
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 19 March 2021
Sec. Microbial Symbioses
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.645070
 Yue Dong1,2
Yue Dong1,2 Yixuan Li1,3
Yixuan Li1,3 Peiqing He1Zongling Wang1,2
Peiqing He1Zongling Wang1,2 Shiliang Fan1,2Zhixin Zhang4
Shiliang Fan1,2Zhixin Zhang4 Xuelei Zhang1,2
Xuelei Zhang1,2 Qinzeng Xu1,2*
Qinzeng Xu1,2*Gut microbiota have important roles in the survival and adaptation of the host. Ophiuroids, as the worldwide dominant benthos, have ecological roles in benthic–pelagic coupling in the sea floor. However, little is known about the composition and diversity of their gut microbiota and its potential functions in benthic ecosystems. In present study, we preformed 16S rRNA sequencing and function analysis in four dominant species (Stegophiura sladeni, Ophiopholis mirabilis, Ophiura sarsii vadicola, and Ophiura kinbergi) with two feeding types (suspension feeding/herbivores and scavenger/carnivores) from the Yellow Sea, China. Results showed that 56 phyla and 569 genera of microbiota were identified among ophiuroid guts. Multivariate and diversity analyses showed that the ophiuroid gut microbiota were independent and have higher biodiversity to the sediment microbial in the Yellow Sea. Phyla Proteobacteria, Firmicutes, Tenericutes, and Bacteroidetes were the dominant bacteria, with more than 80% abundance among the four ophiuroid species. A comparison among the gut microbial compositions among four ophiuroids showed the similarity of two offshore carnivore ophiuroids (S. sladeni and O. sarsii vadicola) and variation in the dominant microbiota types of three nearshore ophiuroids (S. sladeni, O. mirabilis, and O. kinbergi). The functional analysis revealed the significant differences of the environment-related expression in S. sladeni gut microbiota between nearshore and offshore environments. The Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) functional annotation showed the significant divergence of metabolism pathways between two nearshore species, the herbivores O. mirabilis and carnivores S. sladeni, such as the Lipid metabolism, Carbohydrate metabolism, and Metabolism of cofactors and vitamins. The homolog search and phylogenetic analysis identified the first gut symbiotic Candidatus Hepatoplasma in S. sladeni with important roles for the nutrient metabolisms. Overall, our study reported the comprehensive data of ophiuroid gut microbiota, while the functional microbiome provides insight into the physiology and environmental adaptation in ophiuroids.
Ophiuroidea (brittle stars), with 2,064 known species from 16 families, are the largest class of Echinodermata (Geraldi et al., 2017). They play an important ecological role in food webs (Wu and Shin, 1997; Harris et al., 2009; Enoksen and Reiss, 2017) and nutrient recycling in the marine benthic ecosystems (Lebrato et al., 2010; Ravelo et al., 2015). The Ophiuroids possess two feeding types: suspension feeding and scavenger. Brittle stars from families Gorgonocephalidae and Amphiuridae are primarily suspension feeding on organic detritus, plankton, and bacteria (Iken et al., 2001). Scavenger ophiuroids, including Ophiopyrgidae and Ophiacanthidae, feed on dead organisms and small animals (usually dead), including crustaceans, mollusks, and worms (Adarsha et al., 2018). In addition, the assemblage of ophiuroid populations is a common phenomenon in different habitats (Murat et al., 2016), given that they could enhance the process of benthic–pelagic coupling in the sea floor (Geraldi et al., 2017).
The gut microbiota plays key roles in nutrient absorption, environmental adaptation, and anti-pathogens of the host animals (Sugita et al., 1991; Cho and Blaser, 2012; Zhang et al., 2016). The habitat environment (Trabal et al., 2012; Pierce et al., 2016; Nguyen et al., 2020), host feeding habits, and life stages (Tanaka et al., 2004; Li et al., 2019) have effects on the gut microbial community. Studies on the microbiome of echinoderms revealed that gut microbiota in sea cucumber (Zhang et al., 2012, 2013), starfish (Lee et al., 2018), and sea urchin (Hakim et al., 2016) can enhance the digestion and provide missing nutrients from diets to the host. However, the role of gut microbiota still remains unclear, which hinders the understanding of physiologies, ecologies, and life histories of ophiuroids. In addition, the composition of gut microbiota may have divergences among species with different feeding types (Guo et al., 2020). Some echinoids have evolved gut symbiotic bacteria for acquisition of essential nutrients (Temara et al., 1993; Becker et al., 2009). In ophiuroids, symbiotic bacteria have identified on larvae and adult subcuticle to facilitate the uptake of free amino acid from the ambient environment (Walker and Lesser, 1989; Morrow et al., 2018), while the gut symbiont has not been reported yet.
In this study, we focus on the gut microbiota communities of ophiuroids that lived in the Yellow Sea, a marginal sea between China and Korean Peninsula with hydrological phenomena and high biodiversity (Li, 2011). Four dominant microbenthic ophiuroids, including Stegophiura sladeni, Ophiopholis mirabilis, Ophiura sarsii vadicola, and Ophiura kinbergi (Peng et al., 2017), were selected for 16S rRNA sequencing of their gut microbial communities, which covered two feeding types, scavengers (O. sarsii vadicola, O. kinbergi, and S. sladeni; Harris et al., 2009) and suspension feeder (O. mirabilis; Yu et al., 2019). The alpha diversity and multivariate analysis revealed the spatial variation among nearshore and offshore ophiuroids, and their surrounding sediments. The diversity and functional analyses of microbiome indicated significant variation between scavenging and suspension-feeding ophiuroids. Integration of metagenomic sequencing and functional-inference-based approaches provided insight into the metabolic and environmental adaptation from gut communities, which facilitate their survival and organic matters recycling in benthic ecosystems.
Ophiuroid specimens were collected by bottom trawling from the Yellow Sea: offshore (water depth > 50 m) station H (34°00′N, 124°00′E, 80-m depth) for S. sladeni and O. sarsii vadicola in March 2018 and nearshore station Y (35°00′ N, 121°00′ E, 16-m depth) for S. sladeni, O. mirabilis, and O. kinbergi in October 2018 (Figure 1). All specimens were immediately fixed in 95% ethanol and stored at −20°C. After cruises, specimens were transferred to the First Institute of Oceanography, Ministry of Natural Resources, for further analysis. To reduce the contamination by environmental bacteria, the ophiuroids were rinsed with Milli-Q water before dissection. The oral shield was removed, and the gastric contents were sampled under a stereomicroscope. All gut contents were stored in a freezer at −20°C until use for DNA extraction.
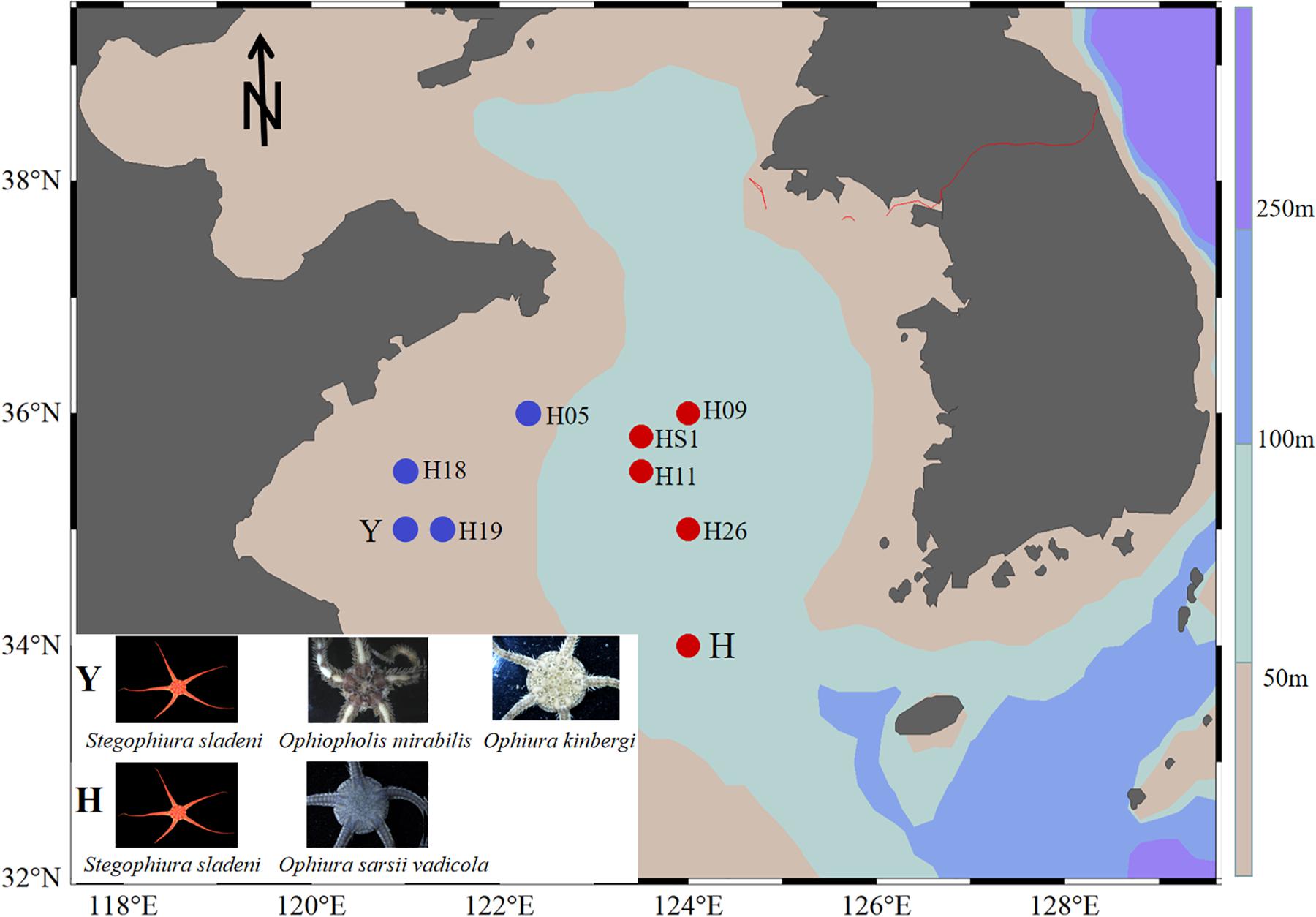
Figure 1. Location of sampling sites in the southern Yellow Sea. Ophiuroids were collected from nearshore (station Y) with blue color and offshore (station H) with red color. Sediment stations were H05, H18, H19, H09, HS1, H11, and H26 (Zhang et al., 2019).
