 Wen Tian
Wen Tian Xing Xiang
Xing Xiang Hongmei Wang
Hongmei Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 28 May 2021
Sec. Terrestrial Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.649981
The level of water table and temperature are two environmental variables shaping soil bacterial communities, particularly in peatland ecosystems. However, discerning the specific impact of these two factors on bacterial communities in natural ecosystems is challenging. To address this issue, we collected pore water samples across different months (August and November in 2017 and May 2018) with a gradient of water table changes and temperatures at the Dajiuhu peatland, Central China. The samples were analyzed with 16S rRNA high-throughput sequencing and Biolog EcoMicroplates. Bacterial communities varied in the relative abundances of dominant taxa and harbored exclusive indicator operational taxonomic units across the different months. Despite these differences, bacterial communities showed high similarities in carbon utilization, with preferences for esters (pyruvic acid methyl ester, Tween 40, Tween 80, and D-galactonic acid γ-lactone), amino acids (L-arginine and L-threonine), and amines (phenylethylamine and putrescine). However, rates of carbon utilization (as indicated by average well-color development) and metabolic diversity (McIntosh and Shannon index) in May and August were higher than those in November. Redundancy analysis revealed that the seasonal variations in bacterial communities were significantly impacted by the level of the water table, whereas the temperature had a fundamental role in bacterial carbon utilization rate. Co-occurrence analysis identified Sphingomonas, Mucilaginibacter, Novosphingobium, Lacunisphaera, Herminiimonas, and Bradyrhizobium as keystone species, which may involve in the utilization of organic compounds such as amino acids, phenols, and others. Our findings suggest that bacterial community functions were more stable than their compositions in the context of water table changes. These findings significantly expand our current understanding of the variations of bacterial community structures and metabolic functions in peatland ecosystems in the context of global warming and fluctuation of the water table.
Peatlands are vast reservoirs of organic carbon, which store approximately 33% of the terrestrial carbon (Yu et al., 2010), and thus play an important role in the global carbon budget. Microbial communities in peatland ecosystems are key players in nutrient cycles, carbon sequestration, and greenhouse gas emissions (Kip et al., 2010; Dedysh, 2011; Yang et al., 2017). Microbial communities in peatlands are sensitive to water table (WT) fluctuations (Jaatinen et al., 2007; Urbanová and Bárta, 2016; Tian et al., 2019) and temperature changes (Fenner et al., 2007; Kolton et al., 2019), which are two major factors of concern in the context of climate change. Understanding the variations of microbial community structures and carbon utilization with WT fluctuations and temperature changes will help us comprehend the dynamics of carbon cycle in peatland ecosystems under climate change scenarios.
Water table fluctuations have been demonstrated to impact microbial diversity and activities. The level of the WT can affect microbial biomass (Mäkiranta et al., 2009) and greenhouse gas emissions (Moore and Dalva, 1993; Strack et al., 2004; Hoyos-Santillan et al., 2019) in peatlands. Short-term drought will increase the bacterial diversity and microbial degradation of phenolic compounds (Kwon et al., 2013). However, long-term drainage could result in the decrease of microbial diversity and activity (Urbanová and Bárta, 2016). Storm water or flooding can cause the loss of microbial diversity and increase the dissolved organic carbon (DOC) in pore water (Fenner et al., 2005), favoring for methanogens (Freitag et al., 2010; Järveoja et al., 2016). In contrast, the increase of the oxic layer promotes aerobic respiration and diminishes substrates for anaerobic degradation and further depresses the activity and diversity of methanogens (Urbanová et al., 2011; Yrjälä et al., 2011).
Besides WT, temperature also influences microbial community structure (Guo et al., 2018) and carbon utilization (Wang R. et al., 2018). Previous studies have demonstrated temperature effects on the quality and quantity of soil organic matter (Fissore et al., 2008), microbial metabolic rates and pathways (Davidson and Janssens, 2006; Dijkstra et al., 2011), and microbial community composition and physiology (Garcia-Pichel et al., 2013). Elevated temperature may increase species diversity, metabolic activity and population growth rate (Zhou et al., 2016), and bacterial biomass (Smoot and Findlay, 2001). It is known to stimulate the biodegradation of organic matter (Kallenbach et al., 2016) via enhancing enzyme activity (Alvarez et al., 2018) and substrate bioavailability (Qin et al., 2019).
In peatland ecosystems, pore water has a vital role in nutrient transport and as a pool of inorganic and organic compounds, and the geochemistry of peat pore water may impact peatland biota (Ulanowski and Branfireun, 2013). Vegetation and sediments are interdependent and connected by the physical, hydrological, biological, and chemical processes in pore water. For instance, fluxes of trace elements and nutrients from pore water to plants are crucial processes (Carling et al., 2013; Kloss et al., 2015). Sediments serve as important sources of trace element and nutrients to pore water via diffusion, dissolution, and bioturbation (Benoit et al., 2009). Previous studies mainly focused on hydrochemical properties and geochemical processes of pore water in peatlands (Ulanowski and Branfireun, 2013; Haapalehto et al., 2014; Juckers and Watmough, 2014). For example, the concentrations of dissolved organic carbon (DOC) in peatlands have increased over last decades due to warming, enhanced precipitation, and changes in atmospheric deposition (Freeman et al., 2001; Kane et al., 2014). DOC is closely linked with the decomposition rate of organic matter, which is strongly affected by changes in the WT (Sapek et al., 2009) and temperature (Jassey et al., 2012). WT and temperature are central, dynamic factors in physicochemical characteristics of peat pore water and accompany methane oxidation (Hornibrook et al., 2009; Putkinen et al., 2012). The responses of microbial communities to changes in peat pore water remain poorly understood. We hypothesize that WT level and temperature may have differential effects on bacterial communities and microbial degradation of organic matters in pore water.
One common approach used to assess the metabolic activities of heterotrophs, using organic carbon as the carbon source, is the community-level physiological profiling analysis via Biolog EcoMicroplates (Garland and Mills, 1991). Although Biolog technology is sensitive to inoculum density and cannot reflect the potential metabolic diversity in situ (Xue et al., 2008), it can detect metabolic activities of culturable microbes and relatively quickly characterize the ecological status of microbial communities (Fisk et al., 2003). This method has been successfully applied to characterize the metabolic activities of heterotrophs in various environmental samples such as sediments (Salomo et al., 2009), wastewater (Zhang et al., 2014), groundwater (Tiquia, 2010), soil (Wang et al., 2019), and compost (Zhou et al., 2019). The next-generation sequencing technology has greatly expanded our knowledge of microbial community composition via 16S rRNA gene sequencing from environmental samples (Thompson et al., 2017). The coupling of high-throughput sequencing and Biolog technologies should provide valuable information about the associations between microbial community structures and their ecological functions in peatland ecosystems.
To test our hypothesis, we combined the high-throughput sequencing technique and Biolog technology to characterize the diversity of bacterial communities in peatland pore water samples. The purpose was to discern between effects brought about by changes in the WT and temperature on bacterial communities and their carbon utilization at the Dajiuhu peatland across spring, summer, and fall. The results provide substantive information on the variation of bacterial communities and the impacts of WT and temperature changes on carbon cycling.
Dajiuhu (1,730 m a.s.l., 31°28′ N, 110°0′ E), a subalpine peatland, is in Shennongjia at the middle reaches of Yangtze River, central China (Supplementary Figure 1), which has developed in the closed intermontane basin since the last deglaciation. The area of peatland is ca. 16 km2, and the mean peat thickness reaches >2 m (Huang et al., 2018). The local climate is dominated by the East Asian monsoon, with a mean annual rainfall of 1,560 mm and temperature of 7.2°C, respectively. Generally, the wet season is from April to September, and dry season is from October to March. The vegetation mainly consists of Carex spp., Sphagnum palustre, Sanguisorba officinalis, and Euphorbia esula accompanying shrubs (Tian et al., 2019). Sphagnum mosses are known as the major charcoal-forming plants in acidic peatlands and greatly contribute to peatland development (Tian et al., 2020). Twelve plots dominated by S. palustre were selected for sampling across three seasons to investigate the impact of WT on bacterial communities in the Dajiuhu peatland (Supplementary Figure 1).
To measure WT, 12 PVC pipes (1.5 m in length and 32 mm in diameter) were perforated with holes (6 mm in diameter) on the wall. The holes were separated with an interval of 20 cm along the entire pipe. The bottom of the perforated pipes was sealed to prevent ingress of sediment, and the top of pipe was covered with a sealed plastic bag. The pipes were drilled into the peat at each plot, and the level of the WT was determined with an Odyssey Logger (Dataflow Systems Ltd., Christchurch, New Zealand). In parallel, another set of 12 pipes was drilled to the same depth with the 12 pipes mentioned above but were perforated according to WT detected. Pore water samples from different WT depths were collected by a peristaltic pump with Teflon tube from the pipes in August and November 2017 and May 2018. Sampling in February was not practical because of the snowy and frozen season. Water samples (2 L in volume for each sampling plot) were immediately filtered through 0.22-μm sterile membranes to collect microbial cells in the field and aliquots of 50 mL water samples were kept in sterile centrifuge tubes at 4°C for the Biolog EcoMicroplate inoculation. The monthly mean temperatures were 18.2, 2.9, and 13.2°C, and monthly precipitations were 195, 63, and 363 mm in August, November 2017, and May 2018, respectively. All samples were transported to the geomicrobiology laboratory at China University of Geosciences within 12 h on ice.
Pore water temperature (PWT), dissolved oxygen (DO), pH, electrical conductivity (EC), and oxidation–reduction potential (ORP) were measured in situ using an HQ40d multiparameter meter (HACH, Loveland, CO, United States). DOC in the water samples was analyzed with an Aurora 1030W TOC Analyzer (OI Analytical, College Station, TX, United States) after filtration with 0.22-μm membrane.
Aliquots of 100-μL peat pore water samples were added into the 96-well EcoMicroplates, which were preheated for 30 min at 25°C. The inoculated microplates were incubated at 25°C in the dark, and the absorbance of microplates was detected at 590 and 750 nm per 12 h for 240 h in total by a Biolog Microstation (Biolog, Hayward, CA, United States).
The 0.22-μm membranes with microbial cells were used to extract total DNA with a PowerWater DNA Isolation Kit (QIAGEN, Düsseldorf). DNA concentration was detected with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States), and 1% agarose gel was used to check the DNA quality. The V4 region of bacterial 16S rRNA gene was amplified with the universal primer set of 515F and 806R (Walters et al., 2016). Polymerase chain reactions (PCRs) contained 25 μL 2× Premix Taq (Takara, Dalian, China), 1 μL each primer (10 mM) and 3 μL DNA (20 ng/μL) template in a volume of 50 μL. Target DNA fragments were amplified by thermocycling: 5 min at 94°C for initialization; 30 cycles of 30-s denaturation at 94°C, 30-s annealing at 52°C, and 30-s extension at 72°C, followed by 10-min final elongation at 72°C. PCR products were purified with EZNA Gel Extraction Kit (Omega Bio-tek, Norcross, GA, United States). Sequencing libraries were generated using NEBNext Ultra DNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, MA, United States) following the manufacturer’s protocols. The acquired libraries were assessed by Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, United States) and then sequenced on an Illumina HiSeq 2,500 platform with 250-bp paired-end reads (Guangdong Magigene Biotechnology Co., Guangzhou, China).
