
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 21 April 2021
Sec. Food Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.649698
 Shuanghong Yang1,2
Shuanghong Yang1,2 Xinqiang Xie2*
Xinqiang Xie2* Jun Ma2Xingxiang He3Ying Li2Mingzhu Du1,2Longyan Li2Lingshuang Yang2
Jun Ma2Xingxiang He3Ying Li2Mingzhu Du1,2Longyan Li2Lingshuang Yang2 Qingping Wu2
Qingping Wu2 Wei Chen1*
Wei Chen1* Jumei Zhang2*
Jumei Zhang2*Bifidobacterium, an important genus for human health, is difficult to isolate. We applied metagenomics, pangenomics, and enzymology to determine the dominant glycoside hydrolase (GH) families of Bifidobacterium and designed selective medium for Bifidobacterium isolation. Pangenomics results showed that the GH13, GH3, GH42, and GH43 families were highly conserved in Bifidobacterium. Metagenomic analysis of GH families in human faecal samples was performed. The results indicated that Bifidobacterium contains core GHs for utilizing raffinose, D-trehalose anhydrous, D(+)-cellobiose, melibiose, lactulose, lactose, D(+)-sucrose, resistant starch, pullulan, xylan, and glucan. These carbohydrates as the main carbon sources were applied for selective media, which were more conducive to the growth of bifidobacteria. In the medium with lactose, raffinose and xylan as the main carbon sources, the ratio of cultivable bifidobacteria to cultivable microorganisms were 89.39% ± 2.50%, 71.45% ± 0.99%, and 53.95% ± 1.22%, respectively, whereas the ratio in the ordinary Gifu anaerobic medium was only 17.90% ± 0.58%. Furthermore, the species significantly (p < 0.05) varied among samples from different individuals. Results suggested that xylan might be a prebiotic that benefits host health, and it is feasible to screen and isolate bifidobacteria using the oligosaccharides corresponding to the specific GHs of bifidobacteria as the carbon sources of the selective media.
Among the numerous microbial communities that colonise the human body, the intestinal microbiome plays a major role in maintaining host health. The intestinal microbiome of the gastrointestinal ecosystem comprises a collective genome of trillions of microorganisms. Interactions between the host and the intestinal microbiota are complex. Changes in the intestinal microbiota might be critical to prevent or treat various intestinal and non-intestinal diseases (Patel and DuPont, 2015; Ghaisas et al., 2016), such as irritable bowel syndrome (Staudacher et al., 2017), ulcerative colitis (UC), central nervous system diseases, Alzheimer’s disease, and Parkinson’s disease (Sochocka et al., 2019). Colonisation by intestinal microorganisms begins due to differences in microflora between the placenta and amniotic fluid; then, it continues post-partum via microorganisms in breast milk (Collado et al., 2016). A key function of the gut microbiota is to facilitate host food digestion, especially that of complex carbohydrates in mammalian diets (Armstrong et al., 2018; Cerqueira et al., 2020). The enzymes involved in polysaccharide decomposition are carbohydrate active enzymes (CAZymes), which consist of carbohydrate-binding modules, carbohydrate esterases, polysaccharide lyases, glycoside transferases, and glycoside hydrolases (GHs), the latter of which are the largest of the CAZymes.
Bifidobacterium, a prevalent genus among intestinal microbiota (Mitsuoka, 2014; O’Callaghan and van Sinderen, 2016), is an important symbiotic group, and Bifidobacterium genus is one of the first to colonise the human gastrointestinal tract (Turroni et al., 2012; Lewis et al., 2015; O’Callaghan and van Sinderen, 2016). It is believed to exert positive health effects on hosts (Hsieh et al., 2015; Matson et al., 2018). The numbers of bifidobacteria in the total colonic microbiota of normally delivered breast-fed infants decrease from 90% to <5% in adults (Riviere et al., 2016). The abundance of bifidobacteria in the gastrointestinal tract is related to some pathologies, such as intestinal (Grazul et al., 2016; Roškar et al., 2017) and mental (Pinto-Sanchez et al., 2017) diseases. Bifidobacteria can also improve the intestinal barrier function by producing salts of short chain fatty acids such as butyrate (Rios-Covian et al., 2015) and acetate (Hsieh et al., 2015) and reducing serum FITC-dextran levels in a mouse model of colitis (Srutkova et al., 2015). Furthermore, Bifidobacterium longum 1714 positively affects cognition in mice (Savignac et al., 2015). In addition to the resistance and regulation of diseases, bifidobacteria can promote nutrient yield to enhance immunity (Kim et al., 2014; Yang et al., 2014; Jaglan et al., 2019).
Tissier isolated the first strain of Bifidobacterium from the faeces of exclusively breastfed infants in 1899 (Riviere et al., 2016), and today, the bifidobacteria group includes 85 species1. However, the isolation of new Bifidobacterium species or their cultivation is difficult because they are strictly anaerobic and easily contaminated by other bacteria. Traditional methods of separation and cultivation no longer meet actual needs, and thus, new technologies are required to explore the taxonomic and functional profiles of microbial DNA extracted from microbial communities, which are constantly increasing (Al-Masaudi et al., 2017). Lugli et al. (2019) proposed that they could mine genetic information of related Bifidobacterium strains based on whole metagenome shotgun sequencing to design a reasonable selective medium based on GHs and obtain target strains when screening new Bifidobacterium strains. However, they only mined GH based on metagenomic data to design the medium. Monosaccharides are limited in the large intestine, which is inhabited by bifidobacteria. Therefore, bifidobacteria possess various glycosidases and sugar transporters that assimilate indigestible polysaccharides, oligosaccharides, and complex carbohydrates (Garrido et al., 2012). The gene content of the Bifidobacterium genome encodes numerous enzymes with predicted roles in carbohydrate modifications (Pokusaeva et al., 2011). To promote the growth of the target genera and inhibit the growth of other microorganisms, selective medium can be designed using omics to identify GHs and their corresponding carbohydrates in samples with unique effects.
We designed 11 selective media based on data derived from pangenomics, metagenomics, and enzymology to promote the proliferation of Bifidobacterium while simultaneously reducing the growth of other intestinal bacteria. Stool samples for faecal bacterial transplantation were collected from the First Affiliated Hospital of Guangdong Pharmaceutical University. We also combined the results of a pangenomic analysis of Bifidobacterium to suitable GHs to select a suitable carbon source for selectively isolating this genus. The growth of Bifidobacterium on different selective media was determined and analysed using 16S amplicon sequencing to determine the optimal carbon source for Bifidobacterium isolation.
We retrieved complete genome sequences for 144 bifidobacteria and 5 other genera (Supplementary Table 2 sheet 6) from the National Center for Biotechnology Information (NCBI) public database. These sequences were used as inputs for the pangenomic analysis to identify specific GHs.
Stool samples were collected from three healthy donors of faecal microbiota transplantation (FMT) at the First Affiliated Hospital of Guangdong Pharmaceutical University. The study was approved by the ethics committee of the First Affiliated Hospital of Guangdong Pharmaceutical University (reference 2017-98). Healthy donors were recommended to not eat spicy and greasy food at least the day before donating faeces. The donors collected the faeces in a sterile container and provided it to a professional for pre-treatment of the FMT. The pretreatment steps were as follows. Install the faecal collection barrel into the automated faecal bacteria separation system GenFMTer (FMT medical, Nanjing, China), add physiological saline to stir, filter, and purify, which can remove faecal residue and large particles. And then collect the faecal bacteria suspension and repeatedly washed three times. The supernatant was removed, and then resuspended in physiological saline.