To understand the gut microbiota, genomic DNA was extracted from the gut contents of 23 ophiuroid specimens (nearshore: four S. sladeni, five O. mirabilis, and five O. kinbergi; offshore: five S. sladeni and four O. sarsii vadicola) and used for sequencing on the V3–V4 region of the microbial 16S rRNA gene. In brief, total microbial community DNA was isolated using the DNeasy Blood & Tissue Kit (Qiagen, Germany) following the manufacturer’s protocol. DNA quality was assessed using agarose gel electrophoresis and a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, United States). The V3–V4 hypervariable region of 16S rRNA gene were amplified using the forward primer 338 F (5′-GTACTCCTACGGGAGGCAGCA-3′) and the reverse primer 806 R (5′-GTGGACTACHVGGGTWTCTAAT-3′). The PCR contained 30 ng of DNA template (30 ng), 1 μl each of 5 mol/L of primers, 3 μl of bovine serum albumin (BSA) (2 ng/μl), 12.5 μl of 2 × Taq Plus Master Mix, and 7.5 μl of ddH2O in a volume of 25 μl. PCR amplification was performed with the following program: pre-denaturation at 94°C for 5 min; 30 cycles of denaturation at 94°C for 30 s, annealing at 50°C for 30 s, and an extension at 72°C for 60 s; and a final extension at 72°C for 7 min. The PCR products were sequenced using an Illumina MiSeq sequencer to produce 300-bp paired-end reads by Allwegene Technology (Beijing, China).
The raw sequencing reads were filtered by removing adapters using Cutadapt software (Marcel, 2011; Callahan et al., 2016). The following taxonomic analyses were performed using the R script implemented in dada2 v1.16.0 packages (Callahan et al., 2016) with the default parameters. The clean reads were further quality filtered, dereplicated, and merged using the R script filterAndTrim, derepFastq, and mergePairs, respectively. The makeSequenceTable command was used to construct an amplicon sequence variant (ASV) table. Potential chimeras were removed using the removeBimeraDenovo command. ASVs were annotated compared with the Silva SSU rRNA database (version 138) for taxonomic classification (Benjamin, 2017) using assignTaxonomy command.
To compare the microbial community in ophiuroid gut and in the sediment, 16S rRNA sequences of the Yellow Sea sediments (three stations near the Y site, four stations near the H site) were downloaded from Zhang et al. (2019; Figure 1). The sequence filtering, clustering, and annotation were performed the same as above. The gut microbiota of ophiuroids and sediment had different ASV numbers and compositions. The results of species-level annotation showed that the proportion of unknown species was 97.5%, so we performed downstream analysis based on the genus and above level. The functional profiles of the gut microbial communities based on 16S rRNA sequences were annotated using Tax4Fun (Aßhauer et al., 2015) and Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) (Langille et al., 2013) with Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) (Minoru et al., 2015). To compare functional differences among samples, abundances of predicted functional pathways were normalized sequencing depth as percentages of the total number of predicted functions from the KO database. Three sets of pairwise comparison analysis were performed for the bacterial function pathways of ophiuroid gut microbial communities, including S. sladeni and O. mirabilis from the nearshore environment, S. sladeni and O. sarsii vadicola from the offshore environment, and S. sladeni from both two environments. Statistical Analysis of Metagenomic Profiles (STAMP) analysis indicated the significant variations in KOs with expression levels above 1% contribution.
To investigate any possible gut symbionts, the ASVs of gut microbiota were BLASTn search against GenBank database with threshold of identity >80% and E-value of 1e−20. Among ASVs, only genus Candidatus Hepatoplasma showed homology to known gut symbionts. To further elucidate the microbial candidates, eight ASVs of Ca. Hepatoplasma, their top BLAST hit sequences, and nine 16S rRNA symbiont sequences [from Cheng et al. (2019) and National Center for Biotechnology Information (NCBI)] were aligned by ClustalW and manually trimmed. Maximum likelihood (ML) and neighbor joining (NJ) were selected for phylogenetic analysis with MEGA v7 (Kumar et al., 2016). The best model of Kimura two-parameter selected by model test in MEGA and 1,000 bootstrap replications were used.
Alpha diversity statistics were calculated for diversity metrics based on Simpson index and Shannon index. The Mann–Whitney U tests were used to evaluate the differences among categories in R. Principal component analysis (PCA) reflects the difference and distance between samples by analyzing ASVs with Origin 2018 software at the genus level (Wang et al., 2012). Analysis of similarity (ANOSIM) was used to evaluate the differences in the microbiota communities among the gut of ophiuroids, and between ophiuroids’ gut and benthic sediment (Clarke, 1993). Similarity percentage analyses (SIMPERs) were used to identify the contribution of each taxon to the community dissimilarity (Clarke, 1993). Permutational multivariate analysis of variance (PERMANOVA) revealed the amount of variation with respect to both the microbiota in the gut of ophiuroids and sediment (Anderson, 2014). Those three analyses were performed using PRIMER version 6 and PERMANOVA + software. The linear discriminant analysis (LDA) effect size (LEfSe) analysis was conducted by the Galaxy website under default parameters1 to evaluate the significance of differences (i.e., biomarkers) at the phylum, class, order, family, and genus levels in five ophiuroid groups. In addition, the abundance of KEGG pathways over 1% was selected to calculate the ANOVA between two selected ophiuroids by using the STAMP software (Parks and Beiko, 2010).
After quality filtering, 586,254 clean reads (9,101–36,825 reads per sample) were clustered in 5,759 ASVs (505–2,706 per sample) from 23 nearshore and offshore ophiuroids (Supplementary Appendix A1). The ASVs were well annotated from the phylum and genus levels (Figures 2A,B). Based on the composition analysis of the gut microbiota at the ASV level (Figure 3A), 31 ASVs were common to ophiuroids’ gut microbiota in different areas. The number of unique ASVs in each community was counted, with 730 unique ASVs in the S. sladeni, 1,567 in the O. kinbergi, and 2,226 in the O. mirabilis from the nearshore environment, and 167 in the S. sladeni and 301 in the O. sarsii vadicola from the offshore environment.
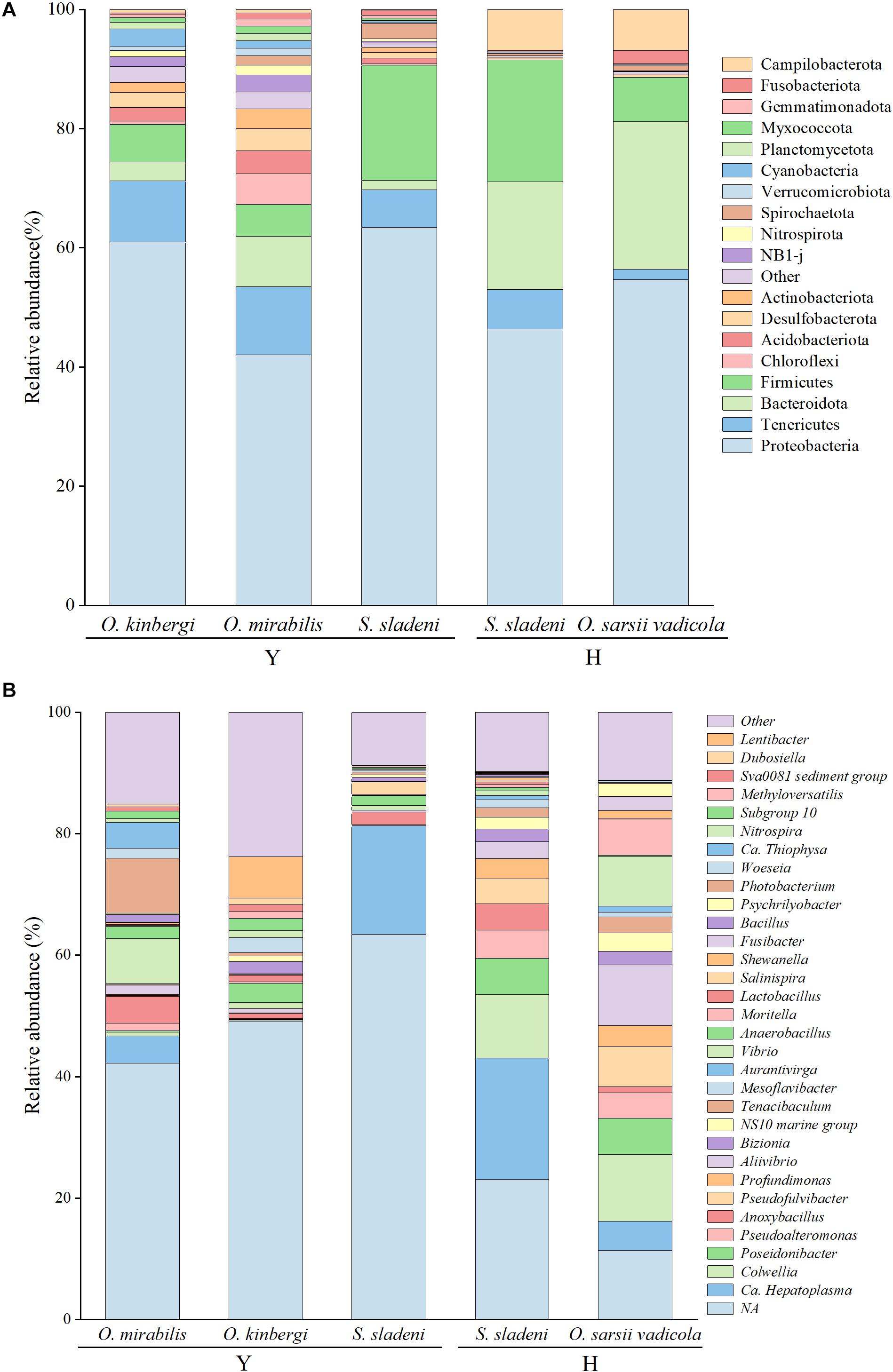
Figure 2. Taxonomic abundance of gut microbiota at the phylum level (A) and genera level (B) in ophiuroid species from the Yellow Sea, China. Other, the taxonomic groups with abundances less than 1%; Y, the nearshore environment; H, the offshore environment.
