
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 06 April 2021
Sec. Microbial Physiology and Metabolism
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.657562
This article is part of the Research Topic Post Translational Modification in Response to Stresses in Bacteria View all 5 articles
 Abida Sultan1
Abida Sultan1 Carsten Jers1
Carsten Jers1 Tariq A. Ganief2
Tariq A. Ganief2 Lei Shi3
Lei Shi3 Meriem Senissar1Julie Bonne Køhler1
Meriem Senissar1Julie Bonne Køhler1 Boris Macek2
Boris Macek2 Ivan Mijakovic1,3*
Ivan Mijakovic1,3*Understanding phosphorylation-mediated regulation of metabolic enzymes, pathways, and cell phenotypes under metabolic shifts represents a major challenge. The kinases associated with most phosphorylation sites and the link between phosphorylation and enzyme activity remain unknown. In this study, we performed stable isotope labeling by amino acids in cell culture (SILAC)-based proteome and phosphoproteome analysis of Escherichia coli ΔyeaG, a strain lacking a poorly characterized serine/threonine kinase YeaG, to decipher kinase-substrate interactions and the effects on metabolic phenotype during shifts from glucose to malate. The starting point of our analysis was the identification of physiological conditions under which ΔyeaG exhibits a clear phenotype. By metabolic profiling, we discovered that ΔyeaG strain has a significantly shorter lag phase than the wild type during metabolic shift from glucose to malate. Under those conditions, our SILAC analysis revealed several proteins that were differentially phosphorylated in the ΔyeaG strain. By focusing on metabolic enzymes potentially involved in central carbon metabolism, we narrowed down our search for putative YeaG substrates and identified isocitrate lyase AceA as the direct substrate of YeaG. YeaG was capable of phosphorylating AceA in vitro only in the presence of malate, suggesting that this phosphorylation event is indeed relevant for glucose to malate shift. There is currently not enough evidence to firmly establish the exact mechanism of this newly observed regulatory phenomenon. However, our study clearly exemplifies the usefulness of SILAC-based approaches in identifying proteins kinase substrates, when applied in physiological conditions relevant for the activity of the protein kinase in question.
Metabolic adaptation is one of the major bacterial responses for coping with the changing environment (Liebeke and Lalk, 2014; Goo et al., 2015). By rearranging their metabolic functions, bacteria can resist antibiotic treatments and evade the host immune response during infection (Sprenger et al., 2018; Jiang et al., 2019), they can adapt to severe hypoxia or starvation (Rittershaus et al., 2018; Gray et al., 2019), or they can effectively switch between alternative carbon or nitrogen sources as they become available (Deutscher, 2008; Esteves-Ferreira et al., 2018). Metabolic adaptation commonly involves changes in transcription of genes encoding metabolic enzymes, an approach that is costly both in terms of time and resources (requirement of building blocks, ATP, etc.; Chubukov et al., 2013). Another, faster mechanism of response is the modification of activity of enzymes already present in the bacterial cell. This is typically achieved by allosteric regulation (Xu et al., 2012; Link et al., 2013) or reversible post-translational modifications (PTMs; Nussinov et al., 2012; Macek et al., 2019) of bacterial proteins.
Among various PTMs known to regulate bacterial metabolism, protein phosphorylation is arguably the most extensively characterized. Phosphorylation and dephosphorylation are a metabolic switch mediated by kinases and phosphatases. Many examples of protein phosphorylation affecting the expression of metabolic genes are known. Notably, bacterial metabolic adaptation processes are known to depend on histidine-kinases and response regulators of the so-called two component systems (Charbonnier et al., 2017; Quiroz-Rocha et al., 2017; Gómez-Mejia et al., 2018). Herein, transient autophosphorylation of bacterial histidine kinases on histidine residues and response regulators on aspartate residues leads to altered expression of metabolic operons. Carbon catabolite repression, a major metabolic switch in bacteria, is known to critically depend on histidine and cysteine phosphorylation of components of the phosphoenolpyruvate:carbohydrate phosphotransferase system (Deutscher et al., 2014). In Firmicutes, carbon catabolite repression is also regulated by serine-phosphorylation of the major transcriptional regulator CcpA (Schumacher et al., 2004). Bacterial protein-tyrosine and -serine/threonine kinases are known to regulate the metabolism by directly phosphorylating transcription regulators and modifying their affinity for binding of target DNA sequences (Derouiche et al., 2013, 2015; Kobir et al., 2014).
While protein phosphorylation is known to play a major role in transcriptional regulation, hitherto, direct regulation of enzyme activity by serine/threonine phosphorylation has been reported for only a handful of bacterial metabolic enzymes. One well-studied example is phosphorylation of isocitrate dehydrogenase (Icd), which controls the branching point between the glyoxylate shunt and the TCA cycle. In Escherichia coli, isocitrate dehydrogenase kinase/phosphatase AceK phosphorylates Icd on a serine residue, thereby inactivating the enzyme and directing the carbon flux through the glyoxylate shunt (LaPorte and Chung, 1985; Thorsness and Koshland, 1987). In the pathogen Mycobacterium tuberculosis, the S-adenosyl-L-homo-cysteine hydrolase (SahH), which plays an important role in regulating cellular methylation processes, gets inactivated by serine/threonine phosphorylation (Corrales et al., 2013). Inhibition of SahH activity by phosphorylation is thought to act by decreasing affinity toward the essential cofactor NAD+ (Singhal et al., 2013). In the same bacterium, phosphorylation of protein GarA abolishes its direct inhibition of metabolic enzymes: α-ketoglutarate dehydrogenase, glutamate dehydrogenase, and glutamate synthase (Ventura et al., 2013). A study by Brunk et al. (2018) demonstrated how phosphorylation-mediated regulation of E. coli enzymes, such as enolase, transaldolase, and serine hydroxymethyltranferase, impacts metabolic pathways and cellular fitness in a changing environment. Virmani et al. (2019) also reported regulation of enolase by Ser/Thr kinase PrkC in sporulating Bacillus anthracis. PrkC was found to augment the germination process by maintaining enolase quantity/expression and activity. In E. coli and Bacillus subtilis, protein-tyrosine kinase-mediated phosphorylation of UDP-glucose dehydrogenases is known to increase their metabolic activity by increasing their affinity for NAD+ (Mijakovic et al., 2003; Lacour et al., 2008; Petranovic et al., 2009). The M. tuberculosis Ser/Thr protein kinase PknJ was shown to phosphorylate the rate-limiting enzyme of glycolysis pyruvate kinase A (Arora et al., 2010). Interestingly, bacterial DNA metabolism is also regulated by direct phosphorylation of the recombinase RecA, demonstrated in B. subtilis (Bidnenko et al., 2013) and Deinococcus radiodurans (Rajpurohit et al., 2016).
Despite there being relatively few cases of well-characterized regulation of bacterial metabolic enzymes via phosphorylation, there is plenty of evidence that metabolic enzymes are extensively phosphorylated in bacterial cells. From the publications of the very first site-specific bacterial phosphoproteomes (Macek et al., 2007, 2008), it became apparent that protein phosphorylation is specifically enriched in metabolic enzymes (Soufi et al., 2008). To the point, a study by Hansen et al. (2013) identified 512 tyrosine-phosphorylation sites on 342 proteins in E. coli K12 MG1655 with roles in glycolysis and the TCA cycle, including fructose-1,6-biphosphate aldolase A and B, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase, enolase, isocitrate lyase, and fumarase. Similarly, phosphoproteome analysis of Streptococcus suis Δstk (Ser/Thr kinase deletion) identified 12 phosphoproteins with differential phosphorylation levels, where glycolytic enzymes FBA, GADPH, and translation associated EF-Tu were found upregulated, and the remaining proteins involved in cell division and purine metabolism were downregulated in phosphorylation levels (Zhang et al., 2017). However, putative phosphorylation-mediated regulation of these metabolic enzymes has not yet been addressed. In that sense, specific follow-up studies are required to establish any links that may exist between enzyme phosphorylation and activity.
One major challenge in establishing a link between enzyme phosphorylation and enzyme activity, or enzyme phosphorylation and the protein kinase that phosphorylates it, is the transient nature of protein phosphorylation. Therefore, as it has been argued before (Macek et al., 2019), it is essential to pick the correct physiological conditions when seeking to establish such links. In this study, we focused on an E. coli serine/threonine protein kinase YeaG (Figueira et al., 2015) that has not been extensively characterized and whose physiological substrates are unknown. As reported by Figueira et al. (2015), following sustained periods of nitrogen starvation, the wild type E. coli displayed metabolic heterogeneity with the metabolically active population displaying impaired viability. By contrast, the ∆yeaG mutant existed as a single metabolically active population. It is well established that bacteria promote survival or adaptation to sustained environmental adversity by generating phenotypically diverse subpopulations. The non-growing phenotype (persisters) typically neglects the available carbon source and enters a dormant state, causing a lag phase (van Heerden et al., 2014). It is recognized that under various stress conditions, E. coli and related bacteria typically increase the accumulation of RpoS sigma factor, which controls the general stress response and increases survival. The reduced lag phase observed for the ∆yeaG mutant in the study by Figueira et al. (2015), was ascribed to indirect transcriptional repression of the toxin-antitoxin modules mqsR/mqsA and dinJ/yafQ, which in turn positively impacts RpoS transcription and translation.