The abundance of bacteria in each sample was determined by quantitative PCR (qPCR) with the primer set of 331F: 5′-TCCTACGGGAGGCAGCAGT-3′ and 797R: 5′-GGACT ACCAGGGTATCTAATCCTGTT-3′ (Nadkarni et al., 2002). The qPCR was run in a 20-μL reaction volume containing 10 μL of TB Green Premix Ex Taq II (Takara Bio, Kusatsu, Japan), 1 μL of each primer (20 μM), 6 μL RNase-free water, 1 μL bovine serum albumin, and 1 μL template by CFX96Touch (Bio-Rad, Hercules, CA, United States). Each qPCR run was conducted with the following procedures: 95°C for 30 s, 40 cycles of 95°C for 1 s, 62°C for 30 s, and 72°C for 1 min (Xiang et al., 2014). Standard curves were constructed with 10-fold serially diluted linear pClone007 plasmids (Tsingke, Wuhan, China) containing targeted fragment from Escherichia coli DH5α (Takara Bio) for bacteria. The amplification efficiency ranged from 90 to 110% and the linear fitting showed a R2 > 0.99.
Utilization of organic carbon substrates in each well of Biolog EcoMicroplate was indicated by the chroma change due to the reduction of tetrazolium dye. The activity of microbial carbon utilization was measured by average well-color development (AWCD) (Garland and Mills, 1991). The functional diversities of microbial communities were determined as described previously (Keylock, 2005). The AWCD was calculated according to the following equations:
In eq. (1), Ci is the absorbance value of an individual reaction well, which is the deference value of between 590 and 750 nm; R is the absorbance value of control well; and n is the number of wells. If the value of (Ci − R) is negative, the absorbance is denoted as 0. The equations for calculating the McIntosh index (U) and Shannon index (H′) are as follows:
In equation (2), ni is (Ci - R) of equation (1). In equations (3) and (4), Pi represents the ratio of the absorbance value of a specific well (1–31) to the total absorbance values of all wells. These indices reflected substrate utilization and functional diversity of microbial communities.
Each Biolog EcoMicroplate contains 31 carbon sources affiliated with six groups including carbohydrates, amino acids, amines, esters, carboxylic acids, and alcohols. For principal component analysis (PCA), redox signal intensities (Rsi) were normalized with AWCD using eq. (5) (Classen et al., 2003):
In this study, optical density at 72-h incubation was used for the calculation and statistical analysis (Garland, 1996).
Paired-end raw sequences were performed according to Trimmomatic (v0.33,1) (Bolger et al., 2014) and assigned to the corresponding samples according to their barcode sequences. Sequences with ambiguous base “N,” quality score <20, and length <100 bp were discarded. High-quality sequences were subsequently merged using FLASH (v1.2.11,2) (Magoè and Salzberg, 2011) according to the overlap >10 bp between R1 and R2 reads and error ratio of the overlap region <0.1. Clean sequences were clustered at a 97% cutoff for each operational taxonomic unit (OTU) by USEARCH (v10,3) (Edgar, 2010), and singleton OTUs were filtered out. The sequences of mitochondria and chloroplast were removed, and the chimera sequences were identified by UCHIME algorithm (Edgar et al., 2011). The representative sequences for each OTU were categorized using Ribosomal Database Project classifier (v11.5,4) (Cole et al., 2014) and annotated referring to the SILVA (v132,5) (Pruesse et al., 2007). All samples were resampled to the same level (40,205 sequences) before further statistical analysis.
The original 16S rRNA gene data are available at the NCBI Sequence Read Archive6 with the accession number of PRJNA593490.
To investigate the impacts of WT and temperature on bacterial communities, nonparametric Kruskal–Wallis tests were conducted to discern the differences of physicochemical properties, alpha and functional diversities, and bacterial copies and taxonomy among samples from different months. Dunn test was used for multiple comparisons of them between bacterial communities collected in different months. The alpha diversity indices [Faith phylogenetic diversity (PD), observed species, Chao1 and Shannon] were conducted, and principal coordinate analysis (PCoA) of 16S rRNA sequences and PCA of carbon utilization data were conducted to visualize bacterial community dissimilarities across 3 months. The permutational multivariate analysis of variance (ADONIS) with the Bray–Curtis, Euclidean, and Jaccard metrics were used to test the structural and functional dissimilarities of bacterial communities across 3 months. The regressions between environmental factors and alpha and beta diversities were evaluated. The changes of community composition were performed by the chord diagram. Heatmaps revealed the carbon source preferences by bacterial communities and correlations between environmental factors, bacterial genus, and carbon sources. These analyses were performed with “vegan 2.5-7,” picante 1.8.2 “ggplot2 3.3.3,” “circlize 0.4.12,” “corrplot 0.84,” and “ggpubr 0.4.0” packages in R 3.5.1 (Oksanen et al., 2007; Kembel et al., 2010; Wickham, 2011; Gu et al., 2014; Wei, 2017; Kassambara, 2018). The relationships between bacterial communities, carbon sources, and environmental factors were performed with Monte-Carlo test of redundancy analysis (RDA) in Canoco 5.
The OTUs with relative abundances greater than 0.1% were selected across all samples, and the correlation matrices between OTUs and environmental factors were calculated. Spearman correlation coefficients (r > 0.7 or r < −0.7) with a significance of P < 0.01 [false discovery rate (FDR) corrected] were integrated into the network analysis. Each node represents one OTU or an environmental variable, and each edge represents a correlation between two nodes in the network. The co-occurrence network was characterized by topology indices including average degree, average path length, clustering coefficient, graph density, modularity, and network diameter. High values of average degree, clustering coefficient, and graph density suggest a more connected network, whereas higher average path length and diameter indicate loose associations in the network (Barberán et al., 2012). The node-level topological features (degree, betweenness, closeness, and eigenvector centrality) were calculated. High values of node topological features suggest the core position of a node in the network, whereas low values suggest a peripheral position (Ma et al., 2016). The real network was compared with 1,000 identical size Erdös-Réyni random networks (Erdös and Réyni, 1960). Nodes with high betweenness centrality values were considered as keystone species (González et al., 2010). These analyses were performed using “psych 2.0.12” and “igraph 1.2.6” packages in R (Csardi and Nepusz, 2006; Revelle, 2020), and network visualization was conducted with Gephi 0.9.2.
Indicator OTUs of microbial communities were determined using “indicspecies 1.7.9” package in R (De Cáceres and Legendre, 2009). In this study, all the OTU nodes of network were compared to determine indicator species in 3 months based on an indicator value >0.85 and P < 0.05 (FDR correction) after 999 permutation tests.
All values of physicochemical parameters showed significant differences among 3 months except EC (P > 0.05) (Table 1). Values for DOC (10.25–22.88 mg/L and 11.28–17.38 mg/L), PWT (19.80–29.20°C and 19.90–26.90°C), and WT (−5.15 to 7.81 cm, −8.07 to 2.65 cm) were greater (P < 0.05) in May and August than in November (5.08–14.34 mg/L, 7.40°C –8.40°C, −14.04 to −1.06 cm, respectively), whereas values for DO (1.12–3.83 mg/L) and ORP (128.10–273.30 mV) were the greatest (P < 0.05) in November. All peat pore water samples were acidic (pH 3.98–5.98) and pH was significantly (P < 0.05) lower in May.

Table 1. Physicochemical properties of the peat pore water samples at the Dajiuhu peatland.
A total of 2,050,535 reads for 36 samples were obtained after quality control, and 12,380 OTUs were determined at a 97% similarity level. The indices of Faith’s PD, observed species, Chao1 and Shannon showed that the richness and diversity of bacterial communities were significantly (P < 0.05) higher in May and August (Table 2). The diversity indices H’ and U of carbon utilization by bacterial communities were also higher (P < 0.05) in May and August than those in November (Table 2). Bacterial 16S rRNA gene copies were significantly (P < 0.05) higher in May and August as indicated by qPCR (Table 2). The alpha diversity indices (Faith’s PD, Chao1 and Shannon) increased (regression model, P < 0.001) with the increase of WT (Figures 1A–C). Similarly, a significant positive relationship between U and PWT was observed (regression model, P < 0.0001) (Figure 1D).
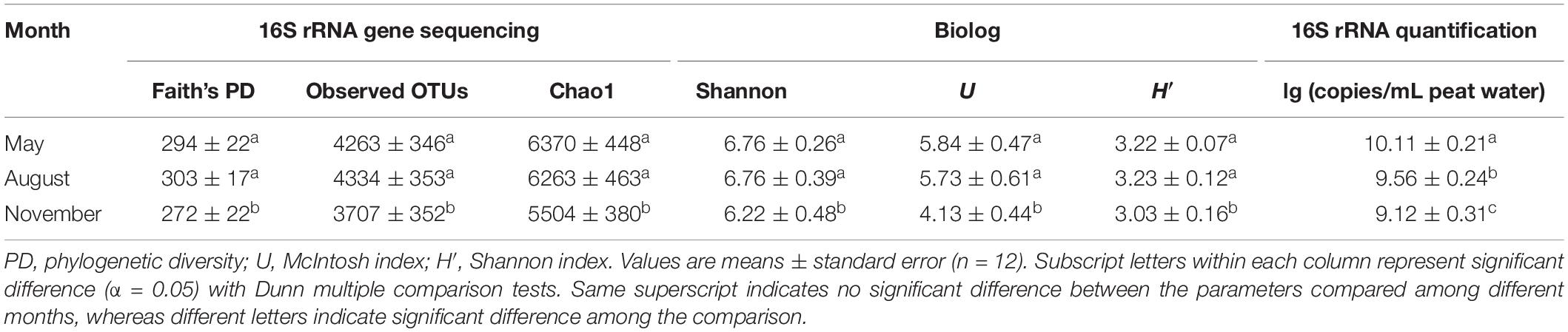
Table 2. Structural and functional diversity indices and bacterial 16S rRNA gene copies of peat pore water samples collected in different months at the Dajiuhu peatland.
