 Mo Jia
Mo Jia Ifigenia Geornaras
Ifigenia Geornaras Jennifer N. Martin
Jennifer N. Martin Keith E. Belk
Keith E. Belk Hua Yang*
Hua Yang*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 01 April 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.647434
A comparative whole genome analysis was performed on three newly sequenced Escherichia coli O157:H7 strains with different stx profiles, previously isolated from feedlot cattle [C1-010 (stx1−, stx2c+), C1-057 (stx−), and C1-067 (stx1+, stx2a+)], as well as five foodborne outbreak strains and six stx-negative strains from NCBI. Phylogenomic analysis demonstrated that the stx2c-carrying C1-010 and stx-negative C1-057 strains were grouped with the six NCBI stx-negative E. coli O157:H7 strains in Cluster 1, whereas the stx2a-carrying C1-067 and five foodborne outbreak strains were clustered together in Cluster 2. Based on different clusters, we selected the three newly sequenced strains, one stx2a-carrying strain, and the six NCBI stx-negative strains and identify their prophages at the stx insertion sites. All stx-carrying prophages contained both the three Red recombination genes (exo, bet, gam) and their repressor cI. On the other hand, the majority of the stx-negative prophages carried only the three Red recombination genes, but their repressor cI was absent. In the absence of the repressor cI, the consistent expression of the Red recombination genes in prophages might result in more frequent gene exchanges, potentially increasing the probability of the acquisition of stx genes. We further investigated each of the 10 selected E. coli O157:H7 strains for their respective unique metabolic pathway genes. Seven unique metabolic pathway genes in the two stx2a-carrying strains and one in the single stx2c-carrying and seven stx-negative strains were found to be associated with an upstream insertion sequence 629 within a conserved region among these strains. The presence of more unique metabolic pathway genes in stx2a-carrying E. coli O157:H7 strains may potentially increase their competitiveness in complex environments, such as feedlot cattle. For the stx2c-carrying and stx-negative E. coli O157:H7 strains, the fact that they were grouped into the same phylogenomic cluster and had the same unique metabolic pathway genes suggested that they may also share closely related evolutionary pathways. As a consequence, gene exchange between them is more likely to occur. Results from this study could potentially serve as a basis to help develop strategies to reduce the prevalence of pathogenic E. coli O157:H7 in livestock and downstream food production environments.
Foodborne pathogenic Escherichia coli O157:H7 causes more than 96,000 cases of diarrheal illness and 3,200 hospitalizations annually in the United States (Scallan et al., 2011). This pathogen can cause severe gastrointestinal illness, including bloody diarrhea, which may progress to more serious illness, such as hemolytic uremic syndrome (HUS) and even death (Karmali, 2004). The eae (encoding intimin) and stx (encoding Shiga toxin) harbored in foodborne pathogenic E. coli O157:H7 strains are central to the pathogenesis of HUS (Paton and Paton, 1998). In addition to causing HUS, Shiga toxin produced by E. coli O157:H7 can enhance the adherence to epithelial cells and colonization in mice intestines (Robinson et al., 2006). Compared with Shiga toxin-producing E. coli O157:H7 that cause severe illness, stx-negative E. coli O157:H7 strains do not produce Shiga toxin. Though they may cause symptoms, such as diarrhea, they are not generally associated with HUS, even though they still carry virulence factors, such as eae and bfpA genes (Black et al., 2010; Ochoa and Contreras, 2011; Ferdous et al., 2015).
For decades, conventional molecular methods, such as polymerase chain reaction (PCR) and Sanger sequencing, have provided valuable information on the potential mechanisms of the acquisition or loss of the stx genes in the chromosome of E. coli O157:H7 cells. A previous PCR-based study found that stx-negative E. coli O157:H7 could acquire the stx genes in food animals (bovine and avian) and in feedlot environments by lysogenization of stx-carrying prophages into the stx insertion sites in bacterial genomes (Wetzel and LeJeune, 2007). Additionally, in humans, stx-positive E. coli O157:H7 may lose their stx-carrying prophages and, as a result, be converted into stx-negative E. coli O157:H7 during the infection process (Bielaszewska et al., 2007; Käppeli et al., 2011; Ferdous et al., 2015).
In addition to the stx genes, studies have also been conducted to compare the antimicrobial resistance of stx-positive and stx-negative E. coli O157:H7 isolates. A recent study compared antibiotic resistance genes in 19 stx-positive and 8 stx-negative E. coli O157:H7 isolates from cattle (Um et al., 2018). These E. coli O157:H7 isolates were screened, using a triplex real-time PCR assay, for 14 resistance genes including blaTEM, strA–strB, addA1, tet(A), tet(B), sulI, sulII, sulIII, cmlA, catI, catII, catIII, and floR. None of the eight stx-negative E. coli O157:H7 strains carried any antibiotic resistance genes, whereas one of the stx-positive strains contained several antibiotic resistance genes including blaTEM, strA–strB, tet(A), and sulII, and another stx-positive strain harbored a single tet(A) gene (Um et al., 2018). Another study compared the resistance to nine antibiotics in 12 stx-positive and 17 stx-negative E. coli O157:H7 isolates from bovine, caprine, and ovine milk in Greece using the disk diffusion method (Solomakos et al., 2009). The nine antibiotics evaluated included amoxicillin + clavulanic acid, ampicillin, cefachlor, cefuroxime, chloramphenicol, gentamicin, streptomycin, tetracycline, and trimethoprim + sulfamethoxazole. On average, the 12 stx-positive isolates were resistant to higher numbers of antibiotics (six of nine) than the 17 stx-negative isolates (four of nine) (Solomakos et al., 2009). These two studies suggest that the presence or absence of the stx genes between stx-positive and stx-negative E. coli O157:H7 may also result in changes in other phenotypic traits, such as antibiotic resistance.
The conventional molecular methods used to compare stx-positive and stx-negative E. coli O157:H7 are limited by their capabilities to study only certain selected DNA sequences from the bacterial genome, such as individual genes and/or short DNA fragments. With the development of next generation sequencing technology and whole genome sequencing (WGS), researchers are now able to compare and analyze DNA sequences at the level of the entire bacterial genome. A whole genome comparison study of E. coli O157 isolates from patients and cattle indicated that stx-negative E. coli O157:H7/NM isolates shared 22 virulence genes, the locus of enterocyte effacement region, and plasmids with stx-positive E. coli O157:H7 isolates (Ferdous et al., 2015). Another study used timed phylogeny analysis of whole genome sequences to investigate the evolution of a clinical stx-positive E. coli O157:H7 strain (Byrne et al., 2018). From this analysis, the authors reported that stx2a-carrying E. coli O157:H7 had evolved from stx-negative E. coli O157:H7 by acquiring a stx2a-carrying prophage, and that the stx-negative E. coli O157:H7 had evolved from a stx2c-carrying E. coli O157:H7 by losing a stx2c-carrying prophage (Byrne et al., 2018). In addition, a recent study used phylogenetic analysis to plot plasmids according to their phylogenetic positions in 14 E. coli O157:H7 and O157:NM strains that emerged stepwise (Nyong et al., 2020). By comparing stx-positive and stx-negative E. coli O157:H7 isolates from patients, cattle feces, and swine feces, they observed a stable evolutionary relationship between the host chromosomes and their respective plasmids. This previous study suggested the coevolution of plasmids and the chromosome in E. coli O157:H7/NM.
Cattle are one of the primary reservoirs of both stx-positive and stx-negative E. coli O157:H7 (Orden et al., 2002; Kuhnert et al., 2005; Keen et al., 2006; Sanchez et al., 2010). While pathogenic stx-positive E. coli O157:H7 are frequently associated with severe disease in humans, food animals are often asymptomatic carriers because they lack vascular Shiga toxin receptors (Pruimboom-Brees et al., 2000; Ferens and Hovde, 2011). Previous studies conducted to compare the whole genome sequences of stx-positive and stx-negative E. coli O157:H7 have mainly focused on human isolates. Similar such studies with stx-positive and stx-negative E. coli O157:H7 isolates of food animal origin are limited.
The current study was conducted to better understand the detailed mechanisms involved in the acquisition or loss of stx genes and the particular genotypes associated with the presence or absence of stx genes in E. coli O157:H7 isolated from feedlot cattle. The feedlot cattle E. coli O157:H7 strains included in the present study were from a previously conducted study (Carlson et al., 2009). The PCR results demonstrated that these E. coli O157:H7 strains displayed different stx profiles. The majority of the E. coli O157:H7 isolates from the study by Carlson et al. (2009) carried both or either stx1 or stx2 genes, but some of the E. coli O157:H7 isolates were stx-negative (i.e., neither stx1 nor stx2 genes present). In the present study, we conducted comparative genomic analysis of three isolates with different stx profiles to investigate their virulence genes, genes involved in the acquisition or loss of stx genes, and genes involved in metabolic pathways. Furthermore, the WGS data of these three strains were compared with those of five foodborne outbreak E. coli O157:H7 strains and six stx-negative E. coli O157:H7 strains from animal and food sources. Comparative whole genome analysis of E. coli O157:H7 strains with different stx gene profiles obtained from animal and food sources will expand our understanding of the mechanisms of exchange of stx genes and other genes associated with the exchange of stx genes between stx-positive and stx-negative E. coli O157:H7. Results from this study could potentially serve as a basis to help develop potential strategies to reduce the prevalence of pathogenic E. coli O157:H7 in livestock and downstream food production environments.
Three E. coli O157:H7 strains with different stx profiles were selected for this study. These included strains C1-057 (stx1−, stx2-), C1-010 (stx1−, stx2c+), and C1-067 (stx1+, stx2a+), and all three strains were previously isolated from fecal samples collected from cattle in a commercial feedlot (Carlson et al., 2009). For the stx-negative E. coli O157:H7 strain C1-057, we have previously reported its draft genome sequence (NCBI accession no. LAZO01000000; Yang et al., 2016). Following publication of the draft genome sequence, further sequencing was conducted by an external laboratory (University of Washington), using the PacBio RSII system. A complete, gapless chromosome and a plasmid of this strain were deposited into NCBI with new NCBI accession number (chromosome CP035366.1; plasmid CP035367.1). Additionally, the two stx-positive E. coli O157:H7 strains, C1-010 and C1-067, were sequenced via the PacBio Sequel system. The two stx-positive E. coli O157:H7 strains were activated from frozen glycerol stocks (−80°C) by two transfers in tryptic soy broth (TSB; Difco, Becton Dickinson and Co., Sparks, MD, United States) at 37°C for 24 h. The genomic DNA of each isolate was extracted using a QIAGEN Genomic DNA kit (QIAGEN, Redwood City, CA, United States). For the PacBio platform, the extracted large genomic DNA needs to be fragmented into smaller size by EcoRI restriction enzyme prior to DNA sequencing. If EcoRI restriction recognition sites were methylated, the restriction enzyme would not be able to cleave the DNA. In our study, a small amount of extracted DNA sample was digested with EcoRI. Then, gel electrophoresis was used to prove that EcoRI restriction recognition sites were not methylated, and the genomic DNA could be fragmented into smaller size. The extracted DNA samples were sent out for sequencing service by Genewiz, Inc. (San Francisco, CA, United States).
