B) The biosynthesis of apicidin and its non-proteinogeneic amino acid building blocks. A, adenylation; C, condensation; E, epimerization.
The biosynthetic gene cluster associated with apicidin F production is localized to the end of chromosome I of F. fujikuroi as determined through analysis of a high quality, 12 scaffold genome that was assembled from pyrosequencing data (N50 = 4.2 Mb) (Wiemann et al., 2013). The position of the gene cluster at the chromosome end suggests it may have been acquired by horizontal gene transfer as end regions are more prone to recombination than telomere-distal regions. Consistent with horizontal acquisition, other Fusarium spp. evolutionarily related to F. fujikuroi do not produce apicidins (Niehaus et al., 2014). Examination of 13 Fusarium incarnatum-equiseti species complex genomes showed the apicidin biosynthetic gene cluster was present in only one member of the Incarnatum clade and two members of the Equiseti clade (Villani et al., 2019). A recent, high-quality genome strongly suggests the apicidin gene cluster resides on an AC in at least one isolate of F. poae (Witte et al., 2021).
Like apicidin, HC-toxin is a cyclic tetrapeptide and contains an oxidized L-Adc residue. It is also a histone deacetylase inhibitor, inhibiting the RPD3 class histone deacetylases, which affects root growth of susceptible maize cultivars (reviewed in Walton, 2006). Highly virulent C. carbonum strains contained a unique genetic locus (collectively designated as TOX2) that spans a 500kb region and is characterized by highly repetitive sequences in which multiple, functional copies of TOX2 reside (Walton, 2006). The TOX2 locus contains the core HC-toxin biosynthetic genes, including the gene encoding the non-ribosomal peptide synthetase HCT1 (Scott-Craig et al., 1992) and shows sequence homology to a number of the apicidin biosynthetic genes (Stergiopoulos et al., 2013).
Characterization of the HC-toxin biosynthetic gene cluster from TOX2 has proven challenging. The biosynthetic genes are sparse in this region of the genome and most occur as multiple functional copies. Furthermore the biosynthetic genes are embedded in highly repetitive sequences (Walton, 2006). While TOX2 has been well mapped in C. carbonum (Ahn and Walton, 1996; Ahn et al., 2002), no complete physical map is available of the HC-toxin gene cluster. However a recent complete genome sequence of A. brassicae revealed a putative HC-toxin biosynthetic gene cluster on an AC (Rajarammohan et al., 2019; Figure 7A). Unfortunately the genome assemblies of both A. jesenskae and C. carbonum are as yet too fragmented to determine whether the TOX2/TOX2-like clusters are localized to ACs or core chromosomes in those species. The detection of a TOX2-like cluster on an AC in A. brassicae has important implications for the evolution of virulence in this genus, particularly since HC-toxin is well established to be an important virulence factor in C. carbonum strains infecting maize genotypes (for review see Walton, 2006).
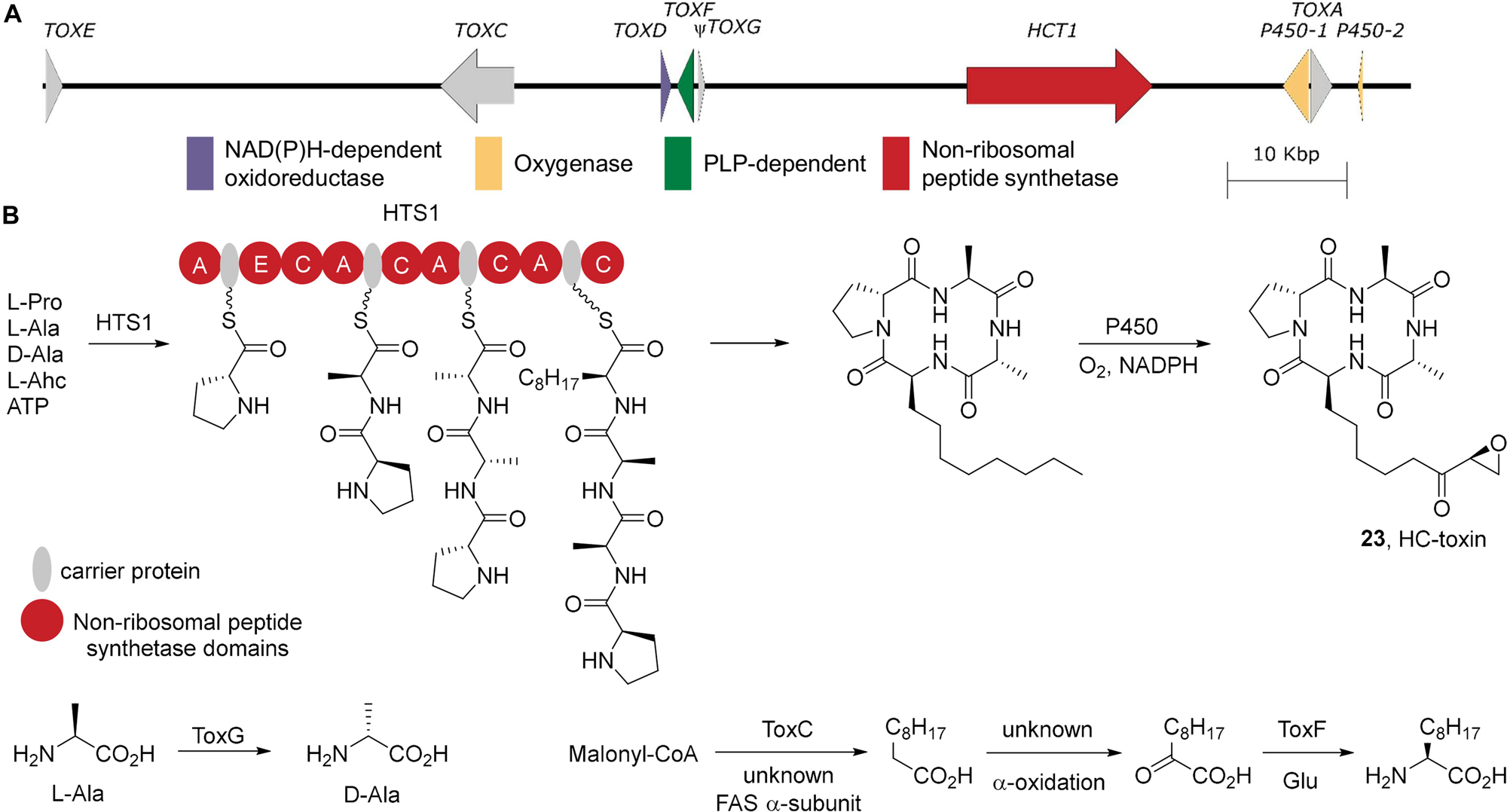
Figure 7. HC-toxin biosynthesis. (A) The biosynthetic gene cluster for HC-toxin from A. brassicae strain J3 scaffold 18 (SMOM01000016.1). (B) The biosynthesis of HC-toxin and its non-proteinogenic building blocks. A, adenylation; C, condensation; E, epimerization.
HC-toxin is produced by the NRPS HCT1 which utilizes L-proline, L-alanine, D-alanine, and L-Adc as building blocks (Figure 7B). The first A domain of the NRPS activates L-proline and loads it onto the carrier protein where it is epimerized generating D-proline. The second A domain loads L-alanine onto the following carrier protein and the C domain condenses with D-proline. The third A domain is presumably D-alanine selective. D-Alanine is generated via TOXG, an alanine racemase, from L-alanine (Cheng and Walton, 2000). Intriguingly, in A. brassicae TOXG is pseudogenized. However it is documented in C. carbonum that TOXG mutants can produce a variant of HC-toxin where D-alanine is replaced by glycine (Cheng and Walton, 2000). Thus, it seems likely that this A. brassicae strain generates the glycine variant of HC-toxin. The growing peptidyl intermediate D-proline-L-alanine is then coupled to a carrier protein bound D-alanine. Lastly, the final A domain likely activates L-Adc and couples this to complete the linear enzyme bound tetrapeptide. Similar to the apicidin biosynthetic gene cluster, the HC-toxin biosynthetic gene cluster contains only a subset of the genes responsible for the formation of L-Adc. TOXC encodes a fatty acid synthase β-subunit which is required for HC-toxin production (Ahn and Walton, 1997) presumably by generation of the medium chain fatty acid for L-Adc formation. However, no corresponding α-subunit has been identified. Oxidation to the 2-ketodecanoate is also required though, as of yet, no genes responsible for the oxidation have been identified. TOXF encodes a putative branched-chain amino acid transaminase (Cheng et al., 1999), which can convert the 2-ketodecanoate into L-Adc. The terminal C domain then completes the cyclization of the linear enzyme-bound tetrapetidyl group. Cytochrome p450-mediated oxidation of the L-Adc side chain then generates the completed, highly bioactive natural product. In A. brassicae, there appear to be multiple cytochrome p450s closely associated with TOXA, which encodes the major facilitator superfamily membrane transporter responsible for secretion of HC-toxin and self-protection (Pitkin et al., 1996).
Koraiol is a tricyclic sesquiterpene alcohol structurally related to caryophyllene. It has been shown to be produced in vitro from the class I terpene synthase STC4 from Fusarium fujikuroi that effects a 1,11 cyclization of the primary metabolite precursor farnesyl diphosphate (Brock et al., 2013; Figure 8). While a variety of fungal sesquiterpene synthases are known, phylogenetic analysis suggest that the synthases that generate the 1,11-cyclized products form a clade that is separate from the synthases that generate 1,6-cyclized sequiterenoids like trichodienes and the 1,10-cyclized sequiterpenes like aristolochene (Wawrzyn et al., 2012; Quin et al., 2014).