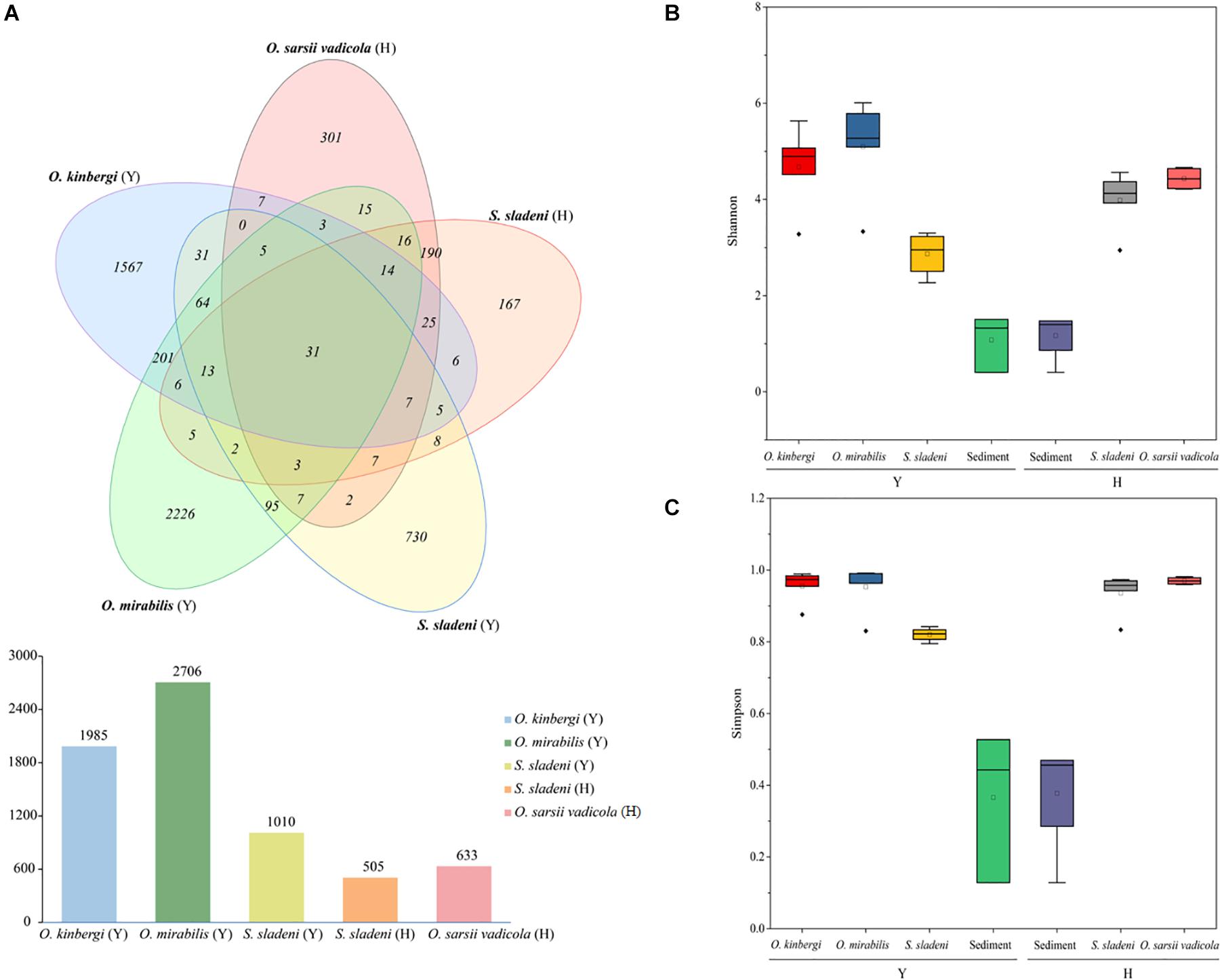
Figure 3. Amplicon sequence variant (ASV) Venn diagram of gut microbiota of ophiuroids and alpha diversity analyses of the ophiuroid gut and sediment microbial communities. (A) ASVs of ophiuroids’ gut microbiota are depicted in a Venn diagram. Alpha diversity was calculated using (B) the Shannon index and (C) the Simpson index. Statistical analysis was conducted on alpha diversity using Mann–Whitney U tests. ns, not significant; p > 0.05. Y, the nearshore environment; H, the offshore environment.
The identified gut microbiota of three ophiuroid species S. sladeni, O. mirabilis, and O. kinbergi, from nearshore included 56 phyla and 540 genera. In the phylum level, Proteobacteria was the highest abundance microbiota among the S. sladeni, O. kinbergi, and O. mirabilis in 63.4%, 60.9%, and 42.0%, respectively (Figure 2A). The phyla Firmicutes, Tenericutes, and Bacteroidetes were also the dominant microbiota in the three species, with various abundance among species. There were some species-specific abundance phyla in three ophiuroids, such as Spirochaetota (2.6%) in S. sladeni, Cyanobacteria (3.0%) in O. kinbergi, and Chloroflexi (5.1%) in O. mirabilis.
At the genus level, more detailed divergences were found in the three species (Figures 2B, 4). The genus Candidatus Hepatoplasma was commonly found in all three ophiuroids with different abundance: 17.9% in S. sladeni, 0.2% in O. mirabilis, and 4.5% in O. kinbergi. Lentibacter was the most dominant group in O. mirabilis (6.9%), while only few (<0.01%) were detected in another two ophiuroids. In the gut of O. kinbergi, Photobacterium (9.1%) and Vibrio (7.5%) were much higher than those in S. sladeni (0.5 and 0.8%) and O. mirabilis (0.6 and 1.0%).
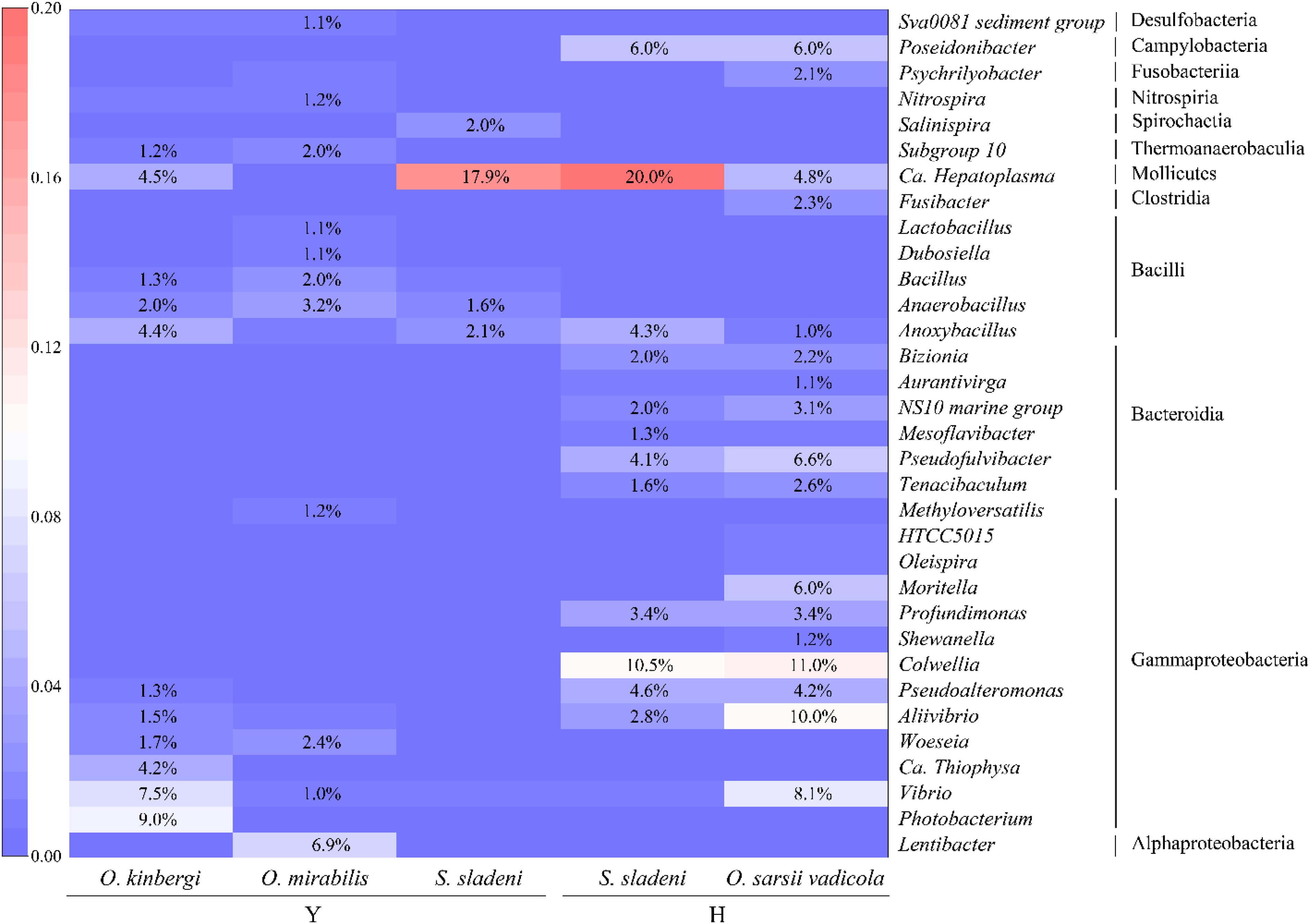
Figure 4. The heatmap of relative abundance of gut microbiota at the genus level (>1%) in ophiuroid species from the Yellow Sea. Y, the nearshore environment; H, the offshore environment.
The gut microbiota of S. sladeni and O. sarsii vadicola from offshore were identified, including 31 phyla and 156 genera. Consistent with the gut microbial composition of ophiuroids from the nearshore area, Proteobacteria was the dominant phylum in S. sladeni and O. sarsii vadicola, accounting for 46.4 and 54.6%, respectively (Figure 2A). Firmicutes (20.5%) was the second highest group in the gut of S. sladeni, followed by Bacteroidota (18.1%), Campilobacterota (6.9%), and Tenericutes (6.7%). For O. sarsii vadicola, the gut microbiota were also dominated with Bacteroidota (24.8%) and Firmicutes (7.4%), Campilobacterota (6.9%), Fusobacteriota (2.1%), and Tenericutes (1.7%).