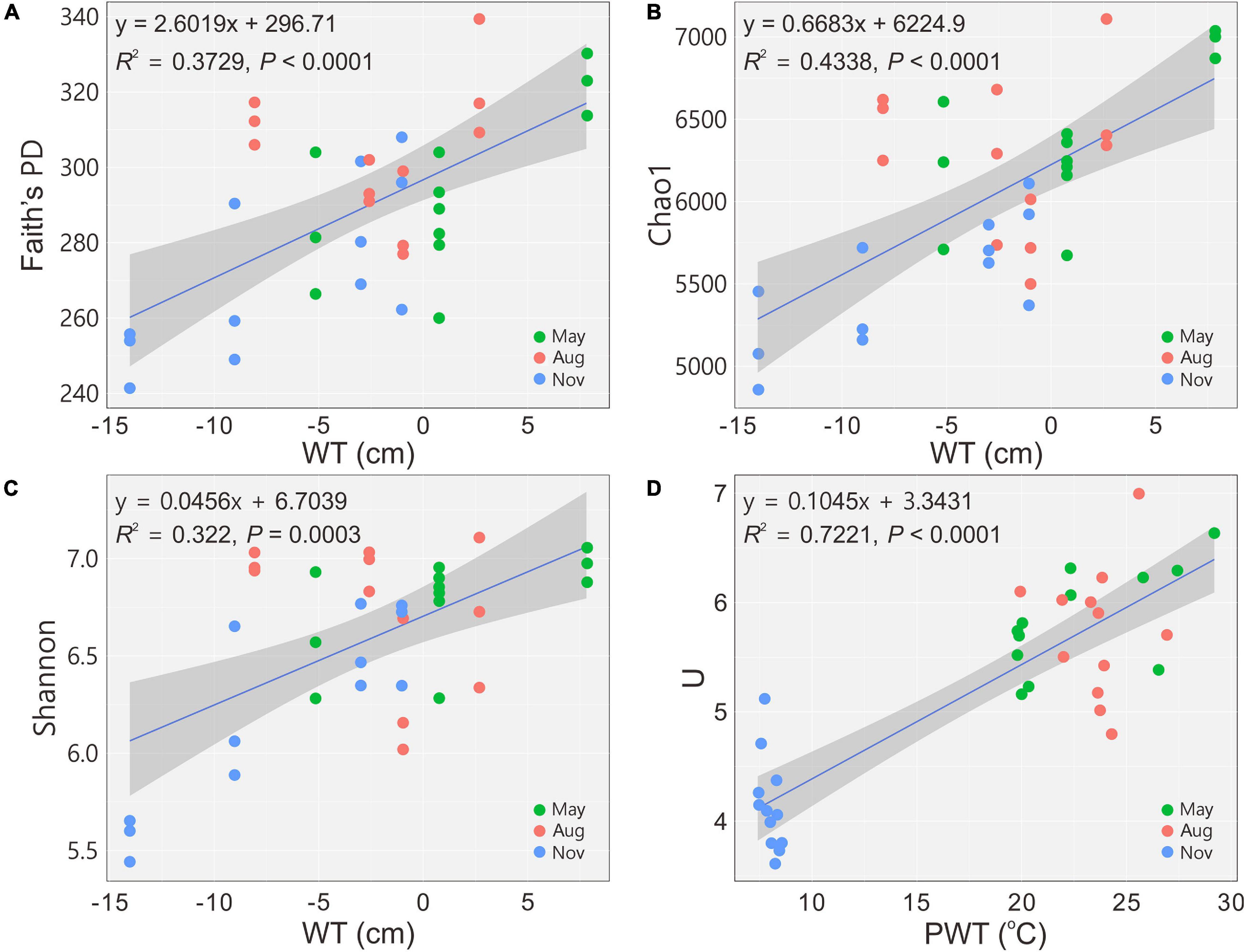
Figure 1. Changes in alpha diversity and functional diversity of bacterial communities with water table (A–C) and pore water temperature (D), respectively. Blue solid lines indicate significant linear regressions (P ≤ 0.05, n = 36); gray background represents 95% confidence level. PD, phylogenetic diversity; U, McIntosh index; WT, water table; PWT, pore water temperature; Aug, August; Nov, November.
The PCoA and PCA indicated that bacterial communities in May and August were closely related to each other but separated from those in November (Figures 2A,C). Beta diversity based on Bray–Curtis, Euclidean, and Jaccard distances of bacterial assemblages were significantly different except those in May versus in August (Supplementary Table 1). Bray–Curtis and Euclidean dissimilarities significantly (regression models, P < 0.0001) decreased as WT (Figure 2B) and PWT increased (Figure 2D), respectively.
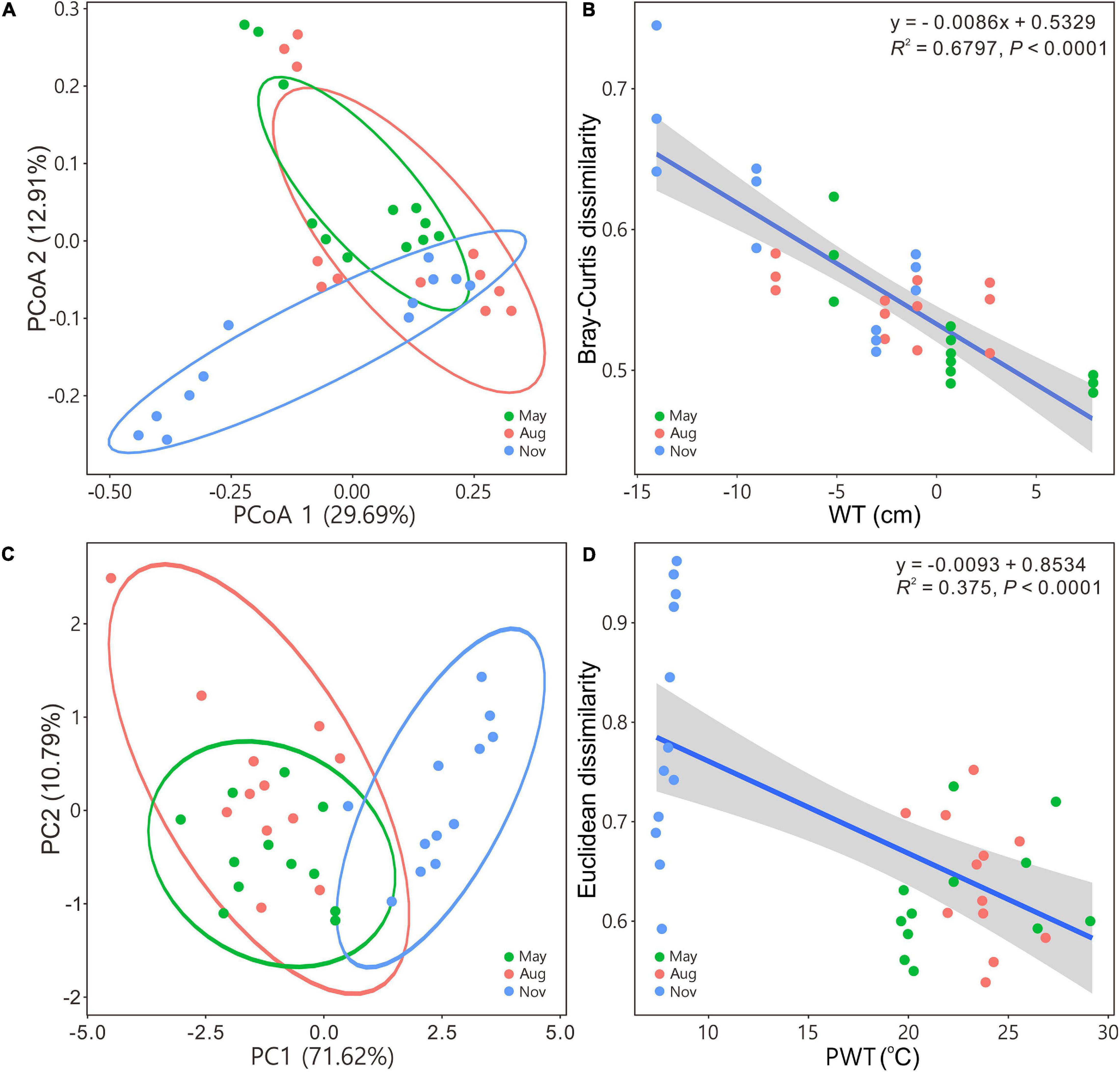
Figure 2. Principal coordinates analysis (A) and principal component analysis (C) illustrating bacterial community beta diversity among seasons. Ellipses represent 95% confidence interval. Variations in Bray–Curtis for bacterial communities and Euclidean dissimilarities for carbon metabolism with water table (B) and pore water temperature (D), respectively. Blue solid lines indicate significant linear regressions (P ≤ 0.05, n = 36); gray background represents the 95% confidence level. The abbreviations are defined in Figure 1.
All high-quality sequences were affiliated with 59 bacterial phyla and the main phyla (relative abundance >5%) were compared among 3 months (Figure 3A). Compared with bacterial communities in November, the relative abundances of Deltaproteobacteria (5.85 ± 1.00%, 7.88 ± 1.60%), Bacteroidetes (14.63 ± 3.33%, 13.67 ± 2.47%), and Acidobacteria (8.43 ± 2.03%, 7.90 ± 2.24%) were significantly (P < 0.05) higher in May and August (Supplementary Table 2), whereas that of Gammaproteobacteria (12.81 ± 2.76%, 10.32 ± 3.03%) were lower (P < 0.05). Among the main orders (relative abundance >1%), relative abundances of Sphingobacteriales (4.56 ± 1.53%, 3.88 ± 1.43%), Acidobacteriales (3.86 ± 1.32%, 2.79 ± 1.04%), Bacteroidales (3.54 ± 1.52%, 3.22 ± 1.66%), Kryptoniales (2.65 ± 1.54%, 3.94 ± 2.30%), Solibacterales (2.72 ± 0.85%, 2.89 ± 1.02%), Syntrophobacterales (1.56 ± 0.63%, 1.76 ± 0.71%), and Myxococcales (1.30 ± 0.38%, 1.19 ± 0.29%) were significantly (P < 0.05) higher in May and August, whereas Betaproteobacteriales (10.78 ± 5.27%), Rhizobiales (6.36 ± 2.29%), and Chlamydiales (5.53 ± 3.26%) dominated in November (Figure 3B and Supplementary Table 3). As for the main genus (relative abundance >0.5%), Candidatus Solibacter (1.62 ± 0.61%, 1.68 ± 0.64%) and Methylocystis (1.50 ± 0.52%, 1.17 ± 0.54%) were significantly (P < 0.05) enriched in May and August, whereas November had more Spirochaeta (1.72 ± 1.38%) (Figure 3C and Supplementary Table 4).
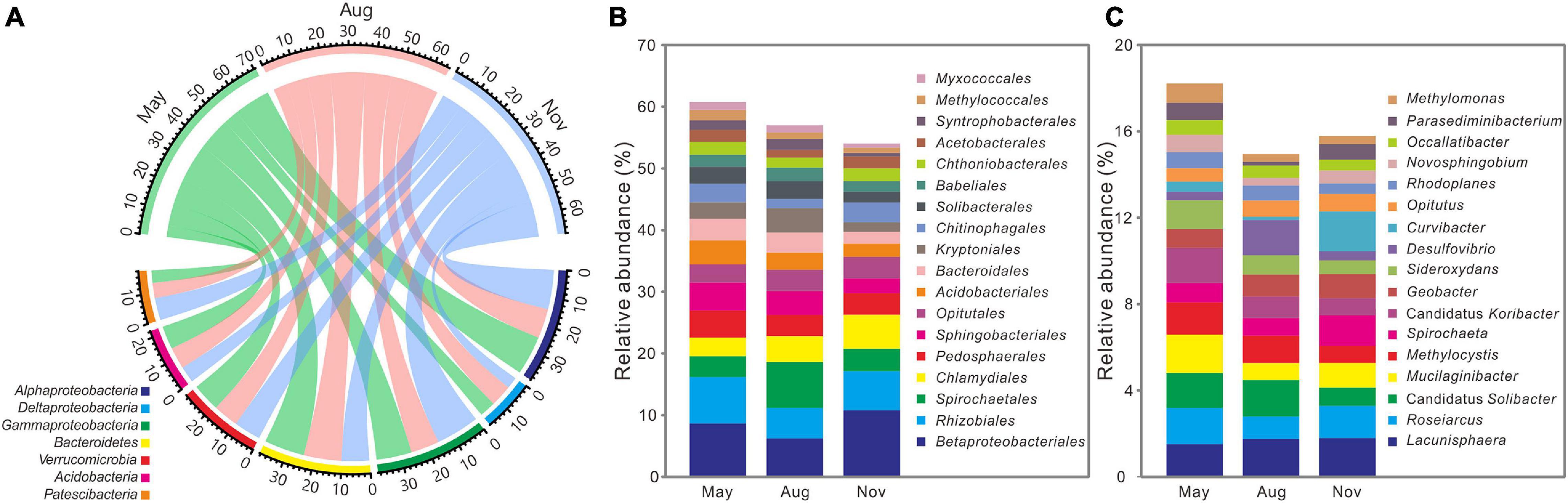
Figure 3. Distributions of the dominant phyla (relative abundance >5%) (A), orders (relative abundance >1%) (B), and genus (relative abundance >0.5%) (C) across 3 months (n = 36) at the Dajiuhu peatland. The numbers on the arc present the relative abundance.