The PacBio Sequel system was used to produce the raw reads with mean genome coverages of 477x for strain C1-010 (stx1−, stx2c+) and 497x for strain C1-067 (stx1+, stx2a+). The lengths of raw reads were between 3 and 40 kb. The raw reads of strains C1-010 and C1-067 were subsequently de novo assembled into contigs separately by Canu (version 1.6). When fully assembled, the genomes of strains C1-010 and C1-067 consisted of 13 and 12 scaffolds, respectively. To identify plasmids, DNA sequences of pO157 (NCBI accession no. NC_002128) and pOsak1 (NCBI accession no. AB011548) of E. coli O157:H7 strain Sakai were used as a reference for Basic Local Alignment Search Tool (BLAST) analyses (Altschul et al., 1990). Strain Sakai was used as it is a well-studied foodborne pathogenic E. coli O157:H7 strain that was responsible for a large outbreak in Japan (Hayashi et al., 2001). Chromosome and plasmid sequences of the three newly sequenced strains were annotated using the NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al., 2016). The sequences of strains C1-057, C1-010, and C1-067 were deposited in the NCBI database, and their NCBI accession numbers are listed in Table 1.
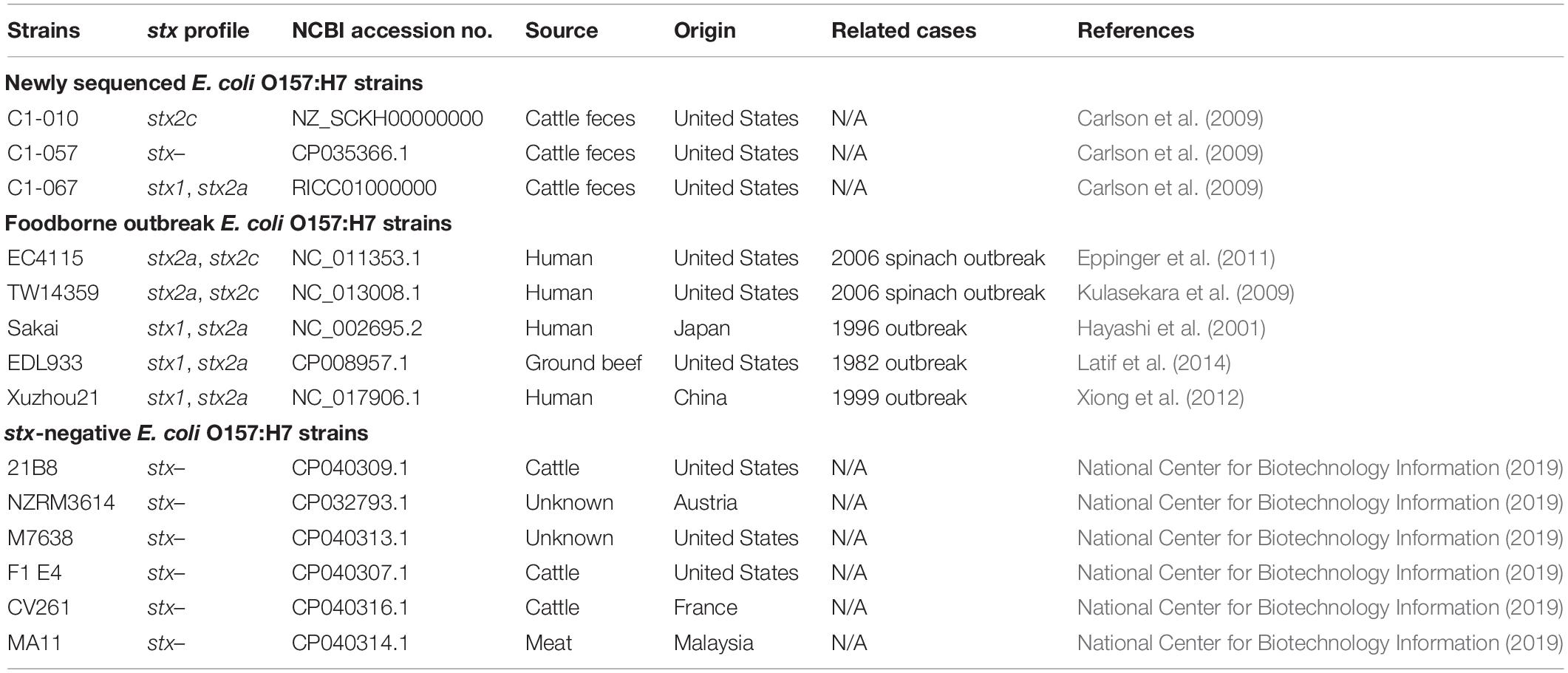
Table 1. The stx gene profiles, accession numbers, sources, and countries of origin of the Escherichia coli O157:H7 strains used in this study.
In addition to the three newly sequenced E. coli O157:H7 strains, we also included five foodborne outbreak E. coli O157:H7 strains and six stx-negative E. coli O157:H7 strains obtained from the NCBI database (Table 1). These strains were selected to represent the different stx profiles, and their genomes were compared with the three newly sequenced strains. The stx gene profiles, origins, and NCBI accession numbers of all 14 E. coli O157:H7 strains used in this study are listed in Table 1.
A phylogenomic analysis was performed on all 14 E. coli O157:H7 strains listed in Table 1. The phylogenomic tree was generated based on the single nucleotide polymorphisms (SNP) matrix between the E. coli O157:H7 strains using kSNP3.1 (available at1) (Gardner et al., 2015). The kSNP3.1 is a non-reference-based SNP analysis software suite that incorporates multiple SNP utility programs on a Linux platform. The parameter k-mer size was determined by Kchooser, a utility program incorporated in kSNP3.1. Chromosome FASTA files of the 14 strains obtained from the NCBI database were imputed in kSNP3.1 to generate the SNP matrix. The SNP matrix output was subsequently used to construct a maximum likelihood (ML) phylogenic tree by MEGA-X (available at2) with the parameter set at 2,000 bootstraps (Kumar et al., 2018).
Based on results from the phylogenomic analysis, 10 E. coli O157:H7 strains were selected for prophage identification. Phage Search Tool Enhanced Release (PHASTER) (available at3) is a web-based service for rapid identification and annotation of prophage sequences within bacterial genomes (Arndt et al., 2016). Prophages in the newly sequenced strains, C1-057 (stx1−, stx2-), C1-010 (stx1−, stx2c+), and C1-067 (stx1+, stx2a+), the reference strain Sakai (stx1+, stx2a+), and the six aforementioned stx-negative strains (Table 1) were identified by PHASTER. GenBank files of these strains were uploaded to the PHASTER server for identifications of intact, questionable, and incomplete prophages harbored in each of the strains. Strain Sakai was used as the stx-positive E. coli O157:H7 reference strain in this analysis since it is a well characterized foodborne pathogenic strain containing 18 prophages (Hayashi et al., 2001).
DNA sequences of six previously reported stx insertion sites (wrbA, sbcB, yehV, argW, yecE, and Z2577) were extracted from the whole genome sequence of E. coli O157:H7 strain Sakai (Yokoyama et al., 2000; Hayashi et al., 2001; Ohnishi et al., 2002; Eppinger et al., 2011). The stx insertion sites in the three newly sequenced strains and six stx-negative strains from NCBI were identified by screening the DNA sequences of the six stx insertion sites from strain Sakai against the chromosomal sequences of each of the nine E. coli O157:H7 strains using BLASTn (Johnson et al., 2008). Each of the six stx insertion sites in all nine E. coli O157:H7 strains was manually checked to determine if these stx insertion sites were occupied by any prophages (stx-carrying or stx-negative) identified by PHASTER.
DNA sequences of three Red recombination genes (exo, bet, and gam) and Red operon repressor gene cI were extracted from the whole genome sequence of E. coli O157:H7 strain Sakai (Hillyar, 2012). These 4 genes were firstly screened against the prophages at the stx insertion sites in the 10 selected strains using BLASTn (Johnson et al., 2008). In addition, we screened all prophages located within in entire genomes of strains Sakai, C1-057 (stx−), C1-010 (stx1−, stx2c+), and C1-067 (stx1+, stx2a+) for these four genes.
The whole genome sequences of E. coli O157:H7 strain C1-057 (stx1−, stx2-) and strain Sakai were aligned by DNASTAR MegAlign Pro 14 to identify large specific sequence regions (SSRs) (> 10 kb) that were located in either strain C1-057 or Sakai. Genes that were harbored in SSRs and the sequence regions (10 kb) downstream or upstream of each SSR in either strain C1-057 or Sakai were mapped against the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database using the Integrated Microbial Genomes and Microbiomes (accessible at4) (Kanehisa et al., 2016; Chen et al., 2018). The unique genes, which were from both SSRs and the sequence regions downstream or upstream of SSRs, with a hit to the genes in the KEGG metabolic pathways were extracted and combined into one FASTA file. Sequences of unique metabolic pathway genes in the FASTA file were mapped against the genomic sequences of the newly sequenced strains, C1-067 (stx1+, stx2+) and C1-010 (stx1−, stx2c+), and the six stx-negative strains from the NCBI database to identify if any of the metabolic pathway genes were present in any of these strains.
Table 2 shows the general features of the three newly sequenced E. coli O157:H7 strains previously isolated from feedlot cattle fecal samples. As already described, each of these three strains had different stx profiles. In the current study, the gapless chromosome of the stx-negative strain, C1-057 (5.45 Mb), was obtained. The chromosomes of the two stx-positive strains, C1-010 (stx1−, stx2c+) (5.57 Mb) and C1-067 (stx1+, stx2a+) (5.56 Mb), were composed of 12 and 11 scaffolds, respectively. In addition, the plasmid identification showed that strains C1-057, C1-010, and C1-067 each contained one pO157-like plasmid with sequence similarity of 97% (97.5 kb), 97% (91.9 kb), and 99% (92.7 kb) to the pO157 (92.7 kb) in strain Sakai, respectively. The DNA sequences of these three strains have been deposited to the NCBI (see Table 2 for their NCBI accession number).
