 Zahra Salehi
Zahra Salehi Masoomeh Shams-Ghahfarokhi
Masoomeh Shams-Ghahfarokhi Mehdi Razzaghi-Abyaneh
Mehdi Razzaghi-Abyaneh- 1Department of Mycology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
- 2Department of Mycology, Pasteur Institute of Iran, Tehran, Iran
Background: Dermatophytes are a homogeneous group of species with low genetic diversity, and there are still many uncertainties about the boundaries among species.
Objectives: Aiming at clarifying the relationships among species in the genus and introducing suitable genes for multilocus sequence typing (MLST), a new MLST scheme approach was developed to characterize the major pathogenic dermatophytes.
Methods: We performed maximum parsimony (MP), MrBayes, RAxML, and eBURST analyses, based on the MLST scheme to scrutinize the evolution within 95 clinical isolates and four reference strains belonging to the four major dermatophytes species. Then, the discriminatory power, pairwise genetic distances, ratio dN/dS, and sequence types (STs) of these isolates were determined. Also, to study taxonomy, sequences of the internal transcribed spacer (ITS), Beta-tubulin (BT2), and translation elongation factor 1-α (TEF-1α) genes of other dermatophytes species available in the GenBank were analyzed.
Results: Findings of the present study indicated that three genes: BT2, ITS, and TEF−1α, which showed the greatest diversity among dermatophyte species, were suitable for MLST. The most prevalent STs were seen among the species of Trichophyton interdigitale. Also, two new genotypes, i.e., XXVII and XXVIII, were introduced for T. interdigitale and Trichophyton mentagrophytes. The least informative sites were found in Epidermophyton floccosum, Trichophyton rubrum, and T. mentagrophytes, while the most informative sites were observed in T. interdigitale. Furthermore, the most informative locus was TEF-1α. The phylogenetic tree, constructed by the combination of the three genes, shows a new topological pattern that confirms the derivation of the anthropophilic and zoophilic genera from the geophilic genus. Also, the phylogenetic analyses and pairwise distances of the combination of the three loci showed that Trichophyton tonsurans and Trichophyton equinum were a species complex, where T. equinum is derived from T. tonsurans.
Conclusions: Results of this study showed that MLST is very effective in determining the boundaries between species and taxonomy. Considering that there is no database for MLST dermatophytes, further studies are needed to determine the suitable genes for MLST. Also, the determination of STs in epidemiological studies and raising epidemiological information are helpful. This study was a new starting point to determine the ST and a foundation for a dermatophyte MLST database.
Introduction
Dermatophytes are a group of related keratinophilic fungi categorized by several genera, namely, Trichophyton, Microsporum, Epidermophyton, Nannizzia, Paraphyton, Lophophyton, and Arthroderma based on the new taxonomy (de Hoog et al., 2017). They play a significant role as pathogens in humans and animals (Summerbell et al., 1999; Cafarchia et al., 2013; Zarrin et al., 2015; Mochizuki et al., 2016). Concerning the recent epidemiologic surveys in Tehran, dermatophytosis was found to be 19.7%, and the major species involved the Trichophyton mentagrophytes/Trichophyton interdigitale species complex, Trichophyton rubrum, Trichophyton tonsurans, and Epidermophyton floccosum (Sadeghi et al., 2011; Zamani et al., 2016). It is believed that the PCR-based assays are quick and useful tools for the diagnosis and differentiation of isolates at the species and subspecies level, especially for the atypical isolates (Kac, 2000; Graser et al., 2008). Gene sequencing is one of the most informative and well-known techniques for fungal diagnosis (Susca et al., 2013). Multilocus sequence typing (MLST) is a very stable and reproducible technique that determines the isolates of the microbial species using the DNA sequences of multiple housekeeping genes (Bougnoux et al., 2002; Bain et al., 2007; Vanhee et al., 2010; Bernhardt et al., 2013). MLST results are unequivocal, and sequence data can be shared and compared between different laboratories (Bernhardt et al., 2013). More specifically, the multilocus sequence can distinguish between the individual strains and other isolates, and evaluate the relationships, recombination, and mutation rates among microorganisms, which are classified under the same genus (Bougnoux et al., 2002). The phylogenetic species concepts and phylogenetic analysis based on the internal transcribed spacer (ITS) regions contribute to the improvement of the taxonomy. At present, the sequence database of the ITS region is considered to be the gold standard for dermatophytes (Graser et al., 2008; Anzawa et al., 2011). Sequencing of the Beta-tubulin (BT2), translation elongation factor 1-α (TEF-1α) genes, calmodulin (CaM), or sequencing the D1/D2 domain of the large-subunit rRNA gene, as well as the ITS region, can reliably identify the species (Li et al., 2008; Anzawa et al., 2011; Mirhendi et al., 2014, 2016; Rezaei-Matehkolaei et al., 2014; Ahmadi et al., 2016). The results of previous studies emphasize that very few genetic changes happened in the evolutionary history of dermatophytes (Makimura et al., 1999; de Hoog et al., 2017; Persinoti et al., 2018; Zhan et al., 2018). Therefore, to attain more accurate results, exploring the SNP analysis of these genes is suggested. Fungal taxonomy development is a controversial issue, and the information in this field is not sufficient for the dermatophyte species. The dermatophyte species has been recently revised based on the MLST (de Hoog et al., 2017). For example, the recent study showed that T. mentagrophytes and T. interdigitale belong to the same phylogenetic species (Pchelin et al., 2016). Yet, there remain many uncertainties concerning the boundaries between dermatophytes species. Data obtained from MLST help to resolve species boundaries (O’Donnell et al., 2004). Therefore, more detailed studies are necessary for determination purposes. These led to the following questions: (i) Are T. tonsurans and Trichophyton equinum distinct species? and (ii) Are T. rubrum and Trichophyton violaceum belong to a complex?
Besides, to the best of the researchers’ knowledge, there are a few studies with the MLST of dermatophytes (Probst et al., 2002; de Hoog et al., 2017; Pchelin et al., 2016; Persinoti et al., 2018; Zhan et al., 2018). In recent years, the trend toward this technique has been increased to investigate dermatophyte species. Moreover, previous studies on MLST have not addressed the genotypes, sequence types (STs) species, discriminatory power, ratio dN/dS, and informative sites (Probst et al., 2002; Pchelin et al., 2016; de Hoog et al., 2017). The present study focuses on the determination of the ST dermatophyte species, and is specially designed for (i) providing a better understanding of the species; (ii) highlighting the molecular diversity and genotypes characterized by major dermatophyte species on seven loci: BT2, TEF-1α, ACT, CaM, HSP70, D1/D2, and ITS; (iii) evaluating the genes used for the MLST approach (iv) specifying the ST dermatophyte species; and (v) investigating the association between genotypes and STs with an antifungal sensitivity profile (vi) offering a closer look at the taxonomy of dermatophytes based on the combination of the three genes: ITS, BT2, and TEF-1α.
Materials and Methods
Fungal Isolates
Clinical samples were selected from the Pathogenic Fungi Culture Collection of Pasteur Institute, Iran. Initially, the phenotypically distinct isolates were identified, followed by re-identification through the use of the sequencing related to the ITS region of rDNA. To this end, a total of 99 strains consisting of 95 dermatophyte clinical isolates, including T. rubrum (n = 19), T. interdigitale (n = 20), T. mentagrophytes (n = 6), T. tonsurans (n = 28), E. floccosum (n = 22), as well as standard strains provided from the Persian Type Culture Collection (PTCC) of T. Rubrum PTCC 5143, T. mentagrophytes PTCC 5054, T. tonsurans CBS 130924, and E. floccosum CBS 767.73 were used in the sequence analysis. Clinical features of these isolates are provided in Supplementary Table 1. All the strains were isolated from the skin and hair specimens. The names of the species were determined according to de Hoog et al. (2017).
