
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 18 February 2021
Sec. Food Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.642999
 Ikram Ali1,2†Ke Liu2,3†Danfeng Long2Shah Faisal1
Ikram Ali1,2†Ke Liu2,3†Danfeng Long2Shah Faisal1 Mian Gul Hilal1Izhar Ali1
Mian Gul Hilal1Izhar Ali1 Xiaodan Huang2*
Xiaodan Huang2* Ruijun Long1*
Ruijun Long1*The structure and diversity of human gut microbiota are directly related to diet, though less is known about the influences of ethnicity and diet-related behaviors, such as fasting (intermittent caloric restriction). In this study, we investigated whether fasting for Ramadan altered the microbiota in Chinese and Pakistani individuals. Using high-throughput 16S rRNA gene sequencing and self-reported dietary intake surveys, we determined that both the microbiota and dietary composition were significantly different with little overlap between ethnic groups. Principal Coordinate Analyses (PCoA) comparison of samples collected from both groups before and after fasting showed partial separation of microbiota related to fasting in the Pakistani group, but not in the Chinese group. Measurement of alpha diversity showed that Ramadan fasting significantly altered the coverage and ACE indices among Chinese subjects, but otherwise incurred no changes among either group. Specifically, Prevotella and Faecalibacterium drove predominance of Bacteroidetes and Firmicutes in the Pakistani group, while Bacteroides (phylum Bacteroidetes) were the most prevalent among Chinese participants both before and after fasting. We observed significant enrichment of some specific taxa and depletion of others in individuals of both populations, suggesting that fasting could affect beta diversity. Notably, Dorea, Klebsiella, and Faecalibacterium were more abundant in the Chinese group after fasting, while Sutterella, Parabacteroides, and Alistipes were significantly enriched after fasting in the Pakistani group. Evaluation of the combined groups showed that genera Coprococcus, Clostridium_XlV, and Lachnospiracea were all significantly decreased after fasting. Analysis of food intake and macronutrient energy sources showed that fat-derived energy was positively associated with Oscillibacter and Prevotella, but negatively associated with Bacteroides. In addition, the consumption of sweets was significantly positively correlated with the prevalence of Akkermansia. Our study indicated that diet was the most significant influence on microbiota, and correlated with ethnic groups, while fasting led to enrichment of specific bacterial taxa in some individuals. Given the dearth of understanding about the impacts of fasting on microbiota, our results provide valuable inroads for future study aimed at novel, personalized, behavior-based treatments targeting specific gut microbes for prevention or treatment of digestive disorders.
The human gut is populated by trillions of microbes which collectively work as a “hidden organ” (Harsch and Konturek, 2018), the composition and structure of which can shift dynamically under the influence of dietary changes, environment, genetics, medication, and lifestyle (Findley et al., 2016; Deschasaux et al., 2018). In addition, several previous studies have identified differences in microbiota profiles across various ethnic groups, potentially driven by host genetics, geographic separation, and markedly distinct lifestyles (Goodrich et al., 2014; Gupta et al., 2017). Yatsunenko et al. (2012) also found that the composition of fecal microbiota differed significantly among United States residents, Amerindian, and Malawian individuals from geographically separated locations, primarily due to distinct lifestyle habits and the contents of their diet.
In fact, diet is well-established as the main factor affecting microbiota structure by enriching for specific species and their metabolic functions (Claesson et al., 2012; Kolodziejczyk et al., 2019). For instance, cultures such as the Hadza people (hunter-gatherers in Peru) consume mostly raw or wild foods and, as a result, show notably higher gut microbe diversity than that observed in Western Urban populations (Obregon-Tito et al., 2015). Typically, rural populations consume greater proportions of foods containing a high fiber content, thus promoting the enrichment of microbial taxa such as Bacteroidetes (e.g., Xylanibacter and Prevotella) and reducing the prevalence of other, such as Firmicutes (De Filippo et al., 2010). Studies in humans have indicated that variations in dietary macro-nutrients, i.e., proteins, fats, and carbohydrates (CHO), can significantly affect the shape of their gut microbiota (Wu et al., 2011; David et al., 2014), while other study have suggested diet-related behaviors, such as fasting for religious purposes, can also contribute to the structure of gut microbiota (Ozkul et al., 2020).
As a widely practiced diet-related behavior, fasting is defined as partial or complete abstention from food intake. Ramadan fasting is a religious practice where practitioners of the Muslim faith undergo a yearly ritual in which no food or beverages are consumed between sunrise to sunset for 29–30 days (Aybak, 1996). In mice given low-fat or high-fat diets, a dietary schedule in which food intake is decreased can trigger changes in microbiota structure as well as urinary and serum metabolic profiles (Zhang et al., 2013). Furthermore, in humans, short-term (28 days) restriction of CHO can result in decreased numbers of butyrate-producing bacteria, as well as butyrate (Duncan et al., 2007). Another study found that calorie restriction over a 10 weeks period can lead to variations in microbiome composition, including enrichment for Bacteroides and decreased abundance of Blautiacoccoides (Santacruz et al., 2009). By contrast, extended caloric restriction (for 12 months) resulted in decreased Actinobacteria and increased Bacteroidetes content in feces (Ruiz et al., 2017).
Based on these reports, we examined the contributions of three factors, including fasting (diet-related behavior), ethnicity, and dietary intake on the structure and diversity of human gut microbiota. We hypothesized that, given a preponderance of evidence showing that diet is the primary driver of microbiota structure, fasting (intermittent caloric restriction) can negatively or positively affect the abundance specific taxa within the gut microbial community, in a manner potentially dependent on ethnicity. To test whether fasting thus deferentially affect the shape of gut microbiota in individuals of different ethnic backgrounds, we used high-throughput metagenomic sequencing and dietary surveys to evaluate the influence of fasting during Ramadan on fecal microbial diversity in two ethnic groups (Pakistani and Chinese). The study participants were not geographically separated and were demographically similar.
All procedures were done under the Medical Ethics Committee approval of the School of Public Health (GW-20171013). Detailed information forms of the survey were signed by all participants before sampling.
The inclusion criteria for participants were living in close regional proximity, practiced fasting, and ranged in age from 18 to 40 years old, furthermore, participants were psychologically and physically healthy. We excluded volunteers in case of (1) diseases, including gastrointestinal diseases, chronic diseases, anorexia nervosa, cachexia, (2) insufficiency of liver and kidney, (3) smoking, (4) drinking alcohol, (5) the use of antibiotics within last 3 months (Mesnage et al., 2019). According to our inclusion and exclusion criteria, 34 healthy adult participants, living in the city of Lanzhou and distributed across two ethnic groups (16 Chinese and 18 Pakistani) were recruited. All participants have attended Ramadan fasting from May 15, 2018, to June 15, 2018; both survey and fecal samples, before and after Ramadan fasting, were collected from the two groups. As a result, participants were distributed into six groups as (i) Chinese before fasting (CBF), (ii) Pakistani before fasting (PBF), (iii) Chinese after fasting (CAF), (iv) Pakistani after fasting (PAF), (v) total before fasting (TBF), and (vi) total after fasting (TAF). Comparisons across ethnic groups were categorized into ethnic groups before fasting (CBF vs. PBF) and ethnic groups after fasting (CAF vs. PAF); while fasting groups were compared among Chinese groups (CBF vs. CAF), Pakistan groups (PBF vs. PAF), and the total subject (TBF vs. TAF).
A questionnaire was established to acquire personal and other measurement information, such as family background and dietary habits, to further collect individualized data. As previously reported, a 3 days 24-h food dietary recall was defined for the consumption of total foods and beverages (Schröder et al., 2001). Selected foods included vegetables, grains, poultry meat, livestock meat, leaf vegetables, eggs, seafood, beans, fruits, dairy, nuts, milk, and sweets. This questionnaire was also used to assess the dietary record of the last 6 months for each individual. Participant background, age, gender, dietary habits and antibiotic usage were all noted before sampling. A software was developed by Peaking Union Medical college and West China Centre of Medical sciences (CDGSS3.0), to be used for nutritional analysis of each subject’s total energy and nutrition. Principal coordinate analysis (PCoA) was used to measure the beta diversity of food components in the diet of each ethnic group by R software (version 3.2.1).
Fecal samples were collected for all participants on the same day. Sample collection before fasting was conducted on the morning of May 15, 2018, while samples after fasting were retrieved on the morning of June 15, 2018. Samples were frozen in liquid nitrogen immediately upon receipt to maintain sample stability (regardless of collection date/time). Samples were further stored at −70°C for future experiments.
DNA extraction was performed according to the manufacturer’s instructions (QIAamp DNA Stool Mini Kit, QIAGEN, Hilden, Germany). An ND-2000 Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States) develop by the United States, was used to determine the DNA content. Electrophoresis on 1% agarose gel was performed to evaluate the size and integrity of DNA. Samples of DNA were stored at −20°C for further analyses.