S171, S181, and S201 were human stool samples (donor S17, S18, and S20) that had not been processed before FMT; the stool samples S171 and S181 were washed to remove food residue (donor S17 and S18), used for FMT in this hospital, and named as S172 and S182. Approximately 2 g of faeces from the original faeces and 2 g of faeces after pre-treatment were taken.
Total faecal DNA and DNA used for 16S rRNA gene sequencing were extracted using QIAamp PowerFecal Pro FNA Kits (50) (Qiagen GmbH, Hilden, Germany), as per the manufacturer’s instructions. We extracted DNA from Bifidobacterium using gram-positive bacterial DNA extraction kits (Magen Biotech, Guangzhou, China). The approximate concentration and purity of all DNA samples were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, United States), and concentrations were accurately measured using a Qubit 3.0 fluorometer (Thermo Fisher Scientific Inc.).
A DNA library was prepared and sequenced using a NovaSeq 6000 system (Illumina, Inc., San Diego, CA, United States) at a commercial laboratory (Novogene Co., Ltd., Beijing, China). Barcodes were removed from the sequencing results using Practical Extraction and Reporting Language (Perl) script, low quality data were removed using Trimmomatic Version 0.39 (Bolger et al., 2014), and host DNA was removed using CLC Genomics Workbench (Qiagen). Thereafter, assembling and announcement were conducted using SPAdes Version 3.12.0 (Nurk et al., 2013) and Prokka (Version 1.13.7) (Seemann, 2014), respectively.
A total of 144 Bifidobacterium genome sequences were downloaded from the NCBI database for pan-genome analysis. The sequences were then annotated and compared them with annotation files through a local server. We also annotated the 144 genomes using the dbCAN-seq database. Relative abundance heat maps were prepared using Prism 8.2.1 (GraphPad Software Inc., San Diego, CA, United States), and stack columns of abundance were drawn using OriginPro® 8.5 (OriginLab Corporation, Northampton, MA, United States).
Traditional GAM (Gifu anaerobic medium) consists of soytone (10 g), proteose peptone (10 g), bovine serum albumin (13.5 g), yeast extract (5 g), beef extract (2.2 g), KH2PO4 (2.5 g), liver extract (1.2 g), NaCl (3.0 g), L-cysteine (0.3 g), sodium thioglycolate (0.3 g), glucose (3 g), and soluble starch (5 g) in 1 L of water. Moreover, for the solid medium plates, agar was added (15 g/L). The selective medium was designed by eliminating glucose and soluble starch and replacing with raffinose, D-trehalose anhydrous, D(+)-cellobiose, melibiose, lactulose, lactose, D(+)-sucrose, resistant starch, pullulan, xylan, and glucan.
A DNA library was prepared and sequenced using a Nova PE250 instrument (Illumina, Inc., San Diego, CA, United States) at a commercial laboratory (Genewiz, Inc., Suzhou, China). The original binary base-calling data obtained by sequencing were converted into PF (pass filtering) or raw sequence data using Illumina bcl2fastq software. Barcodes were removed from raw data using the Perl script and low-quality data were removed using Trimmomatic version. 0.39 (Bolger et al., 2014). Relative abundance heat maps were prepared using Prism 8.2.1 (GraphPad Software Inc., San Diego, CA, United States) and stack columns of abundance were drawn using OriginPro® 8.5 (OriginLab Corporation, Northampton, MA, United States).
The 11 carbohydrates were selected as the main carbon source, based on all analytical findings (Table 1 and Supplementary Table 1). Fresh faeces samples (0.5 g) were diluted 10–1, 10–2, 10–3, 10–4, 10–5, and 10–6 in physiological saline (0.9%), and then, the 10–4, 10–5, and 10–6 dilutions were coated on the different designed medium plates, with each gradient performed in triplicate for each medium. After incubation at 37°C for 48–72 h in the anaerobic workstation, single colonies were inoculated and streaked on plates and cultured in the anaerobic workstation at 37°C for 48–72 h. This step was repeated. Finally, 16S rRNA gene sequences were amplified from DNA extracts of pure cultures using the primer pair 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′), which targets the variable region between the 16S rRNA gene sequences. Bifidobacteria were phylogenetically analysed using MEGAX and iTOL2.
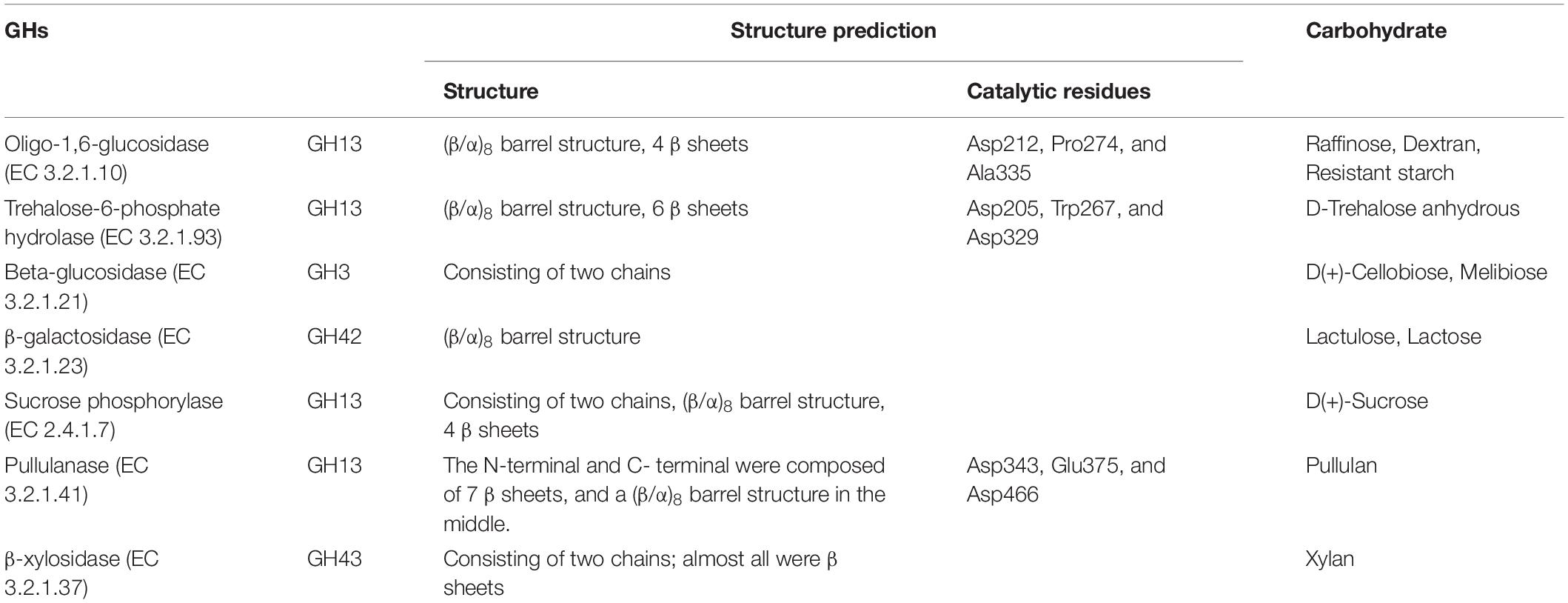
Table 1. Structure prediction of selected GHs and corresponding carbohydrates.