AWCD curves exhibited a lag phase in the first 24 h and increased dramatically from 24 to 72 h (Figure 4A). The increase slowed down afterward, and the carbon metabolic rate of bacterial communities stabilized beyond 168 h. The rate and preference of carbon utilization by bacterial communities were distinctly different among samples collected in different months as indicated by the heatmap (Figure 4B). Bacterial communities in May and August showed significantly higher metabolic rate of carbon utilization than those in November (Supplementary Table 5). However, all bacterial communities showed similar preference for carbon utilization. Overall, esters, amino acids and amines were preferentially utilized, followed by carbohydrates and carboxylic acids, and the alcohols were the lowest (Supplementary Table 6).
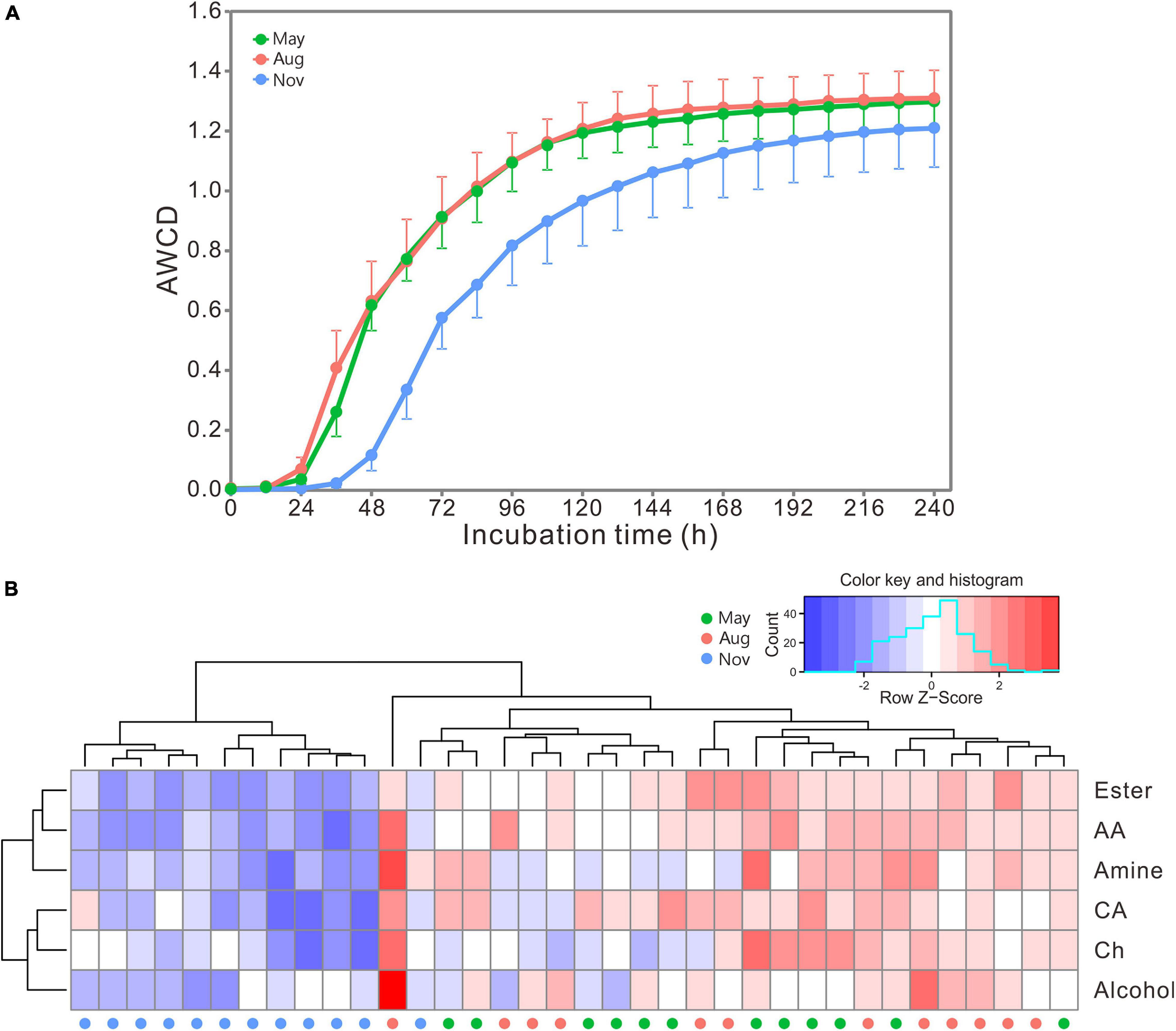
Figure 4. (A) The AWCD changes of carbon sources with incubation time. Bars represent the stand errors (n = 12). (B) Heatmap of bacterial preference for carbon source utilization. The color and histogram account for carbon metabolic rate of bacterial communities. AA, amino acid; CA, carboxylic acid; Ch, carbohydrate; Aug, August; Nov, November.
The loading scores of 31 carbon sources in the first two principal components are shown in Supplementary Table 7. The higher the loading scores were, the larger the effects of carbon sources in the principal components were. Overall, 10 carbon sources including four ester compounds (pyruvic acid methyl ester, Tween 40, Tween 80, and D-galactonic acid γ-lactone), two amino acids (L-arginine and L-threonine) and two amines (phenylethylamine and putrescine), one carboxylic acid substrate (itaconic acid), and one alcohol (D-mannitol) mainly impacted on the PC1. Besides putrescine, itaconic acid, Tween 40, and Tween 80, glucose-1-phosphate, glycyl-L-glutamic acid N-acetyl-D-glucosamine, and L-phenylalanine also impacted PC2 (Supplementary Table 7).
The bacterial network in peat pore water consisted of 142 nodes and 725 edges (Figure 5A). The results showed that more positive interactions were observed in peat pore water bacterial network, and the connections within a module were more intense compared with those among the modules. The degree of integrated network was distributed based on power law in peat pore water (Supplementary Figure 2A), indicating a scale-free distribution and nonrandom co-occurrence pattern compared with Erdös–Réyni random networks (Supplementary Figure 2B). The average path length was 3.62 with a diameter of 12. The average clustering coefficient was 0.556, and the modularity index was 0.547 (>0.4), which suggested a modular structure of the network (Newman, 2006). In the integrated network, WT had high values of degree, betweenness, and eigenvector centralities compared to other environmental factors (Figure 5A).
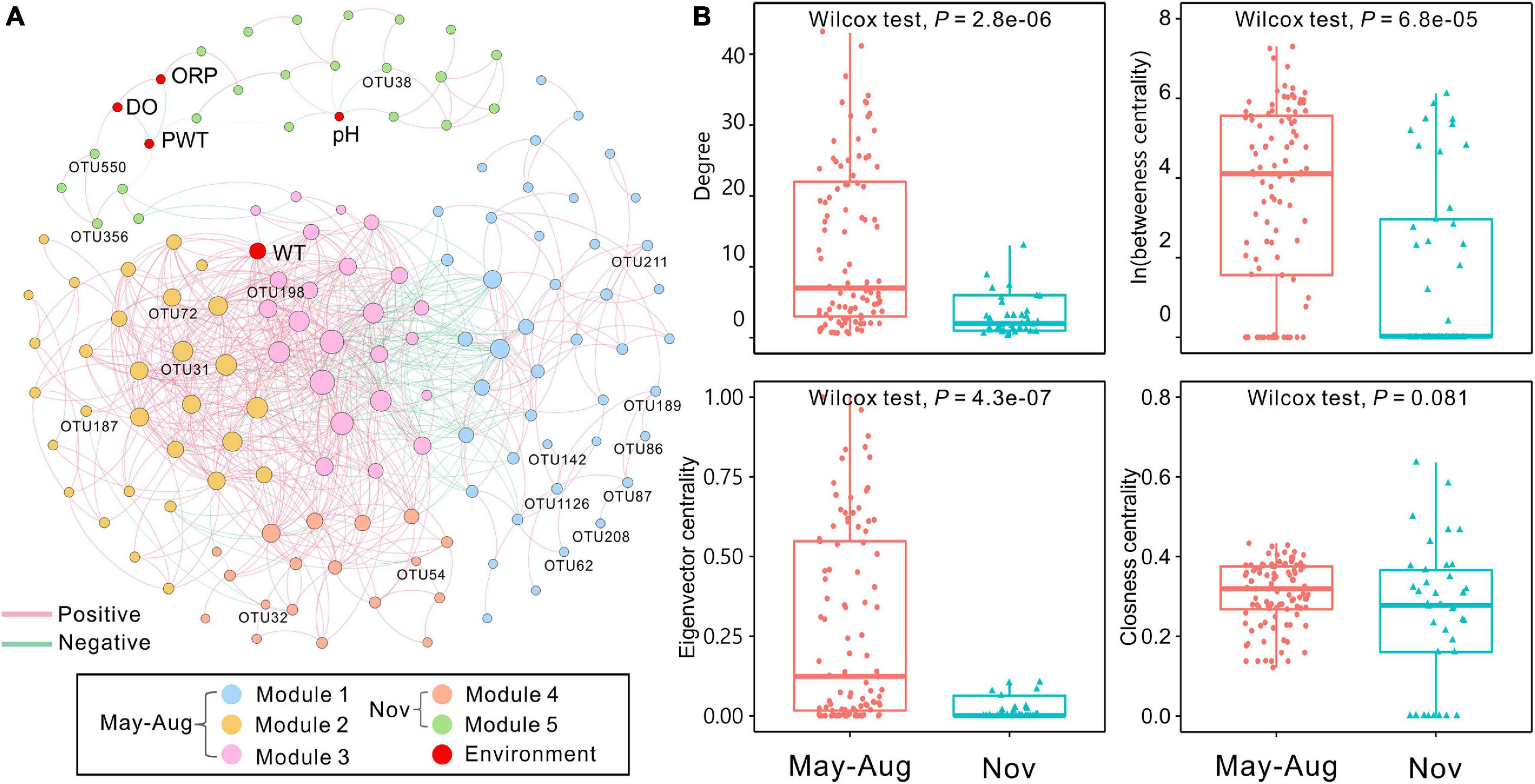
Figure 5. Co-occurrence network of bacterial communities among sampling months based on Spearman correlation analysis. (A) The nodes were colored by modularity. Each edge indicates a strong correlation (Spearman correlation coefficient of >0.7 or <–0.7) with a significance of P < 0.01 (false-discovery rate-corrected). The size of each node is proportional to the degree, and the thickness of edges is proportional to the absolute value of the Spearman correlation coefficient. Environmental variables and indicator OTUs are marked in the network. (B) Comparison of node-level topological features (degree, betweenness, eigenvector, and closeness centrality) of May–August and November bacterial communities. DO, dissolved oxygen; ORP, oxidation–reduction potential; PWT, pore water temperature; WT, water table. The significant differences between May–August and November are determined by Wilcoxon test (α = 0.05).