At the genus level, the high similarity occurred in the gut microbiota of two species (Figures 2B, 4). The genera had the similar abundance in Colwellia (10.5% of S. sladeni and 11.0% of O. sarsii vadicola), Poseidonibacter (6.0 and 6.0%), and Pseudofulvibacter (4.1 and 6.6%). It was worth noting that Ca. Hepatoplasma (20.0%) was the top dominant genus in S. sladeni, while its abundance was lower in O. sarsii vadicola (4.8%, Figure 4).
The above ASV results showed that the gut microbiota in O. mirabilis and O. kinbergi were more specific than those of other species. In terms of alpha diversity (Figures 3B,C), the Simpson index and Shannon index (a measure of richness and evenness) were the highest in O. mirabilis (p > 0.05). In contrast, the Simpson index and Shannon index of nearshore S. sladeni were the lowest among ophiuroids (p > 0.05). And these indexes of sediment communities (nearshore and offshore) were lower than those of ophiuroids (p > 0.05). The alpha diversity metrics of the gut communities were significantly more diverse (Mann–Whitney U tests, p > 0.05) than the sediment microbiome.
Principal component analysis revealed the strong clustering of nearshore ophiuroids, offshore ophiuroids, and sediment microbiomes (Figure 5). The first two axes of PCA accounted for 38.5% of variation with and the gut microbiota of ophiuroids separated from the sediment communities. The difference between offshore ophiuroids and sediment was also statically significant based on the results of ANOSIM and PERMANOVA (Supplementary Appendix A2, p < 0.05). Overall, the results of alpha diversity and multivariate analysis demonstrated the independence of microbial communities between benthic ophiuroids and sediment in the Yellow Sea.
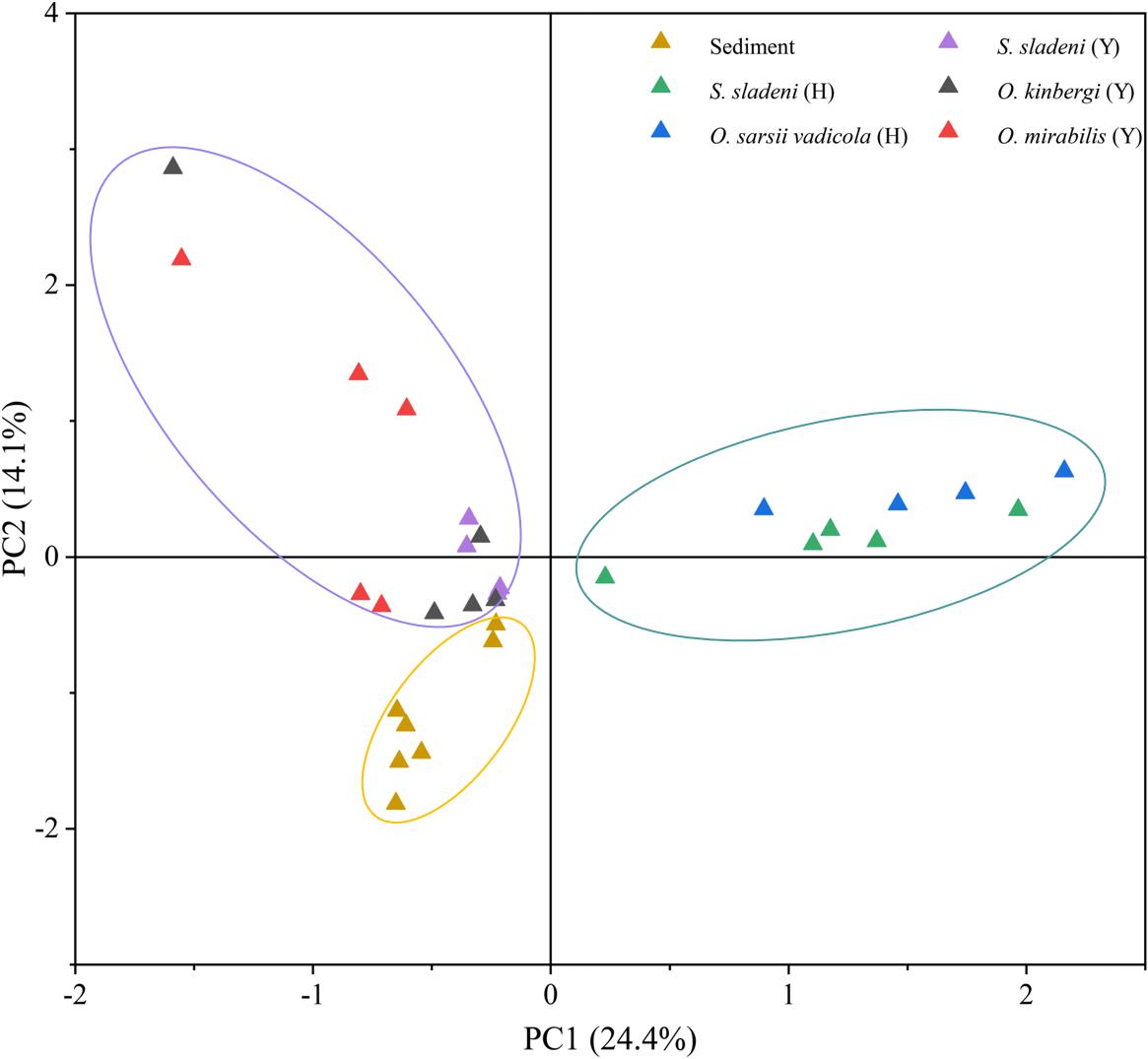
Figure 5. Principal component analysis (PCA) of gut microbiota in ophiuroids and sediment microbiome under genus level. Y, the nearshore environment; H, the offshore environment.
The result of LEfSe analysis showed that the biomarkers in the gut microbial communities were different among five ophiuroid groups (Figure 6; Supplementary Appendix A4). In the nearshore environment, the LDA scores indicated that the class Betaproteobacteria was the largest contributor to intergroup differences in S. sladeni; the phylum Firmicutes and class Bacilli have significant contributions to O. mirabilis; the class Alphaproteobacteria was a prominent contributor to the intergroup differences in O. kinbergi. In the offshore environment, the class Gammaproteobacteria was the main contributor to the difference in O. sarsii vadicola. For S. Sladeni, the genus Ca. Hepatoplasma and higher taxonomy level of this genus were the most prominent difference contributors.
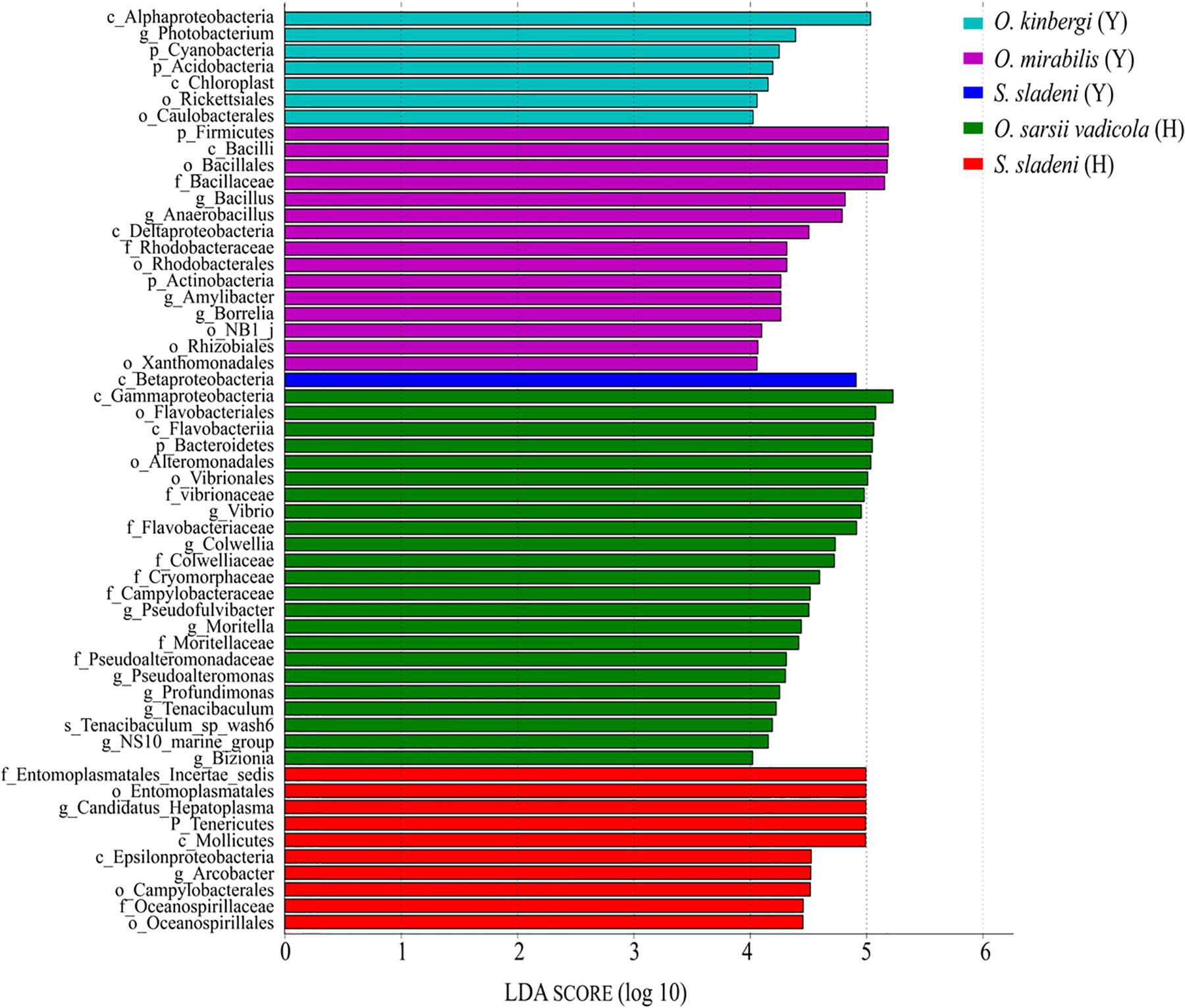
Figure 6. Characterization of gut microbiota in ophiuroids by LEfSe analysis and histogram of the linear discriminant analysis (LDA) scores (log 10). Y, the nearshore environment; H, the offshore environment; g, the genus; f, the family; o, the order; c, the class; p, the phylum.