A total of 144 bifidobacterial genome sequences were downloaded from NCBI for pangenomic analysis (Figure 1A and Supplementary Table 2 sheets 1–3). The total number of genes increased as the genomes increased, whereas conserved genes began to plateau at 20 genomes and remained constant thereafter. We used these whole genomes to find GHs, which are abundant in bifidobacteria species. The glycoside hydrolase annotations of 144 bifidobacteria strains were analysed (Figure 1B and Supplementary Table 2 sheet 5) using the dbCAN-seq database3 (Huang et al., 2018). It was found that the GH13, GH3, GH42, and GH43 families were almost all contained and abundant in the Bifidobacterium genus. These GH families are involved in the hydrolysis of starch, cellulose, and their related hydrolysed derivatives. Starch, cellulose and hemicellulose are the most common plant polysaccharides and are important sources of energy (Brownlee et al., 2018; Soliman, 2019). They are also an important carbon source for bifidobacteria that inhabit the gastrointestinal tract; therefore, related hydrolytic enzymes may be basically shared by bifidobacteria. They were therefore optimal for finding a suitable carbon source. Among the glycoside hydrolase family, GH43 is the largest branch (Matsuzawa et al., 2015), but these GHs were not the dominant GHs in Bifidobacterium, compared with that of other gut microbes. Therefore, we considered the distribution of GHs in other dominant intestinal microorganisms. Based on our previous metagenomic findings of human faeces, we selected 30 complete genomes of five genera, Alistipes, Anaerostipes, Barnesiella, Blautia, and Collinsella genera, as reference genomes for further pangenomic analysis (Supplementary Table 2 sheet 4) of Bifidobacterium. In the previous metagenomic results, the number of this five genera were top few. According to the availability of substrates, economic applicability and the potential as prebiotics, β-galactosidase (EC 3.2.1.23), β-glucosidase (EC 3.2.1.21), oligo-α-glucosidase (EC 3.2.1.10), sucrose phosphorylase (EC 2.4.1.7), trehalose-6-phosphate hydrolase (EC 3.2.1.93), α-amylase (EC 3.2.1.1), pullulanase (EC 3.2.1.41), and β-xylosidase (EC 3.2.1.37) were selected for further analysis. We searched for these enzyme classes in the annotation results in the Bifidobacterium pan-genome, and used the results for comparative analysis (Figure 1C). The proportion of these eight enzyme classes in Bifidobacterium was significantly higher than other five selected genus. The search results of the carbohydrate active enzyme database and UniProt database were combined to analyse the hydrolysis characteristics of these enzymes (Supplementary Table 1). We further combined the metagenomics results to determine the optimal carbon source for growth.
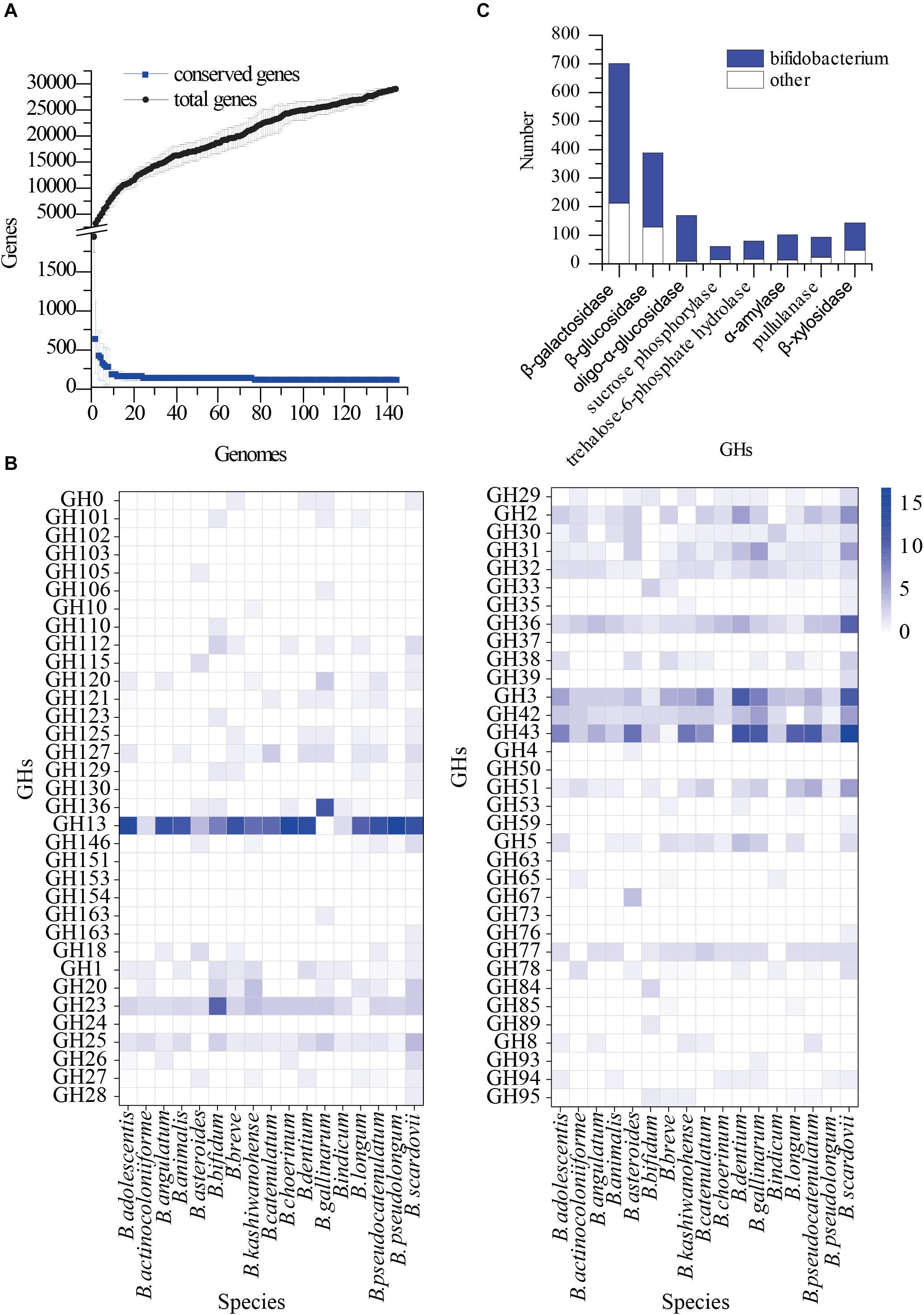
Figure 1. Pangenomics analysis of Bifidobacterium. (A) The number of conserved genes and total genes as the number of genomes increasing; (B) Distribution of glycoside hydrolase families in 144 Bifidobacterium genomes, annotated with the dbCAN-seq database; (C) Distribution of GH13, GH3, GH42, GH43 in Pangenomics of Bifidobacterium and other bacteria.
Whole metagenome shotgun sequencing of faecal bacterial samples from transplanted donor stools produced 32–40 million paired reads, with an average approximate length of 150 bp. Species significantly differed among samples from different individuals. The content of Bifidobacterium in all samples was in the top five (Figure 2A), and the proportion of Bifidobacterium in S201 was the highest, followed by that in S171 and S172, and was lowest in S181 and S182 (Figure 2B). Due to the low abundance of Bifidobacterium in S181 and S182, and no significant difference between S171 and S172, in which S172 is the stool after removing food residue from sample S171, only S201 and S171 were used in the next analysis and experiment to screen for bifidobacteria. The pangenomic results of Bifidobacterium showed that β-galactosidase, β-glucosidase, trehalose-6-phosphate hydrolase, sucrose phosphorylase, α-amylase, pullulanase, β-xylosidase, and oligo-1,6-glucosidase were abundant in Bifidobacterium and the hydrolytic properties of eight specific GHs classes with corresponding organism are shown in Supplementary Table 1. And the amounts of these GHs in the two metagenomic groups were essentially identical (Figure 2C).