DNA Extraction and Gene Amplification
All fungal strains were cultured on a mycobiotic agar (Merck, Germany) and incubated at 28°C for 4–7 days. DNA was extracted using the phenol-chloroform-isoamyl alcohol according to Makimura et al. (1999).
PCRs were carried out in 50 μl reaction volumes for TEF-1α, BT2, CaM, ACT, HSP70, ITS, and the D1/D2 fragments. Each mixture contained 25 μl of Premix (Ampliqon, Denmark), 3 μl of DNA template, and 0.8 μM of each primer, and the remaining volume was filled with water to reach a final volume of 50 μl. Negative controls (water instead of the fungal DNA) were added to each PCR. PCR amplification with different primer pairs was performed for all the isolates, following the seven loci adapted from the earlier genotyping studies (Table 1). The reaction mixture was initially denatured at 95°C for 5 min, followed by 30 cycles of 30 s at 94°C, annealing at changes in temperatures (from 55 to 67°C) for 40 s, 45 s at 72°C, and a terminal extension step of 72°C for 5 min. Five microliters of the PCR products was electrophoresed on 1% agarose gel in TAE buffer (Tris 40 mM, acetic acid 20 mM, and EDTA 1 mM), and photographed under ultraviolet irradiation.
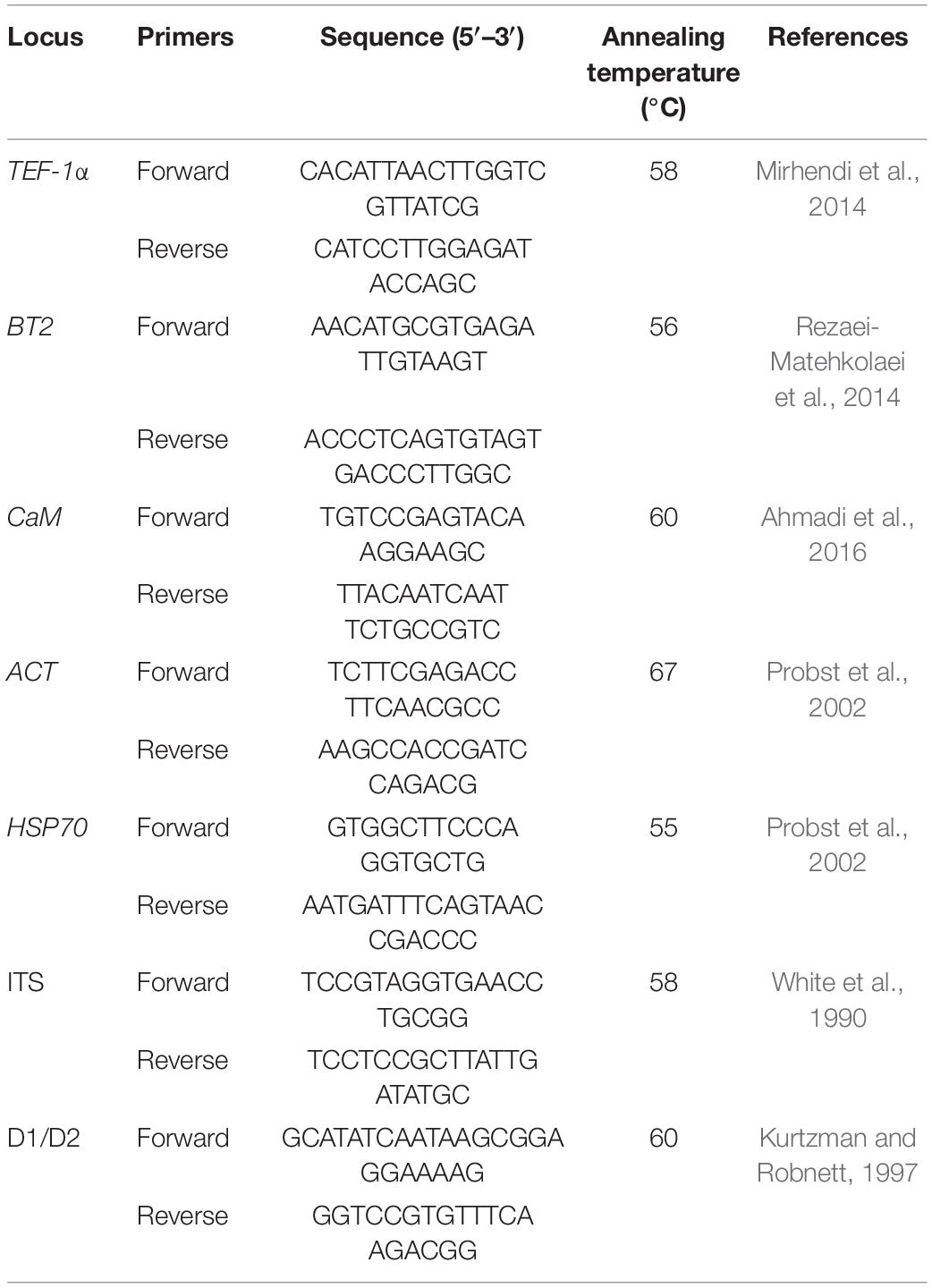
Table 1. Oligonucleotide primers used for the molecular identification of dermatophytes.
Sequencing
All seven genes were successfully amplified and sequenced in forward and reverse for all 99 isolates. The PCR products were subjected to the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA, United States). The forward and reverse sequences of each sample were subjected to ClustalW pairwise alignment using the MEGA7.0.21 software, and edited manually to improve the alignment accuracy (Kumar et al., 2001). Two-web databases of the CBS (for sequencing ITS regions) and BLASTn (for six other gene fragments) were used for identifying and comparing the fungi. All isolates were verified to the species level by sequencing the ITS regions of rDNA.
Phylogenetic Analysis
The bioinformatics data were analyzed and used to diagnose the interspecies and intraspecies nucleotide variation of seven loci in all of the 99 isolates in this study. The sequence data from each gene were aligned using the MEGA7.0.21 software; then, they were manually adjusted and concatenated (4,620 bp). Also, the sequences were analyzed by maximum parsimony (MP) for finding the informative sites of genes of the isolates that belonged to each species and PAUP version 4.0b109 to the Bayesian analysis for drawing the phylogenetic tree (Swofford, 2002). The best-fit model of molecular evolution was estimated in the jModelTest 2.1.10 (Darriba et al., 2012). The programs MrBayes version 3.2 (Ronquist et al., 2012) and RAxML version 8.2 (Miller et al., 2010; Stamatakis, 2014) were run on the CIPRES Science Gateway. Subsequently, two simultaneous analyses with eight Metropolis-coupled Markov chain Monte Carlo (MCMC) chains and incremental heating of 0.2 were run for 20 million generations which were sampled in every 1,000 generations. The convergence of parameter estimates was verified and effective sample sizes were obtained >200 for all parameters using the Tracer version 1.6 (Drummond and Rambaut, 2007). Maximum-likelihood (ML) analyses were performed using the RAxML. Optimization in RAxML was carried out using the GTRCAT option. Bootstrap values for maximum likelihood were 1,000 replicates with one search replicate per bootstrap replicate. This analysis was performed for 99 isolates, an individual gene, and a combination of seven genes. Fusarium solani was used as an outgroup to root the dendrogram.
Phylogenetic Reconstruction
First, species involving all three sequences, BT2, TEF-1α, and ITS, were deposited in the GenBank and identified for phylogenetic reconstruction. Then, these sequences became the datasets. The combined multilocus (BT2, TEF-1α, and ITS) dataset of the 99 isolates of this study along with the published sequences dataset was aligned using the MEGA7.0.21 software. ML analyses of the mentioned sequences were performed using the RAxML as described above. F. solani was used as an outgroup in a phylogenetic tree, constructed with Maximum likelihood using the RAxML v. 8.0.0 under the GTRCAT model and 1,000 bootstrap replications. Bootstrap support above 95% is shown above the branches.