The 142 nodes were grouped into five modules in the network (Figure 5A). Proteobacteria were commonly found in all modules (Supplementary Table 8). Modules 1, 2, and 3 were dominated by Proteobacteria and Bacteroidetes. Nodes of module 4 were mainly belonged to Patescibacteria, Proteobacteria, and Verrucomicrobia. Module 5 was dominated by Proteobacteria. In total, 17 indicator OTUs were identified, and 12 of them were exclusively observed in May–August samples, and the other five indicator OTUs were found in samples of November (Supplementary Table 9). These obligate indicator OTUs were accordingly marked into the network and represented the distributions of bacterial communities in May–August and November. The indicator OTUs in May–August were in modules 1, 2, and 3, whereas those in November were affiliated with modules 4 and 5 (Figure 5A and Supplementary Table 9). Additionally, the node-level topological features (degree, betweenness centrality, and eigenvector centrality) significantly (P < 0.001) differed in May–August and November, whereas closeness centrality showed no significant differences (Figure 5B). According to betweenness centrality values, the top 10 nodes identified as keystone species were Sphingomonas, Mucilaginibacter, Novosphingobium, Lacunisphaera, Herminiimonas, and Bradyrhizobium (Supplementary Table 10).
RDA was applied to examine the effects of environmental factors on abundant bacterial genera (relative abundance >0.5%). The structures of bacterial communities were significantly (P < 0.05) affected by WT and pH (Figure 6A and Supplementary Table 11). Axis 1 and axis 2 explained 32.46 and 9% of the total variances, respectively. Candidatus Koribacter, Candidatus Solibacter, Desulfovibrio, Geobacter, Lacunisphaera, Methylomonas, Opitutus, Sideroxydans, and Spirochaeta were positively related to WT, whereas 10 of the 17 abundant genera negatively correlated with pH (Supplementary Figure 3A). In contrast, the carbon utilization of bacterial communities was significantly (P < 0.05) affected by PWT and DOC. Axis 1 and axis 2 explained 65.87 and 34.91% of the total variances, respectively (Figure 6B). The utilization of all carbon sources was positively related to the PWT and DOC (Supplementary Figure 3B). Altogether, the structure and carbon metabolism of bacterial communities were mainly shaped by WT and PWT, respectively.
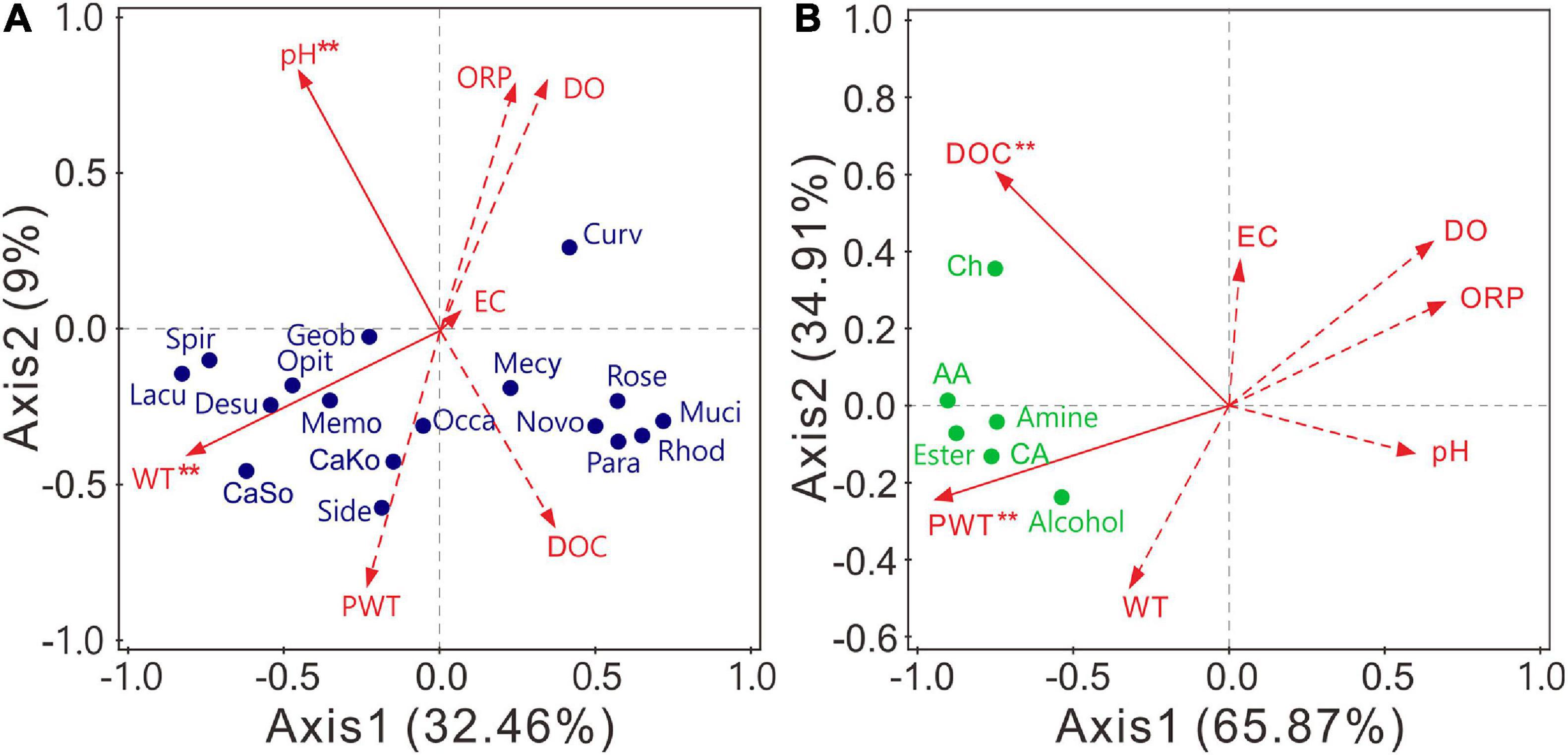
Figure 6. Redundancy analysis (RDA) indicating the influence of environmental variables on bacterial community composition (A) and carbon source utilization (B). Variables with significant impacts (α = 0.05) are marked with red asterisk. Lacu, Lacunisphaera; Rose, Roseiarcus; CaSo, Candidatus Solibacter; Muci, Mucilaginibacter; Mecy, Methylocystis; Spir, Spirochaeta; CaKo, Candidatus Koribacter; Geob, Geobacter; Side, Sideroxydans; Desu, Desulfovibrio; Curv, Curvibacter; Opit, Opitutus; Rhod, Rhodoplanes; Novembero, Novemberosphingobium; Occa, Occallatibacter; Para, Parasediminibacterium; Memo, Methylomonas. DOC, dissolved organic carbon; DO, dissolved oxygen; ORP, oxidation–reduction potential; EC, electrical conductivity; PWT, pore water temperature; WT, water table. AA, amino acid; CA, carboxylic acid; Ch, carbohydrate.
Bacterial communities in natural ecosystems are usually constrained by multiple factors. By combining 16S rRNA sequencing and Biolog techniques, we can demonstrate that WT and pH significantly shaped the compositions of bacterial communities (Figure 6A), whereas temperature and DOC largely impacted the rate of carbon utilization (Figure 6B). Statistically, carbon utilization was dissociated from bacterial community composition, possibly indicating that different taxa in bacterial communities had similar metabolic functions (Louca et al., 2018). Such functional redundancy could be unaffected by variation in taxa with the season of sampling.
Bacterial communities from peat pore water were distinctly diverse between May–August and November in the Dajiuhu peatland. The increase in the WT showed positive correlations with bacterial alpha diversity (Figures 1A–C) and decreased the bacterial community dissimilarities (Figure 2B) based on 16S rRNA gene sequencing. WT fluctuation due to precipitations in May–August and November can directly affect bacterial communities by changing physicochemical properties. For example, DO and ORP (Table 1) were negatively related to the WT (Supplementary Figure 4). WT fluctuation can change the depth of the oxic–anoxic interface (Tian et al., 2019), resulting in the subsequent shifts in electron donors and acceptors (Brune et al., 2000; Daffonchio et al., 2006). High WT facilitates anaerobic processes, and low WT accelerates the biodegradation of organic matters due to the penetration of oxygen and expansion of the oxic zone. Furthermore, WT also impacted dissimilar bacterial communities (Figures 2A,B) and composition (Figures 3A–C) in peat pore water.
Within the ambient environmental range, increasing temperature enhances microbial activities and promotes the decomposition of organic matter (Staley et al., 2015). Low temperature slows down metabolic activity and contributes to organic matter sequestration (Davidson and Janssens, 2006; Sanz-Lázaro et al., 2011). In our results, the decrease in peat PWT from May (22.78 ± 3.49°C) and August (23.55 ± 1.78°C) to November (7.98 ± 0.36°C) substantially decreased bacterial functional diversity (Figure 1D) and the carbon utilization rate (Figure 4A). The decrease in the temperature also shifted functional community structure (Figure metricconverterProductID2C2C) and increased the dissimilarities in metabolic structures of bacterial communities (Figure 2D). DOC was positively related with the PWT (Supplementary Figure 4). The relatively high concentration of DOC (Table 1) in peat pore water samples of May and August may contribute to enhanced heterotrophic respiration and metabolic rates as indicated by RDA between carbon utilization and environmental variables (Figure 6B).
Despite the differences in bacterial communities over the three seasons, the preferences for organic carbon substrates remained similar. Microbial communities in pore water preferred to utilize esters, amino acids, and amines (Supplementary Tables 6, 7), possibly derived from decaying plant debris, microbial biomass, and soil organic matter. The decomposition rate of plant debris particularly in Sphagnum-dominated peatland was extremely low due to the recalcitrance of high-molecular-weight organic compounds (e.g., polyphenols, cellulose, hemicellulose, and lignin derivatives) derived from Sphagnum under acidic conditions (Bragazza et al., 2007; Pankratov et al., 2011; Robroek et al., 2016). Soil-borne fungi have extracellular enzymes that can degrade recalcitrant aromatic and aliphatic compounds such as those in peatlands (Thormann, 2006; Juan-Ovejero et al., 2020). These microbes have peroxidases, laccases, and ligninases, which can produce intermediates that can subsequently serve as carbon sources for heterotrophic bacteria. Fungal biomass outweighs bacteria and upon decay and lysis may also support bacterial growth. The pore water DOC data suggest that it is mainly sourced from microbial metabolites or biodegradation of organic matter pool, with a mean concentration of 18.07 ± 10.60 mg/L. Fractions of this DOC has been identified as humic-like compounds (Wang R. et al., 2018).
Co-occurrence network analyses can illustrate the microbial complexity and interrelationship among community members (Barberán et al., 2012). It is increasingly evident that microbial communities are structured and form complex interconnected networks via modularity (Ma et al., 2016; Jiao et al., 2019). The bacterial network of peat pore water showed a clear modular structure and was grouped into five modules (Figure 5A). Nodes in the modules can perform different functions (Newman, 2006) or prefer different habitats (Tian et al., 2020), as well as respond to environmental variables (de Vries et al., 2012). The WT was strongly linked to bacterial communities in the co-occurrence network (Figure 5A). The WT had remarkably high degrees of betweenness and eigenvector centralities compared with other environmental factors, suggesting a significant influence of WT on bacterial communities.