In order to find physiological substrates of YeaG and hopefully link them to cellular regulation, we focused on growth conditions where ∆yeaG strain has a significant growth phenotype. Given the known involvement of YeaG in metabolic adaptation, we expanded the phenotypic profiling of the ∆yeaG strain focusing on various carbon source shifts. The most striking phenotype was shortening the lag phase in transition from growth on glucose to growth on malate. Focusing on this phenotype, we compared quantitative proteome and phosphoproteome data of ∆yeaG and wild type strains after the shift from glucose to malate, to identify differentially expressed and phosphorylated proteins. By using in vitro phosphorylation assays, we identified isocitrate lyase AceA as the substrate of YeaG. Interestingly, YeaG was capable of phosphorylating AceA only in the presence of malate. Given the known negative impact of AceA on short-term adaptation to glucose starvation (Maharjan et al., 2005), it is probable that YeaG-dependent phosphorylation contributes to this negative effect and is at least in part responsible for the lag phase phenotype of ∆yeaG.
Plasmid and PCR purification kits were purchased from Macherey-Nagel (AH Diagnostics, Aarhus, Denmark). Oligonucleotide primers (Table 1) were purchased from Integrated DNA Technologies (IDT, Leuven, Belgium) and Macrogen (Amsterdam, Holland). Unless specified all the chemicals were purchased from Sigma-Aldrich (Copenhagen, Denmark).

Table 1. List of oligonucleotides used in this study.
Deletions of genes encoding protein kinases were constructed in E. coli K12 MG1655 (genotype: F ̄ λ̄ ilvG rfb-50 rph-1) using λ Red-mediated recombination with temperature sensitive plasmid pSIJ8 (arabinose inducible λ Red recombineering genes and rhamnose inducible flippase recombinase; Datsenko and Wanner, 2000; Thomason et al., 2014). Linear DNA fragments containing a kanamycin resistance gene flanked by FLP recognition target (FRT) sites were amplified from strains in the Keio collection (Baba et al., 2006) by PCR (oligonucleotides are listed in Table 1). The PCR products were introduced by electroporation into cells expressing λ Red by either elevated temperature induction at 42°C or addition of 0.2% L-arabinose, at OD600 of 0.2 at 30°C. Recombinants with insertion in the correct location were confirmed by colony PCR. The plasmid pSIJ8 was cured from cells by incubation at 37°C on plates.
For growth screening experiment, E. coli cells were cultured in complete (LB and Neidhardt EZ) and minimal M9 media [12.8 g/L Na2HPO4·7H2O, 3.0 g/L KH2PO4, 0.5 g/L NaCl, 1.0 g/L NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, and 2000X dilution of trace elements solution (3 g/L FeSO4·7H2O, 4.5 g/L ZnSO4·7H2O, 0.2 g/L CuSO4·2H2O, 0.7 g/L MnCl2·4H2O, 0.3 g/L CoCl2·6H2O, 0.4 g/L Na2MoO4·2H2O, 4.5 g/L CaCl2·2H2O, 15 g/L EDTA, 1 g/L H3BO3, and 0.1 g/L KI)] supplemented with various carbon sources and stressors (i.e., 0.4% glucose, 0.3% malate, 0.4% fructose, 0.4% sorbitol, 0.4% pyruvate, 0.4% succinate, 5 mM EDTA, and 0.5 M NaCl). Briefly, starter cultures of each strain were inoculated from single colonies from agar plates into 300 μl of M9 medium with 0.4% glucose in 96 deepwell plates. Biological triplicates derived from different colonies were cultivated for each strain at 37°C with 300 rpm shaking. After 14–16 h, wells of a 96-well microtiter plates (Enzyscreen) containing M9 medium with 0.4% glucose (total volume 300 μl) were inoculated to an initial OD600 of 0.03 and incubated in a Growth Profiler 960 micro-bioreactor system (System Duetz, EnzyScreen, Heemstede, Netherlands) at 37°C with 250 rpm shaking. When the cultures reached an OD600 of 1, aliquots of cell culture were transferred to fresh M9 medium supplemented with different carbon sources and incubated at 37°C with 250 rpm shaking. Growth was monitored every 20 min by optimal image scanning, and integrated G-values were converted into equivalent OD600 values using the image analysis software (Enzyscreen).
For phosphoproteomics experiments, M9 medium was supplemented with either unlabeled 0.025% “light” L-lysine (12C614N2-lysine, Lys0), or labeled “medium” L-lysine (2H4-lysine, Lys4) and “heavy” L-lysine (13C615N2-lysine, Lys8; Cambridge Isotope Laboratories, Andover, MA). Wild type E. coli K12 and ΔyeaG were cultured in stable isotope labeling by amino acids in cell culture (SILAC) medium supplemented with 0.4% glucose at 37°C to an OD600 of 1.0 and shifted to fresh SILAC medium with either 0.4% glucose or 0.3% malate. After reaching an OD600 of 0.6, the cells were harvested by centrifugation at 7,500 g for 10 min at 4°C and snap-frozen in liquid nitrogen. Cells were lysed in sodium dodecyl sulphate (SDS)-buffer (4% SDS, 100 mM Tris-HCl pH 8.0, and 1 mM EDTA) supplemented with protease inhibitors (Complete protease inhibitor cocktail tablets, Roche Diagnostics) and phosphatase inhibitors (5 mM β-glycerophosphate, 5 mM sodium fluoride, and 1 mM sodium vanadate) for 10 min at room temperature, followed by sonication for 1 min at an amplitude of 40% (pulses of 2 s on and 2 s off). Cell debris was removed by centrifugation at 16,000 g for 30 min. Lysates were precipitated in chloroform/methanol and subsequently re-dissolved in denaturation buffer (6 M urea, 2 M thiourea in 10 mM Tris-HCl, pH 8.0). Protein concentration was estimated by Bradford assay (Bio-Rad). Equal amounts (1:1:1) of protein from the differentially SILAC-encoded cells were pooled for subsequent mass spectrometry (MS) sample preparation. Triple SILAC experiments were performed with wild type grown in M9 medium with glucose labeled with “light” lysine as the common point for integration of all experiments. Biological duplicates derived from different colonies were used for each strain.
To determine complete incorporation of labeled amino acids, an aliquot of 10 μg of medium and heavy labeled protein extracts were digested (as described below) and analyzed on the mass spectrometer. Incorporation of 94% and above was accepted and used for further analysis (Supplementary Figure 1). Four milligrams of protein from each SILAC condition was mixed in a 1:1:1 ratio to a total of 12 mg. A mixing check was performed to check the mixing ratio. Samples were then subjected to in-solution digestion as described previously, with few modifications (Macek et al., 2007). Briefly, protein disulfides were reduced with 1 mM dithiothreitol (DTT) for 1 h at room temperature, and subsequently alkylated with 5.5 mM iodoacetamide (IAA) for 1 h at room temperature in the dark. Proteins were digested with endoproteinase Lys-C (Wako) in an enzyme:protein ratio of 1:100 (w/w) for 3 h, followed by a 4-fold dilution with water, and further digestion overnight with Lys-C 1:100 (w/w). Protease activity was quenched by acidification with trifluoroacetic acid (TFA) to a final concentration of 1–2%, and the resulting peptide mixture was desalted and concentrated on reverse-phase Sep-Pak C18 Cartridge (Waters). Peptides were eluted with 80% acetonitrile (ACN) and 6% TFA. The ACN was removed by vacuum centrifugation for 30 min, and peptide concentration was estimated by measuring the absorbance at 280 nm (Nanodrop 2000C, Thermo Scientific).
An aliquot of the peptide mixture was used for proteome analysis, while the remaining sample was enriched for phosphopeptides using titanium dioxide beads (TiO2, 5 μm Titansphere, GL Sciences, Japan). The TiO2 beads were washed with 80% ACN and 6% TFA prior to incubation with the peptide mixture to a 10:1 peptide-to-beads ratio, with gentle rotation for 10 min. After incubation, the beads were spun down, and the supernatants were incubated with fresh TiO2 beads for a second enrichment round. The procedure was repeated for five consecutive rounds. Beads were loaded onto in-house packed C8 StageTips and washed with 80% ACN and 6% TFA. The phosphopeptides were eluted with 2 × 50 μl 20% NH4OH, pH 10.5, in 60% ACN, and acidified with 20 μl of 20% TFA. Eluted peptides were concentrated and ACN evaporated and loaded onto C18 StageTips (Rappsilber et al., 2007).