A library of the 16S metagenomic sequencing was prepared according to Illumina guidelines. Library construction and amplification of bacterial 16S rRNA gene were performed by metagenomic sequencing. Respective 16S primers (Forward: CCTACGGGNGGCWGCAG; Reverse: GACTACHVGGGTATCTAATCC) were designed for the amplification of bacterial 16S rRNA gene (V3 and V4 variable region). The Thermal Cycler (ABI2720, Thermo Fisher Scientific) was utilized for the amplification of HiFi HotStar ReadyMix (2× KAPA), final reaction (25μl) includes microbial DNA using 2.5 μl of a template (5 ng/μl), 5 μl of respective primers (1 μM each), and 12.5 μl. The cycling parameters were as the following: (i) initial denaturation at 95°C for 3 min, (ii) 25 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and elongation at 72°C for 30 s, and (iii) final extension at 72°C for 5 min. The final PCR products were quantified with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, California, United States) and then purified according to the manufacturer’s protocol (AMPure XP beads, Beckman Coulter, Coulter, Brea, California, United States). DNA sequencing was conducted by the use of MiSeq Reagent Kit v3 (Illumina, San Diego, CA, United States). Raw reads were loaded into the European Nucleotide Archive under the succession number PRJEB382311.
Sequencing data were submitted to quality control and bioinformatics analysis accuracy. The original off-machine data filtering was improved to obtain high-quality data. For this, the software TrimGalore of (version 0.4.2) was used to remove terminal sequences below 20 bp, adapter sequences, and those shorter than 100 bp. FLASH2 software was also used to merge sequences from the splicing of paired-end sequences. Moreover, Mothur (version 1.41.1) was used to find and remove primer sequences. Lastly, USEARCH software (version 10.0) was utilized to remove sequences with less than 100 bp and an error rate higher than 2. Representative sequences were classified according to the Database Project of Ribosome (Cole et al., 2009). Upon improving data quality, more than 97% of similar clusters were added into operational taxonomic units (OTUs). Mothur software was again used for alpha diversity analysis (including Chao1, observed species, ACE, Shannon, Simpson, and coverage) among groups. To assess the beta diversity, principal coordinate analysis (PCoA) based on Bray-Curtis distances was performed by R software (version 3.2.1). VENN analyses and rarefaction curves were calculated at the OTU level using the R (version 3.2.1) software (Colwell et al., 2012). Significant species among the different groups were observed by Linear discriminate analysis effect size (LEfSe) (Segata et al., 2011).
Statistical analysis was performed on alpha diversity, dietary, and taxonomic data. The Wilcoxon signed-rank test was applied to “before vs. after Ramadan fasting” comparisons (CBF vs. CAF, PBF vs. PAF, TBF vs. TAF) and the Mann-Whitney U test was applied to comparisons between ethnic groups (CBF vs. PBF, CAF vs. PAF). The R vegan package (permutations = 9,999) was used for permutational multivariate analysis of variance (PERMANOVA) and, therefore, to verify significant differences in the structure of gut microbiota (McArdle and Anderson, 2001). The R package was assessed to calculate the correlation of microbial genera with diet by Pearson’s correlation analysis. The SPSS version 23.0 was used for all statistical analyses.
PCoA analysis to identify differences in dietary profile revealed divergence among ethnic groups (CBF vs. PBF and CAF vs. PAF, Supplementary Figure S1), but considerable overlap in dietary intake within fasting groups (PBF vs. PAF, CBF vs. CAF, and TBF vs. TAF; Figure 1). The average daily food intake for fasting groups and the respective energy ratios and total energy provided by respective macro-nutrients (i.e., protein, CHO, and fat) are presented in Table 1. Within the Chinese fasting groups (Table 1), we observed a significant decrease in the intake of other vegetables and beans, whereas poultry intake increased after fasting (CBF vs. CAF). Within the Pakistani fasting group, grain intake and consumption of seafood, other vegetables, and vegetable supplements were all significantly increased after fasting (PBF vs. PAF). Regardless of ethnicity, the total population after fasting had a significantly higher poultry intake compared with their intake prior to fasting. The respective energy ratios showed that CHO provides the main source of energy, followed by fats, then proteins comprising the smallest proportion.
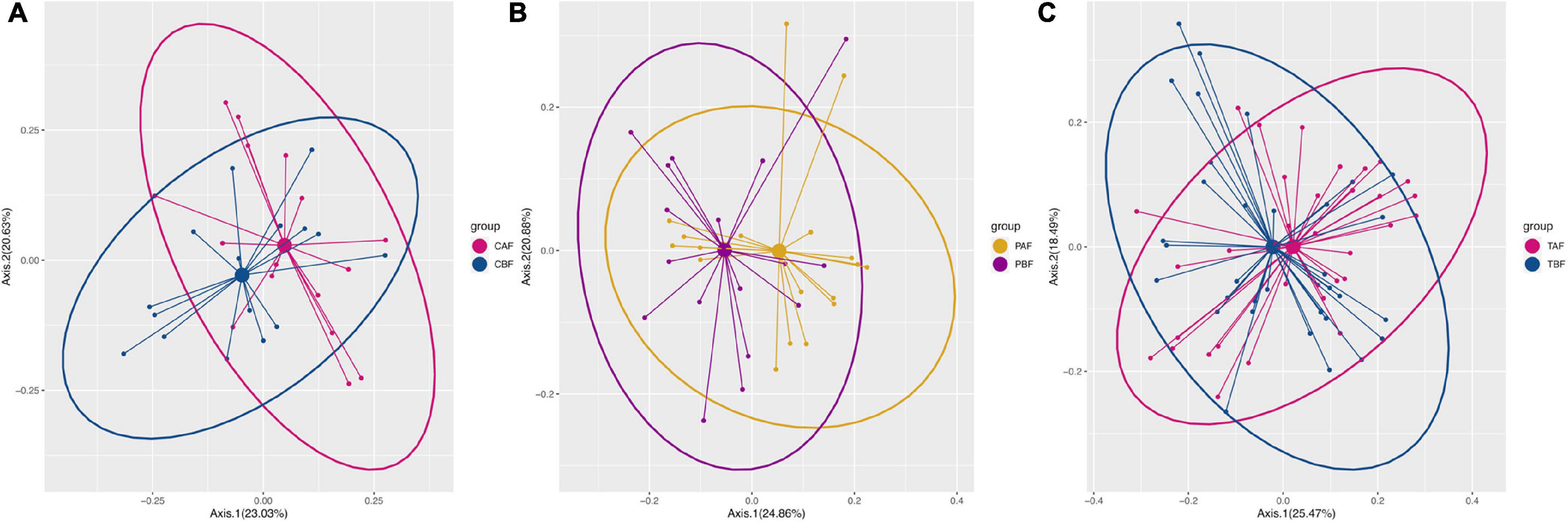
Figure 1. The nutrients intake profile by principal coordinate analysis (PCoA) of (A) Chinese before fasting vs. Chinese after fasting, (B) Pakistani before fasting vs. Pakistani after fasting, and (C) Total before fasting vs. Total after fasting groups. The variance presented by each component is written in brackets using Bray-Curtis.
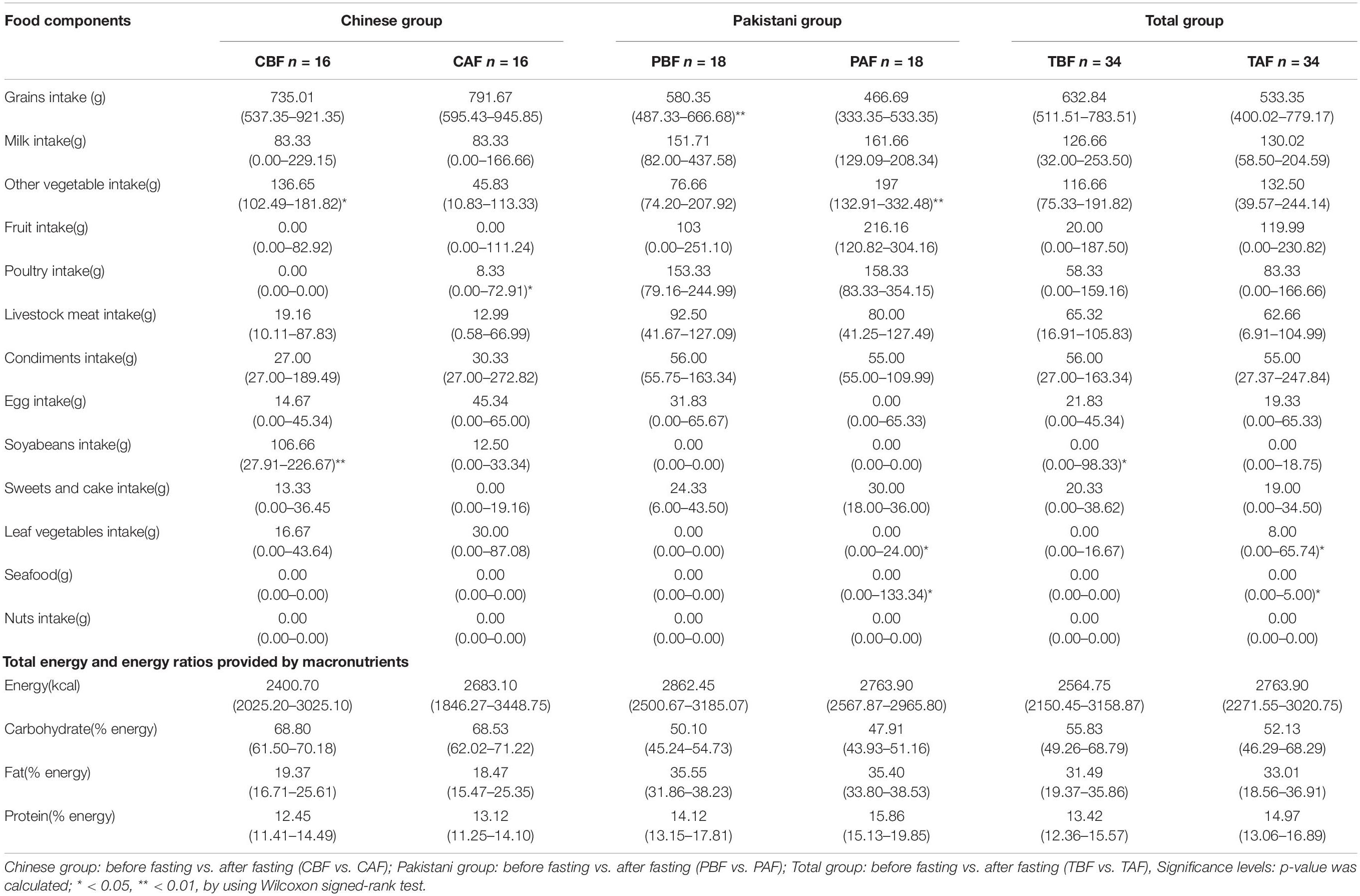
Table 1. Average daily food intake, total energy, and energy ratios provided by macronutrients across different Ramadan fasting groups.