Genotyping
Since ITS-genotyping is only determined for the T. interdigitale/T. mentagrophytes species complex and not for other species and genes, we used the nomenclature from the studies by Heidemann et al. (2010) and Taghipour et al. (2019) for the ITS-genotyping of the T. interdigitale/T. mentagrophytes complex. We chose the borders of the ITS region of T. interdigitale as deposited under the accession number MK312735 for homogeneity, and did not consider the four nucleotides (GGTT) at the 5′−end of our deposited sequences in the GenBank. For other species and genes in this study, the numeration was used, starting from 1.
MLST Analyses
Because an MLST database for dermatophytes does not exist, the ST numbers started from 1. The ST was put in numerical tags as a contract, which was based on the case suggested by Bougnoux et al. (2002).
According to the MLST standards, particular allele sequences for each of the housekeeping genes were labeled as a genotype, and each different combination of genotypes was determined as a distinct ST. STs were also analyzed using the eBURST package1 to determine the possible relationships between isolates based on the single allele differences (Feil et al., 2004; Spratt et al., 2004). The discriminatory power was calculated according to Hunter’s formula (Hunter, 1990). Pairwise genetic distances (p-distances) between species were calculated using the MEGA7.0.21. The possibility of selective pressure at each of the loci was computed by the ratio of non-synonymous nucleotide substitutions (dN/dS) calculated by Nei and Gojobori (1986). Fisher’s exact test was used to determine the associations between the ST and anatomical origin, age, gender, and genotype using the statistical SPSS package (version 19). Values with P < 0.05 were considered significant.
Antifungal Susceptibility Testing
Susceptibility of 95 clinical isolates and four reference strains to griseofulvin, lanoconazole, butenafine (Sigma-Aldrich), and terbinafine (Dr. Reddy’s Laboratories) were determined according to the CLSI M38-A2 broth microdilution method (Wayne, 2008). MIC results were visually read after 4 days of incubation at 35°C. The MIC was defined as the point at which the growth of dermatophyte was inhibited by 80% or more reduction for eight antifungals after a visual inspection in comparison with the control. MIC range, geometric mean, MIC50, and MIC90 were then calculated for the isolates that belonged to each species. All tests were performed in duplicate. Candida krusei (ATCC 6258) and Candida parapsilosis (ATCC 22019) were used as quality controls.
Results
Fungal Isolates
Trichophyton interdigitale and T. mentagrophytes were often isolated from tinea pedis, and E. floccosum was the most common cause of tinea cruris. T. rubrum and T. tonsurans are distributed among different areas (Supplementary Table 1).
Sequencing and Gene Diversity
PCR of the ITS, D1/D2, BT2, TEF-1α, HSP70, ACT, and CaM genes of the 99 isolates and the standard strains showed bands, sizes of which ranged from 350 to 800 bp. The nucleotide substitutions of different species by the gene regions are represented in Table 2. The gene regions without a nucleotide change are not included in Table 2. The HSP70 sequences did not show any intraspecies variation in all species (Figure 1). All the newly generated sequences were deposited in the GenBank (Supplementary Table 2).
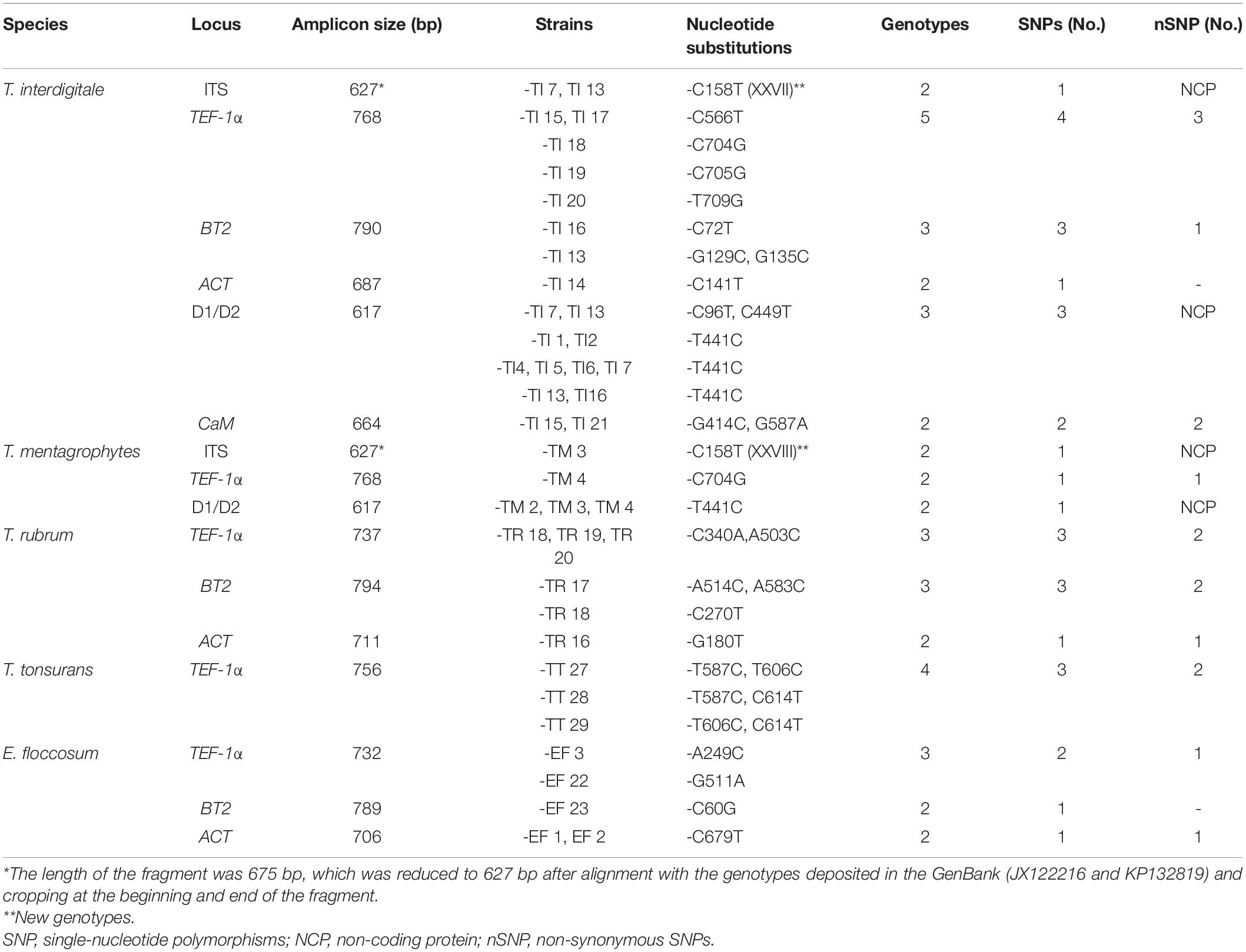
Table 2. Characteristics and the nucleotide substitutions of the housekeeping loci studied.
