 Patricia Saxenborn1
Patricia Saxenborn1 John Baxter1Andreas Tilevik1Magnus Fagerlind1
John Baxter1Andreas Tilevik1Magnus Fagerlind1 Fredrik Dyrkell2
Fredrik Dyrkell2 Anna-Karin Pernestig1Helena Enroth1,3
Anna-Karin Pernestig1Helena Enroth1,3 Diana Tilevik1*
Diana Tilevik1*- 1Systems Biology Research Centre, School of Bioscience, University of Skövde, Skövde, Sweden
- 21928 Diagnostics, Gothenburg, Sweden
- 3Molecular Microbiology, Laboratory Medicine, Unilabs AB, Skövde, Sweden
Klebsiella is a genus of Gram-negative bacteria known to be opportunistic pathogens that may cause a variety of infections in humans. Highly drug-resistant Klebsiella species, especially K. pneumoniae, have emerged rapidly and are becoming a major concern in clinical management. Although K. pneumoniae is considered the most important pathogen within the genus, the true clinical significance of the other species is likely underrecognized due to the inability of conventional microbiological methods to distinguish between the species leading to high rates of misidentification. Bacterial whole-genome sequencing (WGS) enables precise species identification and characterization that other technologies do not allow. Herein, we have characterized the diversity and traits of Klebsiella spp. in community-onset infections by WGS of clinical isolates (n = 105) collected during a prospective sepsis study in Sweden. The sequencing revealed that 32 of the 82 isolates (39.0%) initially identified as K. pneumoniae with routine microbiological methods based on cultures followed by matrix-assisted laser desorption-time of flight mass spectrometry (MALDI-TOF MS) had been misidentified. Of these, 23 were identified as Klebsiella variicola and nine as other members of the K. pneumoniae complex. Comparisons of the number of resistance genes showed that significantly fewer resistance genes were detected in Klebsiella oxytoca compared to K. pneumoniae and K. variicola (both values of p < 0.001). Moreover, a high proportion of the isolates within the K. pneumoniae complex were predicted to be genotypically multidrug-resistant (MDR; 79/84, 94.0%) in contrast to K. oxytoca (3/16, 18.8%) and Klebsiella michiganensis (0/4, 0.0%). All isolates predicted as genotypically MDR were found to harbor the combination of β-lactam, fosfomycin, and quinolone resistance markers. Multi-locus sequence typing (MLST) revealed a high diversity of sequence types among the Klebsiella spp. with ST14 (10.0%) and ST5429 (10.0%) as the most prevalent ones for K. pneumoniae, ST146 for K. variicola (12.0%), and ST176 for K. oxytoca (25.0%). In conclusion, the results from this study highlight the importance of using high-resolution genotypic methods for identification and characterization of clinical Klebsiella spp. isolates. Our findings indicate that infections caused by other members of the K. pneumoniae complex than K. pneumoniae are a more common clinical problem than previously described, mainly due to high rates of misidentifications.
Introduction
The Gram-negative bacteria Klebsiella possess a major threat to public health, causing significant morbidity and mortality worldwide. It is known to be an opportunistic pathogen that can cause a variety of health-care associated and community-acquired infections, such as urinary tract infections, pneumonia, sepsis, meningitis, and pyogenic liver abscesses. The emergence of highly drug-resistant strains as well as the increasing diversity of antibiotic resistance phenotypes observed in Klebsiella spp. are particularly concerning (Jean et al., 2018; Bengoechea and Sa Pessoa, 2019; Heinz et al., 2019; Rodríguez-Medina et al., 2019). Klebsiella infections in humans are caused mainly by Klebsiella pneumoniae, and to a lesser degree by Klebsiella oxytoca (Singh et al., 2016).
Recently, many related Klebsiella spp. have been identified and classified leading to an expansion of the taxonomy of the K. pneumoniae complex. Klebsiella pneumoniae has formerly been divided into three phylogroups (KpI, KpII, and KpIII), later classified as distinct species (K. pneumoniae, Klebsiella quasipneumoniae, and Klebsiella variicola; Brisse and Verhoef, 2001; Brisse et al., 2004, 2014; Rosenblueth et al., 2004). The K. pneumoniae complex currently comprises seven K. pneumoniae-related species: K. pneumoniae, K. quasipneumoniae subsp. quasipneumoniae, K. quasipneumoniae subsp. similipneumoniae, K. variicola subsp. variicola (herein referred to as K. variicola), K. variicola subsp. tropicalensis, K. africanensis, and the recently described K. quasivariicola (Long et al., 2017a). Although K. pneumoniae is the major cause of infections within the complex, the involvement in human infections of the other species is gaining recognition (Seki et al., 2013; Brisse et al., 2014; Maatallah et al., 2014; Holt et al., 2015; Breurec et al., 2016; Chen et al., 2016; Ejaz et al., 2017; Long et al., 2017b; Nicolás et al., 2018). Unfortunately, the inability of conventional microbiological methods to distinguish species within the complex results in high rates of misidentifications (Long et al., 2017b), thus masking the true clinical significance of each phylogroup and their epidemiological features (Seki et al., 2013; Brisse et al., 2014; Long et al., 2017b). Usually, members of the K. pneumoniae complex are misidentified as K. pneumoniae (Rodríguez-Medina et al., 2019). Nowadays, matrix-assisted laser desorption-time of flight mass spectrometry (MALDI-TOF MS) is commonly used for species identification within standard diagnostic routine in clinical laboratories but its ability to discriminate between Klebsiella spp. is highly dependent on the database version used (Long et al., 2017b; Rodrigues et al., 2018). Also identification methods based on PCR have been developed but those are prone to errors or do not distinguish all phylogroups (Bialek-Davenet et al., 2014; Brisse et al., 2014; Garza-Ramos et al., 2015). Reliable identification of the different members of the K. pneumoniae complex can thus only be achieved using whole-genome sequencing (WGS; Long et al., 2017b) or sequencing of specific genetic markers (e.g., blaLEN, blaOKP, blaSHV, rpoB, gyrA, and parC; Brisse et al., 2014; Holt et al., 2015).
In addition to accurate species identification, WGS-based analysis has rendered applicable for other aspects of clinical microbiology, including genotypic resistance prediction, virulence characterization, infectious disease control, and epidemiology of pathogens (Deurenberg et al., 2017; Tagini and Greub, 2017; Rossen et al., 2018). Thereof, WGS has been perceived as one of the most promising techniques in clinical microbiology (Bertelli and Greub, 2013; Deurenberg et al., 2017) and is today widely implemented in research laboratories. The implementation of bacterial WGS in the clinical laboratory has hitherto been hampered by long turn-around time, high cost, and the need for bioinformatics expertise. However, the recent development of long-read sequencers with relatively short runtimes as well as automated user-friendly bioinformatics pipelines may imply means to address these challenges (Petersen et al., 2019). The aim of this study was to characterize the diversity and traits of clinical Klebsiella spp. by WGS-based analysis using bioinformatics tools, and also to compare these results with those obtained by routine microbiological methods. A total of 105 Klebsiella spp. isolates, collected during a prospective, consecutive study in south-west Sweden of adult patients with suspected community-onset sepsis (Ljungström et al., 2019), were included.
Materials and Methods
Bacterial Isolates
From September 2011 to June 2012, a prospective observational study of community-onset severe sepsis and septic shock in adults was conducted at Skaraborg Hospital, a secondary hospital with 640 beds, in the western region of Sweden (Ljungström et al., 2019). All patients ≥18 years consecutively admitted to the emergency department for suspicion of community-onset sepsis were asked to participate in the study. The study was approved by the Regional Ethical Review Board of Gothenburg (376-11). As the present study only focused on bacterial isolates recovered from cultures included in the routine patient care, no individual written consent was needed. During 9 months, approximately 1,800 bacterial isolates from different sample types were recovered from the patients enrolled in the study. These isolates were cryopreserved at the time of recovery by transferring colonial material to Microbank™ vials (Pro-Lab Diagnostics, Ontario, Canada) stored at −80°C. For the present study, all isolates identified as Klebsiella spp. (n = 105) with routine microbiological methods based on cultures followed by MALDI-TOF MS (DB-4110) were selected. The isolates were recovered from 82 patients in samples collected from blood (n = 29), urine (n = 71), nasopharynx (n = 4), and wound (n = 1).
MALDI-TOF MS Identification
Species identification of isolates was performed with MALDI-TOF MS in the clinical laboratory Unilabs at Skaraborg Hospital on a MicroFlex LT mass spectrometer (Bruker Daltonics, Germany) with BioTyper software v2.0 using default parameter settings as part of the routine clinical practice as described elsewhere (Ljungstrom et al., 2015; Enroth et al., 2019). Spectral scores above 2.0 were used as cut-off for correct identification. At the time of the study, the Bruker microorganism database MBT Compass Library DB-4110 (Bruker Daltonics, Germany) released in April 2011 was used.
Phenotypic Antibiotic Susceptibility Testing
Antibiotic susceptibilities were determined by accredited laboratory methods using the disc diffusion method on Mueller-Hinton media according to European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines.1 As the phenotypic antibiotic susceptibility testing (AST) was performed as part of the routine clinical practice, the sample type mainly determined which antibiotics to be tested for each bacterial isolate (Table 1).
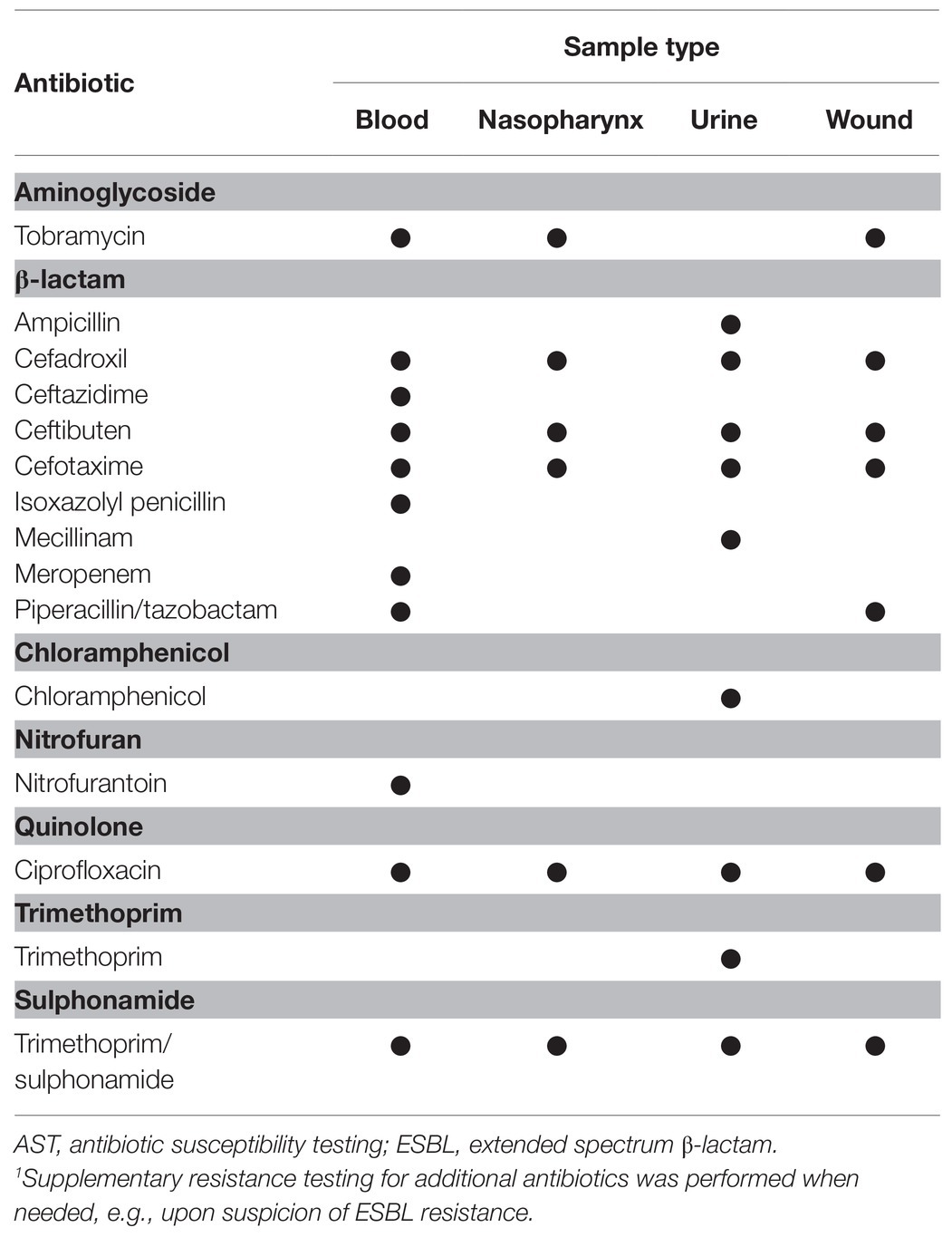
Table 1. Specification of antibiotics used in the phenotypic antibiotic susceptibility testing (AST) for Klebsiella spp. isolates as part of the routine clinical practice.
