 Bin-Bin Chi
Bin-Bin Chi Ya-Nan Lu1,2
Ya-Nan Lu1,2- 1College of Chemistry and Bioengineering, Guilin University of Technology, Guilin, China
- 2Guangxi Key Laboratory of Electrochemical and Magneto-Chemical Functional Materials, College of Chemistry and Bioengineering, Guilin University of Technology, Guilin, China
- 3Hunan Agricultural Product Processing Institute, Hunan Academy of Agricultural Sciences, Changsha, China
Heavy metal pollution is a global knotty problem and fungi hold promising potential for the remediation of wastewater containing heavy metals. Here, a new highly chromium-tolerance species, Penicillium janthinellum P1, is investigated. The genome of P1 was sequenced and assembled into 30 Mb genome size containing 10,955 predicted protein-coding genes with a GC content of 46.16% through an integrated method of Illumina short-read sequencing and single-molecule real-time Pacific Biosciences sequencing platforms. Through a phylogenetic analysis with model species of fungi, the evolutionary divergence time of Penicillium janthinellum P1 and Penicillium oxalicum 114-2 was estimated to be 74 MYA. 33 secondary metabolism gene clusters were identified via antiSMASH software, mainly including non-ribosomal peptide synthase genes and T1 polyketide synthase genes. 525 genes were annotated to encode enzymes that act on carbohydrates, involving 101 glucose-degrading enzymes and 24 polysaccharide synthase. By whole-genome sequence analysis, large numbers of metal resistance genes were found in strain P1. Especially ABC transporter and Superoxide dismutase ensure that the P1 fungus can survive in a chromium-polluted environment. ChrA and ChrR were also identified as key genes for chromium resistance. Analysis of their genetic loci revealed that the specific coding-gene arrangement may account for the fungus’s chromium resistance. Genetic information and comparative analysis of Penicillium janthinellum are valuable for further understanding the mechanism of high resistance to heavy metal chromium, and gene loci analysis provides a new perspective for identifying chromium-resistant strains.
Introduction
The discharge of wastes from increased industrialization and human activities has resulted in negative impacts on the environment (Ayres, 1992). Currently, heavy metals have become a severe environmental pollutant and fungi hold promising potential for the remediation of sewage containing heavy metals (Leitao, 2009; Xu et al., 2020). Compared to physicochemical treatment methods, bioremediation has unique advantages, including adequate availability of materials, low cost, and no secondary pollution (Wingenfelder et al., 2005). Bioremediation continues to attract significant attention as a workable solution (Yin et al., 2019; Pathak et al., 2021). The previous studies showed that fungi have strong heavy metal tolerance (Liaquat et al., 2020) and can remove heavy metals through mechanisms such as cell wall adsorption, extracellular immobilization, intracellular bioaccumulation, and intracellular reduction, etc. (Xu et al., 2020). Filamentous fungi usually accumulate metal ions into their mycelia and spores through a mechanism involving the cell wall of the fungus. This mechanism also plays an important role in the existence and performance of the fungus, as well as energy absorption and valence conversion (Dusengemungu et al., 2020). However, we have not yet fully revealed the mechanism of how these fungi are resistant to heavy metal. Therefore, a finished and accurate reference genome will lay a solid foundation for understanding the genomic evolution and identifying functional genes for heavy-metal resistance features.
In 1996, the genome of the first eukaryotic Saccharomyces cerevisiae was sequenced and genetically annotated (Nies et al., 1990). As progress advances, Saccharomyces cerevisiae, a fungal model organism, has certain limitations (Mattiazzi et al., 2012). In 2000, the United States launched the Fungal Genome Initiative which aimed to promote genome-wide sequencing of important fungal representative species in medicine, agriculture, and industry (Zhou J. F. et al., 2008). Until November 2019, approximately 5,200 fungi genomes had been sequenced throughout the world, with a total of 351 fungal genomic sequences deposited in the NCBI database. A large number of genes in fungi related to characteristic properties, including metal resistance, acid resistance, thermo-resistance, and lignocellulose degradation, etc, have been dissected by genome sequencing and annotation. In recent years, there are an increasing number of studies on the sequencing of the whole genome of Penicillium species. Eberhard Karls Universität Tübingen reported the draft genome sequence of Penicillium islandicum WF-38-12 (Schafhauser et al., 2015). The Institute of Bioengineering of the Russian Academy of Sciences completed the mitochondrial genome sequencing of Penicillium ShG4C acid-resistant strains (Mardanov et al., 2016). The Zhengzhou Tobacco Research Institute depicted the draft genome sequence of Penicillium verruculosum strain TS63-9 (Hu et al., 2016). Utrecht University drafted the genome sequence of Penicillium subrubescens FBCC1632/CBS132785 (Peng et al., 2017). But among those sequenced Penicillium strains, none features a high resistance capability to heavy metals.
ChrA and ChrR genes are well known as the anti-chromium-related genes. ChrA gene is the most widely studied gene encoding Cr efflux protein. In previous studies, researchers believed that ChrA protein, as a membrane protein, could pump out intracellular Cr through proton propulsion to improve Cr tolerance of strains (Nakatsu et al., 2013; Gu et al., 2020). ChrR reduces quinones by simultaneous two-electron transfer, avoiding the formation of highly reactive semiquinone intermediates and producing quinols that promote tolerance of H2O2 (Gonzalez et al., 2005). Enhancing the activity of ChrR in a chromate-remediating bacterial strain may not only increase the rate of chromate transformation (Baldiris et al., 2018), but it may also augment the capacity of these cells to withstand the unavoidable production of H2O2 that accompanies chromate reduction.
Due to the ecological niche, the metabolic pathways and genetic background of marine microorganisms are far more fierce and versatile than those of terrestrial microorganisms (Dash et al., 2013; Lopez-Hernandez and Cortes, 2019). Researches on marine microbial genomes may reveal new metabolic mechanisms.
According to our previous studies, the marine-derived Penicillium janthinellum P1, isolated from sediment samples by our laboratory, shows the high metal-tolerant ability to a variety of heavy metals [survive at below 400 mg/L Cr (VI)], acid resistance, and strong biosorption capacity (Zhou Y. et al., 2008; Chen et al., 2014, 2019; Lu et al., 2017). To gain more genetic information on P. janthinellum to determine the critical genes involved in the metal resistance, we here sequenced and assembled the genome of P1, presenting a high-quality genome of the P. janthinellum P1 using a combined method of sequencing technologies. Then, carbohydrate-active enzyme (CAZyme) genes and secondary metabolism gene clusters were predicted in the assembled genome. Comparative genome analysis of strain P1 with other 24 fungi strains revealed significant genetic variance. Putative genes with respect to chromate resistance such as metal ion transport, metal absorption, and efflux were addressed. The results give a first genetic demonstration of the possible factors that may be responsible for the high levels of chromate resistance in strain P1.
Materials and Methods
Genome Sequencing
Whole-genome sequencing was completed by BGI Genomics Co., Ltd. The genomic DNA was extracted from single spore mycelia of the Penicillium janthinellum P1. Extracted genomic DNA (Grijseels et al., 2016) then was sequenced using high-throughput Illumina Hiseq 2500 sequencing platform for mate paired-end sequencing together with the PacBio RSII single-molecule real-time sequencing platform, respectively.
Data Processing
The Illumina raw reads were evaluated using FastQC v0.11.61, and processed by utilizing the NGS QC Toolkit v2.3.3 (Patel and Jain, 2012) with Phred 20 as the cutoff to delete the low-quality reads. The resulting reads with linker and adaptors, with >10% unknown bases (N) were filtered out for further analysis. FastUniq v1.1 (Xu et al., 2012) was employed to remove PCR repeats and then the high quality paired-end reads were obtained for following assembly. An in-house bash5tools.py command of SMRT Analysis was implemented to filter low-quality reads and obtain clean reads of PacBio sequencing.
Genomic Assembly
The clean reads of the PacBio sequencing were de novo assembled to obtain the original assembly contigs by FALCON, then were optimized and upgraded by FinisherSC (Lam et al., 2015) using the clean reads of the PacBio sequencing. Bowtie2 was conducted to map the clean reads of the Illumina sequence back to the PacBio assembly contigs to obtain the sam file. Use Samtools to convert sam files to bam files, and sort and index them. The upgraded assembly contigs and indexed bam file were used as the input file of Pilon to obtain the final assembly. The quality of the assembly was assessed by QUAST (Niegowski and Eshaghi, 2007).
Gene Prediction
Protein-coding genes in P1 genome were predicted using four softwares including GeneWise2 (Madeira et al., 2019), Augustus3 (Keller et al., 2011), GeneMark-ES4 (Borodovsky and Lomsadze, 2011), and MAKER5 (Cantarel et al., 2008). In this paper, Augustus was trained on gene models for P1 by combining the Coprinus gene model and RNA-Seq data of P1. GeneWise and GeneMark-ES with default parameters achieved different gene prediction sets, respectively. Finally, all prediction gene results were integrated by MAKER.