In the network analysis, samples from May and August were treated as one group due to high similarities based on physicochemical parameters, alpha and beta diversities, community dissimilarities, taxonomic compositions, and carbon utilizations. Distinct indicator species between May–August and November may reflect WT fluctuation. In May–August, 12 indicator OTUs were identified and mapped into modules 1, 2, and 3 (Supplementary Table 9). Among these indicator OTUs or species, Rhodoplanes spp. grow in subsurface anoxic environments (Lodha et al., 2015) and are capable of denitrification (Hiraishi and Ueda, 1994), as well as nitrogen fixation (Too et al., 2018). Members of Desulfomonile, Smithella, and Syntrophobacter affiliated with Deltaproteobacteria are anaerobic and mesophilic bacteria and can potentially degrade hydrocarbons and oxidize propionate in peatlands (Harmsen et al., 1998; Gray et al., 2011; Schmidt et al., 2016). Candidatus Solibacter spp. are capable of utilizing various carbon sources and reducing nitrate and nitrite under acidic, water-logged conditions (Kanokratana et al., 2011). These species may prefer high WT and perform specific functions under anoxic conditions. In contrast, the five obligate indicator OTUs of November were located in modules 4 and 5 (Supplementary Table 9). Members of Reyranella and Aquitalea can decompose polycyclic aromatics and cellulose and contribute to plant litter degradation under aerobic conditions (Woo et al., 2014; Lee et al., 2017). Many Dechloromonas spp. are capable of nitrogen fixation and their relative abundances increase in forest with a low WT (Leppänen et al., 2015). Therefore, functional properties of these indicator species may indicate environmental changes, for example, in the WT and oxygen concentration. Indeed, WT fluctuation (1 cm in May, -2 cm in August, -7 cm in November) and changes in DO concentration (0.34 mg/L in May, 1.34 mg/L in August, and 2.75 mg/L in November) in peat pore water coincided well with the ecological functions of indicator species.
Based on the betweenness centrality scores, Sphingomonas, Mucilaginibacter, Novosphingobium, Lacunisphaera, Herminiimonas, and Bradyrhizobium were identified as keystone species, which were mainly in module 1 (Supplementary Table 10). Acid-tolerant and oligotrophic Sphingomonas spp. participate in microbial communities under extreme environmental conditions (Ogita et al., 2006). Members of Mucilaginibacter isolated in ombrotrophic peatlands are able to utilize a broad range of biopolymers, particularly polysaccharides and proteins (Pankratov et al., 2007). Our results showed that Mucilaginibacter was positively related with carbohydrates and amino acids (Supplementary Table 12). The phenolic compounds from Sphagnum-derived litter could be degraded by several Novosphingobium spp. (DeAngelis et al., 2013). Herminiimonas affiliated with Proteobacteria are capable of degrading aromatic hydrocarbons (Kim et al., 2014). Novosphingobium and Herminiimonas can also utilize aromatics and were positively correlated to amines in the study (Supplementary Table 12). Bradyrhizobium spp. are common nitrogen-fixing soil bacteria (Bragina et al., 2013) and provide nitrogen for microbial communities in the peat pore water. These keystone species play an important role in nutrient cycle and sustain the bacterial co-occurrence network with broad niche and versatile or specific metabolic functions in peatland ecosystems. Keystone OTUs with higher betweenness centrality exhibited much tighter connections with the WT compared to other environmental variables (Figure 5A), which further verifies the large impact of WT on bacterial communities.
Precipitation and temperature are two fundamental factors in the global climate change. In the middle reach of the Yangtze River, high precipitation is usually coupled with high temperature due to the impact of East Asian monsoon. Thus, it is challenging to distinguish how an individual environmental variable impacts bacterial communities. Seasonal sampling strategy and combination of DNA sequencing and Biolog technique enabled us to investigate bacterial communities along gradients of temperature and WT at the level of DNA and organic carbon metabolism. We clearly distinguished the impacts of the WT and temperature on bacterial communities. WT significantly influenced the structures of bacterial community such as composition, relative abundance, and diversity. Carbon utilization rate and metabolic diversity were largely controlled by temperature. The variation in the WT fluctuation without extreme drought and flooding events can change bacterial community structure, but not the bacterial degradation of organic matter (i.e., preference of carbon sources and rate of utilization) in the Dajiuhu peatland. These data indicate that within the time span of this study, microbial functions may be more stable than microbial compositions. This may be attributed to metabolic function redundancy in natural ecosystems. However, the impacts of extreme precipitation events such as the once-in-a-century flood in summer of 2020 and the depth of the WT over a long term on microbial communities remain unknown and merit further investigation.
Water table and temperature impacted bacterial pore water communities at the DNA and organic carbon metabolic levels, respectively, in the Dajiuhu peatland. WT level mainly impacted the structures of bacterial communities (alpha diversity, dissimilarity, relative abundance, and composition) and microbial interactions (co-occurrence pattern). Temperature largely shaped the functional diversity, functional community dissimilarity, and carbon utilization rate. Bacterial groups interacted particularly intensely during May–August with high WT and high temperature. Keystone species were identified that were mainly attributed to the decomposition of organic matter and nitrogen fixation in oligotrophic and acid peatlands. They sustained complex connections in the bacterial network of peat pore water. The combination of 16S rRNA sequencing and Biolog technique can differentiate the impacts of the WT and temperature on bacterial taxa and associated functions. This study substantiates our understanding of microbial community responses to environmental variables in peatland ecosystems. However, long-term series of sampling and in situ/in vivo detection of microbial activity are warranted to corroborate the outcomes of this study.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
WT did the experiments, data analysis, figures drawing, and manuscript drafting. XX conducted the field work and involved in writing. HW designed the study, provided the financial support and wrote up the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by the National Natural Science Foundation of China (Nos. 41572325 and 91951208) and the Fundamental Research Funds for the Central Universities, China University of Geosciences (CUGCJ1703 and CUGQY1922).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Ruicheng Wang and Ying Xu for the sampling in the field. We also thank the editor SD and the three reviewers for constructive suggestions that were helpful in improving the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.649981/full#supplementary-material
Alvarez, G., Shahzad, T., Andanson, L., Bahn, M., Wallenstein, M. D., and Fontaine, S. (2018). Catalytic power of enzymes decreases with temperature: new insights for understanding soil C cycling and microbial ecology under warming. Global. Change Biol. 24, 4238–4250. doi: 10.1111/gcb.14281
Barberán, A., Bates, S. T., Casamayor, E. O., and Fierer, N. (2012). Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6, 343–351. doi: 10.1038/ismej.2011.119
Benoit, J. M., Shull, D. H., Harvey, R. M., and Beal, S. A. (2009). Effect of bioirrigation on sediment-water exchange of methylmercury in Boston Harbor, Massachusetts. Environ. Sci. Technol. 43, 3669–3674. doi: 10.1021/es803552q
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bragazza, L., Siffi, C., Iacumin, P., and Gerdol, R. (2007). Mass loss and nutrient release during litter decay in peatland: the role of microbial adaptability to litter chemistry. Soil Biol. Biochem. 39, 257–267. doi: 10.1016/j.soilbio.2006.07.014
Bragina, A., Berg, C., Müller, H., Moser, D., and Berg, G. (2013). Insights into functional bacterial diversity and its effects on Alpine bog ecosystem functioning. Sci. Rep. 3:1955. doi: 10.1038/srep01955
Brune, A., Frenzel, P., and Cypionka, H. (2000). Life at the oxic–anoxic interface: microbial activities and adaptations. FEMS Microbiol. Rev. 24, 691–710. doi: 10.1016/S0168-6445(00)00054-1
Carling, G., Richards, D., Hoven, H., Miller, T., Fernandez, D., Rudd, A., et al. (2013). Relationships of surface water, pore water, and sediment chemistry in wetlands adjacent to Great Salt Lake, Utah, and potential impacts on plant community health. Sci. Total Environ. 443, 798–811. doi: 10.1016/j.scitotenv.2012.11.063
Classen, A. T., Boyle, S. I., Haskins, K. E., Overby, S. T., and Hart, S. C. (2003). Community-level physiological profiles of bacteria and fungi: plate type and incubation temperature influences on contrasting soils. FEMS Microbiol. Ecol. 44, 319–328. doi: 10.1016/S0168-6496(03)00068-0
Cole, J. R., Wang, Q., Fish, J. A., Chai, B., McGarrell, D. M., Sun, Y., et al. (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. doi: 10.1093/nar/gkt1244
Csardi, G., and Nepusz, T. (2006). The igraph software package for complex network research. InterJournal Complex Syst. 1695, 1–9. doi: 10.1186/1471-2105-12-455
Daffonchio, D., Borin, S., Brusa, T., Brusetti, L., Van Der Wielen, P. W., Bolhuis, H., et al. (2006). Stratified prokaryote network in the oxic–anoxic transition of a deep-sea halocline. Nature 440, 203–207. doi: 10.1038/nature04418
Davidson, E. A., and Janssens, I. A. (2006). Temperature sensitivity of soil carbon decomposition and feedbacks to climate change. Nature 440, 165–173. doi: 10.1038/nature04514
De Cáceres, M., and Legendre, P. (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. doi: 10.1890/08-1823.1
de Vries, F. T., Liiri, M. E., Bjørnlund, L., Bowker, M. A., Christensen, S., Setälä, H. M., et al. (2012). Land use alters the resistance and resilience of soil food webs to drought. Nat. Clim. Change 2, 276–280. doi: 10.1038/nclimate1368
DeAngelis, K. M., Chivian, D., Fortney, J. L., Arkin, A. P., Simmons, B., Hazen, T. C., et al. (2013). Changes in microbial dynamics during long-term decomposition in tropical forests. Soil Biol. Biochem. 66, 60–68. doi: 10.1016/j.soilbio.2013.06.010
Dedysh, S. N. (2011). Cultivating uncultured bacteria from northern wetlands: knowledge gained and remaining gaps. Front. Microbiol. 2:184–198. doi: 10.3389/fmicb.2011.00184
Dijkstra, P., Thomas, S. C., Heinrich, P. L., Koch, G. W., Schwartz, E., and Hungate, B. A. (2011). Effect of temperature on metabolic activity of intact microbial communities: Evidence for altered metabolic pathway activity but not for increased maintenance respiration and reduced carbon use efficiency. Soil Biol. Biochem. 43, 2023–2031. doi: 10.1016/j.soilbio.2011.05.018