Illumina Dye Sequencing and Analysis of Short-Read Data
Genomic DNA was extracted from pure cultures using the MagNA Pure 96 DNA and Viral NA Small Volume Kit (Roche Diagnostics, Switzerland) on a MagNA Pure 96 instrument according to the Pathogen Universal 200 protocol (Roche Diagnostics, Switzerland). NexteraXT libraries were prepared using the manufacturer’s protocol (Illumina, San Diego, CA) and sequenced on an Illumina HiSeq instrument at SciLifeLab, Solna, Sweden.
Primary quality control of the FASTQ files was performed using the FastQC software (v.0.11.5) (Andrews, 2010). Trimmomatic (v.0.36) was used for adapter removal and quality trimming with a sliding window of size 4 and a minimum quality of 20 (Bolger et al., 2014). In addition, the first 12 bases were trimmed by the HEADCROP argument, and reads with a length shorter than 30 bp were removed. FASTQ files were then assembled into contigs using the St. Petersburg genome assembler (SPAdes v.3.11.1; Bankevich et al., 2012). The Quality Assessment Tool for Genome Assemblies (QUAST v.4.6.0) was used to assess the quality of the assembled contigs (Gurevich et al., 2013). Evaluation of the contigs in QUAST was performed using default settings with reference sequences obtained from the National Center for Biotechnology Information (NCBI). The reference sequence for K. pneumoniae subsp. pneumoniae HS11286 (GenBank accession number NC_016845.1) was used for K. pneumoniae and K. variicola isolates, and the reference sequence K. oxytoca CAV1374 (GenBank accession number NZ_CP011636.1) for K. oxytoca isolates (Gurevich et al., 2013). Species identification was performed by calculating the pairwise average nucleotide identity (ANI) based on BLAST+ (ANIb) in JSpeciesWS (Richter and Rosselló-Móra, 2009). The reference genomes for each bacterial species analyzed using ANI were K. pneumoniae HS11286 (GenBank accession number NC_016845.1), K. quasipneumoniae subsp. quasipneumoniae 01A030 (GenBank accession number NZ_CCDF01000032.1), K. quasipneumoniae subsp. similipneumoniae 07A044 (GenBank accession number NZ_CBZR010000026.1), K. variicola At-22 (GenBank accession number NC_013850.1), K. quasivariicola KPN1705 (GenBank accession number NZ_CP022823.1), K. oxytoca CAV1335 (GenBank accession number NZ_CP011618.1), Klebsiella michiganensis M1 (GenBank accession number NZ_CP008841.1), and Escherichia coli ATCC 25922 (GenBank accession number NC_000913.3). An ANI threshold of 96% or greater was considered to delineate species boundaries as a threshold of 96% correlates well to DNA-DNA hybridization (Richter and Rosselló-Móra, 2009; Ciufo et al., 2018).
The presence of plasmid replicons and acquired antibiotic resistance genes, including both chromosomal and plasmid-borne, was detected using free web-based tools hosted by the Center for Genomic Epidemiology (CGE) website2 including PlasmidFinder v.1.3 and ResFinder v.3.0. The tool PlasmidFinder was used with “Enterobacteriaceae” as selected database with default settings for threshold ID (95%) and minimum length (60%; Carattoli et al., 2014). The ResFinder database for detection of acquired antibiotic resistance genes in Klebsiella spp. included markers for 15 antibiotic classes (Table 2), and was used with default settings for threshold ID (90%) and minimum length (60%; Zankari et al., 2012). The presence of resistance genes was further investigated using the commercial cloud-based platform 19283 (1928 Diagnostics, Sweden). The 1928 platform supports a more comprehensive analysis of K. pneumoniae than ResFinder by including markers for additional antibiotic classes (n = 26, Table 2). The FASTQ files for isolates initially reported as K. pneumoniae by MALDI-TOF MS (DB-4110) were uploaded for inferred antibiotic susceptibility based on genotype resistance markers (hereby and later referred to as predicted antibiotic susceptibility). On upload, the average sequence coverage was estimated. A coverage ≥30 was required for analysis to continue. The read data were queried for identifying genes from the genotype resistance database using an assembly-free method based on k-mer counting.
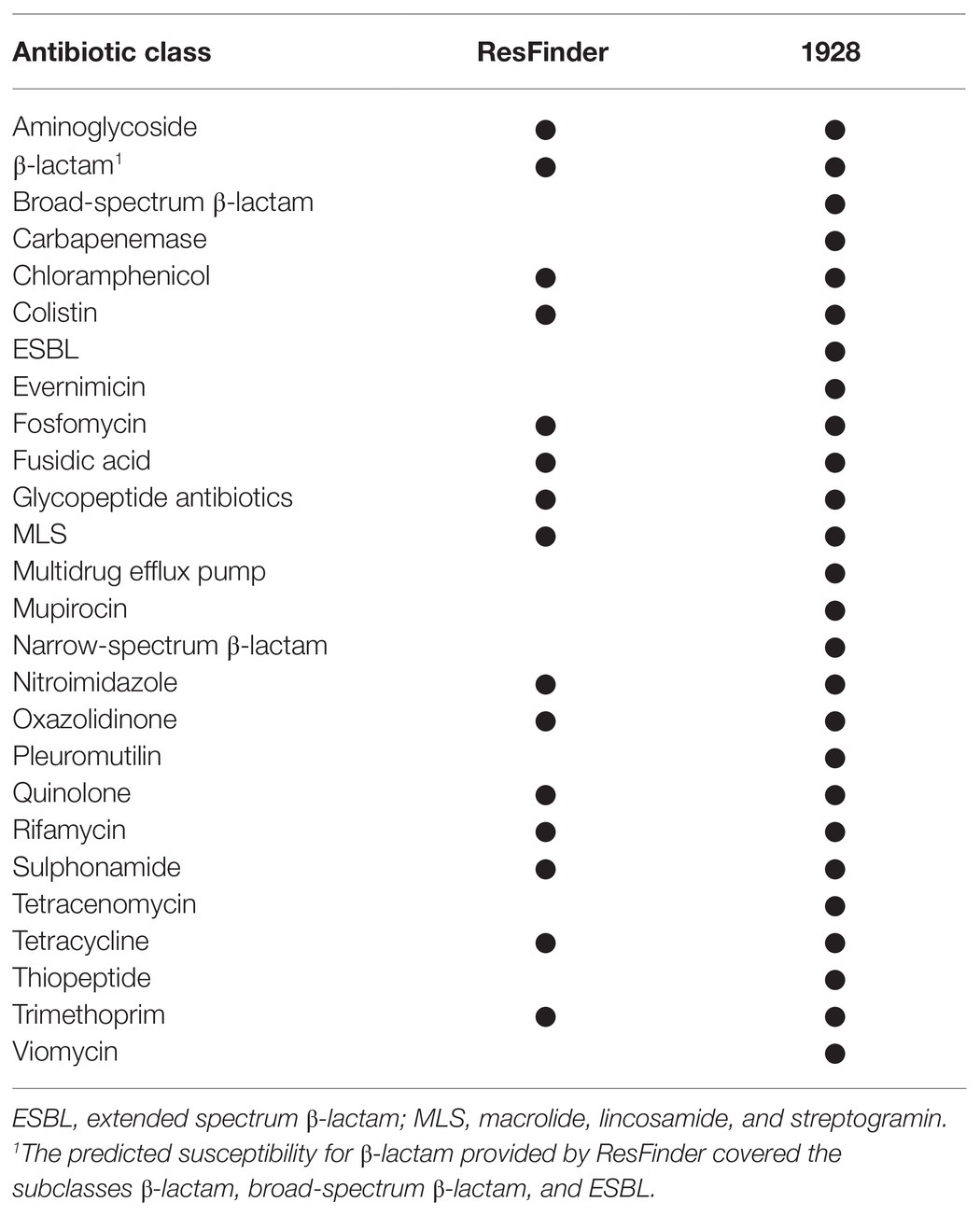
Table 2. Specification of antibiotic classes for which resistance markers were included in respective database.
Multi-locus sequence typing (MLST) analysis was performed using free web-based MLST tools. For isolates genotypically identified as K. michiganensis, K. oxytoca, K. pneumoniae, K. quasipneumoniae subsp. quasipneumoniae, K. quasipneumoniae subsp. similipneumoniae, and K. quasivariicola, the MLST tool v.2.0 hosted by the CGE website4 was used. This MLST scheme consists of alleles from the following seven loci: gapA, infB, mdh, pgi, phoE, rpoB, and tonB. The selected MLST configuration for isolates identified as K. pneumoniae, K. quasipneumoniae subsp. quasipneumoniae, K. quasipneumoniae subsp. similipneumoniae, or K. quasivariicola was “K. pneumoniae,” and “K. oxytoca” for the K. oxytoca and K. michiganensis isolates (Larsen et al., 2012). For one of the K. oxytoca isolates (KLO411), the sequence type (ST) profile could not be determined. Although exact matches for all seven loci were found, they did not match any known ST profile and the sequence data did not meet the quality requirements at the PubMLST website5 to assign a new ST profile. For the isolates genotypically identified as K. variicola, MLST was performed using the online service “MLST K. variicola” hosted by Instituto Nacional de Salud Pública.6 This MLST scheme consists of alleles from the following seven loci: leuS, pgi, pgk, phoE, pyrG, rpoB, and fusA (Barrios-Camacho et al., 2019). For one of the K. variicola isolates (KLP1399), the ST profile could not be determined as the nucleotide sequence for the phoE gene was incomplete.
Nanopore-Based Sequencing and Analysis of Long-Read Data
As a pilot validation study, 12 of the Klebsiella spp. isolates were also subject to nanopore-based sequencing using a MinION instrument (Oxford Nanopore Technology, United Kingdom). The number of isolates to include was set by the number of samples that could be sequenced simultaneously on the MinION device. Six of the isolates were selected based on the high quality of existing Illumina short-read data. The remaining six isolates were chosen due to inconsistent results for species identification with MALDI-TOF MS (DB-4110) compared to the genotypic analysis of Illumina short-read data. Species identification by MALDI-TOF MS was reanalyzed for these 12 isolates in 2019 using the Bruker microorganism database MBT Compass Library DB-7854 (Bruker Daltonics, Germany) released in April 2018.