Gene Function Annotation
The putative proteins of P1 were aligned against the RefSeq non-redundant proteins (NCBI Nr) and the SWISS-PROT Protein Knowledgebase (Swiss-Prot) using BLAST and the HMMER v3.3 (Klingenberg and Marugan-Lobon, 2013) search against the Pfam protein families database (Pfam6) (El-Gebali et al., 2019; Mistry et al., 2021) to assign general protein function profiles. The predicted genes were then aligned against the Conserved Domain Database (CDD7) using RPS-BLAST. Metabolic pathways of the P1 genome were classified based on the KEGG Ortholog results against the Kyoto Encyclopedia of Genes and Genomes (KEGG8). KOG enrichment analysis was assigned based on the best match of alignments against The Clusters of Orthologous Groups of proteins (COG9) through BLASTP. RepeatMasker v4.0.7 and RepeatModeler v1.0.5 software were used to filtrate and annotate the retroelements, DNA transposons, simple repeats and low-complexity DNA sequences. Simple sequence repeats (SSRs) were annotated by MISA. The tRNA and anticodons of the P1 genome were predicted by tRNAscan-SE v1.23 (Schattner et al., 2005). rRNA genes were predicted by Barrnap v0.9. Other non-coding RNAs such as sRNA and snRNA were identified using Rfam v9.1. AntiSMASH was used to predict the putative secondary metabolism gene clusters by using genome files and annotation files.
Phylogenetic Tree Construction and Gene Family Analysis
24 fungal species related to Ascomycota together with P1 were used for phylogenetic analysis (Supplementary Table 7) (Wang et al., 2019). The protein sequences of 25 fungi were compared by BLASTP (e-value < le-5). Then, OrthoMCL under default parameters was used to analyze the blastp results. Batch multiple sequence alignment of these genes were calculated by MAFFT v7.221 software (Katoh and Standley, 2013), subsequently combined into long sequences for each species. The conserved sites of multiple sequence alignment results were extracted using Gblocks v0.91b (parameters -t = p), the best amino acid replacement model was determined by ProTest v3.2. The phylogenetic tree was constructed using RAxML-8.1.24 (bootstraps 1000) by the input of the final alignment sequence. Three pairs of species’ divergence times were estimated by the web10 and added three fossil calibration points to the molecular clock analysis (Floudas et al., 2012). The most recent common ancestor (MRCA) of Aspergillus oryzae and Aspergillus terreus were diverged at 65 MYA. Gloeophyllum trabeum and Saccharomyces cerevisiae were diverged at 628 MYA, while Trichoderma parareesei and Aspergillus clavatus were diverged at 343 MYA (Chen et al., 2016). The divergence time of other nodes was calculated by r8s v1.80 (PL method, TN algorithm and the smoothing parameter value set to through cross-validation). The tree was idealized in iTOL11. The orthologous gene family expansion was analyzed by CAFE v3.1 based on the ultrametric tree (Han et al., 2013). The OCGs undergone expansion/contraction in P1 were predicted by Swiss-Prot. Genome annotation files and protein sequences of Penicillium janthinellum P1, Penicillium oxalicum 114-2 and Penicillium janthinellum ATCC12 were used to perform collinearity analysis via MCScanX software (Wang et al., 2012). Genome comparison was investigated by Mauve v2.3.113.
CAZyme and Heavy Metal Chromium Resistance Gene Analysis
CAZymes were annotated by dbCAN (covered fraction ratio > 0.3, e-value < 1e-5) and BLASTP against CAZyme database (e-value < 1e-5, maximum hit number set 500). The blastp results are integrated with the HMM results to achieve the final CAZyme annotation. Genes related to heavy metal chromium resistance, which are normally regulated by heavy metal stress, were found and analyzed from the proteins encoded by the P1 genome.
Gene Locus Analysis
Through swiss-prot annotation of the whole protein group and gene annotation file, the locations of target encoding genes on the scaffold were determined. Analyses of other key genes adjacent to the target genes were conducted to try to interpret the characteristics of the strain. Meanwhile, the target gene’s locus of other strains with similar genes was analyzed to observe the diversity of genes.
Results
Statistics
As shown in Table 1, a total of 34,520,396 reads were produced using Illumina sequencing technology, thus yielding 5,178 Mb of raw sequencing data. The results of PacBio Sequel platform were shown in Table 2. The total number of bases measured is about 13 GB.

Table 1. Statistics of Illumina sequencing data.

Table 2. Statistics of PacBio Reads sequencing data.
Genome Sequencing and Assembly
The pre-processed Illumina and Pacbio clean data were assembled using SOAPdenovo and FALCON, respectively. The resulting genome size was approximately 31 Mb, resulting in 1,289 contigs with Contig N50 of 50.4 kbp using SOAPdenovo. FALCON pipeline was used for the assembly of PacBio sequencing data, generating 143 contigs with Contig N50 of 3077.5 kbp (Table 3).
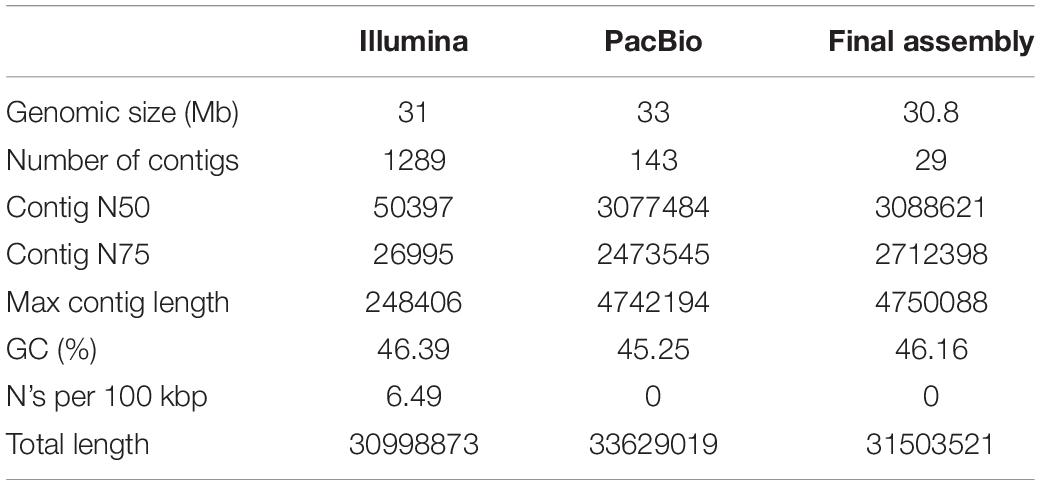
Table 3. The results of the second-generation sequencing, the three- generation sequencing assembly and final Assembly data.
With third-generation data for assembling and second-generation data for evaluation and correction, the de novo genome assembly of P. janthinellum P1 with a total length of 30.8 Mb (Table 3) was yielded. A total of 29 contigs were generated with Contig N50 of 3,088.6 kbp (Table 3). This size was consistent with the estimated genome size of 29–34 Mb for three Penicillium species (Yang et al., 2016; Peng et al., 2020; Zhang et al., 2020).
Gene Prediction
10,955 CDS were identified in strain P1 and the average gene length was 1591 bp using GeneWise, Augustus, GeneMark-ES, and MAKER software for gene prediction.
Genome Annotation
Nr Annotation
Through Nr annotation, 3,737 protein-coding genes were annotated, and the top 20 species were selected as candidate species from the annotated species for the construction of the phylogenetic tree (Supplementary Table 1).
Pfam Annotation
The Pfam database is a collection of a large number of protein families, each of which is represented by multiple sequence alignments and hidden Markov models (HMM). A total of 6,318 protein-coding genes were annotated using the Pfam database.
CDD Annotation
NCBI’s Conservative Domain Database (CDD) is a collection of multiple sequence alignments of ancient domains and full-length proteins, which can provide data on protein retention regions during molecular evolution. Through CDD annotation, a total of 7,618 protein-coding genes were annotated.
KEGG Annotation
KEGG is a database resource for understanding the advanced functions and practicality of biological systems such as cells, organisms, and ecosystems from molecular-level information, especially large-scale molecular data sets generated by genome sequencing and other high-throughput. KEGG is a database with three major categories of comprehensive system information, genomic information and chemical information. A total of 1,341 genes were annotated through the KEGG database, involving 293 metabolic pathways (Supplementary Table 2).
Swiss-Prot Annotation
A total of 7,877 protein-coding genes were predicted in the Swiss-Prot database, including genes with resistance to chromium. The ChrA was annotated in scaffold 14 with function of reducing chromate accumulation, and essential for chromate resistance. The ChrR in scaffold 16 catalyzed the reduction of quinones, and can also reduce toxic chromate to insoluble and less toxic Cr3+ (Cervantes et al., 1990).
KOG/COG Annotation
Functional clustering diagram is shown as below by analysis of KOG: A (RNA processing and modification), J (translation, ribosomal, structure and biogenesis), K (Transcription), O (Posttranslational, modification, protein turnover, chaperones), T (Signal transduction mechanisms), U (Intracellular trafficking, secretion and vesicular transport). These groups are significant enrichment in genes, and are preferentially enriched in transcriptional secretion and transmembrane protein transport functions. The number of genes in the annotated S (Function unknown) and R (General function prediction only) group is enormous, but the current database cannot clarify their biological functions, indicating that there are still many unknown functions worthy of further elucidating in this regard (Figure 1).