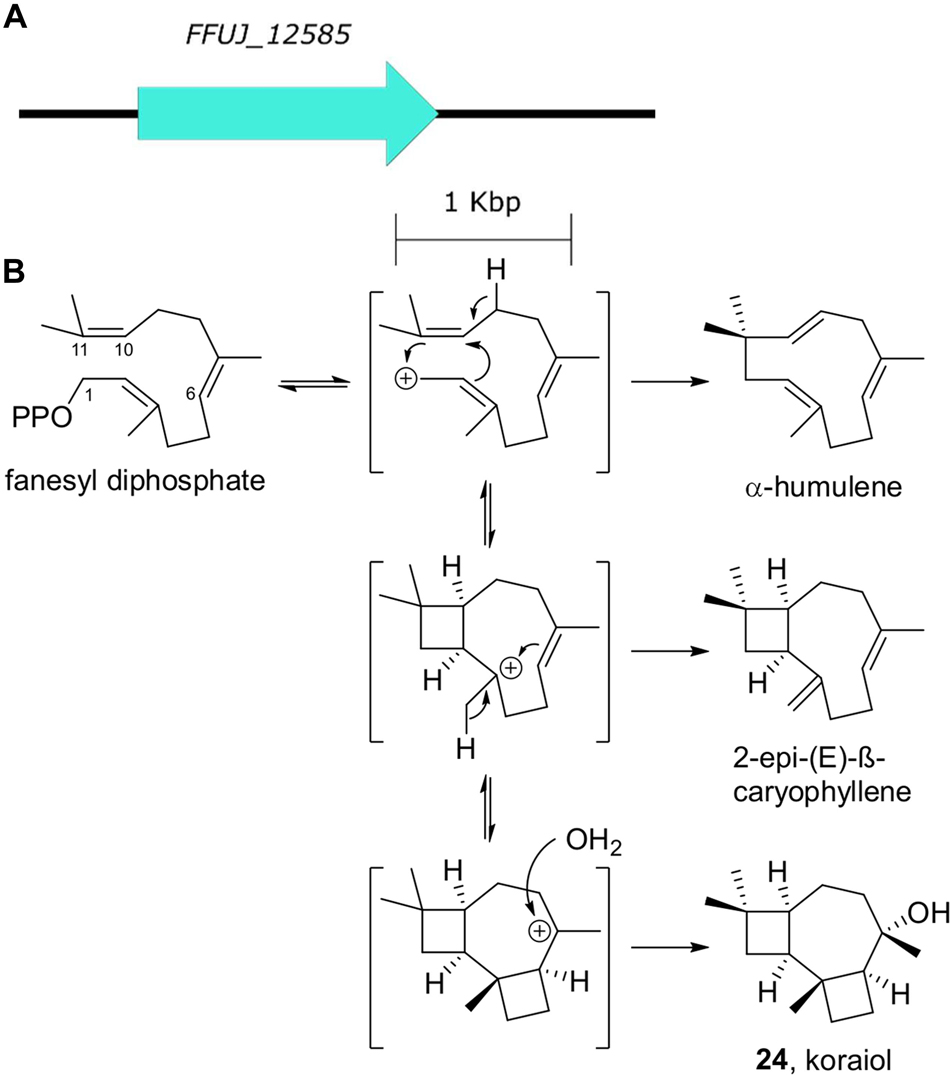
Figure 8. Koraiol biosynthesis. (A) The terpene synthase gene responsible for koraial biosynthesis. (B) Farnesyl diphosphate cyclization generates koraiol as well as additional 1,11-cyclized sequiterpene natural products.
STC4 homologs are commonly detected in Fusarium genomes (Niehaus et al., 2016; Hoogendoorn et al., 2018; Villani et al., 2019). A “multi-omics” study comparing secondary metabolite biosynthetic gene cluster expression and chemical phenotypes of Fusarium species detected STC4 expression in F. mangiferae, F. fujikuroi, F. verticillioides, and F. proliferatum isolates cultured both in vitro and in maize root infection experiments, but only detected koraiol from F. fujikuroi metabolomes (Niehaus et al., 2016). While the effect of koraiol on plant-fungal interactions is unclear, Niehaus et al. speculate that high levels of terpene cyclase expression in all growth conditions tested supports a basic growth- or interaction-associated role of koraiol which is unlikely to be host-specific.
Biosynthetic gene multiplicity associated with ACs drives the evolution of pathogenicity. From our comparison of hybrid long and short read genomes (Table 1), a considerable number of multiplicated genes were found to be associated with ACs, including koraiol synthase, HC-toxin synthetase, AF- and AM-toxin synthetases, and other predicted genes that have yet to be associated with secondary metabolite products. In Alternaria pathotypes, the presence of multiple copies of biosynthetic genes on ACs results in an amplification of host specific toxin production (Izumi et al., 2012a, b). Harimoto et al. (2007, 2008) found that the wild-type AM-toxin producing pathotype of A. alternata (strain IFO8984) likely has 3 or 4 copies of AMT2, AMT3 and AMT4, and observed that mutants containing single and double copies of AMT2 and AMT3 produced less AM-toxin than the wild-type strain. The single and double-mutants of AMT gene knockouts were confirmed as less virulent in plant infection trials compared to wild-type. These observations underline the importance of gene duplication as a potential mechanism to increase host specific toxin production in aggressive pathotypes. Other duplicated Alternaria secondary metabolite biosynthetic genes include those involved in AK-toxin production in A. alternata pear and apple pathotypes (Armitage et al., 2020). Interestingly, biosynthetic gene cluster duplications observed on ACs from A. alternata strains differ from duplications observed in Fusarium spp. In Fusarium, many AC-associated biosynthetic genes are slightly divergent paralogs of core chromosome genes, including synthase/synthetases PKS2, NRPS4, PKS8 (predicted to encode gibepyrone synthase), and PKS22 (for Fusarium biosynthetic enzyme clade nomenclature see Hansen et al., 2015 and Brown and Proctor, 2016). These AC/core chromosome paralogous pairs generally share 70–90% nucleotide identity, suggesting they are either rapidly diverging or are the products of relatively older duplication events.
Not all Fusarium AC-associated biosynthetic gene clusters are mere duplicates of core chromosome-associated biosynthetic gene clusters. The sesquiterpene cyclase gene STC4, associated with koraiol production (Figure 7), is multiplicated six times in F. poae strain 2516 and twice on ACs in F. poae strain Fp157 (Vanheule et al., 2016; Witte et al., 2021). Four of the F. poae 2516 STC4 copies were heterologously expressed in yeast and three were shown to encode functional koraiol synthases, supporting the functionality of the duplicates (Hoogendoorn et al., 2018). Hoogendoorn et al. point to STC4 copies as supporting the classic view of duplication as a means of relaxing evolutionary pressure on unigenes to enable the evolution of novel function (Ohno, 1970; Taylor and Raes, 2004). Another possibility is that more STC4 copies could simply mean more koraiol production, as is the case with duplicated Alternaria biosynthetic genes. Furthermore, the detection of an STC4 paralog in a subtelomeric region of Fp157 core chromosome 1 with a 98% nucleotide identity match to the two AC-associated koraiol synthase gene copies found from the same strain (Witte et al., 2021) indicates that gene transfer is likely happening between core and accessory chromosomes. Inter-chromosomal gene transfer could be an important mechanism by which successful combinations of biosynthetic genes and regulatory elements rapidly evolve on ACs and then move to core genome regions associated with higher levels of meiotic stability.
In addition to duplication, gene disruption via active transposon insertions and rearrangements appears common to both Alternaria and Fusarium biosynthetic gene clusters, where synthase or synthetase copies have missing domains crucial to the production of small molecules (Table 1). In PKSs and terpene synthase/cyclases this disruption likely leads to pseudogenization, however given the modular nature of NRPSs, the potential for truncated—but still functional—NRPS genes producing new molecules is worth investigation. Concrete examples of this are lacking at present, however F. poae NRPS4, the product of which is as yet uncharacterized, illustrates how TE disruption-associated truncation could happen (Figure 9). As more long-read genomes become available, we detect additional variations on truncated NRPSs in F. poae. Truncation followed by rearrangement could, however infrequent, lead to new combinations of domains and be an important process in the evolution of fungal secondary metabolite biosynthesis.
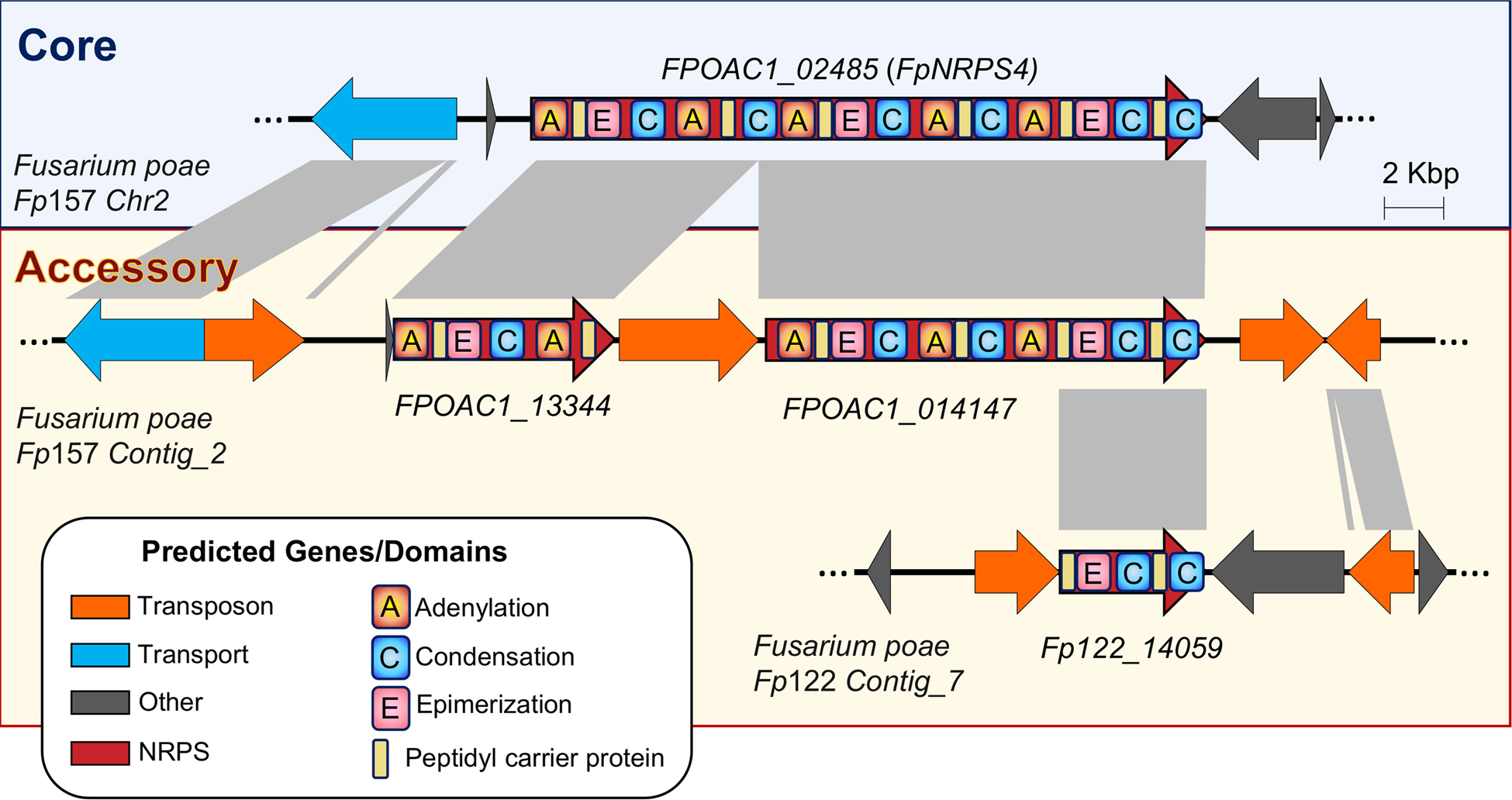
Figure 9. Accessory chromosomes in Fusarium poae illustrate the potential for biosynthetic gene duplication and TE-mediated truncation from which new small molecules could be produced.