For extraction of bacterial DNA for nanopore sequencing, the QIAGEN Genomic-tip 20/G 20/G (QIAGEN, Germany) was used according to the manufacturer’s instructions. Purified DNA was then prepared for nanopore sequencing using the Rapid Barcoding kit SQK-RBK004 (Oxford Nanopore Technologies, United Kingdom) following the manufacturer’s protocol. Sequencing was performed on MinION flow cells (R.1.9 FLO-MIN-106, Oxford Nanopore Technologies, United Kingdom) and data collected using the MinKNOW software v2.0. Automated analysis of the FASTQ files was performed with the cloud-based platform EPI2ME (Oxford Nanopore Technologies, United Kingdom) using What’s-In-My-Pot (WIMP), a Centrifuge-based system for species identification, and Antimicrobial Resistance Mapping Application (ARMA) for detection of antibiotic resistance genes.
Statistical Analysis
Statistical analyses were performed using R v.4.0.3 (R Foundation for Statistical Computing, Austria). All tests were two-sided, and p < 0.05 was considered statistically significant. Poisson and quasi-Poisson regression analysis was performed for comparisons of count data, and adjustment of p-values for multiple comparisons was made using the Holm method. Dispersion test was performed using the R package AER v.1.2-9. Concordances between conventional microbiological methods and bioinformatics analysis tools were tested with Cohen’s kappa statistics for inter-rater agreement using cut-off values for the kappa value as described elsewhere (Landis and Koch, 1977). The kappa values were computed using the R package irr v.0.84.1. For the construction of 95% CI for kappa values, the basic bootstrapping method in the R package boot v.1.3-24 was used. The number of bootstrapping was 1,000. For the construction of 95% CI for proportions of agreement between methods, the Agresti-Coull method was used (Agresti and Coull, 1998). Figures were constructed using R v.4.0.3 (R Foundation for Statistical Computing, Austria) and the data visualization R packages ggplot2 v.3.2.1 and ggsci v.2.9.
Results
Misidentification of Klebsiella spp. by MALDI-TOF MS
During the prospective sepsis study, species identification by MALDI-TOF MS (DB-4110) was performed for all collected bacterial isolates as part of the routine clinical practice. A total of 105 bacterial isolates were identified as Klebsiella spp., whereof 82 isolates were identified as K. pneumoniae, two isolates as K. variicola, and 21 isolates as K. oxytoca (Figure 1). Due to previous reports pointing out the inability of conventional microbiology laboratory techniques to differentiate between Klebsiella spp. (Long et al., 2017a; Rodríguez-Medina et al., 2019), genotypic species identification of the collected isolates was performed by calculating the pairwise ANI on the assembled Illumina short-read data against reference genomes. Out of the 82 isolates initially reported as K. pneumoniae, the species were corrected for 32 (39.0%, 95% CI 29.2–49.9) based on the results from the genotypic species identification. Of these, 23 were genotypically identified as K. variicola, five as K. quasipneumoniae subsp. quasipneumoniae, three as K. quasipneumoniae subsp. similipneumoniae, and one as K. quasivariicola. For both isolates initially reported as K. variicola by MALDI-TOF MS (DB-4110), the genotypic species identification was in agreement. For the 21 isolates initially reported as K. oxytoca, 16 were genotypically identified as K. oxytoca, four as K. michiganensis, and one as E. coli. The isolate identified as E. coli was excluded from further analysis as the current study focuses on Klebsiella spp. The species identification by MALDI-TOF MS (DB-4110) and ANI was discordant for 37.1% (37/105, 95% CI 26.8–44.8) of the isolates (Cohen’s kappa = 0.41, 95% CI 0.28–0.54).
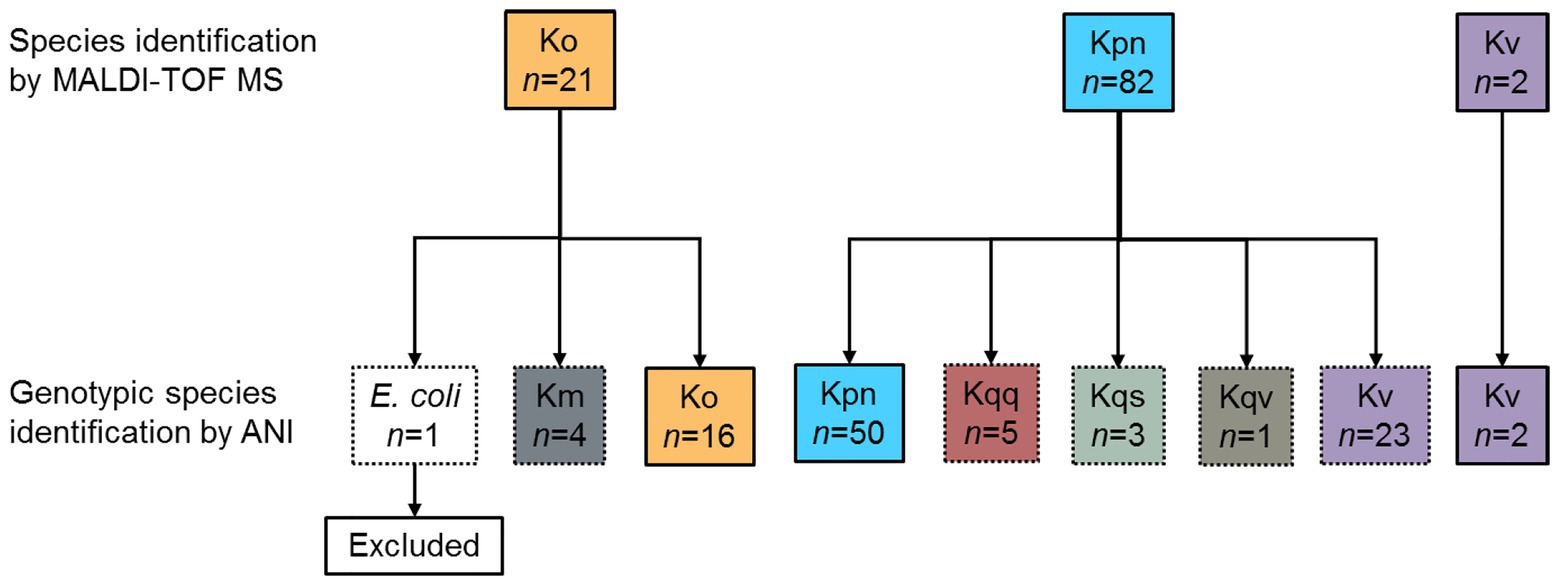
Figure 1. Overview of the results of the species identification. During the prospective sepsis study, species identification by MALDI-TOF MS (DB-4110) was performed for all collected bacterial isolates as part of the routine clinical practices. All isolates initially identified as Klebsiella spp. (n = 105) were subject to whole-genome sequencing (WGS). Genotypic species identification was performed by calculating the pairwise ANI on the assembled Illumina short-read data against reference genomes. The species identification by MALDI-TOF MS (DB-4110) and ANI agreed (solid line boxes) for 68 isolates, whereas the species were corrected based on the ANI results (dotted line boxes) for the remaining 37 isolates. ANI, average nucleotide identity; Km, K. michiganensis; Ko, K. oxytoca; Kpn, K. pneumoniae; Kqq, K. quasipneumoniae subsp. quasipneumoniae; Kqs, K. quasipneumoniae subsp. similipneumoniae; and Kqv, K. quasivariicola, Kv K. variicola.
Genotypic Antibiotic Susceptibility Prediction
The tool ResFinder was used on all Klebsiella spp. isolates to detect the presence of acquired antibiotic resistance genes. ResFinder detected at least one β-lactam resistance gene in all Klebsiella spp. isolates (n = 104, Figure 2). The second most frequently predicted resistance was against fosfomycin (n = 84) followed by quinolone (n = 82). For K. pneumoniae, all isolates were predicted to be resistant against both β-lactam and fosfomycin, and 94.0% (47/50) to quinolone. A similar resistance profile was observed for K. quasipneumoniae subsp. quasipneumoniae, K. quasipneumoniae subsp. similipneumoniae, K. variicola, and K. quasivariicola with 100.0% of the isolates predicted to be resistant to β-lactam, quinolone, and fosfomycin. For K. oxytoca and K. michiganensis, the most commonly predicted resistance was against β-lactam antibiotics (100.0%, Figure 2). Beyond that, quite few resistance markers were detected among the K. oxytoca and K. michiganensis isolates. Comparison of the number of resistance genes showed that significantly fewer resistance genes were detected in K. oxytoca compared to K. pneumoniae and K. variicola (both values of p < 0.001, Figure 3A). Four resistance genes were detected by ResFinder in the single K. quasivariicola isolate, not included in the statistical comparison. No significant differences in the number of detected resistance genes were observed between sample types (all values of p > 0.05, Figure 3B). Seven resistance genes were detected by ResFinder in the only isolate collected from wound, not included in the statistical comparison.
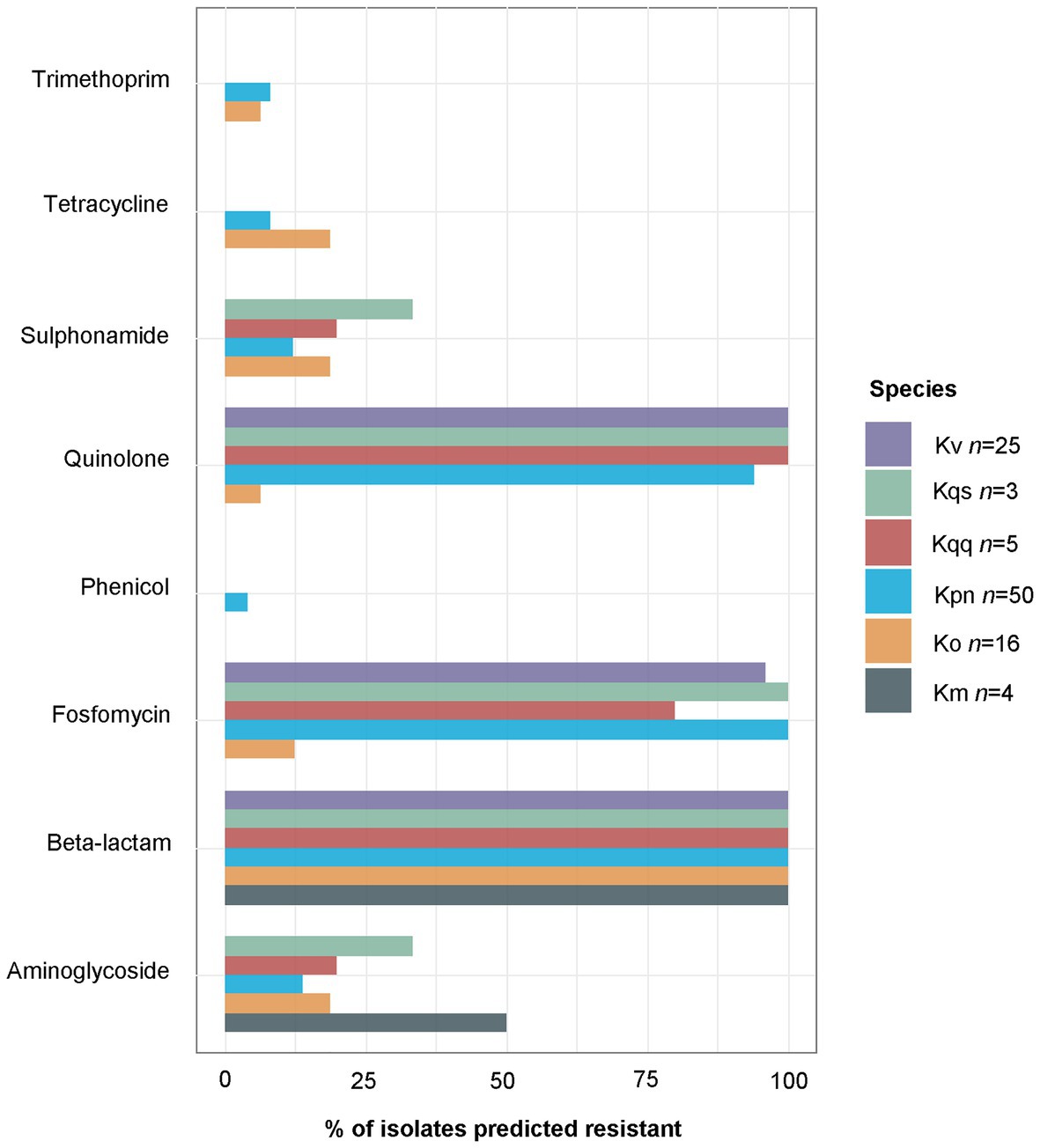
Figure 2. Percentages of isolates per species carrying at least one resistance marker against at least one agent included in the current antibiotic class as predicted by ResFinder. At least one gene conferring resistance against β-lactam antibiotics was predicted for all Klebsiella isolates, and at least one quinolone resistance gene was predicted for all K. variicola, K. quasipneumoniae subsp. quasipneumoniae, and K. quasipneumoniae subsp. similipneumoniae. The single K. quasivariicola isolate was predicted resistant against β-lactam, fosfomycin, and quinolone, data not included in the figure. Km, K. michiganensis; Ko, K. oxytoca; Kpn, K. pneumoniae; Kqq, K. quasipneumoniae subsp. quasipneumoniae; Kqs, K. quasipneumoniae subsp. simili; and Kv K. variicola.