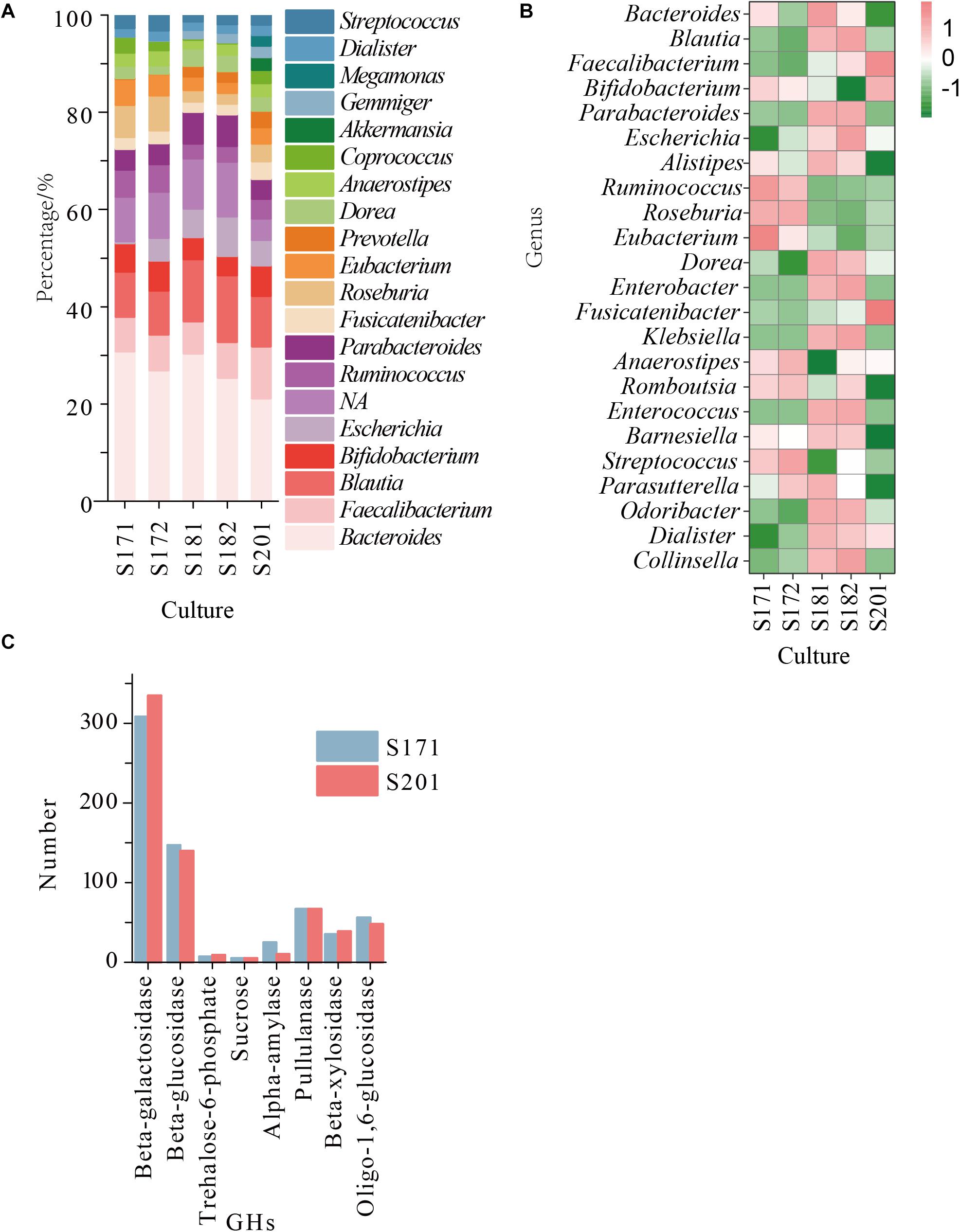
Figure 2. (A) The abundance of the reconstructed bacterial genomic material at genus level obtained from sample 17 (S171 and S172), sample 18 (S181 and S182), and sample 20 (S201); (B) The relative abundance of the reconstructed bacterial genomic material at genus level obtained from sample 17, sample 18, and sample 20; (C) The number of GHs in S171 and S201.
Based on the pangenomic and metagenomic analysis, we found several GHs in Bifidobacterium that were prominent and important for carbohydrate decomposition (Supplementary Table 1). Therefore, we speculated as to whether oligosaccharides or polysaccharides differ in the selective separation and cultivation of Bifidobacterium.
As shown in Table 1, the raffinose, D-trehalose anhydrous, D(+)-cellobiose, melibiose, lactulose, lactose, D(+)-sucrose, resistant starch, pullulan, xylan, and glucan were selected according to the hydrolysis characteristics of the selected GHs. Based on the results of 16S amplicon sequencing of clones collected from sample S171 that were cultured on selective media, in the medium with lactose, raffinose, and xylan as the main carbon sources, the ratio of cultivable bifidobacteria to cultivable microorganisms were 89.39 ± 2.50%, 71.45 ± 0.99%, and 53.95 ± 1.22%, respectively, whereas for the ordinary GAM, it was only 17.90 ± 0.58% (Figure 3, Table 2, and Supplementary Table 2 sheet 7). Except for that in the medium with lactulose as the carbon source, the relative abundance of Bifidobacterium in the remaining 10 media was higher than that in ordinary GAM (Figure 3B). However, only half of S201 achieved this result, and the metagenomic results revealed more Bifidobacteria in S201 than in S171 (Figure 2B), and the abundance of enzymes in GH classes was basically the same (Figure 2C). The heat map (Figure 3D) also showed significantly higher abundances of Bifidobacterium in S171 than in S201, and lower abundances of Shigella in S171. Lactose had the best selectivity for Bifidobacterium, followed by raffinose in S171, and the selectivity of lactulose was the worst. Xylan was more effective than glucan. Raffinose and xylan exhibited the best selectivity for S201.