In level 2 KOs, a total of 38 and 47 pathways were annotated by Tax4Fun and PICRUSt analysis, respectively (Supplementary Appendix A5, A6). Five metabolism pathways were commonly detected in all four ophiuroids, including metabolism of cofactors and vitamins, amino acid metabolism, carbohydrate metabolism, metabolism of terpenoids, and metabolism of other amino acids.
The results of Tax4Fun showed that there was no significant difference within the groups of the nearshore and offshore ophiuroids (Supplementary Appendix A7, A8). The significance of differences in certain expression pathways was found between S. sladeni in the nearshore and offshore environments (p < 0.05, Figure 7A), while the four enriched KOs were Biofilm formation-Vibrio cholerae, Propanoate metabolism, Biofilm formation-Pseudomonas aeruginosa, and Two-component system, which belonged to three metabolic pathways Environmental Information Processing, Environmental Information Processing, and Cellular Processes, suggesting the possible interaction between the gut microbial communities and environment microbiota in S. sladeni. The PICRUSt analysis also showed the enriched KOs that were consistent with those results of Tax4Fun, where the five significantly different KOs were related to environmental signaling and metabolism pathways (p < 0.05, Figure 7B). Using an integrated Tax4Fun and PICRUSt methods, our results indicated the remarkable enrichment of functional pathways under Environmental Information Processing in S. sladeni gut microbiomes.
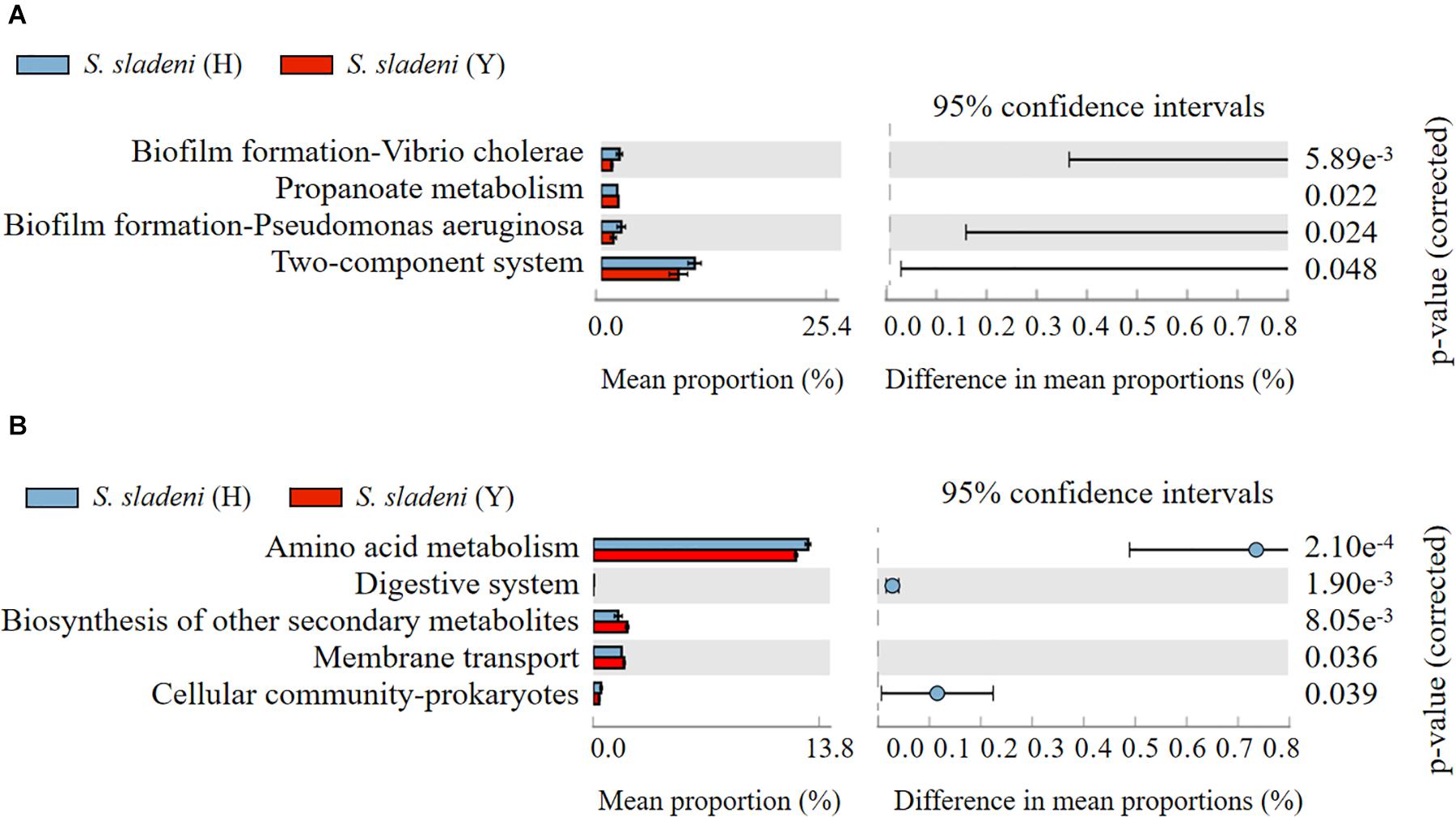
Figure 7. The significant expression pathways based on (A) Tax4Fun and (B) PICRUSt analysis of gut microbial community between the nearshore and offshore Stegophiura sladeni at Kyoto Encyclopedia of Genes and Genomes level 2.
In the nearshore environment, the results of PICRUSt analysis showed that eight KOs were significantly divergent (p < 0.05) between the two species S. sladeni and O. mirabilis, mainly in Metabolism pathways (Figure 8). In the offshore environment, gut microbiota of S. sladeni was statistically enriched in four KOs (p < 0.05) including Folding, sorting, and degradation; Transport and catabolism; and Translation and Endocrine system (Figure 9).
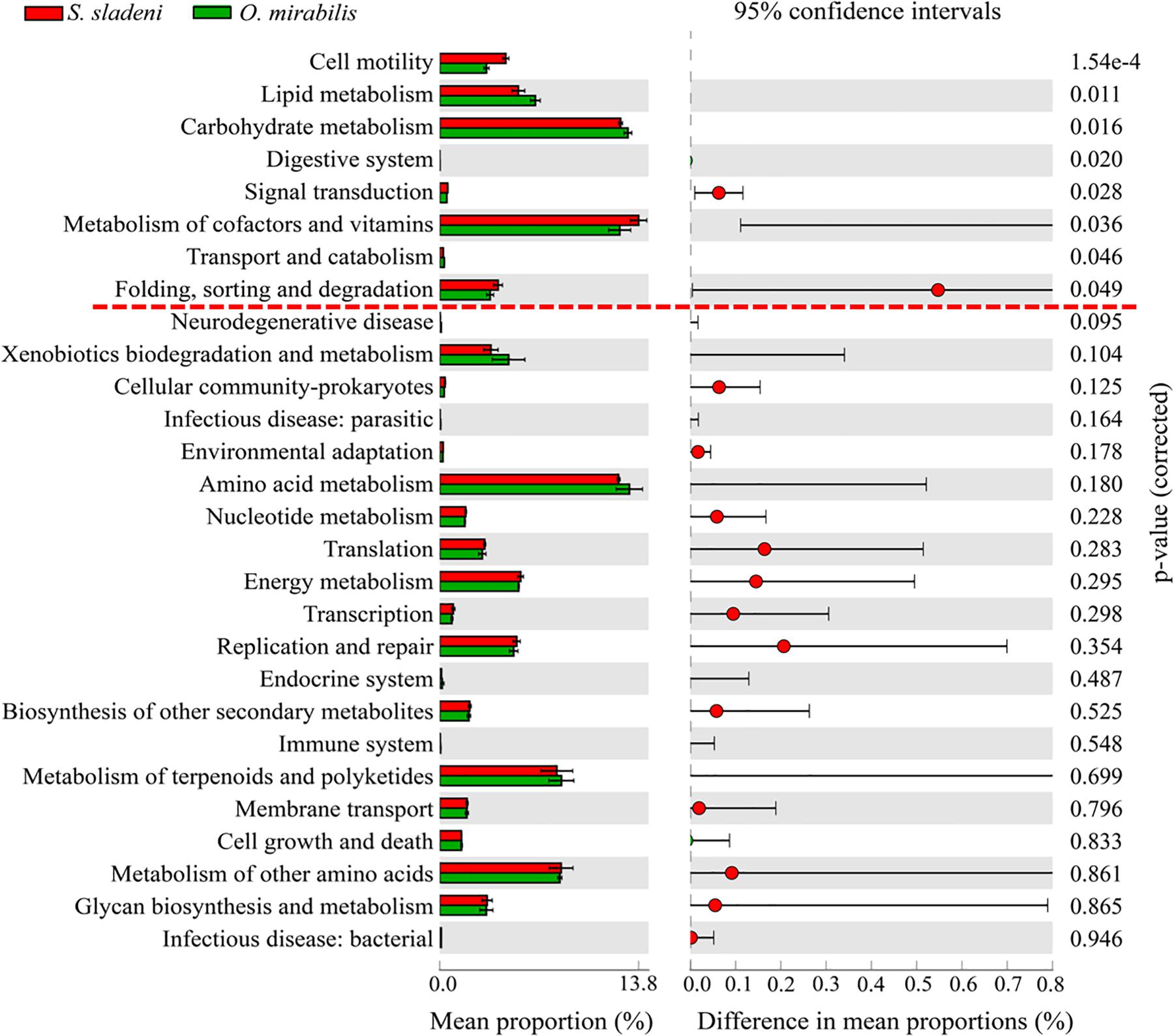
Figure 8. Variation analysis of gut microbial community between nearshore environment ophiuroid species Stegophiura sladeni and Ophiopholis mirabilis at Kyoto Encyclopedia of Genes and Genomes level 2 by PICRUSt. Dotted line: division of p < 0.05.