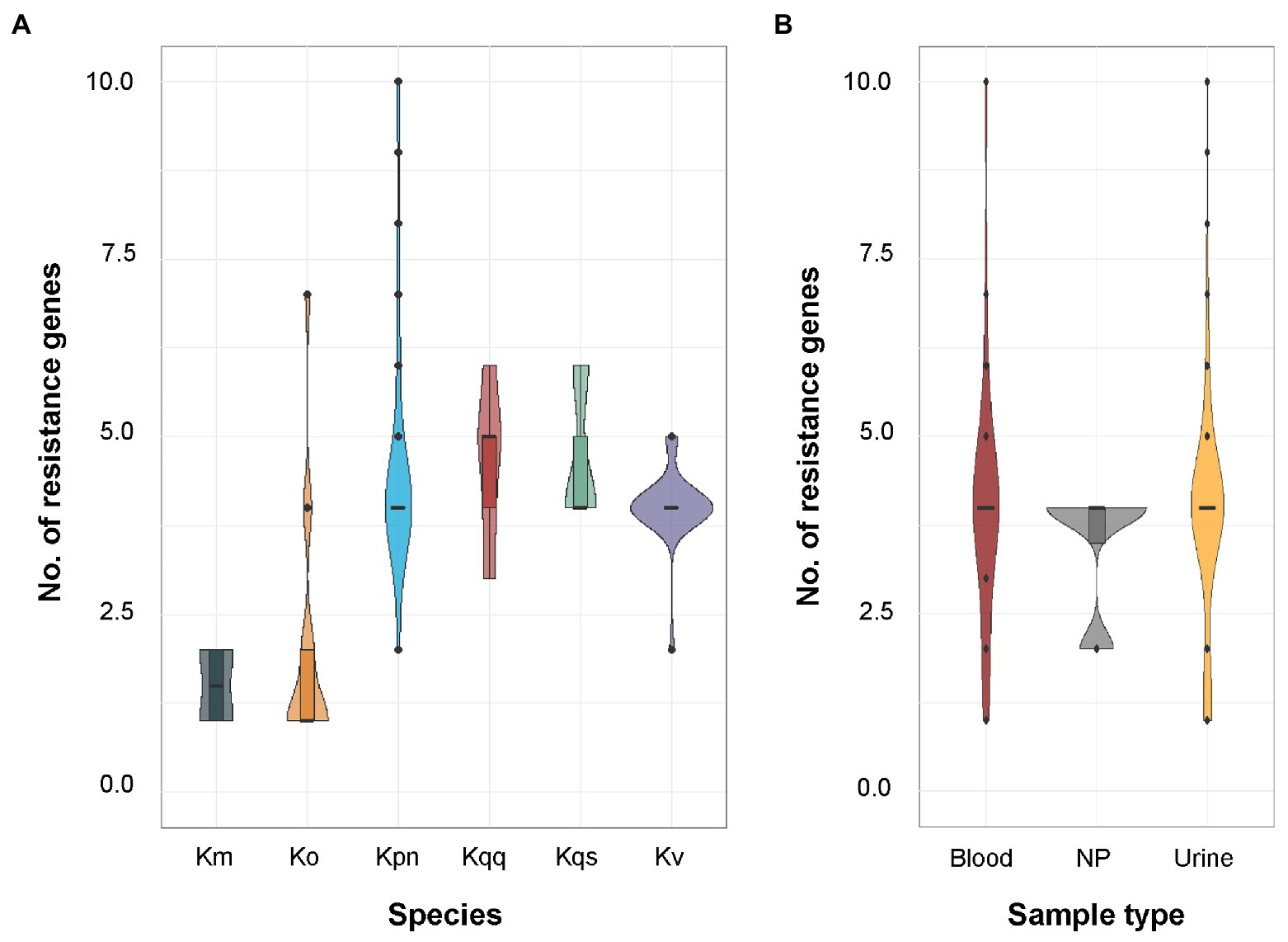
Figure 3. (A) Distribution of the number of resistance genes predicted by ResFinder per species. The sample size for each species was K. oxytoca (n = 16), K. michiganensis (n = 4), K. pneumoniae (n = 50), K. quasipneumoniae subsp. quasipneumoniae (n = 5), K. quasipneumoniae subsp. similipneumoniae (n = 3), K. variicola (n = 25), and K. quasivariicola (n = 1). Significant differences in the number of predicted resistance markers were observed between K. pneumoniae and K. oxytoca (p < 0.001), and between K. oxytoca and K. variicola (p < 0.001) using pairwise quasi-Poisson regression followed by adjustment of the resulting p-values by the Holm method. The single K. quasivariicola isolate was predicted resistant against β-lactam, fosfomycin, and quinolone, data not included in the figure. (B) Distribution of the number of resistance genes predicted by ResFinder per sample type. The sample size for each sample type was blood (n = 29), urine (n = 70), and nasopharynx (n = 4). No significant differences were detected between any sample types using pairwise Poisson regression (all values of p > 0.05). Km, K. michiganensis; Ko, K. oxytoca, Kpn, K. pneumoniae; Kqq, K. quasipneumoniae subsp. quasipneumoniae; Kqs, K. quasipneumoniae subsp. similipneumoniae; Kv, K. variicola; and NP, nasopharynx.
To determine the agreement between phenotypic and genotypic resistance on Illumina short-reads, the results from the phenotypic AST and ResFinder were compared (Table 3). In 65.8% (231/351, 95% CI 60.7–70.6) of the cases, the phenotypic AST and the antibiotic susceptibility predicted by ResFinder agreed (Cohen’s kappa = 0.35, 95% CI 0.27–0.43). Not all isolates were tested for all antibiotics in clinical routine (Table 1), therefore, the number of phenotypic results for each antibiotic varied and were less than the number of resistance genotypes predicted by ResFinder.

Table 3. Comparisons of phenotypic AST and predicted antibiotic susceptibility by ResFinder.
The ResFinder results were also compared to the predicted antibiotic susceptibilities using the 1928 platform for the 82 isolates identified as K. pneumoniae by MALDI-TOF MS (DB-4110). It should be noted that 1928 provided information about β-lactam resistance divided into three subclasses: β-lactam, broad-spectrum β-lactam, and extended spectrum β-lactam (ESBL), whereas the output from ResFinder was only β-lactam thus covering all three sub-classes. To enable a fair comparison, the output data from 1928 were merged into one β-lactam group covering resistance markers detected in all three sub-classes. The agreement between 1928 and ResFinder was 100.0% for all antibiotics, except fosfomycin where 1928 detected the resistance marker fosA in two isolates predicted as susceptible by ResFinder (Table 4). Overall, 99.7% of the predicted antibiotic susceptibilities were identical between ResFinder and 1928, demonstrating high overall agreement (Cohen’s kappa = 0.99, 95% CI 0.99–1.00).
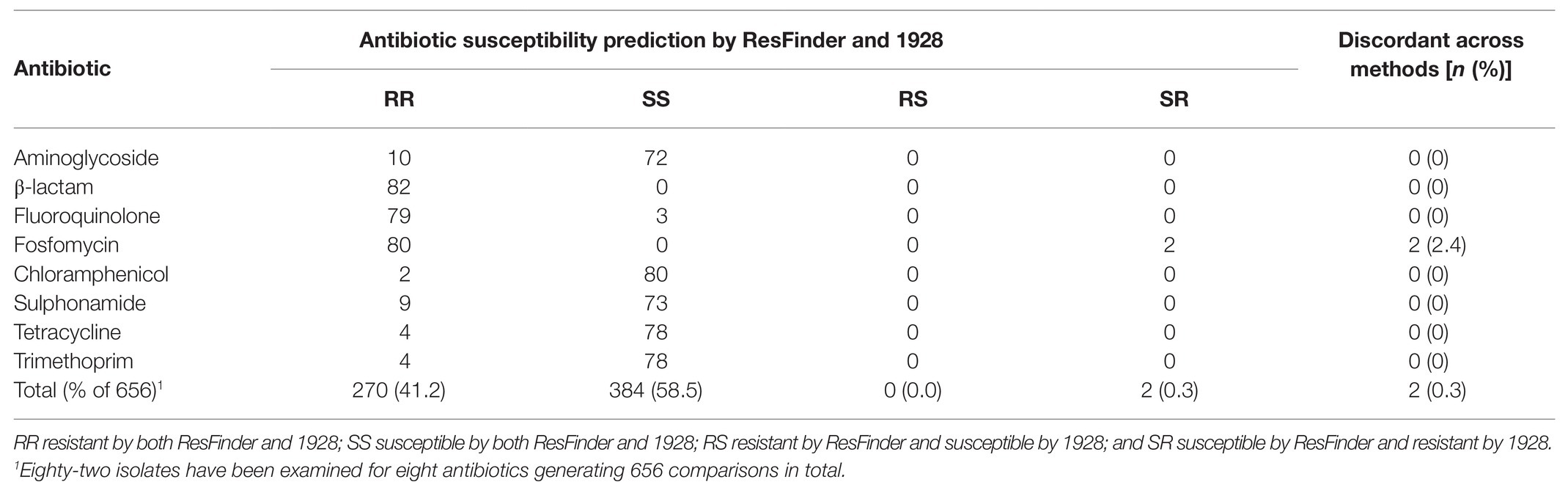
Table 4. Comparisons of predicted antibiotic susceptibility by ResFinder and 1928 for the 82 isolates initially reported as K. pneumoniae by matrix-assisted laser desorption-time of flight mass spectrometry (MALDI-TOF MS; DB-4110).
The predicted antibiotic susceptibilities by 1928 were also compared to the resistance phenotypes (Table 5). In 61.6% (173/281, 95% CI 55.8–67.1%) of the cases, the resistance phenotype and the predicted antibiotic susceptibility by 1928 agreed (Cohen’s kappa = 0.30, 95% CI 0.22–0.38). In 15 isolates, all genotypically identified as K. pneumoniae, an ESBL resistance gene was detected by the 1928 platform (10 blaSHV-1, 3 blaSHV-27, and 2 blaSHV-41), implying that 30.0% (15/50, 95% CI 19.1–43.9) of the K. pneumoniae isolates in this study is genotypically predicted to be ESBL.

Table 5. Comparisons of phenotypic AST and predicted antibiotic susceptibility by 1928.