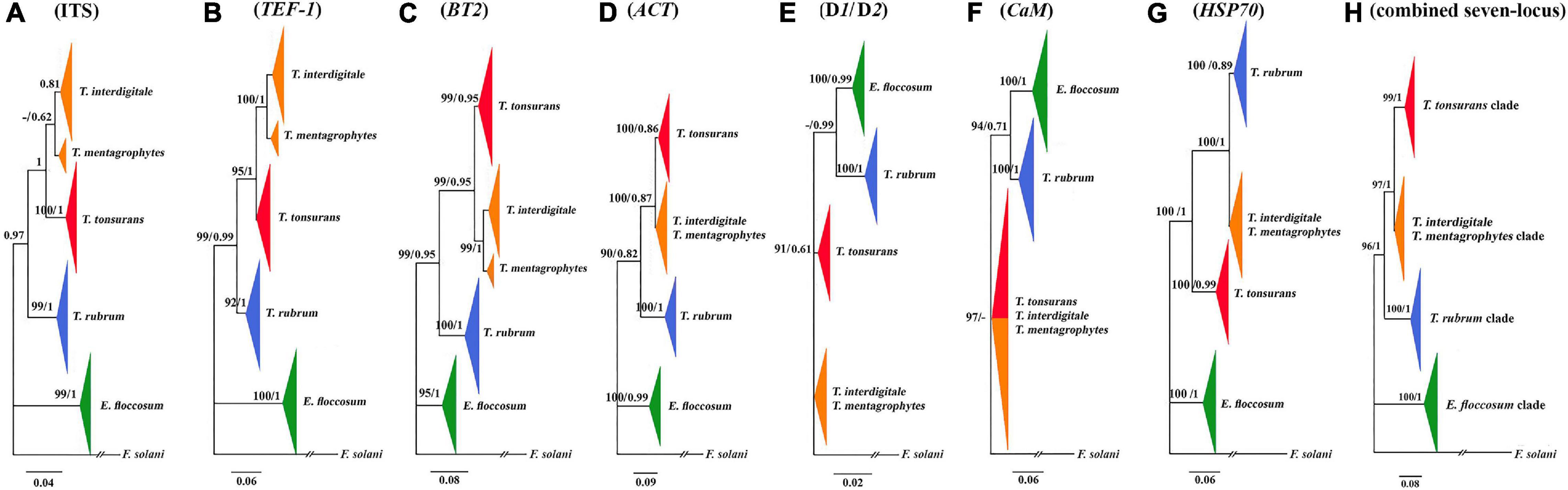
Figure 1. Comparison of seven trees constructed by the Bayesian analysis. Phylogenetic analysis of ITS (A), TEF-1α (B), BT2 (C), ACT (D), D1/D2 (E), CaM (F), HSP70 (G), and the consensus trees constructed by the Bayesian analysis from the combined seven-locus (H) of the 95 clinical isolates, four reference strains, and use of F. solani as the outgroup. The bootstrap percentages of the ML analysis and posterior probabilities values are presented at the node (PP/BS). Values less than 70% bootstrap support in the ML analysis and less than 0.5 posterior possibility in the Bayesian analysis were indicated with a hyphen.
The characteristics of the multilocus obtained from the sequences of the PCR products from T. interdigitale loci were compared to those of the corresponding loci in T. mentagrophytes, T. rubrum, T. tonsurans, and E. floccosum (Table 2). The sequences were obtained from the combination of seven genes which contained 4,620 nucleotides. The HSP70 region of E. floccosum was found to be the shortest gene with only 346 bp, while BT2 was the longest gene of T. rubrum with 794 bp. The number of nucleotide substitutions from the dermatophytes genome sequence was studied, which were not conserved at the amino acid level, and the most amino acid changes were found in T. interdigitale (three) at the locus TEF-1α. For example, the HSP70 sequences failed to reveal intraspecies variation in all species. Also, the ITS, CaM, and D1/D2 sequences did not show intraspecies variation in T. rubrum, adding that the ITS, BT2, ACT, CaM, and D1/D2 sequences did not show intraspecies variation in T. tonsurans. The species with no intraspecies variations (with only one genotype) are excluded from Table 2. In the combination of three loci (ITS, BT2, and TEF-1α), there was a relationship between STs and taxa (except for T. tonsurans that ST1 and 2 were in the same taxa and T. interdigitale that ST1 was distributed among the other STs) (Figure 2), whereas, in the combination of seven genes, this relationship was seen in 21 of the 27 singletons.
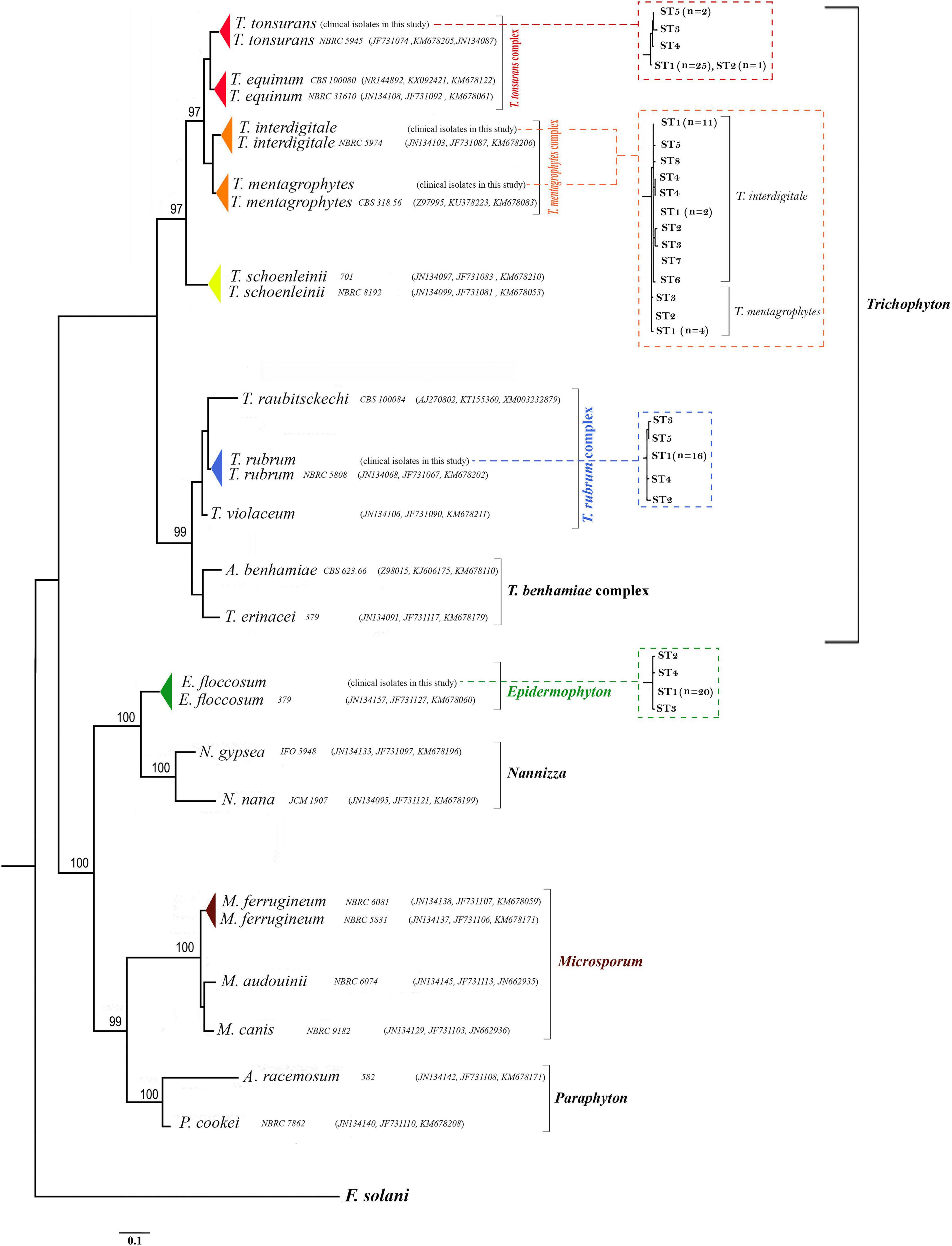
Figure 2. Phylogenetic relationships within dermatophytes. The consensus trees constructed by RAxML analysis from the combined three-locus ITS, BT2, and TEF-1α from clinical isolates and dermatophytes species available in the GenBank. The accession number profile was written in order ITS, BT2, TEF-1α loci. F. solani was used as an outgroup to root the dendrogram. The bootstrap value greater than 95% is shown above the branches. Also, the relationship between STs and taxa in RAxML analyses for the 99 dermatophyte species used in this study from the combined three-locus ITS, BT2, and TEF-1α is shown in the dot-box plots.