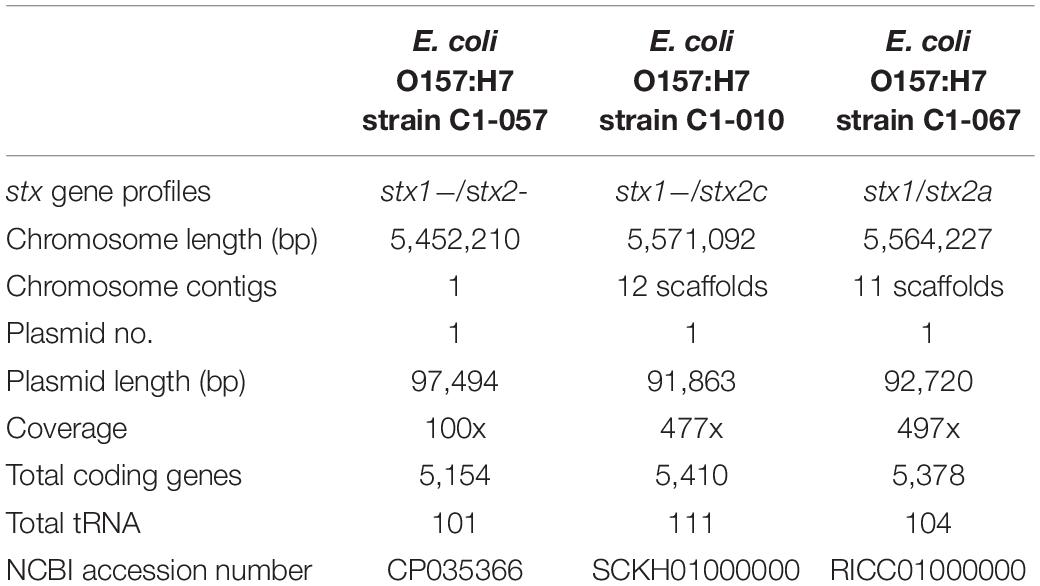
Table 2. General genomic features of the three newly sequenced Escherichia coli O157:H7 strains with different stx profiles.
Single nucleotide polymorphisms analysis was performed to evaluate the phylogenomic relationship among the 14 E. coli O157:H7 strains (Figure 1). The optimal length of k-mer was determined as 21 by Kchooser. A total of 3,581 SNP were identified from the 14 strains. Figure 1 shows the ML phylogenomic tree of the 14 E. coli O157:H7 strains. Overall, C1-057 (stx1−, stx2-) and C1-010 (stx1−, stx2c+), together with the six stx-negative E. coli O157:H7 strains from the NCBI database, were grouped into Cluster 1, whereas C1-067 (stx1+, stx2c+) and the five foodborne outbreak strains were grouped into Cluster 2. More specifically, strains C1-057 and C1-010 were clustered in Cluster 1a with three stx-negative strains from the United States and one stx-negative strain from Malaysia. Strain C1-067 grouped into Cluster 2b with three of the foodborne outbreak strains (Sakai, EDL933, Xuzhou21) had the closest relationship with strain Sakai.
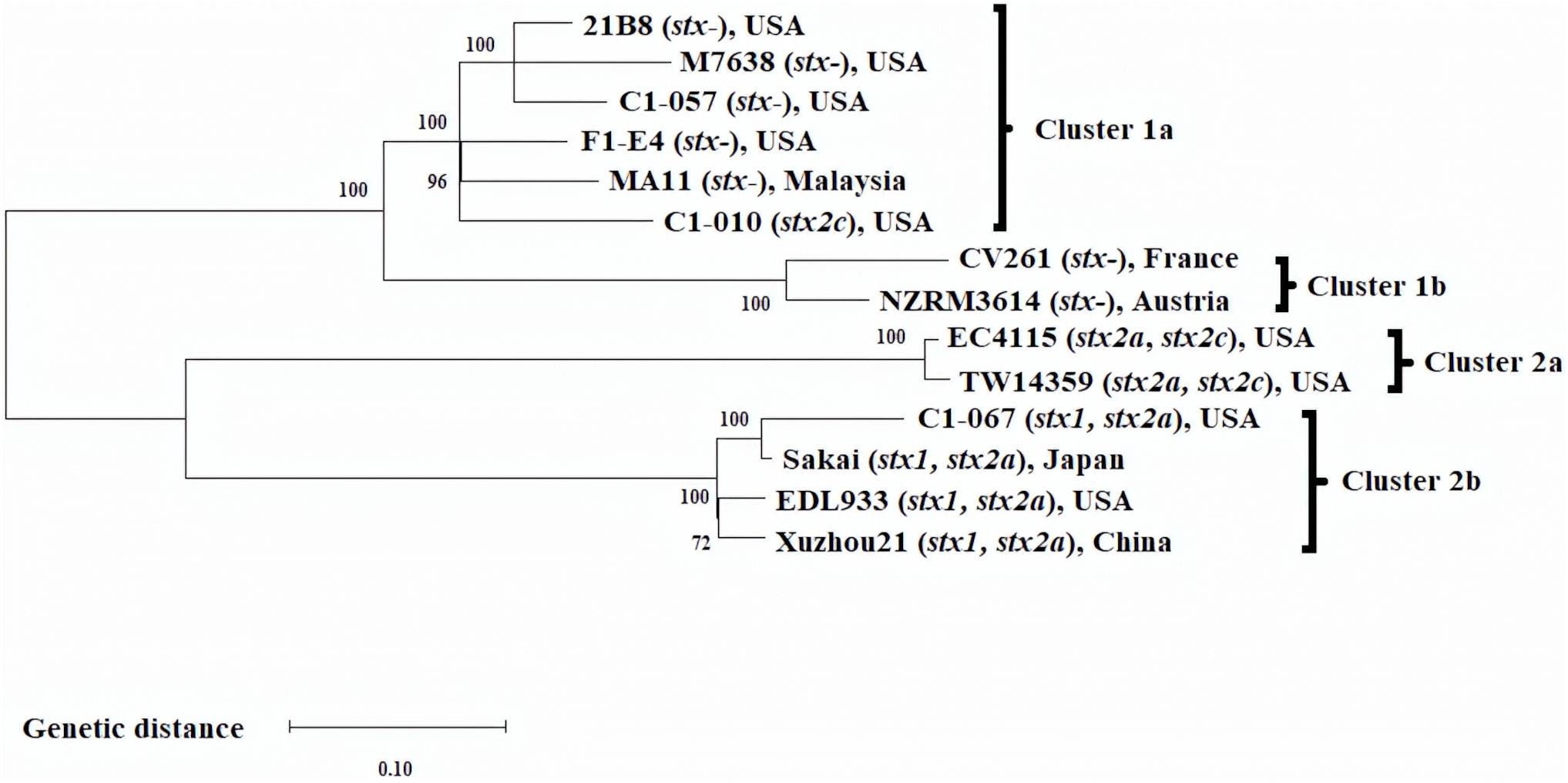
Figure 1. Maximum likelihood phylogenomic tree of 14 Escherichia coli O157:H7 strains. SNP matrix was generated from 3,581 SNP with 2,000 bootstraps, and the phylogenomic tree was visualized using MEGA-X.
Prophage identification, using PHASTER, was conducted on 10 of the 14 E. coli O157:H7 strains. The 10 strains included all the strains that were grouped in Cluster 1 [i.e., C1-010 (stx1−, stx2c+), C1-057 (stx−), and 6 other stx-negative strains] and strains C1-067 (stx1+, stx2a+) and Sakai from Cluster 2b since they had the closest phylogenomic relationship. The results of the identified prophages are shown in Table 3. Overall, 17–32 prophages were identified on the chromosome sequences of the selected E. coli O157:H7 strains. These prophages contained genome sizes ranging from 6.3 to 124.9 kb. Strain Sakai is known to have one P4-like prophage Sp2 (12.9 kb) (Asadulghani et al., 2009). The DNA sequence of prophage Sp2 was screened against the other nine selected E. coli O157:H7 strains. Six stx-negative strains (C1-057, 21B8, M7638, F1 E4, CV261, MA11), strain C1-010 (stx1−, stx2c+), and strain C1-067 (stx1+, stx2a+) harbored the identical Sp2 in their genomes. One stx-negative strain (NZRM3614) contained a Sp2-like prophage that shared 100% coverage and identity to Sp2 but with an insertion sequence of 1.3 kb.
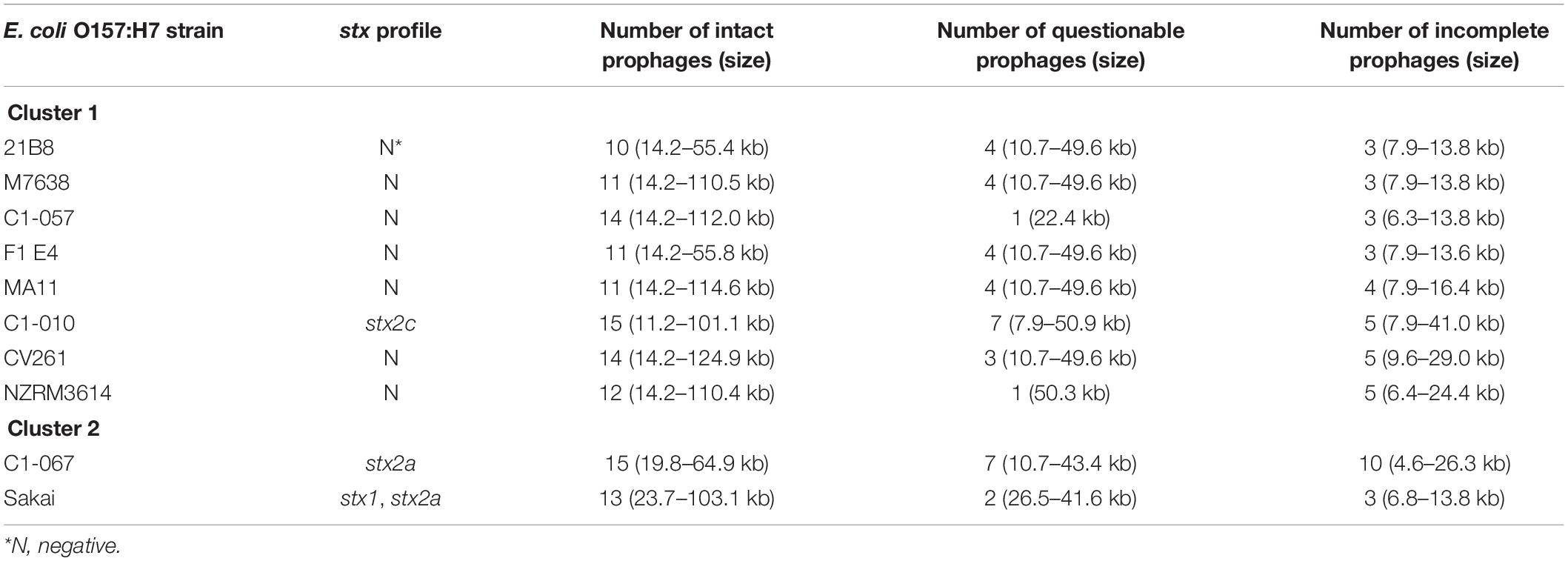
Table 3. Different prophage contents, including intact, questionable, and incomplete prophages, predicted from the Escherichia coli O157:H7 strains belonging to Cluster 1 and Cluster 2 by PHASTER.
Since both strains Sakai and C1-067 (stx1+, stx2a+) were grouped into Cluster 2b (Figure 1), prophages in strain C1-067 (isolated from feedlot cattle) were compared with the prophages in strain Sakai. The chromosome of E. coli O157:H7 strain Sakai was reported by Hayashi et al. (2001) to contain 18 prophages (Sp1–Sp18), and PHASTER identified all these 18 prophages. Sequence alignment between strains Sakai and C1-067 demonstrated that strain C1-067 contained all 18 identical prophages harbored in strain Sakai (data not shown).