By defining genotypically MDR as the detection of at least one resistance gene in at least three of the antibiotic classes included in ResFinder (Table 4), a total of 94.0% (79/84) of the isolates within the K. pneumoniae complex were predicted to be genotypically MDR. More specifically, high percentages of MDR were observed in K. pneumoniae (47/50, 94.0%), K. variicola (24/25, 96.0%), K. quasipneumoniae subsp. quasipneumoniae (4/5, 80.0%), and K. quasipneumoniae subsp. similipneumoniae (3/3, 100.0%) but not in K. oxytoca (3/16, 18.8%) and K. michiganensis (0/4, 0.0%). The single K. quasivariicola isolate was also found to be MDR. All MDR K. pneumoniae, K. variicola, K. quasipneumoniae subsp. quasipneumoniae, and K. quasipneumoniae subsp. similipneumoniae isolates carried the combination of β-lactam, fosfomycin, and quinolone resistance determinants.
High Diversity of Sequence Types
The typing technique MLST was used to further characterize the Klebsiella spp. isolates based on sequence differences in seven house-keeping genes. A high diversity of STs was detected among the Klebsiella spp. isolates. For K. oxytoca, 10 distinct STs were detected with ST176 being the most common one (4/16, 25.0%; Figure 4A), whereas a distinct ST was determined for each of the four K. michiganensis isolates (ST85, ST194, ST231, and ST356). Thirty-five distinct STs were identified for K. pneumoniae with ST14 (5/50, 10.0%; Figure 4B) and ST5429 being the most prevalent ones (5/50, 10.0%; Figure 4B). For the three K. quasipneumoniae subsp. similipneumoniae isolates, two were recognized as ST1647 and one as ST4768. For the five K. quasipneumoniae subsp. quasipneumoniae isolates, two were identified as ST5443, and one each of ST1887, ST4630, and ST5444. The single K. quasivariicola isolate was determined to be ST3497. For K. variicola, 17 distinct STs were identified with ST146 most common (3/25, 12.0%; Figure 4C).
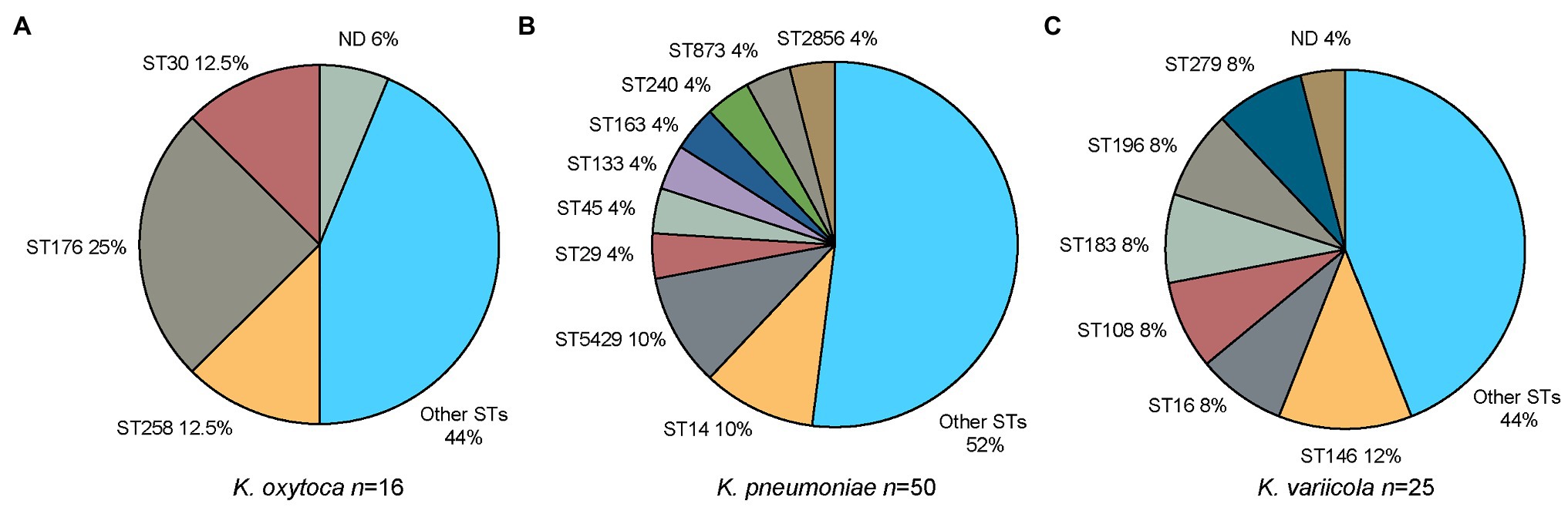
Figure 4. Distribution of sequence types (STs) for the three major groups of Klebsiella spp. in the current study. (A) K. oxytoca. “Other STs” include one isolate each of ST36, ST37, ST201, ST288, ST357, ST358, and ST359. “ND” refers to a single isolate for which the ST profile could not be determined. (B) K. pneumoniae. “Other STs” include one isolate each of ST11, ST20, ST30, ST37, ST39, ST187, ST215, ST220, ST294, ST309, ST381, ST485, ST462, ST678, ST685, ST788, ST870, ST872, ST966, ST1114, ST1948, ST3370, ST4069, ST4631, ST4725, and ST5442. (C) K. variicola. “Other STs” include one isolate each of ST149, ST261, ST280, ST281, ST282, ST283, ST284, ST285, ST286, ST287, and ST288. “ND” refers to a single isolate for which the ST profile could not be determined. MLST, multi-locus sequence typing; ND, not determined; and ST sequence type.
Plasmid Replicons
In total, 16 different plasmid replicon types were detected by PlasmidFinder (Figure 5). The most common plasmid replicon types predicted were IncFIB, whereof 60 were found in 47 isolates, and IncFII, whereof 47 were found in 40 isolates. Three isolates (KLO1516 K. oxytoca, KLP307 K. variicola, and KLP1815 K. pneumoniae) were found to host five plasmid replicons. These isolates were collected from wound, nasopharynx, and blood, respectively. Additionally three isolates (KLP139 K. pneumoniae, KLP188 K. pneumoniae, and KLP189 K. variicola) collected from urine were detected to have four plasmid replicons. The number of plasmid replicons was compared between genotypic species and sample types. No significant differences in the number of plasmid replicons were observed between species (Figure 6A). Comparisons of the number of plasmid replicons between isolates from different sample types were performed for all sample types but wound due to too few isolates collected (n = 1). A significantly higher number of plasmid replicons was detected in isolates from nasopharynx in comparison to blood (p = 0.01), and urine (p = 0.003; Figure 6B).
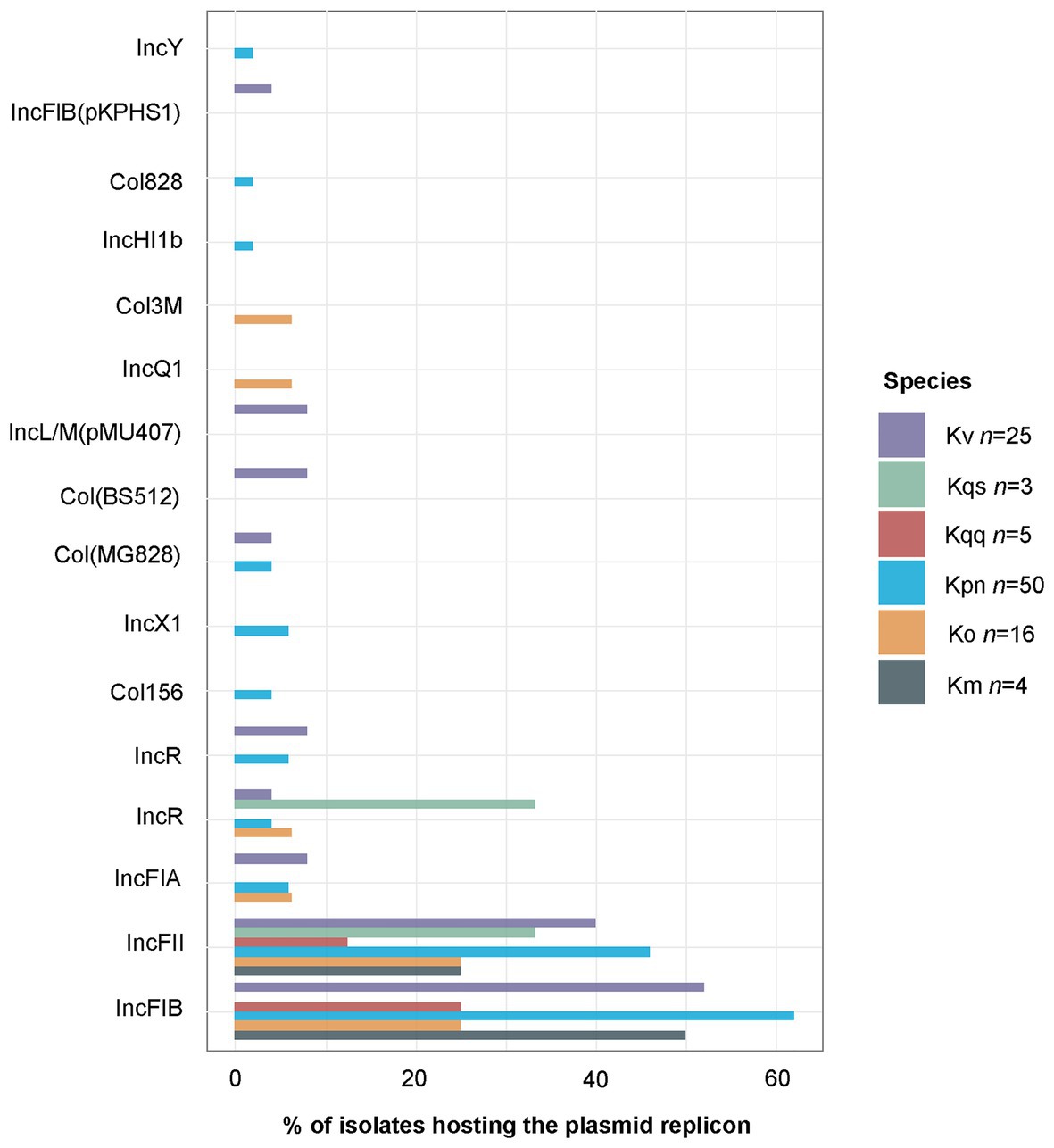
Figure 5. Percentage of isolates hosting each plasmid replicon type as predicted by PlasmidFinder. The total of 142 predicted plasmid replicons were distributed per species as follows: K. michiganensis n = 3, K. oxytoca n = 13, K. pneumoniae n = 84, K. quasipneumoniae subsp. quasipneumoniae n=3, K. quasipneumoniae subsp. similipneumoniae n = 2, K. variicola n = 36, and K. quasivariicola n = 1. The single K. quasivariicola isolate was predicted to host the plasmid replicon Col156, data not included in the figure. Km, K. michiganensis; Ko, K. oxytoca; Kpn, K. pneumoniae; Kqq, K. quasipneumoniae subsp. quasipneumoniae; Kqs, K. quasipneumoniae subsp. similipneumoniae; and Kv, K. variicola.