Phylogenetic Analysis in Dermatophytes
A phylogenetic tree for a combined seven loci is shown in Figure 1. Based on the phylogenetic analysis, all isolated strains studies were placed in four supported clades (pp = 1). E. floccosum clade takes a basal position to the Trichophyton clades (Figure 1).
Individual CaM tree failed to separate the clade of T. mentagrophytes/T. interdigitale complex from the clade of T. tonsurans (Figure 1), while individual ITS, D1/D2, BT2, TEF-1α, HSP70, and ACT trees provided altogether the satisfactory resolution in four clades (Figure 1). The results of individual trees showed that the only individual ITS, BT2, and TEF-1α tree can distinguish between T. mentagrophytes and T. interdigitale in distinct clusters and T. mentagrophytes strains are placed in a basal position to the T. interdigitale (Figure 1).
In the data obtained from the MP analysis of seven loci, no informative site was observed to be HSP70, while the most informative locus was TEF-1α. Overall, these results showed that the most informative sites were found in T. interdigitale isolates (Table 3).
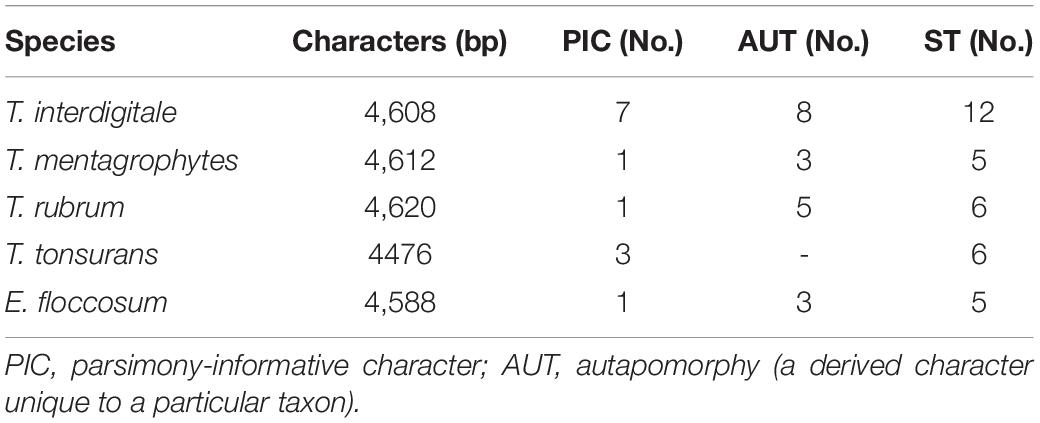
Table 3. The data obtained from the maximum parsimony analysis and phylogenetic analysis of seven loci.
Phylogenetic Reconstruction
Deposited sequence of 22 strains in the GenBank was used for the phylogenetic reconstruction. Twenty-two standard strains were selected in a way that all three BT2, TEF-1α, and ITS sequences were deposited in the GenBank. These 22 sequences along with 99 isolates of this study were subjected to ClustalW pairwise alignment using the MEGA7.0.21 software (Figure 2).
Five genera introduced by the study of de Hoog et al. (2017) (Epidermophyton, Microsporum, Nannizzia, Paraphyton, and Trichophyton) were used in this classification, and other genera were not included in this classification due to the absence of the TEF-1α, BT2, and ITS sequences in the GenBank. The highest ratio of bootstrap-support (100%) was observed in groups Epidermophyton, Microsporum, Nannizzia, and Paraphyton, and the lowest belonged to Trichophyton.
Pairwise distances of the combination of the three loci (based on overall average) were examined, indicating a high degree of overlap (difference 0.001%) between the species T. tonsurans and T. equinum (results are not shown). Also, P-distances of the combination of the three genes showed homology of about 99.994% between T. rubrum and T. violaceum species. On the other hand, P-distances of the combination of the three genes showed significant differences between the species N. nana and N. gypsea with E. floccosum (results are not shown).
Genotyping
A total of 19 out of 21 T. interdigitale isolates were classified in genotype II, and five out of six T. mentagrophytes isolates were classified in genotype II∗. A total of two new genotypes were identified in this study; one genotype was obtained in two T. interdigitale isolates (XXVII) and the other in one isolate of T. mentagrophytes (XXVIII).
Genotypes (except for the ITS-genotyping of T. mentagrophytes/T. interdigitale complex) and STs numerically start from 1 in the contract due to the absence of the MLST database for dermatophyte species. The positions of the single-nucleotide polymorphisms (SNPs), the different genotypes identified at each of the seven loci, and the determination of the genotype, as well as their frequency, are shown in Table 2. Besides, these numbers correlate with the genotype numbers listed in Supplementary Table 2 which represents how ST is determined and shows nucleotide diversity at the level of dermatophytes species. Regarding the genetic variability, the highest number of genotypes was found within the T. interdigitale (five genotypes) species (Supplementary Table 2 and Table 2). The phylogenetic analysis of species with only one genotype is excluded from Table 2. The analysis of 99 isolates yielded 34 STs which are illustrated in Supplementary Table 2. All in all, 27 STs were singletons (represented by a single isolate).
MLST Analysis
The eBURST analysis was performed for five species. The discriminatory power of the MLST for T. interdigitale, T. mentagrophytes, T. rubrum, T. tonsurans, and E. floccosum was estimated to be 0.881, 0.9333, 0.4474, 0.3202, and 0.5217, respectively. The 32 SNPs among the seven loci led to 16 non-synonymous changes in encoding amino acids. Considering the species studied, dS occurred more frequently than dN; thus, the ratio of dN/dS was less than one for five loci (protein-coding): E. floccosum: −1.19, T. interdigitale: −1.18, T. mentagrophytes: −0.98 T. rubrum: −0.78, and T. tonsurans: −0.28. Pairwise distances of the combination of the seven loci (based on an overall average) to resolve the boundaries between the Trichophyton species are shown in Supplementary Table 3, whereas the sequence homology between the T. interdigitale and T. mentagrophytes species was noted to be greater than 99.999%. A comparison of these numbers indicates that these two species belong to a complex. The second-most similarities were observed between the species of T. mentagrophytes/T. interdigitale complex with T. tonsurans, and the most distant species were found to be T. rubrum.
By Fisher’s exact test, no significant association was found between ST and the anatomical source. Also, phylogenetic analysis of each locus did not show any statistically significant variation across clades, anatomical sources, and demographic information.
Antifungal Susceptibility Testing
The results of antifungal susceptibility testing for the 99 isolates of dermatophytes against four antifungals are shown in Table 4. Overall, the lowest MIC were observed for terbinafine > butenafine > lanoconazole > griseofulvin, respectively. Terbinafine and griseofulvin had the lowest and highest geometric mean MICs which were 0.01 and 1.23 μg/ml for T. interdigitale and E. floccosum, respectively.