Genetic duplication is widely held to be a fundamental prerequisite for the evolution of novel function by relaxing selective pressure on individual genes to enable mutation accumulation (Ohno, 1970; Taylor and Raes, 2004). However, duplications involving rapidly multiplying transposable elements can also be disruptive and appear parasitic in nature. “Repeat Induced Point-mutation” (RIP) is a genome defense mechanism that acts to control such parasitic duplications. Activated during the pre-meiotic stage of the sexual cycle, RIP inactivates duplicated genes over a certain length, via induced nucleotide mutation and epigenetic silencing (Selker et al., 1987; Selker and Garrett, 1988; Galagan and Selker, 2004). Since its discovery in Neurospora crassa, RIP has been detected in many filamentous ascomycetes including many important plant pathogens (Ikeda et al., 2002; Idnurm and Howlett, 2003; Cuomo et al., 2007; Clutterbuck, 2011), and is considered a powerful tool to maintain genetic integrity. RIP activity is typically inferred by scanning a genome for the presence of regions over a certain base pair length that are GC-poor compared to background codon frequencies, since RIP converts G:C to T:A dinucleotides in duplicated regions. The scars of disruptive, RIP-subdued transposable elements can be observed as low GC-content sequences inserted into coding regions, as appears to be the case for the pseudogenized chrysogine NRPS present in F. poae genomes (Figure 10).
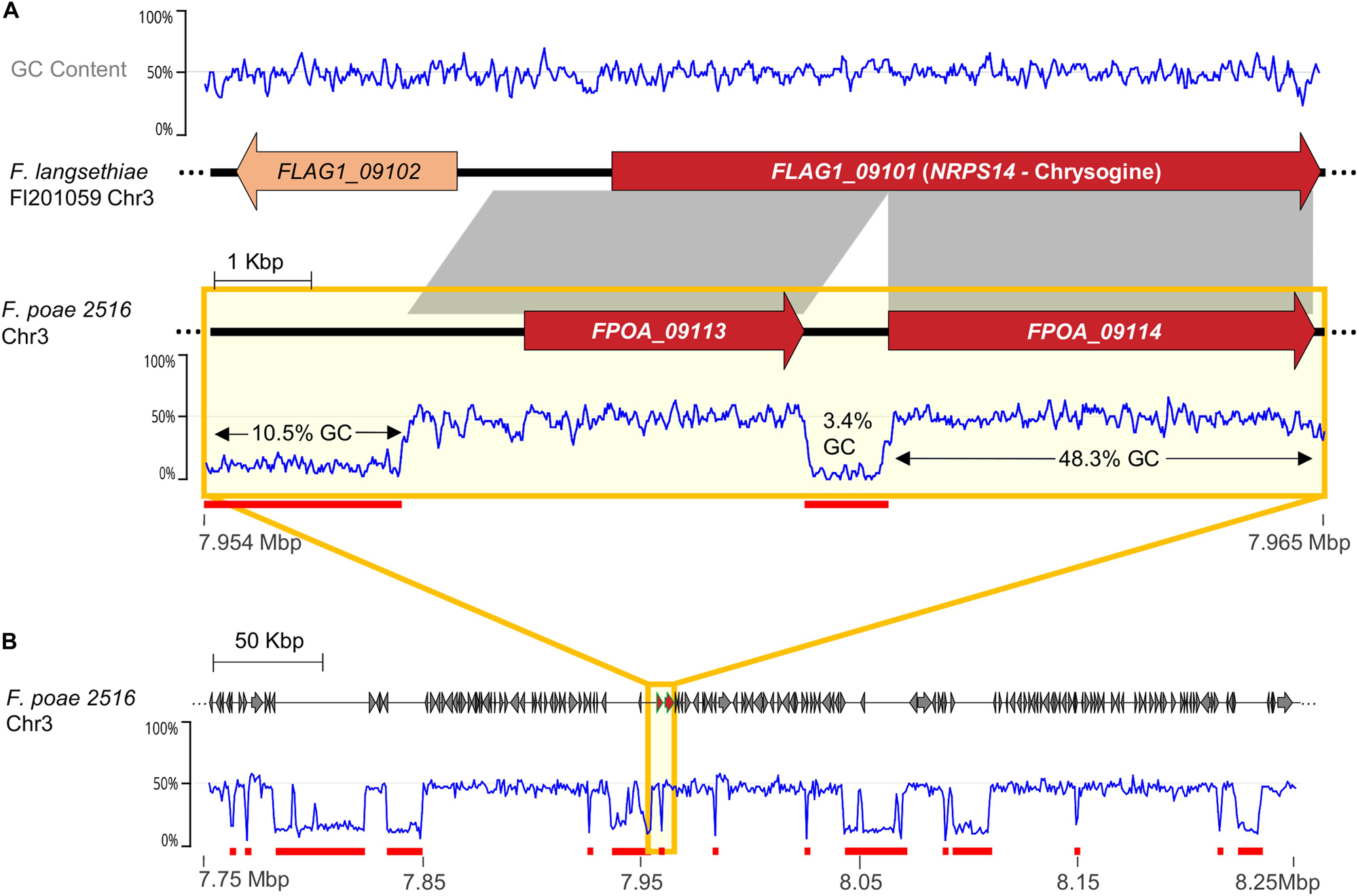
Figure 10. (A) Comparison of chrysogine synthetase NRPS14 homologs in F. langsethiae Fl 201059 and F. poae 2516, illustrating genomic regions predicted to be heavily influenced by repeat-induced point mutation (RIP). Gray blocks indicate syntenic regions (>90% nt identity). F. langsethiae is a known chrysogine producer, whereas F. poae is not, likely because NRPS14 has been pseudogenized via transposable element insertion. The offending transposable element was then likely subject to RIP, leaving an approximately 800 bp long region of very low GC content (3.4%). (B) Overview of surrounding genetic region on Chr3 showing numerous regions of very low GC content. Red underscores indicate sequence blocks with very low average GC content (<20%).
While RIP is clearly advantageous for the defense of genomes, it also likely suppresses gene duplications considered essential for adaptive evolution, including biosynthetic gene cluster duplication (Galagan and Selker, 2004). This presents a paradox: how can plant pathogenic fungi promote adaptation to plant defenses with reduced gene duplication? One solution to the problem could be to stop the sexual cycle and thereby stop RIP. This concept is consistent with the lack of observed sexual states in many plant pathogenic fungi that harbor ACs, including F. poae, F. oxysporum and A. alternata. Alternatively, genomes could be compartmentalized such that RIP is restricted to a subset of chromosomes or chromosome regions. This solution is consistent with the presence of intact, clustered tandem rDNA repeats in genomes of RIP-active species (L. maculans excepted), and is also consistent to F. poae genomes, which have less evidence for RIP in ACs than in core chromosomes (Vanheule et al., 2016; Witte et al., 2021). Little has been reported on RIP in Alternaria, although it has been computationally predicted to occur in at least one species (Clutterbuck, 2011). More research is needed to determine whether the presence of numerous duplicated biosynthetic genes on Alternaria ACs is related to RIP inactivation or genome compartmentalization. Importantly, the mere presence of ACs does not imply RIP activation or inactivation: Zymoseptoria tritici, for example, shows clear evidence for RIP in its ACs (Testa et al., 2015), and Leptosphaeria maculans has an AC predicted to be over 90% affected by RIP (Rouxel et al., 2011). During our long read genome analysis (Table 1), we did not detect secondary metabolite biosynthetic gene cluster duplications in genomes of either species. More research is needed to explore the idea of genetic compartmentalization and ACs in RIP-active species, and could be helpful in defining the functional differences between ACs and accessory regions of core chromosomes.
Our understanding of how ACs originate and affect core chromosome evolution is growing. The potential for gene cluster translocation between core and ACs (often localized in “accessory regions” usually proximal to telomeres) is relevant. Comparative genomic analysis of Zymoseptoria tritici genomes in isolates undergoing sexual reproduction supports an origin for some ACs via chromosome duplication, fusion and degeneration, which involve “breakage-fusion-bridge” cycles between chromosomes followed by mutational decay (Croll et al., 2013). In asexual species, however, the origins of ACs and their relationship to core chromosomes is less resolved. Both types of chromosomes can share genetic content, including transposon “invasions” from accessory chromosomes into core chromosomes (Vanheule et al., 2016), and other gene duplications as described above. Gene cluster translocation from an AC to a core chromosome is an interesting possibility that requires more study. In some F. poae isolates, an accessory region was found to contain lineage-specific, 100 Kb + sequence blocks bordered by unusual transposons (Vanheule et al., 2016). These regions were proposed to represent integration of AC sequences into core chromosomes (Vanheule et al., 2016). Interestingly, genes with “DUF3435” domains, implicated in large sequence translocations in Podospora genomes, have been detected in F. poae 2516 and are a promising avenue for further research of large translocations between chromosomes (Vogan et al., 2020). Understanding how genes move between chromosomes will illuminate the potential structural ramifications of the presence of ACs within a genome.
Horizontal transference of an AC between fungal strains can turn an endophyte or saprophyte into a pathogen (Ma et al., 2010; Vlaardingerbroek et al., 2016a; van Dam et al., 2017), suggesting important implications for the evolution of asexual plant pathogens. Compared to core chromosomes, genomic content of ACs are relatively dynamic (Vlaardingerbroek et al., 2016b; Habig et al., 2017; Möller et al., 2018; Plaumann et al., 2018). ACs can change in size during horizontal chromosome transfer, as higher concentrations of repetitive elements in ACs lead to intra- or interchromosome homologous recombination, resulting in deletions, translocations (Hedges and Deininger, 2007) and multiplication of gene loci (Li et al., 2020). Although exact mechanisms for horizontal chromosome transfer have yet to be fully elucidated, current hypotheses involve AC transfer events through heterokaryosis during hyphal anastomosis (Bertazzoni et al., 2018). In planta, we hypothesize that anastomosis may arise when intracellular hyphae of two strains interact in close proximity in the apoplast of host cells (especially in pathogens that exhibit an endophytic or biotrophic phase). The ability to maintain ACs with virulence factors in a subset of a fungal population could permit the maintenance of different trophic phases within the species, providing an adaptive advantage in periods of time when plant populations develop countermeasures to infection.