The average daily food intake and the respective energy ratios for ethnic groups are presented in Supplementary Table S1, Before fasting, Chinese participants had a significantly higher intake of grains, soy beans, and leaf vegetables with lower intake of livestock meat, fruits, and poultry compared with that of the Pakistani participants. After fasting, the Chinese group presented significantly higher intake of grains and soybeans but lower intake of milk, other vegetables, fruits, livestock meat, poultry, sweets, and seafood compared with the Pakistani group. We also found that the proportions of energy sources consumed by each group were significantly different between the Chinese and Pakistani groups, in that Chinese consumed significantly more CHO, while the Pakistani group consumed significantly more fats and proteins.
A total of 6,944,362 raw reads were derived from 68 fecal samples provided by 34 subjects. Low-quality reads (accounting for 9.94% of raw reads) were filtered, resulting in 6,254,015 high-quality reads with a median length of 415 bp. On average, we obtained 91,970 high-quality reads per sample (minimum: 45,129, maximum: 161,753, SD: 27,905) after processing.
To identify the types and abundance of dominant bacterial taxa among the total cohort, we determined the phylum and genus level taxonomic assignments of OTUs from each sample (Supplementary Figure S2). The results showed that the most dominant phylum across group was Firmicutes (44%), followed by Bacteroidetes (43%), and Proteobacteria (11%) (Supplementary Figure S3A). The predominant genera were Prevotella (18%), Bacteroides (14%), Faecalibacterium (5%), and Dialister (4%) (Supplementary Figure S3B).
We next compared the prevalence of different taxa among fasting groups at the phylum and genus levels, and found a significant shift in these taxa before and after fasting for both ethnic groups. Specifically, within the Chinese cohort, the abundance of Bacteroidetes decreased, while Proteobacteria increased after fasting (CAF vs. CBF) (Figure 2A). Conversely, Bacteroidetes increased after fasting in the Pakistani group, and Firmicutes decreased (PAF vs. PBF) (Figure 2B). Comparisons among the full study cohort revealed a strongly significant increase in the abundance of Proteobacteria after fasting (TAF vs. TBF) (Figure 2C). At the genus level, Dorea, Klebsiella, and Faecalibacterium were all significantly enriched after fasting in comparisons between the CBF with CAF participants (Figure 3A). By contrast, Sutterella, Parabacteroides, and Alistipes were more abundant after fasting in the Pakistani group, while Coprococcus, Blautia, Eubacterium, Streptococcus, Romboutsia, and Dialister were more abundant before fasting (PAF vs. PBF) (Figure 3B). Notably, the shift in Coprococcus could be observed within the full cohort, since this genus, as well as Clostridium_XlVa and Lachnospiracea incertae sedis were all significantly more abundant prior to fasting in comparisons between fasting groups for total participants (TAF vs. TBF) (Figure 3C).
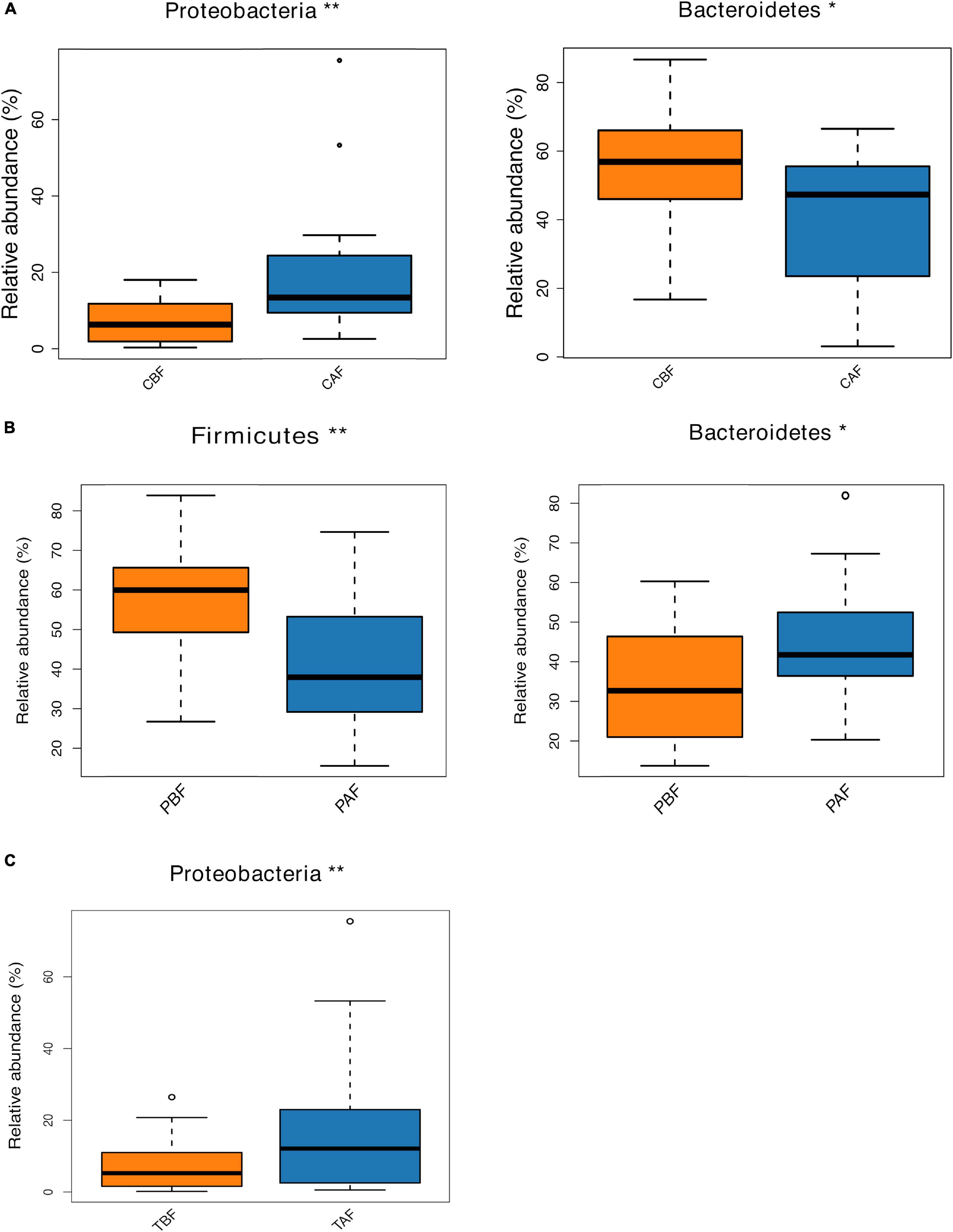
Figure 2. Relative abundances of bacterial phyla analyzed by using a metastats test was varied among each group at phylum level. (A) Chinese before fasting vs. Chinese after fasting, (B) Pakistani before fasting vs. Pakistani after fasting and (C) Total before fasting vs. Total after fasting; *P < 0.05, **P < 0.01.