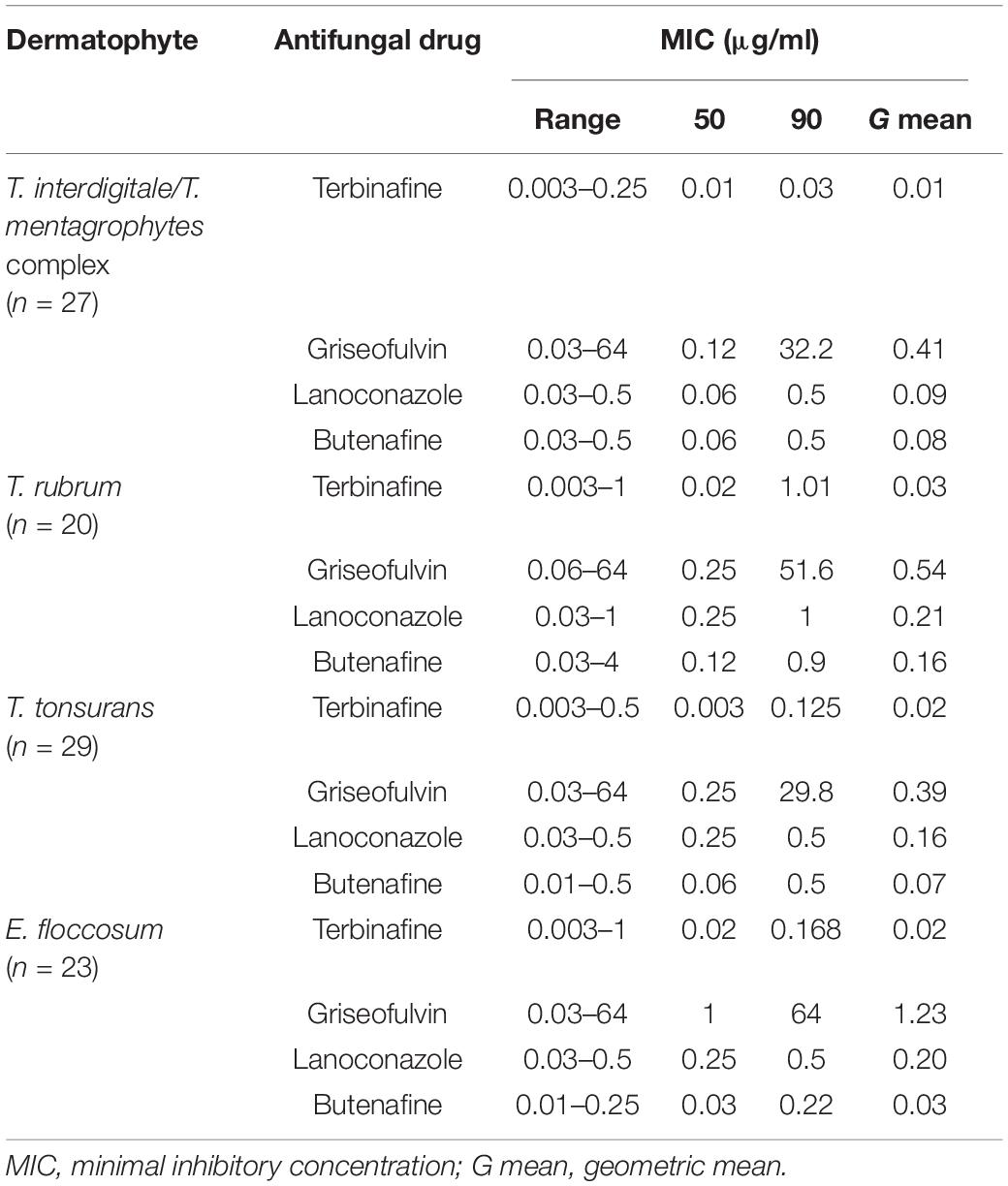
Table 4. Susceptibility of 95 clinical isolates and four reference strains of dermatophytes to four antifungal agents.
Discussion
Multilocus sequence typing is one of the widely used methods for epidemiological studies and the evolution of microorganisms. Accordingly, in this study, a new MLST scheme was developed based on sequence polymorphisms of seven housekeeping genes BT2, TEF-1α, CaM, HSP70, ACT, ITS, and D1/D2 of the five major species of dermatophytes. One of the objectives of this study was to cast a light on the genetic diversity of major species of dermatophytes to use MLST. These techniques have shown a low intraspecific genetic diversity among isolates of T. rubrum, T. tonsurans, and E. floccosum. The 64 genotypes obtained from 99 strains identified by MLST (Supplementary Table 2) were grouped in 34 STs. A few alleles were found in all the genes, except for the HSP70 that had only one genotype. As confirmed by similar studies (Makimura et al., 1999; Persinoti et al., 2018; Zhan et al., 2018), very few genetic changes were observed in the current study. Low levels of polymorphism may be due to the following: (i) the mutation rates in dermatophyte species and (ii) the species recent emergence from a genotype(s).
Species defined by molecular studies do not always correspond to the existing concepts obtained by the ecological and clinical principles. T. interdigitale and T. mentagrophytes share similar morphological characteristics (macro/microscopic features). In the study of de Hoog et al. (2017), four gene regions were sequenced (β-tubulin, ITS, and LSU loci of the rDNA, and the ribosomal 60S protein). Outcomes of the aforementioned study showed a new taxonomy for dermatophytes. T. interdigitale only included the anthropophilic species, and zoophilic T. interdigitale isolates belong to the species T. mentagrophytes. The study of Pchelin et al. (2016) on sequencing four T. mentagrophytes genomes showed that T. mentagrophytes and T. interdigitale belong to the same phylogenetic species. In this study, according to the overall average obtained from intraspecies and interspecies pairwise distances of combination concerning the seven loci (Supplementary Table 3), T. interdigitale and T. mentagrophytes were regarded as species complexes which may be affected by the epigenetic change during the localization on the animal and human body. Furthermore, they likely share a common ancestor and T. interdigitale species are descendants of a T. mentagrophytes species. This study confirms the results of the study undertaken by Pchelin et al. (2016).
Whole-genome sequencing of T. rubrum and T. violaceum showed that a high difference between the two species is found in adhesion genes, which can be the reason for the different localizations on the human body (Zhan et al., 2018). Analysis of sequencing 99 isolates in this study and 22 sequences deposited in the GenBank confirms that these two species belong to the same complex, and T. violaceum is derived from T. rubrum. Also, the phylogenetic analyses and pairwise distances of the combination of the three loci showed that T. tonsurans and T. equinum could be regarded as a species complex, where T. equinum is derived from T. tonsurans.
The phylogenetic tree constructed from the combination of the three genes shows a new topological pattern that confirms the derivation of the anthropophilic and zoophilic genera from the geophilic genus. Besides that, it is possible that Epidermophyton was derived from Nannizzia and has lost the ability to produce microconidia over time. Analysis of multilocus datasets showed that the name change from Microsporum racemosum to Microsporum cookei was performed correctly (high support: 100).
Ahmadi et al. (2016) found one CaM genotype among T. tonsurans and E. floccosum isolates and two genotypes among T. rubrum, while among isolates of T. interdigitale, four genotypes were observed. In the present study, similar to the study of Ahmadi et al. (2016), the most genotype was found in T. interdigitale strains. Also, T. rubrum, T. tonsurans, and E. floccosum species did not follow the intraspecies variation. These results can be affected by the number and isolate examined. In a similar vein, the study conducted by Persinoti et al. (2018) confirms the results of this study. The study led by Li et al. (2008) showed that when the D1/D2 and ITS regions were used, most of the species identified wrongly as the T. mentagrophytes was reported to be T. interdigitale, which supports the findings of the current study.
Another purpose of this study was to evaluate the genes used for the MLST approach. In the present experiment, the housekeeping gene, which has fewer genotypes, is not suitable for MLST, such as the HSP70, D1/D2, and CaM genes. The study of Zhan et al. (2018) showed that TUB2 and RP 60S L1 genes share average performances, while LSU and TEF3 have a very poor performance. The results of the current research work showed that BT2, TEF-1α, and ITS genes are suitable genes for MLST.