In fungi, secondary metabolism is regulated by various, often overlapping mechanisms that include pathway-specific and global regulators, signal transduction pathways, and epigenetic control (Macheleidt et al., 2016). Global regulators translate environmental cues to induce the transcription of multiple secondary metabolite pathways while pathway-specific transcription factors typically only control transcription of their respective cluster biosynthetic genes. Genes for cluster-specific regulators can be located either outside or inside of the cluster that they regulate, a fact that has implications on gene cluster expression or “cross talk” in organisms with ACs. Genes on acquired ACs may not have promoter sequences targeted by global regulators native to the host’s core chromosomes. Therefore, for AC-associated gene cluster expression in the recipient strain to be assured, appropriate transcription factors need to either be encoded on the AC or be sufficiently conserved in their DNA binding domains if encoded on the core genome (van der Does et al., 2016). Additionally, if multiple copies of conserved global regulators reside on ACs, transcriptional control over biosynthetic gene clusters on core chromosomes might also become regulated from the AC. For example, expression of the transcription factor EBR1 is involved with the onset of pathogenicity (as a master regulator of primary and secondary metabolism expression) and typically occurs as a single copy on a core chromosome; however, multiple duplications of EBR1 occur on ACs of F. poae and most F. oxysporum f.sp. pathogenic strains (Jonkers et al., 2014; Vanheule et al., 2016). The occurrence of multiple transcription factor duplications on ACs may offer a selective advantage through increased expression of particular virulence factors during infection. Similar to core chromosomes, expression of AC-associated secondary metabolism is also transcriptionally regulated via epigenetic control over chromatin structure. From studies with F. oxysporum, observations suggest that chromatin-mediated regulation of gene expression occurs on ACs (van der Does et al., 2016).
As described in this review, ACs in fungal plant pathogens harbor a diverse secondary metabolite biosynthetic potential. ACs can be vehicles for transferring biosynthetic gene clusters between isolates, they can influence gene regulatory systems, and they hold the potential to expand secondary metabolite structural diversity. This can lead to important differences in the metabolomes of isolates within the same species, confounding efforts to classify populations within a species based on “core”-associated genotypes—as evidenced by classification within the species A. alternata.
Combining genomics and untargeted metabolomics is a powerful way to screen populations of fungi for secondary metabolite production associated with ACs. Long-read genome sequencing combined with high-fidelity short-read sequencing is a powerful “top down” genomics approach for characterizing and contextualizing fungal genomes at the chromosomal level. In this context, “top down” refers to the ability to detect nearly all secondary metabolite biosynthetic gene clusters in a genome, even if they are silent under laboratory conditions, and derive AC/biosynthetic gene cluster associations. Untargeted metabolomic analysis, however, takes a “bottom up” approach to discovering AC- or accessory region-associated secondary metabolites. Here, chemical extracts of fungal strains are analyzed using high-resolution mass spectrometry to characterize and compare patterns in secondary metabolite production observed within a population to search of unique patterns attributable to sub-sets within the population. Leveraging results obtained from both metabolomics and genomics experiments within a species population allows for the correlation of lineage-specific secondary metabolite products to unique biosynthetic gene clusters residing on ACs.
This “multi-omics” strategy is currently being applied in studies of agriculturally relevant crop pathogens, making use of strain population libraries generated from annual disease surveys on various crops and geographical locations. In such a strategy, results from metabolomics profiling work are used to make selections of the population for full genome sequencing (long and short-read). For pathogens that are known to have ACs, metabolomes derived from extracts of single spore isolates cultured under axenic conditions on multiple media can be combined to generate “consensus” chemical phenotypes (representing the breadth in secondary metabolism expressed by a given strain; Figure 11). Constructing and comparing “consensus” chemical phenotypes from a species population enables the detection of phenotypic anomalies resulting from transcriptionally active biosynthetic gene clusters found on ACs or subtelomeric accessory regions from core chromosomes. Within the “consensus” phenotype, secondary metabolite associations between core and ACs (or accessory regions) are readily apparent. MS2 fragmentation patterns of mass features of interest can be compared to online spectral databases for metabolite identification and molecular networking analysis of MS2 spectra applied to detect deviations in the structures of known secondary metabolite products, indicating if new metabolite analogs are present. Structural information of observed secondary metabolite products can then be correlated with synthase/synthetase motifs from genome predicted secondary metabolite biosynthetic gene clusters and associations inferred. Often these inferences are substantiated by follow-up transcriptomic and synthase/synthetase gene knock-out experiments. Further comparison with associated biotic or abiotic metadata (e.g., host specificity, disease phenotypes or geographic location) can promote possible inference regarding the role that AC-acquired secondary metabolite expression may have on the evolution of strain pathogenicity or virulence within the species population.
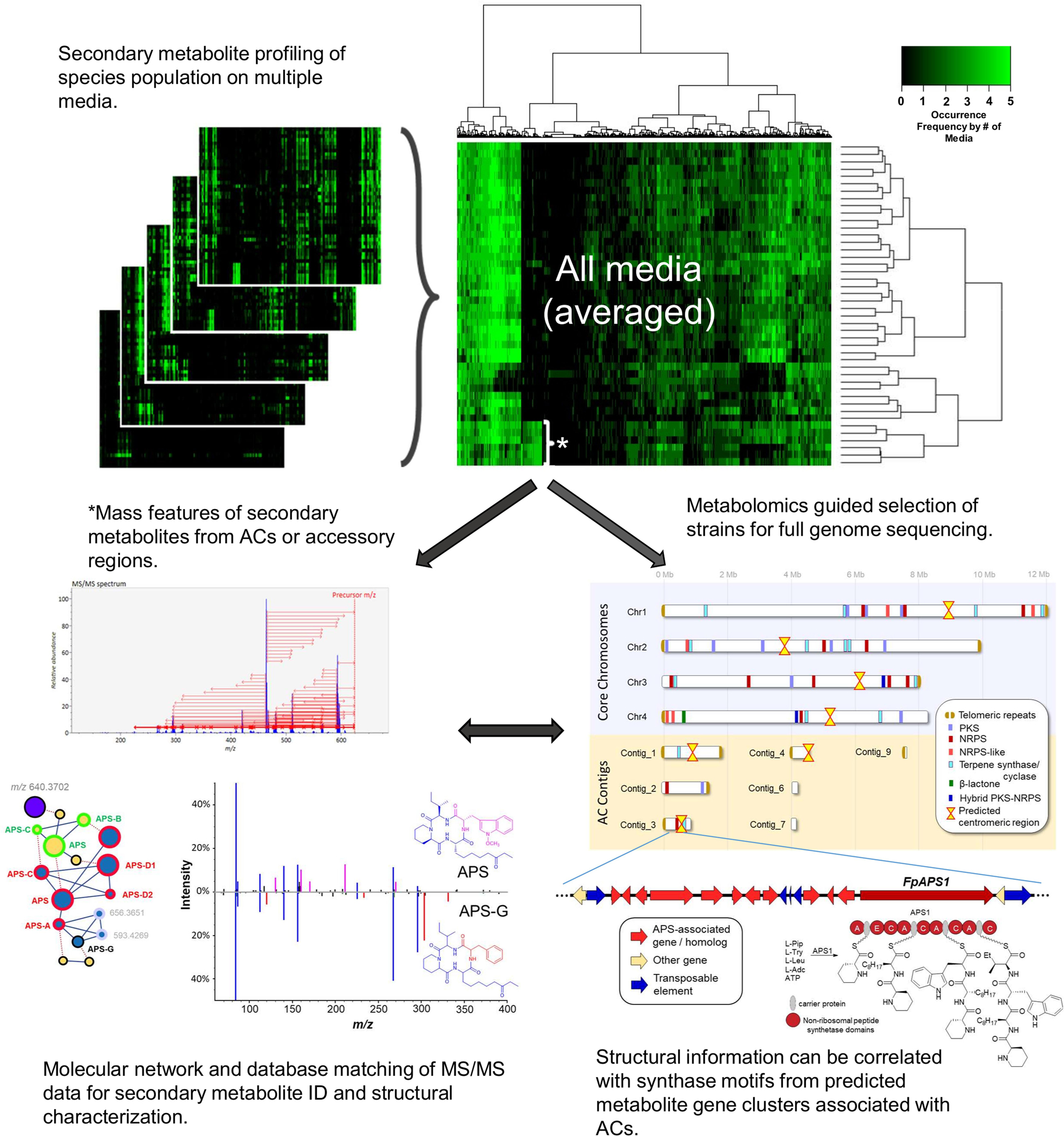
Figure 11. Using metabolomics to identify lineage-specific secondary metabolites produced by active secondary metabolite biosynthetic gene clusters on accessory chromosomes. Extracts from single spore isolates cultured in multiple media conditions are analyzed by high resolution mass spectrometry to produce metabolomes. Metabolomes are averaged across all media to create in vitro consensus chemical phenotypes for each strain. Lineage-specific signals (indicated by the asterisk) can be correlated to secondary metabolite biosynthetic gene clusters detected from isolate genomic analysis. Mass spectra can be further analyzed to compare chemical “fingerprints” (MS2 scans) to online spectral databases or in silico predictions of molecular fragmentation spectra for compound identification.
The study of the accessory genome is the study of the genomic space in which fungi develop novel invasion strategies and adapt to evolving plant defenses. In some Alternaria pathotypes/f.sp., ACs act as a chromosomal “engine” for the horizontal transfer, duplication, and rearrangement of secondary metabolite biosynthetic gene clusters. AC-associated gene cluster expression expands both the diversity and quantity of mycotoxins and other virulence factors produced, enabling isolates to invade and adapt to specific plant hosts. Untargeted metabolomics is a promising tool for exploring the evolving chemical space associated with these adaptations. Metabolomic profiling of Alternaria and Fusarium spp. populations will likely lead to the discovery of more biosynthetically active ACs with potential roles in pathogenicity. Such biosynthetic gene clusters on ACs can also provide insights into the evolution of fungal natural products, as the dynamic nature of ACs can promote diversification of natural product families. Although their effects on pathogenicity are currently unexplored, further study into secondary metabolite biosynthetic gene clusters on Fusarium ACs and their resulting products should provide insight into the evolutionary trajectories of closely-related fungal species with divergent lifestyles and genomic architectures, such as F. poae, F. oxysporum and F. graminearum. The differences between Alternaria and Fusarium genomes reviewed here suggest there are a multiplicity of relationships between ACs and core chromosomes in fungi, and this diversity will likely expand as more fungal taxa are discovered and sequenced.
TW, NV, CB, and DO wrote the manuscript and approved its final version.