Since strains C1-057 (stx1−, stx2-) and C1-010 (stx1−, stx2c+) from feedlot cattle were grouped into Cluster 1 (Figure 1), prophages in strain C1-057 were compared with the prophages in strain C1-010. Strains C1-057 and C1-010 shared 10 intact, 1 questionable, and 2 incomplete prophages. Furthermore, strains C1-057 and C1-010 contained unique prophages that were not present in strains C1-010 and C1-057, respectively. Strain C1-057 harbored four unique intact and one unique incomplete prophages, whereas strain C1-010 had four unique intact and one unique questionable prophages. In addition, we found one, five, and three intact, questionable, and incomplete prophages, respectively, in strain C1-010 that shared similar but shorter sequences with the intact prophages in strain C1-057. It is worth noting that the chromosome of strain C1-057 is fully closed, but strain C1-010 was composed of 12 scaffolds. The shorter prophages observed in strain C1-010 compared with those in strain C1-057 might be due to gaps between scaffolds or the deletion of genes from prophages.
Prophages in the newly sequenced strains C1-057 (stx1−, stx2-) and C1-067 (stx1+, stx2a+) belonging to Clusters 1 and 2, respectively, were also compared. Strains C1-057 and C1-067 shared three intact, one questionable, and two incomplete identical prophages. Strain C1-057 carried eight intact, one questionable, and one incomplete prophages that were not found in strain C1-067, whereas strain C1-067 contained eight intact, one questionable, and three incomplete unique prophages. In addition, in strain C1-067, two intact, one questionable, and five incomplete prophages shared similar but not identical sequences with the corresponding intact prophages in strain C1-057.
A 14.2 kb intact prophage was only found in each of the strains from Cluster 1 [C1-010 (stx1−, stx2c+) and seven stx-negative strains], but not in the two strains within Cluster 2b (Sakai and C1-067). PHASTER results indicated that this prophage was the most similar to a known phage P4 (11.6 kb, NCBI accession no. NC_001609.1) with 61% query coverage and 97.4% identity. It should be noted that when phage P4 infects E. coli cells, it has three known modes of propagation: (1) it can integrate within the host chromosome as a prophage; (2) it can replicate as a plasmid; or (3) it can replicate itself rapidly in its lytic cycle (Briani et al., 2001). Phage P4 contains 19 genes (Briani et al., 2001). The P4-like prophage in the strains from Cluster 1 contained 25 genes, and 10 of which were identical to the genes present in phage P4. It is reasonable to presume that these 10 genes may be involved in one or more of the three modes of propagation.
The chromosome of the 10 E. coli O157:H7 strains selected for prophage identification was further examined for 6 types of previously described stx insertion sites (wrbA, sbcB, yehV, argW, yecE, and Z2577). All 10 strains carried all 6 stx insertion site genes. As shown in Table 4, three of the stx insertion sites of wrbA, yehV, and sbcB were occupied by prophages in 2, 10, and 3 strains, respectively, whereas the other 3 stx insertion sites of argW, yecE, and Z2577 remained accessible and unoccupied in all 10 strains.
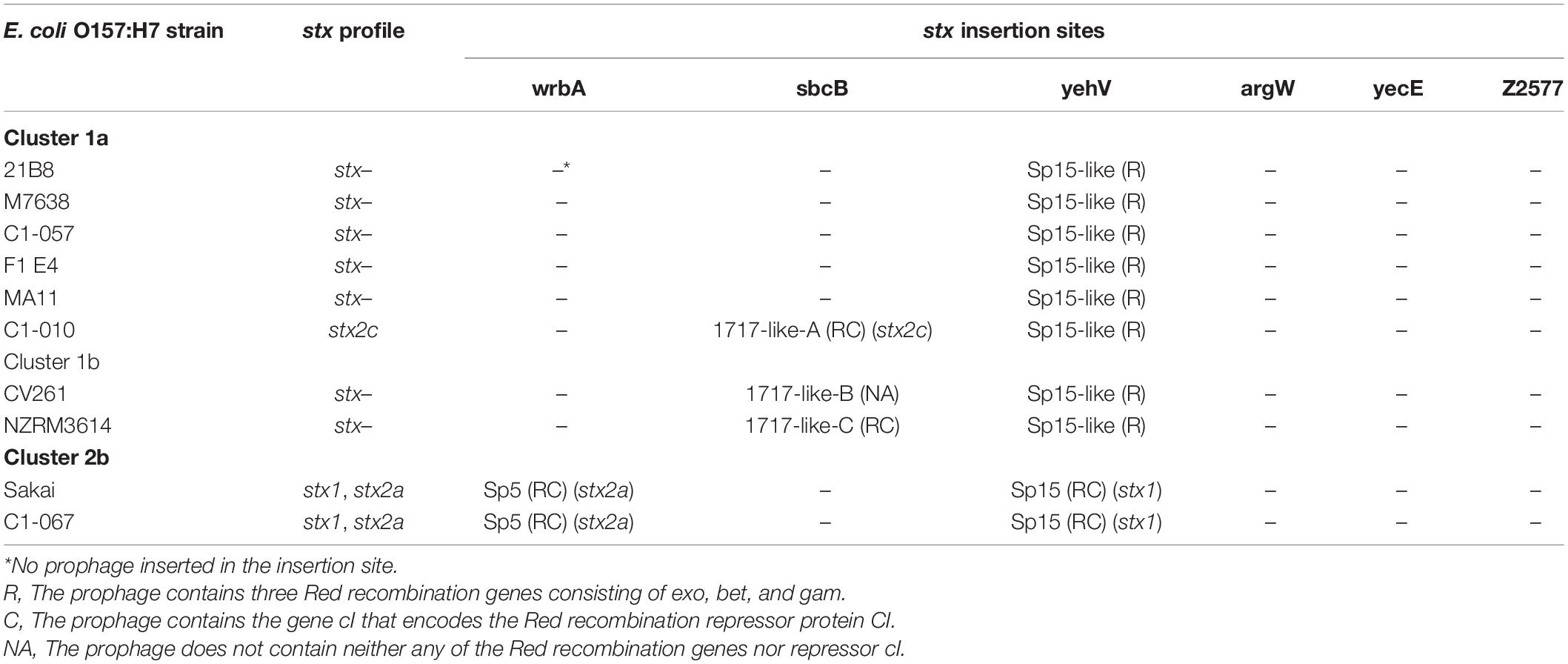
Table 4. Comparison of prophages at the six stx insertion sites in Escherichia coli O157:H7 strains Sakai, the three newly sequenced strains, and six stx-negative strains from the NCBI database.
Among the 10 selected E. coli O157:H7 strains, strains Sakai and C1-067 (stx1+, stx2a+) belonging to Cluster 2b carried 2 identical stx-carrying prophages: 1 stx1-carrying prophage at the yehV stx insertion site and 1 stx2a-carrying prophage at the wrbA stx insertion site. All strains from Cluster 1a, except for strain C1-010 (stx1−, stx2c+), harbored an identical stx-negative prophage at the yehV stx insertion site. The two strains from Cluster 1b and strain C1-010 carried an identical stx-negative prophage at the yehV stx insertion site, whereas strain C1-010 contained a stx2c-carrying prophage, and the two strains from Cluster 1b contained stx-negative prophages with varied DNA sequences at the sbcB stx insertion site.
For all 10 selected E. coli O157:H7 strains, one specific type of prophage with varied DNA sequences was observed at the yehV stx insertion site. The stx1-carrying prophage Sp15 (50.5 kb) was present in strains Sakai and C1-067 (stx1+, stx2a+). An intact stx-negative prophage (47.6 kb), named Sp15-like, in all Cluster 1 strains had 72% query coverage and 95% identity to prophage Sp15. We observed a 10.2 kb DNA fragment without the stx genes of the prophage Sp15-like in the strains from Cluster 1, whereas a 12.8 kb DNA fragment containing the genes of stx1A and stx1B of the stx1-carrying prophage Sp15 was present in each strain of Sakai and C1-067 (Figure 2).
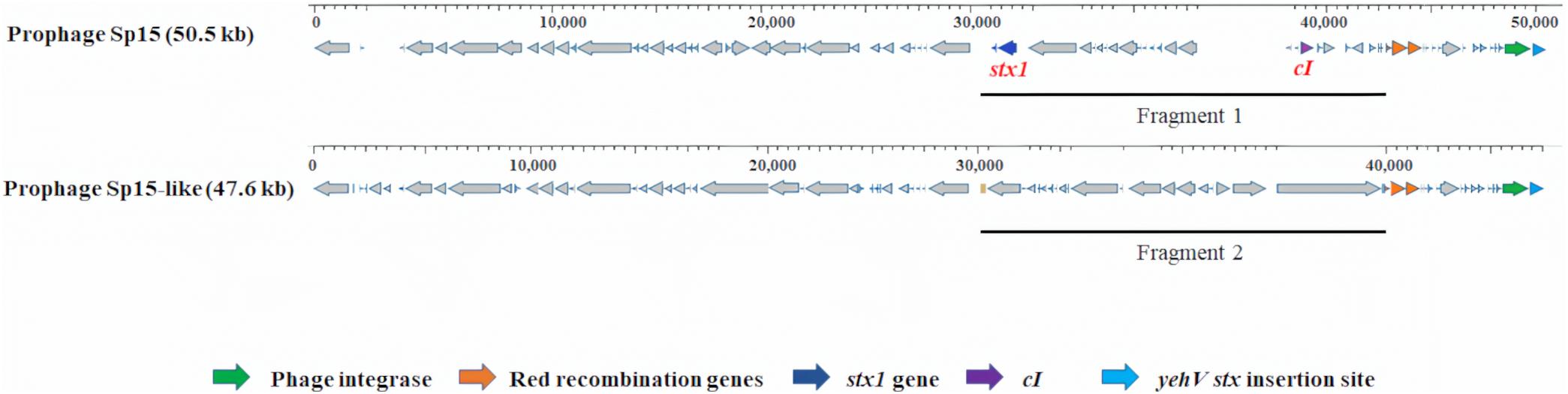
Figure 2. Prophage comparison at yehV stx insertion sites in Escherichia coli O157:H7 strains used in this analysis. The stx1-carrying prophage Sp15 was present in strains Sakai and C1-067 (stx1 +, stx2a +). The stx-negative prophage Sp15-like was present in seven stx-negative strains and strain C1-010 (stx1 +, stx2-). A 12.8 kb DNA fragment of the stx1-carrying prophage Sp15 containing the genes stx1A and stx1B (fragment 1) and a 10.2 kb DNA fragment that does not contain the stx genes of the stx-negative prophage Sp15-like (Fragment 2) are presented.