In the present study, the highest frequency of ITS genotypes was related to genotypes II and II∗ of T. interdigitale and T. mentagrophytes, respectively. Also, two new genotypes XXVII and XXVIII were introduced for T. interdigitale and T. mentagrophytes. Another objective of this study was to determine the ST dermatophyte species. The present study was the first to focus on the determination of the ST dermatophyte species. The determination of genotype and ST is useful and important in epidemiological studies of the dermatophyte species. Considering that the relationship between STs and taxa was seen in the combination of three genes (ITS, BT2, and TEF-1α), it seems that the combination of these three genes to determine ST is more appropriate than the combination of seven genes used in this study. Epidemiology studies to improve the basic knowledge of dermatophyte, knowledge into the how of spreading, and understanding the major clones reported from different parts of the world as well as finding the ancestry of isolates can be put into practice. Previous studies on the fungal species gave conflicting reports about the relationship between the genotype and different anatomical sources of the isolates or genotype and the antifungal susceptibility pattern (Odds et al., 2007; Amanloo et al., 2017; Taghipour et al., 2019). A study conducted by Taghipour concerning the determination of the T. mentagrophytes/T. interdigitale genotype belonging to the rDNA ITS regions showed that there was an association between the geographic locations and clinical presentations (Taghipour et al., 2019). In the present study, such a relationship was not observed due to low genotypes diversity and low number of T. mentagrophytes species. In the current study, according to the Fisher’s exact test, no statistically significant association was found between the anatomical source and demographic information with STs and genotypes. Interestingly, genotypic variation was observed for multiple terbinafine-susceptible isolates; therefore, genotypic variation may not always be accompanied by resistance development. Besides, no statistically significant association was found between the antifungal drug susceptibility profile and demographic information with STs and genotypes. However, these associations may have been influenced by confounding variables, such as the number of species, in the determination of STs in epidemiological studies.
The eBURST analysis was performed for all species. Studies of this type are scarce and there is no database for MLST dermatophytes; thus, it was not feasible to compare the genotypes in the current study with those of the other studies. This study was a new starting point to determine the ST and foundation for a dermatophyte MLST database.
A synonymous substitution is a nucleotide mutation which does not alter the amino acid sequence (silent mutation), while in a non-synonymous substitution, a nucleotide mutation manipulates the amino acid sequence of a protein and biological change in the organism. Low ratio of dN/dS indicates that mutations occur at the critical points and these changes lead to a negative or purifying selection. It is believed that the variability among non-coding sequences was higher than that of the coding sequences, but it is striking to note that in the species that were studied, dS occurred more frequently than dN and the ratio of dN/dS was below 1. Considering that, in this study, half of the nucleotide changes led to amino acid changes (16 of 32), it seems that these numbers are justifiable.
According to our results, the MIC of griseofulvin against 99 dermatophyte isolates was in the range of 0.03–64 μg/ml. T. tonsurans was the most susceptible species to griseofulvin (MIC50 0.25 μg/ml). GRI showed the reduced susceptibility to E. floccosum (MIC50, 1 μg/ml). Among 99 tested isolates, three showed a reduced terbinafine susceptibility. Also, 5% of those isolates were cross-resistant to terbinafine and griseofulvin.
The limitations of our study were the retrospective nature of the analysis followed by the lack which is due to underestimation of fungal-related infections in Iran. Overall, the information gathered from seven genes analysis suggests that they were excellent phylogenetic markers for the species level of dermatophytes, although the CaM gene was unable to separate the T. mentagrophytes/T. interdigitale complex from T. tonsurans species in the dendrogram. The most suitable genes for MLST of major pathogenic dermatophytes are the TEF-1α, BT2, and ITS genes. Also, two new ITS genotypes were found, which had no matches in the GenBank dataset. Besides, data obtained from MLST clarify the status of species: (i) species that are closely related genetically, such as the T. rubrum/T. violaceum and T. equinum/T. tonsurans were regarded as a species complex. (ii) It is possible that Epidermophyton was derived from Nannizzia and then failed to produce microconidia. (iii) The most informative locus was TEF-1α.
Considering that there is no database for MLST dermatophytes, it seems necessary to study another gene with a high polymorphism to select the genes used for MLST.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
MS-G conceived, designed, and supervised the study. ZS performed the experiments and contributed to the analysis of the data. MR-A contributed to the analysis of the data. All authors prepared and wrote the manuscript.
Funding
This work was financially supported by the Research Deputy of Tarbiat Modares University and Iran National Science Foundation: INSF for funding our research (Grant No. 94003277).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.643509/full#supplementary-material
Footnotes
References
Ahmadi, B., Mirhendi, H., Makimura, K., de Hoog, G. S., Shidfar, M. R., Nouripour-Sisakht, S., et al. (2016). Phylogenetic analysis of dermatophyte species using DNA sequence polymorphism in calmodulin gene. Med. Mycol. 54, 500–514. doi: 10.1093/mmy/myw004
Amanloo, S., Shams-Ghahfarokhi, M., Ghahri, M., and Razzaghi-Abyaneh, M. (2017). Genotyping of clinical isolates of Candida glabrata from Iran by multilocus sequence typing and determination of population structure and drug resistance profile. Med. Mycol. 56, 207–215. doi: 10.1093/mmy/myx030
Anzawa, K., Kawasaki, M., Hironaga, M., and Mochizuki, T. (2011). Genetic relationship between Trichophyton mentagrophytes var. interdigitale and Arthroderma vanbreuseghemii. Med. Mycol. J. 52, 223–227. doi: 10.3314/mmj.52.223
Bain, J. M., Tavanti, A., Davidson, A., Jacobsen, M. D., Shaw, D., Gow, N. A., et al. (2007). Multilocus sequence typing of the pathogenic fungus Aspergillus fumigatus. J. Clin. Microbiol. 45, 1469–1477. doi: 10.1128/jcm.00064-07
Bernhardt, A., Sedlacek, L., Wagner, S., Schwarz, C., Würstl, B., and Tintelnot, K. (2013). Multilocus sequence typing of Scedosporium apiospermum and Pseudallescheria boydii isolates from cystic fibrosis patients. J. Cyst. Fibros 12, 592–598. doi: 10.1016/j.jcf.2013.05.007
Bougnoux, M., Morand, S., and d’Enfert, C. (2002). Usefulness of multilocus sequence typing for characterization of clinical isolates of Candida albicans. J. Clin. Microbiol. 40, 1290–1297. doi: 10.1128/jcm.40.4.1290-1297.2002
Cafarchia, C., Iatta, R., Latrofa, M. S., Graser, Y., and Otranto, D. (2013). Molecular epidemiology, phylogeny and evolution of dermatophytes. Infect. Genet. Evol. 20, 361–351.
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–775. doi: 10.1038/nmeth.2109
de Hoog, G. S., Dukik, K., Monod, M., Packeu, A., Stubbe, D., Hendrickx, M., et al. (2017). Toward a novel multilocus phylogenetic taxonomy for the dermatophytes. Mycopathol 182, 5–31. doi: 10.1007/s11046-016-0073-9
Drummond, A. J., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/jb.186.5.1518-1530.2004
Graser, Y., Scott, J., and Summerbell, R. (2008). The new species concept in dermatophytes-a polyphasic approach. Mycopathol 166, 239–256. doi: 10.1007/s11046-008-9099-y
Heidemann, S., Monod, M., and Graser, Y. (2010). Signature polymorphisms in the internal transcribed spacer region relevant for the differentiation of zoophilic and anthropophilic strains of Trichophyton interdigitale and other species of T. mentagrophytes sensu lato. Br. J. Dermatol. 162, 282–295. doi: 10.1111/j.1365-2133.2009.09494.x
Hunter, P. R. (1990). Reproducibility and indices of discriminatory power of microbial typing methods. J. Clin. Microbiol. 28, 1903–1905. doi: 10.1128/jcm.28.9.1903-1905.1990
Kac, G. (2000). Molecular approaches to the study of dermatophytes. Med. Mycol. 38, 329–336. doi: 10.1080/mmy.38.5.329.336
Kumar, S., Tamura, K., Jajobsen, I. B., and Nei, M. (2001). MEGA: molecular evolutionary genetic analysis software. Bioinformatics 17, 1244–1245.