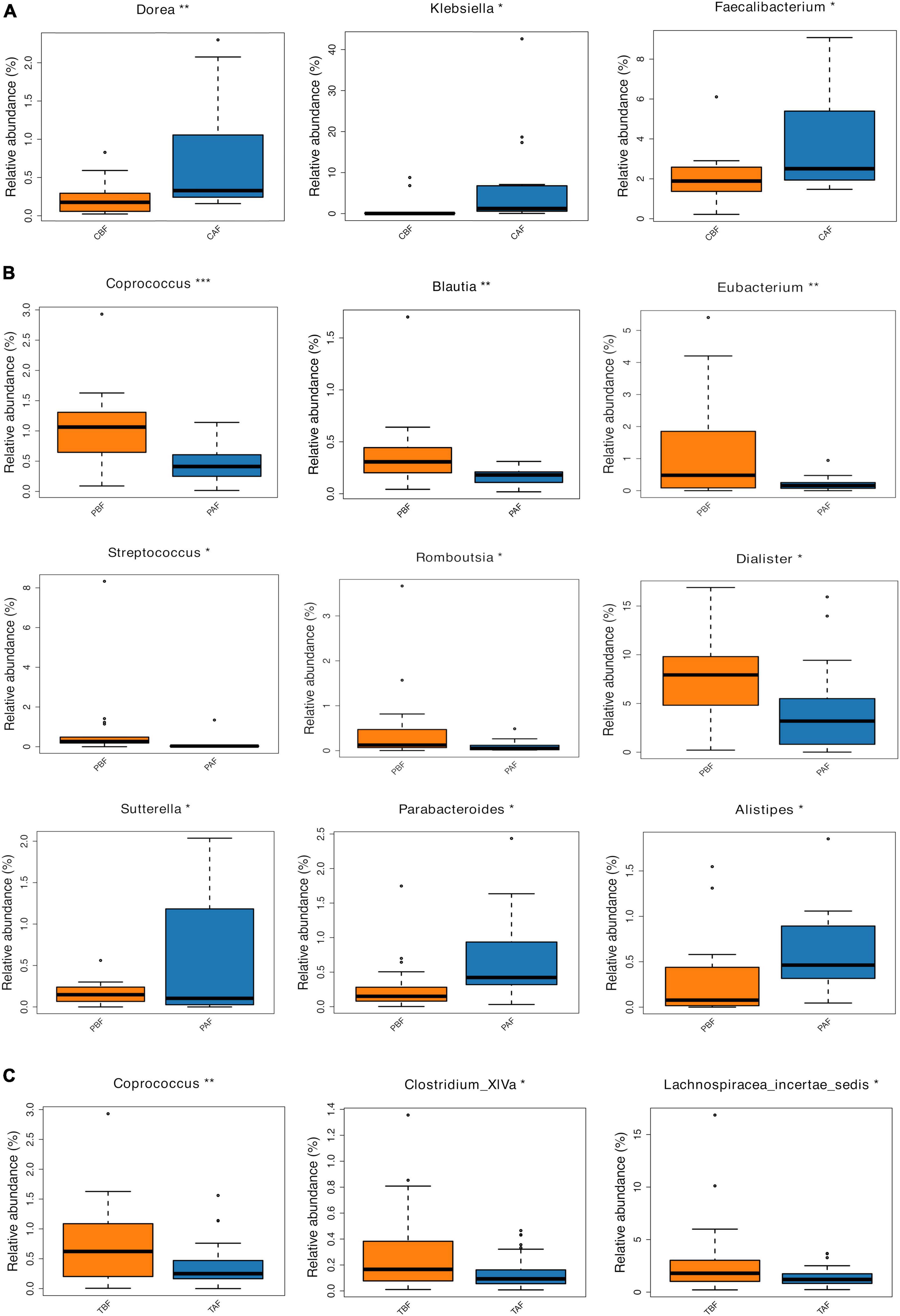
Figure 3. Relative abundances of bacterial taxa analyzed by using a metastats test was varied among each group at genus level. (A) Chinese before fasting vs. Chinese after fasting, (B) Pakistani before fasting vs. Pakistani after fasting, and (C) Total before fasting vs. Total after fasting; *P < 0.05, **P < 0.01, ***P < 0.001.
We next compared differences in the predominant phyla between ethnic groups before and after fasting. The results showed that before fasting, the Pakistani group presented a significantly higher abundance of Firmicutes and Actinobacteria compared to the Chinese group, while the Chinese participants had higher levels Bacteroidetes (Supplementary Figure S4A). Phylum level comparisons after fasting between ethnic groups indicated that Lentisphaerae and Tenericutes were significantly more abundant in the Pakistani samples (Supplementary Figure S4B). At the genus level, Several genera were enriched in Pakistani and Chinese groups, respectively (Supplementary Figures S5A,B).
To account for differences in distribution (species richness) in addition to abundance, we used the Chao1, observed species, ACE, Coverage, Shannon, and Simpson indices to compare alpha diversity of gut microbiota between fasting groups (CBF vs. CAF, PBF vs. PAF, TBF vs. TAF, Supplementary Figure S6) and between ethnic groups (CBF vs. PBF, CAF vs. PAF, Supplementary Figure S7). First, comparisons between fasting groups showed that among Chinese participants, the ACE was higher after fasting, while the coverage index was lower after fasting (CBF vs. CAF; Supplementary Figure S6A); other indices showed no significant differences between fasting groups within either ethnic group (CBF vs. CAF, PBF vs. PAF, TBF vs. TAF). However, comparisons of alpha diversity between ethnic groups revealed significantly higher OTU abundance indices in the Pakistani group (i.e., observed OTUs, Chao1, and ACE) than in the Chinese group, while the coverage index was higher in the Chinese group than in the Pakistani group before fasting, suggesting greater evenness in the distribution of taxa before fasting. However, the Shannon and Simpson indices showed no differences between ethnic groups (CBF vs. PBF and CAF vs. PAF, Supplementary Figure S7). The alpha diversity showed that ethnicity rather than fasting drives differences in alpha diversity as Pakistani group presented significantly higher OTU abundance indices than Chinese group.
To investigate differences in gut microbiota diversity between individual study subjects potentially attributable to ethnicity or fasting practices, we next performed PCoA, PERMANOVA test, Venn, and LEfSe analyses. To identify structural differences in gut microbiota all fasting group samples we conducted PCoA analysis using the Bray-Curtis model. This analysis showed that the microbial community composition shifted only slightly in Chinese fasting group after fasting (Figure 4A), whereas microbiota composition exhibited substantial divergence, with little overlap, between the before and after fasting groups of Pakistani participants (Figure 4B). This stability within the Chinese subjects before and after fasting was potentially reflected in the relative microbiota stability observed in comparisons between the total fasting groups (TAF vs. TBF) (Figure 4C). PCoA analysis comparing ethnic groups showed that ethnicity apparently drives substantial differences in microbiota structure, indicated by the lack of overlap between communities of different ethnicities both before (Supplementary Figure S8A) and after (Supplementary Figure S8B) fasting (CBF vs. PBF and CAF vs. PAF). Subsequent PERMANOVA tests further supported the significant differences between ethnic groups (CBF vs. PBF; CAF vs. PAF; P = 0.0001) and between the Pakistani fasting groups (PBF vs. PAF; P = 0.0129) (Supplementary Figure S9).
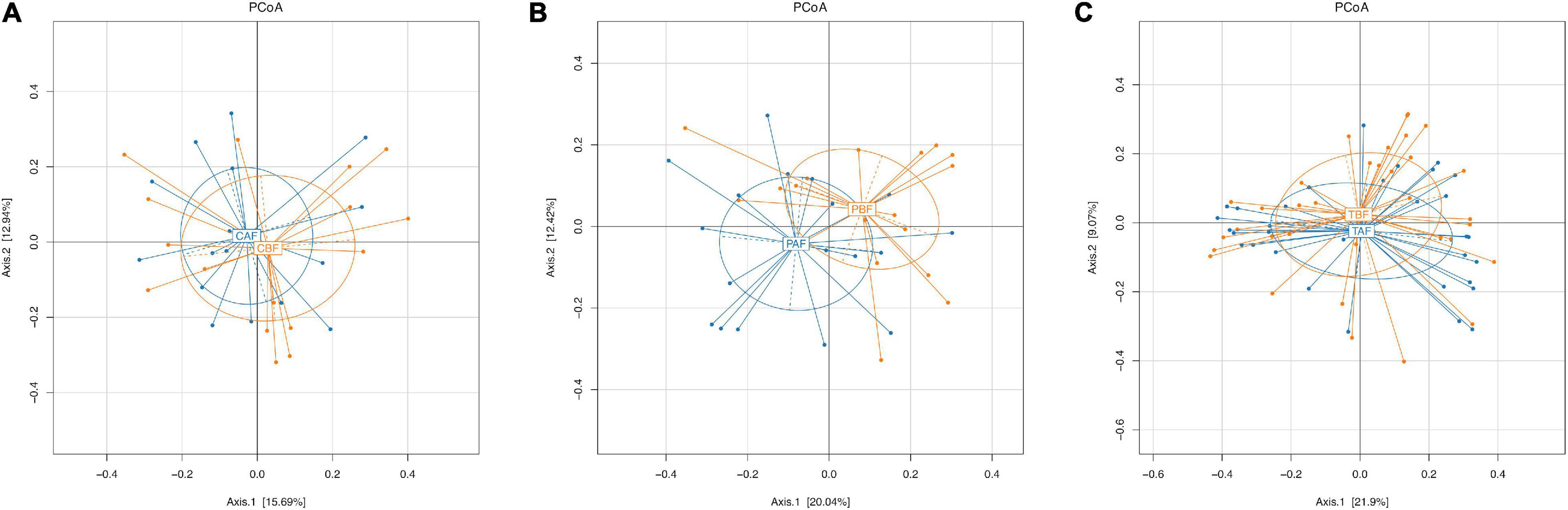
Figure 4. Principle coordinate analysis (PCoA) of the overall composition of the genera communities among the fasting groups. (A) Chinese before fasting vs. Chinese after fasting, (B) Pakistani before fasting vs. Pakistani after fasting, and (C) Total before fasting vs. Total after fasting groups. Each sample of respective groups were represented by a different colors symbol circles like CBF, PBF, TBF (orange circles), CAF, PAF, and TAF (blue circles). The percent of variation for each axis was explained and reported in square brackets using the Bray-Curtis.