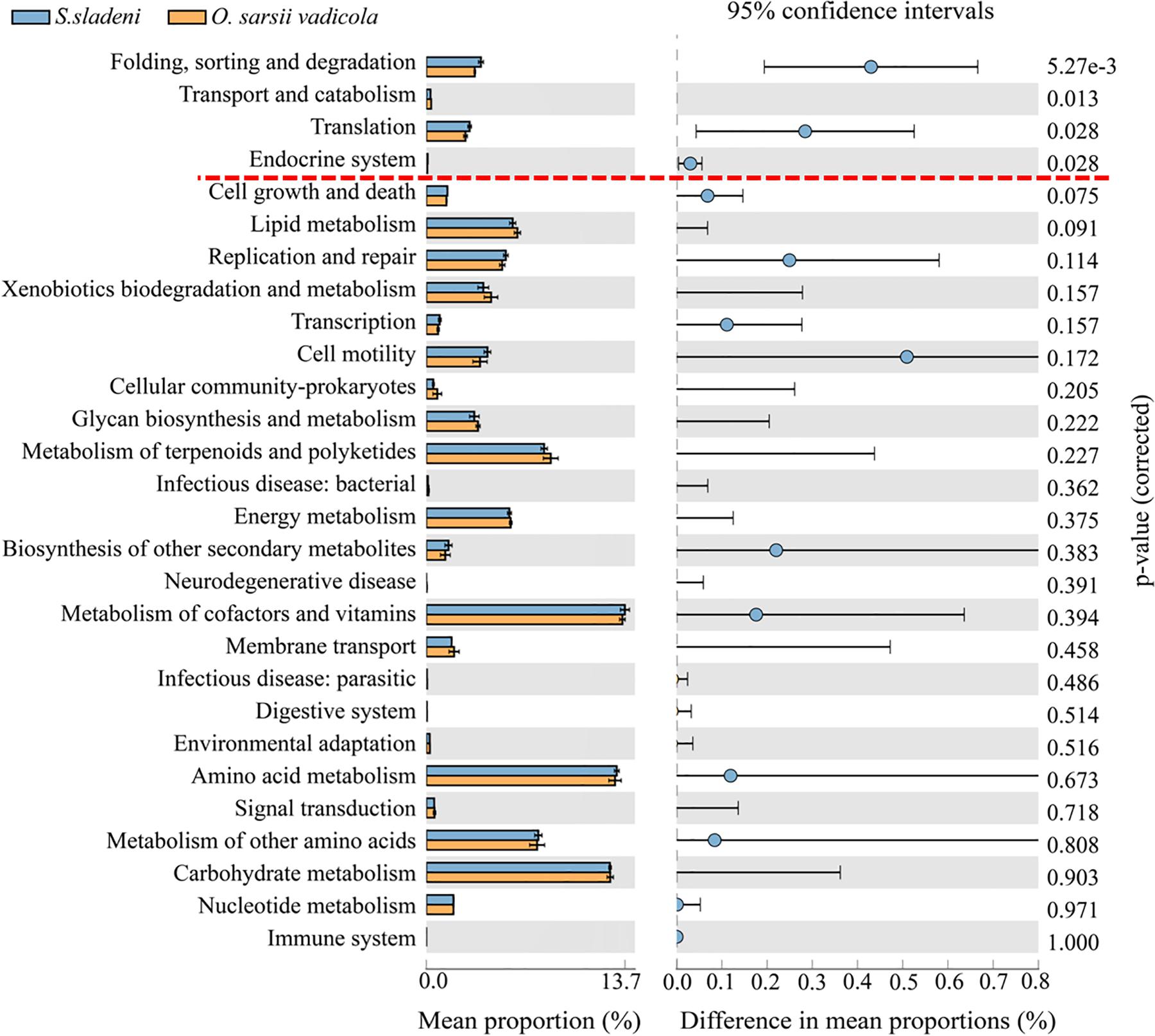
Figure 9. Variation analysis of gut microbial community between offshore environment ophiuroid species Stegophiura sladeni and Ophiura sarsii vadicola at Kyoto Encyclopedia of Genes and Genomes level 2 by PICRUSt. Dotted line: division of p < 0.05.
A BLASTn search indicated that eight ASVs of genus Ca. Hepatoplasma from S. sladeni were close to bacterial symbionts of Ca. Hepatoplasma with identity of >80% (Supplementary Appendix A9, A10). Phylogenetic analysis of Ca. Hepatoplasma and their 16S rRNA homologs revealed the potential symbiotic relationship of all eight ASVs and further clustered them into three groups (Figure 10). Groups A and C, however, were specific to four Ca. Hepatoplasma ASVs from offshore S. sladeni. Given their higher abundance than nearshore S. sladeni (10.2 and 1.8%) and PICRUSt annotation at Amino acid metabolism and Biosynthesis of other secondary metabolites pathways, Groups A and C Ca. Hepatoplasma might play roles in the adaptation of offshore dynamics and oligotrophic environments. Group B comprises four ASVs nested among the uncultured bacterium (Bauermeister et al., 2012; Hewson et al., 2018).

Figure 10. Phylogenetic analysis of Candidatus Hepatoplasma. The percentage represented the relative abundance in Stegophiura sladeni at nearshore and offshore. The bootstrap values (>50%) of relevant nodes are shown based on 1,000 replicates. The two numbers on the branch were the result of the maximum likelihood (ML), with neighbor-joining (NJ) algorithm in brackets. Sequences from this study are shown in bold.
This study characterized the gut microbiota of the Yellow Sea ophiuroids using 16S rRNA sequencing. The taxonomic analysis indicated that the predominant gut microbial phyla were Proteobacteria, Tenericutes, Firmicutes, and Bacteroidetes among the four ophiuroid species. These bacterial phyla have been found in a variety of Echinodermata guts, such as sea cucumbers (Plotieau et al., 2013) and starfish (Certonardoa semiregularis) (Lee et al., 2018), commonly constituting over 93.7% of 16S rRNA reads. In addition, the further classification of the largest phylum of Proteobacteria to the class of Gammaproteobacteria showed that this lineage comprised an average of 43.8% (arranging from 26.6 to 60.1%) of ophiuroid gut communities. This ecologically important class has been reported in both the foregut and hindgut of the sea cucumber Apostichopus japonicus (Gao et al., 2014), with functions of digesting and degrading debris organisms (Kang et al., 2017).
Previous studies showed the multiple associations of gut microflora with the surrounding environments (Fietz et al., 2018) and hosts (Huang et al., 2020), especially for herbivore feeders. The study of the Pacific white shrimps indicated that the bacterial compositions are almost the same between intestine and sediment with different relative abundance (Fan et al., 2019). In ophiuroids, although gut and sediment microbiota shared the same dominant phylum of Proteobacteria, the gut microbiota were relatively independent reflected by the genera-level of PCA and SIMPER analysis (Figure 5 and Supplementary Appendix A3). The species that mainly contributed for diffidence were of genera Candidatus Hepatoplasma, Colwellia, Aliivibrio, Poseidonibacter, and Woeseia in Proteobacteria. The difference of gut and ambient sediment microbial communities was also illustrated in the sea cucumber using the diversity and PCA (Gao et al., 2014). Since ophiuroids are the keystone species in the pelagic–sediment coupling layer, further investigations of gut microbiota relationships among the particle organic matter in benthic column layer and surface sediment are needed for ecological roles and correlation in brittle stars and echinoderms.
Despite that our samples were collected from different seasons and sites in the Yellow Sea, the possible variation caused by sampling methods might have limited influence on the composition of gut microbiome. A previous microbial study of sediments in the Northwest Yellow Sea revealed the stable microbial community structure and its subtle seasonal changes (Li, 2009), indicating the minor influence of seasonal factor on the gut microbiota of benthic organisms in the Yellow Sea. In addition, in this study, the microbes of sediment from nearshore and offshore environments were assembled as a whole group by PCA (Figure 5), showing the slight influence of different locations on sediment microbiota. Considering the feeding behavior and living environment of ophiuroids, the stable community structure of the environmental microbiota may provide a relatively steady microenvironment for ophiuroids in this study. However, further annual observation and sequencing studies are still needed for better understanding of the association and interaction between the gut composition of ophiuroids and their environmental factors.
Host phylogeny and dieting habits all contribute to variation in the gut microbiota composition. Studies have shown that the gut microbiota of animals are species-specific and have various biological functions, depending on the feeding types of hosts, including herbivores, carnivores, and omnivores (Gaulke et al., 2018; Huang et al., 2020; Yukgehnaish et al., 2020). Animals with similar feeding habits have a more similar community structure of their intestinal microbiota (Guo et al., 2020), while the composition and functions of gut microbiomes varied among different feeding types (Liu et al., 2016; Wei et al., 2018). The results of various feeding types of fish showed that certain energy metabolism pathways were enriched in herbivore/omnivore and zooplanktivore/zoobenthivore fishes, whereas Lipid metabolism and glycan metabolism were enriched in zoobenthivore/piscivore fishes (Huang et al., 2020).
In this study, the Tax4fun and PICRUSt were used to analyze the functional annotation for different cohorts of ophiuroids, but the consistent results of two methods were obtained only in S. sladeni from two sampling sites. The remarkably enriched KEGG pathways belonged to Environmental Information Processing. Given that the higher temperature dynamics in nearshore area and stable bottom water temperature (annual < 10°C annual) of the offshore environment, the overexpression of these environmental pathways in S. sladeni might drive their broad-thermal tolerance. The differences of Tax4fun and PICRUSt analysis were commonly reported in microbial studies (Koo et al., 2017; Berlanga et al., 2018), while their variation might be caused by the algorithm for unknown gene prediction (i.e., ancestral state reconstruction algorithm in PICRUSt) and reference database (i.e., Silva-Tax4Fun and Greengenes-PICRUSt; Aßhauer et al., 2015). Further study is essential to integrate the environmental and biological data for refining the microbial functions obtained from analytical methods.
In PICRUSt, server energy metabolism pathways were primarily in abundance in the three scavenger feeders, while the Lipid metabolism and glycan metabolism pathways were enriched in suspension feeder. The KEGG results suggested that the key metabolism pathways associated with growth and development showed high expression in gut microbiota of ophiuroids (Supplementary Appendix A5, A6). The comparison of two nearshore species O. mirabilis and S. sladeni, belonging to different feeding types, showed significant variations in some nutrient metabolism pathways (Figure 8). The suspension feeder O. mirabilis displayed higher-level expression of Carbohydrate metabolism than that in the scavenger feeder S. sladeni (Adarsha et al., 2018). The functional difference was likely caused by the genus Lentibacter and class Bacteroidia (Figure 4). The genus Lentibacter is an aerobic Gram-negative bacterium with association with algae bloom, and their function in hydrocarbon degradation is facilitating the digestion and uptake of organic matter (Angelova, 2017). The class of Bacteroidia, the predominant microbes in O. mirabilis, is a member of polysaccharide-degrading consortia (Flint and Duncan, 2014).