Kurtzman, C. P., and Robnett, C. J. (1997). Identification of clinically important ascomycetous yeasts based on nucleotide divergence in the 59 end of the large-subunit (26s) ribosomal DNA gene. J. Clin. Microbiol. 35, 1216–1223. doi: 10.1128/jcm.35.5.1216-1223.1997
Li, H. C., Bouchara, J. P., Hsu, M. M. L., Barton, R., Su, S., and Chang, T. C. (2008). Identification of dermatophytes by sequence analysis of the rRNA gene internal transcribed spacer regions. J. Med. Microbiol. 57, 592–600. doi: 10.1099/jmm.0.47607-0
Makimura, K., Tamura, Y., Mochizuki, T., Hasegawa, A., Tajiri, Y., Hanazawa, R., et al. (1999). Phylogenetic classification and species identification of dermatophyte strains based on DNA sequences of nuclear ribosomal internal transcribed spacer 1 regions. J. Clin. Microbiol. 37, 920–924. doi: 10.1128/jcm.37.4.920-924.1999
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES science gateway for inference of large phylogenetic trees,” in Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), (New Orleans, LA: IEEE).
Mirhendi, H., Makimura, K., de Hoog, G. S., Rezaei-Matehkolaei, A., Najafzadeh, M. J., Umeda, Y., et al. (2014). Translation elongation factor 1-α gene as a potential taxonomic and identification marker in dermatophytes. Med. Mycol. 53, 215–224. doi: 10.1093/mmy/myu088
Mirhendi, H., Nishiyama, Y., Rezaei-Matehkolaei, A., Satoh, K., and Makimura, K. (2016). The first case of onychomycosis in a koala (Phascolarctos cinereus) due to atypical isolates of Microsporum gypseum, a diagnostic challenge. Curr. Med. Mycol. 2, 45–50.
Mochizuki, T., Takeda, K., and Anzawa, K. (2016). Molecular markers useful for intraspecies subtyping and strain differentiation of dermatophytes. Mycopathol 182, 57–65. doi: 10.1007/s11046-016-0041-4
Nei, M., and Gojobori, T. (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426.
Odds, F. C., Bougnoux, M., Shaw, D., Bain, J. M., Davidson, A. D., Diogo, D., et al. (2007). Molecular phylogenetics of Candida albicans. Eukaryot. Cell 6, 1041–1052.
O’Donnell, K., Sutton, D. A., Rinaldi, M. G., Magnon, K. C., Cox, P. A., Revankar, S. G., et al. (2004). Genetic diversity of human pathogenic members of the Fusarium oxysporum complex inferred from multilocus dna sequence data and amplified fragment length polymorphism analyses: evidence for the recent dispersion of a geographically widespread clonal lineage and nosocomial origin. J. Clin. Microbiol. 42, 5109–5120. doi: 10.1128/jcm.42.11.5109-5120.2004
Pchelin, I. M., Zlatogursky, V. V., and Rudneva, M. V. (2016). Reconstruction of phylogenetic relationships in dermatomycete genus Trichophyton Malmsten 1848 based on ribosomal Internal Transcribed Spacer region, partial 28S rRNA and beta-tubulin genes sequences. Mycoses 59, 566–575. doi: 10.1111/myc.12505
Persinoti, G. F., Martinez, D. A., Li, W., Döðen, A., Billmyre, R. B., Averette, A., et al. (2018). Whole-genome analysis illustrates global clonal population structure of the ubiquitous dermatophyte pathogen Trichophyton rubrum. Genetics 208, 1657–1669. doi: 10.1534/genetics.117.300573
Probst, S., de Hoog, G. S., and Graser, Y. (2002). Development of DNA markers to explore host shifts in dermatophytes. Stud. Mycol. 47, 57–74.
Rezaei-Matehkolaei, A., Mirhendi, H., Makimura, K., de Hoog, G. S., Satoh, K., Najafzadeh, M. J., et al. (2014). Nucleotide sequence analysis of beta tubulin gene in a wide range of dermatophytes. Med. Mycol. 52, 674–688. doi: 10.1093/mmy/myu033
Ronquist, F., Teslenko, M., Mark, P. V. D., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Sadeghi, G., Abouei, M., Alirezaee, M., Toloueia, R., Shams-Ghahfarokhib, M., Mostafavic, E., et al. (2011). A 4-year survey of dermatomycoses in Tehran from 2006 to 2009. J. Mycol. Med. 21, 260–265. doi: 10.1016/j.mycmed.2011.10.001
Spratt, B. G., Hanage, W. P., Li, B., Aanensen, D. M., and Feil, E. J. (2004). Displaying the relatedness among isolates of bacterial species - the eBURST approach. FEMS. Microbiol. Lett. 241, 129–134. doi: 10.1016/j.femsle.2004.11.015
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Summerbell, R. C., Haugland, R. A., Li, A., and Gupta, A. K. (1999). rRNA gene internal transcribed spacer 1 and 2 sequences of asexual, anthropophilic dermatophytes related to Trichophyton rubrum. J. Clin. Microbiol. 37, 4005–4011. doi: 10.1128/jcm.37.12.4005-4011.1999
Susca, A., Perrone, G., Cozzi, G., Stea, G., Logrieco, A. F., and Mulè, G. (2013). Multilocus sequence analysis of Aspergillus Sect. Nigri in dried vine fruits of worldwide origin. Int. J. Food Microbiol. 165, 163–168. doi: 10.1016/j.ijfoodmicro.2013.04.027
Swofford, D. L. (2002). PAUP∗: Phylogenetic Analysis Using Parsimony (∗ and Other Methods), Version 4.0b10. Sunderland, MA: Sinauer Associates.
Taghipour, S., Pchelin, I. M., Zarei-Mahmoudabadi, A., Ansari, S., Katiraee, F., Rafiei, A., et al. (2019). Trichophyton mentagrophytes and T. interdigitale genotypes are associated with particular geographic areas and clinical manifestations. Mycoses 62, 1084–1091. doi: 10.1111/myc.12993
Vanhee, L. M. E., Nelis, N. J., and Coenye, T. (2010). What can be learned from genotyping of fungi? Med. Mycol. 48, 60–69.
Wayne, P. A. (2008). CLSI, Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi. Approved Standard: CLSI document M38–A2, 2nd Edn. Wayne, PA: Clinical and Laboratory Standards Institute.
White, T. J., Bruns, T., Lee, S., and Taylor, W. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: A Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfand, J. J. Sninsky and T. J. White (Cambridge, MA: Academic Press), 18, 315–322. doi: 10.1016/b978-0-12-372180-8.50042-1
Zamani, S., Sadeghi, G., Yazdinia, F., Moosa, H., Pazooki, A., Ghafarinia, Z., et al. (2016). Epidemiological trends of dermatophytosis in Tehran, Iran: a five-year retrospective study. J. Mycol. Med. 26, 351–358. doi: 10.1016/j.mycmed.2016.06.007
Zarrin, M., Salehi, Z., and Zarei Mahmoudabadi, A. (2015). Identification of dermatophytes by arbitrarily primed PCR. Asian Biomed. 9, 291–298.
Keywords: dermatophytes, dermatophytosis, molecular epidemiology, antifungal susceptibility, genetic diversity, multilocus sequence typing
Citation: Salehi Z, Shams-Ghahfarokhi M and Razzaghi-Abyaneh M (2021) Molecular Epidemiology, Genetic Diversity, and Antifungal Susceptibility of Major Pathogenic Dermatophytes Isolated From Human Dermatophytosis. Front. Microbiol. 12:643509. doi: 10.3389/fmicb.2021.643509
Received: 18 December 2020; Accepted: 19 April 2021;
Published: 04 June 2021.
Edited by:
Timoleon-Achilleas Vyzantiadis, Aristotle University of Thessaloniki, GreeceReviewed by:
Michael Arabatzis, Aristotle University of Thessaloniki, GreeceStéphane Ranque, Aix-Marseille Université, France
Copyright © 2021 Salehi, Shams-Ghahfarokhi and Razzaghi-Abyaneh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masoomeh Shams-Ghahfarokhi, shamsm@modares.ac.ir; mshamsgh@yahoo.com