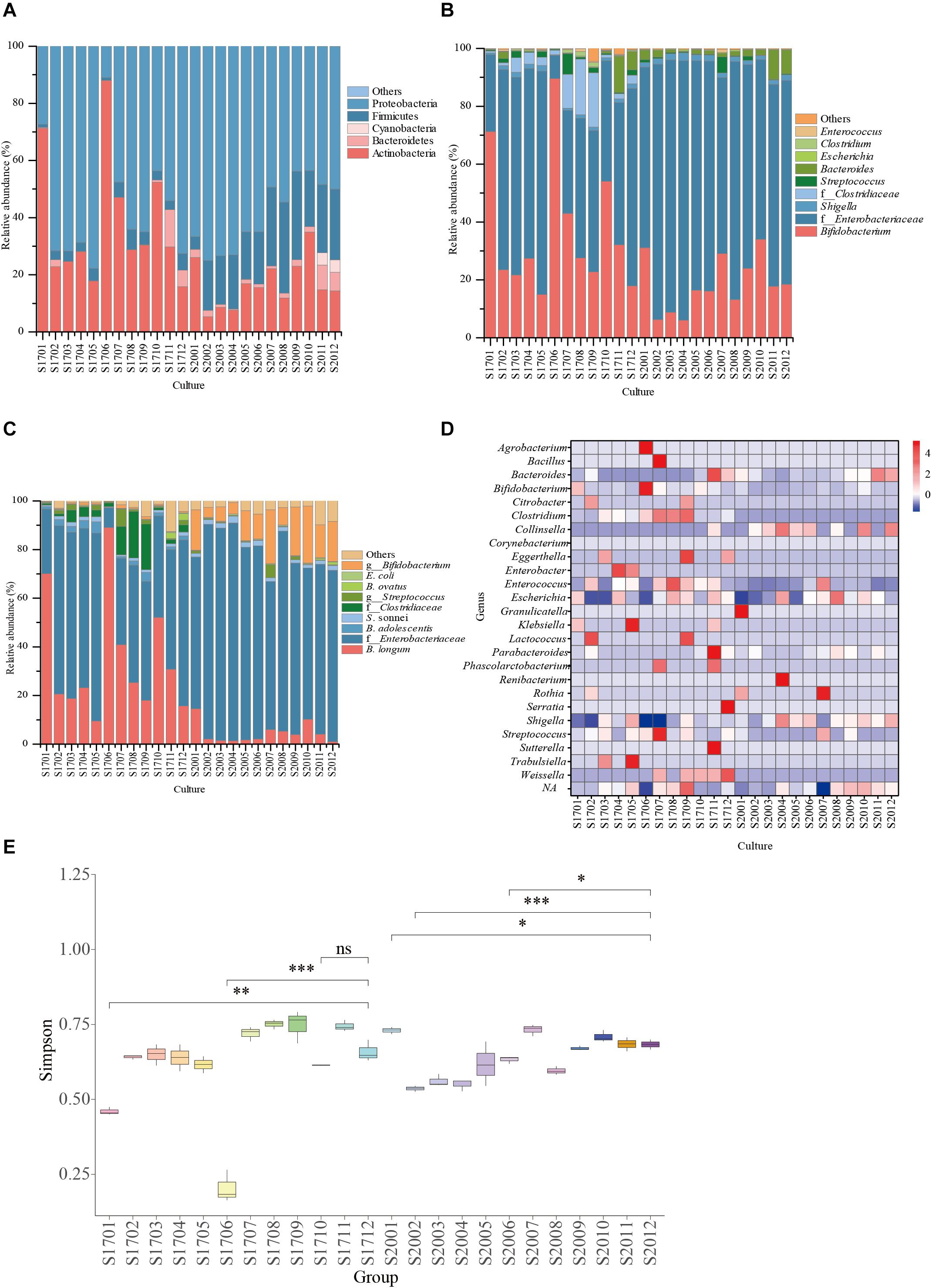
Figure 3. Relative abundance of bacterial community compositions in selective medium plates at family (A), genus (B), or species (C) levels; (D) The genus-level relative abundance hot map of 16S amplicon sequencing. (E) The alpha diversity of 16S amplicon sequencing results. ns, not significant. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001. S1701: S17 represents sample 171 and 01 represents raffinose, 02 represents trehalose, 03 represents cellobiose, 04 represents melibiose, 05 represents lactulose, 06 represents lactose, 07 represents sucrose, 08 represents resistant starch, 09 represents pullulan, 10 represents xylan, 11 represents dextran, and 12 represents the ordinary GAM (glucose and soluble starch). The same is for S2001–S2012, only S20 represents sample 201.
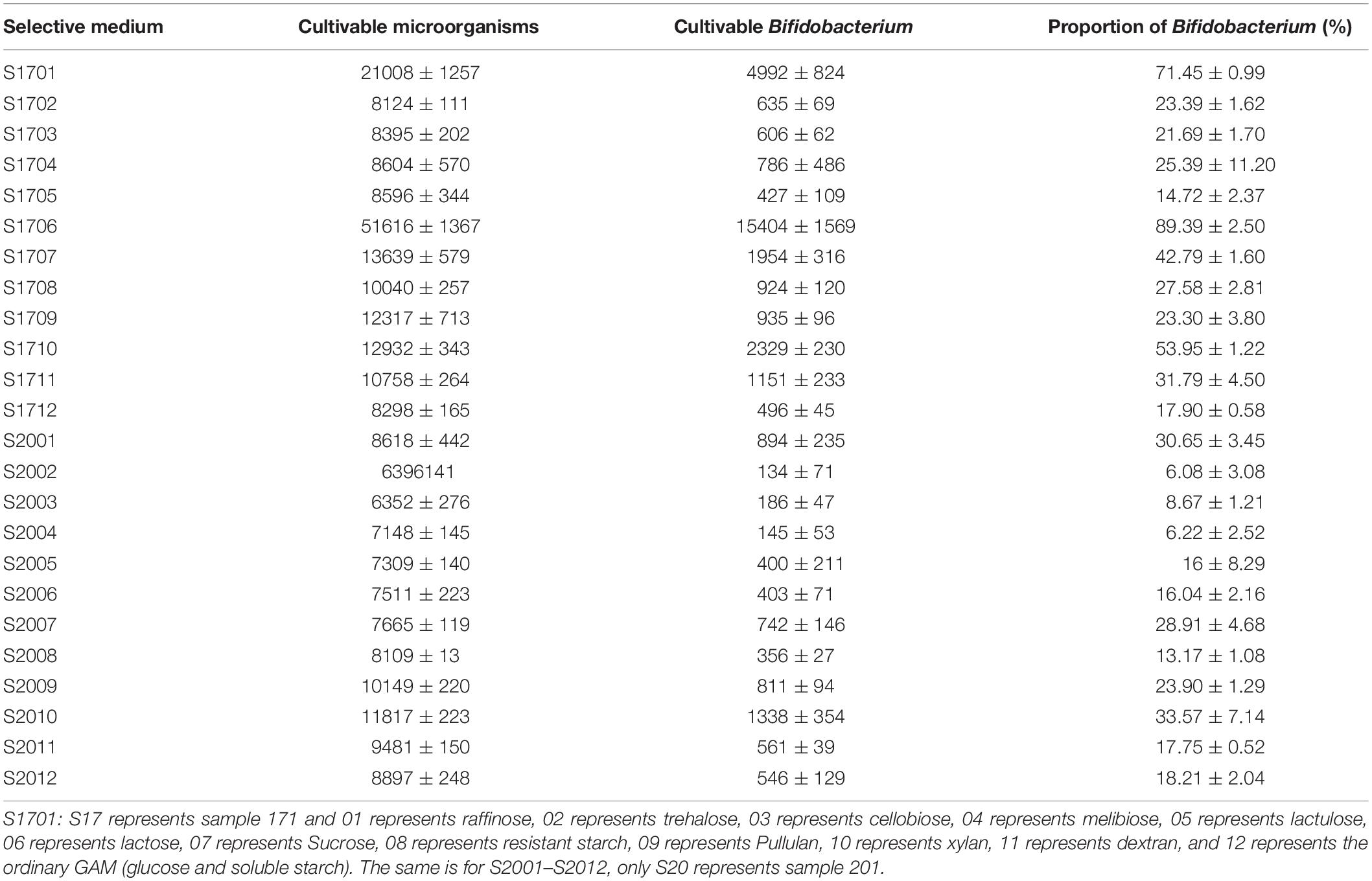
Table 2. Cultivable microorganisms and Bifidobacterium on selective medium.
The results of 16S amplicon sequencing showed that the bacterial composition significantly differed between the S171 and S201 faecal samples. We investigated the details of Bifidobacterium from different sources based on 76 colonies. We then purified these colonies and assessed 16S rDNA. We identified 28 (37%) Bifidobacterium strains (Table 3), and Figure 4 shows the results of the phylogenetic analysis. These strains belong to the three branches of B. longum, Bifidobacterium pseudocatenulatum, and Bifidobacterium bifidum.