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Erdös, P., and Réyni, A. (1960). On the evolution of random graphs. T. Am. Math. Soc. 286, 257–274. doi: 10.2307/1999405
Fenner, N., Freeman, C., Lock, M. A., Harmens, H., Reynolds, B., and Sparks, T. (2007). Interactions between elevated CO2 and warming could amplify DOC exports from peatland catchments. Environ. Sci. Technol. 41, 3146–3152. doi: 10.1021/es061765v
Fenner, N., Freeman, C., and Reynolds, B. (2005). Hydrological effects on the diversity of phenolic degrading bacteria in a peatland: implications for carbon cycling. Soil Biol. Biochem. 37, 1277–1287. doi: 10.1016/j.soilbio.2004.11.024
Fisk, M. C., Ruether, K. F., and Yavitt, J. B. (2003). Microbial activity and functional composition among northern peatland ecosystems. Soil Biol. Biochem. 35, 591–602. doi: 10.1016/S0038-0717(03)00053-1
Fissore, C., Giardina, C. P., Kolka, R. K., Trettin, C. C., King, G. M., Jurgensen, M. F., et al. (2008). Temperature and vegetation effects on soil organic carbon quality along a forested mean annual temperature gradient in North America. Global Change Biol. 14, 193–205. doi: 10.1111/j.1365-2486.2007.01478.x
Freeman, C., Evans, C., Monteith, D., Reynolds, B., and Fenner, N. (2001). Export of organic carbon from peat soils. Nature 412, 785–785. doi: 10.1038/35090628
Freitag, T. E., Toet, S., Ineson, P., and Prosser, J. I. (2010). Links between methane flux and transcriptional activities of methanogens and methane oxidizers in a blanket peat bog. FEMS Microbiol. Ecol. 73, 157–165. doi: 10.1111/j.1574-6941.2010.00871.x
Garcia-Pichel, F., Loza, V., Marusenko, Y., Mateo, P., and Potrafka, R. M. (2013). Temperature drives the continental-scale distribution of key microbes in topsoil communities. Science 340, 1574–1577. doi: 10.1126/science.1236404
Garland, J. L. (1996). Analytical approaches to the characterization of samples of microbial communities using patterns of potential C source utilization. Soil Biol. Biochem. 28, 213–221. doi: 10.1016/0038-0717(95)00112-3
Garland, J. L., and Mills, A. L. (1991). Classification and characterization of heterotrophic microbial communities on the basis of patterns of community-level sole-carbon-source utilization. Appl. Environ. Microb. 57, 2351–2359. doi: 10.1128/AEM.57.8.2351-2359.1991
González, A. M. M., Dalsgaard, B., and Olesen, J. M. (2010). Centrality measures and the importance of generalist species in pollination networks. Ecol. Complex. 7, 36–43. doi: 10.1016/j.ecocom.2009.03.008
Gray, N., Sherry, A., Grant, R., Rowan, A., Hubert, C., Callbeck, C., et al. (2011). The quantitative significance of Syntrophaceae and syntrophic partnerships in methanogenic degradation of crude oil alkanes. Environ. Microbiol. 13, 2957–2975. doi: 10.1111/j.1462-2920.2011.02570.x
Gu, Z., Gu, L., Eils, R., Schlesner, M., and Brors, B. (2014). Circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812. doi: 10.1093/bioinformatics/btu393
Guo, X., Feng, J., Shi, Z., Zhou, X., Yuan, M., Tao, X., et al. (2018). Climate warming leads to divergent succession of grassland microbial communities. Nat. Clim. Change 8, 813–822. doi: 10.1038/s41558-018-0254-2
Haapalehto, T., Kotiaho, J. S., Matilainen, R., and Tahvanainen, T. (2014). The effects of long-term drainage and subsequent restoration on water table level and pore water chemistry in boreal peatlands. J. Hydrol. 519, 1493–1505. doi: 10.1016/j.jhydrol.2014.09.013
Harmsen, H. J., Van Kuijk, B. L., Plugge, C. M., Akkermans, A. D., De Vos, W. M., and Stams, A. J. (1998). Syntrophobacter fumaroxidans sp. nov., a syntrophic propionate-degrading sulfate-reducing bacterium. Int. J. Syst. Evol. Microbiol. 48, 1383–1387. doi: 10.1099/00207713-48-4-1383
Hiraishi, A., and Ueda, Y. (1994). Rhodoplanes gen. nov., a new genus of phototrophic bacteria including Rhodopseudomonas rosea as Rhodoplanes roseus comb. nov. and Rhodoplanes elegans sp. nov. Int. J. Syst. Evol. Microbiol. 44, 665–673. doi: 10.1099/00207713-44-4-665
Hornibrook, E. R. C., Bowes, H. L., Culbert, A., and Gallego-Sala, A. V. (2009). Methanotrophy potential versus methane supply by pore water diffusion in peatlands. Biogeosciences 6, 1491–1504. doi: 10.5194/bg-6-1491-2009
Hoyos-Santillan, J., Lomax, B. H., Large, D., Turner, B. L., Lopez, O. R., Boom, A., et al. (2019). Evaluation of vegetation communities, water table, and peat composition as drivers of greenhouse gas emissions in lowland tropical peatlands. Sci. Total Environ. 688, 1193–1204. doi: 10.1016/j.scitotenv.2019.06.366
Huang, X., Pancost, R. D., Xue, J., Gu, Y., Evershed, R. P., and Xie, S. (2018). Response of carbon cycle to drier conditions in the mid-Holocene in central China. Nat. Commun. 9, 1369–1377. doi: 10.1038/s41467-018-03804-w
Jaatinen, K., Fritze, H., Laine, J., and Laiho, R. (2007). Effects of short-and long-term water-level drawdown on the populations and activity of aerobic decomposers in a boreal peatland. Global Change Biol. 13, 491–510. doi: 10.1111/j.1365-2486.2006.01312.x
Järveoja, J., Peichl, M., Maddison, M., Soosaar, K., Vellak, K., Karofeld, E., et al. (2016). Impact of water table level on annual carbon and greenhouse gas balances of a restored peat extraction area. Biogeosciences 13, 2637–2651. doi: 10.5194/bg-13-2637-2016
Jassey, V. E., Chiapusio, G., Gilbert, D., Toussaint, M.-L., and Binet, P. (2012). Phenoloxidase and peroxidase activities in Sphagnum-dominated peatland in a warming climate. Soil Biol. Biochem. 46, 49–52. doi: 10.1016/j.soilbio.2011.11.011
Jiao, S., Yang, Y., Xu, Y., Zhang, J., and Lu, Y. (2019). Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 14, 202–216. doi: 10.1038/s41396-019-0522-9
Juan-Ovejero, R., Briones, M. J. I., and Öpik, M. (2020). Fungal diversity in peatlands and its contribution to carbon cycling. Appl. Soil Ecol. 146, 103393–103403. doi: 10.1016/j.apsoil.2019.103393
Juckers, M., and Watmough, S. A. (2014). Impacts of simulated drought on pore water chemistry of peatlands. Environ. Pollut. 184, 73–80. doi: 10.1016/j.envpol.2013.08.011
Kallenbach, C. M., Frey, S. D., and Grandy, A. S. (2016). Direct evidence for microbial-derived soil organic matter formation and its ecophysiological controls. Nat. Commun. 7, 13630–13639. doi: 10.1038/ncomms13630
Kane, E. S., Mazzoleni, L. R., Kratz, C. J., Hribljan, J. A., Johnson, C. P., Pypker, T. G., et al. (2014). Peat porewater dissolved organic carbon concentration and lability increase with warming: a field temperature manipulation experiment in a poor-fen. Biogeochemistry 119, 161–178. doi: 10.1007/s10533-014-9955-4
Kanokratana, P., Uengwetwanit, T., Rattanachomsri, U., Bunterngsook, B., Nimchua, T., Tangphatsornruang, S., et al. (2011). Insights into the phylogeny and metabolic potential of a primary tropical peat swamp forest microbial community by metagenomic analysis. Microb. Ecol. 61, 518–528. doi: 10.1007/s00248-010-9766-7
Kassambara, A. (2018). ggpubr: ‘ggplot2’ based Publication Ready Plots. Available online at: https://rpkgs.datanovia.com/ggpubr/ (accessed June 27, 2020).
Kembel, S. W., Cowan, P. D., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., et al. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464. doi: 10.1093/bioinformatics/btq166
Keylock, C. (2005). Simpson diversity and the Shannon–Wiener index as special cases of a generalized entropy. OIKOS 109, 203–207. doi: 10.1111/j.0030-1299.2005.13735.x
Kim, S.-J., Park, S.-J., Jung, M.-Y., Kim, J.-G., Madsen, E. L., and Rhee, S.-K. (2014). An uncultivated nitrate-reducing member of the genus Herminiimonas degrades toluene. Appl. Environ. Microb. 80, 3233–3243. doi: 10.1128/AEM.03975-13
Kip, N., Van Winden, J. F., Pan, Y., Bodrossy, L., Reichart, G.-J., Smolders, A. J., et al. (2010). Global prevalence of methane oxidation by symbiotic bacteria in peat-moss ecosystems. Nat. Geosci. 3, 617–721. doi: 10.1038/ngeo939
Kloss, S., Zehetner, F., Buecker, J., Oburger, E., and Soja, G. (2015). Trace element biogeochemistry in the soil-water-plant system of a temperate agricultural soil amended with different biochars. Environ. Sci. Pollut. R. 22, 4513–4526. doi: 10.1007/s11356-014-3685-y
Kolton, M., Marks, A., Wilson, R., Chanton, J. P., and Kostka, J. E. (2019). Impact of warming on greenhouse gas production and microbial diversity in anoxic peat from a Sphagnum-dominated bog (Grand Rapids, Minnesota, USA): a laboratory incubation study. Front. Microbiol. 10:870–882. doi: 10.3389/fmicb.2019.00870
Kwon, M. J., Haraguchi, A., and Kang, H. (2013). Long-term water regime differentiates changes in decomposition and microbial properties in tropical peat soils exposed to the short-term drought. Soil Biol. Biochem. 60, 33–44. doi: 10.1016/j.soilbio.2013.01.023
Lee, H., Kim, D.-U., Lee, S., Park, S., Yoon, J.-H., Seong, C. N., et al. (2017). Reyranella terrae sp. nov., isolated from an agricultural soil, and emended description of the genus Reyranella. Int. J. Syst. Evol. Microbiol. 67, 2031–2035. doi: 10.1099/ijsem.0.00191
Leppänen, S. M., Rissanen, A. J., and Tiirola, M. (2015). Nitrogen fixation in Sphagnum mosses is affected by moss species and water table level. Plant Soil 389, 185–196. doi: 10.1007/s11104-014-2356-6
Lodha, T. D., Srinivas, A., Sasikala, C., and Ramana, C. V. (2015). Hopanoid inventory of Rhodoplanes spp. Arch. Microbiol. 197, 861–867. doi: 10.1007/s00203-015-1112-5
Louca, S., Polz, M. F., Mazel, F., Albright, M. B. N., Huber, J. A., O’Connor, M. I., et al. (2018). Function and functional redundancy in microbial systems. Nat. Ecol. Evol. 2, 936–943. doi: 10.1038/s41559-018-0519-1
Ma, B., Wang, H., Dsouza, M., Lou, J., He, Y., Dai, Z., et al. (2016). Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME J. 10, 1891–1901. doi: 10.1038/ismej.2015.261
Magoè, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mäkiranta, P., Laiho, R., Fritze, H., Hytönen, J., Laine, J., and Minkkinen, K. (2009). Indirect regulation of heterotrophic peat soil respiration by water level via microbial community structure and temperature sensitivity. Soil Biol. Biochem. 41, 695–703. doi: 10.1016/j.soilbio.2009.01.004
Moore, T. R., and Dalva, M. (1993). The influence of temperature and water table position on carbon dioxide and methane emissions from laboratory columns of peatland soils. J. Soil Sci. 44, 651–664. doi: 10.1111/j.1365-2389.1993.tb02330.x
Nadkarni, M. A., Martin, F. E., Jacques, N. A., and Hunter, N. (2002). Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148, 257–266. doi: 10.1099/00221287-148-1-257
Newman, M. E. (2006). Modularity and community structure in networks. Proc. Natl. Acad. Sci. U.S.A. 103, 8577–8582. doi: 10.1073/pnas.0601602103
Ogita, N., Hashidoko, Y., Limin, S. H., and Tahara, S. (2006). Linear 3-Hydroxybutyrate Tetramer (HB4) produced by Sphingomonas sp. is characterized as a growth promoting factor for some rhizomicrofloal composers. Biosci. Biotech. Bioch. 70, 2325–2329. doi: 10.1271/bbb.60299
Oksanen, J., Kindt, R., Legendre, P., O’Hara, B., Stevens, M. H. H., Oksanen, M. J., et al. (2007). Community Ecology Package. Available online at: https://cran.r-project.org (accessed November 28, 2020).