Samples were analyzed on an EASy-nanoLC 1,000 (Proxeon, Thermo Scientific) coupled to a Q-Exactive orbitrap HF instrument (Thermo Scientific). Peptides were separated on a 15 cm column (75 μm inner diameter) in-house packed with 1.9 μm reverse-phase C18 beads (Reprosil-Pur, Dr. Maisch, Germany) with a maintained column temperature of 40°C. Phosphopeptides were separated over a 49 min gradient ranging from 10 to 33% of the separation buffer (80% ACN and 0.1% formic acid) in 43 min, ramped to 50% B in 3 min, and additionally to 90% B for 3 min at a flow rate of 200 nl/min, followed by a washout at 90% B for 8 min. Protein samples spiked with peptide mixtures (UPS2, Sigma-Aldrich) were separated using a 120 min gradient of 10–30% B. The Q Exactive HF was operated using Xcalibur v. 2.2 in the data dependent mode to automatically switch between full scan MS and MS/MS acquisition as described previously (Michalski et al., 2011). Spray voltage was set to 2 kV, s-lens RF level at 50, and heated capillary temperature at 275°C. Survey full-scans for the MS spectra (300–1,650 m/z) were acquired at a resolution of 120,000 with an ion target value of 3E6 charges with a maximum fill time of 25 ms. Higher-energy collisional dissociation (HCD) fragment scans were acquired with optimal setting for parallel acquisition, using 1.4 m/z isolation width and normalized collision energy of 27. For proteome, a top12 method was employed with fragment scan resolution of 30,000 at 200 m/z and target value of 1E5 with maximum fill time of 45 ms. For phosphoproteomes, a top7 method was employed with fragment scan resolution of 60,000 at 200 m/z and target value of 1E5 with maximum fill time of 120 ms.
The acquired raw data files were processed by MaxQuant v1.6.0.0 and searched against a target-decoy database of E. coli K12 MG1655 (taxonomy ID 83333, last modified November 6, 2016) containing 4,306 protein entries using the integrated Andromeda search engine (Elias and Gygi, 2007; Cox et al., 2011). Carbamidomethylation of cysteines was specified as fixed modification for both protein and phospho-enriched samples. Variable modifications considered were oxidation of methionine and protein N-terminal acetylation. The phospho-enriched dataset was searched using phosphorylation of Ser, Thr, and Tyr residues as variable modification. A false discovery rate (FDR) was set to 1% for both protein and phospho-site, and a minimum peptide length of six amino acids was required. Spectra were searched with a mass tolerance of 6 ppm for precursor ions and 20 ppm for fragment ions. Endoproteinase Lys-C was selected as protease and a maximum of two missed cleavage sites as allowed.
Gene Ontology (GO) annotation of E. coli K12 MG1655 was retrieved from Uniprot-GOA (downloaded 02.06.2018). PANTHER Classification system was used to perform GO term characterization and enrichment analysis according to GO categories for cellular compartment, biological process, and molecular function. Enrichment/fold change analysis of GO categories was done using the Fisher’s exact test. All the GO terms, which were significant with p < 0.05 after correcting for multiple testing by Benferroni FDR, were selected as over- or under-represented.
The genes encoding potential YeaG substrates were amplified from genomic DNA of E. coli K12 MG1655 using specific primers with appropriate restriction sites listed in Table 1. All PCR fragments were ligated in the appropriate restriction sites of pQE30 vector (Qiagen) and propagated in E. coli NM522. After verification of the inserts, the resulting plasmid DNA was used to transform E. coli M15. Cultures were grown in LB medium supplemented with 100 μg/ml ampicillin and 25 μg/ml kanamycin at 37°C with vigorous shaking. Protein expression was induced with addition of 1 mM isopropyl β–D-thiogalactopyranoside (IPTG) at an OD600 of 0.6 and cells were harvested 3 h after induction. Cells were lysed in lysis buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, and 10% glycerol) supplemented with 1 mg/ml lysozyme and 5 μg/ml DNase, followed by sonication (2 min with 2 s on and 2 s off at 30% amplitude) and centrifugation (20,000 g for 20 min). Proteins were purified using nickel-nitrilotriacetic acid (Ni-NTA) beads (Ni-NTA superflow, Qiagen or His HF nickel affinity gel, Sigma-Aldrich) according to the manufacturer’s instructions and desalted on PD-10 columns (GE Healthcare). Proteins were separated by electrophoresis on 12% SDS-polyacrylamide gels and their concentration was estimated by Bradford assay (Bio-Rad).
For each sample, 5 μg of protein was separated on a 4–20% Tris-glycine SDS polyacrylamide gel and transferred onto polyvinylidene difluoride membrane using the iBlot dry blotting system (Invitrogen). The membrane was blocked for 2 h with TBST (10 mM Tris, 150 mM NaCl, 0.1% Tween20, and pH 7.6) containing 5% BSA, washed five times with TBST for 5 min, and probed with appropriate antibody from rabbit/mouse. Anti-phospho-serine antibodies were from Cell Signaling Technology and anti-phospho-threonine antibodies were from Abcam. Both antibodies were used according to the manufacturer’s recommendations. Subsequently, goat anti-rabbit/mouse coupled to horseradish peroxidase antibody (5000x dilution, Bio-Rad) was used and detected using the ECL prime Western blotting detection reagent (GE Healthcare). Exposure time for the film varied between 1 and 5 min.
For in vitro phosphorylation assays, 6 μg YeaG and 5 μg AceA were incubated at 37°C for 2 h in a reaction containing 50 mM HEPES pH 7.4, 10 mM MgCl2, 10 mM MnCl2, 100 mM KCl, 10 mM ATP, and 0.1% triton X-100, with or without 10 mM malate. Reactions were started by adding ATP and stopped by adding sample buffer for sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE; Bio-Rad) and boiling at 100°C. Samples were separated on 10% SDS-PAGE copolymerized with and without Phos-tag acrylamide (Fujifilm). To prepare SDS-PAGE copolymerized with Phos-tag acrylamide, 50 μM Phos-tag acrylamide and 100 μM MnCl2 in final concentration were added into resolving gel. Protein bands were revealed by Coomassie brilliant blue R-250 staining.
YeaG has previously been reported to be involved in metabolic adaptation (Figueira et al., 2015). Hence, we compared the growth of the ∆yeaG strain to that of the wild type E. coli during metabolic shifts from glucose to various other carbon sources. Escherichia coli strains with individual inactivation of all other known serine/threonine/tyrosine kinases (∆aceK, ∆hipA, ∆rdoA, ∆yegI, ∆etk, and ∆wzc) were also used as controls. All the strains were initially grown in M9 medium with glucose as the sole carbon source and were then shifted to fresh medium with different carbon sources (fructose, sorbitol, malate, succinate, and pyruvate). Growth of all strains was monitored in an automated growth profiler. In the control experiment where cells were shifted from glucose to glucose, none of the kinase knockout strains exhibited a growth phenotype (Figure 1A). Among the conditions tested, the most striking phenotype of ∆yeaG was a significant reduction of the duration of lag phase upon transition from growth on glucose to growth on malate (Figure 1B). In order to investigate the mechanism by which YeaG controls the duration of metabolic adaptation to growth on malate, we decided to perform a quantitative MS (phospho)proteomics analysis with SILAC, comparing ∆yeaG to the wild type strain. To do this, we had to scale up the experiment to batch growth and supplement the growth media with different forms of lysine (Lys0, Lys4, and Lys8). In the scaled-up SILAC experiment growth set up, ∆yeaG strain retained its growth phenotype (Figure 1C).
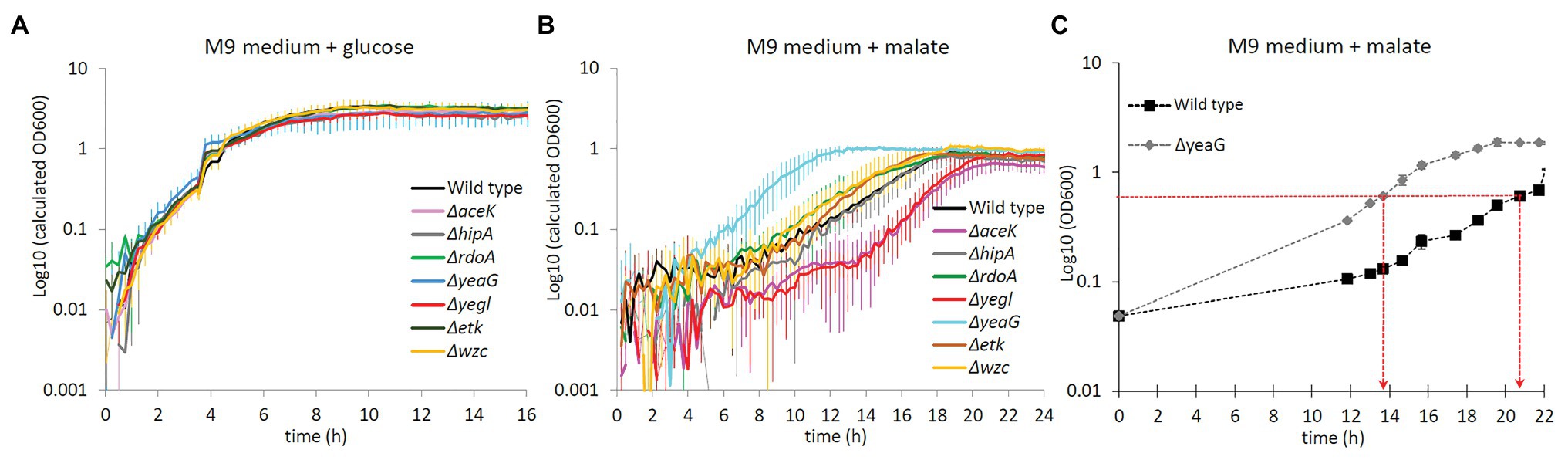
Figure 1. Growth phenotype of ΔyeaG during glucose to malate shift. (A) Growth profiler experiment for different Escherichia coli strains (names indicated in the figure inset) shifted from M9 medium supplemented with glucose to fresh M9 medium supplemented with glucose: control with no metabolic shift (average of biological triplicates). (B) Growth profiler experiment for different E. coli strains (names indicated in the figure inset) shifted from M9 medium supplemented with glucose to fresh M9 medium supplemented with malate: metabolic shift from glucose to malate (average of biological triplicates). (C) Growth profile of ΔyeaG and wild type strains in batch cultures scaled up for the stable isotope labeling by amino acids in cell culture (SILAC) proteomics experiment (average of biological duplicates). Time points when samples were collected for the MS proteomics analysis (at OD600 = 0.6) are indicated with red arrows.