At the sbcB stx insertion site (Figure 3), prophages similar to phage 1717 (62.1 kb, NCBI accession no. FJ188381.1) but with varied sizes and varied query coverages and identities were found in the stx2c-carrying strain C1-010 belonging to Cluster 1a and the two stx-negative strains from Cluster 1b. A stx2c-carrying prophage (61.7 kb), named 1717-like-A, in strain C1-010 had 98% query coverage and 99% identity to phage 1717. A stx-negative prophage (22.0 kb, 34% query coverage and 99% identity to phage 1717), named 1717-like-B, and another stx-negative prophage (30.0 kb, 47% query coverage and 99% identity to phage 1717), named 1717-like-C, were found in the stx-negative strains CV261 and NZRM3614, respectively. Sequence comparisons demonstrated that a 29.2 kb DNA fragment containing the stx2c gene was present in prophage 1717-like-A in strain C1-010 belonging to Cluster 1a, but this DNA fragment was absent in the stx-negative prophages 1717-like-B and 1717-like-C in strains CV261 and NZRM3614 from Cluster 1b, respectively.
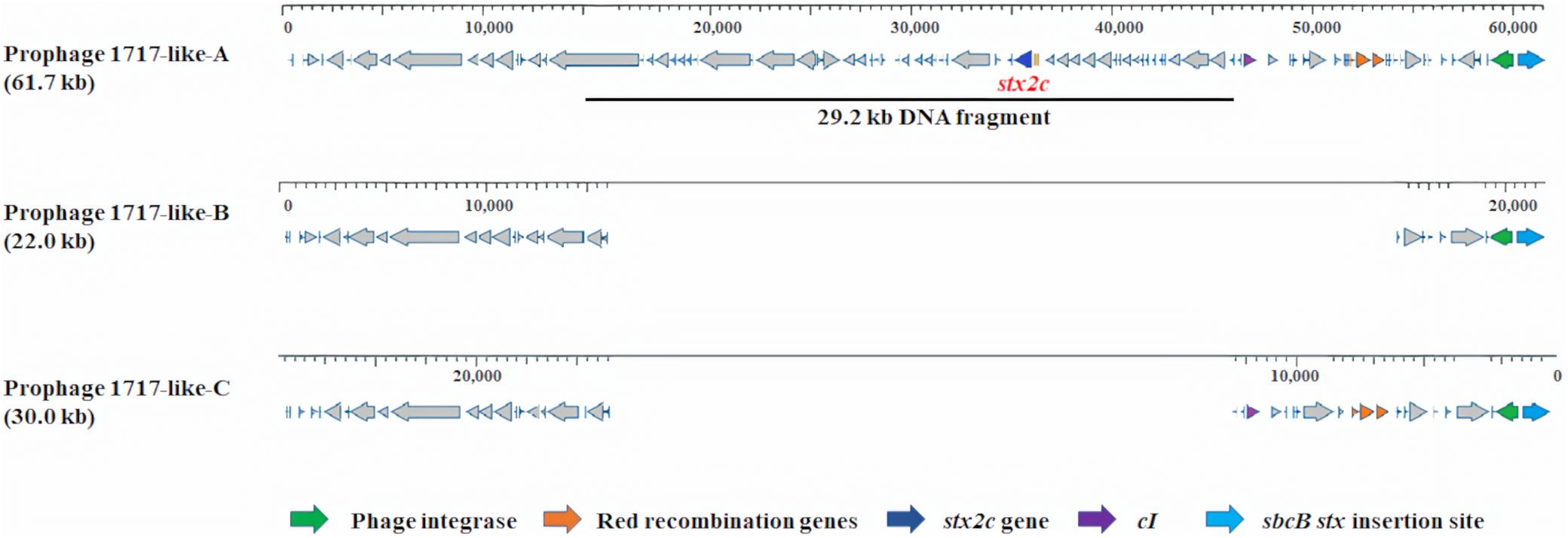
Figure 3. Prophage comparison at sbcB stx insertion sites in Escherichia coli O157:H7 strains used in this analysis. The stx2c-carrying prophages 1717-like-A was present in strain C1-010 (stx1−, stx2c +). The stx-negative prophages 1717-like-B and 1717-like-C were present in strains CV261 and NZRM3614, respectively. A 29.2 kb DNA fragment containing the stx2c gene was present in prophage 1717-like-A in strain C1-010, but this DNA fragment was absent in the stx-negative prophages 1717-like-B and 1717-like-C in strains CV261 and NZRM3614, respectively.
The prophages located at the 3 stx insertion sites in the 10 selected strains were investigated for the presence/absence of Red homologous recombination genes (exo, bet, and gam) and/or their repressor gene cI (Table 4). This analysis showed that three types of stx-carrying prophages (stx2a-carrying prophage Sp5, stx1-carrying prophage Sp15, and stx2c-carrying prophage 1717-like-A) contained both the three Red recombination genes and their repressor cI. Depending on the strain, stx-negative prophages had three situations: (1) prophage 1717-like-C in one strain contained both the three Red recombination genes and their repressor cI; (2) prophage Sp15-like in eight strains harbored only the three Red recombination genes, but the repressor cI was absent; and (3) prophage 1717-like-B in one strain contained neither any of the Red recombination genes nor the repressor cI.
The entire genomes of strains Sakai, C1-057 (stx−), C1-010 (stx1−, stx2c+), and C1-067 (stx1+, stx2a+) were also examined for any other prophages that were not located at the stx insertion sites but also harbored the Red recombination gene set. In addition to the prophages at the stx insertion sites, only one additional prophage (Sp3, 38.6 kb) carrying exo and bet, but not gam and cI, was found in strains Sakai, C1-057, C1-010, and C1-067.
We found that there were nine SSRs and six SSRs uniquely present in strains Sakai (stx1+, stx2a+) and C1-057 (stx−), respectively. The stx2a-carrying E. coli O157:H7 strains Sakai and C1-067 carried eight copies of unique metabolic pathway genes, including sodC, modD, fepC, fecD, fbpA, dhaM, dhaL, and dhaK. On the other hand, the stx2c-carrying E. coli O157:H7 strain C1-010 and seven stx-negative E. coli O157:H7 strains contained one copy of unique metabolic pathway gene, dhaR. Protein functions of the nine copies of unique metabolic pathway genes are listed in Table 5.
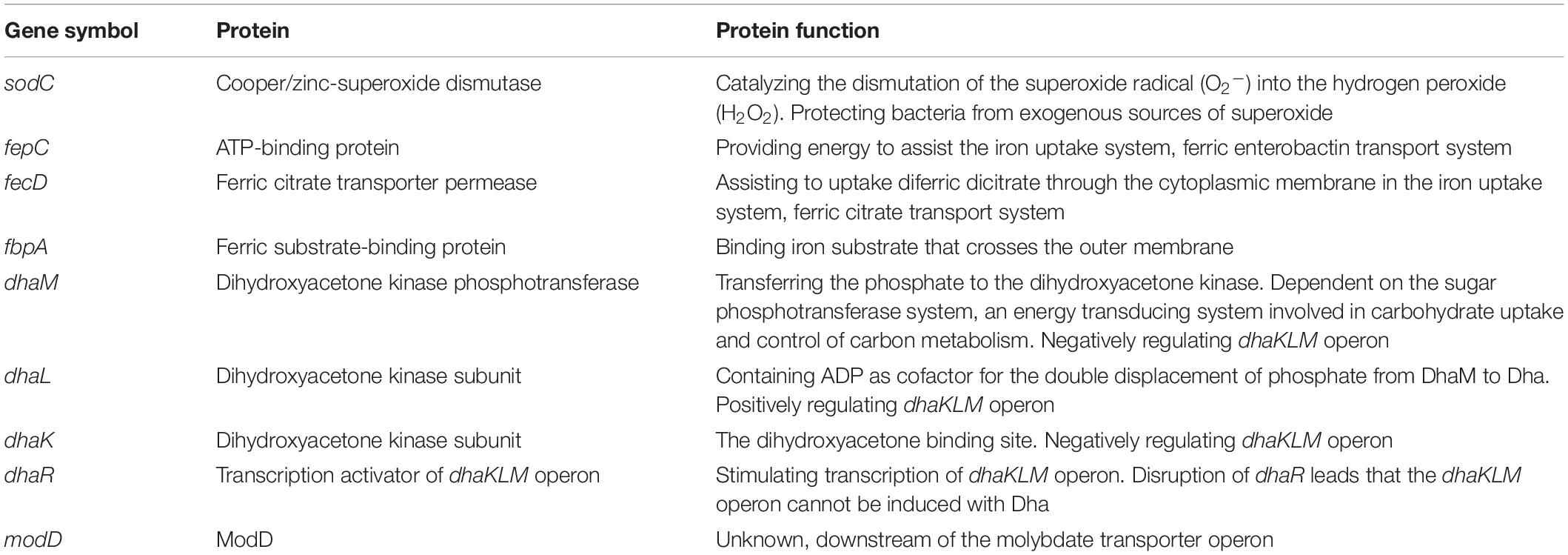
Table 5. The unique genes involved in metabolic pathways harbored in two stx2a-carrying, one stx2c-carrying, and seven stx-negative Escherichia coli O157:H7 strains used in this study.
In stx2a-carrying E. coli O157:H7 strains, one copy of unique metabolic pathway gene sodC was located at prophage Sp10. The other seven unique metabolic pathway genes in two stx2a-carrying strains and one unique metabolic pathway gene in one stx2c-carrying and seven stx-negative strains were found downstream of an insertion sequence 629 (IS629) within a conserved region among these strains (Figures 4A,B). Previous studies reported that E. coli O55:H7 was the ancestor of stx-positive and stx-negative E. coli O157:H7 (Shaikh and Tarr, 2003; Leopold et al., 2009; Byrne et al., 2018). In E. coli O55:H7 strain RM12579, we observed a corresponding conserved region that lacked IS629 but harbored a combination of eight unique metabolic pathway genes, including seven genes from stx2a-carrying strains and one gene from stx2c-carrying and stx-negative strains (Figures 4A,B).

Figure 4. (A) Unique metabolic pathway genes and IS629 in 10 Escherichia coli O157:H7 strains within a conserved region and metabolic pathway genes in 1 Escherichia coli O55:H7 strain within the corresponding conserved region. (B) Enlarged region in (A).
In this study, we performed a whole genome analysis of three E. coli O157:H7 strains with different stx profiles that were previously isolated from feedlot cattle (Carlson et al., 2009). Phylogenomic analysis of the three strains together with 11 other E. coli O157:H7 strains showed that these three strains were grouped into different clusters. The stx-negative strain C1-057 was grouped with four stx-negative strains in Cluster 1a. Strain C1-010, although stx2c positive, was grouped with five stx-negative strains in Cluster 1a. Strain C1-067, carrying both stx1 and stx2a genes, was grouped into Cluster 2b with strain Sakai (stx1+, stx2a+). These results suggested that the three E. coli O157:H7 strains from feedlot cattle may exhibit different levels of pathogenicity.
Wetzel and LeJeune (2007) reported that stx-negative E. coli O157:H7 could potentially be converted to stx-positive E. coli O157:H7 in cattle, swine, and avian production systems. A previous evolutionary model suggested that stx-positive E. coli O157:H7 could have evolved stepwise from an ancestor E. coli O55:H7, following the steps of 1) acquiring stx2-carrying prophages, 2) losing the O55 rfb-gnd gene, acquiring the O157 rfb-gnd gene, and losing the ability to ferment sorbitol, and 3) acquiring stx1-carrying prophage (Shaikh and Tarr, 2003; Leopold et al., 2009). However, the exact mechanisms involved in the acquisition and/or loss of the stx genes in E. coli O157:H7 strains are not fully understood. A greater understanding of the mechanisms involved in the acquisition or loss of stx genes could aid in the development of control strategies to reduce or eliminate highly pathogenic E. coli O157:H7 strains in cattle.