Pankratov, T. A., Ivanova, A. O., Dedysh, S. N., and Liesack, W. (2011). Bacterial populations and environmental factors controlling cellulose degradation in an acidic Sphagnum peat. Environ. Microbiol. 13, 1800–1814. doi: 10.1111/j.1462-2920.2011.02491.x
Pankratov, T. A., Tindall, B. J., Liesack, W., and Dedysh, S. N. (2007). Mucilaginibacter paludis gen. nov., sp. nov. and Mucilaginibacter gracilis sp. nov., pectin-, xylan- and laminarin-degrading members of the family Sphingobacteriaceae from acidic Sphagnum peat bog. Int. J. Syst. Evol. Microbiol. 57, 2349–2354. doi: 10.1099/ijs.0.65100-0
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Putkinen, A., Larmola, T., Tuomivirta, T., Siljanen, H. M., Bodrossy, L., Tuittila, E.-S., et al. (2012). Water dispersal of methanotrophic bacteria maintains functional methane oxidation in Sphagnum mosses. Front. Microbiol. 3:15. doi: 10.3389/fmicb.2012.00015
Qin, S., Chen, L., Fang, K., Zhang, Q., Wang, J., Liu, F., et al. (2019). Temperature sensitivity of SOM decomposition governed by aggregate protection and microbial communities. Sci. Adv. 5:eaau1218. doi: 10.1126/sciadv.aau1218
Revelle, W. (2020). An Overview of the Psych Package. Available online at: https://personality-project.org/r/psych/ (accessed December 16, 2020).
Robroek, B. J., Albrecht, R. J., Hamard, S., Pulgarin, A., Bragazza, L., Buttler, A., et al. (2016). Peatland vascular plant functional types affect dissolved organic matter chemistry. Plant Soil 407, 135–143. doi: 10.1007/s11104-015-2710-3
Salomo, S., Münch, C., and Röske, I. (2009). Evaluation of the metabolic diversity of microbial communities in four different filter layers of a constructed wetland with vertical flow by BiologTM analysis. Water Res. 43, 4569–4578. doi: 10.1016/j.watres.2009.08.009
Sanz-Lázaro, C., Valdemarsen, T., Marín, A., and Holmer, M. (2011). Effect of temperature on biogeochemistry of marine organic-enriched systems: implications in a global warming scenario. Ecol. Appl. 21, 2664–2677. doi: 10.1890/10-2219.1
Sapek, A., Sapek, B., Chrzanowski, S., and Urbaniak, M. (2009). Nutrient mobilisation and losses related to the groundwater level in low peat soils. Int. J. Environ. Pollut. 37, 398–408. doi: 10.1504/IJEP.2009.026057
Schmidt, O., Hink, L., Horn, M. A., and Drake, H. L. (2016). Peat: home to novel syntrophic species that feed acetate-and hydrogen-scavenging methanogens. ISME J. 10, 1954–1966. doi: 10.1038/ismej.2015.256
Smoot, J., and Findlay, R. (2001). Spatial and seasonal variation in a reservoir sedimentary microbial community as determined by phospholipid analysis. Microb. Ecol. 42, 350–358. doi: 10.1007/s002480000102
Staley, C., Gould, T. J., Wang, P., Phillips, J., Cotner, J. B., and Sadowsky, M. J. (2015). Species sorting and seasonal dynamics primarily shape bacterial communities in the Upper Mississippi River. Sci. Total Environ. 505, 435–445. doi: 10.1016/j.scitotenv.2014.10.012
Strack, M., Waddington, J. M., and Tuittila, E. S. (2004). Effect of water table drawdown on northern peatland methane dynamics: implications for climate change. Global Biogeochem. Cycles 18, 2209–2215. doi: 10.1029/2003gb002209
Thompson, L. R., Sanders, J. G., McDonald, D., Amir, A., Ladau, J., Locey, K. J., et al. (2017). A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463. doi: 10.1038/nature24621
Thormann, M. N. (2006). Diversity and function of fungi in peatlands: a carbon cycling perspective. Can. J. Soil Sci. 86, 281–293. doi: 10.4141/S05-082
Tian, W., Wang, H., Xiang, X., Wang, R., and Xu, Y. (2019). Structural variations of bacterial community driven by Sphagnum microhabitat differentiation in a subalpine peatland. Front. Microbiol. 10:1661–1671. doi: 10.3389/fmicb.2019.01661
Tian, W., Xiang, X., Ma, L., Evers, S., Wang, R., Qiu, X., et al. (2020). Rare species shift the structure of bacterial communities across Sphagnum compartments in a subalpine peatland. Front. Microbiol. 10:3138–3150. doi: 10.3389/fmicb.2019.03138
Tiquia, S. (2010). Metabolic diversity of the heterotrophic microorganisms and potential link to pollution of the Rouge River. Environ. Pollut. 158, 1435–1443. doi: 10.1016/j.envpol.2009.12.035
Too, C. C., Keller, A., Sickel, W., Lee, S. M., and Yule, C. M. (2018). Microbial community structure in a Malaysian tropical peat swamp forest: the influence of tree species and depth. Front. Microbiol. 9:2859–2871. doi: 10.3389/fmicb.2018.02859
Ulanowski, T., and Branfireun, B. (2013). Small-scale variability in peatland pore-water biogeochemistry, Hudson Bay Lowland, Canada. Sci. Total Environ. 454, 211–218. doi: 10.1016/j.scitotenv.2013.02.087
Urbanová, Z., and Bárta, J. (2016). Effects of long-term drainage on microbial community composition vary between peatland types. Soil Biol. Biochem. 92, 16–26. doi: 10.1016/j.soilbio.2015.09.017
Urbanová, Z., Picek, T., and Bárta, J. (2011). Effect of peat re-wetting on carbon and nutrient fluxes, greenhouse gas production and diversity of methanogenic archaeal community. Ecol. Eng. 37, 1017–1026. doi: 10.1016/j.ecoleng.2010.07.012
Walters, W., Hyde, E. R., Berg-Lyons, D., Ackermann, G., Humphrey, G., Parada, A., et al. (2016). Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1, e00009-15. doi: 10.1128/mSystems.00009-15
Wang, D., Zhang, Y., Wang, R., Zhao, B., Zhang, Z., and Huang, X. (2018). Characteristics of dissolved organic matter in pore water from the Dajiuhu Peatland, Central China. Resour. Environ. Yangtze Basin 27, 2568–2577. doi: 10.11870/cjlyzyyhj201811018
Wang, R., Wang, H., Xiang, X., Gao, Y., Song, Q., and Gong, L. (2018). Temporal and spatial variations of microbial carbon utilization in water bodies from the Dajiuhu Peatland, central China. J. Earth Sci. China 29, 969–976. doi: 10.1007/s12583-017-0818-5
Wang, S., Li, T., Zheng, Z., and Chen, H. Y. (2019). Soil aggregate-associated bacterial metabolic activity and community structure in different aged tea plantations. Sci. Total Environ. 654, 1023–1032. doi: 10.1016/j.scitotenv.2018.11.032
Wei, T. (2017). An Introduction to Matrix Visualization & corrplot Package. Available online at: https://github.com/taiyun/corrplot (accessed October 16, 2017).
Woo, H. L., Hazen, T. C., Simmons, B. A., and DeAngelis, K. M. (2014). Enzyme activities of aerobic lignocellulolytic bacteria isolated from wet tropical forest soils. Syst. Appl. Microbiol. 37, 60–67. doi: 10.1016/j.syapm.2013.10.001
Xiang, X., Wang, H., Gong, L., and Liu, Q. (2014). Vertical variations and associated ecological function of bacterial communities from Sphagnum to underlying sediments in Dajiuhu Peatland. Sci. China Earth Sci. 57, 1013–1020. doi: 10.1007/s11430-013-4752-9
Xue, D., Yao, H., Ge, D., and Huang, C. (2008). Soil microbial community structure in diverse land use systems: a comparative study using Biolog, DGGE, and PLFA analyses. Pedosphere 18, 653–663. doi: 10.1016/S1002-0160(08)60060-0
Yang, G., Mei, W., Chen, H., Liu, L., Wu, N., Zhu, D., et al. (2017). Responses of CO2 emission and pore water DOC concentration to soil warming and water table drawdown in Zoige Peatlands. Atmos. Environ. 152, 323–329. doi: 10.1016/j.atmosenv.2016.12.051
Yrjälä, K., Tuomivirta, T., Juottonen, H., Putkinen, A., Lappi, K., Tuittila, E. S., et al. (2011). CH4 production and oxidation processes in a boreal fen ecosystem after long-term water table drawdown. Global Change Biol. 17, 1311–1320. doi: 10.1111/j.1365-2486.2010.02290.x
Yu, Z., Loisel, J., Brosseau, D. P., Beilman, D. W., and Hunt, S. J. (2010). Global peatland dynamics since the Last Glacial Maximum. Geophys Res. Lett. 37, L13402–L13406. doi: 10.1029/2010GL043584
Zhang, T., Wu, Y., Zhuang, L., Wang, X., and Hu, H. (2014). Screening heterotrophic microalgal strains by using the Biolog method for biofuel production from organic wastewater. Algal Res. 6, 175–179. doi: 10.1016/j.algal.2014.10.003
Zhou, G., Qiu, X., Chen, L., Zhang, C., Ma, D., and Zhang, J. (2019). Succession of organics metabolic function of bacterial community in response to addition of earthworm casts and zeolite in maize straw composting. Bioresource Technol. 280, 229–238. doi: 10.1016/j.biortech.2019.02.015
Keywords: bacterial structure, metabolic diversity, bacterial phenotype, co-occurrence network, water table, temperature
Citation: Tian W, Xiang X and Wang H (2021) Differential Impacts of Water Table and Temperature on Bacterial Communities in Pore Water From a Subalpine Peatland, Central China. Front. Microbiol. 12:649981. doi: 10.3389/fmicb.2021.649981
Received: 06 January 2021; Accepted: 01 April 2021;
Published: 28 May 2021.
Edited by:
Svetlana N. Dedysh, Winogradsky Institute of Microbiology, Russian Academy of Sciences (RAS), RussiaReviewed by:
Carl-Eric Wegner, Friedrich Schiller University Jena, GermanyCopyright © 2021 Tian, Xiang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongmei Wang, d2FuZ2htZWkwNEAxNjMuY29t; aG13YW5nQGN1Zy5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.