Cell cultures (∆yeaG and wild type) labeled with the different forms of lysine (Lys0, Lys4, and Lys8) for SILAC labeling were initially grown on glucose. At an OD600 of 1, cells were shifted to malate as the sole carbon source, or back to glucose as a control experiment. After the shift, cell cultures were harvested when they reached an OD600 of 0.6 (late exponential phase). Proteins extracted from the corresponding SILAC cultures were combined in equal amounts and digested to peptides using endoprotease Lys-C. The wild type grown on glucose labeled with Lys0 was used as the common point in the two SILAC experiments, thus enabling integration of all datasets. In the first experiment, wild type shifted to glucose (Lys0) was combined with wild type shifted to malate (Lys4). In the second experiment, wild type shifted to glucose (Lys0) was combined with ∆yeaG shifted to glucose (Lys4) and ∆yeaG shifted to malate (Lys8). Incorporation level of the SILAC amino acids (Lys4 and Lys8) in all the samples was found to be |>94% (Supplementary Figure 1). Analysis of all the fractions (proteomes and phosphoproteomes) resulted in identification and quantification of 1,772 proteins (Supplementary Table 1). The estimated FDR was 0.29% at peptide level and 1.45% at protein level, respectively. Phosphoproteome analysis resulted in identification of 127 phosphorylation events (Supplementary Table 1). A majority of the localized phosphorylation events were observed on serine (67.7%), followed by threonine (28.31%) and tyrosine (3.94%). This agrees with a previous study in E. coli (Macek et al., 2008), which reported serine, threonine, and tyrosine phosphoproteome fractions of 67.9, 23.5, and 8.6%, respectively. Pearson correlation coefficients between the two biological replicates for differentially regulated proteome and phosphoproteome are provided in Supplementary material (Supplementary Figures 2, 3).
The SILAC approach enabled us to perform a quantitative comparison of the differentially expressed proteome between cells shifted from growth on glucose to a fresh glucose-containing medium and cells shifted from glucose to malate. A GO terms analysis of the differentially expressed proteome indicated that no specific category of biological processes, molecular functions, or cellular components was particularly affected in the ∆yeaG strain (Supplementary Figure 4). In Figure 2A, we attempted to visualize the main differences between ∆yeaG and the wild type strain proteomes during glucose to malate shift. On the “y” axis are expression ratios for individual proteins in ∆yeaG grown on malate over ∆yeaG grown on glucose – thus representing the expression change during glucose-to-malate shift. On the “x” axis, the same glucose-to-malate shift ratios are plotted for individual proteins in the wild type strain (a cutoff of two SDs from the dataset mean was applied). A large majority of proteins that are differentially expressed during the glucose-to-malate shift were not significantly affected by inactivation of YeaG. Among the proteins more strongly expressed in ΔyeaG upon shift to malate, there were proteins related to outer membrane-bound periplasmic space and monosaccharide transmembrane transport. In the wild type strain, cytosolic proteins related to stress response were more strongly expressed during shift to malate. In order to examine the possible impact of these proteome changes on the key phenotype of ΔyeaG, the reduced lag phase during glucose to malate shift, in Figure 2B, we mapped the changes in differential expression onto the map of E. coli central carbon metabolism. One category of proteins that was largely affected was the transporters. In particular, the kinase mutant overexpressed the aerobic C4 transporter DctA, capable of importing L-malate, alongside fumarate and succinate (Lukas et al., 2010). Stronger expression of DctA in ΔyeaG is consistent with the shorter lag phase observed for this strain during the shift to malate. Metabolic enzymes that were overexpressed only in the wild type during the shift included the 6-phospho-gluconate dehydratase, alcohol dehydrogenase, and catalase. ΔyeaG differentially overexpressed phosphoenolpyruvate carboxykinase, acetyl-CoA synthase, and aldehyde dehydrogenase. Based on the presented differences in the expressed proteome during glucose to malate shift, it seems plausible to presume that the YeaG protein kinase does not exert its metabolic control only by direct phosphorylation of target proteins/enzymes. It seems to also affect the expression of several genes, by an unknown mechanism that is not fully accounted for by the available literature. Bacterial serine/threonine kinases are known to phosphorylate transcription regulators and affect gene expression (Kalantari et al., 2015), so it is plausible to presume that YeaG might act by a similar mechanism.
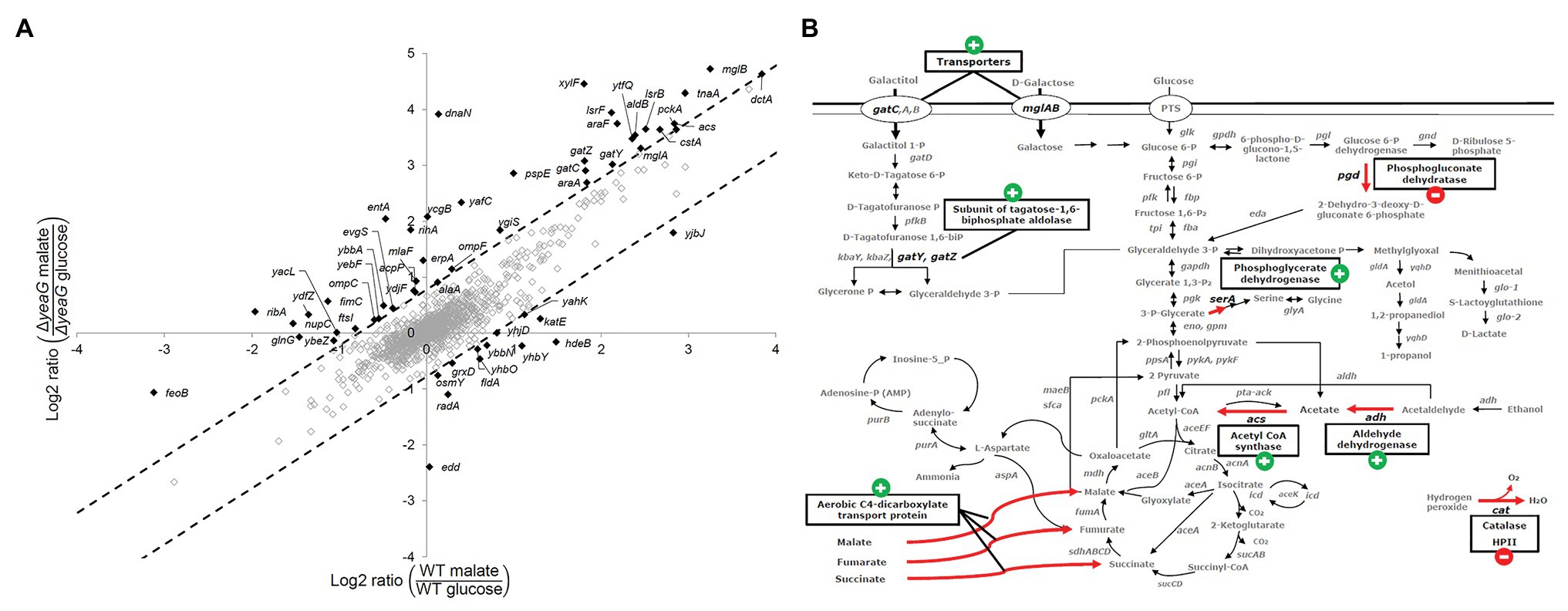
Figure 2. Differential expression of proteins during glucose to malate shift. (A) Plot of quantified protein ratios (in response to shift in carbon source from glucose to malate) for the ΔyeaG vs. the wild type strain. As a cutoff for assigning proteins differentially regulated in the two strains, we chose to use two SDs from the mean of the dataset (indicated by dotted lines). Proteins that were differentially upregulated or downregulated are marked in black and gene names are shown. (B) Proteins that are differently regulated during malate shift in the wild type and ΔyeaG strains mapped on the central carbon metabolism of E. coli K-12 MG1655. Red arrows are used to highlight metabolic and transport reactions catalyzed by the affected enzymes. The expression change during glucose to malate shift in ΔyeaG compared to the wild type strain is indicated next to the enzyme name by a plus/minus sign.