We then performed LEfSe analysis further explore the structure of microbiota and found that several specific taxa were significantly different in their distribution between fasting groups (LDA > 3, P < 0.05, Figure 5). In particular, Bacteroidetes were higher in abundance before fasting among Chinese participants (LDA = 4.85), while Proteobacteria were enriched after fasting (LDA = 4.79), possibly reflecting the significantly higher abundance of Klebsiella after fasting (LDA = 4.42). By contrast, Firmicutes were enriched before fasting among Pakistani subjects (LDA = 4.91), while Bacteroidetes were significantly higher in abundance after fasting (LDA = 4.74). At the genus level, these changes were indicated by the significant prevalence of Dialister and Faecalibacterium OTUs before fasting (LDA = 4.23 and 4.31, respectively). The substantial shifts observed in the Chinese group appeared to be reflected in the total cohort, since Proteobacteria (LDA = 4.69), and Klebsiella in particular (LDA = 4.18), were significantly more abundant after fasting in TBF vs. TAF comparisons. Interestingly, genus Stomatobaculum was significantly enriched in the TBF group (LDA = 3.63).
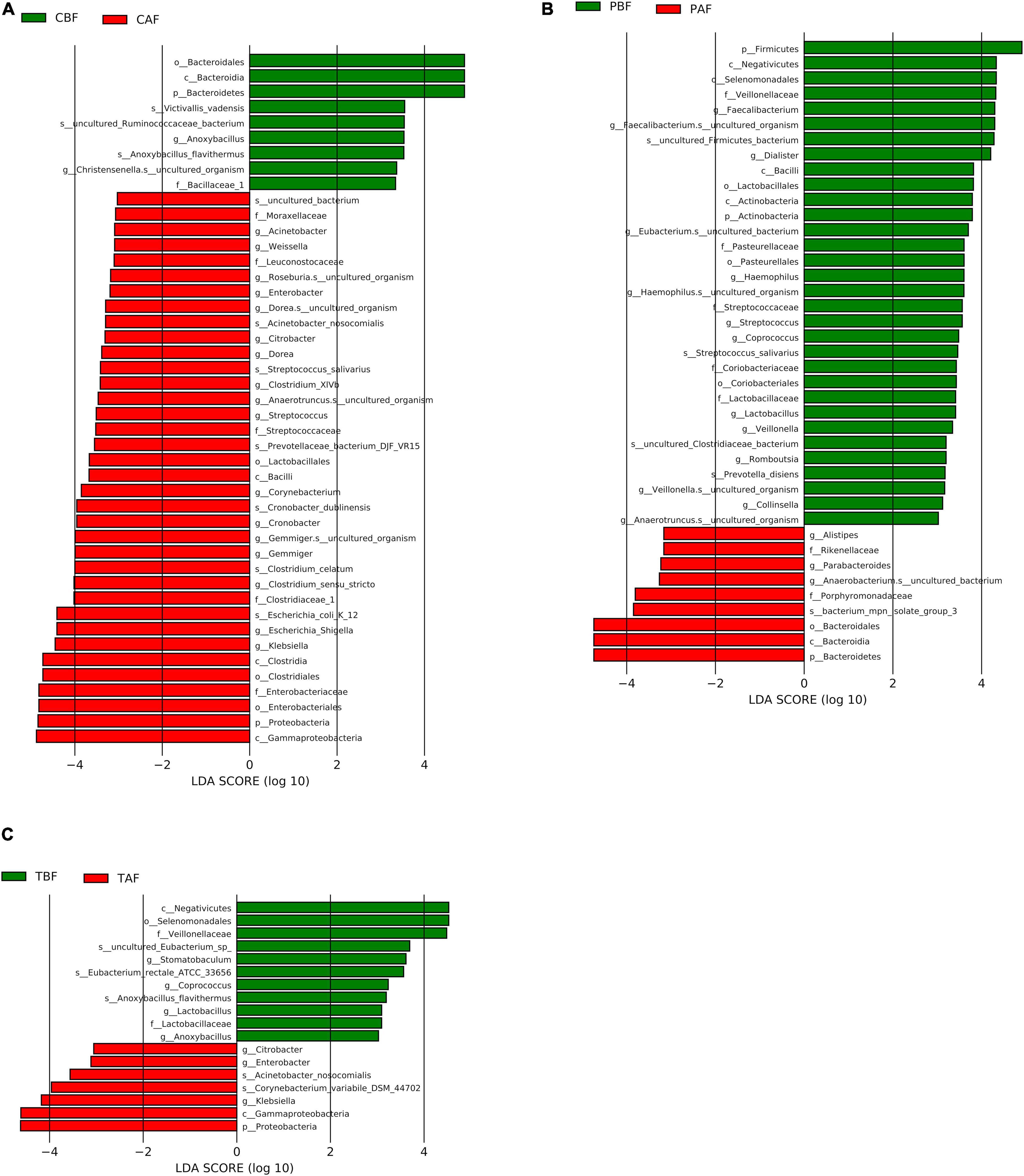
Figure 5. LEfSe analysis of signature taxa in the (A) Chinese before fasting vs. Chinese after fasting, (B) Pakistani before fasting vs. Pakistani after fasting, and (C) Total before fasting vs. Total after fasting groups. Linear discriminant analysis report represents the Prefixes abbreviations for the taxonomic rank of each taxon: phylum (p), class (c), order (o), family (f), genus (g), and species (s).
Further LEfSe analysis comparing ethnic groups showed the significant enrichment of several taxa (LDA > 3) (Supplementary Figures S10, S11). Before fasting, Firmicutes (LDA = 5.04), and especially Prevotella (LDA = 4.84) were found in high abundance among Pakistani subjects, and Bacteroidetes were significantly higher in the Chinese group (LDA = 5.03) potentially due to the high abundance of genus Bacteroides (LDA = 5.13). After fasting, Bacteroidetes remained dominant in the Chinese group (LDA = 4.98), while Prevotella and Succinivibrio were significantly enriched in the Pakistani group (LDA = 5.08 and 4.61, respectively).
Venn analysis showed that among the total 1,051 OTUs identified in all samples (Figure 6), 631 OTUs were shared between the Chinese before and after fasting groups, with 85 and 107 unique OTUs in the CBF and CAF groups, respectively. Similarly, 678 OTUs were shared between both Pakistani fasting groups, with 116 OTUs unique to the PBF group, and 80 OTUs unique to the PAF group. Venn comparisons of the total fasting groups showed that 871 OTUs were shared in between samples collected before and after fasting, with 105 OTUs unique to the TBF group and 75 OTUs unique to the TAF group. Venn analysis of ethnic differences showed that 534 OTUs were shared between the Chinese and Pakistani groups prior to fasting, while 182 were unique to the CBF group and 260 were unique to the PBF group (Supplementary Figure S12A). Similar overlap was observed in comparisons of ethnic groups after fasting, with 550 commonly shared OTUs, 188 unique to CAF, and 208 unique to PAF (Supplementary Figure S12B).
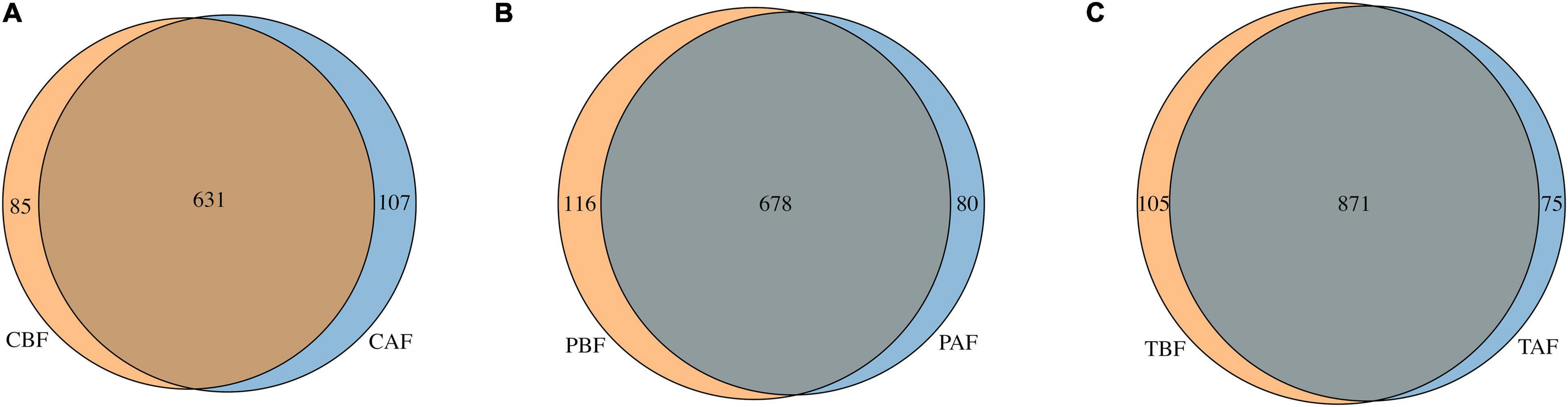
Figure 6. Venn diagram of unique and shared OTUs (operational taxonomic units) in different fasting groups. (A) Chinese before fasting vs. Chinese after fasting, (B) Pakistani before fasting vs. Pakistani after fasting and (C) Total before fasting vs. Total after fasting. The overlaps represent the common taxa between groups, and the non-overlapping portions represent unique taxa in each group.