In addition to acquiring the stx-carrying prophage to become stx-positive E. coli O157:H7 strains, the results of this study suggested a potential alternative mechanism that could result in the acquisition of stx genes via the Red homologous recombination system. According to Yu et al. (2000), the Red homologous recombination system, which is usually found in lambda prophages, consists of three Red recombination genes, exo, bet, and gam, and the expression of these genes is regulated by their repressor CI (Table 6). The binding of repressor protein CI to the promoter pL in lambda prophages inhibits the expression of the Red recombination genes (Yu et al., 2000). We compared the presence or absence of Red recombination genes and the repressor gene cI between stx-negative and stx-positive prophages (Figures 2, 3). Overall, all stx-carrying prophages contained both the three Red recombination genes and their repressor cI, whereas the majority of the stx-negative prophages at the stx insertion sites carried only the three Red recombination genes but not the repressor cI gene. Our findings are consistent with the findings of previous studies conducted to examine stx2-carrying prophages in stx-positive E. coli O157:H7 strains (Smith et al., 2012; Tozzoli et al., 2014). These studies also demonstrated that both the three Red recombination genes and their repressor gene cI were present in stx2-carrying prophages (Smith et al., 2012; Tozzoli et al., 2014).
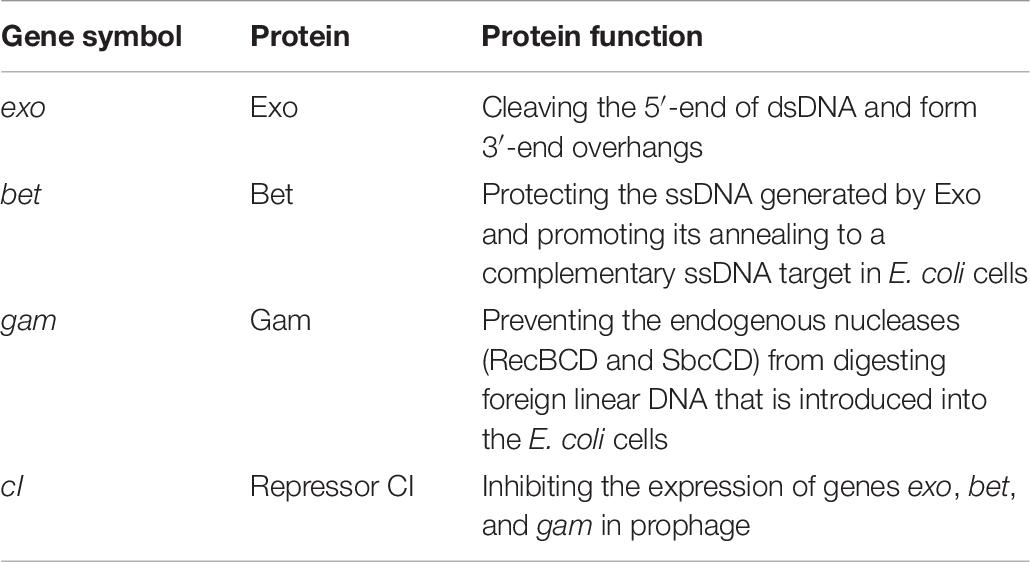
Table 6. Genes involved in the Red homologous recombination system in lambda prophages.
As previously mentioned, the presence of repressor CI in stx-carrying prophages could inhibit the activity of Red homologous recombination, and consequently, the stability of the stx genes in stx-positive E. coli O157:H7 could be increased. On the other hand, the majority of the stx-negative prophages at the stx insertion sites in this study carried only the three Red recombination genes, but their repressor cI was absent. In the absence of the repressor cI in stx-negative prophages, the consistent expression of the Red recombination genes may result in more frequent gene exchange, potentially contributing to the acquisition of stx genes. Overall, our results indicate that the presence or absence of repressor cI might result in different levels of activity of Red recombination in stx-positive and stx-negative prophages. For the stx-carrying prophages containing both the Red recombination genes and their repressor cI, the potential of losing stx genes is relatively low due to the inhibition of the activity of Red homologous recombination by their repressor CI. However, for the stx-negative prophages containing only Red recombination genes but not repressor cI, the potential of acquiring stx genes is relatively high due to the consistent expression of Red recombination genes, resulting in frequent gene exchanges. To our knowledge, this is the first report to suggest the possible functions of Red recombination genes and their repressor relating to the stability and acquisition of stx genes. Further studies are required to verify this proposed function associated with Red recombination genes and their repressor cI in more stx-negative and stx-positive E. coli O157:H7 strains under laboratory conditions and in the cattle environment.
The presence or absence of stx genes may also result in changes in other genotypic traits between stx-positive and stx-negative E. coli O157:H7, such as antibiotic resistance genes (Garcia-Aljaro et al., 2009; Colavecchio et al., 2017). In this study, we identified eight copies of unique metabolic pathway genes in two stx2a-carrying E. coli O157:H7 strains and one copy of a unique metabolic pathway gene in one stx2c-carrying and seven stx-negative E. coli O157:H7 strains. Among these nine metabolic pathway genes, gene sodC might contribute to the increased survival of stx-positive E. coli O157:H7 in complex microbial communities (Battistoni, 2003). Genes fepC, fecD, and fbpA could potentially be associated with improved iron uptake ability (Staudenmaier et al., 1989; Chen et al., 1993; Raymond et al., 2003). Genes dhaK, dhaL, dhaM, and dhaR are associated with glycerol metabolism (Bächler et al., 2005). The presence of more unique metabolic pathway genes in stx-positive (stx1+, stx2a+) strains may potentially lead to an increase in their competitiveness in complex environments, such as feedlot cattle.
The sodC gene encodes copper/zinc-superoxide dismutase (Cu–Zn SOD) that catalyzes the dismutation of the superoxide radical (O2–) into hydrogen peroxide (H2O2) (Fridovich, 1981). The sodC is considered a virulence gene since the Cu–Zn SOD protects bacteria from exogenous sources of superoxide. A previous study indicated that Cu–Zn SOD protected E. coli strains from superoxide generated by macrophages and enhanced bacterial survival (Battistoni, 2003). We found one copy of sodC harbored on the chromosomes of all 10 strains used in this analysis. However, an additional copy of sodC harbored in the Sp10 prophage was found only in Sakai and C1-067 (stx1+, stx2a+) but was absent in strains C1-057 (stx1−, stx2-), C1-010 (stx1−, stx2c+), and the six stx-negative strains from the NCBI database. The second copy of sodC may provide additional protection from the toxicity of superoxide radical. Thus, the stx-positive strains Sakai and C1-067 may be more resistant to exogenous superoxide radical than strains C1-057, C1-010, and the six stx-negative strains from the NCBI database.
The fepC, fecD, and fbpA genes encode proteins that are involved in three separate iron uptake systems, respectively. The fepC, from the ferric enterobactin transport system FepABCDG, encodes an ATP-binding protein transport that provides energy to assist iron uptake from the environment (Raymond et al., 2003). The fecD, from the ferric citrate transport system FecABCDE, encodes a ferric citrate transporter permease that assists to uptake diferric dicitrate through the cytoplasmic membrane (Staudenmaier et al., 1989). The fbpA, from the iron uptake gene cluster fbpABC, encodes ferric substrate-binding protein (Chen et al., 1993). Although this transposable element (TE) did not harbor any of the intact iron uptake gene cluster, the presence of genes fepC, fecD, and fbpA in strains Sakai and C1-067 (stx1+, stx2a+) can potentially increase iron uptake ability compared with strains C1-057 (stx1−, stx2-), C1-010 (stx1−, stx2c+), and the six stx-negative strains from the NCBI database.
Escherichia coli can metabolize glycerol via two microaerobic pathways: the dihydroxyacetone kinase (DAK) pathway and the glycerol pathway (Durnin et al., 2009). DhaK, DhaL, and DhaM, encoded by the dha operon, are proteins to phosphorylate DHA in the DAK pathway. The dha operon is controlled by DhaR, a transcriptional activator (Bächler et al., 2005). All four proteins are essential for the DAK pathway. Based on the O serotype and stx-profiles, we observed four dha operons (Figure 4B): (1) E. coli O55:H7 strain RM12579 harbored an intact dha operon consisting of dhaK, dhaL, dhaM, and dhaR; (2) E. coli O157:H7 stx-positive strains Sakai and C1-067 (stx1+, stx2a+) harbored a dha operon containing dhaK, dhaL, and dhaM, but not dhaR; (3) E. coli O157:H7 stx-positive strain C1-010 (stx1−, stx2c+) harbored another dha operon comprising only dhaR but not dhaK, dhaL, or dhaM; and (4) seven E. coli O157:H7 stx-negative strains harbored the same disrupted dha operon as strain C1-010. Additionally, both two types of disruptions of dha systems as shown in strains C1-067 and C1-010 inactivated the metabolism of glycerol via the DAK pathway. Durnin et al. (2009) reported that the deletion of dhaK led to a reduction in the efficacy of glycerol utilization by approximately 50%, resulting in a reduction in cell growth rate by 70%, in a minimal medium supplemented with glycerol as the sole carbon source. Another study also indicated that adding three levels of crude glycerin (0, 4, and 8%) in corn silage diets decreased the prevalence levels of E. coli O157 to 4.4, 3.2, and 1.8% in fecal samples from growing cattle, respectively (P < 0.01) (Aperce et al., 2013). Our results provided evidence that the disruption of the DAK pathway in both stx-positive and stx-negative E. coli O157:H7 strains from this study might contribute to a decrease in the rate of glycerol metabolism. Therefore, the disruption of the DAK pathways can potentially decrease the competitiveness and survival of E. coli O157:H7 in growing cattle fed diets that include crude glycerin. However, the addition of glycerol as a supplement may result in varying effects on the growth and activity of rumen bacteria. For example, high levels of glycerol (72–108 g/kg dry matter) in feed decreased (P < 0.05) the DNA concentrations of fiber-fermenting bacteria Butyrivibrio fibrisolvens and Selenomonas ruminantium in rumen, potentially resulting in the reduction of fiber digestibility (AbuGhazaleh et al., 2011). However, glycerol had no effect on fiber-fermenting bacteria Ruminococcus albus and starch-digesting bacteria Succinivibrio dextrinosolvens (El-Nor et al., 2010; AbuGhazaleh et al., 2011). In addition, the numbers of Enterobacteriaceae and Lactobacillus in rumen were not affected when cattle were fed oral rehydration solution containing glycerol (Omazic et al., 2013). The addition of glycerol in animal feed to manipulate rumen microflora for reducing pathogenic bacteria while at the same time retaining beneficial microflora could be complicated and, therefore, requires careful consideration and further investigation.