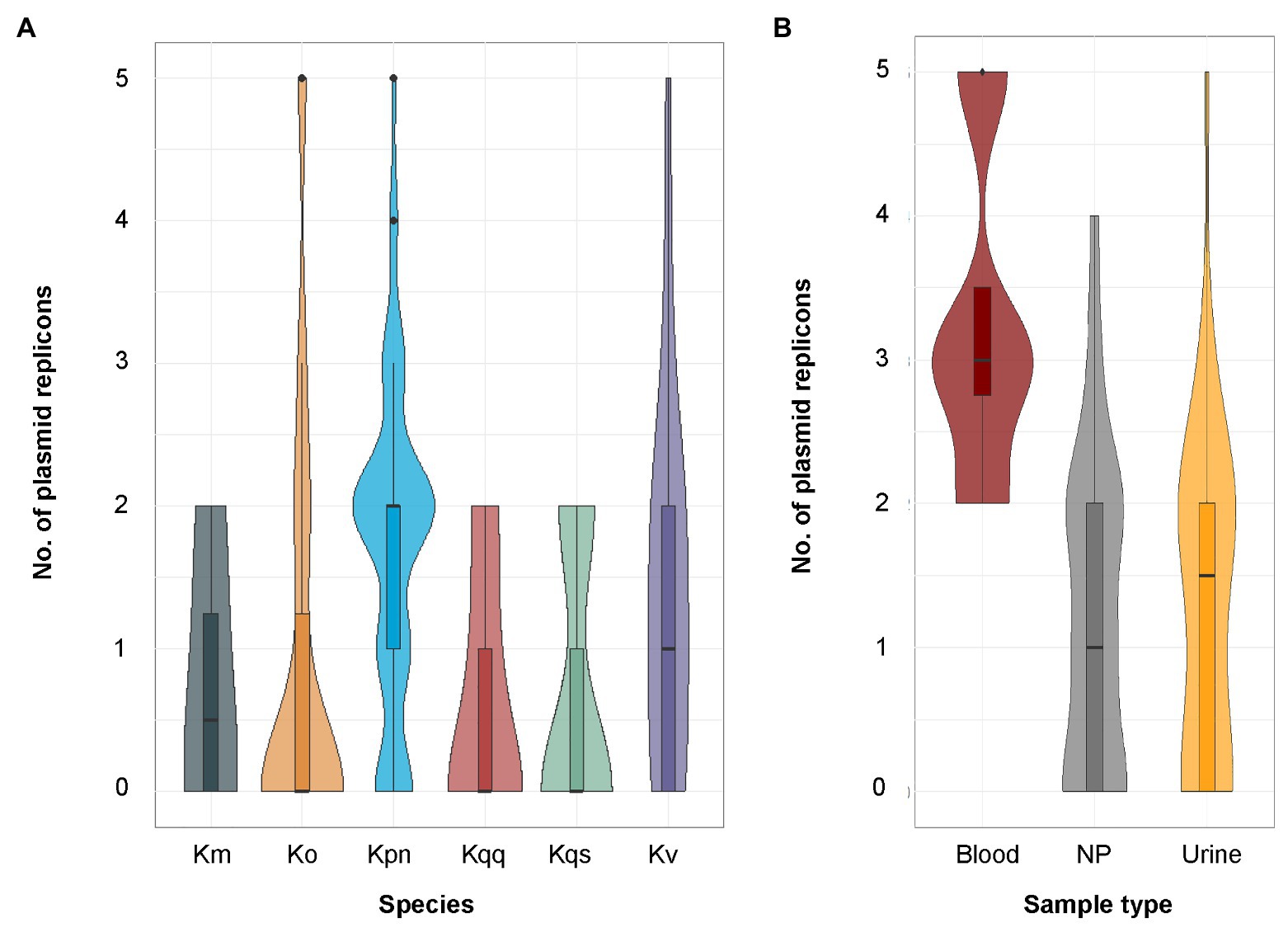
Figure 6. (A) Distribution of the number of plasmid replicons predicted by PlasmidFinder per species. The sample size for each species was K. oxytoca (n = 16), K. michiganensis (n = 4), K. pneumoniae (n = 50), K. quasipneumoniae subsp. quasipneumoniae (n = 5), K. quasipneumoniae subsp. similipneumoniae (n = 3), K. variicola (n = 25), and K. quasivariicola (n = 1, data not included in the figure). No significant differences in the number of predicted plasmid replicons were observed between any of the species using pairwise Poisson regression (all values of p > 0.05). (B) Distribution of the number of plasmid replicons predicted by PlasmidFinder per sample type. The sample size for each sample type was blood (n = 29), nasopharynx (n = 4), and urine (n = 70). Significant differences in the number of predicted plasmid replicons were observed between blood and nasopharynx (p = 0.01), and between urine and nasopharynx (p = 0.003), but not between blood and urine (p = 0.60) using pairwise Poisson regression followed by adjustment of the resulting p-values by the Holm method. Km, K. michiganensis; Ko, K. oxytoca; Kpn, K. pneumoniae; Kqq, K. quasipneumoniae subsp. quasipneumoniae; Kqs, K. quasipneumoniae subsp. similipneumoniae; Kv, K. variicola; and NP, nasopharynx.
Nanopore-Based Sequencing
To evaluate long-reads for microbial profiling, 12 isolates were sequenced using the MinION device and also re-analyzed using an updated database version of MALDI-TOF MS (DB-7854). For only six of the isolates, the species identification using MALDI-TOF MS (DB-7854) matched the results of ANI (50.0%, 95% CI 25.5–74.5), whereas the species identification with WIMP on MinION long-read data was in agreement with ANI for nine isolates (75.0%, 95% CI 46.0–91.5; Table 6). All 12 isolates were predicted to be resistant to β-lactam antibiotics with ARMA on MinION long-read data consistent with ResFinder on Illumina short-read data. Interestingly, all 12 isolates were as well predicted to be resistant to colistin by ARMA, whereas no colistin resistance genes were detected by ResFinder. Otherwise, the antibiotic susceptibility prediction by ARMA and ResFinder found generally consistent matches.
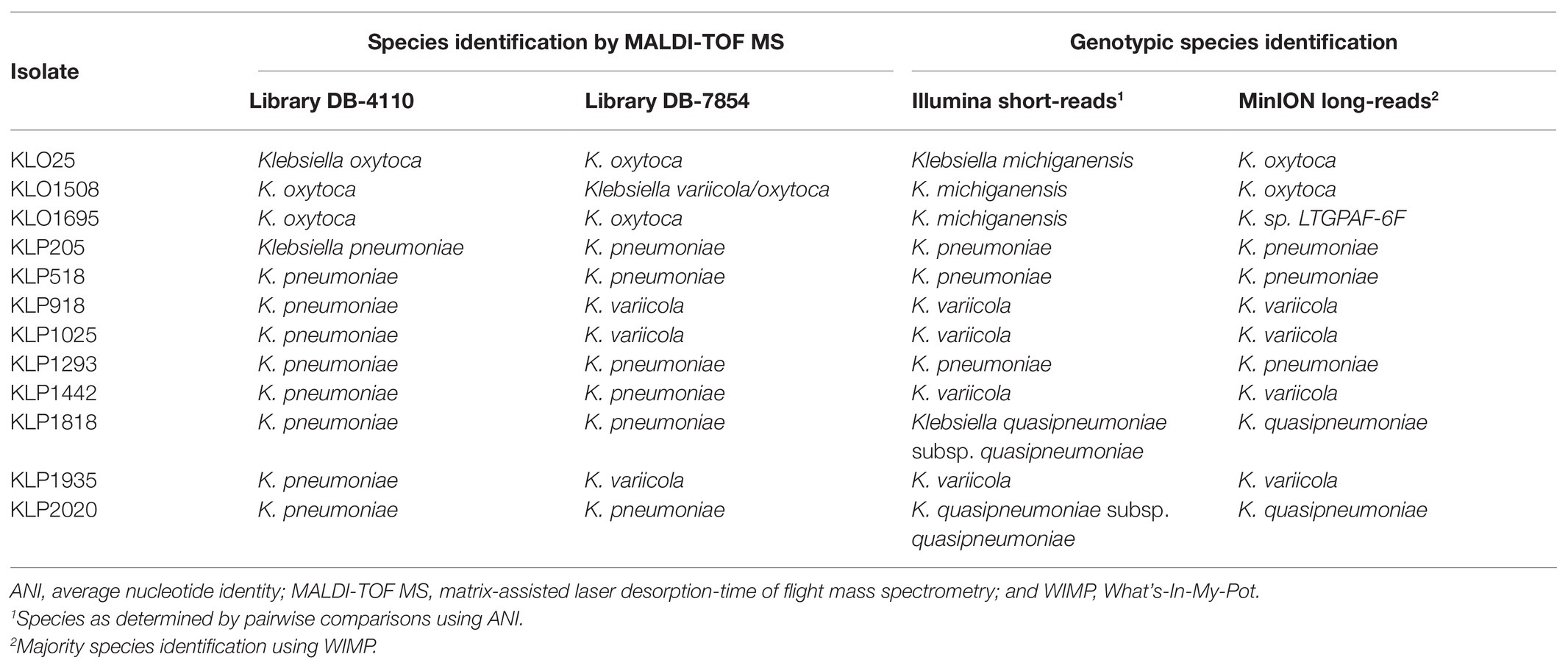
Table 6. Comparisons of species identification by MALDI-TOF MS and genotypic species identification by bioinformatics tools.
Discussion
In this study, we have characterized a collection of clinical Klebsiella spp. isolates using WGS-based analysis. Species identification based on Illumina short-read data was performed by calculating the pairwise ANI against reference genomes, and the results were compared to those obtained by routine microbiological methods. Among the 82 isolates identified as K. pneumoniae with MALDI-TOF MS (DB-4110), as many as 32 were genotypically identified as another Klebsiella spp. (39.0%, Figure 1) by ANI. This is in agreement with previous studies reporting that around 30% of clinical isolates initially identified as K. pneumoniae in clinical routine were later recognized as K. variicola or K. quasipneumoniae based on genotypic identification (Maatallah et al., 2014; Imai et al., 2019). The extensive misidentification of species within the Klebsiella complex has likely led to an underestimation of the clinical significance of other species than K. pneumoniae. A previous report from Stockholm, Sweden (Maatallah et al., 2014) suggests these organisms as a probable underrecognized cause of bacteremia, including fatal infections. Concerning the ability of MALDI-TOF MS to differentiate the species constituting the K. pneumoniae complex, it should be noted that recent updates of the Bruker reference database now should allow for identification of K. variicola from K. pneumoniae but not for the rest of the members of K. pneumoniae complex (Long et al., 2017a; Rodrigues et al., 2018). Our results for the 12 isolates re-analyzed with MALDI-TOF MS using an updated reference database (DB-7854) partially support this as three out of four K. variicola isolates then were correctly identified (Table 6). In addition to the misidentification of members of the K. pneumoniae complex, the species for five of 21 isolates initially reported as K. oxytoca with MALDI-TOF MS (DB-4110) were corrected when using ANI on the Illumina short-read data (Figure 1). Four isolates were genotypically identified as the closely related K. michiganensis as reported previously (Founou et al., 2019; Chapman et al., 2020), and one isolate as E. coli.
A correlation between β-lactamase genes and species has formerly been reported where K. pneumoniae is associated with blaSHV, K. quasipneumoniae with blaOKP, and K. variicola with blaLEN (Haeggman et al., 2004). This is consistent with our results from ResFinder since all K. variicola, as well as the K. quasivariicola isolate, harbored the blaLEN gene, all K. quasipneumoniae subsp. quasipneumoniae and K. quasipneumoniae subsp. similipneumoniae had the blaOKP, and all K. pneumoniae isolates carried the blaSHV. Multiplex PCR assays targeting specific core chromosomal β-lactamase genes have previously been proposed as potential methods for differentiation between the Klebsiella spp. (Fonseca et al., 2017). However, although there is an association between the above mentioned β-lactamase genes with the chromosomes of K. pneumoniae, K. quasipneumoniae, and K. variicola, several studies have reported findings contradicting the use of such lab-developed multiplex PCR methods for species identification. For example, the presence of the gene blaSHV on plasmids in isolates belonging to all three species has been revealed previously (Garza-Ramos et al., 2007; Long et al., 2017a). In addition, there are reports describing K. variicola isolates for which the chromosomal blaLEN gene has not been identified (Long et al., 2017a; Barrios-Camacho et al., 2019).