As both S. sladeni and O. sarsii vadicola share close food diets and feeding type as scavenger (Adarsha et al., 2018), the PICRUSt predicted similar expression profiles of metabolism pathways in their gut microbiota. Variation analysis mainly found the significant variations in the environmental adaptation pathways, i.e., Cellular Processes, Genetic Information Processing, and Organismal Systems in two species (Figure 9; Supplementary Appendix A6).
Symbiotic bacteria widely identified in the gut microflora from varied marine invertebrates (Carrier and Reitzel, 2020) Ca. Hepatoplasma, as a symbiotic bacterium, are comparatively stable intestinal members, which are reported in a variety of the isopod species (Sebastian and Zimmer, 2008; Cheng et al., 2019), sea urchin (Li et al., 2018), and shrimps (Dong et al., 2019). In our study, Ca. Hepatoplasma was abundant in the gut of S. sladeni from nearshore and offshore environments and existed in O. kinbergi and O. sarsii vadicola. The BLAST search revealed the low similarity (<50%) of Ca. Hepatoplasma between ophiuroids and identified sequences in NCBI. The phylogenetic analysis further revealed the sister relationship between Ca. Hepatoplasma in ophiuroids and gut symbionts, such as the genus in amphipods (Figure 10). In amphipods, the genus Ca. Hepatoplasma plays an important role in improving growth rate and survival of host under low nutrient conditions (Sebastian and Zimmer, 2008; Horváthová et al., 2015). Due to the high abundance of Ca. Hepatoplasma in ophiuroid guts, the association of symbiotic bacteria may improve the nutrition metabolism and survival of brittle stars. Further studies are needed to characterize these symbiotic bacteria and their potential roles of the metabolism and environmental adaptation in ophiuroids.
Here, we report the gut microbiota of four dominant ophiuroids in the Yellow Sea, and we enhance our understanding of diversity and association of host–microbiomes in the environmental adaptation of echinoderms. Despite the limitations (different times and different locations) of sampling efforts, some interesting phenomena were found in the gut microbiota of ophiuroids between two different feeding types, such as scavenger and suspension feeders. The composition of the gut microbiota exhibited divergent bacterial profiles at higher taxonomic levels in the four ophiuroid species. Functional analysis revealed the significant difference in some metabolism-related pathways, such as Amino acid metabolism and Carbohydrate metabolism, in the herbivores O. mirabilis and carnivores S. sladeni. Moreover, the putative symbiotic bacteria Ca. Hepatoplasma was found in ophiuroid guts, which may facilitate the host’ nutritional metabolism and life-history adaptation. Our work contributed a comprehensive set of data for further understanding the diversity and function of gut microbiota. Further analysis by metagenome and other relative technologies of ophiuroids and their symbiotic bacteria should be conducted to elucidate the host-microbiome interaction and their implication in benthic–pelagic coupling.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SUB8304440.
QX, ZW, and XZ contributed to the conception and design of the study. SF and ZZ collected the samples. YD extracted the DNA. YD and YL performed the statistical analysis. YD, YL, PH, and QX drafted the manuscript. All authors contributed to the manuscript revision and approval of the submitted version.
This study was supported by the Basic Scientific Fund for National Public Research Institutes of China (2018Q09), the National Natural Science Foundation of China (41706190 and 4181101341), and the Key Laboratory for Ecological Environment in Coastal Areas, State Oceanic Administration (201609).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank the NSFC Open Research Cruise (Cruise No. NORC2018-01), funded by Shiptime Sharing Project of NSFC, which supported the data acquisition and sample collections. This cruise was conducted on board R/V Dongfanghong 2 by Ocean University of China. The authors also thank Dr. Yuan Chao, Miao Xiaoxiang, and Huang Wenhao from the First Institute of Oceanography for technical assistance, Jack Chi-Ho Ip from the Hong Kong Baptist University, and Fuad Tarif from the Ocean University of China for language polishing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.645070/full#supplementary-material
Adarsha, J., Shibsankar, S., Priti, S., Subhamita, S., Anuradha, S., Nilanjana, M., et al. (2018). Echinodermata Larva, Their Distribution, Habits, Characters and Phylogeny. West Bengal: Science Bulletin.
Anderson, M. J. (2014). Permutational Multivariate Analysis of Variance (PERMANOVA). Hoboken, NJ: Wiley.
Angelova, A. G. (2017). The Hydrocarbon Biodegradation Potential of Faroe-Shetland Channel Bacterioplankton. Edinburgh: Heriot-Watt University.
Aßhauer, K. P., Bernd, W., Rolf, D., and Peter, M. (2015). Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31, 2882–2884. doi: 10.1093/bioinformatics/btv287
Bauermeister, J., Ramette, A., and Dattagupta, S. (2012). Repeatedly evolved host-specific ectosymbioses between sulfur-oxidizing bacteria and amphipods living in a cave ecosystem. PLoS One 7:e50254. doi: 10.1371/journal.pone.0050254
Becker, P. T., Samadi, S., Zbinden, M., Hoyoux, C., Compère, P., and Ridder, C. D. (2009). First insights into the gut microflora associated with an echinoid from wood falls environments. Cah. Biol. Mar. 50:343.
Benjamin, C. (2017). Silva Taxonomic Training Data Formatted For Dada2 (Silva Version 128). Geneva: Zenodo.
Berlanga, M., Palau, M., Guerrero, R., and Wilson, B. A. (2018). Gut microbiota dynamics and functionality in Reticulitermes grassei after a 7-day dietary shift and ciprofloxacin treatment. PLoS One 13:e0209789. doi: 10.1371/journal.pone.0209789
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Carrier, T. J., and Reitzel, A. M. (2020). Symbiotic life of echinoderm larvae. Front. Ecol. Evol. 7:509. doi: 10.3389/fevo.2019.00509
Cheng, X., Wang, Y., Li, J., Yan, G., and He, L. (2019). Comparative analysis of the gut microbial communities between two dominant amphipods from the challenger deep, mariana trench. Deep Sea Res. Part I: Oceanographic Res. Papers 151:103081. doi: 10.1016/j.dsr.2019.103081
Cho, I., and Blaser, M. J. (2012). The human microbiome: at the interface of health and disease. Nat. Rev. Genet. 13, 260–270. doi: 10.1038/nrg3182
Clarke, K. R. (1993). Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
Dong, X., Lv, L., Zhao, W., Yu, Y., and Liu, Q. (2019). Effects of different cultural patterns on microbial communities in the intestine of Macrobrachium rosenbergii and interactions with environment factors. J. Shanghai Ocean University. 28, 501–510.
Enoksen, S. E., and Reiss, H. (2017). Diet of Norwegian coastal cod (Gadus morhua) studied by using citizen science. J. Mar. Systems 180, 246–254. doi: 10.1016/j.jmarsys.2017.06.006
Fan, L., Wang, Z., Chen, M., Qu, Y., Li, J., Zhou, A., et al. (2019). Microbiota comparison of Pacific white shrimp intestine and sediment at freshwater and marine cultured environment. Sci. Total Environ. 657, 1194–1204. doi: 10.1016/j.scitotenv.2018.12.069
Fietz, K., Hintze, C. O. R., Skovrind, M., Nielsen, T. K., Limborg, M. T., Krag, M. A., et al. (2018). Mind the gut: genomic insights to population divergence and gut microbial composition of two marine keystone species. Microbiome 6:82. doi: 10.1186/s40168-018-0467-7
Flint, H. J., and Duncan, S. H. (2014). Bacteroides and Prevotella. Encyclopedia of Food Microbiology. Amsterdam: Elsevier. doi: 10.1016/B978-0-12-384730-0.00031-8
Gao, F., Li, F., Tan, J., Yan, J., and Sun, H. (2014). Bacterial community composition in the gut content and ambient sediment of sea cucumber apostichopus japonicus revealed by 16S rRNA gene pyrosequencing. PLoS One 9:e100092. doi: 10.1371/journal.pone.0100092
Gaulke, C. A., Arnold, H. K., Humphreys, I. R., Kembel, S. W., O’Dwyer, J. P., and Sharpton, T. J. (2018). Ecophylogenetics clarifies the evolutionary association between mammals and their gut microbiota. mBio 9:e01348-18. doi: 10.1128/mBio.01348-18
Geraldi, N. R., Bertolini, C., Emmerson, M. C., Roberts, D., Sigwart, J. D., and O’Connor, N. E. (2017). Aggregations of brittle stars can perform similar ecological roles as mussel reefs. Mar. Ecol. Prog. Ser. 563, 157–167. doi: 10.3354/meps11993
Guo, X., Lei, H., Zhang, K., Ke, F., and Song, C. (2020). Diversification of animal gut microbes and NRPS gene clusters in some carnivores, herbivores and omnivores. Biotechnol. Biotechnol. Equip. 34, 1280–1287. doi: 10.1080/13102818.2020.1835536
Hakim, J. A., Koo, H., Kumar, R., Lefkowitz, E. J., Morrow, C. D., Powell, M. L., et al. (2016). The gut microbiome of the sea urchin, Lytechinus variegatus, from its natural habitat demonstrates selective attributes of microbial taxa and predictive metabolic profiles. FEMS Microbiol. Ecol. 92:fiw146. doi: 10.1093/femsec/fiw146
Harris, J. L., MacIsaac, K., Gilkinson, K. D., and Kenchington, E. L. (2009). Feeding biology of Ophiura sarsii Lütken, 1855 on Banquereau bank and the effects of fishing. Mar. Biol. 156, 1891–1902. doi: 10.1007/s00227-009-1222-1
Hewson, I., Bistolas, K. S., Cardé, E. M. Q., Button, J. B., Foster, P. J., Flanzenbaum, J. M., et al. (2018). Investigating the complex association between viral ecology, environment, and northeast Pacific sea star wasting. Front. Mar. Sci. 5:77. doi: 10.3389/FMARS.2018.00077
Horváthová, T., Kozłowski, J., and Bauchinger, U. (2015). Growth rate and survival of terrestrial isopods is related to possibility to acquire symbionts. Eur. J. Soil Biol. 69, 52–56.