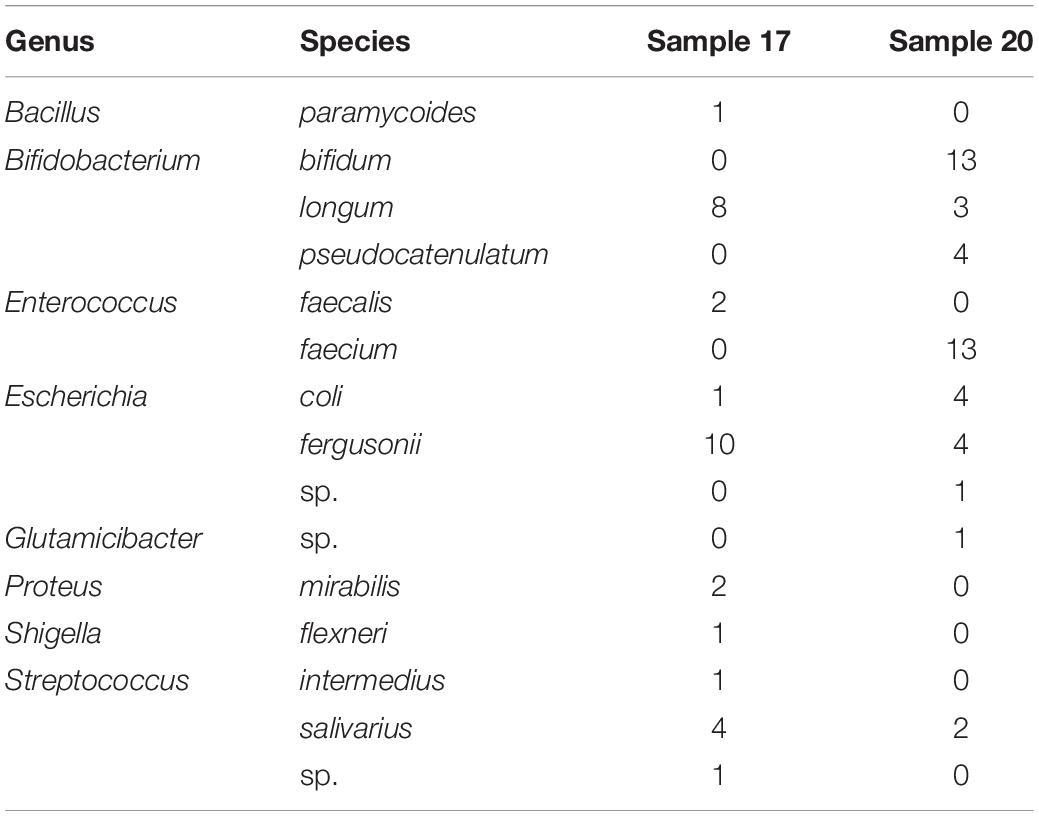
Table 3. The 16S rDNA identification of selected and predicated colonies.
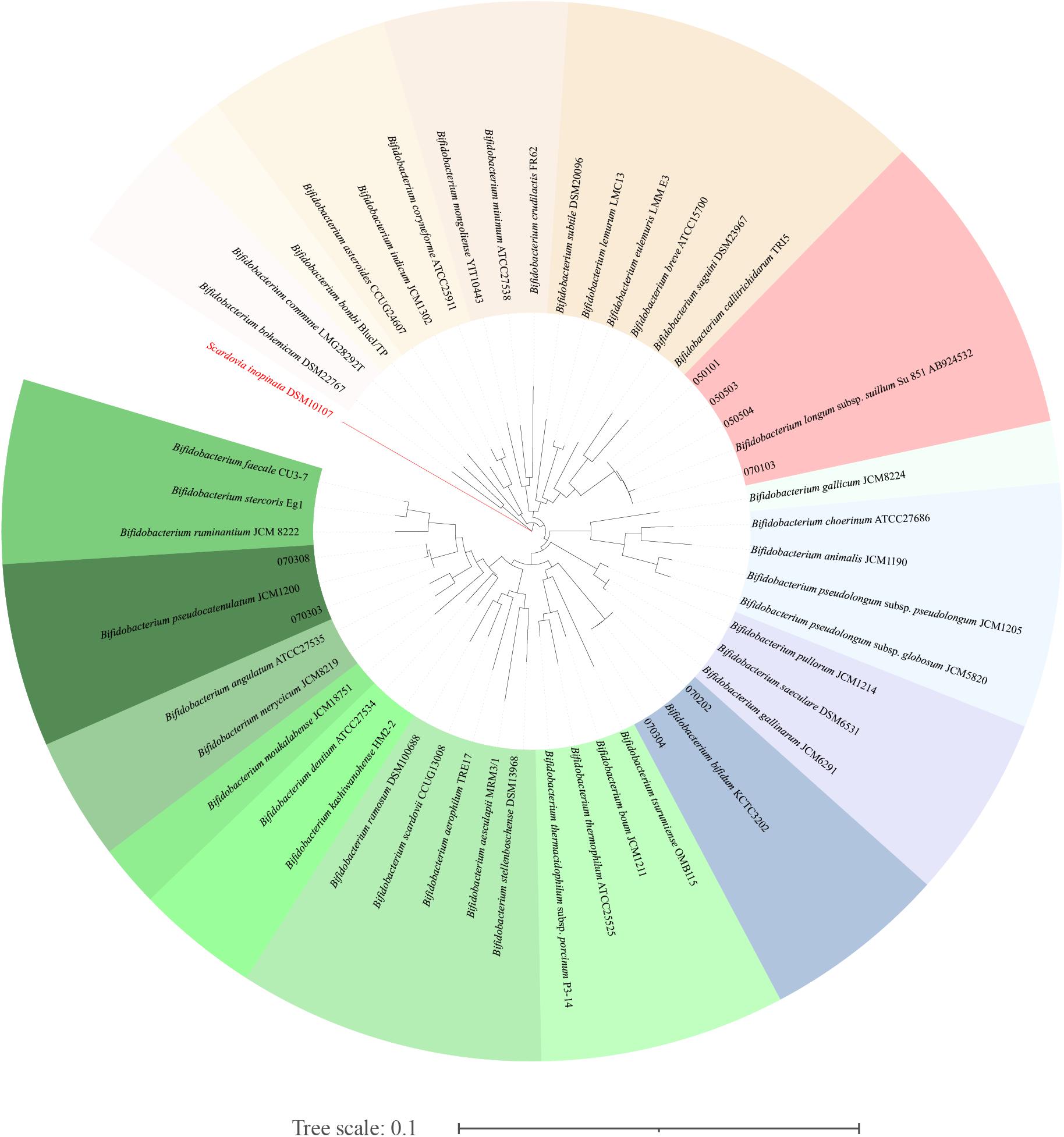
Figure 4. The phylogenetic analysis of the obtained Bifidobacterium. The phylogenetic tree was constructed by the neighbour-joining method, with the genome sequence of Scardovia inopinata JCM 12537 as outgroup, based on 1000 replicates of the phylogenetic tree.
Many studies have associated gut microbes with human health. Bifidobacteria among intestinal microbes are beneficial microorganisms (Jaglan et al., 2019) and have attracted the most attention. However, strict requirements for growth have caused difficulties with their isolation and purification. The carbon source is an indispensable factor for the growth of microorganisms, and it can be used to screen these bacteria. The distribution of glycoside hydrolases in Bifidobacterium as the key enzymes for carbohydrate hydrolysis is important for bacterial culture and functional analysis. Therefore, pangenomic analysis of the whole genome sequence of 144 Bifidobacterium species downloaded from NCBI and the dbCAN-seq database revealed the common GH families, GH13, GH3, GH42, and GH43, in Bifidobacterium.