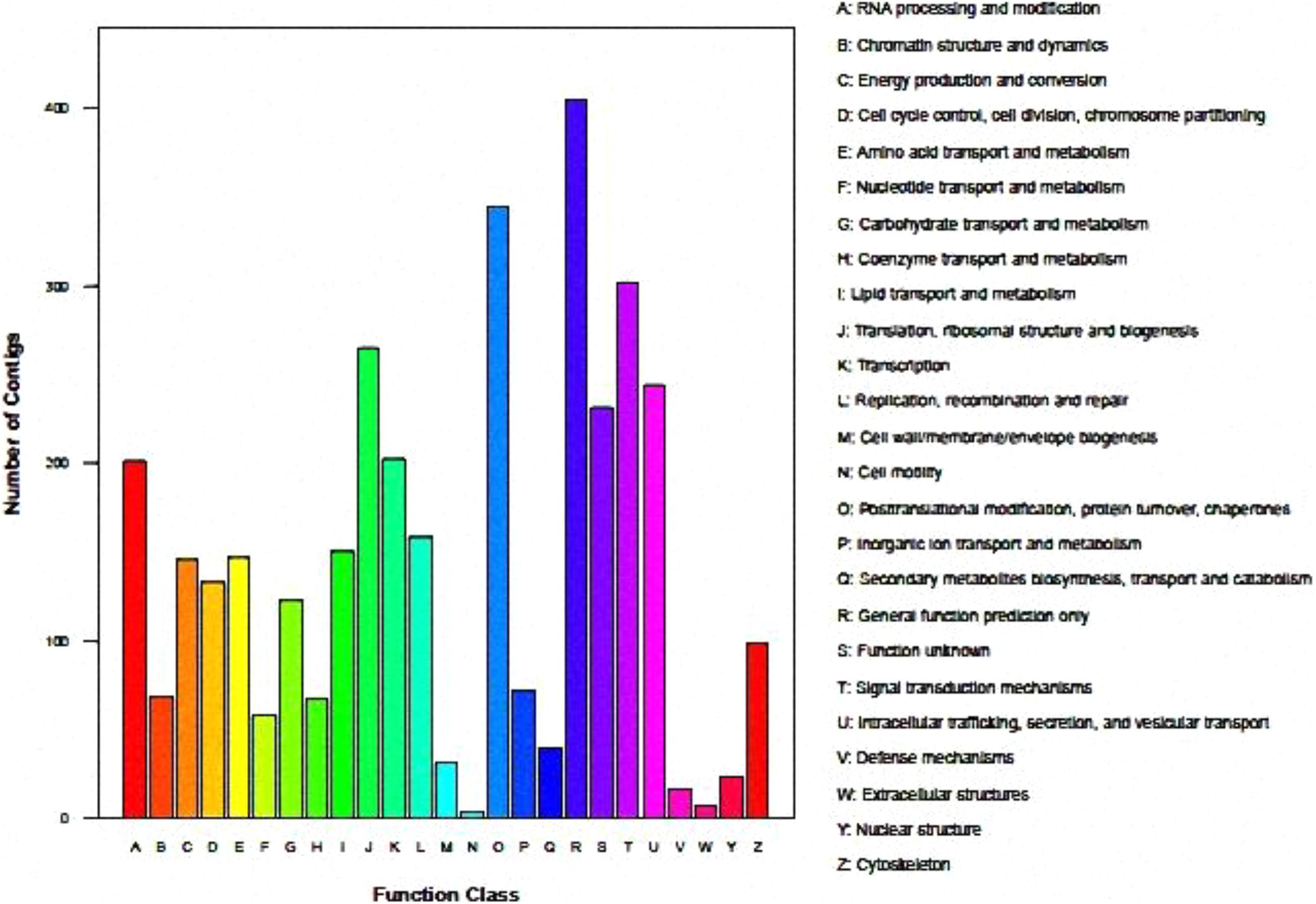
Figure 1. KOG function classification of P1.
Repeated Sequence Annotation
Through repeated sequence analysis, there were about 1.90% of the repeated sequences (Supplementary Table 3). Among DNA transposons, Tc1-IS630-Pogo accounts for the largest proportion, amounting to 0.36% of the genome. Among the retrotransposons, LTR transposons are the majority, accounting for 0.18% of the genome.
Simple Sequence Repeats (SRRs) Annotation
Although simple sequence repeats are usually more likely to occur in non-coding regions, tandem repeats in the coding region of fungal genes cannot be ignored. Research by relevant scholars has shown that SSR occurs in the coding region, which will cause repeated patterns in protein sequences, thus quickly increasing the speed of protein evolution and make fungi adapt to environmental changes faster (Clayton et al., 2017). Therefore, MISA was employed to annotate simple sequence repeats of the coding region of the P1 genome, and the SRR annotation results were shown in Supplementary Table 4.
Non-coding RNA Annotation
Through analyzing the distribution of tRNA genes, codon usage patterns and amino acid composition at the genomic level is an effective method to decipher the biologically important characteristics of intracellular tRNA and the bias of codon and amino acid usage. A total of 274 tRNAs were annotated, of which there were no false tRNAs (Supplementary Table 5). The number of tRNAs recognizing the same amino acid varied. There were six tRNAs over 20, which were tRNA-Val (27) carrying valine, tRNA-Arg (26) carrying arginine, and glutamic acid. TRNA-Glu (25), tRNA-Ile carrying isoleucine (24), tRNA-Phe carrying phenylalanine (22), tRNA-His carrying glutamic acid (21). The frequency of use of tRNAs carrying anticodons was also more scattered, with 39 groups of tRNAs that recognize the same amino acid carrying the same anticodons.
rRNA is the structural and functional core of the ribosome, which has peptidyl transferase activity. rRNA mainly serves as a binding site for tRNA and a variety of protein synthesis factors, and often binds to mRNA in the extension of peptide chains (Supplementary Table 6).
Comparative Analysis of P. janthinellum P1 With Other Strains
Total 19150 Ortholog Cluster Groups (OCGs) were constructed, 8764 of which contained 9776 P1 proteins through OrthoMCL analysis. Approximately 20.2% of predicted proteins in P1 had orthotics in all species, whereas 10.8% of proteins were not orthologous to other fungi, and 19.9% of proteins had at least ten paralogs (Figure 2 and Supplementary Table 8), indicating that the P1 strain shares high genetic similarity with the selected Aspergillus and Penicillium strains.
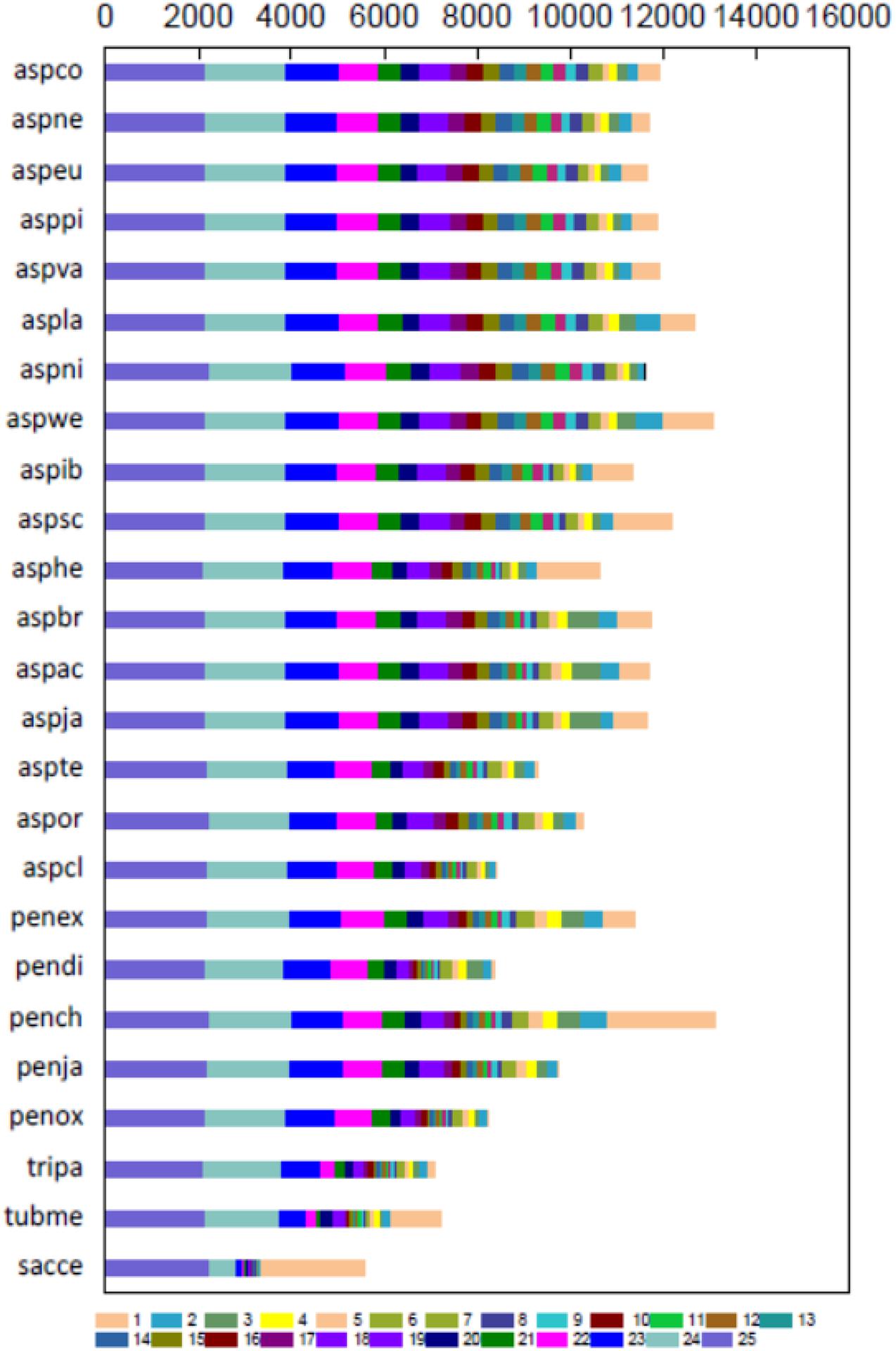
Figure 2. Gene cluster map. Orthologous gene numbers of Penicillium janthinellum P1 with other 24 fungal species. The smaller the number of column 1 of a certain species, the higher the degree of similarity with these 25 species.