Funding for this review was provided by the Science and Technology Branch of Agriculture and Agri-Food Canada (Project J-002071 “The population structure of Fusarium pathogens of small grain cereals, their distribution and relationship to mycotoxins” and Project J-002272 “Fungal and Bacterial Biosystematics—bridging taxonomy and “omics” technology in agricultural research and regulation”) to DO. Funding was also awarded via the Tito Scaiano Scholarship and the Keith Fagnou memorial scholarships from the University of Ottawa Chemistry and Biomolecular Sciences department to TW. CB was supported by the NSERC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.664276/full#supplementary-material
ACs, Accessory chromosomes; NRPS, Non-ribosomal peptide synthetase; PKS, Polyketide synthase; RIP, Repeat-induced point mutation.
Abbas, H. K., Gronwald, J. W., Plaisance, K. L., Paul, R. N., and Lee, Y. W. (2001). Histone deacetylase activity and phytotoxic effects following exposure of duckweed (Lemna pausicostata L.) to apicidin and HC-toxin. Phytopathology 91, 1141–1148. doi: 10.1094/PHYTO.2001.91.12.1141
Ahn, J. H., Cheng, Y. Q., and Walton, J. D. (2002). An extended physical map of the TOX2 locus of Cochliobolus carbonum required for biosynthesis of HC-toxin. Fungal Genet. Biol. 35, 31–38. doi: 10.1006/fgbi.2001.1305
Ahn, J. H., and Walton, J. D. (1996). Chromosomal organization of TOX2, a complex locus controlling host-selective toxin biosynthesis in Cochliobolus carbonum. Plant Cell 8, 887–897. doi: 10.1105/tpc.8.5.887
Ahn, J. H., and Walton, J. D. (1997). A fatty acid synthase gene in Cochliobolus carbonum required for production of HC-toxin, cyclo(D-prolyl-L-alanyl-D-alanyl-L-2-amino-9,10-epoxi-8-oxodecanoyl). Mol. Plant Microbe Interact. 10, 207–214. doi: 10.1094/MPMI.1997.10.2.207
Akagi, Y., Akamatsu, H., Otani, H., and Kodama, M. (2009). Horizontal chromosome transfer, a mechanism for the evolution and differentiation of a plant-pathogenic fungus. Eukaryot. Cell 8, 1732–1738. doi: 10.1128/EC.00135-09
Akamatsu, H., Taga, M., Kodama, M., Johnson, R., Otani, H., and Kohmoto, K. (1999). Molecular karyotypes for Alternaria plant pathogens known to produce host-specific toxins. Curr. Genet. 35, 647–656. doi: 10.1007/s002940050464
Akimitsu, K., Kohmoto, K., Otani, H., and Nishimura, S. (1989). Host-specific effects of toxin from the rough lemon pathotype of Alternaria alternata on mitochondria. Plant Physiol. 89, 925–931. doi: 10.1104/pp.89.3.925
Akimitsu, K., Peever, T. L., and Timmer, L. W. (2003). Molecular, ecological and evolutionary approaches to understanding Alternaria diseases of citrus. Mol. Plant Pathol. 4, 435–446. doi: 10.1046/j.1364-3703.2003.00189.x
Akimitsu, K., Tsuge, T., Kodama, M., Yamamoto, M., and Otani, H. (2014). Alternaria host-selective toxins: determinant factors of plant disease. J. Gen. Plant Pathol. 80, 109–122. doi: 10.1007/s10327-013-0498-7
Andersen, B., Smedsgaard, J., Jørring, I., Skouboe, P., and Pedersen, L. H. (2006). Real-time PCR quantification of the AM-toxin gene and HPLC qualification of toxigenic metabolites from Alternaria species from apples. Int. J. Food Microbiol. 111, 105–111. doi: 10.1016/j.ijfoodmicro.2006.04.021
Armitage, A. D., Cockerton, H. M., Sreenivasaprasad, S., Woodhall, J., Lane, C. R., Harrison, R. J., et al. (2020). Genomics evolutionary history and diagnostics of the Alternaria alternata species group including apple and asian pear pathotypes. Front. Microbiol. 10:3124. doi: 10.3389/fmicb.2019.03124
Bertazzoni, S., Williams, A. H., Jones, D. A., Syme, R. A., Tan, K. C., and Hane, J. K. (2018). Accessories make the outfit: accessory chromosomes and other dispensable DNA regions in plant-pathogenic fungi. Mol. Plant Microbe Interact. 31, 779–788. doi: 10.1094/MPMI-06-17-0135-CR
Bills, G., Martin, J., Collado, J., Platas, G., Overy, D., Tormo, J. R., et al. (2009). Measuring the distribution and diversity of antibiosis and secondary metabolites in filamentous fungi. SIMB News 59, 133–147.
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Bojja, R. S., Cerny, R. L., Proctor, R. H., and Du, L. (2004). Determining the biosynthetic sequence in the early steps of the fumonisin Pathway by use of three gene-disruption mutants of Fusarium verticillioides. J. Agric. Food Chem. 52, 2855–2860. doi: 10.1021/jf035429z
Brandenburger, E., Gressler, M., Leonhardt, R., Lackner, G., Habel, A., Hertweck, C., et al. (2017). A highly conserved basidiomycete peptide synthetase produces a trimeric hydroxamate siderophore. Appl. Environ. Microbiol. 83, e1478-17. doi: 10.1128/AEM.01478-17
Brock, N. L., Huss, K., Tudzynski, B., and Dickschat, J. S. (2013). Genetic dissection of sesquiterpene biosynthesis by Fusarium fujikuroi. Chembiochem 14, 311–315. doi: 10.1002/cbic.201200695
Brown, D. W., and Proctor, R. H. (2016). Insights into natural products biosynthesis from analysis of 490 polyketide synthases from Fusarium. Fungal Genet. Biol. 89, 37–51. doi: 10.1016/j.fgb.2016.01.008
Butchko, R. A. E., Plattner, R. D., and Proctor, R. H. (2003). FUM13 encodes a short chain dehydrogenase/reductase required for C-3 carbonyl reduction during fumonisin biosynthesis in Gibberella moniliformis. J. Agric. Food Chem. 51, 3000–3006. doi: 10.1021/jf0262007
Caldas, E. D., Sadilkova, K., Ward, B. L., Jones, A. D., Winter, C. K., and Gilchrist, D. G. (1998). Biosynthetic studies of fumonisin B1 and AAL Toxins. J. Agric. Food Chem. 46, 4734–4743. doi: 10.1021/jf980210j
Cheng, Y. Q., Ahn, J. H., and Walton, J. D. (1999). A putative branched-chain-amino-acid transaminase gene required for HC-toxin biosynthesis and pathogenicity in Cochliobolus carbonum. Microbiology 145, 3539–3546. doi: 10.1099/00221287-145-12-3539
Cheng, Y. Q., and Walton, J. D. (2000). A eukaryotic alanine racemase gene involved in cyclic peptide biosynthesis. J. Biol. Chem. 275, 4906–4911. doi: 10.1074/jbc.275.7.4906
Clutterbuck, A. J. (2011). Genomic evidence of repeat-induced point mutation (RIP) in filamentous ascomycetes. Fungal Genet. Biol. 48, 306–326. doi: 10.1016/j.fgb.2010.09.002
Covert, S. F. (1998). Supernumerary chromosomes in filamentous fungi. Curr. Genet. 33, 311–319. doi: 10.1007/s002940050342
Croll, D., and McDonald, B. A. (2012). The accessory genome as a cradle for adaptive evolution in pathogens. PLoS Pathog. 8:e1002608. doi: 10.1371/journal.ppat.1002608
Croll, D., Zala, M., and McDonald, B. A. (2013). Breakage-fusion-bridge cycles and large insertions contribute to the rapid evolution of accessory chromosomes in a fungal pathogen. PLoS Genet. 9:e1003567. doi: 10.1371/journal.pgen.1003567
Cuomo, C. A., Güldener, U., Xu, J. R., Trail, F., Turgeon, B. G., Di Pietro, A., et al. (2007). The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317, 1400–1402. doi: 10.1126/science.1143708
Darkin-Rattray, S. J., Gurnett, A. M., Myers, R. W., Dulski, P. M., Crumley, T. M., Allocco, J. J., et al. (1996). Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. U.S.A. 93, 13143–13147. doi: 10.1073/pnas.93.23.13143
Ding, Y., Bojja, R. S., and Du, L. (2004). Fum3p, a 2-ketoglutarate-dependent dioxygenase required for C-5 hydroxylation of fumonisins in Fusarium verticillioides. Appl. Environ. Microbiol. 70, 1931–1934. doi: 10.1128/aem.70.4.1931-1934.2004
Du, L., Zhu, X., Gerber, R., Huffman, J., Lou, L., Jorgenson, J., et al. (2008). Biosynthesis of sphinganine-analog mycotoxins. J. Ind. Microbiol. Biotechnol. 35, 455–464. doi: 10.1007/s10295-008-0316-y
Filajdic, N., and Sutton, T. (1991). Identification and distribution of Alternaria mali on apples in North Carolina and susceptibility of different varieties of apples to Alternaria blotch. Plant Dis. 75, 1045. doi: 10.1094/pd-75-1045
Fokkens, L., Guo, L., Dora, S., Wang, B., Ye, K. Sánchez-Rodríguez, C., et al. (2020). A chromosome-scale genome assembly for the fusarium oxysporum strain Fo5176 to establish a model arabidopsis-Fungal pathosystem. G3 (Bethesda). 10, 3549–3555. doi: 10.1534/g3.120.401375
Galagan, J. E., and Selker, E. U. (2004). RIP: the evolutionary cost of genome defense. Trends Genet. 20, 417–423. doi: 10.1016/j.tig.2004.07.007
Gao, X., Haynes, S. W., Ames, B. D., Wang, P., Vien, L. P., Walsh, C. T., et al. (2012). Cyclization of fungal nonribosomal peptides by a terminal condensation-like domain. Nat. Chem. Biol. 8, 823–830. doi: 10.1038/nchembio.1047
Gardner, J. M., Kono, Y., Tatum, J. H., Suzuki, Y., and Takeuchi, S. (1985). Plant pathotoxins from Alternaria citri: the major toxin specific for rough lemon plants. Phytochemistry 24, 2861–2867. doi: 10.1016/0031-9422(85)80015-7
Gault, C. R., Obeid, L. M., and Hannun, Y. A. (2010). An overview of sphingolipid metabolism: from synthesis to breakdown. Adv. Exp. Med. Biol. 688, 1–23. doi: 10.1007/978-1-4419-6741-1_1
Gechev, T. S., Gadjev, I. Z., and Hille, J. (2004). An extensive microarray analysis of AAL-toxin-induced cell death in Arabidopsis thaliana brings new insights into the complexity of programmed cell death in plants. Cell. Mol. Life Sci. 61, 1185–1197. doi: 10.1007/s00018-004-4067-2
Gilchrist, D. G. (1998). Programmed cell death in plant disease: the purpose and promise of cellular suicide. Annu. Rev. Phytopathol. 36, 393–414. doi: 10.1146/annurev.phyto.36.1.393
Habig, M., Kema, G. H. J., and Stukenbrock, E. H. (2018). Meiotic drive of female-inherited supernumerary chromosomes in a pathogenic fungus. Elife 7:e40251. doi: 10.7554/eLife.40251
Habig, M., Quade, J., and Stukenbrock, E. H. (2017). Forward genetics approach reveals host genotype-dependent importance of accessory chromosomes in the fungal wheat pathogen Zymoseptoria tritici. MBio 8:e1919-17. doi: 10.1128/mBio.01919-17
Han, Y., Liu, X., Benny, U., Corby Kistler, H., and VanEtten, H. D. (2001). Genes determining pathogenicity to pea are clustered on a supernumerary chromosome in the fungal plant pathogen Nectria haematococca. Plant J. 25, 305–314. doi: 10.1046/j.1365-313X.2001.00969.x
Hansen, F. T., Gardiner, D. M., Lysøe, E., Fuertes, P. R., Tudzynski, B., Wiemann, P., et al. (2015). An update to polyketide synthase and non-ribosomal synthetase genes and nomenclature in Fusarium. Fungal Genet. Biol. 75, 20–29. doi: 10.1016/j.fgb.2014.12.004
Harimoto, Y., Hatta, R., Kodama, M., Yamamoto, M., Otani, H., and Tsuge, T. (2007). Expression profiles of genes encoded by the supernumerary chromosome controlling AM-toxin biosynthesis and pathogenicity in the apple pathotype of Alternaria alternata. Mol. Plant Microbe Interact. 20, 1463–1476. doi: 10.1094/MPMI-20-12-1463
Harimoto, Y., Tanaka, T., Kodama, M., Yamamoto, M., Otani, H., and Tsuge, T. (2008). Multiple copies of AMT2 are prerequisite for the apple pathotype of Alternaria alternata to produce enough AM-toxin for expressing pathogenicity. J. Gen. Plant Pathol. 74, 222–229. doi: 10.1007/s10327-008-0089-1
Hatta, R., Ito, K., Hosaki, Y., Tanaka, T., Tanaka, A., Yamamoto, M., et al. (2002). A conditionally dispensable chromosome controls host-specific pathogenicity in the fungal plant pathogen Alternaria alternata. Genetics 161, 59–70.