Since YeaG is a poorly characterized protein kinase (Figueira et al., 2015), we next asked what are the proteins that are differentially phosphorylated in the ΔyeaG. The SILAC approach has been previously used to quantitatively compare the phosphoproteome of a kinase knockout and wild type strain, in order to identify substrates of a protein kinase; proteins whose phosphorylation is attenuated in the kinase knockout (Shi et al., 2016). In this study, our approach was a bit more complex, since we compared the ratios of occupancy of individual phosphorylation sites during the glucose to malate shift, first individually for the wild type and ΔyeaG strains, and then made a strain-to-strain comparison. The results of differentially phosphorylated phosphorylation sites are shown in Table 2. Here, one can distinguish three scenarios. The first can be exemplified by the protein Hfq, phosphorylated at threonine 49. The occupancy of this phosphorylation site increases by almost the same factor during glucose to malate shift in the wild type and ΔyeaG strain. This means that YeaG exerts no influence, direct (phosphorylation), or indirect (e.g., affecting another kinase, which is directly responsible for phosphorylation) on the occupancy of this phosphorylation site. Another case, exemplified by phosphorylation of YjhQ at threonine 11, phosphorylation during glucose to malate shift is significantly reduced in the ΔyeaG strain compared to the wild type. Here, the most logical explanation is that YeaG is the kinase directly responsible for phosphorylating this amino acid. Having said that, other explanations are possible, e.g., an indirect effect, where YeaG activates another kinase, which is directly responsible for this phosphorylation. Therefore, further evidence would be required before YjhQ could be declared to be a direct substrate of YeaG. Finally, a third scenario can be observed, for example with phosphorylation of GlyA on tyrosine 55. Here, the phosphorylation during glucose to malate shift increases in occupancy in the ΔyeaG strain. In this case, the most logical explanation is that YeaG inactivates another kinase, which is directly responsible for phosphorylating this tyrosine residue. Such regulatory interactions among serine/threonine and tyrosine kinases have been documented in bacteria (Shi et al., 2014) and are most likely at play here. In addition to the proteins with altered SILAC ratios (Table 2), our phosphoproteome study also identified 13 phosphorylation sites detectable in the wild type strain (some of them differentially phosphorylated during the glucose to malate shift) that were no longer phosphorylated at all in the ΔyeaG strain (Table 3). Based on the argumentation presented above, these proteins are also very likely to be direct phosphorylation substrates of YeaG.
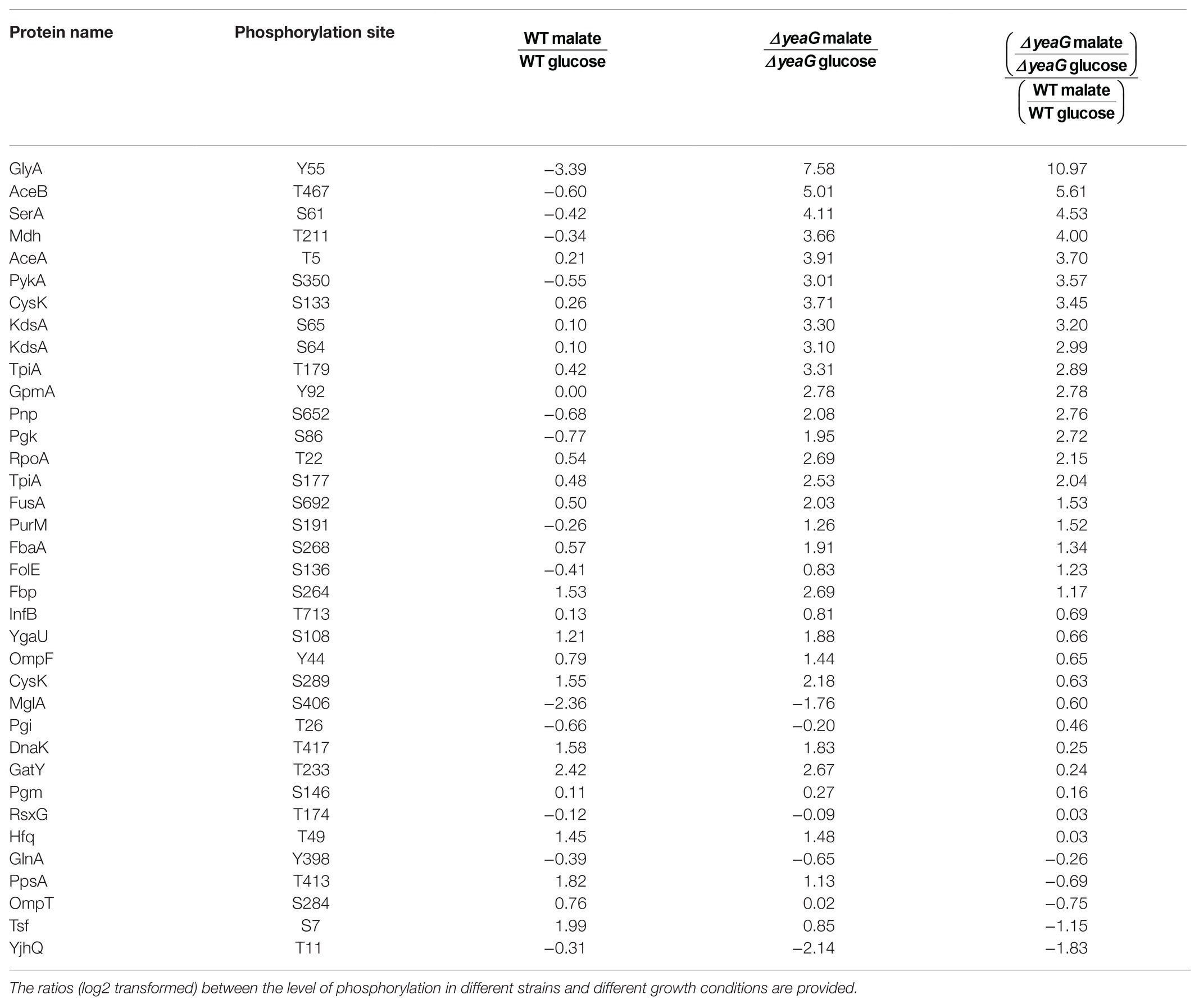
Table 2. List of phosphorylation sites quantified in both strains in both conditions.
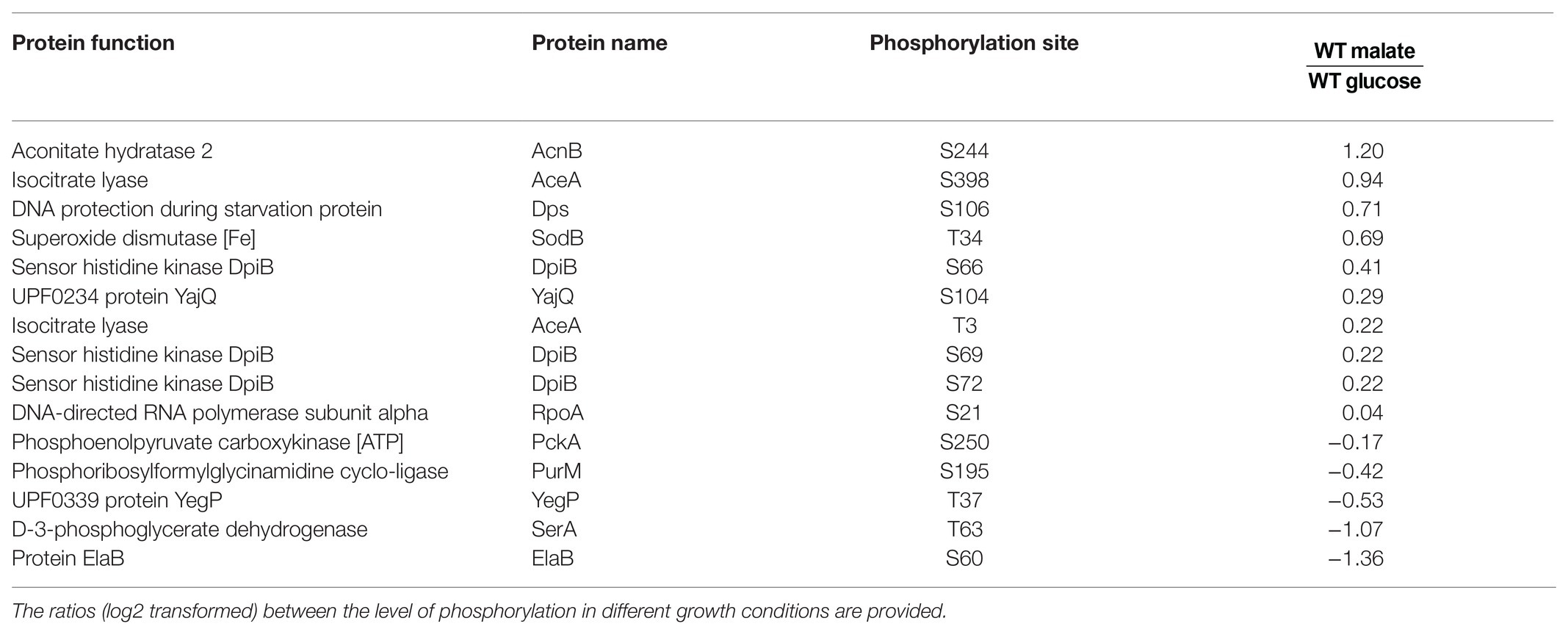
Table 3. List of proteins identified as phosphorylated in both conditions in the wild type strain but not in the ΔyeaG strain.