Among the metabolic pathway genes discussed above, seven unique metabolic pathway genes in the two stx2a-carrying strains and one in the one stx2c-carrying and seven stx-negative strains were found downstream of an IS629 within a conserved region (Figure 4A). Previous studies reported that E. coli O157:H7 could have evolved from an ancestor E. coli O55:H7 (Shaikh and Tarr, 2003; Leopold et al., 2009; Byrne et al., 2018). Sequence alignment demonstrated that a corresponding conserved region was also observed in the E. coli O55:H7 strain. Unlike the 10 E. coli O157:H7 strains, the corresponding conserved region in the E. coli O55:H7 strain lacked IS629, but it contained all eight metabolic pathway genes, including seven from stx2a-carrying E. coli O157:H7 strains and one from stx2c-carrying and stx-negative E. coli O157:H7 strains. Researchers used to believe that excision of IS elements rarely occurred in bacteria due to no regeneration of donor DNA by end-joining activity after IS excision (Synder and Champness, 2003). However, a study reported that excision of IS629 could occur frequently in E. coli O157, causing the diversity of genomic structure in E. coli O157 (Kusumoto et al., 2011). IS629 has been reported to be associated with a variety of genomic arrangements, including gene deletions, inversions, and duplications in Shigella flexneri (Wei et al., 2003) and E. coli O157:H7 (Iguchi et al., 2006; Ooka et al., 2009; Kusumoto et al., 2011). Our results indicated that insertion of IS629 may lead to the different presence of unique metabolic pathway genes between E. coli O157:H7 strains with different stx profiles. Additionally, for the stx2c-carrying and stx-negative E. coli O157:H7 strains, the fact that they shared the same unique metabolic pathway genes within the conserved region and they were grouped into the same phylogenomic cluster demonstrated that they had a closer genomic relationship, indicating that they may share more closely related genomic evolutionary pathways. Gene exchange between stx-negative and stx2c-carrying E. coli O157:H7 was more likely to occur.
In summary, our study firstly reported the potential functions of Red recombination genes and their repressor cI associating with the maintenance of stx genes in stx-carrying prophages and the acquisition of stx genes in stx-negative prophages. The presence of repressor CI in stx-carrying prophages could inhibit the activity of Red homologous recombination. Consequently, the absence of repressor cI in stx-negative prophages at the stx insertion sites could potentially lead to the consistent expression of Red recombination genes. As a result, gene exchange might occur more frequently, potentially contributing to the acquisition of stx genes in the stx-negative prophages at the stx insertion sites. Additionally, stx2a-carrying E. coli O157:H7 contained more copies of unique metabolic pathway genes than stx2c-carrying and stx-negative E. coli O157:H7. The presence of more unique metabolic pathway genes in stx2a-carrying E. coli O157:H7 may potentially lead to an increase in their competitiveness and survival in complex environments, such as feedlot cattle. Furthermore, within a conserved region, we observed seven unique metabolic pathway genes in stx2a-carrying E. coli O157:H7 and one unique metabolic pathway gene in stx2c-carrying and stx-negative E. coli O157:H7 strains downstream of an IS629. The difference in the presence of metabolic pathway genes of E. coli O157:H7 with different stx profiles may be associated with insertion of IS629 from their ancestor E. coli O55:H7. The fact that stx2c-carrying and stx-negative E. coli O157:H7 strains were grouped into the same cluster and they shared unique metabolic pathway genes within the conserved region suggested that stx2c-carrying and stx-negative E. coli O157:H7 had closely related evolutionary pathways. Gene exchange between stx-negative and stx2c-carrying E. coli O157:H7 was more likely to occur. Our comparative genomic analysis also revealed that E. coli O157:H7, regardless of carrying stx genes or not, did not contain a functional dha operon. This result indicated that neither stx-positive nor stx-negative E. coli O157:H7 could not utilize glycerol via the DAK pathway. Adding glycerol as a feed supplement may be the potential strategy to reduce pathogenic E. coli O157:H7 in livestock and downstream food production processing environments.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm. nih.gov/, CP035366.1, https://www.ncbi.nlm.nih.gov/, CP0353 67.1, https://www.ncbi.nlm.nih.gov/, NZ_SCKH00000000, https://www.ncbi.nlm.nih.gov/, RICC01000000.
MJ conceived the presented idea and carried out the experiment and wrote the manuscript with the support from HY. HY helped supervise the project. All authors provided critical feedback and contributed to the final version of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AbuGhazaleh, A. A., Abo El-Nor, S., and Ibrahim, S. A. (2011). The effect of replacing corn with glycerol on ruminal bacteria in continuous culture fermenters. J. Animal Physiol. Animal Nutr. 95, 313–319. doi: 10.1111/j.1439-0396.2010.01056.x
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Aperce, C. C., Heidenreich, J. M., Schneider, C. S., and Drouillard, J. S. (2013). Effect of crude glycerin on fecal shedding of Escherichia coli O157 in growing and finishing cattle. J. Appl. Res. Vet. Med. 11, 202–211.
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Asadulghani, M., Ogura, Y., Ooka, T., Itoh, T., Sawaguchi, A., Iguchi, A., et al. (2009). The defective prophage pool of Escherichia coli O157: prophage–prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog. 5:e1000408. doi: 10.1371/journal.ppat.1000408
Bächler, C., Schneider, P., Bähler, P., Lustig, A., and Erni, B. (2005). Escherichia coli dihydroxyacetone kinase controls gene expression by binding to transcription factor DhaR. EMBO J. 24, 283–293. doi: 10.1038/sj.emboj.7600517
Battistoni, A. (2003). Role of prokaryotic Cu, Zn superoxide dismutase in pathogenesis. Biochem. Soc. Trans. 31(Pt 6), 1326–1329. doi: 10.1042/bst0311326
Bielaszewska, M., Köck, R., Friedrich, A. W., Von Eiff, C., Zimmerhackl, L. B., Karch, H., et al. (2007). Shiga toxin-mediated hemolytic uremic syndrome: time to change the diagnostic paradigm? PLoS One 2:e1024. doi: 10.1371/journal.pone.0001024
Black, R. E., Cousens, S., Johnson, H. L., Lawn, J. E., Rudan, I., Bassani, D. G., et al. (2010). Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet 375, 1969–1987. doi: 10.1016/S0140-6736(10)60549-1
Briani, F., Dehò, G., Forti, F., and Ghisotti, D. (2001). The plasmid status of satellite bacteriophage P4. Plasmid 45, 1–17. doi: 10.1006/plas.2000.1497
Byrne, L., Dallman, T. J., Adams, N., Mikhail, A. F., McCarthy, N., and Jenkins, C. (2018). Highly pathogenic clone of Shiga toxin–producing Escherichia coli O157: H7, England and Wales. Emerg. Infect. Dis. 24:2303. doi: 10.3201/eid2412.180409
Carlson, B. A., Nightingale, K. K., Mason, G. L., Ruby, J. R., Choat, W. T., Loneragan, G. H., et al. (2009). Escherichia coli O157: H7 strains that persist in feedlot cattle are genetically related and demonstrate an enhanced ability to adhere to intestinal epithelial cells. Appl. Environ. Microbiol. 75, 5927–5937. doi: 10.1128/AEM.00972-09
Chen, C. Y., Berish, S. A., Morse, S. A., and Mietzner, T. A. (1993). The ferric iron-binding protein of pathogenic Neisseria spp. functions as a periplasmic transport protein in iron acquisition from human transferrin. Mol. Microbiol. 10, 311–318. doi: 10.1111/j.1365-2958.1993.tb01957.x
Chen, I.-M. A., Chu, K., Palaniappan, K., Pillay, M., Ratner, A., Huang, J., et al. (2018). IMG/M v. 5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 47, D666–D677. doi: 10.1093/nar/gky901
Colavecchio, A., Cadieux, B., Lo, A., and Goodridge, L. D. (2017). Bacteriophages contribute to the spread of antibiotic resistance genes among foodborne pathogens of the Enterobacteriaceae family–a review. Front. Microbiol. 8:1108. doi: 10.3389/fmicb.2017.01108
Durnin, G., Clomburg, J., Yeates, Z., Alvarez, P. J., Zygourakis, K., Campbell, P., et al. (2009). Understanding and harnessing the microaerobic metabolism of glycerol in Escherichia coli. Biotechnol. Bioeng. 103, 148–161. doi: 10.1002/bit.22246
El-Nor, S. A., AbuGhazaleh, A. A., Potu, R. B., Hastings, D., and Khattab, M. S. A. (2010). Effects of differing levels of glycerol on rumen fermentation and bacteria. Animal Feed Sci. Technol. 162, 99–105. doi: 10.1016/j.anifeedsci.2010.09.012
Eppinger, M., Mammel, M. K., Leclerc, J. E., Ravel, J., and Cebula, T. A. (2011). Genomic anatomy of Escherichia coli O157: H7 outbreaks. Proc. Natl. Acad. Sci. U.S.A. 108, 20142–20147. doi: 10.1073/pnas.1107176108
Ferdous, M., Zhou, K., Mellmann, A., Morabito, S., Croughs, P. D., de Boer, R. F., et al. (2015). Is Shiga toxin-negative Escherichia coli O157: H7 enteropathogenic or enterohemorrhagic Escherichia coli? Comprehensive molecular analysis using whole-genome sequencing. J. Clin. Microbiol. 53, 3530–3538. doi: 10.1128/JCM.01899-15
Ferens, W. A., and Hovde, C. J. (2011). Escherichia coli O157: H7: animal reservoir and sources of human infection. Foodborne Pathog. Dis. 8, 465–487. doi: 10.1089/fpd.2010.0673
Fridovich, I. (1981). “Superoxide radical and superoxide dismutases,” in Oxygen and Living Processes. Topics in Environmental Physiology and Medicine, ed. D. L. Gilbert (New York, NY: Springer), 250–272. doi: 10.1007/978-1-4612-5890-2_13
Garcia-Aljaro, C., Moreno, E., Andreu, A., Prats, G., and Blanch, A. R. (2009). Phylogroups, virulence determinants and antimicrobial resistance in stx2 gene-carrying Escherichia coli isolated from aquatic environments. Res. Microbiol. 160, 585–591. doi: 10.1016/j.resmic.2009.08.004
Gardner, S. N., Slezak, T., and Hall, B. G. (2015). kSNP3. 0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878. doi: 10.1093/bioinformatics/btv271
Hayashi, T., Makino, K., Ohnishi, M., Kurokawa, K., Ishii, K., Yokoyama, K., et al. (2001). Complete genome sequence of enterohemorrhagic Eschelichia coli O157: H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8, 11–22. doi: 10.1093/dnares/8.1.11
Hillyar, C. R. (2012). Genetic recombination in bacteriophage lambda. Biosci. Horizons 5:hzs001. doi: 10.1093/biohorizons/hzs001
Iguchi, A., Iyoda, S., Terajima, J., Watanabe, H., and Osawa, R. (2006). Spontaneous recombination between homologous prophage regions causes large-scale inversions within the Escherichia coli O157: H7 chromosome. Gene 372, 199–207. doi: 10.1016/j.gene.2006.01.005
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., and Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36(Suppl. 2), W5–W9. doi: 10.1093/nar/gkn201
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Käppeli, U., Hächler, H., Giezendanner, N., Cheasty, T., and Stephan, R. (2011). Shiga toxin-producing Escherichia coli O157 associated with human infections in Switzerland, 2000–2009. Epidemiol. Infect. 139, 1097–1104. doi: 10.1017/S0950268810002190
Karmali, M. A. (2004). Infection by Shiga toxin-producing Escherichia coli. Mol. Biotechnol. 26, 117–122. doi: 10.1385/MB:26:2:117
Keen, J. E., Wittum, T. E., Dunn, J. R., Bono, J. L., and Durso, L. M. (2006). Shiga-toxigenic Escherichia coli O157 in agricultural fair livestock, United States. Emerg. Infect. Dis. 12:780. doi: 10.3201/eid1205.050984
Kuhnert, P., Dubosson, C. R., Roesch, M., Homfeld, E., Doherr, M. G., and Blum, J. W. (2005). Prevalence and risk-factor analysis of Shiga toxigenic Escherichia coli in faecal samples of organically and conventionally farmed dairy cattle. Vet. Microbiol. 109, 37–45. doi: 10.1016/j.vetmic.2005.02.015
Kulasekara, B. R., Jacobs, M., Zhou, Y., Wu, Z., Sims, E., Saenphimmachak, C., et al. (2009). Analysis of the genome of the Escherichia coli O157: H7 2006 spinach-associated outbreak isolate indicates candidate genes that may enhance virulence. Infect. Immun. 77, 3713–3721. doi: 10.1128/IAI.00198-09
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kusumoto, M., Ooka, T., Nishiya, Y., Ogura, Y., Saito, T., Sekine, Y., et al. (2011). Insertion sequence-excision enhancer removes transposable elements from bacterial genomes and induces various genomic deletions. Nat. Commun. 2:152. doi: 10.1038/ncomms1152
Latif, H., Li, H. J., Charusanti, P., Palsson, B. Ø, and Aziz, R. K. (2014). A gapless, unambiguous genome sequence of the enterohemorrhagic Escherichia coli O157: H7 strain EDL933. Genome Announc. 2:e00821-14. doi: 10.1128/genomeA.00821-14
Leopold, S. R., Magrini, V., Holt, N. J., Shaikh, N., Mardis, E. R., Cagno, J., et al. (2009). A precise reconstruction of the emergence and constrained radiations of Escherichia coli O157 portrayed by backbone concatenomic analysis. Proc. Natl. Acad. Sci. 106, 8713–8718. doi: 10.1073/pnas.0812949106
National Center for Biotechnology Information (2019). Impact of Shiga Toxin Bacteriophage Carriage in EHEC O157:H7. Available online at: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA530317/ (accessed April 01, 2019).