The abundance of Bifidobacterium obviously differed among samples, and it was the most abundant in S201. The pangenomic findings of Bifidobacterium showed that GH13, GH3, GH42, and GH43 were abundant, and the abundance of enzymes in GH classes was basically the same in the metagenomic results of S171 and S201. Based on the hydrolysis characteristics of these enzymes, the corresponding carbohydrates (Table 1) were selected as main carbon source of the selective medium. The abundance of bifidobacteria in sample S201 is higher, so the abundance of bifidobacteria on the solid medium of S201 should be higher, but in fact sample S171 has a higher cultivable Bifidobacterium abundance on almost selective plates.
The selectivity of raffinose and xylan for bifidobacteria in both samples was effective based on the 16S amplicon sequencing analysis. Lactose was significantly selective for S171. Not only did the number of bifidobacteria surpass that of the control group (the ordinary GAM), but few other bacterial strains proliferated. Raffinose is an established prebiotic (Ose et al., 2018). Adding raffinose to the medium increases the production of short-chain fatty acids and carbon dioxide, and reduces the final pH and ammonia concentration in the medium (Amorim et al., 2020). In addition, raffinose can increased the relative abundance of probiotics (p < 0.05), and decreased that of pathogenic bacteria (Pacifici et al., 2017). Therefore, xylan might also have similar or even more powerful prebiotic effects, but this needs to be further verified by animal experiments and high-performance liquid chromatography (HPLC). Animal experiments were done to verify whether xylan can regulate the intestinal microbiota of mice, i.e., whether there is a difference between the intestinal flora of mice on a diet containing xylan and a diet without xylan and whether mice fed on a xylan-containing diet have more probiotics in their gut microbiota than mice on a xylan-free diet (Pacifici et al., 2017). HPLC was used to determine short-chain fatty acids in the mouse faeces (Li et al., 2020). GH3 and GH43 families containing xylanases and xylan was easily used, thus GH43 or GH3 glycoside hydrolases might be functioning as xylanases in Bifidobacterium. The results from the present study were obtained using a limited number of human faecal samples. This work on selective media could benefit further from testing on a larger number of human faecal samples.
Glycoside hydrolase 13, GH3, GH42, and GH43 are prevalent in Bifidobacterium, and the corresponding carbohydrates substrate can serve as the main carbon source in medium to selectively isolate and cultivate Bifidobacterium. Xylan might be a prebiotic that benefits host health, but its effects might differ among individuals. Metagenomics and pangenomics allow the accumulation of more information that can facilitate the isolation and cultivation of Bifidobacterium. The abundance of Bifidobacterium in samples might not mean that more bifidobacteria can be isolated, but it also depends on the characteristics of the Bifidobacterium species in samples. In the future, we will further verify the properties of specific GHs and the prebiotic properties of related oligosaccharides.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI: BioProject ID PRJNA695860 for 16S rDNA amplicon sequencing and PRJNA695407 for Metagenome.
The studies involving human participants were reviewed and approved by the Ethics Committee of The First Affiliated Hospital of Guangdong Pharmaceutical University (reference 2017-98). The patients/participants provided their written informed consent to participate in this study.
JZ, WC, XX, and QW conceived and designed the experiments. XX, SY, MD, LL, and LY conducted the experiments. SY, JM, XH, and YL participated in data collection and processing. SY and XX drafted the manuscript. JZ and XX reviewed the manuscript. All authors read and approved submission of the final version of the manuscript for publication.
This study was supported by research grants from the Guangdong Province Key R&D Program (2018B020205002), Project by the Department of Science and Technology of Guangdong Province (2019QN01N107), and the Guangdong Province Academy of Sciences Special Project for Capacity Building of Innovation Driven Development (2020GDASYL-20200301002, 2018GDASCX-0102, and 2020GDASYL-20200 102003).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Yanbin Yin and his team, who built the dbCAN-seq database and opened it to the public free of charge for the annotation of carbohydrate-active enzymes.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.649698/full#supplementary-material
Al-Masaudi, S., El Kaoutari, A., Drula, E., Al-Mehdar, H., Redwan, E. M., Lombard, V., et al. (2017). A metagenomics investigation of carbohydrate-active enzymes along the gastrointestinal tract of saudi sheep. Front. Microbiol. 8:666.
Amorim, C., Silvério, S. C., Cardoso, B. B., Alves, J. I., Pereira, M. A., and Rodrigues, L. R. (2020). In vitro fermentation of raffinose to unravel its potential as prebiotic ingredient. LWT 126:109322. doi: 10.1016/j.lwt.2020.109322
Armstrong, Z., Mewis, K., Liu, F., Morgan-Lang, C., Scofield, M., Durno, E., et al. (2018). Metagenomics reveals functional synergy and novel polysaccharide utilization loci in the Castor canadensis fecal microbiome. ISME J. 12, 2757–2769. doi: 10.1038/s41396-018-0215-9
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brownlee, I. A., Gill, S., Wilcox, M. D., Pearson, J. P., and Chater, P. I. J. S. S. (2018). Starch digestion in the upper gastrointestinal tract of humans. Starch Strke 70:1700135.
Cerqueira, F. M., Photenhauer, A. L., Pollet, R. M., Brown, H. A., and Koropatkin, N. M. (2020). Starch digestion by gut bacteria: crowdsourcing for carbs. Trends Microbiol. 28, 95–108. doi: 10.1016/j.tim.2019.09.004
Collado, M. C., Rautava, S., Aakko, J., Isolauri, E., and Salminen, S. (2016). Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Reprod. Sci. 6:23129.
Garrido, D., Barile, D., and Mills, D. A. (2012). A molecular basis for bifidobacterial enrichment in the infant gastrointestinal tract. Adv. Nutr. Res. 3, 415S–421S.
Ghaisas, S., Maher, J., and Kanthasamy, A. (2016). Gut microbiome in health and disease: linking the microbiome-gut-brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol. Ther. 158, 52–62. doi: 10.1016/j.pharmthera.2015.11.012
Grazul, H., Kanda, L. L., and Gondek, D. (2016). Impact of probiotic supplements on microbiome diversity following antibiotic treatment of mice. Gut Microbes 7, 101–114. doi: 10.1080/19490976.2016.1138197
Hsieh, C. Y., Osaka, T., Moriyama, E., Date, Y., Kikuchi, J., and Tsuneda, S. (2015). Strengthening of the intestinal epithelial tight junction by Bifidobacterium bifidum. Physiol. Rep. 3:e12327. doi: 10.14814/phy2.12327
Huang, L., Zhang, H., Wu, P., Entwistle, S., Li, X., Yohe, T., et al. (2018). dbCAN-seq: a database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 46, D516–D521.
Jaglan, N., Kumar, S., Choudhury, P. K., Tyagi, B., and Tyagi, A. K. (2019). Isolation, characterization and conjugated linoleic acid production potential of bifidobacterial isolates from ruminal fluid samples of Murrah buffaloes. Anaerobe 56, 40–45. doi: 10.1016/j.anaerobe.2019.02.001
Kim, L., Park, Y., and Park, Y. (2014). trans-10, cis-12 CLA promotes osteoblastogenesis via SMAD mediated mechanism in bone marrow mesenchymal stem cells. J. Funct. Foods 8, 367–376. doi: 10.1016/j.jff.2014.04.006
Lewis, Z. T., Totten, S. M., Smilowitz, J. T., Popovic, M., Parker, E., Lemay, D. G., et al. (2015). Maternal fucosyltransferase 2 status affects the gut bifidobacterial communities of breastfed infants. Microbiome 3:13.