Huang, Q., Sham, R. C., Deng, Y., Mao, Y., Wang, C., Zhang, T., et al. (2020). Diversity of gut microbiomes in marine fishes is shaped by host-related factors. Mol. Ecol. 29, 5019–5034. doi: 10.1111/mec.15699
Iken, K., Brey, T., Wand, U., Voigt, J., and Junghans, P. (2001). Food web structure of the benthic community at the porcupine abyssal plain (NE Atlantic): a stable isotope analysis. Prog. Oceanography 50, 383–405. doi: 10.1016/S0079-6611(01)00062-3
Kang, I., Lim, Y., and Cho, J. C. (2017). Complete genome sequence of Granulosicoccus antarcticus type strain IMCC3135T, a marine gammaproteobacterium with a putative dimethylsulfoniopropionate demethylase gene. Mar. Genom. 37, 176–181. doi: 10.1016/j.margen.2017.11.005
Koo, H., Hakim, J. A., Morrow, C. D., Eipers, P. G., Davila, A., Andersen, D. T., et al. (2017). Comparison of two bioinformatics tools used to characterize the microbial diversity and predictive functional attributes of microbial mats from Lake Obersee. Antarctica. J. Microbiol. Methods 140, 15–22. doi: 10.1016/j.mimet.2017.06.017
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., Mcdonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Lebrato, M., Iglesias-Rodriguez, D., Feely, R. A., Greeley, D., Jones, D. O. B., Suarez-bosche, N., et al. (2010). Global contribution of echinoderms to the marine carbon cycle: CaCO3 budget and benthic compartments. Ecol. Monographs 80, 441–467. doi: 10.1890/09-0553.1
Lee, H., Park, S., Kim, D., Moon, K., and Heo, M. (2018). Microbial community analysis isolated from red starfish (Certonardoa semiregularis) gut. J. Life Sci. 28, 955–961.
Li, G., Li, S., Li, M., Lyn, D., Ding, J., Chang, Y., et al. (2018). Effects of dietary arachidonic acid on survival and growth performance and the intestinal bacterial profile of adult sea urchin (Strongylocentrotus intermedius). Chinese Fish. Qual. Stand. 3, 42–51.
Li, H. (2009). Study of the Eubacterial Community Structure and Diversity in the Northwest of Yellow Sea. Postgraduate: Ocean University of China.
Li, X. (2011). An overview of studies on marine macrobenthic biodiversity from Chinese waters: principally from the Yellow Sea. Biodiversity Sci. 19, 676–684. doi: 10.3724/SP.J.1003.2011.09126
Li, Z., Zhang, X., Aweya, J. J., Wang, S., Hu, Z., Li, S., et al. (2019). Formulated diet alters gut microbiota compositions in marine fish Nibea coibor and Nibea diacanthus. Aquacul. Res. 50, 126–138. doi: 10.1111/are.13874
Liu, H., Guo, X., Gooneratne, R., Lai, R., Zeng, C., Zhan, F., et al. (2016). The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci. Rep. 6:24340. doi: 10.1038/srep24340
Marcel, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Minoru, K., Yoko, S., Masayuki, K., Miho, F., and Mao, T. (2015). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Morrow, K. M., Tedford, A. R., Pankey, M. S., and Lesser, M. P. (2018). A member of the Roseobacter clade, Octadecabacter sp., is the dominant symbiont in the brittle star Amphipholis squamata. FEMS Microbiol. Ecol. 94:fiy030. doi: 10.1093/femsec/fiy030
Murat, A., Méar, Y., Poizot, E., Jean-Claude Dauvin, and Beryouni, K. (2016). Silting up and development of anoxic conditions enhanced by high abundance of the geoengineer species Ophiothrix fragilis. Cont. Shelf Res. 118, 11–22. doi: 10.1016/j.csr.2016.01.003
Nguyen, V. K., King, W. L., Siboni, N., Mahbub, K. R., Dove, M., O’Connor, W., et al. (2020). The Sydney rock oyster microbiota is influenced by location, season and genetics. Aquaculture 527:735472. doi: 10.1016/j.aquaculture.2020.735472
Parks, D. H., and Beiko, R. G. (2010). Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26, 715–721. doi: 10.1093/bioinformatics/btq041
Peng, S., Li, X., Xu, Y., Wang, H., and Zhang, B. (2017). Variation of macrobenthos in yellow sea in past 10 years. Oceanol. Limnol. Sinica. 48, 536–542.
Pierce, M. L., Ward, J. E., Holohan, B. A., Zhao, X., and Hicks, R. E. (2016). The influence of site and season on the gut and pallial fluid microbial communities of the eastern oyster, Crassostrea virginica (Bivalvia, Ostreidae): community-level physiological profiling and genetic structure. Hydrobiologia 765, 97–113. doi: 10.1007/s10750-015-2405-z
Plotieau, T., Lavitra, T., Gillan, D. C., and Eeckhaut, I. (2013). Bacterial diversity of the sediments transiting through the gut of Holothuria scabra (Holothuroidea; Echinodermata). Mar. Biol. 160, 3087–3101. doi: 10.1007/s00227-013-2297-2
Ravelo, A. M., Konar, B., and Bluhm, B. A. (2015). Spatial variability of epibenthic communities on the Alaska Beaufort Shelf. Polar Biol. 38, 1783–1804. doi: 10.1007/s00300-015-1741-9
Sebastian, F., and Zimmer, M. (2008). Host-specificity of environmentally transmitted Mycoplasma-like isopod symbionts. Environ. Microbiol. 10, 2497–2504. doi: 10.1111/j.1462-2920.2008.01672.x
Sugita, H., Miyajima, C., and Deguchi, Y. (1991). The vitamin B12-producing ability of the intestinal microflora of freshwater fish. Aquaculture 92, 267–276. doi: 10.1016/0044-8486(91)90028-6
Tanaka, R., Ootsubo, M., Sawabe, T., Ezura, Y., and Tajima, K. (2004). Biodiversity and in situ abundance of gut microflora of abalone (Haliotis discus hannai) determined by culture-independent techniques. Aquaculture 241, 453–463. doi: 10.1016/j.aquaculture.2004.08.032
Temara, A., Ridder, C. D., Kuenen, J., and Robertson, L. (1993). Sulfide-oxidizing bacteria in the burrowing echinoid, Echinocardium cordatum (Echinodermata). Mar. Biol. 115, 179–185. doi: 10.1007/BF00346333
Trabal, N., Mazón-Suástegui, J. M., Vázquez-Juárez, R., Asencio-Valle, F., Morales-Bojórquez, E., and Romero, J. (2012). Molecular analysis of bacterial microbiota associated with oysters (Crassostrea gigas and Crassostrea corteziensis) in different growth phases at two cultivation sites. Microbial Ecol. 64, 555–569. doi: 10.1007/s00248-012-0039-5
Walker, C., and Lesser, M. (1989). Nutrition and development of brooded embryos in the brittlestar Amphipholis squamata: do endosymbiotic bacteria play a role? Mar. Biol. 103, 519–530. doi: 10.1007/BF00399584
Wang, Y., Sheng, H. F., He, Y., Wu, J. Y., Jiang, Y. X., Tam, N. F. Y., et al. (2012). Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl. Environ. Microbiol. 78, 8264–8271. doi: 10.1128/AEM.01821-12
Wei, J., Guo, X., Liu, H., Chen, Y., and Wang, W. (2018). The variation profile of intestinal microbiota in blunt snout bream (Megalobrama amblycephala) during feeding habit transition. BMC Microbiol. 18:99. doi: 10.1186/s12866-018-1246-0
Wu, R. S. S., and Shin, P. K. S. (1997). Food segregation in three species of portunid crabs. Hydrobiologia 362, 107–113. doi: 10.1023/A:1003126415990
Yu, N., Sun, S., Wang, S., Liu, Q., Zhang, G., Zhang, F., et al. (2019). An enhanced underwater camera apparatus for seabed observation of megabenthic epifauna in the northern Yellow Sea. J. Oceanol. Limnol. 38, 1799–1810. doi: 10.1007/s00343-019-9122-9
Yukgehnaish, K., Kumar, P., Sivachandran, P., Marimuthu, K., Arshad, A., Paray, B. A., et al. (2020). Gut microbiota metagenomics in aquaculture: factors influencing gut microbiome and its physiological role in fish. Rev. Aquacult. 12, 1903–1927. doi: 10.1111/raq.12416
Zhang, J., Chen, M., Huang, J., Guo, X., Zhang, Y., Liu, D., et al. (2019). Diversity of the microbial community and cultivable protease-producing bacteria in the sediments of the Bohai Sea, Yellow Sea and South China Sea. PLoS One 14:e0215328. doi: 10.1371/journal.pone.0215328
Zhang, M., Sun, Y., Liu, Y., Qiao, F., Chen, L., Liu, W. T., et al. (2016). Response of gut microbiota to salinity change in two euryhaline aquatic animals with reverse salinity preference. Aquaculture 454, 72–80. doi: 10.1016/j.aquaculture.2015.12.014
Zhang, X., Nakahara, T., Miyazaki, M., Nogi, Y., Taniyama, S., Arakawa, O., et al. (2012). Diversity and function of aerobic culturable bacteria in the intestine of the sea cucumber Holothuria leucospilota. J. General Appl. Microbiol. 58, 447–456. doi: 10.2323/jgam.58.447
Keywords: gut microbiota, ophiuroids, feeding type, symbiotic bacteria, functional microbiomes, Yellow Sea
Citation: Dong Y, Li Y, He P, Wang Z, Fan S, Zhang Z, Zhang X and Xu Q (2021) Gut Microbial Composition and Diversity in Four Ophiuroid Species: Divergence Between Suspension Feeder and Scavenger and Their Symbiotic Microbes. Front. Microbiol. 12:645070. doi: 10.3389/fmicb.2021.645070
Received: 07 January 2021; Accepted: 19 February 2021;
Published: 19 March 2021.
Edited by:
Lifeng Zhu, Nanjing Normal University, ChinaCopyright © 2021 Dong, Li, He, Wang, Fan, Zhang, Zhang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qinzeng Xu, eHF6MDhAMTYzLmNvbQ==; eHVxaW56ZW5nQGZpby5vcmcuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.