He, C., Rusu, A. G., Poplawski, A. M., Irwin, J. A. G., and Manners, J. M. (1998). Transfer of a supernumerary chromosome between vegetatively incompatible biotypes of the fungus Colletotrichum gloeosporioides. Genetics 150, 1459–1466.
Hedges, D. J., and Deininger, P. L. (2007). Inviting instability: transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res. 616, 46–59.
Hoogendoorn, K., Barra, L., Waalwijk, C., Dickschat, J. S., van der Lee, T. A. J., and Medema, M. H. (2018). Evolution and diversity of biosynthetic gene clusters in Fusarium. Front. Microbiol. 9:1158. doi: 10.3389/fmicb.2018.01158
Houterman, P. M., Speijer, D., Dekker, H. L., De Koster, C. G., Cornelissen, B. J. C., and Rep, M. (2007). The mixed xylem sap proteome of Fusarium oxysporum-infected tomato plants: short communication. Mol. Plant Pathol. 8, 215–221. doi: 10.1111/j.1364-3703.2007.00384.x
Idnurm, A., and Howlett, B. J. (2003). Analysis of loss of pathogenicity mutants reveals that repeat-induced point mutations can occur in the Dothideomycete Leptosphaeria maculans. Fungal Genet. Biol. 39, 31–37. doi: 10.1016/S1087-1845(02)00588-1
Ikeda, K., Nakayashiki, H., Kataoka, T., Tamba, H., Hashimoto, Y., Tosa, Y., et al. (2002). Repeat-induced point mutation (RIP) in Magnaporthe grisea: implications for its sexual cycle in the natural field context. Mol. Microbiol. 45, 1355–1364. doi: 10.1046/j.1365-2958.2002.03101.x
Ito, K., Tanaka, T., Hatta, R., Yamamoto, M., Akimitsu, K., and Tsuge, T. (2004). Dissection of the host range of the fungal plant pathogen Alternaria alternata by modification of secondary metabolism. Mol. Microbiol. 52, 399–411. doi: 10.1111/j.1365-2958.2004.04004.x
Izumi, Y., Kamei, E., Miyamoto, Y., Ohtani, K., Masunaka, A., Fukumoto, T., et al. (2012a). Role of the pathotype-specific ACRTS1 gene encoding a hydroxylase involved in the biosynthesis of host-selective ACR-toxin in the rough lemon pathotype of Alternaria alternata. Phytopathology 102, 741–748. doi: 10.1094/PHYTO-02-12-0021-R
Izumi, Y., Ohtani, K., Miyamoto, Y., Masunaka, A., Fukumoto, T., Gomi, K., et al. (2012b). A polyketide synthase gene, ACRTS2, is responsible for biosynthesis of host-selective ACR-toxin in the rough lemon pathotype of Alternaria alternata. Mol. Plant Microbe Interact. 25, 1419–1429. doi: 10.1094/MPMI-06-12-0155-R
Jin, J., Baek, S.-R., Lee, K.-R., Lee, J., Yun, S.-H., Kang, S.-C., et al. (2008). Purification and phytotoxicity of apicidins produced by the Fusarium semitectum KCTC16676. Plant Pathol. J. 24, 417–422. doi: 10.5423/PPJ.2008.24.4.417
Jin, J.-M., Lee, S., Lee, J., Baek, S.-R., Kim, J.-C., Yun, S.-H., et al. (2010). Functional characterization and manipulation of the apicidin biosynthetic pathway in Fusarium semitectum. Mol. Microbiol. 76, 456–466. doi: 10.1111/j.1365-2958.2010.07109.x
Johnson, L. J., Johnson, R. D., Akamatsu, H., Salamiah, A., Otani, H., Kohmoto, K., et al. (2001). Spontaneous loss of a conditionally dispensable chromosome from the Alternaria alternata apple pathotype leads to loss of toxin production and pathogenicity. Curr. Genet. 40, 65–72. doi: 10.1007/s002940100233
Johnson, R. D., Johnson, L., Itoh, Y., Kodama, M., Otani, H., and Kohmoto, K. (2000). Cloning and characterization of a cyclic peptide synthetase gene from Alternaria alternata apple pathotype whose product is involved in AM-toxin synthesis and pathogenicity. Mol. Plant Microbe Interact. 13, 742–753. doi: 10.1094/MPMI.2000.13.7.742
Jonkers, W., Xayamongkhon, H., Haas, M., Olivain, C., van der Does, H. C., Broz, K., et al. (2014). EBR1 genomic expansion and its role in virulence of Fusarium species. Environ. Microbiol. 16, 1982–2003. doi: 10.1111/1462-2920.12331
Kautsar, S. A., Blin, K., Shaw, S., Navarro-Muñoz, J. C., Terlouw, B. R., van der Hooft, J. J. J., et al. (2020). MIBiG 2.0: a repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 48, D454–D458. doi: 10.1093/nar/gkz882
Kheder, A. A., and Akagi, Y. (2012). Functional analysis of the ceramide synthase gene ALT7, a homolog of the disease resistance gene Asc1, in the plant pathogen Alternaria alternata. J. Plant Pathol. Microbiol. 01:001. doi: 10.4172/2157-7471.s2-001
Kohlhaw, G. B. (2003). Leucine biosynthesis in fungi: entering metabolism through the back door. Microbiol. Mol. Biol. Rev. 67, 1–15. doi: 10.1128/mmbr.67.1.1-15.2003
Kohmoto, K. (1993). Isolation and biological activities of two host-specific toxins from the tangerine pathotype of Alternaria alternata. Phytopathology 83:495. doi: 10.1094/phyto-83-495
Kohmoto, K., Kondoh, Y., and Kohguchi, T. (1984). Ultrastructural changes in host leaf cells caused by host-selective toxin of Alternaria alternata from rough lemon. Can. J. Bot. 62, 2485–2492. doi: 10.1139/b84-339
Kohmoto, K., Nishimura, S., and Osaki, K. (1985). Isolation and structures of AK-Toxin I and II, host-specific phytotoxic metabolites -produced by Alternaria alternata japanese pear pathotype. Agric. Biol. Chem. 49, 807–815. doi: 10.1271/bbb1961.49.807
Kohmoto, K., Nishimura, S., and Otani, H. (1983). “Action sites for AM-toxins produced by the apple pathotype of Alternaria alternata,” in Plant Infection: the Physiological and Biochemical Basis, eds Y. Asada, W. R. Bushnell, S. Ouchi, and C. P. Vance (Berlin: Springer-Verlag), 253–263.