The phylogenetic tree was constructed based on 1423 selected single-copy orthologous genes presented in ≥70% of species, revealing the evolutionary history of the P1 strain. The topological structure of the tree was consistent with the taxonomic classification of species (Figure 3). The molecular clock analysis showed that Penicillium oxalicum 114-2 had the closest divergence time, which was estimated to be 74 million years ago (MYA). The long divergence time with other strain may be the isolation effect brought by the living environment (Nayfa et al., 2020).
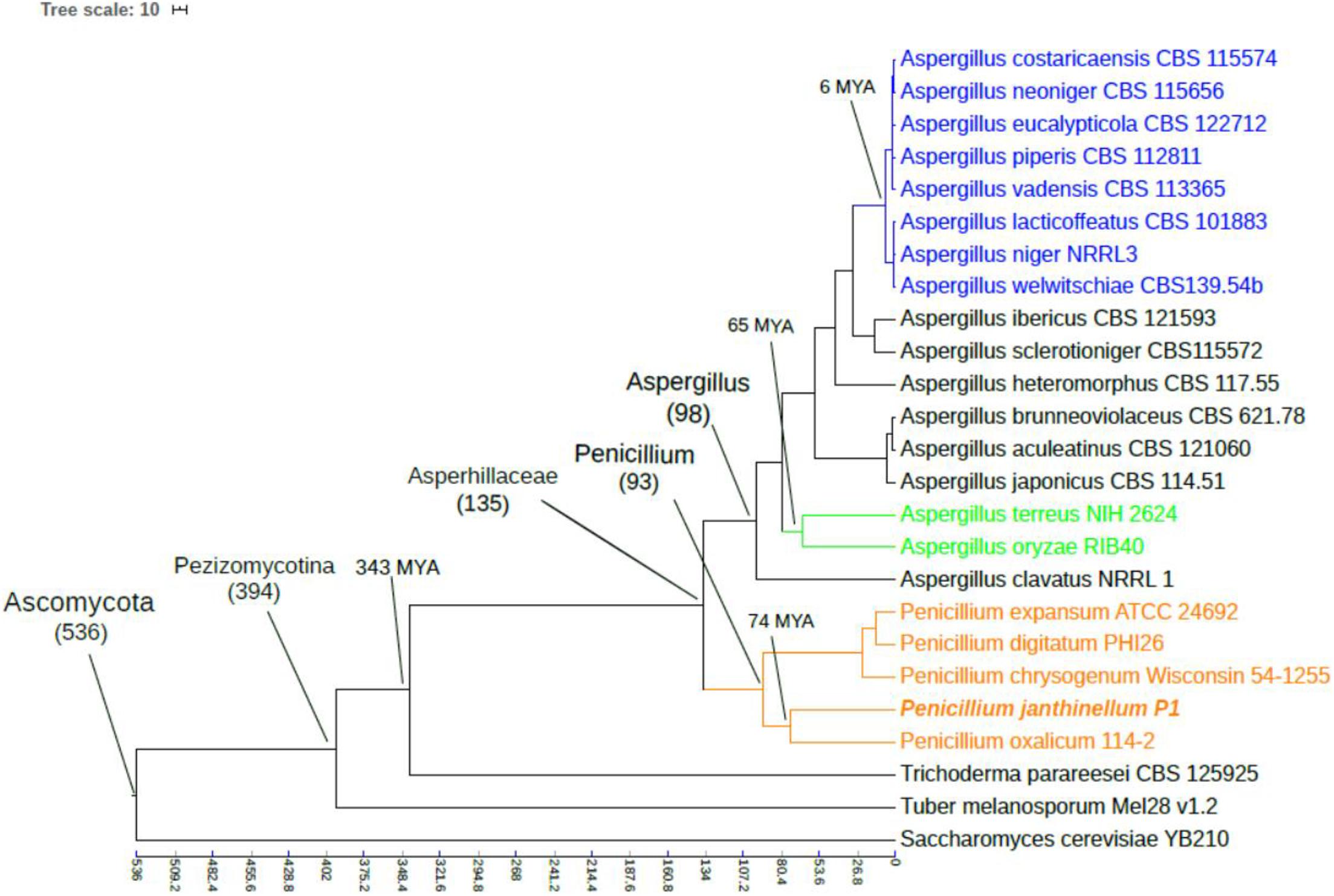
Figure 3. Phylogenetic tree of Penicillium janthinellum P1 with other 24 fungal species. The topology of the phylogenetic tree was constructed by the maximum likelihood method (bootstrap = 1000), and all bootstrap values were 100%. Time scale was shown by MYA (million years ago).
A total of 1183 expansion/contractions family numbers were identified and 102 OCGs were found to have expansion/contraction in P1. After annotation and analysis of those OCGs, it was confirmed that the largest expanded OCG, which encodes Malformin synthetase mlfA, harbors seven genes in P1. The expanded mlfA may indicate that P1 has the potential for biosynthesis of secondary metabolites. Other 17 OCGs including Dynein assembly factor, Aconitate hydratase, Autoinducer 2 sensor kinase/phosphatase, iron transporter, Tol-Pal system protein, oxygen-dependent choline dehydrogenase, high-affinity glucose transporter, L-threonine 3-dehydrogenase, serine/threonine-protein kinase were also expanded in P1. Some enzymes related to respiration have also expanded, which may facilitate strains to resist heavy metal toxicity. Additionally, 306 and 431 syntenic blocks were explored on the basis of the conserved gene order between P1 and Penicillium oxalicum 114-2, P1 and Penicillium janthinellum ATCC, respectively (Supplementary Figure 1). Average protein similarity between the proteome predicted by P1 with Penicillium oxalicum 114-2 and Penicillium janthinellum ATCC was evident (Supplementary Figure 2).
Secondary Metabolism Gene Clusters
A wealth of varied secondary metabolites were found in P1 using the AntiSMASH. A total of 33 secondary metabolism gene clusters were identified. The predicted putative products of 24 secondary metabolism gene clusters were classified into eight different types: seven T1 polyketide synthase (T1pks) gene clusters, seven non-ribosomal peptide synthase (Nrps) gene clusters, two terpene gene clusters, three T1pks-Nrps gene clusters, two fatty acid gene cluster, one Indole-Nrps gene cluster, one saccharide gene cluster, and one Lantipeptide gene cluster; the remaining nine gene clusters synthesized other unknown secondary metabolites (Supplementary Table 12).
The Secretory System and Transporter
Some microorganisms can secrete extracellular polymeric substances (EPS) such as glycoproteins and soluble amino acids which have the effect of complexing or precipitating metal ions (Naveed et al., 2020). The release of EPS by microorganisms needs the aids of secretory systems and transporters, therefore signal recognition particle (SRP) plays a critical role in the targeting of secretory proteins to cellular membranes (Gowda et al., 1998), and Sec secretion system is responsible for the transport of proteins on the plasma membrane (Fagan and Fairweather, 2011). Within P1 genomic information, 22 proteins were predicted to be components of the eukaryotic Sec-SRP secretion systems (Supplementary Table 9).
CAZymes
Carbohydrate and energy metabolism acted as key attribution in response to heavy metal adaptation (Kan et al., 2019). Comparing carbohydrate active enzymes of similar fungi may reveal the anti-heavy metal properties of P1. The CAZy database14 contains a series of enzymatic catalytic modules and sugar binding modules that degrade, modify or create glycosidic bonds. A total of 525 encoding CAZymes were annotated using dbCAN. The putative CAZymes included 92 Auxiliary Activities (AAs), 44 Carbohydrate-Binding Modules (CBMs), 27 Carbohydrate Esterases (CEs), 250 Glycoside Hydrolases (GHs), 104 GlycosylTransferases (GTs), 8 Polysaccharide Lyases (PLs), of which carbohydrate active enzymes such as glycoside hydrolase were most annotated (Supplementary Table 10). Compared with P. janthinellum ATCC 10455 and P. oxalicum 114-2, families of AAs in P1 expanded while families of CBMs contracted. AAs acts as redox enzymes that act in conjunction with CAZymes (Supplementary Table 11). This reveals a high oxidoreductase-coding activity in P1, which may be more conducive to the reduction of toxic heavy metals. Cytochrome-c peroxidase of AA2 (EC 1.11.1.5) and p-benzoquinone reductase (NADPH) of AA6 (EC 1.6.5.6) were identified within the P1 genome which were anti-chromium associated proteins. CBM1 as an important cellulose-binding domain family in cellulose degradation was rare in the P1 fungal genome, inferring that it may be related to marine living environment.
Further analyses revealed that 190 CAZymes were predicted to be sugar metabolism enzymes, involving 68 polysaccharide degrading enzymes, 100 glucose degrading enzymes and 12 polysaccharide synthases. The P1 genome was enriched in glucose degrading enzymes assigned to 10 predicted CAZyme families. This finding was compared to 99 genes in P. janthinellum ATCC 10455 and 54 genes in P. oxalicum 114-2. Compared with the above two strains, larger numbers of genes encoding the important glucose oxidase were classified into AA3 in the P1 genome. This result supports the high sugar metabolism activity of P1, and provides a certain insight into anti-metal activity.