Li, Y., Dong, J., Xiao, H., Zhang, S., Wang, B., Cui, M., et al. (2020). Gut commensal derived-valeric acid protects against radiation injuries. Gut Microbes 11, 789–806. doi: 10.1080/19490976.2019.1709387
Lugli, G. A., Milani, C., Duranti, S., Alessandri, G., Turroni, F., Mancabelli, L., et al. (2019). Isolation of novel gut bifidobacteria using a combination of metagenomic and cultivation approaches. Genome Biol. 20:96.
Matson, V., Fessler, J., Bao, R., Chongsuwat, T., Zha, Y., Alegre, M. L., et al. (2018). The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359:104. doi: 10.1126/science.aao3290
Matsuzawa, T., Kaneko, S., and Yaoi, K. (2015). Screening, identification, and characterization of a GH43 family beta-xylosidase/alpha-arabinofuranosidase from a compost microbial metagenome. Appl. Microbiol. Biotechnol. 99, 8943–8954. doi: 10.1007/s00253-015-6647-5
Mitsuoka, T. (2014). Establishment of intestinal bacteriology. Biosci. Microbiota Food Health 33, 99–116. doi: 10.12938/bmfh.33.99
Nurk, S., Antipov, D., Gurevich, A., Korobeynikov, A., Lapidus, A., Prjibelsky, A., et al. (2013). “Assembling genomes and mini-metagenomes from highly chimeric reads,” in Research in Computational Molecular Biology. RECOMB 2013. Lecture Notes in Computer Science, Vol. 7821, eds M. Deng, R. Jiang, F. Sun, and X. Zhang (Berlin: Springer), 158–170. doi: 10.1007/978-3-642-37195-0_13
O’Callaghan, A., and van Sinderen, D. (2016). Bifidobacteria and their roles as members of the human gut microbiota. Front. Microbiol. 7:925. doi: 10.3389/fmicb.2016.00925
Ose, R., Hirano, K., Maeno, S., Nakagawa, J., Salminen, S., Tochio, T., et al. (2018). The ability of human intestinal anaerobes to metabolise different oligosaccharides: a novel means for microbiota modulation? Anaerobe 51, 110–119. doi: 10.1016/j.anaerobe.2018.04.018
Pacifici, S., Song, J., Zhang, C., Wang, Q., Glahn, R. P., Kolba, N., et al. (2017). Intra amniotic administration of raffinose and stachyose affects the intestinal brush border functionality and alters gut microflora populations. Nutrients 9:304. doi: 10.3390/nu9030304
Patel, R., and DuPont, H. L. (2015). New approaches for bacteriotherapy: prebiotics, new-generation probiotics, and synbiotics. Clin. Infect. Dis. 60(Suppl. 2), S108–S121.
Pinto-Sanchez, M. I., Hall, G. B., Ghajar, K., Nardelli, A., Bolino, C., Lau, J. T., et al. (2017). Probiotic Bifidobacterium longum NCC3001 reduces depression scores and alters brain activity: a pilot study in patients with irritable bowel syndrome. Gastroenterology 153, 448–459. doi: 10.1053/j.gastro.2017.05.003
Pokusaeva, K., Fitzgerald, G. F., and Van Sinderen, D. (2011). Carbohydrate metabolism in bifidobacteria. Genes Nutr. 6, 285–306. doi: 10.1007/s12263-010-0206-6
Rios-Covian, D., Gueimonde, M., Duncan, S. H., Flint, H. J., and De Los Reyes-Gavilan, C. G. (2015). Enhanced butyrate formation by cross-feeding between Faecalibacterium prausnitzii and Bifidobacterium adolescentis. FEMS Microbiol. Lett. 21:21.
Riviere, A., Selak, M., Lantin, D., Leroy, F., and De Vuyst, L. (2016). Bifidobacteria and butyrate-producing colon bacteria: importance and strategies for their stimulation in the human gut. Front. Microbiol. 7:1030.
Roškar, I., Švigelj, K., Štempelj, M., Volfand, J., Štabuc, B., and Malovrh, Š, et al. (2017). Effects of a probiotic product containing Bifidobacterium animalis subsp. animalis IM386 and Lactobacillus plantarum MP2026 in lactose-intolerant individuals: randomised, placebo-controlled clinical trial. J. Funct. Foods 35, 1–8. doi: 10.1016/j.jff.2017.05.020
Savignac, H. M., Tramullas, M., Kiely, B., Dinan, T. G., and Cryan, J. F. (2015). Bifidobacteria modulate cognitive processes in an anxious mouse strain. Behav. Brain Res. 287, 59–72. doi: 10.1016/j.bbr.2015.02.044
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sochocka, M., Donskow-Lysoniewska, K., Diniz, B. S., Kurpas, D., Brzozowska, E., and Leszek, J. (2019). The gut microbiome alterations and inflammation-driven pathogenesis of Alzheimer’s disease is a critical review. Cell. Mol. Neurobiol. 56, 1841–1851. doi: 10.1007/s12035-018-1188-4
Soliman, G. A. (2019). Dietary fiber, atherosclerosis, and cardiovascular disease. Nutrients 11:1155. doi: 10.3390/nu11051155
Srutkova, D., Schwarzer, M., Hudcovic, T., Zakostelska, Z., Drab, V., Spanova, A., et al. (2015). Bifidobacterium longum CCM 7952 promotes epithelial barrier function and prevents acute DSS-induced colitis in a strictly strain-specific manner. PLoS One 10:e0134050. doi: 10.1371/journal.pone.0134050
Staudacher, H. M., Lomer, M. C. E., Farquharson, F. M., Louis, P., Fava, F., Franciosi, E., et al. (2017). A diet low in FODMAPs reduces symptoms in patients with irritable bowel syndrome and a probiotic restores Bifidobacterium species: a randomised controlled trial. Gastroenterology 153, 936–947. doi: 10.1053/j.gastro.2017.06.010
Turroni, F., Peano, C., Pass, D. A., Foroni, E., Severgnini, M., Claesson, M. J., et al. (2012). Diversity of bifidobacteria within infant gut microbiota. PLoS One 7:e36957. doi: 10.1371/journal.pone.0036957
Keywords: Bifidobacterium, glycoside hydrolase, metagenomics, pangenomics, enzymology
Citation: Yang S, Xie X, Ma J, He X, Li Y, Du M, Li L, Yang L, Wu Q, Chen W and Zhang J (2021) Selective Isolation of Bifidobacterium From Human Faeces Using Pangenomics, Metagenomics, and Enzymology. Front. Microbiol. 12:649698. doi: 10.3389/fmicb.2021.649698
Received: 05 January 2021; Accepted: 19 March 2021;
Published: 21 April 2021.
Edited by:
Eugenia Bezirtzoglou, Democritus University of Thrace, GreeceReviewed by:
Elisabeth Laville, Institut National de la Recherche Agronomique (INRA), FranceCopyright © 2021 Yang, Xie, Ma, He, Li, Du, Li, Yang, Wu, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinqiang Xie, d29zaGl4aW5xaWFuZ0AxMjYuY29t; Wei Chen, Y2hlbndlaTY2QGppYW5nbmFuLmVkdS5jbg==; Jumei Zhang, emhhbmdqbTkyNkAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.