To uncover whether latent relationships exist between the intake of specific nutrients sources and the composition of gut microbiota, we constructed a heatmap to illustrate correlations between bacterial taxa and diet. Pearson’s correlation test was used to examine the relationship between specific bacteria and respective nutrients (Figure 7).
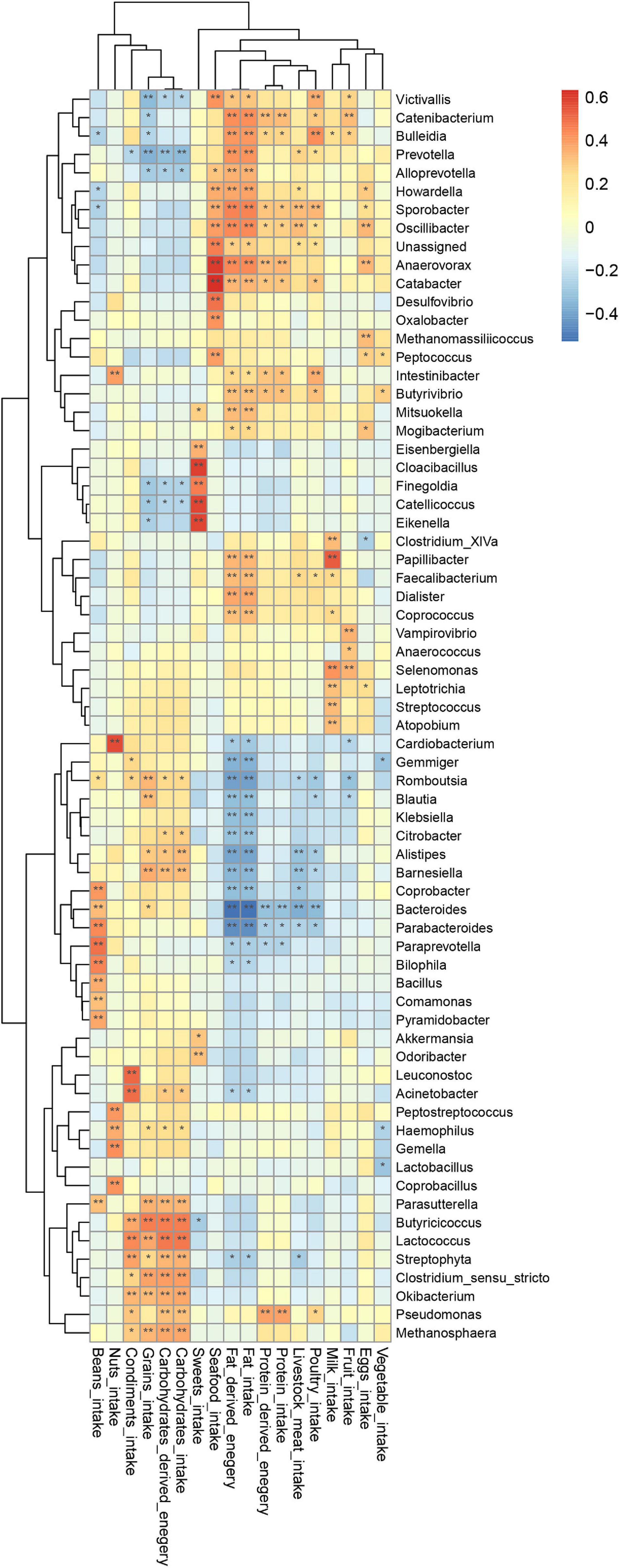
Figure 7. Correlation of genus with dietary intake; p-value was calculated using Pearson’s correlation; *P < 0.05, **P < 0.01.
This heatmap analysis showed that different microbial taxa were significantly associated with dietary intake, indicated by a Pearson’s correlation > 0.3 (Figure 5). In particular, CHO-derived energy was positively correlated with the abundance of Alistipes, Barnesiella, Clostridium sensu stricto, Parasutterella, Butyricicoccus, Streptophyta, Lactococcus, Okibacterium, Pseudomonas, and Methanosphaera (r = 0.31–0.46), but negatively correlated with Prevotella (r = −0.32). Fat-derived energy was positively associated with Victivallis, Coprococcus, Butyrivibrio, Faecalibacterium, Mitsuokella, Papillibacter, Catabacter, Alloprevotella, Dialister, Howardella, Bulleidia, Catenibacterium, Oscillibacter, Anaerovorax, Sporobacter, and Prevotella (r = 0.30–0.45), but was negatively associated with Bacteroides, Parabacteroides, Romboutsia, Alistipes, Gemmiger, Barnesiella, Coprobacter, Klebsiella, Blautia, and Citrobacter (r = −0.31 to (−0.52). Protein-derived energy was positively correlated with Sporobacter, Intestinibacter, Catabacter, Butyrivibrio, Anaerovorax, Catenibacterium, and Pseudomonas (r = 0.30–0.39), but negatively associated with Bacteroides (r = 0.31, P < 0.05). Notably, consumption of sweets was positively correlated with Akkermansia, Odoribacter, Eisenbergiella, Catellicoccus, Eikenella, Cloacibacillus, and Finegoldia (r = 0.30–0.59).
In this study, we investigated whether fasting-associated dietary behavior affected shifts in gut microbiota composition among 34 healthy participants originating from China and Pakistan, using high-throughput 16S rRNA gene sequencing. To avoid any geographic bias, study participants from different ethnic groups were all recruited from the same city of Lanzhou (Gansu, China). Previous studies have reported that gut microbiota can be influenced by environment, host genetic background and ethnicity, drug intake, diet, and dietary behavior (Lloyd-Price et al., 2016; Gupta et al., 2017; Liao et al., 2018; Li et al., 2020; Liu et al., 2020). Zeb et al. (2020) reported that time-restricted feeding could regulate circadian rhythms associated with modulation of gut microbiota. However, among the changes in gut microbiota incurred by dietary behavioral patterns, the impact of fasting on gut microbiota has not been well investigated.
One previous study of the effects of fasting for Ramadan on gut microbiota revealed that fasting behaviors can lead to shifts in in gut microbial community composition (Ozkul et al., 2020). However, this study was unable to capture the influence of dietary composition on these shifts, only the practice of fasting, itself, thus leaving the effects of several, potentially major contributing factors unresolved. Furthermore, the inclusion of participants from two distinct ethnic groups can provide deeper insight into the drivers of gut microbiota dynamics.
Dietary composition and behaviors have been shown to serve as strong influences that can shift gut microbiota structure (David et al., 2014; Mesnage et al., 2019). Since short-term alterations in dietary behavior, such as fasting, may alter the mealtimes and meal sizes, these behaviors may thus lead to rapid metabolic changes and consequent modifications in the proportions of Firmicutes and Bacteroidetes (Jumpertz et al., 2011; Li et al., 2017). At the phylum level, an increased proportion of Bacteroidetes and decreased Firmicutes were observed after fasting in the Pakistani group (Figure 2B), which is in agreement with previously described effects of caloric restriction on gut microbiota in a mouse model (Wang et al., 2018). Bacteroidetes were also significantly increased in the Chinese participants after fasting. This could be at least partially explained by enrichment for anaerobic fermenting taxa capable of degrading recalcitrant substrates and converting complex polysaccharides to simple sugars for ATP production (Dahiya et al., 2017). Shifts in the ratio of Firmicutes to Bacteroidetes has been previously described as part of a response to dietary changes, genetic and age variations in the host, and disease (Turnbaugh et al., 2006; Mariat et al., 2009; Jumpertz et al., 2011; Kwok et al., 2014; Lan et al., 2017). Our results indicate that fasting for Ramadan can also affect this ratio, suggesting that fasting can have implications on health. In addition, Proteobacteria were significantly enriched after fasting in the total group comparison (TBF vs. TAF), which is noteworthy because enrichment of Proteobacteria has been suggested as a gut microbial signature of dysbiosis (Shin et al., 2015).
In this study, we found that, at the genus level, Sutterella and Parabacteroides increased in abundance in the Pakistani group after fasting (Figure 3B). The prevalence of Sutterella suggested potential metabolic benefits, in light of previous reports indicating positive impacts by this genus on glucose levels, while Parabacteroides has been described as a potential factor associated with inhibition of weight gain (Zeng et al., 2019). Within the Chinese group, Faecalibacterium and Butyricicoccus increased after fasting, which aligned with the findings of Ozkul et al. (2020), suggesting that caloric restriction can enrich for beneficial bacteria (Ruiz et al., 2017). A study by Li et al. (2017) also found a higher abundance of Butyricicoccus after restricting food intake to simulate fasting in mice.
It warrants mention that in comparisons of total fasting groups, the taxa enriched prior to Ramadan fasting are primarily butyrate-producing bacteria, such as Clostridium_XlVa and Lachnospiracea incertae sedis, which can perform necessary functions in maintaining intestinal mucosa, and thus contribute to health (Peng et al., 2009; Louis et al., 2010). In addition, the significant enrichment of Klebsiella in the total cohort after fasting (TAF) suggests that gut health could also be negatively affected by fasting since the high abundance of Klebsiella has been associated with various diseases, such as neonatal necrotizing enterocolitis, multiple myeloma, and virus infections (Qin et al., 2015; Olm et al., 2019). However, we found significantly lower levels of genus Coprococcus after fasting in total group comparisons. This finding is also notable since previous studies have reported an association between high abundance of this genus and obesity (Kasai et al., 2015).