Heavy Metal Chromium Resistance Gene
The maximum Cr (VI) biosorption capacity for living fungus P1 pellets was about 87% at the optimum condition (Chen et al., 2019), demonstrating strong chromium removal ability (Sriharsha et al., 2020). By analyzing the genes annotated in the P1 genome, the genes that may be related to their high resistance to heavy metal chromium are listed below (Table 4). As we can see, cytochrome P450s (CYPs) were heavily annotated in P1. As a multicomponent electron transport chain system, CYPs were critical in degradation, detoxification, and syntheses of life-critical compounds in organisms (Deng et al., 2007). In addition, CYPs were also found in Phanerochaete chrysosporium and the unique P450s had a potential for the production of useful flavonoids (Kasai et al., 2010). Superoxide dismutase can eliminate radicals (Liu et al., 2020), which may be involved in the process of reducing Cr (VI) by fungi.
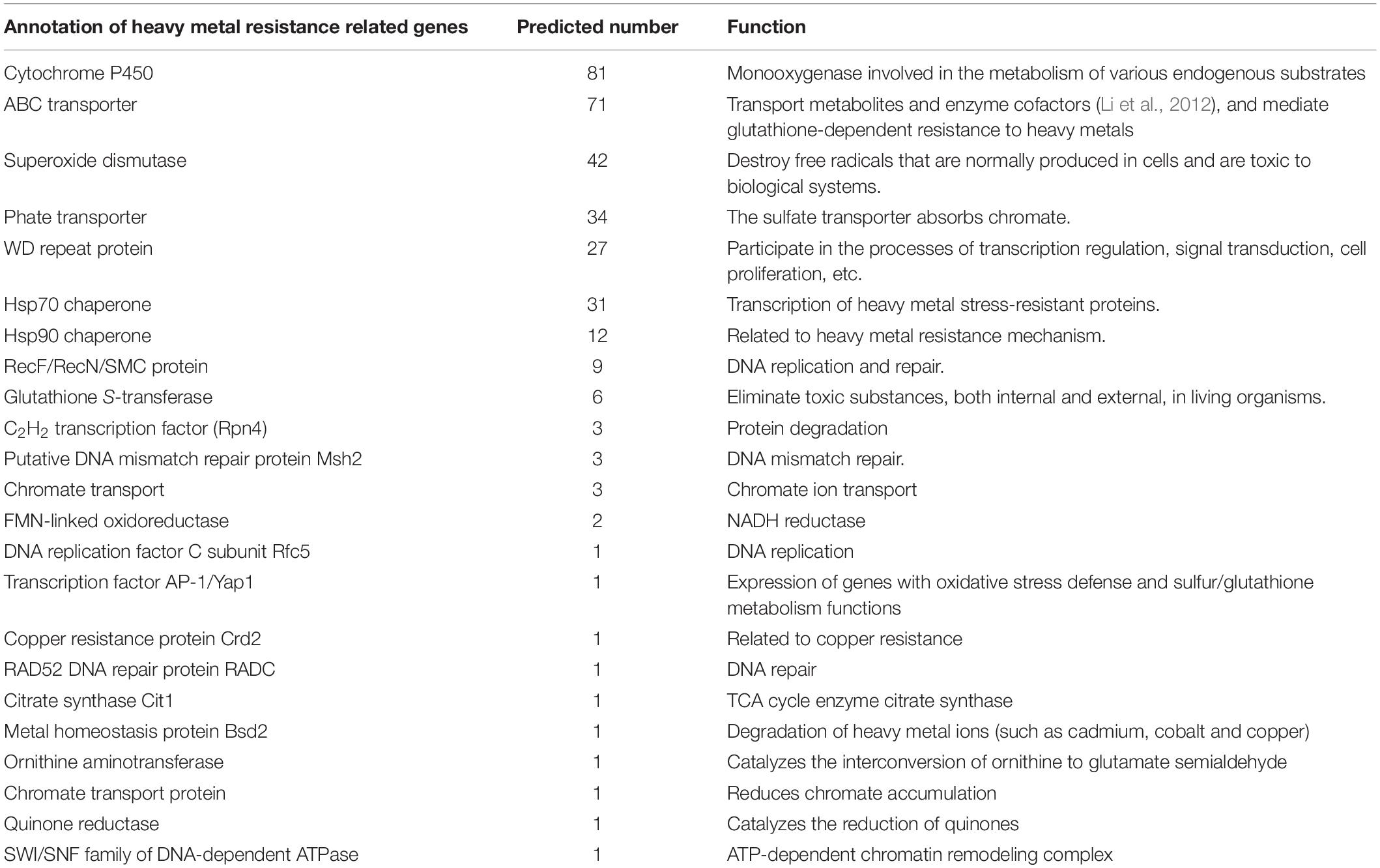
Table 4. Putative genes with heavy metal chromium resistance in P1.
ChrA and ChrR Gene Loci Analysis
The encoding genes flanking to the ChrA and ChrR gene loci were studied in detail, seen in Figure 4. ChrA gene located on scaffold 14 can encode chromate transport protein. OpS5 acted as an oxidoreductase is attributed to secondary metabolite biosynthesis. Its physical distance from ChrA gene is about 5.6 Kb. ChrA gene is also present in other similar fungi, and a number of oxidoreductases are present around it. ChrAs in P. aeruginosa (Cervantes et al., 1990), A. eutrophus (Nies et al., 1990), Shewanella sp. (Aguilar-Barajas et al., 2008) have been studied in detail. These studies examined the resistance of direct homologous microorganisms carrying the ChrA gene to different Cr (VI) concentrations, and found that the ChrA gene alone could not explain the different Cr (VI) resistance of different microorganisms.
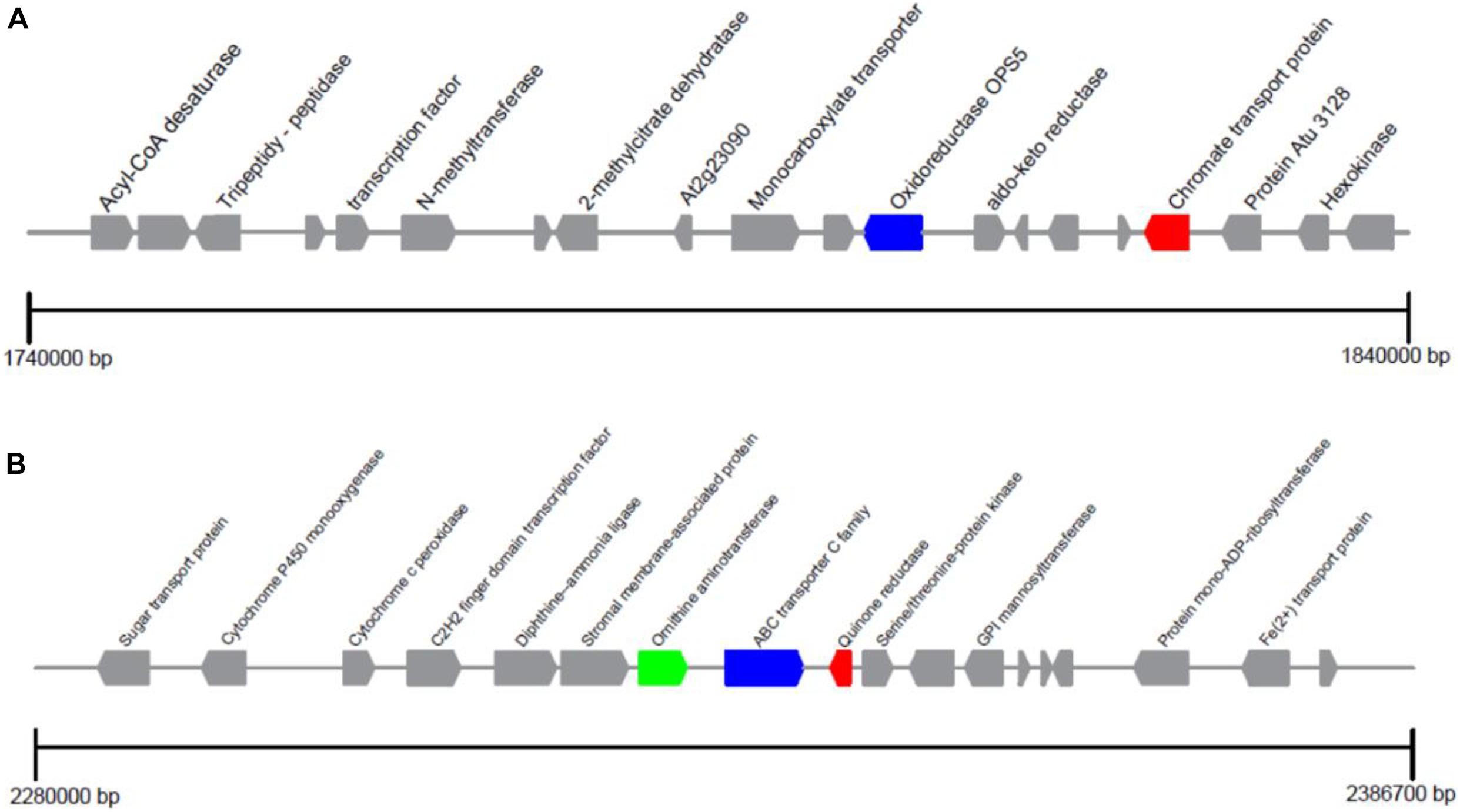
Figure 4. Distribution of genes in the ChrA and ChrB loci of Penicillium janthinellum P1. (A) ChrA encoding chromate transport protein and (B) ChrR encoding quinone reductase loci are positioned on scaffold 14 and 16, respectively.