Kohmoto, K., Taniguchi, T., and Nishimura, S. (1977). Correlation between the susceptibility of apple cultivars to Alternaria mali and their sensitivity to AM-toxin I. Jpn. J. Phytopathol. 43, 65–68. doi: 10.3186/jjphytopath.43.65
Kwon, S. H., Ahn, S. H., Kim, Y. K., Bae, G. U., Yoon, J. W., Hong, S., et al. (2002). Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J. Biol. Chem. 277, 2073–2080. doi: 10.1074/jbc.M106699200
Li, J., Fokkens, L., Conneely, L. J., and Rep, M. (2020). Partial pathogenicity chromosomes in Fusarium oxysporum are sufficient to cause disease and can be horizontally transferred. Environ. Microbiol. 22, 4985–5004. doi: 10.1111/1462-2920.15095
Lia, Y., Lou, L., Cerny, R. L., Butchko, R. A. E., Proctor, R. H., Shen, Y., et al. (2013). Tricarballylic ester formation during biosynthesis of fumonisin mycotoxins in Fusarium verticillioides. Mycology 4, 179–186. doi: 10.1080/21501203.2013.874540
Ma, H., Zhang, B., Gai, Y., Sun, X., Chung, K. R., and Li, H. (2019). Cell-wall-degrading enzymes required for virulence in the host selective toxin-producing necrotroph Alternaria alternata of citrus. Front. Microbiol. 10:2514. doi: 10.3389/fmicb.2019.02514
Ma, L.-J., van der Does, H. C., Borkovich, K. A., Coleman, J. J., Daboussi, M.-J., Di Pietro, A., et al. (2010). Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464, 367–373. doi: 10.1038/nature08850
Macheleidt, J., Mattern, D. J., Fischer, J., Netzker, T., Weber, J., Schroeckh, V., et al. (2016). Regulation and role of fungal secondary metabolites. Annu. Rev. Genet. 50, 371–392. doi: 10.1146/annurev-genet-120215-035203
Mao, D., Okada, B. K., Wu, Y., Xu, F., and Seyedsayamdost, M. R. (2018). Recent advances in activating silent biosynthetic gene clusters in bacteria. Curr. Opin. Microbiol. 45, 156–163. doi: 10.1016/j.mib.2018.05.001
Masunaka, A., Ohtani, K., Peever, T. L., Timmer, L. W., Tsuge, T., Yamamoto, M., et al. (2005). An isolate of Alternaria alternata that is pathogenic to both tangerines and rough lemon and produces two host-selective toxins, ACT- and ACR-toxins. Phytopathology 95, 241–247. doi: 10.1094/PHYTO-95-0241
Meena, M., Gupta, S. K., Swapnil, P., Zehra, A., Dubey, M. K., and Upadhyay, R. S. (2017). Alternaria toxins: potential virulence factors and genes related to pathogenesis. Front. Microbiol. 8:1451. doi: 10.3389/fmicb.2017.01451
Miyamoto, Y., Ishii, Y., Honda, A., Masunaka, A., Tsuge, T., Yamamoto, M., et al. (2009). Function of genes encoding acyl-CoA synthetase and enoyl-CoA hydratase for host-selective ACT-toxin biosynthesis in the tangerine pathotype of Alternaria alternata. Phytopathology 99, 369–377. doi: 10.1094/PHYTO-99-4-0369
Miyamoto, Y., Masunaka, A., Tsuge, T., Yamamoto, M., Ohtani, K., Fukumoto, T., et al. (2008). Functional analysis of a multicopy host-selective ACT-Toxin biosynthesis gene in the Tangerine pathotype of Alternaria alternata using RNA silencing. Mol. Plant Microbe Interact. 21, 1591–1599. doi: 10.1094/MPMI-21-12-1591
Miyamoto, Y., Masunaka, A., Tsuge, T., Yamamoto, M., Ohtani, K., Fukumoto, T., et al. (2010). ACTTS3 encoding a polyketide synthase is essential for the biosynthesis of ACT-Toxin and pathogenicity in the tangerine pathotype of Alternaria alternata. Mol. Plant Microbe Interact. 23, 406–414. doi: 10.1094/MPMI-23-4-0406
Möller, M., Habig, M., Freitag, M., and Stukenbrock, E. H. (2018). Extraordinary genome instability and widespread chromosome rearrangements during vegetative growth. Genetics 210, 517–529. doi: 10.1534/genetics.118.301050
Nakatsuka, S., Feng, B., Goto, T., Tsuge, T., and Nishimura, S. (1990). Biosynthetic origin of (8R,9S)-9,10-epoxy-8-hydroxy-9-methyl-deca-(2E,4Z,6E)-trienoic acid, a precursor of AK-toxins produced by Alternaria alternata. Phytochemistry 29, 1529–1531. doi: 10.1016/0031-9422(90)80114-V
Nakatsuka, S. i, Ueda, K., Goto, T., Yamamoto, M., Nishimura, S., and Kohmoto, K. (1986). Structure of AF-toxin II, one of the host-specific toxins produced by Alternaria alternata strawberry pathotype. Tetrahedron Lett. 27, 2753–2756. doi: 10.1016/S0040-4039(00)84635-3
Niehaus, E. M., Janevska, S., Von Bargen, K. W., Sieber, C. M. K., Harrer, H., Humpf, H. U., et al. (2014). Apicidin F: characterization and genetic manipulation of a new secondary metabolite gene cluster in the rice pathogen Fusarium fujikuroi. PLoS One 9:e103336. doi: 10.1371/journal.pone.0103336
Niehaus, E.-M., Münsterkötter, M., Proctor, R. H., Brown, D. W., Sharon, A., Idan, Y., et al. (2016). Comparative “omics” of the Fusarium fujikuroi species complex highlights differences in genetic potential and metabolite synthesis. Genome Biol. Evol. 8, 3574–3599. doi: 10.1093/gbe/evw259
O’Connell, R. J., Thon, M. R., Hacquard, S., Amyotte, S. G., Kleemann, J., Torres, M. F., et al. (2012). Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 44, 1060–1065. doi: 10.1038/ng.2372
Ohno, S. (1970). The enormous diversity in genome sizes of fish as a reflection of nature’s extensive experiments with gene duplication. Trans. Am. Fish. Soc. 99, 120–130. doi: 10.1577/1548-8659197099<120:TEDIGS<2.0.CO;2
Park, J. S., Lee, K. R., Kim, J. C., Lim, S. H., Seo, J. A., and Lee, Y. W. (1999). A hemorrhagic factor (apicidin) produced by toxic Fusarium isolates from soybean seeds. Appl. Environ. Microbiol. 65, 126–130. doi: 10.1128/aem.65.1.126-130.1999
Park, P., and Ikeda, K. I. (2008). Ultrastructural analysis of responses of host and fungal cells during plant infection. J. Gen. Plant Pathol. 74, 2–14. doi: 10.1007/s10327-007-0042-8
Peever, T. L., Su, G., Carpenter-Boggs, L., and Timmer, L. W. (2004). Molecular systematics of citrus-associated Alternaria species. Mycologia 96, 119–134. doi: 10.1080/15572536.2005.11833002
Pitkin, J. W., Panaccione, D. G., and Walton, J. D. (1996). A putative cyclic peptide efflux pump encoded by the TOXA gene of the plant-pathogenic fungus Cochliobolus carbonum. Microbiology 142, 1557–1565. doi: 10.1099/13500872-142-6-1557
Plaumann, P. L., Schmidpeter, J., Dahl, M., Taher, L., and Koch, C. (2018). A dispensable chromosome is required for virulence in the hemibiotrophic plant pathogen Colletotrichum higginsianum. Front. Microbiol. 9:1005. doi: 10.3389/fmicb.2018.01005
Quin, M. B., Flynn, C. M., and Schmidt-Dannert, C. (2014). Traversing the fungal terpenome. Nat. Prod. Rep. 31, 1449–1473. doi: 10.1039/c4np00075g
Raffaele, S., Farrer, R. A., Cano, L. M., Studholme, D. J., MacLean, D., Thines, M., et al. (2010). Genome evolution following host jumps in the irish potato famine pathogen lineage. Science 330, 1540–1543. doi: 10.1126/science.1193070
Rajarammohan, S., Paritosh, K., Pental, D., and Kaur, J. (2019). Comparative genomics of Alternaria species provides insights into the pathogenic lifestyle of Alternaria brassicae – a pathogen of the Brassicaceae family. BMC Genomics 20:1036. doi: 10.1186/s12864-019-6414-6
Ray, L., and Moore, B. S. (2016). Recent advances in the biosynthesis of unusual polyketide synthase substrates. Nat. Prod. Rep. 33, 150–161. doi: 10.1039/c5np00112a
Rep, M., Van Der Does, H. C., Meijer, M., Van Wijk, R., Houterman, P. M., Dekker, H. L., et al. (2004). A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol. Microbiol. 53, 1373–1383. doi: 10.1111/j.1365-2958.2004.04177.x
Rouxel, T., Grandaubert, J., Hane, J. K., Hoede, C., Van De Wouw, A. P., Couloux, A., et al. (2011). Effector diversification within compartments of the Leptosphaeria maculans genome affected by repeat-induced point mutations. Nat. Commun. 2:202. doi: 10.1038/ncomms1189
Ruswandi, S., Kitani, K., Akimitsu, K., Tsuge, T., Shiraishi, T., and Yamamoto, M. (2005). Structural analysis of cosmid clone pcAFT-2 carrying AFT10-1 encoding an acyl-CoA dehydrogenase involved in AF-toxin production in the strawberry pathotype of Alternaria alternata. J. Gen. Plant Pathol. 71, 107–116. doi: 10.1007/s10327-004-0170-3
Schmidt, S. M., Houterman, P. M., Schreiver, I., Ma, L., Amyotte, S., Chellappan, B., et al. (2013). MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genomics 14:119. doi: 10.1186/1471-2164-14-119
Scott-Craig, J. S., Panaccione, D. G., Pocard, J. A., and Walton, J. D. (1992). The cyclic peptide synthetase catalyzing HC-toxin production in the filamentous fungus Cochliobolus carbonum is encoded by a 15.7-kilobase open reading frame. J. Biol. Chem. 267, 26044–26049. doi: 10.1016/S0021-9258(18)35714-4
Selker, E. U., Cambareri, E. B., Jensen, B. C., and Haack, K. R. (1987). Rearrangement of duplicated DNA in specialized cells of Neurospora. Cell 51, 741–752. doi: 10.1016/0092-8674(87)90097-3
Selker, E. U., and Garrett, P. W. (1988). DNA sequence duplications trigger gene inactivation in Neurospora crassa. Proc. Natl. Acad. Sci. U.S.A. 85, 6870–6874. doi: 10.1073/pnas.85.18.6870
Shahi, S., Beerens, B., Bosch, M., Linmans, J., and Rep, M. (2016). Nuclear dynamics and genetic rearrangement in heterokaryotic colonies of Fusarium oxysporum. Fungal Genet. Biol. 91, 20–31. doi: 10.1016/j.fgb.2016.03.003
Shimomura, N., Otani, H., Tabira, H., Kodama, M., and Kohmoto, K. (1991). Two primary action sites for AM-toxin produced by Alternaria alternata apple pathotype and their pathological significance. Jpn. J. Phytopathol. 57, 247–255. doi: 10.3186/jjphytopath.57.247
Singh, S. B., Zink, D. L., Liesch, J. M., Dombrowski, A. W., Darkin-Rattray, S. J., Schmatz, D. M., et al. (2001). Structure, histone deacetylase, and antiprotozoal activities of apicidins B and C, congeners of apicidin with proline and valine substitutions. Org. Lett. 3, 2815–2818. doi: 10.1021/ol016240g
Singh, S. B., Zink, D. L., Liesch, J. M., Mosley, R. T., Dombrowski, A. W., Bills, G. F., et al. (2002). Structure and chemistry of apicidins, a class of novel cyclic tetrapeptides without a terminal alpha-keto epoxide as inhibitors of histone deacetylase with potent antiprotozoal activities. J. Org. Chem. 67, 815–825.