In the present study, Chinese and Pakistani participants self-reported the consumption of grains, vegetables, milk, livestock meat, poultry, sweets, beans, seafood, and fruits and the correlation between diet and gut microbiota showed that specific taxa were correlated with dietary intake, such as Alloprevotella, Dialister, and Oscillibacter with fat-derived energy (r = 0.37–0.44, P < 0.05), whereas other studies have reported a decreased of the Alloprevotella abundance in mice following a high calorie diet (Kong et al., 2019), while Oscillibacter and Akkermansia were negatively associated with obesity (Thingholm et al., 2019). Routy et al. (2018) studied patients with lung, renal, and other cancers, and found that patients with intestinal enrichment for Akkermansia muciniphila had better outcomes of immunotherapy. The results of other studies have suggested that Akkermansia can also help inhibit obesity and ameliorate alcohol-associated cirrhosis (Grander et al., 2018; Depommier et al., 2019). In our study, we observed that the consumption of sweets was positively correlated with the prevalence of Akkermansia. However, further study is required to clearly resolve how Akkermansia is affected by dietary intake.
Further PCoA analysis of dietary composition indicated that diets differed slightly across fasting groups, but dramatically differed (i.e., no overlap) across ethnic groups. This result was highly similar to the results of PCoA analysis for gut microbiota across the respective ethnic groups. Taken together, these findings highlight the clear contribution of diet on gut microbiota during fasting and across ethnic groups. Moreover, this study confirms the occurrence of shifts in gut microbial community triggered by dietary changes (e.g., including dietary habits such as fasting). Based on these results, we thus propose that ethnicity and fasting for Ramadan can alter the gut microbiota of humans, strongly mediated by changes in diet.
In agreement with other studies in humans, here we found that the dominant phyla in all groups were Firmicutes, Bacteroidetes, and Proteobacteria (Schnorr et al., 2014; Zhang et al., 2015). We observed that phylum-level structure was distinct among ethnic groups, with a higher abundance of Firmicutes in Pakistani participants than in Chinese before fasting, which instead had a higher prevalence of Bacteroidetes. Different relative proportions of Firmicutes and Bacteroidetes were previously reported by several studies, suggesting the potential influence of the host (Mariat et al., 2009; Kwok et al., 2014).
The Faecalibacterium and Roseburia genera belonging to Firmicutes phylum are well-established butyrate producers, which is necessary for maintaining intestinal mucosa (Peng et al., 2009; Louis et al., 2010). Metabolites such as butyrate, short-chain fatty acids, and acetate have been reported to improve gut barrier function (Peng et al., 2009), suppress the accumulation of insulin-mediated fat in adipose tissue (Kimura et al., 2013), and also contribute to prevention of colonic diseases in the host.
Schnorr et al. (2014) found significant changes in the structure of the gut community in comparisons of Italian and Hadza individuals. Despite the well-established, significant contribution of the ethnic background toward gut microbial alpha diversity (but not beta diversity) (Deschasaux et al., 2018), here we observed that both alpha and beta diversity were influenced by ethnicity independently of geographic location. This observation is possibly dependent on dietary intake and diet-related behavior. In particular, ethnic groups showed significant differences in several alpha diversity indices and clear separation in beta diversity. Pakistani group presented significantly higher OTU abundance indices (i.e., observed OTUs, Chao1, and ACE) than Chinese group, while the coverage index was higher in the Chinese group than in the Pakistani group before fasting. The alteration of alpha diversity among ethic groups were also reported by Liu et al. (2020), however beta diversity was not shifted according to ethnicity in their study. Future and ongoing studies will further clarify how ethnicity, which has many associated genetic, behavioral, and geographic factors, can contribute to driving the structure of microbiota in the context of regional diets and intermittent or stochastic changes in nutrient inputs.
In the light of several other studies that provide evidence of differences in microbiota diversity and composition associated with distinct ethnic groups, and under the influences of nutritional intake and environment, here we endeavor to resolve the impact of dietary behavior (i.e., intermittent caloric restriction associated with Ramadan fasting) on gut microbiota by controlling geographic factors, and by addressing differences in diet and ethnic origin through the inclusion of participants from two distinct ethnic groups (Pakistani and Chinese) from the same city, socio-economic status, and presumably, similar environmental exposure.
Although the results of the study were significant but limited by the number of participants, it was necessary to further examined the influences of dietary behaviors such as fasting in a larger study cohort. Furthermore, future projects using high-throughput metabolomics, in conjunction with sequencing techniques, will provide remarkably higher resolution to our current understanding of the tripartite contributions of diet, microbiota, and their metabolites to human health.
To our knowledge, this study provides the first investigation into the influence of Ramadan fasting on gut microbiota in either Chinese or Pakistani individuals. Our results demonstrate that, in the absence of geographic separation, significant differences exist in the structure, composition, and alpha- and beta diversities of gut microbiota in Chinese and Pakistani individuals, largely attributable to significant variations in diet. In addition, intermittent caloric restriction associated with fasting for Ramadan only appeared to affect beta diversity and the prevalence of some signature taxa. This work provides inroads to understanding how modification to dietary behavioral patterns can influence the generally diet-driven microbiota, whose further study can potentially guide behavior- or lifestyle-based treatments for targeted enrichment of specific taxa beneficial to gut health.
The datasets presented in this article are available with reasonable requests. Requests to access the datasets should be directed to XH, School of Public Health, Lanzhou University, Lanzhou China.
The studies involving human participants were reviewed and approved by Medical Ethics Committee approval of the School of Public Health (GW-20171013), Lanzhou University, China. The patients/participants provided their written informed consent to participate in this study.
XH and RL conceived the study and designed the experiments. IkA and KL performed DNA extraction and drafted the manuscript. IkA, KL, SF, MH, and IzA coordinated in selecting field sampling sites and sample collection. IkA and KL analyzed the data and contributed to data interpretation. All authors contributed to the critical revision of the manuscript, read, and approved the final manuscript.
Funding support: The ‘111’ Program (BP0719040); The fundamental research funded for the Central University (lzujbky-2019-48).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge Mr. Qi Wanpeng and Miss. Zheng Xueqin for their technical assistance and Dr. Isaac V. Greenhut, Ph.D., ELS for advice with language and editorial assistance in manuscript preparation.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.642999/full#supplementary-material
Aybak, M. (1996). Effect of Ramadan fasting on platelet aggregation in healthy male subjects. Eur. J. Appl. Physiol. Occup. Physiol. 73, 552–556. doi: 10.1007/BF00357678
Claesson, M. J., Jeffery, I. B., Conde, S., Power, S. E., O’Connor, E. M., Cusack, S., et al. (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184. doi: 10.1038/nature11319
Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., et al. (2009). The Ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, 141–145. doi: 10.1093/nar/gkn879
Colwell, R. K., Chao, A., Gotelli, N. J., Lin, S. Y., Mao, C. X., Chazdon, R. L., et al. (2012). Models and estimators linking individual-based and sample-based rarefaction, extrapolation and comparison of assemblages. J. Plant Ecol. 5, 3–21. doi: 10.1093/jpe/rtr044
Dahiya, D. K., Renuka, Puniya, M., Shandilya, U. K., Dhewa, T., Kumar, N., et al. (2017). Gut microbiota modulation and its relationship with obesity using prebiotic fibers and probiotics: a review. Front. Microbiol. 8:563. doi: 10.3389/fmicb.2017.00563
David, L. A., Maurice, C. F., Carmody, R. N., Gootenberg, D. B., Button, J. E., Wolfe, B. E., et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. doi: 10.1038/nature12820
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696. doi: 10.1073/pnas.1005963107
Depommier, C., Everard, A., Druart, C., Plovier, H., Van Hul, M., Vieira-Silva, S., et al. (2019). Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat. Med. 25, 1096–1103. doi: 10.1038/s41591-019-0495-2