The ChrR gene is located on scaffold 16 and can encode quinone reductase. Adjacent to the ChrR, the Ornithine aminotransferase RocD and ABC Transporter C family (ABCC) were identified. Interestingly, the same genetic arrangement was found in Penicillium oxalicum 114-2, Penicillium expansum ATCC 24692, and Penicillium chrysogenum Wisconsin 54-1255, and these three genes were arranged in the same order. Part of Penicillium species are resistant to chromium (Fukuda et al., 2008; Zhang et al., 2011; Long et al., 2020), which may be explained by the special gene arrangement to a certain extent. In addition, Ornithine aminotransferase catalyzes the conversion of Ornithine into glutamic semialdehyde which subsequently can catalyze the reduction of NADP+ to NADPH. The generated NADP+ and NADPH will then serve as cofactors of ChrR to catalyze the reduction of Cr (VI). Quinone reductase acted as Cr (VI) reductase was confirmed to be an NADH-dependent reductase and also found in L. fusiformis ZC1 (He et al., 2011). ABCC mediates the transport of glutathione (GSH), also be known as an important antioxidant, which can eliminate the ROS produced by the ChrR enzyme during the reduction of Cr ions. GSH is crucial for chromium resistance to microorganisms. Glutathione synthase and glutathione s-transferase were up-regulated under chromium pressure in P. aeruginosa (Kılıç et al., 2010). Also the increase of Cr (VI) toxicity in E. coli was reported due to the absence of GSH (Helbig et al., 2008). ChrR gene, RocD and ABCC also exist in Trichoderma parareesei CBS 125925 and Saccharomyces cerevisiae YB210, but they are not on the same scaffold and far apart. We hypothesis this potentially may be a reason for differences in chromium resistance. ABCC alone was not related to the presence of heavy metal-related genes. In summary, it is noteworthy that whether the target resistance gene has such a genetic arrangement may be used as a basis for judging whether the strain is resistant to heavy-metal chromium at the genetic level.
Discussion
All life phenomena are intimately connected with genetic material. The high quality and accuracy genetic information of filamentous fungi can reveal the mechanism of heavy-metal resistance, and contribute to genetic manipulation of diverse industrial microbial strains for improving the efficiency of bioremediation sewage treatment. Here, we evaluated factors related to chromium resistance at the genomic level. Illumina and PacBio sequencing technologies are now widely used to assemble genomes due to their economy and efficiency (Ma et al., 2021; Yan et al., 2021). In this study, the Contig N50 length of Illumina sequencing technology was relatively short, which was probably related to the high percentage of repetitive sequences, whereas the third-generation sequencing technology can be more efficient in solving the high repeat ratio problem. After comprehensively considering the pros and cons of different sequencing technologies, we adopted an integrated strategy of hybrid assembly that the third-generation sequencing data was mainly utilized to assemble into chromosome sequence, while the second-generation sequencing data for error correction. Thus, the total de novo assembled genome size was 30.8 Mb, including 29 contigs with Contig N50 of 3088.6 kbp. By comparing different sets of assembly results, it was found that the hybrid assembly result was significantly better than any sole assembly method. To our knowledge, this is the first report of genome assembly of P. janthinellum using combined Illumina and PacBio sequencing platforms.
Genome annotation was implemented through a series of various databases, so that the characteristics of the genome could be precisely evaluated from all different aspects. In the end, a total of 10,955 protein-coding genes within P1 genome was annotated, about 1.90% of which were repetitive sequences, mainly involving simple repetitive sequences, together with 503 tRNA and 274 rRNA.
Metal exposure can lead to a significant inhibition in respiratory enzyme activities as well as corresponding transcript level (Samanta et al., 2020). A large inventory of CAZymes was found, including sugar metabolism enzymes which may ensure respiratory metabolism under heavy metal stress, supporting the high energy metabolism activity of this species. P. janthinellum P1 had more T1PKs than P. oxalicum sp. And many Sec-SRP secretion proteins were identified in the analysis of the secretory system and transporter. These results suggested that P1 has potential for producing bioactive secondary metabolites.
Our research confirms that P1 possesses the genes encoding proteins required for chromate reduction and efflux, namely ChrR, ChrA, respectively, which could be responsible for chromate resistance in this fungus. Through the analyses of the loci of ChrA and ChrR and the special genes in the vicinity of these loci, together with the annotation of other non-chromium resistant strains, it is found out that chromium resistance of microorganisms may be related to the gene arrangement around the anti-chromium genes. Heavy metal can induce mRNA transcription of Cytochrome P450s (CYPs) enzymes (Sun et al., 2020) and CYPs play diverse roles in metabolism including the synthesis of secondary metabolites as well as the degradation of recalcitrant organic substrates (Linder, 2019). The large amount of CYPs might be essential for the life cycle of P. janthinellum P1 and the synthesis of the metabolic products. The microorganism exhibited high oxidative stress supported by a high concentration of reactive oxygen species (ROS) and low levels of reduced glutathione (GSH) in the presence of heavy metals (Caldeira et al., 2021). Biological cells improved the antioxidant capacity by increasing the activities of superoxide dismutase, catalase, and glutathione production (Qi et al., 2021). In addition, Heavy metals can participate in Fenton-like reactions to generate more ROS, which is a direct factor in inducing oxidative stress (Liochev, 1999) and can interact directly with antioxidant enzymes (Gurer-Orhan et al., 2004). In summary, large numbers of identified superoxide dismutases may directly participate in the reduction of heavy metal chromium and reduce toxicity.
Conclusion
In conclusion, we studied in detail the putative genes in P. janthinellum P1 isolated from the ocean that may be involved in important traits of resistance to heavy metals using whole-genome sequencing and comparative genome analysis. The results of this study can facilitate us to further understand the genetic diversity of P. janthinellum and shed light on its chromium resistance. The follow-up research work will analyze these in depth from the perspective of transcriptome and proteome level, and further explore mechanism related to heavy metal chromium resistance.
Data Availability Statement
The datasets presented in this study can be found in online repositories, SRA SRR13016573 and SRR13016574.
Author Contributions
H-YC and P-CY conceived and designed the experiments. B-BC analyzed the data. H-YC contributed materials and analysis tools. B-BC and Y-NL wrote the manuscript. H-YL and YS revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The present study was financially supported by the National Natural Science Foundation of China (Grant Nos. 21762015 and 31860251), the Natural Science Foundation of Guangxi Province (Grant No. 2020GXNSFAA238037), the Guangxi Key Laboratory of Electrochemical and Magneto-chemical Functional Materials (Grant No. EMFM20162203), and the Research Program of Guangxi Specially-invited Experts (Key technologies for intensive processing and quality safety of agricultural products; Grant No. TingFa[2018]39).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.663217/full#supplementary-material
Footnotes
- ^ https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ http://www.ebi.ac.uk/Tools/psa/genewise/
- ^ http://bioinf.uni-greifswald.de/augustus/
- ^ http://exon.gatech.edu/GeneMark/
- ^ http://gmod.org.wiki.MAKER
- ^ https://pfam.xfam.org
- ^ http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
- ^ http://www.genome.jp/kegg/
- ^ http://www.ncbi.nlm.nih.gov/COG/
- ^ http://www.timetree.org/
- ^ https://itol.embl.de/
- ^ https://mycocosm.jgi.doe.gov/Penja1/Penja1.home.html
- ^ http://darlinglab.org/mauve/mauve.html
- ^ http://www.cazy.org/
References