In order to assess which potential substrates of YeaG could be involved in the ΔyeaG phenotype during glucose to malate shift, in Figure 3A, we mapped them onto the map of E. coli central carbon metabolism. Four enzymes feature in this analysis: phosphoenolpyruvate synthase PpsA (phosphorylated at threonine 413), isocitrate lyase AceA (phosphorylated at threonine 3), aconitate hydratase AcnB (phosphorylated at serine 244), and superoxide dismutase SodB (phosphorylated atthreonine 34). To verify the mass spectrometry results that indicated these proteins to be phosphorylated, we first performed a Western blot analysis of the four putative YeaG substrates purified directly from E. coli, by using anti-phospho-serine and anti-phospho-threonine antibodies (Figure 3B). All four proteins were confirmed as being phosphorylated to some extent in vivo, with somewhat variable intensity of Western signals. Next, we asked whether YeaG is directly responsible for phosphorylating any of these proteins. To verify that, we used Phos-tag, a reagent that can specifically trap phosphorylated proteins, resulting in their lower mobility in SDS-PAGE. In this assay, only AceA tested positive for direct YeaG-dependent phosphorylation (Figure 3C) and all other putative substrates, SodA, PpsA, and AcnB tested negative (Supplementary Figure 5). Interestingly, YeaG phosphorylated AceA only in the presence of malate. This finding suggests that, during the glucose to malate shift, increasing intracellular concentration of malate can lead to activation of the kinase function of YeaG, which in turn phosphorylates AceA. AceA has been shown to have a negative influence on initial metabolic adaptation to glucose starvation and shift to another carbon source (Maharjan et al., 2005). Interestingly, in a long-term adaptation experiment, this influence of AceA was reverted by extragenic changes (mutations in other genes), rather than mutations of aceA itself, suggesting regulatory interactions with other proteins. Our data indicate that YeaG has a similarly negative influence on short-term metabolic adaptation to glucose starvation during the shift to malate. Also, our results indicate that YeaG phosphorylates AceA in the presence of malate. Based on this, one could hypothesize that YeaG exerts its negative influence, at least partly, via phosphorylation of AceA, which results in prolonging the lag phase during glucose to malate shift. This negative influence can be alleviated either by knocking out the kinase gene itself, as reported in our study, or by inactivating the gene encoding its substrate AceA (Maharjan et al., 2005). During metabolic adaptation to glucose starvation, AceA is known to work in tandem with malate synthase AceB (Maharjan et al., 2005), which is consistent with malate being the intracellular signal for activating the kinase function of YeaG. However, examination of the primary structure of YeaG indicated no known malate binding site. Therefore, the mechanism of this putative interaction of the kinase with malate remains elusive.
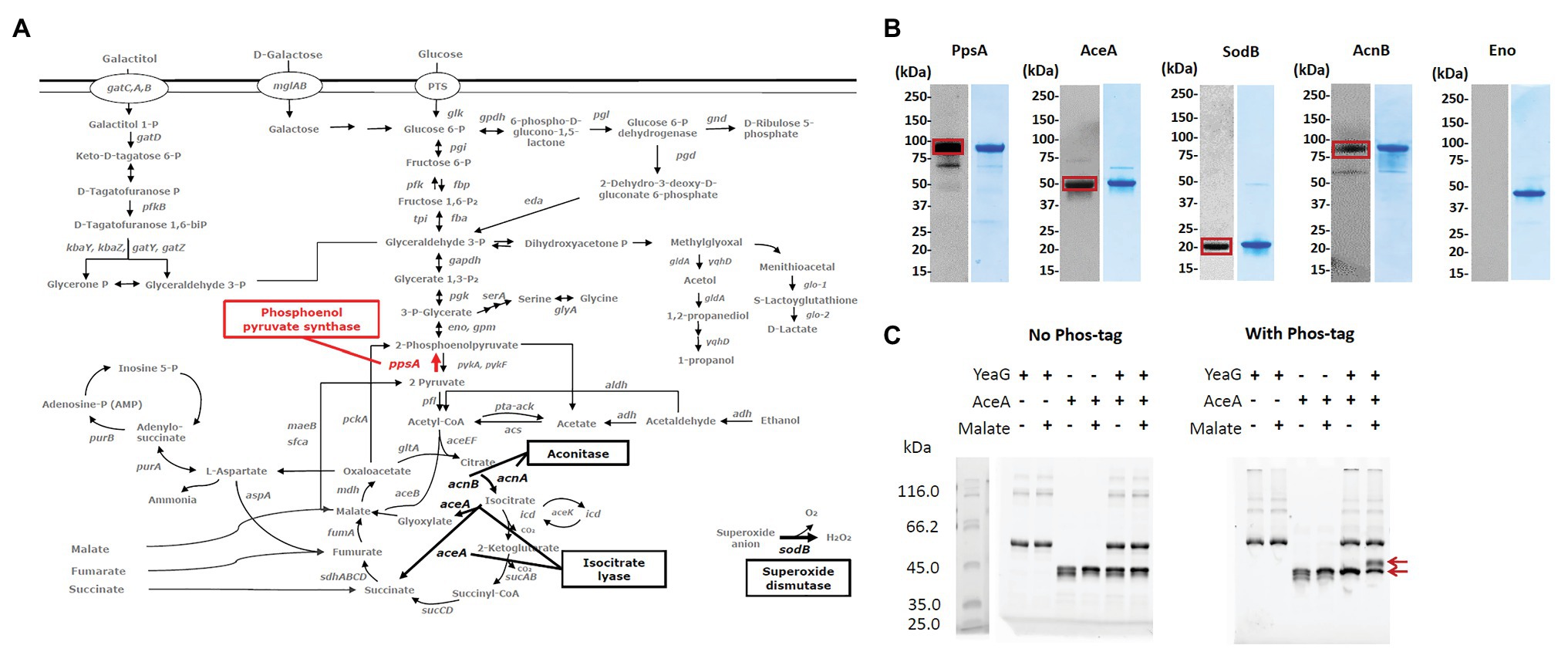
Figure 3. Putative substrates of YeaG that are potentially involved in the ΔyeaG phenotyope during glucose to malate shift. (A) Proteins that are either less phosphorylated or completely dephosphorylated in ΔyeaG strain during glucose to malate shift, mapped on the central carbon metabolism of E. coli K-12 MG1655. Red arrows signify phosphorylation events differentially downregulated in ΔyeaG, while black arrows indicate phosphorylation events detected only in the wild type strain and undetected in ΔyeaG. (B) Sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot images of proteins purified from E. coli. Purified N-terminally His-tagged proteins were probed with anti-phospho-serine and anti-phospho-threonine antibodies, according the phosphorylated residue identified in the SILAC experiment. Positive Western signals are highlighted with red squares. Sizes of examined proteins are: PpsA 87.4 kDa, AceA 48.9 kDA, AcnB 93.5 kDa, SodB 21.3 kDa, and Eno (45.7 kDa). AceA, PpsA, SodB, and Eno were probed with the anti-phospho-threonine antibody, while AcnB was probed with the anti-phospho-serine antibody. Eno was used as a negative control. (C) AceA is phosphorylated by YeaG in vitro. In vitro phosphorylation assay was performed with 6 μg of YeaG (74.5 kDa) and 5 μg of AceA (48.9 kDa), in a 20 μl reaction containing 50 mM HEPES pH 7.4, 10 mM MgCl2, 10 mM MnCl2, 100 mM KCl, 10 mM ATP, 0.1% triton-100, and 10 mM malate, and incubated at 37°C for 2 h. Presence of key components (YeaG, AceA, and malate) in the reactions is indicated with +/− above each lane. After the reaction, proteins were separated by SDS-PAGE, with and without Phos-tag (Fujifilm). Bands corresponding to AceA non-phosphorylated and phosphorylated forms are indicated by red arrows.