Although the emergence of highly drug-resistant K. pneumoniae is particularly concerning, the true clinical significance of the other Klebsiella spp. has not yet been fully elucidated due to the species misidentification. This addresses the need to investigate the prevalence of MDR on species level. MDR is usually defined as acquired non-susceptibility to at least one agent in three or more classes of antimicrobial agents among those that are considered as potentially effective against wild-type pathogens (Magiorakos et al., 2012). Currently there is no definition of genotypically MDR; therefore, we defined it as the detection of at least one resistance gene in at least three of the antibiotic classes included in ResFinder (Table 2). A total of 94.0% of the isolates within the K. pneumoniae complex were found to be genotypically MDR, whereas the proportion of MDR isolates was low in both K. oxytoca (18.8%) and K. michiganensis (0.0%). We detected significantly fewer resistance genes in K. oxytoca compared to K. pneumoniae and K. variicola (p < 0.001, Figure 3A) but not any significant differences in the number of plasmids between species (all values of p > 0.05, Figure 6A). The most frequently predicted plasmid replicons among all species in our study were the IncFIB (60 plasmid replicons in 47 isolates) and IncFII (47 plasmid replicons in 40 isolates; Figure 5). The IncFIB and IncFII plasmids have previously been reported to be the predominant plasmids found in Klebsiella (Cao et al., 2014; Long et al., 2017a).
A relatively high discordance for antibiotic susceptibility results was observed between phenotypic AST and WGS-AST, regardless of the bioinformatics tool used. This is mainly due to the fewer cases of resistance observed by phenotypic AST compared to the predictions made by ResFinder and 1928 (Tables 3, 5), also when excluding the cases when disc diffusion testing was not performed. However, a principal limitation of genotypic methods is that detected genetic markers are not necessarily expressed and translated into phenotypic resistance. False resistance can thus be expected for genetic markers that are tightly regulated, such as efflux pumps or inducible β-lactamases (Ferreira et al., 2020). Thereof, the high rate false-resistant results observed for quinolone (Tables 3, 5) for example, is most likely due to that the WGS-AST detected the presence of the gene oqxA, encoding an efflux pump conferring resistance to fluoroquinolones. In contrast, a few cases were determined as resistant by phenotypic AST but not by ResFinder nor 1928. Possible explanations for this discordance include the occurrence of natural resistance not found genotypically or the presence of a currently undescribed allelic variant related to resistance.
The misidentification of isolates within the K. pneumoniae complex has led to the deposition of misidentified reference strains in public repositories, including the public K. pneumoniae MLST scheme. Therefore, the MLST scheme for K. pneumoniae has previously been applied for typing other species within the K. pneumoniae complex. This may limit the use of MLST as a typing tool for molecular epidemiology in Klebsiella spp. and may further contribute to the misidentification problems within the K. pneumoniae complex (Long et al., 2017a). Consequently, the K. variicola MLST was recently developed allowing for molecular epidemiological analysis of K. variicola isolates by assignation of ST (Barrios-Camacho et al., 2019).
The Klebsiella isolates were found to be highly diverse comprising of e.g., 35 distinct STs for K. pneumoniae, 17 for K. variicola, and 10 for K. oxytoca. Earlier studies have also reported high genetic diversity among clinical Klebsiella spp. isolates (Wang et al., 2013; Ejaz et al., 2017; Wyres et al., 2020). For example, Wyres et al. (2020) observed a high diversity in their study of clinical Klebsiella spp. isolates in South and Southeast Asia with ST14 (16.0%) and ST231 (17.0%) as the dominating STs for the K. pneumoniae isolates from South Asia sites (Wyres et al., 2020). The most common ST among the K. pneumoniae isolates in this study was ST14 (10.0%), which has been associated with clonal spread of resistant K. pneumoniae (Giske et al., 2012; Mshana et al., 2013; Moubareck et al., 2018; Wyres et al., 2020).
Although MinION does not suffer from PCR bias introduction (Shagin et al., 2017) as found in Illumina, it does have a higher proportion of errors due to poor quality and issues sequencing homopolymer regions (Istace et al., 2017). As such, MinION long-reads can detect antimicrobial resistance mediated by acquired resistance genes, but it does not provide enough accuracy to identify resistances from chromosomal point mutations (Judge et al., 2016). In our study, the MinION long-reads identified similar genotypic findings, e.g., the species identification with WIMP on MinION long-reads confirmed the results found from the analysis of Illumina short-reads with a few exceptions. The automated workflow, EPI2ME, on the MinION long-reads rapidly provided clinically relevant information (~6 h.) and required minimal bioinformatics know-how, which seems promising for nanopore-based sequencing for future applications in clinical microbiology.
Conclusion
The obtained results emphasize the importance of high-resolution genomic methods for identification and characterization of Klebsiella spp. For 39% of the isolates reported as K. pneumoniae by routine microbiological methods, the species were corrected based on the results from the genotypic species identification by ANI. Moreover, a very high prevalence (94%) of genotypically MDR isolates was observed among the species of the K. pneumoniae complex. These results together indicate an underrecognized clinical importance of other members of the K. pneumoniae complex than K. pneumoniae.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA606666.
Ethics Statement
The study was approved by the Regional Ethical Review Board of Gothenburg (376-11). As the present study only focused on bacterial isolates recovered from cultures included in the routine patient care, no individual written consent was needed.
Author Contributions
The conception and design of the study were done by all authors. PS performed the bioinformatics analyses of Illumina short-read data. JB performed the experimental work and bioinformatics analyses related to the MinION long-read data. AT, HE, and MF supervised PS and JB. DT wrote the original draft of the manuscript, curated the data, and performed downstream sequence analyses using ANI, MLST, and 1928, statistical analyses, and visualizations. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by external research grants from the Swedish Knowledge Foundation (BioMine grant no. 206/0330), Stiftelsen Tornspiran, and internal research fund Unilabs AB.
Conflict of Interest
FD was employed by the company 1928 Diagnostics. HE was employed by the company Unilabs AB.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ANI, Average nucleotide identity; ARMA, Antimicrobial resistance mapping application; AST, Antibiotic susceptibility testing; CGE, Center for genomic epidemiology; ESBL, Extended spectrum β-lactam; EUCAST, European committee on antimicrobial susceptibility testing; MALDI-TOF MS, Matrix-assisted laser desorption-time of flight mass spectrometry; MDR, Multidrug-resistance; MLST, Multilocus sequence typing; NCBI, National center for biotechnology information; PCR, Polymerase chain reaction; QUAST, Quality assessment tool for genome assemblies; SPAdes, St. Petersburg genome assembler; ST, Sequence type; WGS, Whole-genome sequencing; WIMP, What’s-In-My-Pot.
Footnotes
2. ^https://cge.cbs.dtu.dk/services/
3. ^https://www.1928diagnostics.com/
References
Agresti, A., and Coull, B. A. (1998). Approximate is better than “exact” for interval estimation of binomial proportions. Am. Stat. 52, 119–126.
Andrews, S.. (2010). FastQC: a quality control tool for high throughput sequence data. Available at. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (Accessed February 27, 2018).
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barrios-Camacho, H., Aguilar-Vera, A., Beltran-Rojel, M., Aguilar-Vera, E., Duran-Bedolla, J., Rodriguez-Medina, N., et al. (2019). Molecular epidemiology of Klebsiella variicola obtained from different sources. Sci. Rep. 9, 1–10. doi: 10.1038/s41598-019-46998-9
Bengoechea, J. A., and Sa Pessoa, J. (2019). Klebsiella pneumoniae infection biology: living to counteract host defences. FEMS Microbiol. Rev. 43, 123–144. doi: 10.1093/femsre/fuy043
Bertelli, C., and Greub, G. (2013). Rapid bacterial genome sequencing: methods and applications in clinical microbiology. Clin. Microbiol. Infect. 19, 803–813. doi: 10.1111/1469-0691.12217
Bialek-Davenet, S., Criscuolo, A., Ailloud, F., Passet, V., Nicolas-Chanoine, M.-H., Decré, D., et al. (2014). Development of a multiplex PCR assay for identification of Klebsiella pneumoniae hypervirulent clones of capsular serotype K2. J. Med. Microbiol. 63, 1608–1614. doi: 10.1099/jmm.0.081448-0
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Breurec, S., Melot, B., Hoen, B., Passet, V., Schepers, K., Bastian, S., et al. (2016). Liver abscess caused by infection with community-acquired Klebsiella quasipneumoniae subsp. quasipneumoniae. Emerg. Infect. Dis. 22:529. doi: 10.3201/eid2203.151466
Brisse, S., Passet, V., and Grimont, P. A. (2014). Description of Klebsiellaquasipneumoniae sp. nov., isolated from human infections, with two subspecies, Klebsiellaquasipneumoniae subsp. quasipneumoniae subsp. nov. and Klebsiellaquasipneumoniae subsp. similipneumoniae subsp. nov., and demonstration that Klebsiella singaporensis is a junior heterotypic synonym of Klebsiella variicola. Int. J. Syst. Evol. Microbiol. 64, 3146–3152. doi: 10.1099/ijs.0.062737-0
Brisse, S., Van Himbergen, T., Kusters, K., and Verhoef, J. (2004). Development of a rapid identification method for Klebsiella pneumoniae phylogenetic groups and analysis of 420 clinical isolates. Clin. Microbiol. Infect. 10, 942–945. doi: 10.1111/j.1469-0691.2004.00973.x
Brisse, S., and Verhoef, J. (2001). Phylogenetic diversity of Klebsiella pneumoniae and Klebsiella oxytoca clinical isolates revealed by randomly amplified polymorphic DNA, gyrA and parC genes sequencing and automated ribotyping. Int. J. Syst. Evol. Microbiol. 51, 915–924. doi: 10.1099/00207713-51-3-915
Cao, X., Xu, X., Zhang, Z., Shen, H., Chen, J., and Zhang, K. (2014). Molecular characterization of clinical multidrug-resistant Klebsiella pneumoniae isolates. Ann. Clin. Microbiol. Antimicrob. 13, 1–5. doi: 10.1186/1476-0711-13-16
Carattoli, A., Zankari, E., García-Fernández, A., Larsen, M. V., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Chapman, P., Forde, B. M., Roberts, L. W., Bergh, H., Vesey, D., Jennison, A. V., et al. (2020). Genomic investigation reveals contaminated detergent as the source of an ESBL-producing Klebsiella michiganensis outbreak in a neonatal unit. J. Clin. Microbiol. 58, e01980–e01919. doi: 10.1128/JCM.01980-19