Aguilar-Barajas, E., Paluscio, E., Cervantes, C., and Rensing, C. (2008). Expression of chromate resistance genes from Shewanella sp. strain ANA-3 in Escherichia coli. FEMS Microbiol. Lett. 285, 97–100. doi: 10.1111/j.1574-6968.2008.01220.x
Ayres, R. U. (1992). Toxic Heavy Metals: Materials Cycle Optimization. PNAS 89, 815–820. doi: 10.1073/pnas.89.3.815
Baldiris, R., Acosta-Tapia, N., Montes, A., Hernandez, J., and Vivas-Reyes, R. (2018). Reduction of Hexavalent Chromium and Detection of Chromate Reductase (ChrR) in Stenotrophomonas maltophilia. Molecules 23:23020406. doi: 10.3390/molecules23020406
Borodovsky, M., and Lomsadze, A. (2011). Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Curr. Protoc. Bioinformat. 6, 1–10. doi: 10.1002/0471250953.bi0406s35
Caldeira, J. B., Chung, A. P., Morais, P. V., and Branco, R. (2021). Relevance of FeoAB system in Rhodanobacter sp. B2A1Ga4 resistance to heavy metals, aluminium, gallium, and indium. Appl. Microbiol. Biotechnol. 14, 3301–3314. doi: 10.1007/s00253-021-11254-6
Cantarel, B. L., Korf, I., Robb, S. M., Parra, G., Ross, E., et al. (2008). MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196. doi: 10.1101/gr.6743907
Cervantes, C., Ohtake, H., Chu, L., Misra, T. K., and Silver, S. (1990). Cloning, nucleotide sequence, and expression of the chromate resistance determinant of Pseudomonas aeruginosa plasmid pUM505. J. Bacteriol. 172, 287–291. doi: 10.1128/jb.172.1.287-291.1990
Chen, H. Y., Guan, Y. X., and Yao, S. J. (2014). A novel two-species whole-cell immobilization system composed of marine-derived fungi and its application in wastewater treatment. J. Chemical Technol. Biotechnol. 89, 1733–1740. doi: 10.1002/jctb.4253
Chen, H. Y., Lu, Y. N., Yin, P. C., Li, X., and Shan, Y. (2019). Exploring the Mechanisms of Biosorption of Cr(VI) by Marine-Derived Penicillium janthinellum P1. Int. J. Agricult. Biol. 22, 913–920. doi: 10.17957/ijab/15.1148
Chen, L. F., Gong, Y. H., Cai, Y. L., Liu, W., Zhou, Y., Xiao, Y., et al. (2016). Genome Sequence of the Edible Cultivated Mushroom Lentinula edodes (Shiitake) Reveals Insights into Lignocellulose Degradation. PLoS One 11:20. doi: 10.1371/journal.pone.0160336
Clayton, W., Eaton, C. J., Dupont, P. Y., Gillanders, T., Cameron, N., Saikia, S., et al. (2017). Analysis of simple sequence repeat (SSR) structure and sequence within Epichloe endophyte genomes reveals impacts on gene structure and insights into ancestral hybridization events. PLoS One 12:183748. doi: 10.1371/journal.pone.0183748
Dash, H. R., Mangwani, N., Chakraborty, J., Kumari, S., and Das, S. (2013). Marine bacteria: potential candidates for enhanced bioremediation. Appl. Microbiol. Biotechnol. 97, 561–571. doi: 10.1007/s00253-012-4584-0
Deng, J. X., Carbone, L., and Dean, R. A. (2007). The evolutionary history of Cytochrome P450 genes in four filamentous Ascomycetes. BMC Evol. Biol. 7:30. doi: 10.1186/1471-2148-7-30
Dusengemungu, L., Kasali, G., Gwanama, C., and Ouma, K. O. (2020). Recent Advances in Biosorption of Copper and Cobalt by Filamentous Fungi. Front. Microbiol. 11:582016. doi: 10.3389/fmicb.2020.582016
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Fagan, R. P., and Fairweather, N. F. (2011). Clostridium difficile Has Two Parallel and Essential Sec Secretion Systems. J. Biol. Chem. 286, 27483–27493. doi: 10.1074/jbc.M111.263889
Floudas, D., Binder, M., Riley, R., Barry, K., Blanchette, R. A., Henrissat, B., et al. (2012). The Paleozoic Origin of Enzymatic Lignin Decomposition Reconstructed from 31 Fungal Genomes. Science 336, 1715–1719. doi: 10.1126/science.1221748
Fukuda, T., Ishino, Y., Ogawa, A., Tsutsumi, K., and Morita, H. (2008). Cr(VI) reduction from contaminated soils by Aspergillus sp N2 and Penicillium sp N3 isolated from chromium deposits. J. Gen. Appl. Microbiol. 54, 295–303. doi: 10.2323/jgam.54.295
Gonzalez, C. F., Ackerley, D. F., Lynch, S. V., and Matin, A. (2005). ChrR, a soluble quinone reductase of Pseudomonas putida that defends against H2O2. J. Biol. Chem. 280, 22590–22595. doi: 10.1074/jbc.M501654200
Gowda, K., Black, S. D., Moller, I., Sakakibara, Y., Liu, M. C., and Zwieb, C. (1998). Protein SRP54 of human signal recognition particle: cloning, expression, and comparative analysis of functional sites. Gene 207, 197–207. doi: 10.1016/s0378-1119(97)00627-6
Grijseels, S., Nielsen, J. C., Randelovic, M., Nielsen, J., Nielsen, K. F., Workman, M., et al. (2016). Penicillium arizonense, a new, genome sequenced fungal species, reveals a high chemical diversity in secreted metabolites. Sci. Rep. 6:35112. doi: 10.1038/srep35112
Gu, R. J., Gao, J. Y., Dong, L. L., Liu, Y., Li, X. L., Bai, Q. H., et al. (2020). Chromium metabolism characteristics of coexpression of ChrA and ChrT gene. Ecotoxicol. Environ. Saf. 204:111060. doi: 10.1016/j.ecoenv.2020.111060
Gurer-Orhan, H., Sabir, H. U., and Ozgunes, H. (2004). Correlation between clinical indicators of lead poisoning and oxidative stress parameters in controls and lead-exposed workers. Toxicology 195, 147–154. doi: 10.1016/j.tox.2003.09.009
Han, M. V., Thomas, G. W., Lugo-Martinez, J., and Hahn, M. W. (2013). Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997. doi: 10.1093/molbev/mst100
He, M. Y., Li, X. Y., Liu, H. L., Miller, S. J., Wang, G. J., and Rensing, C. (2011). Characterization and genomic analysis of a highly chromate resistant and reducing bacterial strain Lysinibacillus fusiformis ZC1. J. Hazard. Mater. 185, 682–688. doi: 10.1016/j.jhazmat.2010.09.072
Helbig, K., Bleuel, C., Krauss, G. J., and Nies, D. H. (2008). Glutathione and Transition-Metal Homeostasis in Escherichia coli. J. Bacteriol. 190, 5431–5438. doi: 10.1128/jb.00271-08
Hu, L., Taujale, R., Liu, F., Song, J., Yin, Q., Zhang, Y., et al. (2016). Draft genome sequence of Talaromyces verruculosus (”Penicillium verruculosum”) strain TS63-9, a fungus with great potential for industrial production of polysaccharide-degrading enzymes. J. Biotechnol. 219, 5–6. doi: 10.1016/j.jbiotec.2015.12.017
Kan, G. F., Wang, X. F., Jiang, J., Zhang, C. S., Chi, M. L., Ju, Y., et al. (2019). Copper stress response in yeast Rhodotorula mucilaginosa AN 5 isolated from sea ice, Antarctic. MicrobiologyOpen 8:657. doi: 10.1002/mbo3.657
Kasai, N., Ikushiro, S., Hirosue, S., Arisawa, A., Ichinose, H., Uchida, Y., et al. (2010). Atypical kinetics of cytochromes P450 catalysing 3’-hydroxylation of flavone from the white-rot fungus Phanerochaete chrysosporium. J. Biochem. 147, 117–125. doi: 10.1093/jb/mvp155
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keller, O., Kollmar, M., Stanke, M., and Waack, S. (2011). A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763. doi: 10.1093/bioinformatics/btr010
Kılıç, N. K., Stensballe, A., Otzen, D. E., and Dönmez, G. (2010). Proteomic changes in response to chromium(VI) toxicity in Pseudomonas aeruginosa. Bioresour. Technol. 101, 2134–2140. doi: 10.1016/j.biortech.2009.11.008
Klingenberg, C. P., and Marugan-Lobon, J. (2013). Evolutionary Covariation in Geometric Morphometric Data: Analyzing Integration, Modularity, and Allometry in a Phylogenetic Context. Syst. Biol. 62, 591–610. doi: 10.1093/sysbio/syt025
Lam, K. K., LaButti, K., Khalak, A., and Tse, D. (2015). FinisherSC: a repeat-aware tool for upgrading de novo assembly using long reads. Bioinformatics 31, 3207–3209. doi: 10.1093/bioinformatics/btv280
Leitao, A. L. (2009). Potential of Penicillium Species in the Bioremediation Field. Int. J. Env. Res. Public Health 6, 1393–1417. doi: 10.3390/ijerph6041393
Li, W. L., Cong, Q., Pei, J. M., Kinch, L. N., and Grishin, N. V. (2012). The ABC transporters in Candidatus Liberibacter asiaticus. Proteins Struct. Funct. Bioinformat. 80, 2614–2628. doi: 10.1002/prot.24147
Liaquat, F., Munis, M. F. H., Haroon, U., Arif, S., Saqib, S., Zaman, W., et al. (2020). Evaluation of Metal Tolerance of Fungal Strains Isolated from Contaminated Mining Soil of Nanjing. China. Biology-Basel 9:12. doi: 10.3390/biology9120469
Linder, T. (2019). Taxonomic Distribution of Cytochrome P450 Monooxygenases (CYPs) among the Budding Yeasts (Sub-Phylum Saccharomycotina). Microorganisms 7:20. doi: 10.3390/microorganisms7080247
Liochev, S. I. (1999). “The mechanism of “Fenton-like” reactions and their importance for biological systems. A biologist’s view,” in Metal Ions in Biological Systems, Vol 36: Interrelations between Free Radicals and Metal Ions in Life Processes, eds A. Sigel and H. Sigel (New York, NY: Marcel Dekker), 1–39.