In this study, we have demonstrated that a SILAC-based proteome and phosphoproteome analysis, when implemented under proper physiological conditions, can be a powerful tool in identifying relevant substrates for bacterial protein kinases. The key to success is choosing conditions in which a kinase knockout has a significant phenotype. Herein, we demonstrated that inactivation of the serine/threonine kinase YeaG leads to shortening the lag phase of E. coli growth during transition from glucose to malate as the main carbon source. Under those conditions, several proteins were found to be differentially phosphorylated in the ΔyeaG strain. By focusing on metabolic enzymes potentially involved in central carbon metabolism, we narrowed down our search for putative YeaG substrates. Then, by using in vitro phosphorylation assays, we identified AceA as the substrate of YeaG, which gets phosphorylated specifically in the presence of malate. There is currently not enough evidence to firmly establish the exact mechanism of this newly observed regulatory phenomenon. Further biochemical studies will be needed to clarify the structural and functional aspects of YeaG activation by malate, and the impact of YeaG-dependent phosphorylation on AceA activity. Moreover, physiological studies, e.g., with phospho-mimetic and phospho-ablative mutants of aceA will be required to establish a clear link between YeaG-dependent phosphorylation of AceA and the observed lag phase phenotype. Before such studies are carried out, it is hard to make any definite conclusions as to the physiological relevance of YeaG-dependent phosphorylation of AceA.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
AS performed the strain construction and phenotyping experiments. AS and TG performed and analyzed the proteomics experiments. AS, CJ, MS, JK, and LS performed the protein purification, Western blotting, and phosphorylation experiments. BM assisted in analysis of proteomics experiments. IM supervised the whole study, assisted with data analysis, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Novo Nordisk Foundation (grant number NNF10CC1016517), the Independent Research Fund Denmark (grant number FSS 4183-00252A), and the Swedish Research Council (grant number 2020–03176) to IM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Rebecca M. Lennen, Ksenia Chekina, and Markus J. Herrgård for helpful discussion and advice on strain construction.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.657562/full#supplementary-material
Arora, G., Sajid, A., Gupta, M., Bhaduri, A., Kumar, P., Basu-Modak, S., et al. (2010). Understanding the role of PknJ in Mycobacterium tuberculosis: biochemical characterization and identification of novel substrate pyruvate kinase A. PLoS One 5:e10772. doi: 10.1371/journal.pone.0010772
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006–0008. doi: 10.1038/msb4100050
Bidnenko, V., Shi, L., Kobir, A., Ventroux, M., Pigeonneau, N., Henry, C., et al. (2013). Bacillus subtilis serine/threonine protein kinase YabT is involved in spore development via phosphorylation of a bacterial recombinase. Mol. Microbiol. 88, 921–935. doi: 10.1111/mmi.12233
Brunk, E., Chang, R. L., Xia, J., Hefzi, H., Yurkovich, J. T., Kim, D., et al. (2018). Characterizing posttranslational modifications in prokaryotic metabolism using a multiscale workflow. Proc. Natl. Acad. Sci. U. S. A. 115, 11096–11101. doi: 10.1073/pnas.1811971115
Charbonnier, T., Le Coq, D., McGovern, S., Calabre, M., Delumeau, O., Aymerich, S., et al. (2017). Molecular and physiological logics of the pyruvate-induced response of a novel transporter in Bacillus subtilis. mBio 8, e00976–e00917. doi: 10.1128/mBio.00976-17
Chubukov, V., Uhr, M., Le Chat, L., Kleijn, R. J., Jules, M., Link, H., et al. (2013). Transcriptional regulation is insufficient to explain substrate-induced flux changes in Bacillus subtilis. Mol. Syst. Biol. 9:709. doi: 10.1038/msb.2013.66
Corrales, R. M., Leiba, J., Cohen-Gonsaud, M., Molle, V., and Kremer, L. (2013). Mycobacterium tuberculosis S-adenosyl-l-homocysteine hydrolase is negatively regulated by Ser/Thr phosphorylation. Biochem. Biophys. Res. Commun. 430, 858–864. doi: 10.1016/j.bbrc.2012.11.038
Cox, J., Neuhauser, N., Michalski, A., Scheltema, R. A., Olsen, J. V., and Mann, M. (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805. doi: 10.1021/pr101065j
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Derouiche, A., Bidnenko, V., Grenha, R., Pigonneau, N., Ventroux, M., Franz-Wachtel, M., et al. (2013). Interaction of bacterial fatty-acid-displaced regulators with DNA is interrupted by tyrosine phosphorylation in the helix-turn-helix domain. Nucleic Acids Res. 41, 9371–9381. doi: 10.1093/nar/gkt709
Derouiche, A., Shi, L., Bidnenko, V., Ventroux, M., Pigonneau, N., Franz-Wachtel, M., et al. (2015). Bacillus subtilis SalA is a phosphorylation-dependent transcription regulator that represses scoC and activates the production of the exoprotease AprE. Mol. Microbiol. 97, 1195–1208. doi: 10.1111/mmi.13098
Deutscher, J. (2008). The mechanisms of carbon catabolite repression in bacteria. Curr. Opin. Microbiol. 11, 87–93. doi: 10.1016/j.mib.2008.02.007
Deutscher, J., Aké, F. M., Derkaoui, M., Zébré, A. C., Cao, T. N., Bouraoui, H., et al. (2014). The bacterial phosphoenolpyruvate:carbohydrate phosphotransferase system: regulation by protein phosphorylation and phosphorylation-dependent protein-protein interactions. Microbiol. Mol. Biol. Rev. 78, 231–256. doi: 10.1128/MMBR.00001-14
Elias, J. E., and Gygi, S. P. (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214. doi: 10.1038/nmeth1019
Esteves-Ferreira, A. A., Inaba, M., Fort, A., Araújo, W. L., and Sulpice, R. (2018). Nitrogen metabolism in cyanobacteria: metabolic and molecular control, growth consequences and biotechnological applications. Crit. Rev. Microbiol. 44, 541–560. doi: 10.1080/1040841X.2018.1446902
Figueira, R., Brown, D. R., Ferreira, D., Eldridge, M. J. G., Burchell, L., Pan, Z., et al. (2015). Adaptation to sustained nitrogen starvation by Escherichia coli requires the eukaryote-like serine/threonine kinase YeaG. Sci. Rep. 5:17524. doi: 10.1038/srep17524
Gómez-Mejia, A., Gámez, G., Hirschmann, S., Kluger, V., Rath, H., Böhm, S., et al. (2018). Pneumococcal metabolic adaptation and colonization are regulated by the two-component regulatory system 08. mSphere 3, e00165–e00118. doi: 10.1128/mSphere.00165-18
Goo, E., An, J. H., Kang, Y., and Hwang, I. (2015). Control of bacterial metabolism by quorum sensing. Trends Microbiol. 23, 567–576. doi: 10.1016/j.tim.2015.05.007
Gray, D. A., Dugar, G., Gamba, P., Strahl, H., Jonker, M. J., and Hamoen, L. W. (2019). Extreme slow growth as alternative strategy to survive deep starvation in bacteria. Nat. Commun. 10:890. doi: 10.1038/s41467-019-08719-8
Hansen, A. M., Chaerkady, R., Sharma, J., Díaz-Mejía, J. J., Tyagi, N., Renuse, S., et al. (2013). The Escherichia coli phosphotyrosine proteome relates to core pathways and virulence. PLoS Pathog. 9:e1003403. doi: 10.1371/journal.ppat.1003403
Jiang, J. H., Bhuiyan, M. S., Shen, H. H., Cameron, D. R., Rupasinghe, T. W. T., Wu, C. M., et al. (2019). Antibiotic resistance and host immune evasion in Staphylococcus aureus mediated by a metabolic adaptation. Proc. Natl. Acad. Sci. U. S. A. 116, 3722–3727. doi: 10.1073/pnas.1812066116
Kalantari, A., Derouiche, A., Shi, L., and Mijakovic, I. (2015). Serine/threonine/tyrosine phosphorylation regulates DNA-binding of bacterial transcription regulators. Microbiology 161, 1720–1729. doi: 10.1099/mic.0.000148