Deschasaux, M., Bouter, K. E., Prodan, A., Levin, E., Groen, A. K., Herrema, H., et al. (2018). Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 24, 1526–1531. doi: 10.1038/s41591-018-0160-1
Duncan, S. H., Belenguer, A., Holtrop, G., Johnstone, A. M., Flint, H. J., and Lobley, G. E. (2007). Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl. Environ. Microbiol. 73, 1073–1078. doi: 10.1128/AEM.02340-06
Findley, K., Williams, D. R., Grice, E. A., and Bonham, V. L. (2016). Health disparities and the microbiome. Trends Microbiol. 24, 847–850. doi: 10.1016/j.tim.2016.08.001
Goodrich, J. K., Waters, J. L., Poole, A. C., Sutter, J. L., Koren, O., Blekhman, R., et al. (2014). Human genetics shape the gut microbiome. Cell 159, 789–799. doi: 10.1016/j.cell.2014.09.053
Grander, C., Adolph, T. E., Wieser, V., Lowe, P., Wrzosek, L., Gyongyosi, B., et al. (2018). Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 67, 891–901. doi: 10.1136/gutjnl-2016-313432
Gupta, V. K., Paul, S., and Dutta, C. (2017). Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front. Microbiol. 8:1162. doi: 10.3389/fmicb.2017.01162
Harsch, I., and Konturek, P. (2018). The role of gut microbiota in obesity and Type 2 and Type 1 diabetes mellitus: new insights into “Old” diseases. Med. Sci. 6:32. doi: 10.3390/medsci6020032
Jumpertz, R., Le, D. S., Turnbaugh, P. J., Trinidad, C., Bogardus, C., Gordon, J. I., et al. (2011). Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 94, 58–65. doi: 10.3945/ajcn.110.010132
Kasai, C., Sugimoto, K., Moritani, I., Tanaka, J., Oya, Y., Inoue, H., et al. (2015). Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 15:100. doi: 10.1186/s12876-015-0330-2
Kimura, I., Ozawa, K., Inoue, D., Imamura, T., Kimura, K., Maeda, T., et al. (2013). The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 4, 1–12. doi: 10.1038/ncomms2852
Kolodziejczyk, A. A., Zheng, D., and Elinav, E. (2019). Diet-microbiota interactions and personalized nutrition. Nat. Rev. Microbiol. 17, 742–753. doi: 10.1038/s41579-019-0256-8
Kong, C., Gao, R., Yan, X., Huang, L., and Qin, H. (2019). Probiotics improve gut microbiota dysbiosis in obese mice fed a high-fat or high-sucrose diet. Nutrition 60, 175–184. doi: 10.1016/j.nut.2018.10.002
Kwok, L., Zhang, J., Guo, Z., Gesudu, Q., Zheng, Y., Qiao, J., et al. (2014). Characterization of fecal microbiota across seven chinese ethnic groups by quantitative polymerase chain reaction. PLoS One 9:e93631. doi: 10.1371/journal.pone.0093631
Lan, D., Ji, W., Lin, B., Chen, Y., Huang, C., Xiong, X., et al. (2017). Correlations between gut microbiota community structures of Tibetans and geography. Sci. Rep. 7:16982. doi: 10.1038/s41598-017-17194-4
Li, G., Xie, C., Lu, S., Nichols, R. G., Tian, Y., Li, L., et al. (2017). Intermittent fasting promotes white adipose browning and decreases obesity by shaping the gut microbiota. Cell Metab. 26:672–685.e4. doi: 10.1016/j.cmet.2017.08.019
Li, Q., Chen, S., Liu, K., Long, D., Liu, D., Jing, Z., et al. (2020). Differences in gut microbial diversity are driven by drug use and drug cessation by either compulsory detention or methadone maintenance treatment. Microorganisms 8:411. doi: 10.3390/microorganisms8030411
Liao, M., Xie, Y., Mao, Y., Lu, Z., Tan, A., Wu, C., et al. (2018). Comparative analyses of fecal microbiota in Chinese isolated Yao population, minority Zhuang and rural Han by 16sRNA sequencing. Sci. Rep. 8, 1–10. doi: 10.1038/s41598-017-17851-8
Liu, K., Zhang, Y., Li, Q., Li, H., Long, D., Yan, S., et al. (2020). Ethnic differences shape the alpha but not beta diversity of gut microbiota from school children in the absence of environmental differences. Microorganisms 8:254. doi: 10.3390/microorganisms8020254
Lloyd-Price, J., Abu-Ali, G., and Huttenhower, C. (2016). The healthy human microbiome. Genome Med. 8:51. doi: 10.1186/s13073-016-0307-y
Louis, P., Young, P., Holtrop, G., and Flint, H. J. (2010). Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ. Microbiol. 12, 304–314. doi: 10.1111/j.1462-2920.2009.02066.x
Mariat, D., Firmesse, O., Levenez, F., Guimarães, V., Sokol, H., Doré, J., et al. (2009). The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 9:123. doi: 10.1186/1471-2180-9-123
McArdle, B. H., and Anderson, M. J. (2001). Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82, 290–297.
Mesnage, R., Grundler, F., Schwiertz, A., Le Maho, Y., and Wilhelmi de Toledo, F. (2019). Changes in human gut microbiota composition are linked to the energy metabolic switch during 10 d of Buchinger fasting. J. Nutr. Sci. 8:e36. doi: 10.1017/jns.2019.33
Obregon-Tito, A. J., Tito, R. Y., Metcalf, J., Sankaranarayanan, K., Clemente, J. C., Ursell, L. K., et al. (2015). Subsistence strategies in traditional societies distinguish gut microbiomes. Nat. Commun. 6, 1–9. doi: 10.1038/ncomms7505
Olm, M. R., Bhattacharya, N., Crits-Christoph, A., Firek, B. A., Baker, R., Song, Y. S., et al. (2019). Necrotizing enterocolitis is preceded by increased gut bacterial replication, Klebsiella, and fimbriae-encoding bacteria that may stimulate TLR4 receptors. bioRxiv [Preprint], doi: 10.1101/558676
Ozkul, C., Yalinay, M., and Karakan, T. (2020). Structural changes in gut microbiome after Ramadan fasting: a pilot study. Benef. Microb. 11, 227–233. doi: 10.3920/BM2019.0039
Peng, L., Li, Z. R., Green, R. S., Holzman, I. R., and Lin, J. (2009). Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 139, 1619–1625. doi: 10.3945/jn.109.104638
Qin, N., Zheng, B., Yao, J., Guo, L., Zuo, J., Wu, L., et al. (2015). Influence of H7N9 virus infection and associated treatment on human gut microbiota. Sci. Rep. 5:14771. doi: 10.1038/srep14771
Routy, B., Le Chatelier, E., Derosa, L., Duong, C. P. M., Alou, M. T., Daillère, R., et al. (2018). Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 359, 91–97. doi: 10.1126/science.aan3706
Ruiz, A., Cerdó, T., Jáuregui, R., Pieper, D. H., Marcos, A., Clemente, A., et al. (2017). One-year calorie restriction impacts gut microbial composition but not its metabolic performance in obese adolescents. Environ. Microbiol. 19, 1536–1551. doi: 10.1111/1462-2920.13713
Santacruz, A., Marcos, A., Wärnberg, J., Martí, A., Martin-Matillas, M., Campoy, C., et al. (2009). Interplay between weight loss and gut microbiota composition in overweight adolescents. Obesity 17, 1906–1915. doi: 10.1038/oby.2009.112
Schnorr, S. L., Candela, M., Rampelli, S., Centanni, M., Consolandi, C., Basaglia, G., et al. (2014). Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 5:3654. doi: 10.1038/ncomms4654
Schröder, H., Covas, M. I., Marrugat, J., Vila, J., Pena, A., Alcántara, M., et al. (2001). Use of a three-day estimated food record, a 72-hour recall and a food-frequency questionnaire for dietary assessment in a Mediterranean Spanish population. Clin. Nutr. 20, 429–437. doi: 10.1054/clnu.2001.0460
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Shin, N.-R., Whon, T. W., and Bae, J.-W. (2015). Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 33, 496–503. doi: 10.1016/j.tibtech.2015.06.011
Thingholm, L. B., Rühlemann, M. C., Koch, M., Fuqua, B., Laucke, G., Boehm, R., et al. (2019). Obese individuals with and without Type 2 diabetes show different gut microbial functional capacity and composition. Cell Host Microb. 26, 252–264.e10. doi: 10.1016/j.chom.2019.07.004
Turnbaugh, P., Ley, R., Mahowald, M., Magrini, V., Mardis, E. R., Gordon, J. I., et al. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031.
Wang, S., Huang, M., You, X., Zhao, J., Chen, L., Wang, L., et al. (2018). Gut microbiota mediates the anti-obesity effect of calorie restriction in mice. Sci. Rep. 8:13037. doi: 10.1038/s41598-018-31353-1
Wu, G. D., Chen, J., Hoffmann, C., Bittinger, K., Chen, Y., Keilbaugh, S., et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108. doi: 10.1126/science.1208344
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. doi: 10.1038/nature11053
Zeb, F., Wu, X., Chen, L., Fatima, S., Haq, I., Chen, A., et al. (2020). Effect of time-restricted feeding on metabolic risk and circadian rhythm associated with gut microbiome in healthy males. Br. J. Nutr. 123, 1216–1226. doi: 10.1017/S0007114519003428
Zeng, Q., Li, D., He, Y., Li, Y., Yang, Z., Zhao, X., et al. (2019). Discrepant gut microbiota markers for the classification of obesity-related metabolic abnormalities. Sci. Rep. 9, 1–10. doi: 10.1038/s41598-019-49462-w
Zhang, C., Li, S., Yang, L., Huang, P., Li, W., Wang, S., et al. (2013). Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat. Commun. 4:2163. doi: 10.1038/ncomms3163
Keywords: ethnicity, Ramadan fasting, dietary habit, gut microbial diversity, MiSeq
Citation: Ali I, Liu K, Long D, Faisal S, Hilal MG, Ali I, Huang X and Long R (2021) Ramadan Fasting Leads to Shifts in Human Gut Microbiota Structured by Dietary Composition. Front. Microbiol. 12:642999. doi: 10.3389/fmicb.2021.642999
Received: 17 December 2020; Accepted: 01 February 2021;
Published: 18 February 2021.
Edited by:
Lin Lin, Jiangsu University, ChinaReviewed by:
Marcello Abbondio, University of Sassari, ItalyCopyright © 2021 Ali, Liu, Long, Faisal, Hilal, Ali, Huang and Long. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaodan Huang, SHVhbmd4aWFvZGFuQGx6dS5lZHUuY24=; Ruijun Long, bG9uZ3JqQGx6dS5lZHUuY24=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.