Nyong, E. C., Zaia, S. R., Allue-Guardia, A., Rodriguez, A. L., Irion-Byrd, Z., Koenig, S. S., et al. (2020). Pathogenomes of atypical non-shigatoxigenic Escherichia coli NSF/SF O157: H7/NM: comprehensive phylogenomic analysis using closed genomes. Front. Microbiol. 11:619. doi: 10.3389/fmicb.2020.00619
Ochoa, T. J., and Contreras, C. A. (2011). Enteropathogenic E. coli (EPEC) infection in children. Curr. Opin. Infect. Dis. 24:478. doi: 10.1097/QCO.0b013e32834a8b8b
Ohnishi, M., Terajima, J., Kurokawa, K., Nakayama, K., Murata, T., Tamura, K., et al. (2002). Genomic diversity of enterohemorrhagic Escherichia coli O157 revealed by whole genome PCR scanning. Proc. Natl. Acad. Sci. U.S.A. 99, 17043–17048. doi: 10.1073/pnas.262441699
Omazic, A. W., Tråvén, M., Roos, S., Mellgren, E., and Holtenius, K. (2013). Oral rehydration solution with glycerol to dairy calves: effects on fluid balance, metabolism, and intestinal microbiota. Acta Agric. Scand. A Animal Sci. 63, 47–56. doi: 10.1080/09064702.2013.785585
Ooka, T., Ogura, Y., Asadulghani, M., Ohnishi, M., Nakayama, K., Terajima, J., et al. (2009). Inference of the impact of insertion sequence (IS) elements on bacterial genome diversification through analysis of small-size structural polymorphisms in Escherichia coli O157 genomes. Genome Res. 19, 1809–1816. doi: 10.1101/gr.089615.108
Orden, J., Cid, D., Ruiz-Santa-Quiteria, J., Garcia, S., Martinez, S., and De La Fuente, R. (2002). Verotoxin-producing Escherichia coli (VTEC), enteropathogenic E. coli (EPEC) and necrotoxigenic E. coli (NTEC) isolated from healthy cattle in Spain. J. Appl. Microbiol. 93, 29–35. doi: 10.1046/j.1365-2672.2002.01649.x
Paton, J. C., and Paton, A. W. (1998). Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 11, 450–479. doi: 10.1128/CMR.11.3.450
Pruimboom-Brees, I. M., Morgan, T. W., Ackermann, M. R., Nystrom, E. D., Samuel, J. E., Cornick, N. A., et al. (2000). Cattle lack vascular receptors for Escherichia coli O157: H7 Shiga toxins. Proc. Natl. Acad. Sci. U.S.A. 97, 10325–10329. doi: 10.1073/pnas.190329997
Raymond, K. N., Dertz, E. A., and Kim, S. S. (2003). Enterobactin: an archetype for microbial iron transport. Proc. Natl. Acad. Sci. U.S.A. 100, 3584–3588. doi: 10.1073/pnas.0630018100
Robinson, C. M., Sinclair, J. F., Smith, M. J., and O’Brien, A. D. (2006). Shiga toxin of enterohemorrhagic Escherichia coli type O157: H7 promotes intestinal colonization. Proc. Natl. Acad. Sci. U.S.A. 103, 9667–9672. doi: 10.1073/pnas.0602359103
Sanchez, S., Martinez, R., Rey, J., Garcia, A., Blanco, J., Blanco, M., et al. (2010). Pheno-genotypic characterisation of Escherichia coli O157: H7 isolates from domestic and wild ruminants. Vet. Microbiol. 142, 445–449. doi: 10.1016/j.vetmic.2009.10.009
Scallan, E., Hoekstra, R. M., Angulo, F. J., Tauxe, R. V., Widdowson, M.-A., Roy, S. L., et al. (2011). Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 17:7. doi: 10.3201/eid1701.P11101
Shaikh, N., and Tarr, P. I. (2003). Escherichia coli O157: H7 Shiga toxin-encoding bacteriophages: integrations, excisions, truncations, and evolutionary implications. J. Bacteriol. 185, 3596–3605. doi: 10.1128/JB.185.12.3596-3605.2003
Smith, D. L., Rooks, D. J., Fogg, P. C., Darby, A. C., Thomson, N. R., McCarthy, A. J., et al. (2012). Comparative genomics of Shiga toxin encoding bacteriophages. BMC Genomics 13:311. doi: 10.1186/1471-2164-13-311
Solomakos, N., Govaris, A., Angelidis, A. S., Pournaras, S., Burriel, A. R., Kritas, S. K., et al. (2009). Occurrence, virulence genes and antibiotic resistance of Escherichia coli O157 isolated from raw bovine, caprine and ovine milk in Greece. Food Microbiol. 26, 865–871. doi: 10.1016/j.fm.2009.06.002
Staudenmaier, H., Van Hove, B., Yaraghi, Z., and Braun, V. (1989). Nucleotide sequences of the fecBCDE genes and locations of the proteins suggest a periplasmic-binding-protein-dependent transport mechanism for iron (III) dicitrate in Escherichia coli. J. Bacteriol. 171, 2626–2633. doi: 10.1128/jb.171.5.2626-2633.1989
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Tozzoli, R., Grande, L., Michelacci, V., Fioravanti, R., Gally, D., Xu, X., et al. (2014). Identification and characterization of a peculiar vtx2-converting phage frequently present in verocytotoxin-producing Escherichia coli O157 isolated from human infections. Infect. Immun. 82, 3023–3032. doi: 10.1128/IAI.01836-14
Um, M. M., Brugere, H., Kérourédan, M., Oswald, E., and Bibbal, D. (2018). Antimicrobial resistance profiles of Enterohemorrhagic and Enteropathogenic Escherichia coli of serotypes O157: H7, O26: H11, O103: H2, O111: H8, O145: H28 compared to Escherichia coli isolated from the same adult cattle. Microb. Drug Resist. 24, 852–859. doi: 10.1089/mdr.2017.0106
Wei, J., Goldberg, M. B., Burland, V., Venkatesan, M. M., Deng, W., Fournier, G., et al. (2003). Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect. Immun. 71, 2775–2786. doi: 10.1128/iai.71.5.2775-2786.2003
Wetzel, A., and LeJeune, J. (2007). Isolation of Escherichia coli O157: H7 strains that do not produce Shiga toxin from bovine, avian and environmental sources. Lett. Appl. Microbiol. 45, 504–507. doi: 10.1111/j.1472-765X.2007.02228.x
Xiong, Y., Wang, P., Lan, R., Ye, C., Wang, H., Ren, J., et al. (2012). A novel Escherichia coli O157: H7 clone causing a major hemolytic uremic syndrome outbreak in China. PLoS One 7:e36144. doi: 10.1371/journal.pone.0036144
Yang, H., Carlson, B., Geornaras, I., Woerner, D., Sofos, J., and Belk, K. (2016). Draft genome sequence of Shiga toxin-negative Escherichia coli O157: H7 strain C1-057, isolated from feedlot cattle. Genome Announc. 4:e0049-16. doi: 10.1128/genomeA.00049-16
Yokoyama, K., Makino, K., Kubota, Y., Watanabe, M., Kimura, S., Yutsudo, C. H., et al. (2000). Complete nucleotide sequence of the prophage VT1-Sakai carrying the Shiga toxin 1 genes of the enterohemorrhagic Escherichia coli O157: H7 strain derived from the Sakai outbreak. Gene 258, 127–139. doi: 10.1016/S0378-1119(00)00416-9
Keywords: whole genome analysis, Shiga toxin-producing and stx-negative E. coli O157:H7, cattle, acquisition and loss of stx genes, Red homologous recombination, IS629, metabolic pathway genes, evolutionary pathways
Citation: Jia M, Geornaras I, Martin JN, Belk KE and Yang H (2021) Comparative Whole Genome Analysis of Escherichia coli O157:H7 Isolates From Feedlot Cattle to Identify Genotypes Associated With the Presence and Absence of stx Genes. Front. Microbiol. 12:647434. doi: 10.3389/fmicb.2021.647434
Received: 29 December 2020; Accepted: 28 February 2021;
Published: 01 April 2021.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Atsushi Iguchi, University of Miyazaki, JapanCopyright © 2021 Jia, Geornaras, Martin, Belk and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Yang, aHVhLnlhbmdAY29sb3N0YXRlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.