Liu, L. Y., Cui, H. N., and Xu, Y. (2020). Quantitative Estimation of Oxidative Stress in Cancer Tissue Cells Through Gene Expression Data Analyses. Front. Genet. 11:494. doi: 10.3389/fgene.2020.00494
Long, B. B., Ye, J. E., Ye, Z., He, J. Y., Luo, Y. T., Zhao, Y. G., et al. (2020). Cr(VI) removal by Penicillium oxalicum SL2: Reduction with acidic metabolites and form transformation in the mycelium. Chemosphere 253:126731. doi: 10.1016/j.chemosphere.2020.126731
Lopez-Hernandez, F., and Cortes, A. J. (2019). Last-Generation Genome-Environment Associations Reveal the Genetic Basis of Heat Tolerance in Common Bean (Phaseolus vulgaris L.). Front. Genet. 10:954. doi: 10.3389/fgene.2019.00954
Lu, T., Zhang, Q. L., and Yao, S. J. (2017). Efficient decolorization of dye-containing wastewater using mycelial pellets formed of marine-derived Aspergillus niger. Chin. J. Chem. Eng. 25, 330–337. doi: 10.1016/j.cjche.2016.08.010
Ma, D. N., Guo, Z. J., Ding, Q. S., Zhao, Z. Z., Shen, Z. J., Wei, M. Y., et al. (2021). Chromosome-level assembly of the mangrove plant Aegiceras corniculatum genome generated through Illumina, PacBio and Hi-C sequencing technologies. Mol. Ecol. Resour. 15:13347. doi: 10.1111/1755-0998.13347
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. doi: 10.1093/nar/gkz268
Mardanov, A. V., Glukhova, L. B., Gruzdev, E. V., Beletsky, A. V., Karnachuk, O. V., and Ravin, N. V. (2016). The complete mitochondrial genome of the acid-tolerant fungus Penicillium ShG4C. Genomics Data 10, 141–143. doi: 10.1016/j.gdata.2016.11.009
Mattiazzi, M., Petrovic, U., and Krizaj, I. (2012). Yeast as a model eukaryote in toxinology: a functional genomics approach to studying the molecular basis of action of pharmacologically active molecules. Toxicon 60, 558–571. doi: 10.1016/j.toxicon.2012.03.014
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi: 10.1093/nar/gkaa913
Nakatsu, C. H., Barabote, R., Thompson, S., Bruce, D., Detter, C., Brettin, T., et al. (2013). Complete genome sequence of Arthrobacter sp strain FB24. Stand. Genomic Sci. 9, 106–116. doi: 10.4056/sigs.4438185
Naveed, S., Li, C. H., Zhang, J. Y., Zhang, C. H., and Ge, Y. (2020). Sorption and transformation of arsenic by extracellular polymeric substances extracted from Synechocystis sp. PCC6803. Ecotoxicol. Environ. Saf. 206:111200. doi: 10.1016/j.ecoenv.2020.111200
Nayfa, M. G., Jones, D. B., Benzie, J. A. H., Jerry, D. R., and Zenger, K. R. (2020). Comparing Genomic Signatures of Selection Between the Abbassa Strain and Eight Wild Populations of Nile Tilapia (Oreochromis niloticus) in Egypt. Front. Genet. 11:567969. doi: 10.3389/fgene.2020.567969
Niegowski, D., and Eshaghi, S. (2007). The CorA family: structure and function revisited. Cell. Mol. Life Sci. CMLS 64, 2564–2574. doi: 10.1007/s00018-007-7174-z
Nies, A., Nies, D. H., and Silver, S. (1990). Nucleotide sequence and expression of a plasmid-encoded chromate resistance determinant from Alcaligenes eutrophus. J. Biol. Chem. 265, 5648–5653.
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: A Toolkit for Quality Control of Next Generation Sequencing Data. PLoS One 7:30619. doi: 10.1371/journal.pone.0030619
Pathak, A., Kothari, R., Vinoba, M., Habibi, N., and Tyagi, V. V. (2021). Fungal bioleaching of metals from refinery spent catalysts: A critical review of current research, challenges, and future directions. J. Environ. Manage. 280:111789. doi: 10.1016/j.jenvman.2020.111789
Peng, L., Li, L. W., Liu, X. C., Chen, J. W., Shi, C. C., Guo, W. J., et al. (2020). Chromosome-Level Comprehensive Genome of Mangrove Sediment-Derived Fungus Penicillium variabile HXQ-H-1. J. Fungi 6:12. doi: 10.3390/jof6010007
Peng, M., Dilokpimol, A., Makela, M. R., Hilden, K., Bervoets, S., Riley, R., et al. (2017). The draft genome sequence of the ascomycete fungus Penicillium subrubescens reveals a highly enriched content of plant biomass related CAZymes compared to related fungi. J. Biotechnol. 246, 1–3. doi: 10.1016/j.jbiotec.2017.02.012
Qi, C. L., Wang, X. D., Han, F. L., Chen, X. F., Li, E. C., Zhang, M. L., et al. (2021). Dietary arginine alleviates the oxidative stress, inflammation and immunosuppression of juvenile Chinese mitten crab Eriocheir sinensis under high pH stress. Aquacult. Rep. 19:8. doi: 10.1016/j.aqrep.2021.100619
Samanta, S., Singh, A., Banerjee, A., and Roychoudhury, A. (2020). Exogenous supplementation of melatonin alters representative organic acids and enzymes of respiratory cycle as well as sugar metabolism during arsenic stress in two contrasting indica rice cultivars. J. Biotechnol. 324, 220–232. doi: 10.1016/j.jbiotec.2020.10.013
Schafhauser, T., Wibberg, D., Ruckert, C., Winkler, A., Flor, L., van Pee, K. H., et al. (2015). Draft genome sequence of Talaromyces islandicus (”Penicillium islandicum”) WF-38-12, a neglected mold with significant biotechnological potential. J. Biotechnol. 211, 101–102. doi: 10.1016/j.jbiotec.2015.07.004
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Sriharsha, D. V., Kumar, R. L., and Janakiraman, S. (2020). Absorption and Reduction of Chromium by Fungi. Bull. Environ. Contam. Toxicol. 105, 645–649. doi: 10.1007/s00128-020-02979-7
Sun, J. X., Wang, S. C., Cao, Y. R., Wang, S. T., and Li, S. (2020). Cadmium exposure induces apoptosis, inflammation and immunosuppression through CYPs activation and antioxidant dysfunction in common carp neutrophils. Fish Shellf. Immunol. 99, 284–290. doi: 10.1016/j.fsi.2020.02.015
Wang, Y. P., Tang, H. B., DeBarry, J. D., Tan, X., Li, J. P., Wang, X. Y., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:gkr1293. doi: 10.1093/nar/gkr1293
Wang, Y. S., Liang, H. L., Chen, G. P., Liao, C. G., Wang, Y. C., Hu, Z. L., et al. (2019). Molecular and Phylogenetic Analyses of the Mediator Subunit Genes in Solanum lycopersicum. Front. Genet. 10:01222. doi: 10.3389/fgene.2019.01222
Wingenfelder, U., Hansen, C., Furrer, G., and Schulin, R. (2005). Removal of heavy metals from mine waters by natural zeolites. Environ. Sci. Technol. 39, 4606–4613. doi: 10.1021/es048482s
Xu, H. B., Luo, X., Qian, J., Pang, X. H., Song, J. Y., Qian, G. R., et al. (2012). FastUniq: A Fast De Novo Duplicates Removal Tool for Paired Short Reads. PLoS One 7:6. doi: 10.1371/journal.pone.0052249
Xu, X. Y., Hao, R. X., Xu, H., and Lu, A. H. (2020). Removal mechanism of Pb(II) by Penicillium polonicum: immobilization, adsorption, and bioaccumulation. Sci. Rep. 10:9079. doi: 10.1038/s41598-020-66025-6
Yan, W. Z., Wang, N., Wei, D., Liang, C. Y., Chen, X. M., Liu, L., et al. (2021). Bacterial community compositions and nitrogen metabolism function in a cattle farm wastewater treatment plant revealed by Illumina high-throughput sequencing. Environ. Sci. Pollut. Res. 13, [preprint]. doi: 10.1007/s11356-021-13570-w
Yang, Y., Chen, M., Li, Z. W., Al-Haimi, A. M. S., de Hoog, S., Pan, W. H., et al. (2016). Genome Sequencing and Comparative Genomics Analysis Revealed Pathogenic Potential in Penicillium capsulatum as a Novel Fungal Pathogen Belonging to Eurotiales. Front. Microbiol. 7:01541. doi: 10.3389/fmicb.2016.01541
Yin, K., Wang, Q. N., Lv, M., and Chen, L. X. (2019). Microorganism remediation strategies towards heavy metals. Chem. Eng. J. 360, 1553–1563. doi: 10.1016/j.cej.2018.10.226
Zhang, L. F., Chen, Y. Y., and Zhang, W. J. (2011). Removal of Cr(VI) from Aqueous Solution by Acid Treated Fungal Biomass. Adv. Mater. Res. 197-198, 131–135. doi: 10.4028/www.scientific.net/AMR.197-198.131
Zhang, L. L., Huang, W., Zhang, Y. Y., Fan, G., He, J., Ren, J. N., et al. (2020). Genomic and Transcriptomic Study for Screening Genes Involved in the Limonene Biotransformation of Penicillium digitatum DSM 62840. Front. Microbiol. 11:744. doi: 10.3389/fmicb.2020.00744
Zhou, J. F., Zeng, Z. Q., Gao, Y. G., and Yu, Z. H. (2008). The Summary of the Database of Fungal Genomes Publicly Available. Instit. Microbiol. 35, 1311–1318. doi: 10.13344/j.microbiol.china.2008.08.008
Keywords: Penicillium janthinellum, chromate resistance, whole genome sequence, comparative genomic, gene loci analysis
Citation: Chi B-B, Lu Y-N, Yin P-C, Liu H-Y, Chen H-Y and Shan Y (2021) Sequencing and Comparative Genomic Analysis of a Highly Metal-Tolerant Penicillium janthinellum P1 Provide Insights Into Its Metal Tolerance. Front. Microbiol. 12:663217. doi: 10.3389/fmicb.2021.663217
Received: 02 February 2021; Accepted: 14 May 2021;
Published: 04 June 2021.
Edited by:
Daniel Yero, Autonomous University of Barcelona, SpainReviewed by:
Li Xiong, Central China Normal University, ChinaRenato Chavez, University of Santiago, Chile
Copyright © 2021 Chi, Lu, Yin, Liu, Chen and Shan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui-Ying Chen, aHljaGVuQGdsdXQuZWR1LmNu