Chen, M., Li, Y., Li, S., Tang, L., Zheng, J., and An, Q. (2016). Genomic identification of nitrogen-fixing Klebsiella variicola, K. pneumoniae and K. quasipneumoniae. J. Basic Microbiol. 56, 78–84. doi: 10.1002/jobm.201500415
Ciufo, S., Kannan, S., Sharma, S., Badretdin, A., Clark, K., Turner, S., et al. (2018). Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 68:2386. doi: 10.1099/ijsem.0.002809
Deurenberg, R. H., Bathoorn, E., Chlebowicz, M. A., Couto, N., Ferdous, M., García-Cobos, S., et al. (2017). Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 243, 16–24. doi: 10.1016/j.jbiotec.2016.12.022
Ejaz, H., Wang, N., Wilksch, J. J., Page, A. J., Cao, H., Gujaran, S., et al. (2017). Phylogenetic analysis of Klebsiella pneumoniae from hospitalized children, Pakistan. Emerg. Infect. Dis. 23:1872. doi: 10.3201/eid2311.170833
Enroth, H., Retz, K., Andersson, S., Andersson, C., Svensson, K., Ljungstrom, L., et al. (2019). Evaluation of QuickFISH and maldi Sepsityper for identification of bacteria in bloodstream infection. Infect. Dis. 51, 249–258. doi: 10.1080/23744235.2018.1554258
Ferreira, I., Beisken, S., Lueftinger, L., Weinmaier, T., Klein, M., Bacher, J., et al. (2020). Species identification and antibiotic resistance prediction by analysis of whole-genome sequence data by use of ARESdb: an analysis of isolates from the Unyvero lower respiratory tract infection trial. J. Clin. Microbiol. 58, e00273–e00220. doi: 10.1128/JCM.00273-20
Fonseca, E. L., Da Veiga Ramos, N., Andrade, B. G. N., Morais, L. L., Marin, M. F. A., and Vicente, A. C. P. (2017). A one-step multiplex PCR to identify Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae in the clinical routine. Diagn. Microbiol. Infect. Dis. 87, 315–317. doi: 10.1016/j.diagmicrobio.2017.01.005
Founou, R. C., Founou, L. L., Allam, M., Ismail, A., and Essack, S. Y. (2019). Genomic characterisation of Klebsiella michiganensis co-producing OXA-181 and NDM-1 carbapenemases isolated from a cancer patient in uMgungundlovu district, KwaZulu-Natal Province, South Africa. S. Afr. Med. J. 109, 7–8. doi: 10.7196/SAMJ.2018.v109i1.13696
Garza-Ramos, U., Martínez-Romero, E., and Silva-Sánchez, J. (2007). SHV-type extended-spectrum β-lactamase (ESBL) are encoded in related plasmids from enterobacteria clinical isolates from Mexico. Salud Publica Mex. 49, 415–421. doi: 10.1590/S0036-36342007000600008
Garza-Ramos, U., Silva-Sánchez, J., Martínez-Romero, E., Tinoco, P., Pina-Gonzales, M., Barrios, H., et al. (2015). Development of a multiplex-PCR probe system for the proper identification of Klebsiella variicola. BMC Microbiol. 15, 1–4. doi: 10.1186/s12866-015-0396-6
Giske, C. G., Fröding, I., Hasan, C. M., Turlej-Rogacka, A., Toleman, M., Livermore, D., et al. (2012). Diverse sequence types of Klebsiella pneumoniae contribute to the dissemination of blaNDM-1 in India, Sweden, and the United Kingdom. Antimicrob. Agents Chemother. 56, 2735–2738. doi: 10.1128/AAC.06142-11
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haeggman, S., Löfdahl, S., Paauw, A., Verhoef, J., and Brisse, S. (2004). Diversity and evolution of the class A chromosomal beta-lactamase gene in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 48, 2400–2408. doi: 10.1128/AAC.48.7.2400-2408.2004
Heinz, E., Ejaz, H., Scott, J. B., Wang, N., Gujaran, S., Pickard, D., et al. (2019). Resistance mechanisms and population structure of highly drug resistant Klebsiella in Pakistan during the introduction of the carbapenemase NDM-1. Sci. Rep. 9, 1–13. doi: 10.1038/s41598-019-38943-7
Holt, K. E., Wertheim, H., Zadoks, R. N., Baker, S., Whitehouse, C. A., Dance, D., et al. (2015). Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. U. S. A. 112, E3574–E3581. doi: 10.1073/pnas.1501049112
Imai, K., Ishibashi, N., Kodana, M., Tarumoto, N., Sakai, J., Kawamura, T., et al. (2019). Clinical characteristics in blood stream infections caused by Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae: a comparative study, Japan, 2014–2017. BMC Infect. Dis. 19, 1–10. doi: 10.1186/s12879-019-4498-x
Istace, B., Friedrich, A., D'agata, L., Faye, S., Payen, E., Beluche, O., et al. (2017). de novo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. Gigascience 6:giw018. doi: 10.1093/gigascience/giw018
Jean, S.-S., Lee, N.-Y., Tang, H.-J., Lu, M.-C., Ko, W.-C., and Hsueh, P.-R. (2018). Carbapenem-resistant Enterobacteriaceae infections: Taiwan aspects. Front. Microbiol. 9:2888. doi: 10.3389/fmicb.2018.02888
Judge, K., Hunt, M., Reuter, S., Tracey, A., Quail, M. A., Parkhill, J., et al. (2016). Comparison of bacterial genome assembly software for MinION data and their applicability to medical microbiology. Microb. Genom. 2:e000085. doi: 10.1099/mgen.0.000085
Landis, J. R., and Koch, G. G. (1977). The measurement of observer agreement for categorical data. Biometrics 33, 159–174. doi: 10.2307/2529310
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Ljungström, L., Andersson, R., and Jacobsson, G. (2019). Incidences of community onset severe sepsis, Sepsis-3 sepsis, and bacteremia in Sweden–A prospective population-based study. PLoS One 14:e0225700. doi: 10.1371/journal.pone.0225700
Ljungstrom, L., Enroth, H., Claesson, B. E., Ovemyr, I., Karlsson, J., Froberg, B., et al. (2015). Clinical evaluation of commercial nucleic acid amplification tests in patients with suspected sepsis. BMC Infect. Dis. 15, 1–10. doi: 10.1186/s12879-015-0938-4
Long, S. W., Linson, S. E., Saavedra, M. O., Cantu, C., Davis, J. J., Brettin, T., et al. (2017a). Whole-genome sequencing of a human clinical isolate of the novel species Klebsiella quasivariicola sp. nov. Genome Announc. 5, e01057–e01017. doi: 10.1128/genomeA.01057-17
Long, S. W., Linson, S. E., Saavedra, M. O., Cantu, C., Davis, J. J., Brettin, T., et al. (2017b). Whole-genome sequencing of human clinical Klebsiella pneumoniae isolates reveals misidentification and misunderstandings of Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae. Msphere 2, e00290–e00217. doi: 10.1128/mSphereDirect.00290-17
Maatallah, M., Vading, M., Kabir, M. H., Bakhrouf, A., Kalin, M., Nauclér, P., et al. (2014). Klebsiella variicola is a frequent cause of bloodstream infection in the Stockholm area, and associated with higher mortality compared to K. pneumoniae. PLoS One 9:e113539. doi: 10.1371/journal.pone.0113539
Magiorakos, A. P., Srinivasan, A., Carey, R., Carmeli, Y., Falagas, M., Giske, C., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Moubareck, C. A., Mouftah, S. F., Pál, T., Ghazawi, A., Halat, D. H., Nabi, A., et al. (2018). Clonal emergence of Klebsiella pneumoniae ST14 co-producing OXA-48-type and NDM carbapenemases with high rate of colistin resistance in Dubai, United Arab Emirates. Int. J. Antimicrob. Agents 52, 90–95. doi: 10.1016/j.ijantimicag.2018.03.003
Mshana, S. E., Hain, T., Domann, E., Lyamuya, E. F., Chakraborty, T., and Imirzalioglu, C. (2013). Predominance of Klebsiella pneumoniae ST14 carrying CTX-M-15 causing neonatal sepsis in Tanzania. BMC Infect. Dis. 13, 1–8. doi: 10.1186/1471-2334-13-466
Nicolás, M. F., Ramos, P. I. P., Marques De Carvalho, F., Camargo, D. R., De Fátima Morais Alves, C., Loss De Morais, G., et al. (2018). Comparative genomic analysis of a clinical isolate of Klebsiella quasipneumoniae subsp. similipneumoniae, a KPC-2 and OKP-B-6 beta-lactamases producer harboring two drug-resistance plasmids from Southeast Brazil. Front. Microbiol. 9:220. doi: 10.3389/fmicb.2018.00220
Petersen, L. M., Martin, I. W., Moschetti, W. E., Kershaw, C. M., and Tsongalis, G. J. (2019). Third-generation sequencing in the clinical laboratory: exploring the advantages and challenges of nanopore sequencing. J. Clin. Microbiol. 58, e01315–e01319. doi: 10.1128/JCM.01315-19
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U. S. A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rodrigues, C., Passet, V., Rakotondrasoa, A., and Brisse, S. (2018). Identification of Klebsiella pneumoniae, Klebsiella quasipneumoniae, Klebsiella variicola and related phylogroups by MALDI-TOF mass spectrometry. Front. Microbiol. 9:3000. doi: 10.3389/fmicb.2018.03000
Rodríguez-Medina, N., Barrios-Camacho, H., Duran-Bedolla, J., and Garza-Ramos, U. (2019). Klebsiella variicola: an emerging pathogen in humans. Emerg. Microbes Infect. 8, 973–988. doi: 10.1080/22221751.2019.1634981
Rosenblueth, M., Martínez, L., Silva, J., and Martínez-Romero, E. (2004). Klebsiella variicola, a novel species with clinical and plant-associated isolates. Syst. Appl. Microbiol. 27, 27–35. doi: 10.1078/0723-2020-00261
Rossen, J. W., Friedrich, A., and Moran-Gilad, J. (2018). Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin. Microbiol. Infect. 24, 355–360. doi: 10.1016/j.cmi.2017.11.001
Seki, M., Gotoh, K., Nakamura, S., Akeda, Y., Yoshii, T., Miyaguchi, S., et al. (2013). Fatal sepsis caused by an unusual Klebsiella species that was misidentified by an automated identification system. J. Med. Microbiol. 62, 801–803. doi: 10.1099/jmm.0.051334-0
Shagin, D. A., Shagina, I. A., Zaretsky, A. R., Barsova, E. V., Kelmanson, I. V., Lukyanov, S., et al. (2017). A high-throughput assay for quantitative measurement of PCR errors. Sci. Rep. 7, 1–11. doi: 10.1038/s41598-017-02727-8
Singh, L., Cariappa, M., and Kaur, M. (2016). Klebsiella oxytoca: an emerging pathogen? Med. J. Armed Forces India 72, S59–S61. doi: 10.1016/j.mjafi.2016.05.002
Tagini, F., and Greub, G. (2017). Bacterial genome sequencing in clinical microbiology: a pathogen-oriented review. Eur. J. Clin. Microbiol. Infect. Dis. 36, 2007–2020. doi: 10.1007/s10096-017-3024-6
Wang, Q., Li, B., Tsang, A. K., Yi, Y., Woo, P. C., and Liu, C. H. (2013). Genotypic analysis of Klebsiella pneumoniae isolates in a Beijing hospital reveals high genetic diversity and clonal population structure of drug-resistant isolates. PLoS One 8:e57091. doi: 10.1371/journal.pone.0057091
Wyres, K. L., Nguyen, T. N., Lam, M. M., Judd, L. M., Van Vinh Chau, N., Dance, D. A., et al. (2020). Genomic surveillance for hypervirulence and multi-drug resistance in invasive Klebsiella pneumoniae from south and Southeast Asia. Genome Med. 12, 1–16. doi: 10.1186/s13073-019-0706-y
Keywords:Klebsiella, whole-genome sequencing, antimicrobial susceptibility, clinical microbiology, multidrug resistance, nanopore-based sequencing, Illumina sequencing
Citation: Saxenborn P, Baxter J, Tilevik A, Fagerlind M, Dyrkell F, Pernestig A-K, Enroth H and Tilevik D (2021) Genotypic Characterization of Clinical Klebsiella spp. Isolates Collected From Patients With Suspected Community-Onset Sepsis, Sweden. Front. Microbiol. 12:640408. doi: 10.3389/fmicb.2021.640408
Edited by:
Karsten Becker, University Medicine Greifswald, GermanyReviewed by:
Hasan Ejaz, Al Jouf University, Saudi ArabiaUlises Garza-Ramos, National Institute of Public Health (Mexico), Mexico
Copyright © 2021 Saxenborn, Baxter, Tilevik, Fagerlind, Dyrkell, Pernestig, Enroth and Tilevik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Tilevik, ZGlhbmEudGlsZXZpa0BoaXMuc2U=