Singh, S. B., Zink, D. L., Polishook, J. D., Dombrowski, A. W., Darkin-Rattray, S. J., Schmatz, D. M., et al. (1996). Apicidins: novel cyclic tetrapeptides as coccidiostats and antimalarial agents from Fusarium pallidoroseum. Tetrahedron Lett. 37, 8077–8080. doi: 10.1016/0040-4039(96)01844-8
Sørensen, J. L., Sondergaard, T. E., Covarelli, L., Fuertes, P. R., Hansen, F. T., Frandsen, R. J. N., et al. (2014). Identification of the biosynthetic gene clusters for the lipopeptides fusaristatin A and W493 B in Fusarium graminearum and F. pseudograminearum. J. Nat. Prod. 77, 2619–2625. doi: 10.1021/np500436r
Spassieva, S. D., Markham, J. E., and Hille, J. (2002). The plant disease resistance gene Asc-1 prevents disruption of sphingolipid metabolism during AAL-toxin-induced programmed cell death. Plant J. 32, 561–572. doi: 10.1046/j.1365-313X.2002.01444.x
Stergiopoulos, I., Collemare, J., Mehrabi, R., and De Wit, P. J. G. M. (2013). Phytotoxic secondary metabolites and peptides produced by plant pathogenic Dothideomycete fungi. FEMS Microbiol. Rev. 37, 67–93. doi: 10.1111/j.1574-6976.2012.00349.x
Takaoka, S., Kurata, M., Harimoto, Y., Hatta, R., Yamamoto, M., Akimitsu, K., et al. (2014). Complex regulation of secondary metabolism controlling pathogenicity in the phytopathogenic fungus Alternaria alternata. New Phytol. 202, 1297–1309. doi: 10.1111/nph.12754
Tanaka, A., and Tsuge, T. (2000). Structural and functional complexity of the genomic region controlling AK-toxin biosynthesis and pathogenicity in the Japanese pear pathotype of Alternaria alternata. Mol. Plant Microbe Interact. 13, 975–986. doi: 10.1094/MPMI.2000.13.9.975
Taylor, J. S., and Raes, J. (2004). Duplication and divergence: the evolution of new genes and old ideas. Annu. Rev. Genet. 38, 615–643. doi: 10.1146/annurev.genet.38.072902.092831
Testa, A., Oliver, R., and Hane, J. (2015). Overview of genomic and bioinformatic resources for Zymoseptoria tritici. Fungal Genet. Biol. 79, 13–16. doi: 10.1016/j.fgb.2015.04.011
Thomma, B. P. H. J., Seidl, M. F., Shi-Kunne, X., Cook, D. E., Bolton, M. D., van Kan, J. A. L., et al. (2016). Mind the gap; seven reasons to close fragmented genome assemblies. Fungal Genet. Biol. 90, 24–30. doi: 10.1016/j.fgb.2015.08.010
Timmer, L. W., Peever, T. L., Solel, Z., and Akimitsu, K. (2003). Alternaria diseases of citrus – Novel pathosystems. Phytopathol. Mediterr. 42, 99–112. doi: 10.14601/Phytopathol_Mediterr-1710
Tsuge, T. (2019). Exploring the origin of crop pathogens: host-specific toxin-producing pathogens as a case study. J. Gen. Plant Pathol. 85, 458–462. doi: 10.1007/s10327-019-00874-6
Tsuge, T., Harimoto, Y., Akimitsu, K., Ohtani, K., Kodama, M., Akagi, Y., et al. (2013). Host-selective toxins produced by the plant pathogenic fungus Alternaria alternata. FEMS Microbiol. Rev. 37, 44–66. doi: 10.1111/j.1574-6976.2012.00350.x
Ueno, T., Nakashima, T., Hayashi, Y., and Fukami, H. (1975). Structures of AM-Toxin I and II, host specific phytotoxic metabolites produced by Alternaria Mali. Agric. Biol. Chem. 39, 1115–1122. doi: 10.1271/bbb1961.39.1115
van Dam, P., Fokkens, L., Ayukawa, Y., van der Gragt, M., Ter Horst, A., Brankovics, B., et al. (2017). A mobile pathogenicity chromosome in Fusarium oxysporum for infection of multiple cucurbit species. Sci. Rep. 7:9042. doi: 10.1038/s41598-017-07995-y
van der Does, H. C., Fokkens, L., Yang, A., Schmidt, S. M., Langereis, L., Lukasiewicz, J. M., et al. (2016). Transcription factors encoded on core and accessory chromosomes of Fusarium oxysporum induce expression of effector genes. PLoS Genet. 12:e1006401. doi: 10.1371/journal.pgen.1006401
Vanheule, A., Audenaert, K., Warris, S., van de Geest, H., Schijlen, E., Höfte, M., et al. (2016). Living apart together: crosstalk between the core and supernumerary genomes in a fungal plant pathogen. BMC Genomics 17:670. doi: 10.1186/s12864-016-2941-6
Villani, A., Proctor, R. H., Kim, H. S., Brown, D. W., Logrieco, A. F., Amatulli, M. T., et al. (2019). Variation in secondary metabolite production potential in the Fusarium incarnatum-equiseti species complex revealed by comparative analysis of 13 genomes. BMC Genomics 20:314. doi: 10.1186/s12864-019-5567-7
Vlaardingerbroek, I., Beerens, B., Rose, L., Fokkens, L., Cornelissen, B. J. C., and Rep, M. (2016a). Exchange of core chromosomes and horizontal transfer of lineage-specific chromosomes in Fusarium oxysporum. Environ. Microbiol. 18, 3702–3713. doi: 10.1111/1462-2920.13281
Vlaardingerbroek, I., Beerens, B., Schmidt, S. M., Cornelissen, B. J. C., and Rep, M. (2016b). Dispensable chromosomes in Fusarium oxysporum f. sp. lycopersici. Mol. Plant Pathol. 17, 1455–1466. doi: 10.1111/mpp.12440
Vogan, A. A., Ament-Velásquez, S. L., Bastiaans, E., Wallerman, O., Saupe, S. J., Suh, A., et al. (2020). The enterprise: a massive transposon carrying Spok meiotic drive genes. bioRxiv [Preprint]. doi: 10.1101/2020.03.25.007153
Von Bargen, K. W., Niehaus, E. M., Bergander, K., Brun, R., Tudzynski, B., and Humpf, H. U. (2013). Structure elucidation and antimalarial activity of Apicidin F: an apicidin-like compound produced by Fusarium fujikuroi. J. Nat. Prod. 76, 2136–2140. doi: 10.1021/np4006053
Walker, P. D., Weir, A. N. M., Willis, C. L., and Crump, M. P. (2020). Polyketide β-branching: diversity, mechanism and selectivity. Nat. Prod. Rep. doi: 10.1039/d0np00045k
Wang, M., Fu, H., Shen, X., Ruan, R., Rokas, A., and Li, H. (2019). Genomic features and evolution of the conditionally dispensable chromosome in the tangerine pathotype of Alternaria alternata. Mol. Plant Pathol. 20, 1425–1438. doi: 10.1111/mpp.12848
Wang, M., Sun, X., Yu, D., Xu, J., Chung, K., and Li, H. (2016). Genomic and transcriptomic analyses of the tangerine pathotype of Alternaria alternata in response to oxidative stress. Sci. Rep. 6:32437. doi: 10.1038/srep32437
Wawrzyn, G. T., Quin, M. B., Choudhary, S., López-Gallego, F., and Schmidt-Dannert, C. (2012). Draft genome of omphalotus olearius provides a predictive framework for sesquiterpenoid natural product biosynthesis in basidiomycota. Chem. Biol. 19, 772–783. doi: 10.1016/j.chembiol.2012.05.012
Wiemann, P., Sieber, C. M. K., von Bargen, K. W., Studt, L., Niehaus, E.-M., Espino, J. J., et al. (2013). Deciphering the cryptic genome: genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathog. 9:e1003475. doi: 10.1371/journal.ppat.1003475
Witte, T., Harris, L., Nguyen, H., Hermans, A., Johnston, A., Sproule, A., et al. (2021). Apicidin biosynthesis is linked to accessory chromosomes in Fusarium poae isolates. BMC Genomics (in press). doi: 10.21203/rs.3.rs-116075/v1
Wolpert, T. J., Dunkle, L. D., and Ciuffetti, L. M. (2002). Host-selective toxins and avirulence determinants: what’s in a name? Annu. Rev. Phytopathol. 40, 251–285. doi: 10.1146/annurev.phyto.40.011402.114210
Woudenberg, J. H. C., Seidl, M. F., Groenewald, J. Z., de Vries, M., Stielow, J. B., Thomma, B. P. H. J., et al. (2015). Alternaria section Alternaria: species, formae speciales or pathotypes? Stud. Mycol. 82, 1–21. doi: 10.1016/j.simyco.2015.07.001
Xie, X., Meehan, M. J., Xu, W., Dorrestein, P. C., and Tang, Y. (2009). Acyltransferase mediated polyketide release from a fungal megasynthase. J. Am. Chem. Soc. 131, 8388–8389. doi: 10.1021/ja903203g
Yang, H., Yu, H., and Ma, L.-J. (2020). Accessory chromosomes in Fusarium oxysporum. Phytopathology 110, 1488–1496. doi: 10.1094/PHYTO-03-20-0069-IA
Yi, H., Bojja, R. S., Fu, J., and Du, L. (2005). Direct evidence for the function of FUM13 in 3-ketoreduction of mycotoxin fumonisins in Fusarium verticillioides. J. Agric. Food Chem. 53, 5456–5460. doi: 10.1021/jf050062e
Yue, Q., Chen, L., Zhang, X., Li, K., Sun, J., Liu, X., et al. (2015). Evolution of chemical diversity in echinocandin lipopeptide antifungal metabolites. Eukaryot. Cell 14, 698–718. doi: 10.1128/EC.00076-15
Zhang, C. X., Tian, Y., and Cong, P. H. (2015). Proteome analysis of pathogen-responsive proteins from apple leaves induced by the Alternaria blotch Alternaria alternata. PLoS One 10:e0122233. doi: 10.1371/journal.pone.0122233
Keywords: accessory chromosomes, secondary metabolites, biosynthetic gene clusters, Alternaria host specific toxins, metabolomics, RIP, Alternaria pathotypes, Fusarium
Citation: Witte TE, Villeneuve N, Boddy CN and Overy DP (2021) Accessory Chromosome-Acquired Secondary Metabolism in Plant Pathogenic Fungi: The Evolution of Biotrophs Into Host-Specific Pathogens. Front. Microbiol. 12:664276. doi: 10.3389/fmicb.2021.664276
Received: 04 February 2021; Accepted: 09 March 2021;
Published: 23 April 2021.
Edited by:
Rosa Durán-Patrón, University of Cádiz, SpainReviewed by:
Slavica Janevska, University of Amsterdam, NetherlandsCopyright © 2021 Witte, Villeneuve, Boddy and Overy. Boddy and Her Majesty the Queen in Right of Canada, as represented by the Minister of Agriculture and Agri-Food Canada for the contribution of Witte, Villeneuve and Overy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher N. Boddy, Y2JvZGR5QHVvdHRhd2EuY2E=; David P. Overy, ZGF2aWQub3ZlcnlAY2FuYWRhLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.