Kobir, A., Poncet, S., Bidnenko, V., Delumeau, O., Jers, C., Zouhir, S., et al. (2014). Phosphorylation of Bacillus subtilis gene regulator AbrB modulates its DNA-binding properties. Mol. Microbiol. 92, 1129–1141. doi: 10.1111/mmi.12617
Lacour, S., Bechet, E., Cozzone, A. J., Mijakovic, I., and Grangeasse, C. (2008). Tyrosine phosphorylation of the UDP-glucose dehydrogenase of Escherichia coli is at the crossroads of colanic acid synthesis and polymyxin resistance. PLoS One 3:e3053. doi: 10.1371/journal.pone.0003053
LaPorte, D. C., and Chung, T. (1985). A single gene codes for the kinase and phosphatase which regulate isocitrate dehydrogenase. J. Biol. Chem. 260, 15291–15297. doi: 10.1016/S0021-9258(18)95734-0
Liebeke, M., and Lalk, M. (2014). Staphylococcus aureus metabolic response to changing environmental conditions - a metabolomics perspective. Int. J. Med. Microbiol. 304, 222–229. doi: 10.1016/j.ijmm.2013.11.017
Link, H., Kochanowski, K., and Sauer, U. (2013). Systematic identification of allosteric protein-metabolite interactions that control enzyme activity in vivo. Nat. Biotechnol. 31, 357–361. doi: 10.1038/nbt.2489
Lukas, H., Reimann, J., Kim, O. B., Grimpo, J., and Unden, G. (2010). Regulation of aerobic and anaerobic D-malate metabolism of Escherichia coli by the LysR-type regulator DmlR (YeaT). J. Bacteriol. 192, 2503–2511. doi: 10.1128/JB.01665-09
Macek, B., Forchhammer, K., Hardouin, J., Weber-Ban, E., Grangeasse, C., and Mijakovic, I. (2019). Protein post-translational modification in bacteria. Nat. Rev. Microbiol. 17, 651–664. doi: 10.1038/s41579-019-0243-0
Macek, B., Gnad, F., Soufi, B., Kumar, C., Olsen, J. V., Mijakovic, I., et al. (2008). Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell. Proteomics 7, 299–307. doi: 10.1074/mcp.M700311-MCP200
Macek, B., Mijakovic, I., Olsen, J. V., Gnad, F., Kumar, C., Jensen, P. R., et al. (2007). The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol. Cell. Proteomics 6, 697–707. doi: 10.1074/mcp.M600464-MCP200
Maharjan, R. P., Yu, P.-L., Seeto, S., and Ferenci, T. (2005). The role of isocitrate lyase and the glyoxylate cycle in Escherichia coli growing under glucose limitation. Res. Microbiol. 156, 178–183. doi: 10.1016/j.resmic.2004.09.004
Michalski, A., Damoc, E., Hauschild, J.-P., Lange, O., Wieghaus, A., Makarov, A., et al. (2011). Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole orbitrap mass spectrometer. Mol. Cell. Proteomics 10:M111.011015. doi: 10.1074/mcp.M111.011015
Mijakovic, I., Poncet, S., Boël, G., Mazé, A., Gillet, S., Jamet, E., et al. (2003). Transmembrane modulator-dependent bacterial tyrosine kinase activates UDP-glucose dehydrogenases. EMBO J. 22, 4709–4718. doi: 10.1093/emboj/cdg458
Nussinov, R., Tsai, C. J., Xin, F., and Radivojac, P. (2012). Allosteric post-translational modification codes. Trends Biochem. Sci. 37, 447–455. doi: 10.1016/j.tibs.2012.07.001
Petranovic, D., Grangeasse, C., Macek, B., Abdillatef, M., Gueguen-Chaignon, V., Nessler, S., et al. (2009). Activation of Bacillus subtilis Ugd by the BY-kinase PtkA proceeds via phosphorylation of its residue tyrosine 70. J. Mol. Microbiol. Biotechnol. 17, 83–89. doi: 10.1159/000206635
Quiroz-Rocha, E., Bonilla-Badía, F., García-Aguilar, V., López-Pliego, L., Serrano-Román, J., Cocotl-Yañez, M., et al. (2017). Two-component system CbrA/CbrB controls alginate production in Azotobacter vinelandii. Microbiology 163, 1105–1115. doi: 10.1099/mic.0.000457
Rajpurohit, Y. S., Bihani, S. C., Waldor, M. K., and Misra, H. S. (2016). Phosphorylation of Deinococcus radiodurans RecA regulates its activity and may contribute to radioresistance. J. Biol. Chem. 291, 16672–16685. doi: 10.1074/jbc.M116.736389
Rappsilber, J., Mann, M., and Ishihama, Y. (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906. doi: 10.1038/nprot.2007.261
Rittershaus, E. S. C., Baek, S. H., Krieger, I. V., Nelson, S. J., Cheng, Y. S., Nambi, S., et al. (2018). A lysine acetyltransferase contributes to the metabolic adaptation to hypoxia in Mycobacterium tuberculosis. Cell. Chem. Biol. 25, 1495–1505. doi: 10.1016/j.chembiol.2018.09.009
Schumacher, M. A., Allen, G. S., Diel, M., Seidel, G., Hillen, W., and Brennan, R. G. (2004). Structural basis for allosteric control of the transcription regulator CcpA by the phosphoprotein HPr-Ser46-P. Cell 118, 731–741. doi: 10.1016/j.cell.2004.08.027
Shi, L., Pigeonneau, N., Ravikumar, V., Dobrinic, P., Macek, B., Franjevic, D., et al. (2014). Cross-phosphorylation of bacterial serine/threonine and tyrosine protein kinases on key regulatory residues. Front. Microbiol. 5:495. doi: 10.3389/fmicb.2014.00495
Shi, L., Ravikumar, V., Derouiche, A., Macek, B., and Mijakovic, I. (2016). Tyrosine 601 of Bacillus subtilis DnaK undergoes phosphorylation and is crucial for chaperone activity and heat shock survival. Front. Microbiol. 7:533. doi: 10.3389/fmicb.2016.00533
Singhal, A., Arora, G., Sajid, A., Maji, A., Bhat, A., Virmani, R., et al. (2013). Regulation of homocysteine metabolism by Mycobacterium tuberculosis S-adenosylhomocysteine hydrolase. Sci. Rep. 3:2264. doi: 10.1038/srep02264
Soufi, B., Gnad, F., Jensen, P. R., Petranovic, D., Mann, M., Mijakovic, I., et al. (2008). The Ser/Thr/Tyr phosphoproteome of Lactococcus lactis IL1403 reveals multiply phosphorylated proteins. Proteomics 8, 3486–3493. doi: 10.1002/pmic.200800069
Sprenger, M., Kasper, L., Hensel, M., and Hube, B. (2018). Metabolic adaptation of intracellular bacteria and fungi to macrophages. Int. J. Med. Microbiol. 308, 215–227. doi: 10.1016/j.ijmm.2017.11.001
Thomason, L. C., Sawitzke, J. A., Li, X., Costantino, N., and Court, D. L. (2014). Recombineering: genetic engineering in bacteria using homologous recombination. Curr. Protoc. Mol. Biol. 106, 1.16.1–1.16.39. doi: 10.1002/0471142727.mb0116s106
Thorsness, P. E., and Koshland, D. E. (1987). Inactivation of isocitrate dehydrogenase by phosphorylation is mediated by the negative charge of the phosphate. J. Biol. Chem. 262, 10422–10425. doi: 10.1016/S0021-9258(18)60975-5
van Heerden, J. H., Wortel, M. T., Bruggeman, F. J., Heijnen, J. J., Bollen, Y. J. M., Planqué, R., et al. (2014). Lost in transition: start-up of glycolysis yields subpopulations of nongrowing cells. Science 343:1245114. doi: 10.1126/science.1245114
Ventura, M., Rieck, B., Boldrin, F., Degiacomi, G., Bellinzoni, M., Barilone, N., et al. (2013). GarA is an essential regulator of metabolism in Mycobacterium tuberculosis. Mol. Microbiol. 90, 356–366. doi: 10.1111/mmi.12368
Virmani, R., Sajid, A., Singhal, A., Gaur, M., Joshi, J., Bothra, A., et al. (2019). The Ser/Thr protein kinase PrkC imprints phenotypic memory in Bacillus anthracis spores by phosphorylating the glycolytic enzyme enolase. J. Biol. Chem. 294, 8930–8941. doi: 10.1074/jbc.RA118.005424
Xu, Y. F., Amador-Noguez, D., Reaves, M. L., Feng, X. J., and Rabinowitz, J. D. (2012). Ultrasensitive regulation of anapleurosis via allosteric activation of PEP carboxylase. Nat. Chem. Biol. 8, 562–568. doi: 10.1038/nchembio.941
Keywords: phosphoproteome, protein kinase, SILAC, metabolic adaptation, kinase-substrate relationship
Citation: Sultan A, Jers C, Ganief TA, Shi L, Senissar M, Køhler JB, Macek B and Mijakovic I (2021) Phosphoproteome Study of Escherichia coli Devoid of Ser/Thr Kinase YeaG During the Metabolic Shift From Glucose to Malate. Front. Microbiol. 12:657562. doi: 10.3389/fmicb.2021.657562
Edited by:
Hari S. Misra, Bhabha Atomic Research Centre (BARC), IndiaReviewed by:
Andaleeb Sajid, Yale University, United StatesCopyright © 2021 Sultan, Jers, Ganief, Shi, Senissar, Køhler, Macek and Mijakovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivan Mijakovic, aXZhbi5taWpha292aWNAY2hhbG1lcnMuc2U=; aXZtaUBiaW9zdXN0YWluLmR0dS5kaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.